

Abstract
Oncolytic herpes simplex virus (oHSV) was one of the first genetically-engineered oncolytic viruses. Because HSV is a natural human pathogen that can cause serious disease, it is incumbent that it can be genetically-engineered or significantly attenuated for safety. Here, we present a detailed explanation of the functions of HSV-1 genes frequently mutated to endow oncolytic activity. These genes are nonessential for growth in tissue culture cells but are important for growth in postmitotic cells, interfering with intrinsic antiviral and innate immune responses or causing pathology, functions dispensable for replication in cancer cells. Understanding the function of these genes leads to informed creation of new oHSVs with better therapeutic efficacy. Virus infection and replication can also be directed to cancer cells through tumor-selective receptor binding and transcriptional- or post-transcriptional miRNA-targeting, respectively. In addition to the direct effects of oHSV on infected cancer cells and tumors, oHSV can be “armed” with transgenes that are: reporters, to track virus replication and spread; cytotoxic, to kill uninfected tumor cells; immune modulatory, to stimulate antitumor immunity; or tumor microenvironment altering, to enhance virus spread or to inhibit tumor growth. In addition to HSV-1, other alphaherpesviruses are also discussed for their oncolytic activity.
Today there are many therapies to address the malady of cancer, although often with incomplete success. A confounding issue with conventional therapies is that they do not specifically target tumor cells, often destroying the normal untransformed cells of the patient in the process of treatment. Chemotherapeutics and radiation generally disrupt and kill any cell that is rapidly synthesizing DNA, tumor or not. Surgery removes tissues that appear to be tumors and often leaves behind malignant cells. These therapies lack the targeted and continual killing of tumor cells which oncolytic virus therapy offers. Oncolytic viruses (OV) selectively replicate in and kill tumor cells while sparing normal cells.1 Oncolytic activity is endowed through multiple mechanisms: viruses with a natural selectivity for tumor cells (i.e., myxoma and Newcastle disease viruses), vaccine vectors that have been genetically manipulated or attenuated (i.e., measles and vaccinia viruses), and genetically-engineered viruses with mutations in pathogenic genes, genes required for replication in normal cells, and/or retargeted to tumor cell receptors (i.e., adenovirus and herpes simplex virus (HSV)). Infecting a tumor with an OV leads to a positive feedback system, whereby an OV infects a tumor cell and produces more therapeutic virus to infect and kill more tumor cells, thereby spreading throughout the tumor while sparing normal tissues. OVs have advantages over chemotherapy, antibody, and radiation therapies because they are biological agents capable of renewing themselves whilst spreading throughout the tumor.
Most viruses are very small 10–200 nm particles and all reproduce their genetic material during infection of a host cell. A virus is unable to replicate by itself and thus hijacks the host’s transcriptional and translational machinery in order to create more virions before exiting and/or killing the host cell. Tumors are made up of dividing cells with diminished control of normal cell cycle/cell death and biosynthesis regulation, allowing them to endlessly proliferate. The increased levels of DNA and protein synthesis required for dividing tumor cells, compared to normal nondividing cells, makes them more permissive for productive viral replication.
The HSV-1 virion is composed of an envelope, a tegument, and a viral capsid core. The envelope is studded with viral glycoproteins responsible for grasping onto host attachment factors heparin sulfate, nectin-1, PILRα, and herpesvirus entry mediator (HVEM), and inducing membrane fusion.2 The tegument is made up of multiple HSV-1 proteins, including ICP0, ICP4, and Us11, which are freed into the cytosol to prepare the cell for virus replication.2 The capsid is transported to the cell’s nucleus where viral replication will begin. HSV-1’s lytic lifecycle is composed of three sequential steps. First, α or immediate early (IE) genes are transcribed into mRNAs and translated. The five IE proteins (ICP0, ICP4, ICP22, ICP27, and ICP47) are responsible for gene regulation, disabling certain innate and adaptive immune functions, as well as enabling the virus to move to the next step in its lifecycle.2 Once the IE ICP4 protein is recruited to the virus genome, β or Early genes (E) are transcribed. Many of the E genes are involved in the replication of the virus’s DNA genome. Discrete compartments form in the nucleus of infected cells where viral DNA replication occurs. The final step in the virus lifecycle is the Late (L) step, whereby the L or γ genes are transcribed and translated after viral DNA synthesis. The new viral genomes are packaged in virions that are exported from the nucleus to the cell membrane in order to spread to new cells. The entire lytic cycle takes between 8 and 24 hours from infection to exit. HSV-1 is also capable of establishing a lifelong latent (nonlytic) infection in peripheral neurons. Latency is defined by the absence of viral protein production, until a reactivation event occurs causing the virus to rouse itself from dormancy and spread to other cells.2
The HSV-1 genome is around 150 kilobases in length and encodes about 84 genes.2 Unlike smaller viruses, HSV-1 encodes many genes that are not essential for its productive growth in cultured cells. This allows researchers to manipulate the HSV-1 genome to enhance oncolytic activity without critically damaging the ability of the virus to replicate. In order to create oncolytic HSVs (oHSVs), it is important to understand the functions of the gene(s) to be altered; their contributions toward cell specificity, replication efficiency and pathology, and whether or not removing them will fundamentally alter the virus’s ability to replicate. In this review we discuss the function of viral genes that have been altered to create oHSVs, and additional strategies to target tumor cells and enhance oncolytic activity.
Gene Mutations in HSV-1 Endowing Oncolytic Activity
The HSV-1 genome has many genes that can be deleted or mutated to confer safety and/or tumor targeting specificity (Figure 1). In this section, we will discuss the function of genes mutated in oHSV, what roles they play in the replication cycle of the virus, and how mutations in these genes confer oncolytic activity. The genetic alterations of oHSVs are described in Table 1. The parenthesized letters (IE, E, L) indicate when the gene is expressed during the HSV-1 lifecycle. The key distinction to be made when designing an oHSV is: what is the difference between the tumor cells to be destroyed and the normal cells to be spared?
Figure 1.
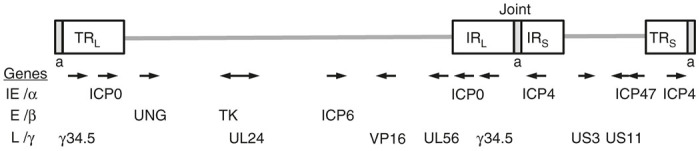
Schematic map of herpes simplex virus (HSV) genome illustrating the position of genes (IE, E, L) deleted or mutated in oHSVs. The genome consists of unique long (L) and short (S) sequences bracketed by terminal (TR) and internal (IR) inverted repeat sequences and separated by the joint region. The number of a sequence repeats is variable. Arrows indicate direction of transcription.
Table 1. Oncolytic HSV constructs.
| Oncolytic virus | HSV gene mutations (deletion: Δ; inactivating mutation: -) | Transgenes | Reference |
|---|---|---|---|
| 1716* | γ34.5Δ | 182 | |
| 1716-6 | γ34.5Δ, ICP6- | ICP6-LacZ fusion in ICP6 | 23 |
| 3616UB | UNG (UL2)-, γ34.5Δ | UNG-LacZ fusion in UNG | 27 |
| C134 | γ34.5Δ | CMVpro-CMV IRS1 | 52 |
| Δ68H-6 | γ34.5 BBDΔ, ICP6- | ICP6-LacZ fusion in ICP6 | 23 |
| ΔPK | HSV-2; ICP10-PKΔ | 96 | |
| Dl8.36tk | TK- | LacZ | 7 |
| Dlsptk | TKΔ | 6 | |
| FusOn-H2 | HSV-2: ICP10-PKΔ | CMVpro-eGFP-ICP10 RR fusion | 97 |
| G207* | γ34.5Δ, ICP6- | ICP6-LacZ fusion in ICP6 | 22 |
| G47Δ* | γ34.5Δ, ICP6-, ICP47Δ, ICP47pro-Us11 | ICP6-LacZ fusion in ICP6 | 55 |
| G47ΔUs11fluc | γ34.5Δ, ICP6-, ICP47Δ, ICP47pro-Us11 | Us11pro-fluc, ICP6pro-LacZ in ICP6 | 183 |
| HF10* | IRLΔ, UL56-LATΔ, gBsyn, UL53-55 duplicated | 85 | |
| hrR3 | ICP6- | ICP6-LacZ fusion in ICP6 | 25 |
| JD0G | ICP0Δ, Joint deletion (1 copy of ICP4, γ34.5) | CMVpro-eGFP in ICP0 | 83 |
| KM100 | ICP0n212, VP16in1814 | 88 | |
| KM110 | ICP0n212, VP16V422 | 88 | |
| L1BR1 | HSV-2; Us3Δ | ICP8pro-LacZ in Us3 | 77 |
| MG18L | Us3Δ, ICP6- | ICP6-LacZ fusion in ICP6 | 24 |
| NV1020* (R7020) | UL24Δ, UL5, 6 (duplicated), TKΔ, UL56Δ, Joint deletion (1 copy of ICP0, ICP4, γ34.5) | ICP4pro-TK, HSV-2: gG, PK, in joint | 87 |
| NV1023 | UL24+, UL5, 6 (duplicated), TK+, UL56Δ, Joint deletion (1 copy of ICP0, ICP4, γ34.5), ICP47Δ, Us11Δ, Us10Δ | ICP47pro-LacZ | 87 |
| NV1066 | Joint deletion (1 copy of ICP0, ICP4, γ34.5) copy), TKΔ | CMVpro-GFP in Joint, BAC in TK | 184 |
| R3616 | γ34.5Δ | 42 | |
| R7041 | Us3Δ | 69 | |
| SUP1 | γ34.5Δ, ICP47Δ, ICP47pro-Us11 | γ34.5pro-Gluc | 57 |
|
Targeted vectors | |||
| bM24-TE | ICP4Δ, US3-, ICP6- | 4F2 enh-TRE-CMVminpro-ICP4, ICP6-LacZ fusion in ICP6 | 107 |
| CMV-ICP4-143T /-145T (amplicon plasmid) | CgalΔ3 (ICP4Δ) helper HSV | miR 143 or 145 targeting sequence in ICP4 | 106 |
| D12.CALP | ICP4Δ, TK-, UL24Δ, US3- | TKpro-LacZ, 4F2 enh-Calponin pro-ICP4 in TK | 108 |
| G92A | ICP4Δ, TK-, UL24Δ, US3- | TKpro-LacZ, Albumin enh-pro-ICP4 in TK | 90 |
| KeM34.5 | γ34.5Δ, ICP6- | Msi pro-γ34.5 in ICP6 | 118 |
| KGE4:T124 | Deletion of residues 2-24 and amino acid substitution, Y38C, in gD. gB:NT | αhuEGFR scFv in gD2-24Δ, gC-GFP fusion, miR-124 target in ICP4 3’UTR | 120 |
| KNC | Deletion of residues 2-24 and amino acid substitution, Y38C, in gD. gB:NT | αhuCEA scFv inserted in gD2-24Δ | 127 |
| KNE | Deletion of residues 2-24 and amino acid substitution, Y38C, in gD. gB:NT | αEGFRscFv inserted in gD2-24Δ | 127 |
| LCSOV | gHΔ | ApoE-AATpro-gH-miR112a, miR124a, miRlet7 targets in gH | 121 |
| Myb34.5 | ICP6Δ, γ34.5Δ | Myb pro-γ34.5 in ICP6 | 113 |
| oHSV-MDK-34.5 | γ34.5Δ, ICP6Δ | Mdk pro-γ34.5 and ICP6-GFP fusion in ICP6 | 119 |
| R5141 | Deletion of residues 68-78 in gB, 1-140 in gC, and 1-33 in gD. gC V34S substitution | IL13 fused to gD and gC | 125 |
| R-LM113 | Deletion of residues 6-38 in gD | scHER2 fused to gD, ICP27pro-EGFP between UL3 and UL4 | 130 |
| R-LM249 | Deletion of residues 61-218 in gD | scHER2 fused to gD, ICP27pro-EGFP between UL3 and UL4 | 126 |
| rQnestin34.5 | γ34.5Δ, ICP6Δ | Nestin enh-Hsp68pro-γ34.5 and ICP6-GFP fusion in ICP6 | 48 |
|
Armed vectors | |||
| bPΔ6-hPAP | ICP6Δ | CMVpro-hPAP, ICP6-LacZ in ICP6 | 136 |
| 34.5ENVE | γ34.5Δ, ICP6Δ | ICP6-GFP fusion, IE4/5pro-Vstat120, Nestin enh-Hsp68pro-γ34.5 in ICP6 | 117 |
| G47Δ-IL18/B7 | γ34.5Δ, ICP6-, ICP47Δ, ICP47pro-Us11 | CMVpro-mIL-18-IRES-B7-1-Ig, ICP6-LacZ fusion in ICP6 | 133 |
| G47Δ-mIL12 | γ34.5Δ, ICP6-, ICP47Δ, ICP47pro-Us11 | CMVpro-mIL-12, ICP6-LacZ fusion in ICP6 | 140 |
| G47Δ-mAngio | γ34.5Δ, ICP6-, ICP47Δ, ICP47pro-Us11 | CMVpro-smAngio, ICP6-LacZ fusion in ICP6 | 173 |
| G47Δ-PF4 | γ34.5Δ, ICP6-, ICP47Δ, ICP47pro-Us11 | CMVpro-PF4, ICP6-LacZ fusion in ICP6 | 175 |
| HSV1790 | γ34.5Δ | CMVpro-NTR-IRES-eGFP | 151 |
| HSV-1γCD | ICP6- | CMVpro-yeast CD, CMVpro-AFP in ICP6 | 147 |
| HSV-PNP (M016) | γ34.5Δ | Egr1pro-PNP in γ34.5 | 152 |
| M002 | γ34.5Δ | Egr1pro-mIL-12 in γ34.5 | 166 |
| M012 | γ34.5Δ | Egr1pro-bacterial CD in γ34.5 | 148 |
| M032* | γ34.5Δ | Egr1pro-hIL-12 in γ34.5 | 169 |
| MGH2 | γ34.5Δ, ICP6- | HSVIE4/5pro-CYP2B1, CMVpro-shiCE, ICP6-GFP fusion in ICP6 | 150 |
| NV1034 | UL5, 6 (duplicated), UL56Δ, Joint deletion (1 copy of ICP0, ICP4, γ34.5), ICP47Δ, Us11Δ, Us10Δ | ICP47pro-LacZ in ICP47, ICP4 enh-TKpro-GM-CSF, HSV-2 gG, PK in joint | 87 |
| NV1042 | UL5, 6 (duplicated), UL56Δ, Joint deletion (1 copy of ICP0, ICP4, γ34.5), ICP47Δ, Us11Δ, Us10Δ | ICP47pro-LacZ in ICP47, ICP4 enh-TKpro-mIL-12, HSV-2 gG, PK in joint | 87 |
| oHSV-NIS | γ34.5Δ, ICP6- | CMVpro-NIS in ICP6, ICP6-GFP fusion | 145 |
| oHSV-TRAIL | γ34.5Δ, ICP6-, ICP47Δ, ICP47pro-Us11 | IE4/5pro-sTRAIL in ICP6, ICP6-LacZ fusion | 155 |
| OncoVexGMCSF* (Talimogene laherparepvec) | γ34.5Δ, ICP47Δ | CMVpro-GM-CSF in γ34.5 | 56 |
| OV-Chase | γ34.5Δ, ICP6- | IE4/5pro-Chase, ICP6-GFP fusion in ICP6 | 179 |
| R8308 | γ34.5Δ | Egr1pro-mIL-10 in γ34.5 | 171 |
| RAMBO | γ34.5Δ, ICP6- | ICP6-GFP fusion, IE4/5pro-Vstat120 in ICP6 | 176 |
| rQT3 | γ34.5Δ, ICP6- | IE4/5pro-TIMP3, ICP6-GFP fusion in ICP6 | 180 |
| rRp450* | ICP6Δ | ICP6pro-CYP2B1 in ICP6 | 149 |
| Synco-B18R | γ34.5Δ, syn-, | UL38pro-B18R, CMVpro-eGFP in joint | 53 |
| T-TSP-1 | γ34.5Δ, ICP6-, ICP47Δ, ICP47pro-Us11 | CMVpro-TSP1, ICP6-LacZ fusion in ICP6 | 177 |
| VAE | γ34.5Δ, ICP6- | ICP6pro-endo-angio fusion in ICP6 | 174 |
AFP, aequorea fluorescent protein; BAC, bacterial artificial chromosome; BBD, beclin binding domain; CEA, carcinoembryonic antigen; CD, cytosine deaminase; Chase, chondroitinase ABC; CYP2B1, cytochrome P450; eGFP, enhanced green fluorescent protein; endo-angio, endostatin-angiostatin; enh, enhancer; fluc, firefly luciferase; gD, HSV glycoprotein D; Gluc, β-glucuronidase; h, human; IE4/5, immediate-early gene 4/5; IL, interleukin; LacZ, β-galactosidase; m, mouse; minpro, minimal promoter; Msi, musashi1; NIS, sodium/iodide symporter; NTR, E. coli nitroreductase; PNP, E. coli purine nucleoside phosphorylase; PK, protein kinase; pro, promoter; RR, ribonucleotide reductase; TIMP3, tissue inhibitor of metalloproteinases 3; TK, thymidine kinase; shiCE, secreted human intestinal carboxylesterase; smAngio, secreted mouse angiostatin; sTRAIL, secreted TNF-related apoptosis-inducing ligand; TRE, Tcf response element; TSP1, thrombospondin-1; Vstat120, vasculostatin.
oHSVs in clinical trial are marked with *.
Nucleotide Metabolism
One general feature of cancer cells that distinguishes them from the majority of normal cells, which are postmitotic, is that they proliferate, and thus maintain sufficient nucleotide pools for DNA replication. HSV-1 harbors several genes involved in nucleotide metabolism (thymidine kinase, ribonucleotide reductase, uracil DNA glycosylase) that permit virus replication in nondividing cells lacking sufficient nucleotide pools. Mutations in these genes confer specificity for dividing cells and often attenuate viral pathogenicity as well.
Thymidine kinase/UL23 (E)
HSV-1 thymidine kinase (TK) is a multifunctional enzyme that catalyzes the creation of deoxythymidine 5’-phosphate (dTMP) from deoxythymidine, as well as phosphorylating deoxycytidine and nucleoside analogs, creating precursors for viral DNA synthesis.2 Loss of TK activity halts HSV-1 replication in nondividing cells, making TK mutants safer than wild type HSV-1.3 However, the antiherpetic nucleoside analog drugs, i.e., acyclovir and ganciclovir, depend upon TK for inhibition of DNA replication. Therefore, oHSVs without TK are more difficult to treat should a dangerous expansion of the virus occur in a patient undergoing oHSV therapy. The availability of antivirals that do not depend upon TK, but directly target HSV DNA polymerase, like foscarnet and cidofovir, even with their intravenous administration provides a basis for reconsidering TK- oHSV.4 Conversely, TK has been used as a “suicide gene” in oHSV by adding ganciclovir during viral infection (see cytotoxic transgenes).5
Dlsptk, a HSV-1 lacking TK, was the first genetically-engineered oncolytic virus devised to kill tumor cells while sparing normal cells.6 Originally created to study TK function in HSV-1, dlsptk selectively killed U87 glioblastoma cells and increased the survival of nude mice harboring U87 orthotopic gliomas, validating the hypothesis that mutations in viral nucleotide metabolism could endow HSV-1 with tumor selectivity.6 Other TK-deleted oHSVs (Dl8.36tk, KOS-SB) were found to be efficacious in immunocompetent rat tumor models.7,8 One drawback of dlsptk, was that it was insufficiently attenuated in the brain.6
ICP6/UL39 (E)
ICP6 is the large subunit of the viral ribonucleotide reductase, which transforms ribonucleotides into deoxyribonucleotides (dNMP) by reducing the 2’-COH group.2 HSV-1 requires an abundant supply of dNMPs to synthesize new viral genomes. ICP6 is essential for HSV replication in noncycling cells, making ICP6-null oHSVs selective for dividing cells.9 Quiescent cancer cells that are mutated for p16INK4a are also permissive for ICP6- HSV-1, suggesting ICP6- oHSVs could target quiescent tumor stem cell populations with mutations in their cell cycle control.10 In relation to safety, ICP6 deleted HSV-1 is attenuated in mice compared to wild type HSV-1.11,12 This is attributed to the inability of the virus to replicate in and kill mature neurons and other postmitotic cells in the brain, which lack ribonucleotide reductase expression and sufficient pools of dNTPs for virus replication.13 However, it has not been reported how large a dNTP pool is needed to support HSV-1 replication. Interestingly, even in cancer cells, increasing cellular ribonucleotide reductase activity, through the use of ENT1 inhibitors, or 5FU or FUdR, can increase the replication of ICP6- oHSV.14–16 For example, an approximate 30% increase in ribonucleotide reductase activity after ENT1 inhibition with dilazep resulted in a greater than 10-fold increase in virus yield.14 Mutating ICP6 in oHSV restricts the virus to cycling cells, thus giving the virus selectivity for tumor cells. Likewise, mutations within the ICP6 gene also prevent oHSV from harming normal tissue.
ICP6, through its N-terminus directly binds eIF4G, expediting the formation of the eIF4 protein translation complex via eIF4E phosphorylation, and stabilizes interactions between eIF4G and eIF4E.17 However, the N-terminal domain alone, as in hrR3, does not promote complex formation.17 EIF4 complex formation is often reduced in growth-arrested cells so capped mRNAs, including HSV transcripts, are poorly translated. Thus, tumor cells lacking negative regulation of their initiation complexes will be susceptible to ICP6 mutant oHSV, while normal cells will shut down viral protein production. In addition, the ICP6 N-terminal RHIM domain binds and activates RIP3, triggering necrosis.18 ICP6 also plays a role in blocking TNFα- and FasL-induced apoptosis, likely through interactions with caspase 8 and RIP1.19,20 This antiapoptotic effect was not seen with hrR3, suggesting the inhibitory domain may be in the C-terminus.20 These actions confer further specificity to tumor cells, which tend to be resistant to apoptosis. As additional safety features, ICP6 mutants are somewhat temperature-sensitive and hypersensitive to antiviral nucleoside analogs and DNA replication inhibitors.9,21
Aside from the colon crypts, epidermal and bone marrow progenitors, and activated splenic immune cells, most normal tissues in humans are not actively replicating. Removing ICP6 limits the ability of the HSV-1 to replicate in postmitotic cells and directs the virus toward dividing tumor cell populations. For these reasons, deletion/inactivation of ICP6 creates a safer oHSV while targeting actively growing tumors. ICP6-deleted rRp450 is currently in a clinical trial for liver tumors (see http://clinicaltrials.gov/show/NCT01071941). ICP6 mutations have been combined with other single mutations in order to enhance safety and tumor specificity; G207 from R3616,22 1716-6 from 1716,23 MG18L from R7041,24 and Δ68H-6 from Δ68H23 (Table 1). The C-terminus of ICP6 is responsible for its ribonucleotide reductase activity, which is why many oHSVs have inserted a LacZ fusion within this region of the gene to inactivate it.22–25 The N-terminus of ICP6 is responsible for expediting the eIF4G interaction with eIF4E and binding RIP3, and is present in several oHSVs. Going forward, it will be important to ascertain which functions of ICP6 are related to oncolytic activity.
UL2/UNG (E)
UL2, uracil DNA glycosylase, is another HSV-1 gene involved in nucleotide metabolism, which when removed attenuates neurovirulence, neuroinvasiveness, and reactivation.26 Uracil-DNA glycosylases prevent mutagenesis by excising uracil from DNA molecules and signaling the base excision repair pathway. OHSV 3616UB lacks γ34.5 and UL2, and is hypersensitive to ganciclovir.27 It demonstrated oncolytic efficacy in six brain tumor cell lines as well as in xenografts, and could not replicate in mature neurons in vitro or in vivo.27
Pathogenicity and Antiviral Suppression
Mammalian organisms have evolved multiple mechanisms for detecting and disabling intracellular pathogens. In order for HSV-1 to replicate its genome and create the next generation of virus, it must cloak itself or actively disrupt host responses to foreign pathogens. Many of the HSV-1 genes which inhibit the host cell’s ability to initiate apoptosis or hamper viral replication are nonessential for viral replication in cancer cells, and therefore provide mutation targets for creating oHSV.
γ34.5/RL1 (L)
γ34.5, one of the most frequently mutated genes for creating oHSVs, has many functions. HSV-1 is diploid for the γ34.5 gene, and mature virions contain the protein in their tegument in order to deliver it upon entry.2 γ34.5 promotes the rapid dephosphorylation of the translational initiation factor eIF2α by activating protein phosphatase 1 (PP1α).28 When eIF2α is phosphorylated, eIF2β is unable to exchange GDP for GTP and methionine-tRNA is not loaded into the ribosome, preventing translation from initiating.29 Dephosphorylation of eIF2α prevents host translational shutoff induced by virus infection, thereby allowing the continued production of viral proteins.28 EIF2α is phosphorylated by several kinases (PKR, PERK, GCN2, HRI) activated by environmental stresses or upon detection of foreign pathogen associated molecular patterns (PAMPs) such as viral DNA and double-stranded RNA, and dephosphorylated by PP1α bound to GADD34.29 The C-terminus of γ34.5 is homologous to GADD34 and can replace GADD34 for PP1α activation.30 Thus, treatment with chemotherapeutics that induce GADD34 expression can enhance γ34.5-deficient oHSV replication and cytotoxicity.31–33 Tumor cells often lack the meticulous control of translation or antiviral/stress responses observed in nontransformed cells, allowing oHSV lacking γ34.5 to replicate. Many tumors also overexpress eIF2α, coinciding with the down regulation of the eIF2 kinases, effectively disabling the tumor’s ability to halt viral protein translation, even in the absence of γ34.5.29
In addition to disrupting the translational shutoff pathway, γ34.5 interrupts autophagic vesicle maturation,34 blocks the tank binding kinase (TBK-1)-mediated interferon-stimulated gene (ISG) signaling pathway,35 interacts with proliferating cell nuclear antigen (PCNA),36 and relocates p32 (HABP1/gC1qR) for nuclear egress.37 Autophagic vesicles, or autophagosomes, are created by a collection of autophagy factors and kinases supported by the mitochondrial protein Beclin-1 (BCN1; yeast homologue Atg6).38 BCN1 binds to PI3 kinase class III (PI3KcIII) and other ATG co-factors, to convert LC3-I into LC3-II and recruit ATG components into forming autophagic vesicles.38 γ34.5 disrupts autophagosome formation by binding to BCN1 via its BCN1 binding domain (BBD; amino acids 68-87), preventing BCN1 binding to ATG/PI3KcIII cofactors.39 Autophagy has both positive and negative impacts on tumorigenesis and maintenance,38 but γ34.5 BBDΔ oHSV (Δ68H-6) replicates in and kills glioma cells similarly to γ34.5+ oHSV.23 Deletion of the γ34.5 BBD somewhat attenuated neuropathogenicity.39,40 Therefore, ICP6 inactivation was combined with BBD deletion in Δ68H-6 to eliminate pathogenicity.23 TBK-1 normally functions as a messenger protein to activate IRF3, which then activates interferon α/β and ISGs. γ34.5 binds TBK-1, preventing it from activating the IRF3 complex.35 Thus, HSV lacking γ34.5 are highly susceptible to interferon (IFN) inhibition. The function of γ34.5 binding to PCNA and its shuttling between the nucleus and cytoplasm is unknown.36,41 P32 relocalizes to the nuclear envelope upon binding γ34.5 to facilitate nuclear egress, and knockdown of p32 decreased the replication of the virus.37
The γ34.5 gene is the major determinant of HSV neuropathogenicity, although the exact mechanism(s) by which γ34.5 contributes to herpes simplex encephalitis is unclear.42 MHC class II cell surface complexes are increased after infection of glioma cells with γ34.5-deleted R3616, as compared to wild-type or mock infection.43 γ34.5-deleted HSV-1 regains neurovirulence in IFNα/βR- or PKR-deficient mice.44 Thus, many oHSVs being clinically evaluated have both copies of γ34.5-deleted: G207, G47Δ, 1716, OncoVEXGMCSF, and M032 (Table 1). Unfortunately, γ34.5-deleted oHSVs (1716, G207, R3616), but not G47Δ or Δ68H-6, replicate poorly, if at all, in glioblastoma stem cells.23,45 oHSVs containing only 1 copy of γ34.5 have been constructed (i.e., NV1020), but the contribution of the γ34.5 deletion is unclear because of other mutations present in these oHSVs. To improve safety and reduce the chance of second-site suppressors, deletion of γ34.5 was combined with ICP6 inactivation to create G207, the first oHSV to enter clinical trial in the US.22,46,47 Because deleting γ34.5 attenuates virus replication even in cancer cells, a strategy to confine γ34.5 expression to cancer cells was developed. OHSV rQnestin34.5, with the endogenous γ34.5 genes deleted, has a γ34.5 gene cassette controlled by a nestin promoter so it is only expressed in cancer cells expressing nestin48 (see Transcriptional Targeting). An unbiased strategy has been to select more effective oHSV by passaging virus in tumors or tumor cells. The ICP47 deletion (see below) was isolated by passaging γ34.5-deficient HSV-1 in non-permissive SK-N-SH tumor cells.49 This second-site suppressor of γ34.5 was due to immediate-early expression of Us11 because of deletion of its late promoter.50 A similar strategy was followed by passaging M002 in D54MG glioma cells in culture or glioma xengrafts. The in vivo passaged virus was more efficacious in both immune-deficient D54MG and syngeneic Neuro2a brain tumor models, while the in vitro passaged virus had increased neurovirulence.51 However, no analysis was performed to identify the genetic alterations.
An alternate strategy to complement the loss of γ34.5 is to express genes from other viruses that evade PKR-mediated protein shutoff or IFN-mediated antiviral responses. CMV IRS1, which prevents PKR activation, has been inserted into γ34.5Δ R3616 to generate C134.52 C134 infection of glioma cells, compared to its γ34.5Δ parent, overcame the block in protein synthesis, replicated better in tumors, and was more efficacious in extending survival of glioma-bearing mice, but maintained safety after intracranial injection.52 Vaccinia virus B18R, a secreted IFNα/β decoy receptor, is expressed in Synco-B18R under control of the Late UL38 promoter, so it is only expressed in tumor cells after oHSV replication.53 Synco-B18R was able to replicate in tumor cell lines poorly permissive to oHSV and was somewhat more efficacious in vivo, but was safe after intravenous administration.53
ICP47/Us12 (IE) + Us11 (L→IE)
ICP47 blocks the TAP protein channel preventing peptide loading onto MHC I protein and subsequent MHC I expression.2 This shrouds virus-infected cells from HSV-1-specific CD8+ T-cells, but can trigger an NK cell response due to the lack of MHC 1 on the infected cell’s surface.54 The TAP channel blocking activity of ICP47 is species-specific, making studies of ICP47 activity in mice not representative of the protein’s activity in humans. ICP47 is deleted in G47Δ and several other oHSVs (OncoVEXGMCSF, SUP-1 (Table 1)) to enhance immune responses against virus-infected tumor cells and antitumor immunity.55–57 Deletion of the ICP47 ORF and Us11 promoter places Us11 under control of the ICP47 IE promoter. Us11 is a true L gene that binds dsRNA-dependent eIF2α kinase (PKR), precluding it from binding dsRNA and phosphorylating eIF2α.58,59 IE expression of Us11 complements the deletion of γ34.5 in nonpermissive cells by keeping eIF2α dephosphorylated and protein translation unhindered. G47Δ and SUP-1 replicate in cells nonpermissive to γ34.5-deleted HSV and better in permissive cells, yet retain a lack of pathogenicity.55,60
RIGI/MDA5 are sensors that detect RNA in the cytoplasm and activate IRF3, leading to induction of the ISG pathway. Us11 directly binds RIGI and MDA5 preventing activation of IRF3 and production of IFNβ.61 This interaction is RNA independent and mediated by the C-terminus of Us11, which is also where the PKR and dsRNA binding pockets of the protein are located.61,62 Us11 excels at deactivating RNA-mediated immune responses, so that expression of Us11 rescues Sindbis virus replication, which is very sensitive to the RNA sensor activated ISGs.61 Us11 also directly binds ISG 2′5′ oligoadenylate synthase (2′5′OAS) to prevent it from recognizing dsRNA and subsequently activating RNaseL.63 Interruption of 2′5′OAS signaling allows the virus to express its mRNAs without being shut off by host defenses. Us11 also prevents cell death from staurosporine, associates with nucleolin within the nucleus, interacts with nuclear transport machinery during viral replication, and moderately inhibits autophagy.62,64,65 How or whether these other functions of Us11 enhance the replication of γ34.5-deficient oHSV is unclear, however, IE expression of Us11 rescues the growth of γ34.5-deficient virus in nonpermissive tumor cells, while still being safe in mice.55,60
Us3 (L)
Us3 is a serine-threonine protein kinase with substrate specificity overlapping PKA and Akt,66,67 affecting a multitude of antiviral host responses. One of the earliest functions identified was prevention of apoptosis in infected cells, possibly by phosphorylating PKA substrates, blocking proapoptotic BAD protein activation, and/or eliciting a prosurvival signaling pathway.68 Defects in apoptotic pathways are common in tumor cells, while inhibition of apoptosis is important for virus replication in normal cells. Thus, Us3-deleted R7041 induced much greater apoptosis in normal HUVEC than its wild-type revertant, while inducing similar low levels in cancer cells.69 Us3 phosphorylates a large number of viral and cellular proteins, affecting many pathways in infected cells: dUTPase (UL50) to increase enzymatic activity70; gB to enhance virus replication in vivo71; p65 to inhibit NF-κB activity and innate immune responses72; IRF3 to inhibit IFNβ expression73; IFNγRα to abrogate expression of IFNγ-dependent genes74; and TSC2 to activate mTORC1 and stimulate translation.67 It also indirectly downregulates cell surface expression of MHC 1, increasing HSV-specific CD8+ T cells in mice.75 These activities promote Us3-deleted oHSVs tumor specificity due to deficiencies in innate immune responses, as well as contribute to reducing neuroinvasiveness, neuropathogenicity, and peripheral pathogenicity of Us3-deleted oHSV.76
Us3-deleted HSV-1 (R7041) and HSV-2 (L1BR1) were shown to have oncolytic activity, with greatly reduced replication compared to wild-type HSV in normal cells in vitro, and elevated replication in tumors in vivo compared to normal tissue.69,77 Chemotherapy increased apoptosis in L1BR1, but not wild-type, infected cancer cells.77 R7041 was safe after intravenous injection in immune-deficient mice and intraperitoneal injection in immune-competent mice.69 However, it was insufficiently safe in the brain, in contrast to γ34.5Δ virus, so MG18L containing an additional inactivating LacZ insertion in ICP6 was constructed.24 MG18L induces increased levels of apoptosis in glioblastoma stem cells (GSCs) in vitro and in vivo.24 Because Akt activation is elevated after infection with Us3-deleted HSV, the combination with PI3K/Akt inhibitors was evaluated and found to synergize in cancer cell lines and GSCs, through increased apoptosis.24,69
ICP0 (IE)
ICP0 encodes a RING finger ubiquitin E3 ligase that targets cellular proteins.78 ICP0 targets PML or ND10 bodies and other host factors designed to silence HSV-1 genomes and induce IFN signaling for ubiquitination and degradation.78 Thus, ICP0-null HSV-1 is extremely sensitive to IFN and PML-mediated disruption of the viral lifecycle.78 In multiple tumors, PML is downregulated, making them permissive for ICP0-null oHSV.79 ICP0 is also involved in 4E-BP1 degradation and promoting eIF4E phosphorylation, facilitating eIF4F complex formation in quiescent cells, important for HSV replication.80
oHSV KM100 has mutations which disable the ICP0 gene, as well as VP16.81 It is able to grow in IFN-deficient breast cancer cell lines and demonstrated therapeutic efficacy using the immunocompetent mouse FVB breast cancer model.81 ICP0 truncation mutant HSV-1 (n212) replicated better in mouse cancer cells in vitro than KM100, while the efficacy of the two was the same in an immunocompetent mouse tumor model.82 Mice receiving KM100 that survived the initial tumor were protected from tumor rechallenge, demonstrating that the virus elicits a memory immune response to the tumor.81,82 Another ICP0-null oHSV, JD0G replicates in osteosarcoma U2OS and glioblastoma U251 cells.83 The ability of ICP0 mutants to replicate in a variety of human and mouse tumor cell lines suggests that transformed cells complement ICP0 defects, while untransformed cells remain nonpermissive, making ICP0 a good target for designing oHSVs with specificity for tumor cells.
UL56 (L)
UL56 is another gene associated with pathogenicity and neuroinvasiveness, as removing it attenuates the virus.84 oHSV HF10 has a number of deletions and insertions in the genome, resulting in the lack of expression of UL43, UL49.5, UL55, and UL56.85 HF10 has demonstrated efficacy in a variety of preclinical models and entered early phase clinical trials for breast and head and neck cancer.86 NV1020, which was initially developed as a vaccine (R7020), and NV1023 are also deleted for UL56, along with ICP47 and a single copy of ICP0, ICP4, and γ34.5.87 Whether UL56 contributes to tumor selectivity is unknown, but it is a good candidate for mutation when creating a safer oHSV.
Other HSV-1 Functions
The proteome of HSV-1 is rather well defined due to extensive study; however, the functions of many proteins still remain unknown. Because of the large genome size and number of nonessential viral genes, the opportunities for identifying additional genes endowing oncolytic activity remains. Some mutations have been incorporated into oHSV, without their contribution being defined. The following are other mutated genes present in oHSVs, whose roles are not well defined in relationship to oncolytic activity.
VP16/UL48 (L)
VP16 is delivered during entry of HSV-1 via the tegument. It plays a role in aiding the transcription of IE genes during initial infection and during reactivation from latency.2 In KM100, VP16 is mutated along with ICP0.88 Interestingly, combining a VP16 mutant lacking the C-terminus with ICP0-null (KM110), produces a less cytopathic, more debilitated virus than with VP16 mutation in1814 (in KM100), which removes the trans-activating activity of VP16.88 This difference was also exhibited in cancer cell lines.81
UL24 (L)
The UL24 gene overlaps TK, so some TK deletions also contain UL24 mutations (i.e., RH105),89 although not dlsptk, and G92A.90 UL24 causes the dispersal of nucleolin throughout the nucleus.91 UL24 single mutants are defective in establishing latency and reactivation, replicating in the eye, and causing ocular disease.92 UL24 mutants can also have a syn phenotype, whereby infected cells form syncytia.2,92 NV1020 contains a UL24 deletion as a result of removing TK, as well as deletion of UL56.87 NV1020 is efficacious in a variety of colon, gastric, and prostate cancer models and safe in patients, although the role of the UL24 deletion in its oncolytic activity is unknown.93
Other Oncolytic Herpesviruses
HSV-2
HSV-2, another human alphaherpesvirus primarily associated with genital disease, has a genome that is very similar to HSV-1, both in genomic organization and protein coding sequences.2 In a comparison between ICP0Δ HSV-1 and HSV-2, HSV-2 dICP0 was more cytopathic in mouse breast cancer cells than HSV-1 dICP0, but had much lower virus burst sizes.94 In vivo, in an immunocompetent mouse breast tumor model, only HSV-1 dICP0 significantly extended survival.94
The HSV-2 UL39 gene encodes ICP10, a homolog of HSV-1 ICP6 that contains a unique N-terminal domain proposed to encode a serine/threonine protein kinase (PK).95 Thus, in addition to ribonucleotide reductase activity, ICP10 is a constitutively active growth factor receptor that directly interacts with RAS-GAP to activate RAS-GTP and stimulate the RAF/MEK/ERK signaling pathway, which prevents apoptosis in infected cells.95 HSV-2 ICP10 PK deletions have been tested for oncolytic efficacy due to the inability of HSV-2 to replicate in noncycling cells without ICP10/PK, and the presence of constitutive RAS signaling and defective apoptosis in many tumors. FusOn-H2 and DeltaPK (HSV2ΔPK) are oHSV2 which have deletions of the PK domain of ICP10.96,97 Both do not replicate in noncycling normal cells, induce apoptosis, and engender stalwart therapeutic efficacy in melanoma, esophageal, and breast cancer orthotopic tumor models.96,97 FusOn-H2 exhibited no toxicity after i.v. injection in BALB/c mice at a dose 3-logs higher than the wild-type lethal dose, but one out of five mice died after intracerebral injection at 107 pfu.98 HSV2ΔPK was safe after foodpad injection and in a clinical vaccine trial where three injections of 2 × 105 pfu were administered subcutaneously.99,100
HSV-2 Us3 has many of the same functions as HSV-1 Us3, including preventing apoptosis, but lacks some of the HSV protein kinase substrates. Thus, HSV-2 Us3 deletions are only minimally attenuated for neurovirulence compared to HSV-1 Us3 deletions.101 HSV-2 L1BR1 was safe when injected into mouse footpads, but not for intracranial or corneal injections, and induces apoptosis in neurons.102 In SW1990 pancreatic tumor implanted mice, L1BR1 was significantly better than oHSVs R3616 and hrR3 in inhibiting tumor growth.77
Bovine herpesvirus-1
BHV-1, an alphaherpesvirus that affects cattle, causes fever and respiratory disease. BHV-1 has a very restricted host range, likely regulated by IFN. This suggested that it might be inherently oncolytic, like myxoma virus, killing human tumor cells and not normal human cells.103 The majority of the NCI panel of established human tumor cell lines are permissive to BHV-1 infection and cytotoxicity, with mutations in KRAS associated with virus replication.104 Direct exposure to BHV-1 seems to be sufficient to induce cell death in many cancer cells, even when replication does not occur.104
Equine herpesvirus type 1
EHV-1 is an alphaherpesvirus mainly associated with equine disease that does not infect humans, but does infect human cells in culture. It uses the equine MHCI protein as its entry receptor, which has regions highly conserved across species, including humans.105 An attenuated EHV-1, deleted for glycoproteins gI and gE (L11ΔgIΔgE), was found to replicate in and kill a number of human glioma cell lines.105 Given that many tumors, and especially glioblastoma, often downregulate the expression of MHC 1, it is unclear whether this oncolytic herpes virus is capable of delivering an in vivo response.
Transcriptional and Micro-RNA Targeting
Understanding the lifecycle of HSV-1 allows researchers to target tumor cell populations by altering expression of viral genes to control productive infection in select cell types. ICP4, one of five IE genes, is an essential gene and the major virus trans-activator responsible for activating transcription of the E and L genes.2 Because ICP4 is essential, regulating its expression with cell-specific transcriptional regulatory sequences can restrict virus replication and oncolytic activity to cells expressing those transcription factors.90 Thus, oHSV can be targeted to cancer cells based on transcription factor expression in those cells. Alternatively, ICP4 expression can be silenced in normal cells by cell-specific miRNAs targeting ICP4 mRNA containing miRNA target sequences.106
Several oHSVs have been constructed with their endogenous ICP4 genes deleted and an ICP4 coding sequence controlled by a cell- or tissue-specific promoter inserted, which confers specificity to a tumor tissue.90,107–109 These are transcriptionally-targeted oHSVs because they are only able to replicate in cells where expression of a tumor-specific transcription factor drives ICP4 transcription. G92A is the prototypic vector, targeting liver cancer via insertion of ICP4 under control of the albumin promoter into the TK locus.90 G92A replicates in albumin-expressing Hep3B hepatoma cells, but not the albumin-negative HT29 colon carcinoma cells. In nude mice, G92A completely inhibited the growth of Hep3B, but not prostrate PC3, tumors.110 D12.CALP was similarly constructed, but contains the calponin promoter driving ICP4.108 Calponin is expressed in many postmitotic cells, but overexpressed in several sarcomas. Thus, d12.CALP was efficacious against human leiomyosarcama, but not calponin-negative osteosarcoma xenografts.108 The Wnt/β-catenin pathway is upregulated in the majority of colorectal cancers and many cancer stem cells, and leads to transcriptional activation of Wnt response elements containing Tcf binding sites.107 In bM24-TE, a synthetic enhancer containing multiple Tcf binding sites, a 4F2 enhancer, and a CMV minimal promoter was used to drive ICP4 expression after insertion into the ICP6 locus.107 BM24-TE selectively replicated in and killed cancer cells with strong β-catenin/Tcf signaling (APC or β-catenin mutants), but not cancer cells with low signaling, with similar inhibition of tumor growth.107
There have also been several unsuccessful attempts to use cellular promoters to target oHSVs to tumors. Human carcinoembryonic antigen (CEA) is overexpressed in several epithelial tumors. CEAICP4 was created using the CEA promoter to drive ICP4.111 Unfortunately, it replicated poorly in CEA-expressing cells and not at all in some high-expressing tumors cells,111 demonstrating that not all tumor associated promoters are good candidates for creating transcriptionally-targeted oHSV. Another problem with transcriptionally targeted oHSV is that the promoter may not behave as expected in HSV. For example, HIF-V6R-HSV, with a HIF-1α-responsive promoter driving ICP4, was active under both normoxic and hypoxic conditions.112 Confounding the strategy, it was found that HSV-1 infection increased transcription from the HIF responsive promoter in normoxia. In these studies, HIF-V6R-HSV was also found to have reverted back to the wt ICP4 configuration, likely through recombination in complementing cells during its isolation.112
Rather than targeting expression of ICP4, a number of oHSV employ regulated expression of γ34.5 to enhance virus replication in tumor cells. This strategy was initially described with Myb34.5, which lacks ICP6 and expresses γ34.5 under a cellular E2F-responsive B-myb promoter inserted into ICP6 in γ34.5Δ MGH1 (similar construct to G207).113 This transcriptional targeting increased the growth of Myb34.5 in human colon cancer cells, but not hepatocytes, and extended survival of mice with liver metastases compared to parental MGH1.114 The nestin enhancer/Hsp78 minimal promoter drives γ34.5 expression in rQnestin34.5 and 34.5ENVE,48,115 in order to selectively target tumor cells overexpressing nestin, such as glioblastoma, some peripheral tumors, and cancer stem-like cells.116 Unfortunately, nestin is also expressed in neural stem/progenitor cells and other tissue stem cells, creating potential safety concerns for nestin-targeted oHSV.116 RQnestin34.5 and 34.5ENVE replicate well in glioblastoma cell lines and confer a survival benefit in immune-deficient mice implanted intracerebrally with glioblastoma cell lines.48,117 34.5ENVE was also effective against patient ascites-derived ovarian cancer cells, which have increased nestin expression.115 Musashi1 is another gene selectively expressed in glioblastoma. Its promoter was used to drive γ34.5 expression after recombination in G207 to create KeM34.5.118 KeM34.5 replicated much better in glioma cells, but not musashi1-negative lung or colon cancer cells, than G207, and not in astrocytes.118 Midkine, upregulated in a number of tumors was used in place of the nestin enhancer to drive γ34.5 expression in oHSV-MDK-34.5 in MPNST.119
Several researchers have tested a different method for targeting expression of oHSV genes, in this case silencing expression with microRNAs. MicroRNAs (miR) are small 22-28nt RNAs that signal the destruction of complementary mRNAs via the RISC complex. There are many miRNAs expressed in normal but not tumor cells, so miRNA target sequences inserted into essential HSV-1 genes can selectively inhibit virus replication and/or gene expression in normal cells, leading to selective replication in tumor cells. For example, miR-143 and -145 are expressed in most human tissues but not in prostate cancer cells. MiR-143 and -145 target sequences inserted into the 3’UTR of ICP4 (142T and 145T) blocked its expression in normal but not prostrate cancer cells.106 The replication of CMV-ICP4-143T, an amplicon plasmid with ICP4Δ helper HSV, in normal tissues was much more restricted than CMV-ICP4-145T.106 KG4:T124, containing tandem copies of a miR-124 target sequence within the 3’UTR of ICP4, was safe after intracranial injection compared to parent oHSV KG, deleted for the internal repeat region.120 ICP4-T124 was recombined into KGE, with an EGFR-targeted gD fusion (see Receptor Targeting), and found to be similarly effective in treating mice harboring intracranial glioblastoma stem cell GBM30 tumors as KGE.120 In LCSOV, miR targets to miR-122a, mir-124a, and miR-let-7a, were used to repress liver-specific apoE-AAT promoter driven gH expression in liver tissue, but not hepatocellular carcinoma.121 LCSOV was able to replicate in miR122-negative Hep3B cells and inhibit tumor growth.121
Receptor Targeting
The HSV glycoproteins stud the envelope and are responsible for binding to viral attachment factors; heparan sulfate, HVEM (HveA), and nectin-1 (HveC).122 Upon binding their attachment factors, the glycoproteins aid fusion of the HSV envelope with the plasma membrane. It is possible to modify one or more of the glycoproteins to create a virus that infects cells expressing a targeted receptor and/or detargets cells expressing the normal HSV-1 attachment factors. Glycoprotein C has been used as a site to insert tumor-specific receptor binding ligands or single-chain antibodies (scFv) to retarget virus, but this only removes the heparan sulfate binding domain.123,124 Glycoprotein D (gD) binds to the major HSV cell surface receptors, and thus deleting gD binding domains (N-terminus (aa7-32) for HVEM and Ig-core (aa61-218) for nectin 1) and introducing ligands for tumor-specific receptors is an attractive strategy to retarget HSV. Two basic strategies have been used: insert a tumor-specific receptor ligand or scFv into a deletion of a gD-binding domain or detargeted gD mutant125–127; or use a recombinant bispecific adapter protein containing the gD-binding domain of nectin 1 or HVEM fused to a tumor-specific receptor scFv with a HVEM or nectin 1 detargeted gD mutant, respectively.128 HER2 is overexpressed in a number of tumors, including breast and ovarian cancer, and glioblastoma. A HER2 targeted oHSV (R-LM249) was constructed by inserting scFv human HER2 (trastuzumab) into a gD Ig-folded core deletion.126 R-LM249 inhibited the growth of HER2+ ovarian and breast subcutaneous or metastatic tumors, but not HER2- SJ-Rh4 rabdomyosarcoma tumors after intraturmoral or intraperitoneal injection.126,129 A similar HER2-targeted oHSV, R-LM113, replicated in and was efficacious against a PDGF-induced mouse glioma overexpressing HER2 in both immunodeficient and immunocompetent mice, and was safe after intracerebral injection.130,131 An important caveat to this approach is that HER2 scFv is human specific and does not bind mouse HER2. The use of scFv allows one to potentially target any cell surface molecule, as illustrated by other receptor-targeted oHSVs binding to human EGFR (KNE) or CEA (KNC),127 or IL13Rα2 (R5141), upregulated in glioblastoma.125 A confounding feature of this strategy is that all cancer cells must express high levels of the targeted receptor, while cancers are typically heterogeneous for receptor expression.
Genetic Engineering of oHSVs
Many researchers now use bacterial artificial chromosome (BAC) systems to quickly swap in transgenes or create specific mutations more easily than classical homologous recombination in tissue culture.126,132–136 HSV BACs are large circular plasmids (>150 kb), which replicate in bacteria as an artificial genome. BACs are useful for cloning DNAs much larger than is possible using conventional plasmids. For example, the pM24-BAC contains the entire HSV genome with a bacterial origin of replication flanked by Cre/loxP and Flp/Frt sites inserted into the ICP6 gene.134 To create a new virus, a shuttle vector containing the transgene flanked by Cre/LoxP sites and a stuffer sequence flanked by a Frt/Flp site is recombined into the HSV-BAC with Cre recombinase. The recombined HSV-BAC plasmid is harvested from bacteria and cotransfected into mammalian cells with a Flp recombinase plasmid to remove the BAC and stuffer sequence, and infectious recombinant virus harvested.134 This method results in a high frequency of recombinant virus. HSV-BACs can also be used to introduce specific mutations in genes, for example through the use of lambda phage Red recombinase.137
Aside from BACs, the very recent development of the CRISPR/Cas mutagenesis system allows researchers to quickly insert or remove genes/sequences into the HSV genome without cloning into BAC plasmids, decreasing the time it takes to create a new HSV.138,139 The CRISPR/Cas9 system is a defense platform for bacteria to target the DNA genomes of bacteriophages. Cas9 is a bacterial nuclease complex, which recognizes a single-stranded guide RNA complementary to a region of dsDNA. The Cas9 enzyme uses the guide RNA to cleave complementary DNA. However, the accuracy of this cleavage is not perfect and mismatches of the guide RNA sequence with the targeted DNA can result in aberrant DNA cleavage. Recently, the CRISPR/Cas9 system was used to insert green fluorescent protein (GFP) into HSV-1, and to introduce gene-specific mutations or repair mutations.138,139 The CRISPR/Cas9 system is rapidly evolving beyond mutagenesis to gene regulation, with nuclease-deficient Cas9 fused to functional effectors such as transcriptional activators/repressors. CRISPR/Cas9 could also be used to direct proteins to DNA sequences within cancer cells to disrupt oncogenic super-enhancers, epigenetic modifications, or transcriptional complexes.
oHSV-1 Expressing Reporter or Therapeutic Genes
Transgenes come in several flavors for creating vectors. Genes are added to visualize the virus’ spread, make the virus more cytotoxic to a specific tumor cell population, elicit a targeted immune response, and/or modify the tumor microenvironment. To this end, researchers have outfitted multiple oHSV vectors with imaginative genes to create better vectors. Vectors which express cytotoxic or immune-stimulatory transgenes are usually termed “armed” oHSV,135 and in the following, we will discuss some of the genes and control mechanisms inserted into several armed vectors.
Reporter genes
The Escherichia coli LacZ gene encodes β-galactosidase (β-gal), which cleaves β-bonded sugars, like lactose, into single moieties. A chromogenic substrate of β-gal, termed X-gal, turns blue upon cleavage of its β-bonded sugar, allowing researchers to visualize cells expressing β-gal. LacZ can be inserted into oHSVs in order to monitor their infection of cells in vitro and in vivo.22 Several oHSVs have LacZ inserted into the ICP6 gene, derived from hrR3, in order to disrupt ICP6 gene activity and allow for easy monitoring of viral spread (i.e., G207, G47Δ, MG18L (Table 1)).22,24,25,55 Bioluminescence imaging of luciferase is a noninvasive method for observing viral spread in vivo.119,140–142 If the Luc gene is expressed under a HSV-1 late promoter (i.e., G47ΔUs11fluc), it is possible to monitor the replication of the virus in vivo over time, without having to sacrifice the animal.140,142 GFP expression, for example in NV1066, can also be imaged in vivo using fluorescent endoscopy.143,144 The sodium iodide symporter (NIS) can be monitored noninvasively by CT/SPECT after infected cell uptake of 99mTcO4.145 In addition, oHSV-NIS can be combined with cytotoxic 131I radiotherapy to enhance antitumor efficacy.145 The ability to monitor virus biodistribution and replication in live animals and potentially patients provides an important avenue to follow oHSV therapy.
Cytotoxic transgenes
Theoretically, the most straightforward way to increase the efficacy of an oHSV is to arm it with a transgene that is toxic to nearby noninfected tumor cells, in addition to the virus’ own cytotoxicity. Expression of prodrug-activating enzymes, suicide gene therapy, is the most common strategy. HSV-TK is the prototypic “suicide gene”, converting GCV to GCV-monophosphate, however the toxic metabolite also blocks HSV replication, thereby ablating further virus spread.5,146 Cytosine deaminase (CD), not found in humans, converts 5-fluorocytosine (5-FC) into 5-fluorouracil (5-FU), a highly toxic pyrimidine antimetabolite chemotherapeutic. HSV-1yCD and M012 express yeast and bacterial CD, respectively, and the combination with 5-FC significantly increased tumor cell killing, including of noninfected cells (bystander effect), and tumor growth, but not with CD- parental viruses.147,148 Other “suicide genes” successfully utilized in oHSV include: rat cytochrome P450 (CYP2B1) in rRp450 and MGH2, converting cyclophosphamide (CPA) into toxic phosphoramide mustard149,150; secreted intestinal carboxylesterase (CE) in MGH2, for the conversion of irinotecan (CPT-11) to SN-38150; bacterial nitroreductase (NTR) in HSV1790, for the conversion of BC1954 to DNA crosslinker 4-hydroxylamine151; and E. coli purine nucleoside phosphorylase (PNP) in HSV-PNP, for the conversion of 6-methypurine-2’-deoxyriboside to 6-methylpurine.152 It is important to bear in mind that some of the toxic metabolites, such as 5-FU and SN-38, can also inhibit HSV replication.153,154 MGH2 contains two suicide genes, CYP2B1 and CE, and the combination of MGH2 + CPA + CPT-11 was much more effective than MGH2 with either prodrug alone, in an intracerbral glioma model.150 Another cytotoxic strategy is to express secreted TRAIL, which induces apoptosis after binding to death receptor complexes that are often upregulated in tumor cells. oHSV-TRAIL extended the survival of mice bearing TRAIL- and oHSV-resistant intracerebral gliomas, and was associated with greatly increased apoptosis.155
Immune stimulatory transgenes
Although a lively debate is still ongoing, more and more data suggests that a driving force behind oncolytic virotherapy’s success is due to immune responses elicited against the tumor.156 Whether stimulation of the immune system benefits or hinders oHSV therapy is debated due to the efficacy observed in immunodeficient models, which have no adaptive immune system, and the deleterious effects of the innate immune system.157,158 However, it is clear that robust antitumor adaptive immune responses are elicited by oHSV, as well as protective immune memory, suggesting that activation of the immune system is ultimately responsible for destroying the tumor and preventing its reoccurrence.81,159,160 Currently, there are several oHSVs that take advantage of stimulating the immune system by expressing immune-stimulatory factors to create a robust T-cell-mediated clearance of the tumor.
Granulocyte macrophage colony stimulating factor has been cloned into several oHSVs (NV1034 and OncoVEXGMCSF).56,87 Granulocyte macrophage colony stimulating factor stimulates myeloid lineage precursor cells to differentiate, as well as recruiting and activating macrophages and dendritic cells. In immunocompetent mouse models, the expression of granulocyte macrophage colony stimulating factor had only small or no enhanced efficacy over nonarmed oHSV.56,161,162 OncoVEXGMCSF or currently named Talimogene laherparepvec (T-Vec), the first “armed” oHSV to enter clinical trial, has demonstrated promising efficacy in patients with metastatic melanoma after intratumoral injection, with induction of local and systemic melanoma-specific T cells.163 In the pivotal phase III OPTiM trial, T-Vec-treated patients had a significant improvement in the durable response rate (16 versus 2% for controls) and a trend toward improved overall median survival.164
Using the Flip-Flop HSV-BAC system, a number of immune modulatory transgenes have been inserted into G47Δ (IL-12, IL18, soluble B7.1, and Flt3L), including multiple transgenes in G47Δ-IL18/B7.133,135,140 IL-12, a heterodimeric cytokine, acts directly to enhance Th1 and cytotoxic T-cell responses, and stimulates IFNγ, with its associated immune effects.165 IL-12 has been one of the most effective antitumor cytokines expressed from oHSV; M002 in brain tumors,166 NV1042 in squamous cell carcinoma and metastatic prostate cancer,87,167 and G47Δ-mIL12 in glioblastoma and malignant peripheral nerve sheath tumors.140,168 With this success, an oHSV expressing human IL-12 has been constructed, M032, that was safe after intracranial injection in nonhuman primates169 and is currently in a phase 1 clinical trial for recurrent glioblastoma (http://clinicaltrials.gov/show/NCT02062827). IL-12 also has antiangiogenic activity, which enhances oHSV efficacy in human glioblastoma models in immunodeficient mice.170 In contrast, treatment of intracerebral gliomas with oHSV expressing immunosuppressive IL-10 (R8308) was no better than mock treatment.171 In order to prime the adaptive immune response, xenogeneic tumor antigens can be expressed in the tumor, such as prostatic acid phosphatase (PAP) in bPΔ6-hPAP for prostate cancer.136
Tumor microenvironment modifying transgenes
Many tumors create a special environment allowing them to grow faster and invade nearby tissues. Growing tumors require new blood vessel formation, angiogenesis, to supply their growing peripheries with nutrients and oxygen. These new vessels also allow infiltration of tumor-associated macrophages, which benefit tumor growth.172 Neovascularization is promoted by secretion of vascular endothelial growth factor (VEGF) and other angiogenic factors. G47Δ-mAngio encodes the antiangiogenic peptide angiostatin inserted into the ICP6 locus.173 Infection with G47Δ-mAngio reduced angiogenesis in glioblastoma stem cell and orthopic tumor models, as well as conferring a survival advantage over the empty control virus.170 The VAE virus is very similar, but expresses a fusion endostatin-angiostatin peptide to dampen angiogenesis.174 These viruses work under the principle that inhibiting angiogenesis will prevent tumor growth by blocking new blood vessel formation. Other oHSVs expressing antiangiogenic factors include platelet factor-4, G47Δ-PF4, to inhibit endothelial cell growth and migration175; Vstat120, extracellular fragment of brain-specific angiogenesis inhibitor 1, expressed by RAMBO and 34.5ENVE117,176; thrombospondin-1, a naturally occurring antiangiogenic factor and matrix metalloproteinase (MMP)-9 inhibitor, by T-TSP-1177; and endostatin, by HSV-endo.178
The tumor extracellular matrix poses a problem for oHSV therapy. Many tumors create an extensive extracellular matrix that limits cell-to-cell interactions within a tumor, and thus cell-to-cell spread of oHSV. MMPs chew through this environment, which opens space for virus transfer. However, MMPs are known to increase the proliferation of many tumors and often lead to metastasis. To avoid this, OV-Chase expresses the bacterial chondroitinase-ABC, which increases interstitial diffusion while maintaining the structural composition of the extracellular matrix.179 OV-Chase was shown to spread better in glioblastoma spheroids in vitro, and increased the survival over nonchondroitinase-ABC expressing rHSVQ.179 In a reverse approach, rQT3 virus expresses an inhibitor of matrix metalloproteinases, tissue inhibitor of metalloproteinases 3 (TIMP3).180 rQT3 inhibited the growth of MPNST tumors in mice and MMP activity, and reduced vascular density.180
Conclusion
There has been an explosion of research on oHSV in the last 25 years, since genetically-engineered HSV was first tested as an oncolytic virus.6 During that time, eight different oHSVs and “armed” oHSVs have entered clinical trial (G207, 1716, NV1020, OncoVEXGMCSF, HF-10, G47Δ, rRp450, M032 (Table 1)), and OncoVEXGMCSF or T-Vec has successfully completed a pivotal phase 3 clinical trial for recurrent melanoma. Host/tumor interactions that limit virus replication and activity, be they intrinsic cellular responses, innate, or adaptive immunity, will need to be better understood in order to develop strategies to improve efficacy. The potential for developing new oHSV vectors is great, as well as strategies for combining oHSV with other therapeutic approaches to maximize outcomes.181 The next big step in the field will likely come from more in depth use of clinical tumor isolates to discover patterns and biomarkers associated with certain cancers. Identifying biomarkers, which distinguish cancer subtypes from one another, will allow for the development of more targeted oHSV, either by transcriptional or receptor targeting. Advances in basic tumor biology to determine what makes a tumor cell or tumor tissue different from normal tissues will allow the creation of oHSV that act like molecular scalpels, infecting and removing only the tumorigenic tissues. When designing or using an oHSV, it is important to closely study host:virus interactions in order to discover new peculiarities within the tumor that can be exploited by the next generation of vectors. Now that CRISPR/Cas9 and BACs are reliably available, it is faster and easier to create new vectors than ever before. Understanding the cellular factors that antagonize virus replication and spread throughout the tumor, as well as contribute to oncolytic activity will create opportunities to improve oHSV therapy, while the clinical trial results will provide insights into the limitations of oHSV and illuminate pathways to success.
Acknowledgments
The authors would like to thank the members of the Brain Tumor Research Center who have contributed to our research. Our studies have been supported in part by grants from the National Institutes of Health (R01CA160762, R01NS032677, and R01CA102139) and the Thomas A. Pappas Chair in Neurosciences.
Footnotes
SDR is a named inventor on patents relating to oncolytic herpes simplex viruses, which were filed by Georgetown University and Massachusetts General Hospital.
References
- Russell, SJ, Peng, KW and Bell, JC (2012). Oncolytic virotherapy. Nat Biotechnol 30: 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman, B, Knipe, DM and Whitley, RJ. Herpes simplex viruses. In: Knipe DM and Howley PM (eds). Fields Virology. Lippincott Williams &Wilkins, Philadelphia, 2013. pp 1823–1897. [Google Scholar]
- Field, HJ and Wildy, P (1978). The pathogenicity of thymidine kinase-deficient mutants of herpes simplex virus in mice. J Hyg (Lond) 81: 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert, A, Gentner, E, Bohn, K, Schwarz, M, Mertens, T and Sauerbrei, A (2014). Single nucleotide polymorphisms of thymidine kinase and DNA polymerase genes in clinical herpes simplex virus type 1 isolates associated with different resistance phenotypes. Antiviral Res 107: 16–22. [DOI] [PubMed] [Google Scholar]
- Luo, C, Mori, I, Goshima, F, Ushijima, Y, Nawa, A, Kimura, H et al. (2007). Replication-competent, oncolytic herpes simplex virus type 1 mutants induce a bystander effect following ganciclovir treatment. J Gene Med 9: 875–883. [DOI] [PubMed] [Google Scholar]
- Martuza, RL, Malick, A, Markert, JM, Ruffner, KL and Coen, DM (1991). Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 252: 854–856. [DOI] [PubMed] [Google Scholar]
- Kaplitt, MG, Tjuvajev, JG, Leib, DA, Berk, J, Pettigrew, KD, Posner, JB et al. (1994). Mutant herpes simplex virus induced regression of tumors growing in immunocompetent rats. J Neurooncol 19: 137–147. [DOI] [PubMed] [Google Scholar]
- Jia, WW, McDermott, M, Goldie, J, Cynader, M, Tan, J and Tufaro, F (1994). Selective destruction of gliomas in immunocompetent rats by thymidine kinase-defective herpes simplex virus type 1. J Natl Cancer Inst 86: 1209–1215. [DOI] [PubMed] [Google Scholar]
- Goldstein, DJ and Weller, SK (1988). Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 166: 41–51. [DOI] [PubMed] [Google Scholar]
- Aghi, M, Visted, T, Depinho, RA and Chiocca, EA (2008). Oncolytic herpes virus with defective ICP6 specifically replicates in quiescent cells with homozygous genetic mutations in p16. Oncogene 27: 4249–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, Y, Kimura, H, Morishima, T, Daikoku, T, Maeno, K and Nishiyama, Y (1991). The pathogenicity of ribonucleotide reductase-null mutants of herpes simplex virus type 1 in mice. J Infect Dis 164: 1091–1097. [DOI] [PubMed] [Google Scholar]
- Cameron, JM, McDougall, I, Marsden, HS, Preston, VG, Ryan, DM and Subak-Sharpe, JH (1988). Ribonucleotide reductase encoded by herpes simplex virus is a determinant of the pathogenicity of the virus in mice and a valid antiviral target. J Gen Virol 69 (Pt 10): 2607–2612. [DOI] [PubMed] [Google Scholar]
- Zhu, H, Dahlström, A and Hansson, HA (2005). Characterization of cell proliferation in the adult dentate under normal conditions and after kainate induced seizures using ribonucleotide reductase and BrdU. Brain Res 1036: 7–17. [DOI] [PubMed] [Google Scholar]
- Passer, BJ, Cheema, T, Zhou, B, Wakimoto, H, Zaupa, C, Razmjoo, M et al. (2010). Identification of the ENT1 antagonists dipyridamole and dilazep as amplifiers of oncolytic herpes simplex virus-1 replication. Cancer Res 70: 3890–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrowsky, H, Roberts, GD, Kooby, DA, Burt, BM, Bennett, JJ, Delman, KA et al. (2001). Functional interaction between fluorodeoxyuridine-induced cellular alterations and replication of a ribonucleotide reductase-negative herpes simplex virus. J Virol 75: 7050–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, K, Todo, T, Zhao, G, Yamaguchi, K, Kuroki, S, Cohen, JB et al. (2005). Enhanced efficacy of conditionally replicating herpes simplex virus (G207) combined with 5-fluorouracil and surgical resection in peritoneal cancer dissemination models. J Gene Med 7: 638–648. [DOI] [PubMed] [Google Scholar]
- Walsh, D and Mohr, I (2006). Assembly of an active translation initiation factor complex by a viral protein. Genes Dev 20: 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Li, Y, Liu, S, Yu, X, Li, L, Shi, C et al. (2014). Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA 111: 15438–15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour, F, Sasseville, AM, Chabaud, S, Massie, B, Siegel, RM and Langelier, Y (2011). The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFa- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis 16: 256–271. [DOI] [PubMed] [Google Scholar]
- Langelier, Y, Bergeron, S, Chabaud, S, Lippens, J, Guilbault, C, Sasseville, AM et al. (2002). The R1 subunit of herpes simplex virus ribonucleotide reductase protects cells against apoptosis at, or upstream of, caspase-8 activation. J Gen Virol 83(Pt 11): 2779–2789. [DOI] [PubMed] [Google Scholar]
- Mineta, T, Rabkin, SD and Martuza, RL (1994). Treatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res 54: 3963–3966. [PubMed] [Google Scholar]
- Mineta, T, Rabkin, SD, Yazaki, T, Hunter, WD and Martuza, RL (1995). Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med 1: 938–943. [DOI] [PubMed] [Google Scholar]
- Kanai, R, Zaupa, C, Sgubin, D, Antoszczyk, SJ, Martuza, RL, Wakimoto, H et al. (2012). Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol 86: 4420–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai, R, Wakimoto, H, Martuza, RL and Rabkin, SD (2011). A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res 17: 3686–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, DJ and Weller, SK (1988). Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol 62: 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyles, RB and Thompson, RL (1994). Evidence that the herpes simplex virus type 1 uracil DNA glycosylase is required for efficient viral replication and latency in the murine nervous system. J Virol 68: 4963–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyles, RB, Warnick, RE, Chalk, CL, Szanti, BE and Parysek, LM (1997). A novel multiply-mutated HSV-1 strain for the treatment of human brain tumors. Hum Gene Ther 8: 533–544. [DOI] [PubMed] [Google Scholar]
- He, B, Gross, M and Roizman, B (1997). The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA 94: 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvera, D, Formenti, SC and Schneider, RJ (2010). Translational control in cancer. Nat Rev Cancer 10: 254–266. [DOI] [PubMed] [Google Scholar]
- He, B, Chou, J, Liebermann, DA, Hoffman, B and Roizman, B (1996). The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the gamma(1)34.5 gene of herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. J Virol 70: 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, JJ, Adusumilli, P, Petrowsky, H, Burt, BM, Roberts, G, Delman, KA et al. (2004). Up-regulation of GADD34 mediates the synergistic anticancer activity of mitomycin C and a gamma134.5 deleted oncolytic herpes virus (G207). FASEB J 18: 1001–1003. [DOI] [PubMed] [Google Scholar]
- Aghi, M, Rabkin, S and Martuza, RL (2006). Effect of chemotherapy-induced DNA repair on oncolytic herpes simplex viral replication. J Natl Cancer Inst 98: 38–50. [DOI] [PubMed] [Google Scholar]
- Aghi, MK, Liu, TC, Rabkin, S and Martuza, RL (2009). Hypoxia enhances the replication of oncolytic herpes simplex virus. Mol Ther 17: 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallóczy, Z, Jiang, W, Virgin, HW 4th, Leib, DA, Scheuner, D, Kaufman, RJ et al. (2002). Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 99: 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpooten, D, Ma, Y, Hou, S, Yan, Z and He, B (2009). Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J Biol Chem 284: 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland, J, Dunn, P, Cameron, E, Conner, J and Brown, SM (2003). The herpes simplex virus (HSV) protein ICP34.5 is a virion component that forms a DNA-binding complex with proliferating cell nuclear antigen and HSV replication proteins. J Neurovirol 9: 477–488. [DOI] [PubMed] [Google Scholar]
- Wang, Y, Yang, Y, Wu, S, Pan, S, Zhou, C, Ma, Y, et al. (2014). P32 is a novel target for ICP34.5 of herpes simplex virus type 1, and facilitates viral nuclear egress. J Biol Chem 289: 35795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutia, SK, Mukhopadhyay, S, Sinha, N, Das, DN, Panda, PK, Patra, SK et al. (2013). Autophagy: cancer’s friend or foe? Adv Cancer Res 118: 61–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl, A, Alexander, D, Tallóczy, Z, Sun, Q, Wei, Y, Zhang, W et al. (2007). HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1: 23–35. [DOI] [PubMed] [Google Scholar]
- Leib, DA, Alexander, DE, Cox, D, Yin, J and Ferguson, TA (2009). Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J Virol 83: 12164–12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, G, Brett, ME and He, B (2002). Signals that dictate nuclear, nucleolar, and cytoplasmic shuttling of the gamma(1)34.5 protein of herpes simplex virus type 1. J Virol 76: 9434–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, J, Kern, ER, Whitley, RJ and Roizman, B (1990). Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250: 1262–1266. [DOI] [PubMed] [Google Scholar]
- Trgovcich, J, Johnson, D and Roizman, B (2002). Cell surface major histocompatibility complex class II proteins are regulated by the products of the gamma(1)34.5 and U(L)41 genes of herpes simplex virus 1. J Virol 76: 6974–6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib, DA, Machalek, MA, Williams, BR, Silverman, RH and Virgin, HW (2000). Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc Natl Acad Sci USA 97: 6097–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto, H, Kesari, S, Farrell, CJ, Curry, WT Jr, Zaupa, C, Aghi, M et al. (2009). Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res 69: 3472–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert, JM, Medlock, MD, Rabkin, SD, Gillespie, GY, Todo, T, Hunter, WD et al. (2000). Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther 7: 867–874. [DOI] [PubMed] [Google Scholar]
- Markert, JM, Razdan, SN, Kuo, HC, Cantor, A, Knoll, A, Karrasch, M et al. (2014). A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther 22: 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambara, H, Okano, H, Chiocca, EA and Saeki, Y (2005). An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res 65: 2832–2839. [DOI] [PubMed] [Google Scholar]
- Mohr, I and Gluzman, Y (1996). A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J 15: 4759–4766. [PMC free article] [PubMed] [Google Scholar]
- Mulvey, M, Poppers, J, Ladd, A and Mohr, I (1999). A herpesvirus ribosome-associated, RNA-binding protein confers a growth advantage upon mutants deficient in a GADD34-related function. J Virol 73: 3375–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, AC, Price, KH, Parker, JN, Samuel, SL, Meleth, S, Cassady, KA et al. (2006). Serial passage through human glioma xenografts selects for a Deltagamma134.5 herpes simplex virus type 1 mutant that exhibits decreased neurotoxicity and prolongs survival of mice with experimental brain tumors. J Virol 80: 7308–7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, AC, Parker, JN, Gillespie, GY, Lakeman, FD, Meleth, S, Markert, JM et al. (2007). Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther 14: 1045–1054. [DOI] [PubMed] [Google Scholar]
- Fu, X, Rivera, A, Tao, L and Zhang, X (2012). Incorporation of the B18R gene of vaccinia virus into an oncolytic herpes simplex virus improves antitumor activity. Mol Ther 20: 1871–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard, B and Früh, K (2000). A role for MHC class I down-regulation in NK cell lysis of herpes virus-infected cells. Eur J Immunol 30: 509–515. [DOI] [PubMed] [Google Scholar]
- Todo, T, Martuza, RL, Rabkin, SD and Johnson, PA (2001). Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci USA 98: 6396–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, BL, Robinson, M, Han, ZQ, Branston, RH, English, C, Reay, P et al. (2003). ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther 10: 292–303. [DOI] [PubMed] [Google Scholar]
- Taneja, S, MacGregor, J, Markus, S, Ha, S and Mohr, I (2001). Enhanced antitumor efficacy of a herpes simplex virus mutant isolated by genetic selection in cancer cells. Proc Natl Acad Sci USA 98: 8804–8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassady, KA, Gross, M and Roizman, B (1998). The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J Virol 72: 8620–8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppers, J, Mulvey, M, Khoo, D and Mohr, I (2000). Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J Virol 74: 11215–11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr, I, Sternberg, D, Ward, S, Leib, D, Mulvey, M and Gluzman, Y (2001). A herpes simplex virus type 1 gamma34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J Virol 75: 5189–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, J, Wang, S, Lin, R, Mossman, KL and Zheng, C (2012). Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol 86: 3528–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussignol, M, Queval, C, Bernet-Camard, MF, Cotte-Laffitte, J, Beau, I, Codogno, P et al. (2013). The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol 87: 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sànchez, R and Mohr, I (2007). Inhibition of cellular 2’-5’ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J Virol 81: 3455–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javouhey, E, Gibert, B, Arrigo, AP, Diaz, JJ and Diaz-Latoud, C (2008). Protection against heat and staurosporine mediated apoptosis by the HSV-1 US11 protein. Virology 376: 31–41. [DOI] [PubMed] [Google Scholar]
- Greco, A, Arata, L, Soler, E, Gaume, X, Couté, Y, Hacot, S et al. (2012). Nucleolin interacts with US11 protein of herpes simplex virus 1 and is involved in its trafficking. J Virol 86: 1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benetti, L and Roizman, B (2004). Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci USA 101: 9411–9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuluunbaatar, U, Roller, R, Feldman, ME, Brown, S, Shokat, KM and Mohr, I (2010). Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev 24: 2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, ML and Blaho, JA (2007). Apoptosis during herpes simplex virus infection. Adv Virus Res 69: 67–97. [DOI] [PubMed] [Google Scholar]
- Liu, TC, Wakimoto, H, Martuza, RL and Rabkin, SD (2007). Herpes simplex virus Us3(-) mutant as oncolytic strategy and synergizes with phosphatidylinositol 3-kinase-Akt targeting molecular therapeutics. Clin Cancer Res 13: 5897–5902. [DOI] [PubMed] [Google Scholar]
- Kato, A, Tsuda, S, Liu, Z, Kozuka-Hata, H, Oyama, M and Kawaguchi, Y (2014). Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J Virol 88: 655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai, T, Sagou, K, Arii, J and Kawaguchi, Y (2010). Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J Virol 84: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K, Ni, L, Wang, S and Zheng, C (2014). Herpes simplex virus 1 protein kinase US3 hyperphosphorylates p65/RelA and dampens NF-?B activation. J Virol 88: 7941–7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S, Wang, K, Lin, R and Zheng, C (2013). Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta interferon production. J Virol 87: 12814–12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, L and Roizman, B (2008). Expression of gamma interferon-dependent genes is blocked independently by virion host shutoff RNase and by US3 protein kinase. J Virol 82: 4688–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai, T, Koyanagi, N, Ogawa, R, Shindo, K, Suenaga, T, Sato, A et al. (2013). Us3 kinase encoded by herpes simplex virus 1 mediates downregulation of cell surface major histocompatibility complex class I and evasion of CD8+ T cells. PLoS ONE 8: e72050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi, N, Imai, T, Arii, J, Kato, A and Kawaguchi, Y (2014). Role of herpes simplex virus 1 Us3 in viral neuroinvasiveness. Microbiol Immunol 58: 31–37. [DOI] [PubMed] [Google Scholar]
- Kasuya, H, Nishiyama, Y, Nomoto, S, Goshima, F, Takeda, S, Watanabe, I et al. (2007). Suitability of a US3-inactivated HSV mutant (L1BR1) as an oncolytic virus for pancreatic cancer therapy. Cancer Gene Ther 14: 533–542. [DOI] [PubMed] [Google Scholar]
- Boutell, C and Everett, RD (2013). Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94(Pt 3): 465–481. [DOI] [PubMed] [Google Scholar]
- Sobol, PT, Hummel, JL, Rodrigues, RM and Mossman, KL (2009). PML has a predictive role in tumor cell permissiveness to interferon-sensitive oncolytic viruses. Gene Ther 16: 1077–1087. [DOI] [PubMed] [Google Scholar]
- Walsh, D and Mohr, I (2004). Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev 18: 660–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel, JL, Safroneeva, E and Mossman, KL (2005). The role of ICP0-Null HSV-1 and interferon signaling defects in the effective treatment of breast adenocarcinoma. Mol Ther 12: 1101–1110. [DOI] [PubMed] [Google Scholar]
- Sobol, PT, Boudreau, JE, Stephenson, K, Wan, Y, Lichty, BD and Mossman, KL (2011). Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol Ther 19: 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart, B, Mazzacurati, L, Forero, A, Hong, CS, Eguchi, J, Okada, H et al. (2012). Inhibition of Indoleamine-2,3-dioxygenase (IDO) in Glioblastoma Cells by Oncolytic Herpes Simplex Virus. Adv Virol 2012: 815465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehm, R, Rösen-Wolff, A and Darai, G (1996). Restitution of the UL56 gene expression of HSV-1 HFEM led to restoration of virulent phenotype; deletion of the amino acids 217 to 234 of the UL56 protein abrogates the virulent phenotype. Virus Res 40: 17–31. [DOI] [PubMed] [Google Scholar]
- Ushijima, Y, Luo, C, Goshima, F, Yamauchi, Y, Kimura, H and Nishiyama, Y (2007). Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect 9: 142–149. [DOI] [PubMed] [Google Scholar]
- Nawa, A, Luo, C, Zhang, L, Ushjima, Y, Ishida, D, Kamakura, M et al. (2008). Non-engineered, naturally oncolytic herpes simplex virus HSV1 HF-10: applications for cancer gene therapy. Curr Gene Ther 8: 208–221. [DOI] [PubMed] [Google Scholar]
- Wong, RJ, Patel, SG, Kim, S, DeMatteo, RP, Malhotra, S, Bennett, JJ et al. (2001). Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum Gene Ther 12: 253–265. [DOI] [PubMed] [Google Scholar]
- Mossman, KL and Smiley, JR (1999). Truncation of the C-terminal acidic transcriptional activation domain of herpes simplex virus VP16 renders expression of the immediate-early genes almost entirely dependent on ICP0. J Virol 73: 9726–9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boviatsis, EJ, Scharf, JM, Chase, M, Harrington, K, Kowall, NW, Breakefield, XO et al. (1994). Antitumor activity and reporter gene transfer into rat brain neoplasms inoculated with herpes simplex virus vectors defective in thymidine kinase or ribonucleotide reductase. Gene Ther 1: 323–331. [PubMed] [Google Scholar]
- Miyatake, S, Iyer, A, Martuza, RL and Rabkin, SD (1997). Transcriptional targeting of herpes simplex virus for cell-specific replication. J Virol 71: 5124–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand, L, Leiva-Torres, GA, Hyjazie, H and Pearson, A (2010). Conserved residues in the UL24 protein of herpes simplex virus 1 are important for dispersal of the nucleolar protein nucleolin. J Virol 84: 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiva-Torres, GA, Rochette, PA and Pearson, A (2010). Differential importance of highly conserved residues in UL24 for herpes simplex virus 1 replication in vivo and reactivation. J Gen Virol 91(Pt 5): 1109–1116. [DOI] [PubMed] [Google Scholar]
- Kelly, KJ, Wong, J and Fong, Y (2008). Herpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancy. Expert Opin Investig Drugs 17: 1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workenhe, ST, Simmons, G, Pol, JG, Lichty, BD, Halford, WP and Mossman, KL (2014). Immunogenic HSV-mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol Ther 22: 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, CC (2005). The herpes simplex virus type 2 protein ICP10PK: a master of versatility. Front Biosci 10: 2820–2831. [DOI] [PubMed] [Google Scholar]
- Colunga, AG, Laing, JM and Aurelian, L (2010). The HSV-2 mutant DeltaPK induces melanoma oncolysis through nonredundant death programs and associated with autophagy and pyroptosis proteins. Gene Ther 17: 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, X, Tao, L, Cai, R, Prigge, J and Zhang, X (2006). A mutant type 2 herpes simplex virus deleted for the protein kinase domain of the ICP10 gene is a potent oncolytic virus. Mol Ther 13: 882–890. [DOI] [PubMed] [Google Scholar]
- Fu, X, Tao, L and Zhang, X (2007). An oncolytic virus derived from type 2 herpes simplex virus has potent therapeutic effect against metastatic ovarian cancer. Cancer Gene Ther 14: 480–487. [DOI] [PubMed] [Google Scholar]
- Aurelian, L (2004). Herpes simplex virus type 2 vaccines: new ground for optimism? Clin Diagn Lab Immunol 11: 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurelian, L, Kokuba, H and Smith, CC (1999). Vaccine potential of a herpes simplex virus type 2 mutant deleted in the PK domain of the large subunit of ribonucleotide reductase (ICP10). Vaccine 17: 1951–1963. [DOI] [PubMed] [Google Scholar]
- Morimoto, T, Arii, J, Tanaka, M, Sata, T, Akashi, H, Yamada, M et al. (2009). Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83: 11624–11634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, I (2012). Herpes simplex virus US3 protein kinase regulates host responses and determines neurovirulence. Microbiol Immunol 56: 351–355. [DOI] [PubMed] [Google Scholar]
- Rodrigues, R, Cuddington, B and Mossman, K (2010). Bovine herpesvirus type 1 as a novel oncolytic virus. Cancer Gene Ther 17: 344–355. [DOI] [PubMed] [Google Scholar]
- Cuddington, BP and Mossman, KL (2014). Permissiveness of human cancer cells to oncolytic bovine herpesvirus 1 is mediated in part by KRAS activity. J Virol 88: 6885–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne, MJ, White, MC, Stanfield, BA and Frampton, AR Jr (2012). Equine herpesvirus type 1-mediated oncolysis of human glioblastoma multiforme cells. J Virol 86: 2882–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, CY, Rennie, PS and Jia, WW (2009). MicroRNA regulation of oncolytic herpes simplex virus-1 for selective killing of prostate cancer cells. Clin Cancer Res 15: 5126–5135. [DOI] [PubMed] [Google Scholar]
- Kuroda, T, Rabkin, SD and Martuza, RL (2006). Effective treatment of tumors with strong beta-catenin/T-cell factor activity by transcriptionally targeted oncolytic herpes simplex virus vector. Cancer Res 66: 10127–10135. [DOI] [PubMed] [Google Scholar]
- Yamamura, H, Hashio, M, Noguchi, M, Sugenoya, Y, Osakada, M, Hirano, N et al. (2001). Identification of the transcriptional regulatory sequences of human calponin promoter and their use in targeting a conditionally replicating herpes vector to malignant human soft tissue and bone tumors. Cancer Res 61: 3969–3977. [PubMed] [Google Scholar]
- Pan, W, Bodempudi, V, Esfandyari, T and Farassati, F (2009). Utilizing ras signaling pathway to direct selective replication of herpes simplex virus-1. PLoS ONE 4: e6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake, SI, Tani, S, Feigenbaum, F, Sundaresan, P, Toda, H, Narumi, O et al. (1999). Hepatoma-specific antitumor activity of an albumin enhancer/promoter regulated herpes simplex virus in vivo. Gene Ther 6: 564–572. [DOI] [PubMed] [Google Scholar]
- Mullen, JT, Kasuya, H, Yoon, SS, Carroll, NM, Pawlik, TM, Chandrasekhar, S et al. (2002). Regulation of herpes simplex virus 1 replication using tumor-associated promoters. Ann Surg 236: 502–12; discussion 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo, SL, Griffith, C, Glass, A, Shillitoe, EJ and Post, DE (2011). Development of an oncolytic herpes simplex virus using a tumor-specific HIF-responsive promoter. Cancer Gene Ther 18: 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, RY, Saeki, Y and Chiocca, EA (1999). B-myb promoter retargeting of herpes simplex virus gamma34.5 gene-mediated virulence toward tumor and cycling cells. J Virol 73: 7556–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, H, Kasuya, H, Mullen, JT, Yoon, SS, Pawlik, TM, Chandrasekhar, S et al. (2002). Regulation of herpes simplex virus gamma(1)34.5 expression and oncolysis of diffuse liver metastases by Myb34.5. J Clin Invest 109: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyard, C, Yoo, JY, Wang, PY, Saini, U, Rath, KS, Cripe, TP et al. (2014). Doxorubicin synergizes with 34.5ENVE to enhance antitumor efficacy against metastatic ovarian cancer. Clin Cancer Res 20: 6479–6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupkova, O Jr, Loja, T, Zambo, I and Veselska, R (2010). Nestin expression in human tumors and tumor cell lines. Neoplasma 57: 291–298. [DOI] [PubMed] [Google Scholar]
- Yoo, JY, Haseley, A, Bratasz, A, Chiocca, EA, Zhang, J, Powell, K et al. (2012). Antitumor efficacy of 34.5ENVE: a transcriptionally retargeted and “Vstat120”-expressing oncolytic virus. Mol Ther 20: 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai, R, Tomita, H, Hirose, Y, Ohba, S, Goldman, S, Okano, H et al. (2007). Augmented therapeutic efficacy of an oncolytic herpes simplex virus type 1 mutant expressing ICP34.5 under the transcriptional control of musashi1 promoter in the treatment of malignant glioma. Hum Gene Ther 18: 63–73. [DOI] [PubMed] [Google Scholar]
- Maldonado, AR, Klanke, C, Jegga, AG, Aronow, BJ, Mahller, YY, Cripe, TP et al. (2010). Molecular engineering and validation of an oncolytic herpes simplex virus type 1 transcriptionally targeted to midkine-positive tumors. J Gene Med 12: 613–623. [DOI] [PubMed] [Google Scholar]
- Mazzacurati, L, Marzulli, M, Reinhart, B, Miyagawa, Y, Uchida, H, Goins, WF et al. (2015). Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol Ther 23: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, X, Rivera, A, Tao, L, De Geest, B and Zhang, X (2012). Construction of an oncolytic herpes simplex virus that precisely targets hepatocellular carcinoma cells. Mol Ther 20: 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campadelli-Fiume, G, Menotti, L, Avitabile, E and Gianni, T (2012). Viral and cellular contributions to herpes simplex virus entry into the cell. Curr Opin Virol 2: 28–36. [DOI] [PubMed] [Google Scholar]
- Grandi, P, Fernandez, J, Szentirmai, O, Carter, R, Gianni, D, Sena-Esteves, M et al. (2010). Targeting HSV-1 virions for specific binding to epidermal growth factor receptor-vIII-bearing tumor cells. Cancer Gene Ther 17: 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, G and Roizman, B (2005). Characterization of a recombinant herpes simplex virus 1 designed to enter cells via the IL13Ralpha2 receptor of malignant glioma cells. J Virol 79: 5272–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, G and Roizman, B (2006). Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc Natl Acad Sci USA 103: 5508–5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menotti, L, Nicoletti, G, Gatta, V, Croci, S, Landuzzi, L, De Giovanni, C et al. (2009). Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc Natl Acad Sci USA 106: 9039–9044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida, H, Marzulli, M, Nakano, K, Goins, WF, Chan, J, Hong, CS et al. (2013). Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther 21: 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek, H, Uchida, H, Jun, K, Kim, JH, Kuroki, M, Cohen, JB et al. (2011). Bispecific adapter-mediated retargeting of a receptor-restricted HSV-1 vector to CEA-bearing tumor cells. Mol Ther 19: 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanni, P, Gatta, V, Menotti, L, De Giovanni, C, Ianzano, M, Palladini, A et al. (2013). Preclinical therapy of disseminated HER-2? ovarian and breast carcinomas with a HER-2-retargeted oncolytic herpesvirus. PLoS Pathog 9: e1003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambini, E, Reisoli, E, Appolloni, I, Gatta, V, Campadelli-Fiume, G, Menotti, L et al. (2012). Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol Ther 20: 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisoli, E, Gambini, E, Appolloni, I, Gatta, V, Barilari, M, Menotti, L et al. (2012). Efficacy of HER2 retargeted herpes simplex virus as therapy for high-grade glioma in immunocompetent mice. Cancer Gene Ther 19: 788–795. [DOI] [PubMed] [Google Scholar]
- Terada, K, Wakimoto, H, Tyminski, E, Chiocca, EA and Saeki, Y (2006). Development of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther 13: 705–714. [DOI] [PubMed] [Google Scholar]
- Fukuhara, H, Ino, Y, Kuroda, T, Martuza, RL and Todo, T (2005). Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7-1 constructed by bacterial artificial chromosome-mediated system. Cancer Res 65: 10663–10668. [DOI] [PubMed] [Google Scholar]
- Kuroda, T, Martuza, RL, Todo, T and Rabkin, SD (2006). Flip-Flop HSV-BAC: bacterial artificial chromosome based system for rapid generation of recombinant herpes simplex virus vectors using two independent site-specific recombinases. BMC Biotechnol 6: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyaretna, DS and Kuroda, T (2007). Recent advances in the development of oncolytic HSV-1 vectors: ‘arming’ of HSV-1 vectors and application of bacterial artificial chromosome technology for their construction. Curr Opin Mol Ther 9: 447–466. [PubMed] [Google Scholar]
- Castelo-Branco, P, Passer, BJ, Buhrman, JS, Antoszczyk, S, Marinelli, M, Zaupa, C et al. (2010). Oncolytic herpes simplex virus armed with xenogeneic homologue of prostatic acid phosphatase enhances antitumor efficacy in prostate cancer. Gene Ther 17: 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer, BK and Kaufer, BB (2012). Viral bacterial artificial chromosomes: generation, mutagenesis, and removal of mini-F sequences. J Biomed Biotechnol 2012: 472537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, Y, Sun, L, Gao, D, Ding, C, Li, Z, Li, Y et al. (2014). High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog 10: e1004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga, T, Kohyama, M, Hirayasu, K and Arase, H (2014). Engineering large viral DNA genomes using the CRISPR-Cas9 system. Microbiol Immunol 58: 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema, TA, Wakimoto, H, Fecci, PE, Ning, J, Kuroda, T, Jeyaretna, DS et al. (2013). Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci USA 110: 12006–12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argnani, R, Marconi, P, Volpi, I, Bolanos, E, Carro, E, Ried, C et al. (2011). Characterization of herpes simplex virus 1 strains as platforms for the development of oncolytic viruses against liver cancer. Liver Int 31: 1542–1553. [DOI] [PubMed] [Google Scholar]
- Yamamoto, S, Deckter, LA, Kasai, K, Chiocca, EA and Saeki, Y (2006). Imaging immediate-early and strict-late promoter activity during oncolytic herpes simplex virus type 1 infection and replication in tumors. Gene Ther 13: 1731–1736. [DOI] [PubMed] [Google Scholar]
- Adusumilli, PS, Stiles, BM, Chan, MK, Eisenberg, DP, Yu, Z, Stanziale, SF et al. (2006). Real-time diagnostic imaging of tumors and metastases by use of a replication-competent herpes vector to facilitate minimally invasive oncological surgery. FASEB J 20: 726–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter, S and Fong, Y (2012). Real-time fluorescence imaging of abdominal, pleural, and lymphatic metastases. Methods Mol Biol 872: 141–157. [DOI] [PubMed] [Google Scholar]
- Li, H, Nakashima, H, Decklever, TD, Nace, RA and Russell, SJ (2013). HSV-NIS, an oncolytic herpes simplex virus type 1 encoding human sodium iodide symporter for preclinical prostate cancer radiovirotherapy. Cancer Gene Ther 20: 478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todo, T, Rabkin, SD and Martuza, RL (2000). Evaluation of ganciclovir-mediated enhancement of the antitumoral effect in oncolytic, multimutated herpes simplex virus type 1 (G207) therapy of brain tumors. Cancer Gene Ther 7: 939–946. [DOI] [PubMed] [Google Scholar]
- Nakamura, H, Mullen, JT, Chandrasekhar, S, Pawlik, TM, Yoon, SS and Tanabe, KK (2001). Multimodality therapy with a replication-conditional herpes simplex virus 1 mutant that expresses yeast cytosine deaminase for intratumoral conversion of 5-fluorocytosine to 5-fluorouracil. Cancer Res 61: 5447–5452. [PubMed] [Google Scholar]
- Guffey, MB, Parker, JN, Luckett, WS Jr, Gillespie, GY, Meleth, S, Whitley, RJ et al. (2007). Engineered herpes simplex virus expressing bacterial cytosine deaminase for experimental therapy of brain tumors. Cancer Gene Ther 14: 45–56. [DOI] [PubMed] [Google Scholar]
- Chase, M, Chung, RY and Chiocca, EA (1998). An oncolytic viral mutant that delivers the CYP2B1 transgene and augments cyclophosphamide chemotherapy. Nat Biotechnol 16: 444–448. [DOI] [PubMed] [Google Scholar]
- Tyminski, E, Leroy, S, Terada, K, Finkelstein, DM, Hyatt, JL, Danks, MK et al. (2005). Brain tumor oncolysis with replication-conditional herpes simplex virus type 1 expressing the prodrug-activating genes, CYP2B1 and secreted human intestinal carboxylesterase, in combination with cyclophosphamide and irinotecan. Cancer Res 65: 6850–6857. [DOI] [PubMed] [Google Scholar]
- Braidwood, L, Dunn, PD, Hardy, S, Evans, TR and Brown, SM (2009). Antitumor activity of a selectively replication competent herpes simplex virus (HSV) with enzyme prodrug therapy. Anticancer Res 29: 2159–2166. [PubMed] [Google Scholar]
- Bharara, S, Sorscher, EJ, Gillespie, GY, Lindsey, JR, Hong, JS, Curlee, KV et al. (2005). Antibiotic-mediated chemoprotection enhances adaptation of E. coli PNP for herpes simplex virus-based glioma therapy. Hum Gene Ther 16: 339–347. [DOI] [PubMed] [Google Scholar]
- Cheema, TA, Kanai, R, Kim, GW, Wakimoto, H, Passer, B, Rabkin, SD et al. (2011). Enhanced antitumor efficacy of low-dose Etoposide with oncolytic herpes simplex virus in human glioblastoma stem cell xenografts. Clin Cancer Res 17: 7383–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulu, Y, Kawasaki, H, Donahue, JM, Kasuya, H, Cusack, JC, Choi, EW et al. (2013). Concurrent chemotherapy inhibits herpes simplex virus-1 replication and oncolysis. Cancer Gene Ther 20: 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K, Wakimoto, H, Agarwal, AS, Rabkin, SD, Bhere, D, Martuza, RL et al. (2013). Multimechanistic tumor targeted oncolytic virus overcomes resistance in brain tumors. Mol Ther 21: 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichty, BD, Breitbach, CJ, Stojdl, DF and Bell, JC (2014). Going viral with cancer immunotherapy. Nat Rev Cancer 14: 559–567. [DOI] [PubMed] [Google Scholar]
- Fulci, G, Breymann, L, Gianni, D, Kurozomi, K, Rhee, SS, Yu, J et al. (2006). Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci USA 103: 12873–12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Breckenridge, CA, Yu, J, Price, R, Wojton, J, Pradarelli, J, Mao, H et al. (2012). NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med 18: 1827–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda, M, Rabkin, SD, Kojima, H and Martuza, RL (1999). Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther 10: 385–393. [DOI] [PubMed] [Google Scholar]
- Li, H, Dutuor, A, Fu, X and Zhang, X (2007). Induction of strong antitumor immunity by an HSV-2-based oncolytic virus in a murine mammary tumor model. J Gene Med 9: 161–169. [DOI] [PubMed] [Google Scholar]
- Varghese, S, Rabkin, SD, Liu, R, Nielsen, PG, Ipe, T and Martuza, RL (2006). Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther 13: 253–265. [DOI] [PubMed] [Google Scholar]
- Malhotra, S, Kim, T, Zager, J, Bennett, J, Ebright, M, D’Angelica, M et al. (2007). Use of an oncolytic virus secreting GM-CSF as combined oncolytic and immunotherapy for treatment of colorectal and hepatic adenocarcinomas. Surgery 141: 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, HL, Kim, DW, DeRaffele, G, Mitcham, J, Coffin, RS and Kim-Schulze, S (2010). Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol 17: 718–730. [DOI] [PubMed] [Google Scholar]
- Hersey, P and Gallagher, S (2014). Intralesional immunotherapy for melanoma. J Surg Oncol 109: 320–326. [DOI] [PubMed] [Google Scholar]
- Lasek, W, Zagozdzon, R and Jakobisiak, M (2014). Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother 63: 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, JN, Gillespie, GY, Love, CE, Randall, S, Whitley, RJ and Markert, JM (2000). Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci USA 97: 2208–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese, S, Rabkin, SD, Nielsen, PG, Wang, W and Martuza, RL (2006). Systemic oncolytic herpes virus therapy of poorly immunogenic prostate cancer metastatic to lung. Clin Cancer Res 12: 2919–2927. [DOI] [PubMed] [Google Scholar]
- Antoszczyk, S, Spyra, M, Mautner, VF, Kurtz, A, Stemmer-Rachamimov, AO, Martuza, RL et al. (2014). Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro-oncology 16: 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, JC, Cassady, KA, Cody, JJ, Parker, JN, Price, KH, Coleman, JM et al. (2014). Evaluation of the safety and biodistribution of M032, an attenuated herpes simplex virus type 1 expressing hIL-12, after intracerebral administration to aotus nonhuman primates. Hum Gene Ther Clin Dev 25: 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W, Fulci, G, Wakimoto, H, Cheema, TA, Buhrman, JS, Jeyaretna, DS et al. (2013). Combination of oncolytic herpes simplex viruses armed with angiostatin and IL-12 enhances antitumor efficacy in human glioblastoma models. Neoplasia 15: 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreansky, S, He, B, van Cott, J, McGhee, J, Markert, JM, Gillespie, GY et al. (1998). Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther 5: 121–130. [DOI] [PubMed] [Google Scholar]
- Pyonteck, SM, Akkari, L, Schuhmacher, AJ, Bowman, RL, Sevenich, L, Quail, DF et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19: 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W, Fulci, G, Buhrman, JS, Stemmer-Rachamimov, AO, Chen, JW, Wojtkiewicz, GR et al. (2012). Bevacizumab with angiostatin-armed oHSV increases antiangiogenesis and decreases bevacizumab-induced invasion in U87 glioma. Mol Ther 20: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G, Jin, G, Nie, X, Mi, R, Zhu, G, Jia, W et al. (2014). Enhanced antitumor efficacy of an oncolytic herpes simplex virus expressing an endostatin-angiostatin fusion gene in human glioblastoma stem cell xenografts. PLoS ONE 9: e95872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, TC, Zhang, T, Fukuhara, H, Kuroda, T, Todo, T, Martuza, RL et al. (2006). Oncolytic HSV armed with platelet factor 4, an antiangiogenic agent, shows enhanced efficacy. Mol Ther 14: 789–797. [DOI] [PubMed] [Google Scholar]
- Hardcastle, J, Kurozumi, K, Dmitrieva, N, Sayers, MP, Ahmad, S, Waterman, P et al. (2010). Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol Ther 18: 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji, T, Nakamori, M, Iwahashi, M, Nakamura, M, Ojima, T, Iida, T et al. (2013). An armed oncolytic herpes simplex virus expressing thrombospondin-1 has an enhanced in vivo antitumor effect against human gastric cancer. Int J Cancer 132: 485–494. [DOI] [PubMed] [Google Scholar]
- Goodwin, JM, Schmitt, AD, McGinn, CM, Fuchs, BC, Kuruppu, D, Tanabe, KK et al. (2012). Angiogenesis inhibition using an oncolytic herpes simplex virus expressing endostatin in a murine lung cancer model. Cancer Invest 30: 243–250. [DOI] [PubMed] [Google Scholar]
- Dmitrieva, N, Yu, L, Viapiano, M, Cripe, TP, Chiocca, EA, Glorioso, JC et al. (2011). Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin Cancer Res 17: 1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahller, YY, Vaikunth, SS, Ripberger, MC, Baird, WH, Saeki, Y, Cancelas, JA et al. (2008). Tissue inhibitor of metalloproteinase-3 via oncolytic herpesvirus inhibits tumor growth and vascular progenitors. Cancer Res 68: 1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai, R, Wakimoto, H, Cheema, T and Rabkin, SD (2010). Oncolytic herpes simplex virus vectors and chemotherapy: are combinatorial strategies more effective for cancer? Future Oncol 6: 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean, AR, ul-Fareed, M, Robertson, L, Harland, J and Brown, SM (1991). Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J Gen Virol 72 (Pt 3): 631–639. [DOI] [PubMed] [Google Scholar]
- Sgubin, D, Wakimoto, H, Kanai, R, Rabkin, SD and Martuza, RL (2012). Oncolytic herpes simplex virus counteracts the hypoxia-induced modulation of glioblastoma stem-like cells. Stem Cells Transl Med 1: 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, RJ, Joe, JK, Kim, SH, Shah, JP, Horsburgh, B and Fong, Y (2002). Oncolytic herpesvirus effectively treats murine squamous cell carcinoma and spreads by natural lymphatics to treat sites of lymphatic metastases. Hum Gene Ther 13: 1213–1223. [DOI] [PubMed] [Google Scholar]