

ABSTRACT
Bacterial SH3 (SH3b) domains are commonly fused with papain-like Nlp/P60 cell wall hydrolase domains. To understand how the modular architecture of SH3b and NlpC/P60 affects the activity of the catalytic domain, three putative NlpC/P60 cell wall hydrolases were biochemically and structurally characterized. These enzymes all have γ-d-Glu-A2pm (A2pm is diaminopimelic acid) cysteine amidase (or dl-endopeptidase) activities but with different substrate specificities. One enzyme is a cell wall lysin that cleaves peptidoglycan (PG), while the other two are cell wall recycling enzymes that only cleave stem peptides with an N-terminal l-Ala. Their crystal structures revealed a highly conserved structure consisting of two SH3b domains and a C-terminal NlpC/P60 catalytic domain, despite very low sequence identity. Interestingly, loops from the first SH3b domain dock into the ends of the active site groove of the catalytic domain, remodel the substrate binding site, and modulate substrate specificity. Two amino acid differences at the domain interface alter the substrate binding specificity in favor of stem peptides in recycling enzymes, whereas the SH3b domain may extend the peptidoglycan binding surface in the cell wall lysins. Remarkably, the cell wall lysin can be converted into a recycling enzyme with a single mutation.
IMPORTANCE
Peptidoglycan is a meshlike polymer that envelops the bacterial plasma membrane and bestows structural integrity. Cell wall lysins and recycling enzymes are part of a set of lytic enzymes that target covalent bonds connecting the amino acid and amino sugar building blocks of the PG network. These hydrolases are involved in processes such as cell growth and division, autolysis, invasion, and PG turnover and recycling. To avoid cleavage of unintended substrates, these enzymes have very selective substrate specificities. Our biochemical and structural analysis of three modular NlpC/P60 hydrolases, one lysin, and two recycling enzymes, show that they may have evolved from a common molecular architecture, where the substrate preference is modulated by local changes. These results also suggest that new pathways for recycling PG turnover products, such as tracheal cytotoxin, may have evolved in bacteria in the human gut microbiome that involve NlpC/P60 cell wall hydrolases.
INTRODUCTION
Peptidoglycan (PG) forms a meshlike protective layer that envelopes bacteria and fortifies the cell wall, thereby maintaining structural integrity and internal osmotic pressure. PG consists of long glycan chains interconnected by short stem peptides (1). The glycan chains contain two alternating amino sugars, N-acetylmuramic acid (MurNAc) and N-acetylglucosamine (GlcNAc), that are connected by a β-1→4 glycosidic bond. The stem peptides typically contain 4 or 5 amino acid residues, such as the tetrapeptide l-Ala-γ-d-Glu-meso-A2pm-d-Ala (A2pm is diaminopimelic acid) in the case of the Gram-negative bacterium Escherichia coli, which are attached to the MurNAc d-lactoyl moiety. Cross-linking of the glycan chains generally occurs between the carboxyl group of d-Ala at position 4 and the amino group of the residue at position 3 (most often A2pm or Lys), either directly or through a short peptide bridge (see Fig. S1 in the supplemental material for the E. coli PG structure) (1). The PG network is dynamic and is constantly broken down by enzymes in a process known as cell wall turnover to accommodate cell expansion during growth (2–5), such that up to 50% of this polymer is degraded during each generation (4, 6, 7). The turnover products are typically recovered and eventually recycled for de novo PG biosynthesis in a process that involves multiple dedicated enzymes (7). These recycling enzymes generally convert the larger turnover products into incrementally smaller fragments, which are eventually fed back to energy or peptidoglycan synthesis pathways. As PG recycling enzymes target the same chemical linkages present in PG, as well as in intermediates of peptidoglycan synthesis, they have to be highly specific toward substrates, highly regulated at the expression level (8), and/or compartmentalized (e.g., E. coli localizes its recycling enzymes in the cytoplasm [7]), so as not to compromise PG integrity or biosynthesis. The evolution and mechanisms for maintaining the substrate specificity of these enzymes are currently not well understood.
NlpC/P60 proteins (9) are a major class of cell wall hydrolases that typically cleave the linkage between d-Glu and A2pm (or Lys) within PG stem peptides. These enzymes have prototypical papain-like folds and active sites, consisting of a Cys-His dyad and a third polar residue (His, Asn, or Gln) (9–11). Our previous structural analysis of three NlpC/P60 amidases (also known as endopeptidases), PCP (PG cysteine peptidase) from cyanobacterium Anabaena variabilis (AvPCP) (11), YkfC from Bacillus cereus (BcYkfC) (12), and the bifunctional cell wall hydrolase CwlT (13), revealed the structural basis and sequence motifs for recognition of l-Ala and d-Glu. Whereas the NlpC/P60 domain of CwlT is capable of cleaving PG (13, 14), BcYkfC (12) and AvPCP (11), which have additional N-terminal bacterial SH3 (SH3b) domains, are proposed to be hydrolases that have greater specificity for stem peptides. SH3b domains (15) are ubiquitous in bacteria and are often fused to NlpC/P60 domains and other cell wall related modules (9), presumably as auxiliary modules that may bind PG and facilitate enzyme function. Indeed, it was recently shown that SH3b domains of the pneumococcal murein hydrolase (lysin) CbpD specifically recognized and bound PG (16). However, the proposed role of SH3b in YkfC, which functions as a specificity determinant for stem peptides (11, 12), was unexpected. Thus, the mechanisms by which SH3b domains modulate the catalytic activity of NlpC/P60 domains had not yet been explored.
To probe the roles of auxiliary domains in modular cell wall hydrolases and, more broadly, how substrate specificity for lysins and recycling enzymes evolved, NlpC/P60 enzymes with SH3b domains were selected and studied using the JCSG structural biology pipeline (17, 18). Here, we present biochemical and structural characterizations of three SH3b-NlpC/P60 fusion proteins consisting of a new cell wall lysin from Desulfovibrio vulgaris Hildenborough (DvLysin) and two putative YkfC orthologs from members of the gut microbiome, Bacteroides thetaiotaomicron (BtYkfC) and Bacteroides ovatus (BoYkfC). DvLysin is active on PG, as well as on a wide array of PG fragments, while the two distant YkfC orthologs are only active toward stem peptides with a free N-terminal l-Ala. We also biochemically characterized BcYkfC, whose structure we reported previously (12), and confirmed that it was indeed specific for stem peptides, as we had predicted. Surprisingly, the structure of DvLysin is similar overall to YkfC proteins, except for an additional N-terminal domain. Comparison of the active sites of DvLysin and YkfC orthologs revealed that changes in substrate specificity are mainly due to only a few changes at the domain interface. These results provide new insights into the evolution of enzyme specificity and the roles of SH3b domains in bacteria.
RESULTS
DvLysin and YkfC are γ-d-Glu-A2pm amidases with different substrate specificities.
Sequence analysis predicted that DvLysin is a lipoprotein and the three YkfC orthologs (BcYkfC, BoYkfC, and BtYkfC) are extracellular proteins. Thus, the predicted signal peptides were removed from expression constructs to improve solubility and the likelihood of crystallization. Selenomethionine derivatives of these proteins were expressed in E. coli with an N-terminal, tobacco etch virus (TEV)-cleavable His tag and purified by metal affinity chromatography. The purification tag was removed prior to biochemical and crystallographic characterization.
We first tested the activities of the four enzymes using PG and various PG biosynthesis or degradation fragments as the substrates. DvLysin exhibited γ-d-Glu-A2pm amidase activity for all the substrates tested, including PG, while YkfC proteins showed γ-d-Glu-A2pm amidase activities only toward tri-, tetra-, and pentapeptides (Table 1; see also Fig. S2 in the supplemental material). Therefore, DvLysin and YkfC proteins both have amidase activity, but they have different substrate specificities. BoYkfC and BtYkfC, with 86% sequence identity, had similar activities and preferred tetrapeptides (>10-fold over other substrates tested), while BcYkfC preferred tripeptides. YkfC proteins could not hydrolyze the dipeptide γ-d-Glu-A2pm, indicating that the presence of l-Ala on the substrate is essential for their activities. DvLysin was active toward most substrates tested (Table 1), except for the γ-d-Glu-A2pm dipeptide. MurNAc-tripeptide(Lys) and pentapeptide(Lys), in which A2pm is replaced by Lys, were cleaved at much lower rates by DvLysin, indicating a clear preference for A2pm-containing substrates.
TABLE 1 .
Specific activities of DvLysin and YkfC orthologs
| Substrate | Sp act (nmol/min/mg)a: |
|||
|---|---|---|---|---|
| DvLysin | BoYkfC | BtYkfCb | BcYkfC | |
| GlcNAc-1,6-anhydro-MurNAc-tetrapeptide (TCT) | 5500 | NA | NA | NA |
| TCT dimer | 5700 | NA | NA | NA |
| GlcNAc-MurNAc-tetrapeptide | 3700 | NA | NA | NA |
| Dimer of GlcNAc-MurNAc-tetrapeptide | 3800 | NA | NA | NA |
| MurNAc-pentapeptide | 2200 | NA | NA | NA |
| MurNAc-tetrapeptide | 5400 | NA | NA | NA |
| MurNAc-tripeptide | 2200 | NA | NA | NA |
| MurNAc-tripeptide(Lys) | 90 | NA | NA | NA |
| UDP-MurNAc-tripeptide | 1000 | NA | NA | NA |
| Lactoyl-pentapeptide | 2300 | 1.5 | 0.5 | NA |
| Pentapeptide | 2400 | 900 | 130 | 4450 |
| Pentapeptide(Lys) | 300 | 150 | 70 | 300 |
| Tetrapeptide | 6100 | 25,800 | 5750 | 5550 |
| Tripeptide | 3950 | 1750 | 260 | 8900 |
| Dipeptidec | 0.5 | NA | NA | NA |
| Peptidoglycan | ≥35d | NA | NA | NA |
Each value represents the mean of at least two independent determinations; the standard deviation was less than 10% in all cases. NA, no activity detected using up to 20 µg of protein per assay.
BtYkfC has the same substrate specificity as BoYkfC. However, a portion of BtYkfC precipitated and may have lost activity during the assay.
The dipeptide γ-d-Glu-meso-A2pm used here was from InvivoGen (mixture of two diastereoisomers in which meso-A2pm is bound to d-Glu by either its L or its D carbon).
Purified E. coli peptidoglycan polymer (60 nmol in terms of A2pm content) was incubated for 30 min with 15 µg of DvLysin, and the soluble peptides released (A2pm-d-Ala and its dimer) were purified by HPLC and quantified with the amino acid analyzer.
Structure determination of DvLysin and YkfC proteins.
To explore the structure and function relationships of these NlpC/P60 hydrolases, we determined the crystal structures of DvLysin, BoYkfC, and BtYkfC using Se–multiple-wavelength anomalous diffraction (MAD) or Se–single-wavelength anomalous diffraction (SAD) methods. The crystal structures were determined for DvLysin (PDB accession number 3M1U) in space group P21 to a resolution of 1.75 Å with Rcryst of 13.9% and Rfree of 17.3%, for BoYkfC (PDB accession number 3NPF) in space group P212121 to a resolution of 1.72 Å with Rcryst of 14.1% and Rfree of 17.1%, and for BtYkfC in space group P21 to resolutions of 2.1 Å (crystal 1; PDB accession number 3PVQ) and 1.75 Å (crystal 2; PDB accession number 4R0K) with Rcryst/Rfree of 16.5%/19.8% and 15.4%/17.7%, respectively. The two structures of BtYkfC from different crystallization conditions are almost identical, except for differences at their N termini and the catalytic cysteine (see below). The geometric qualities of all final models are excellent, with overall Molprobity scores (19) that rank in the 99th to 100th percentile compared to other structures at similar resolutions. Summaries of data collection and processing and model refinement statistics are shown in Table S1 in the supplemental material.
Overall structural description.
Analysis of intermolecule packing interfaces in the crystals suggests that DvLysin and YkfC proteins function as monomers. The DvLysin monomer consists of four domains (Fig. 1A): the N-terminal “c-clip” domain (residues 32 to 144), two SH3b domains, SH3b1 (residues 145 to 223) and SH3b2 (residues 224 to 304), and the C-terminal NlpC/P60 catalytic domain (residues 305 to 461). The hoof-shaped molecule has dimensions of 76 Å by 58 Å by 45 Å, with the c-clip, SH3b1, and NlpC/P60 catalytic domains located at the front and SH3b2 at the back of the molecule. The c-clip domain is in an extended helical conformation (αA to αE) that wraps across and stabilizes SH3b1 and NlpC/P60. The active site containing the catalytic Cys333 sits at the center of the hoof and is located near the interface defined by the SH3b1 and NlpC/P60 domains (Fig. 1B).
FIG 1 .

Crystal structures of DvLysin and BoYkfC. (A) Front and back view of a ribbon representation of the DvLysin monomer, colored by domain as indicated by the domain architecture schematic below. The catalytic Cys333 is shown as red spheres. Secondary structures for each domain are numbered separately: c-clip domain (αA to αE), SH3b1 and SH3b2 (βA to βE), and NlpC/P60 (β1 to β6 and α1 to α5). (B) Surface representation of DvLysin in the same front view and coloring scheme indicated in panel A. The modeled substrate is shown as sticks. (C) Structure of BoYkfC, shown in an orientation and coloring scheme similar to those used for DvLysin. (D) Surface representation of BoYkfC with a modeled tetrapeptide. (E) Catalytic site of BoYkfC. Density-modified SAD experimental map for the covalently modified cysteine is shown as light blue mesh (contoured at 1 σ). Hydrogens bonds are shown as dashed lines.
The two SH3b domains in DvLysin are similar in structure despite low sequence similarity (root mean square deviation [RMSD] of 2.5 Å for 56 Cα atoms and 11% sequence identity). Both SH3b domains consist of seven conserved strands (βA-βA1-βA2-βB-βC-βD-βE). Five of these strands (βA to βE) are also structurally conserved in eukaryotic SH3 domains, while the βA1-βA2 β-hairpins correspond to the RT loops of eukaryotic SH3 domains. SH3b1 contains a much longer RT loop than SH3b2, which extends the domain interface where the PG cross-link is expected to bind (Fig. 1B).
The catalytic domain of DvLysin has a prototypical papain-like α/β/α sandwich fold with a topology of α1-α2-α3-β1-α4-β2-β3-β4-β5-α5-β6, where α1-α2-α3 and α4-α5 protect either side of the central β-sheet (in the order 162345) (Fig. 1A). The catalytic domain is very similar in structure to other NlpC/P60 proteins, such as the NlpC/P60 domain of CwlT (PDB accession number 4FDY, RMSD of 1.3 Å for 107 aligned Cα atoms and 20% sequence identity). Interestingly, the catalytic domain of DvLysin contains a long β4-β5 β-hairpin, which is stabilized by a longer loop insertion between β5 and α5 and the RT loop of SH3b1 (Fig. 1A).
As BoYkfC and BtYkfC have almost identical structures (RMSD of ~0.5 Å for 295 Cα atoms), we focused our analysis on BoYkfC unless specified otherwise. BoYkfC has the same SH3b1-SH3b2-NlpC/P60 arrangement as DvLysin despite limited sequence similarity (RMSD of 2.7 Å for 247 aligned Cα atoms and 19% sequence identity) but has no c-clip domain (Fig. 1C). Also, BoYkfC is similar to BcYkfC (RMSD of 2.2 Å for 252 aligned Cα atoms and 28% sequence identity). The active-site cavity of BoYkfC, containing the catalytic cysteine (Cys203), is formed by residues that reside mostly on the catalytic domain (Fig. 1D).
Active site and inhibition.
Residues that are potentially important for catalysis, including the Cys-His-His triad and a nearby tyrosine, are strictly conserved in DvLysin and YkfCs. The arrangement of these active-site residues in BoYkfC is shown in Fig. 1E. Interestingly, the catalytic Cys203 of BoYkfC is covalently modified, by what we infer to be an acetonyl group, based on well-resolved density and the chemical environment. The Tyr191 hydroxyl forms a hydrogen bond with the carbonyl group of the acetonyl-cysteine. In contrast, the catalytic cysteine is not modified in DvLysin (see Fig. S3 in the supplemental material), while oxidized cysteine (crystal 1) and acetonyl-cysteine (crystal 2) are found in BtYkfC. Since the enzymes used for crystallization were active (i.e., the same protein stocks were used for the biochemical assays) and the types of the modification are influenced by the crystallization conditions, as observed in the BtYkfC crystals, we concluded that these modifications most likely occurred during crystallization.
The presence of S-acetonyl-cysteine in the BoYkfC structure raised the possibility that the enzyme could be inhibited by chloroacetone. Thus, we tested the inhibition of BoYkfC activity by various reagents that react with the thiol group (see Fig. S4 in the supplemental material). Indeed, BoYkfC activity was completely abolished in the presence of 2,4-dinitrothiocyanobenzene (DTNB), para-hydroxymercuribenzoate (pHMB), and chloroacetone. The activity was also greatly reduced but not totally abolished in the presence of 2-nitro-5-thiocyanobenzoic acid (NTCB) and was partially inhibited by N-ethylmaleimide (NEM). However, iodoacetamide did not markedly affect the enzyme activity. The inhibitory effect of DTNB can be reversed by reducing agents, such as 2-mercaptoethanol (see Fig. S4), consistent with the formation of a disulfide bond upon inhibition.
Model of substrate recognition.
The crystal structure of BcYkfC in complex with the dipeptide l-Ala-d-Glu (12) provided insights into recognition of the P2 and P1 moieties of the substrate (the Schechter and Berger nomenclature for peptidases [20] is used here; see Fig. S1 in the supplemental material). The S2 and S1 sites are highly conserved in DvLysin, BoYkfC, and BtYkfC, supporting a common mode of substrate recognition at the nonprime sites (see Fig. S3). Cryoprotectant molecules (glycerol or ethylene glycol) are found in many of the S2 subsites (see Fig. S3). In addition, for DvLysin, the S1, S1′, and S2′ subsites are occupied by acetate, glycerol, and 2-(N-morpholino)ethanesulfonic acid (MES) molecules, respectively (see Fig. S3). These solvent molecules bear similarity to components of the substrate (see Fig. S3). Thus, we were able to model a portion of PG [lactoyl-l-Ala-γ-d-Glu-A2pm(-d-Ala′)-d-Ala] into the active site of DvLysin, based on information gleaned from the BcYkfC-dipeptide complex structure and its relation to the bound solvent molecules. The BoYkfC-acetonyl-cysteine complex structure possibly resembles the transition state, and the bound solvent molecules may mimic substrate binding in DvLysin and, hence, were used as guides for placement of functional groups (see Fig. S5).
In our model (Fig. 2A), the substrate is complementary to the active site in both shape and electrostatic potential. DvLysin recognizes the tetrapeptide portion of the substrate through extensive hydrogen bonding interactions, as well as hydrophobic contacts for aliphatic portions of the substrate. l-Ala (P2) is sandwiched between Arg350 and Trp324, with its NH group forming a hydrogen bond with the carbonyl group of SH3b1 Arg183. d-Glu (P1) forms hydrogen bonds with Ser334, Ser352, and Tyr322. The main-chain NH and carbonyl groups of A2pm (P1′) each form a hydrogen bond, with the carbonyl group of Gly391 and the NH group of Trp411, respectively, while the carboxylate group of A2pm is stabilized by the Arg423 side chain. The carboxylate of d-Ala (P2′) is stabilized by Arg414 and Arg452, while its Cβ points to a hydrophobic hole formed by His392, Met410, Trp411, and Gly412. Moreover, two additional subsites can be identified for moieties extended from the tetrapeptide, the lactoyl group, and d-Ala′. The lactoyl group (P3) is located between Ala211 and Asn351, with its methyl group pointing toward Ala211 and its carbonyl group interacting with the Asn351 side chain, while d-Ala´ is stabilized by interactions with Gly173, Phe178, and Trp324.
FIG 2 .
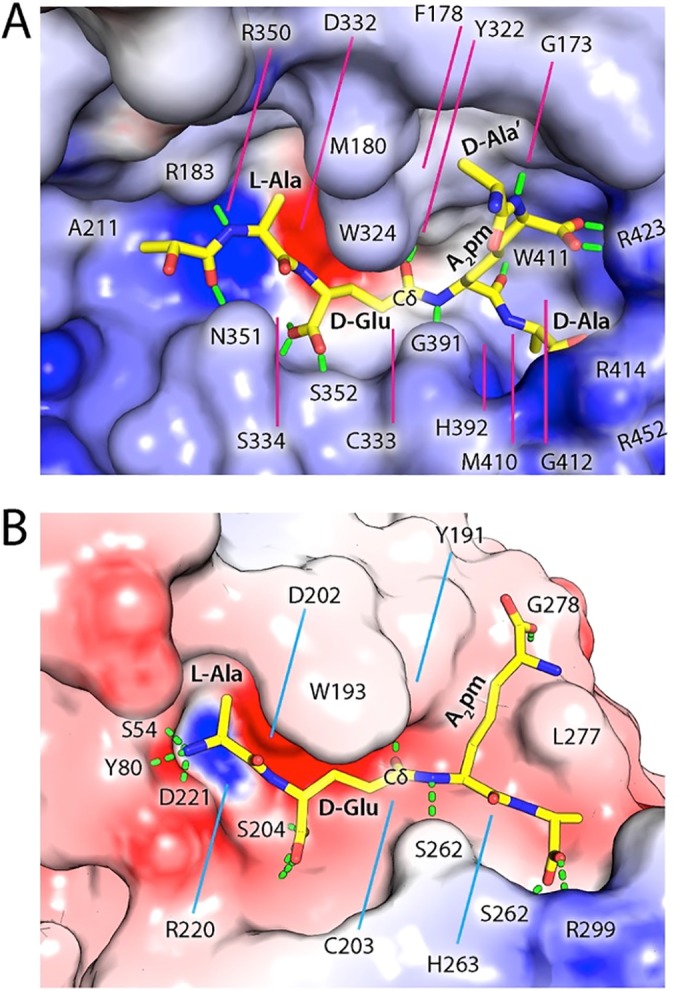
Models of substrate recognition by DvLysin and BoYkfC. (A) Interactions between the modeled substrate (shown as sticks) and the active site cavity of DvLysin. The molecular surface is colored by electrostatic potential, scaled from to −15 (red; negative) to 15 kT/e (blue; positive). Predicted hydrogen bonds between protein and substrate are shown as green dashes. Residues involved in binding the substrate are labeled. (B) A modeled tetrapeptide (sticks) in the active site of BoYkfC (colored by electrostatic potential using the same coloring scale described for panel A).
Our model is consistent with our experimental data described above. Arg423 is strategically located to recognize the carboxylate of A2pm (Fig. 2A) but not the positively charged NH3 group of a Lys. Thus, our model readily explains the dramatic reduction in enzymatic activity when A2pm in the substrate is replaced by Lys. Furthermore, the presence of d-Ala at S2′ allows increased interaction with Arg414 (Fig. 2A), explaining the better activity of DvLysin toward substrates with a tetrapeptide than to those with a tripeptide.
A tetrapeptide can be accommodated similarly by the active site of BoYkfC (Fig. 2B). It is interesting to note that the DvLysin substrate binding site has an overall positive electrostatic potential, in contrast to that of BoYkfC, which is more negative. Moreover, the binding groove of DvLysin is more restrictive than that of BoYkfC on the prime side. The more restricted prime subsites can explain the difference in the activities of DvLysin (weak activity) and BoYkfC (no activity) toward the γ-d-Glu-A2pm dipeptide. The longer RT loops in DvLysin and BcYkfC play an important role in forming the prime side substrate binding groove, thereby affecting substrate preference, whereas the short RT loops in AvPCP and BoYkfC correspond to more accessible prime subsites (see Fig. S6 in the supplemental material).
A conserved SH3b and NlpC/P60 domain interface.
We have now determined five structures containing an NlpC/P60 domain: the prototypical NlpC/P60 domain of the cell wall lysin CwlT (13), DvLysin, and 3 cell wall recycling enzymes, AvPCP (11), BoYkfC (12), and BcYkfC. Due to differences in domain architecture and limited sequence similarity (the pairwise sequence identity is typically ~20%), we analyzed their interrelationships by clustering analysis based on structure (Fig. 3; see also Fig. S7 in the supplemental material). The three proteins containing two SH3b domains (BoYkfC, BcYkfC, and DvLysin) are more similar to each other than to AvPCP (Fig. 3A). The first three proteins share similar SH3b1-SH3b2-NlpC/P60 interdomain configurations (average RMSD of 2.3 Å for 234 aligned Cα atoms), while all four proteins share similar SH3b1-NlpC/P60 interdomain configurations (average RMSD of 2.1 Å for 175 aligned Cα atoms) (Fig. 4B and C). Thus, AvPCP essentially represents a substructure of YkfC structures with a missing SH3b2 domain, while YkfC structures represent a substructure of the DvLysin structure with a missing c-clip domain (Fig. 3). Overall, a highly conserved two-domain core was identified that consists of SH3b1 and NlpC/P60 domains in both cell wall lysins and recycling enzymes, suggesting that these four SH3b-NlpC/P60 fused proteins may share an ancestor.
FIG 3 .

Structure comparisons of a prototypic NlpC/P60 domain and SH3b-NlpC/P60 fusion proteins (DvLysin, AvPCP, BoYkfC, and BcYkfC). (A) Tree representation of relationships based on structural similarity. (B) Superimposed structures. Equivalent residues are colored by domain (magenta, SH3b1; orange, SH3b2; green, NlpC/P60), while nonsuperimposed residues are shown in gray, except that the c-clip domain of DvLysin is in blue. The Cα atoms of catalytic cysteines are shown as green spheres. (C) Side-by-side comparison of the domain architecture of lysins and cell wall recycling enzymes.
FIG 4 .
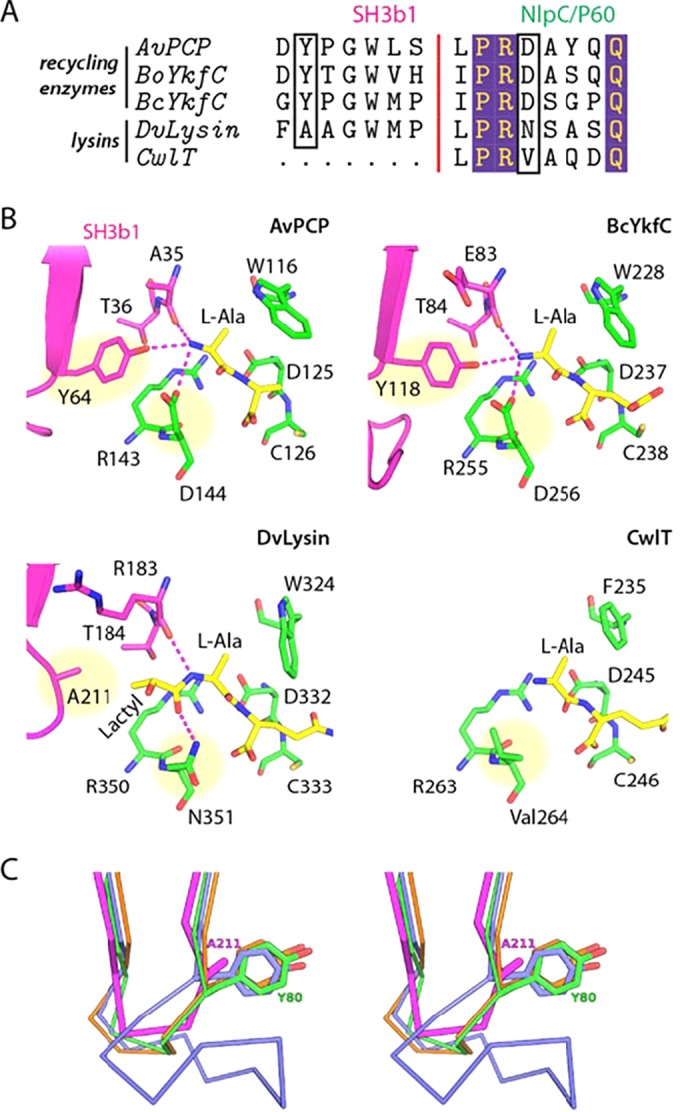
Residues at the SH3b1-NlpC/P60 domain interface affect substrate specificity in cell wall recycling enzymes and lysins. (A) Sequence alignment of the S3 and S2 subsites in CwlT, DvLysin, YkfCs, and AvPCP. The residues that we propose are the key determinants of substrate specificity for cell wall lysins and recycling enzymes are highlighted by black boxes. (B) Close-up views of the S3 and S2 subsites in lysins and recycling enzymes. Residues are colored by domain (magenta, SH3b1; green, NlpC/P60), while substrates (modeled or observed) are colored in yellow. The key residues indicated in panel A are highlighted by yellow circles. Hydrogen bonds are shown as dashed lines. (C) Comparison of the distal loops of DvLysin (magenta), BoYkfC (green), BcYkfC (blue), and AvPCP (orange), shown in stereoview. Side chains at the position equivalent to Ala211 of DvLysin are shown as sticks.
Differences at the SH3b1 and NlpC/P60 interface alter substrate specificity.
Given the presence of highly conserved active sites (Fig. 2) and a common core structure (Fig. 3), it is intriguing to explore how AvPCP, YkfC, and DvLysin achieve different substrate specificities. To answer this question, we examined the S3 (binds lactoyl acid group) and S2 (binds l-Ala) subsites (Fig. 4). The S2 sites of YkfC and AvPCP are identical, such that free amine groups are stabilized by three hydrogen bond interactions with a tyrosine hydroxyl (e.g., Tyr64 of AvPCP) and a main-chain carbonyl (e.g., Ala35 of AvPCP, Fig. 4), both of which are from the SH3b1 domain, plus an aspartate side chain from the NlpC/P60 domain (e.g., Asp144 of AvPCP). These residues are strictly conserved in both sequence and structure among cell wall recycling enzymes active only toward substrates with a free N-terminal l-Ala. However, these Tyr and Asp residues are not conserved in lysins. In DvLysin, an alanine takes the place of the tyrosine and an asparagine that of aspartate (Fig. 4A). In CwlT, a valine replaces aspartate, while there is no equivalent residue for the tyrosine. Modeling studies indicate that the tyrosine side chain would sterically clash with any additional nonhydrogen atoms that extend from the l-Ala, while the lactoyl group (and additional moieties) can be accommodated when the tyrosine (for DvLysin) (Fig. 2) or the entire SH3b1 (for CwlT) is absent. Thus, the tyrosine/aspartate pair in stem peptide recycling enzymes plays a dual role. First, they optimize the recognition of the positively charged N-terminal amine group of l-Ala. Second, the Tyr side chain functions as a steric barrier and prevents the binding of compounds containing a stem peptide as a substructure, such as PG or intermediates of PG biosynthesis. Thus, we have identified the local structural differences that are responsible for different substrate specificities in DvLysin, YkfC, and AvPCP.
Mutational analysis.
To confirm that Cys333 is indeed critical for catalysis, we created a C333A DvLysin mutant and tested its specific activity (Table 2). The C333A mutation greatly reduced the enzyme activity but did not totally abolish it, especially for free tetrapeptide or lactoyl-pentapeptide substrates.
TABLE 2 .
Specific activities of DvLysin and BoYkfC mutants
| Substrate | Sp act (nmol/min/mg) of WT or mutant forma: |
|||||
|---|---|---|---|---|---|---|
| DvLysin |
BoYkfC |
|||||
| WT | C333A | A211Y | WT | Y80A | Y80A D221N | |
| Peptidoglycan | >35 | NA | NA | NA | NA | NA |
| TCT | 5500 | 0.7 | 90 | NA | NA | 0.6 |
| MurNAc-tetrapeptide | 5400 | 0.7 | 60 | NA | NA | 1.1 |
| Lactoyl-pentapeptide | 2300 | 10 | 1100 | 1.5 | NA | NA |
| Tetrapeptide | 6100 | 30 | 4900 | 25,800 | 3500 | 3300 |
NA, no activity detected with up to 20 µg of protein under the assay conditions used.
Given the above observations that substrate specificity could be dictated by only a few interfacial residues (Fig. 4), we reasoned that it might be possible to modify the substrate specificity of these enzymes through site-directed mutagenesis. We first mutated Ala211 of DvLysin into a tyrosine at the equivalent position of the recycling enzymes (Fig. 4A). Indeed, this mutation greatly reduced the activity of DvLysin toward larger substrates (PG, GlcNAc-1,6-anhydro-MurNAc-l-Ala-γ-d-Glu-meso-A2pm-d-Ala [TCT], and MurNAc-tetrapeptide), while its activity for free tetrapeptide was the same as for the wild type (WT) (Table 2).
To check whether the more specific recycling enzymes can be turned into a lysin in a similar manner, we mutated the Tyr80 and Asp221 residues of BoYkfC into alanine and asparagine, respectively, as observed at the equivalent positions in DvLysin (Fig. 4A), to generate the Y80A mutant and the Y80A D221N double mutant (Table 2). The Y80A mutation significantly reduced the enzyme activity toward tetrapeptide compared to that of the WT, supporting its role in substrate binding. However, both mutants retained substrate preference similar to those of the WT, although the double mutant showed very weak activity toward larger substrates, TCT, and MurNAc-tetrapeptide.
DISCUSSION
Rational design of substrate specificity of a cell wall lysin.
In this study, we performed comparative biochemical and structural studies of a novel lysin (DvLysin) and several YkfC orthologs that have specific activity toward PG stem peptides with a free N-terminal l-Ala (Table 1 and Fig. 1). To our surprise, DvLysin has a very similar overall structure and active site to those of the YkfC proteins, despite significant sequence divergence (Fig. 2 and 3). Furthermore, we identified only a few local differences in the active sites that appear to play a significant role in substrate specificity (Fig. 4), raising the possibility of redesigning the enzyme substrate specificity with site-directed mutagenesis. For DvLysin, the idea was to introduce bulky side chains to disrupt the upstream binding sites, as observed in the recycling enzymes. Remarkably, DvLysin was converted into a specific and efficient cell wall recycling enzyme with a single mutation (A211Y) (Table 2).
Similarly, one might expect that we could also turn the YkfC enzymes into lysins by creating space at the upstream sites. However, we were unable to alter the substrate specificity using limited site-directed mutagenesis of BoYkfC (Y80A and Y80A/D221N) (Table 2). The results suggest that BoYkfC may have evolved additional structural features to safeguard its stricter substrate preference. For example, the distal loops of SHb1 of the recycling enzymes are generally longer than that of DvLysin (Fig. 4C; see also Fig. S6 in the supplemental material), resulting in a more crowded constellation of atoms proximal to the S2 site. Moreover, the electrostatic potential of the active site of DvLysin differs significantly from those of YkfC homologs (Fig. 2 and 5B).
SH3b1 and c-clip domains may extend the PG recognition surface of DvLysin.
D. vulgaris is ubiquitous in nature and an opportunistic pathogen. DvLysin orthologs are widely distributed in other pathogens, for example, Helicobacter pylori (accession number HP0087), Salmonella enterica (STM1940), and Xanthomonas campestris (XCC3806). The active site of DvLysin is highly conserved among its orthologs. The S2 and S1 subsites for binding l-Ala and d-Glu consist of several conserved regions with motifs RDC333S, GH392, Y322GWG, and PR350NS (Fig. 5A). DvLysin orthologs also preserve Trp411 and Arg423 in the long β4-β5 hairpins that are involved in the binding of A2pm. Two other conserved regions, R87XFXPW from the c-clip domain and G173EGYPFD from the SH3b1 domain, are located near the domain interfaces. These regions are likely important for structural integrity and may also provide extended binding surfaces for PG due to their proximity to the active site (Fig. 5A). A shallow surface groove with positive electrostatic potential, located between α-helix D of the c-clip domain and the NlpC/P60 domain, extends on the SH3b1 surface from α-helix C (at Arg87) toward the S2 site (Fig. 5B), while Gly173 is adjacent to the S1′ site (Fig. 2A). These arrangements suggest that SH3b1 could contribute to binding other moieties on PG, such as the lactoyl group and the cross-linked d-Ala′ (Fig. 2A).
FIG 5 .

SH3b1 extends the putative substrate recognition surface of DvLysin. (A) Sequence motifs (conserved among DvLysin orthologs) at or near the active site. The domains are colored as in Fig. 1, and conserved residues or regions are highlighted in lavender and annotated with the corresponding consensus sequence motifs. (B) A surface groove at the junction of the c-clip, SH3b1, and NlpC/P60 domain with positive electrostatic potential (same scale and coloring scheme as in Fig. 2) may extend nonprime binding sites. Domain boundaries are outlined by dashed lines. (C) Comparison of the NlpC/P60 domain in DvLysin (decorated by SH3b and c-clip domains) and the NlpC/P60 domain of CwlT shows that auxiliary domains alter the chemical and structural environments at the perimeter of the substrate recognition sites. The NlpC/P60 domains are shown as molecular surfaces (green). The modeled substrates are shown as sticks (yellow, red, blue), while residues from the SH3b1 and c-clip domains are shown as ribbons and sticks. NAM, N-acetylmuramic acid.
It is also informative to examine how SH3b1 may affect PG binding in the context of single-domain NlpC/P60 cell wall lysin, such as that of CwlT (Fig. 5C). CwlT has an open-ended active site groove that narrows at the top of the catalytic center. PG can be accommodated in a manner similar that of DvLysin, but specific recognition of the PG is mainly achieved in the middle part of the groove. In contrast, both ends of the substrate binding site in DvLysin are more well defined due to the contribution of SH3b1. In addition, the glycan orientation and binding could be affected by SH3b1. Thus, the SH3b1 domain could result in more specific recognition of the cross-linked stem peptide and enhance affinity toward PG by providing a more extended recognition surface.
Evolution of SH3b domains and SH3b-NlpC/P60 fusion proteins.
Although the SH3b domains studied here are extremely diverse in sequence (<10% pairwise sequence identity), they all share a core consisting of seven β-strands (average RMSD of 2.1 Å for 54 aligned Cα atoms) (see Fig. S8 in the supplemental material). Structurally, SH3b1 domains and SH3b2 domains can be clustered into two separate groups. SH3b1 domains share more conserved sequence features, such as the GW motif located on strand βD (see Fig. S8). Profile-based sequence similarity analysis (21) suggests that bacterial and eukaryotic SH3 domains are evolutionarily related. Here, we provide evidence that the RT loop and distal loop of SH3b1 could play an important role in defining substrate specificity. Interestingly, the RT loop regions of eukaryotic SH3 domains are also important for recognition of proline-rich peptides (22). However, the protein surfaces utilized by SH3b1 differ from those of eukaryotic SH3 domains, such as Abl-SH3 (PDB accession number 2JMA) (23). SH3b2 domains are not needed for the catalytic function, as they are distal to the active sites. By interacting with both SH3b1 and NlpC/P60, they could provide additional support to maintain SH3b1-NlpC/P60 interdomain conformation. However, the same function could be accomplished by other structural adaptions, such as the c-clip domain in DvLysin or C-terminal appendages in AvPCP and BoYkfC that extend from NlpC/P60 to interact with SH3b1.
Our studies of NlpC/P60 proteins have uncovered the core structural features of a prototypical NlpC/P60 domain and a series of SH3b-NlpC/P60 fusion proteins. Unexpectedly, these SH3b-containing proteins all share a two-domain core (SH3b1-NlpC/P60), despite differences in their domain architectures and substrate specificities (Fig. 3). Conserved sequence features in the core suggest that DvLysin, YkfC, and AvPCP share an evolutionary origin, with each structure representing a snapshot in the evolution of SH3b-containing NlpC/P60 proteins from cell wall lysins to more specialized enzymes. Although accurate reconstruction of the evolutionary history of these related proteins is difficult given the low sequence similarity, a plausible evolutionary path could be proposed based on our analysis (Fig. 6A). DvLysin could have evolved from gene fusion between SH3b domains and the NlpC/P60 catalytic domain. SH3b1 and SH3b2 domains have likely originated from gene duplication, although it is hard to say whether this event happened pre- or postfusion. YkfC-like cell wall recycling enzymes could have evolved from a DvLysin-like ancestor with local changes to give rise to substrate specificity toward stem peptides, while AvPCP could have evolved from a YkfC-like ancestor by shedding of the SH3b2 domain. Indeed, our mutagenesis data suggest that it is relatively straightforward to evolve a more specific enzyme from the lysin scaffold.
FIG 6 .

Protein evolution and PG recycling pathways. (A) Proposed evolutionary pathway of the NlpC/P60 cell wall lysins and recycling enzymes. (B) Proposed pathway for PG recycling in B. thetaiotaomicron (left) in comparison with that of E. coli (right). The two NlpC/P60 proteins (AmiA and YkfC) in B. thetaiotaomicron (blue) are cysteine amidases, while the corresponding E. coli proteins (AmpD and MpaA) are both zinc amidases.
NlpC/P60 amidases in cell wall recycling.
The study of PG fragments released by the human gut microbiome is of great biological interest since PG fragments can mediate a range of microbe-host interactions (24, 25). Here, we demonstrate that YkfC orthologs are γ-d-Glu-A2pm (or Lys) amidases with specificity for stem peptides with a free N-terminal l-Ala. Although the physiological role of these enzymes is not yet known, it is likely that they are involved in cell wall recycling, as they are functionally equivalent to the E. coli MpaA zinc amidase and have the same substrate specificity (26, 27). Interestingly, we recently identified another NlpC/P60-related N-acetylmuramoyl-l-alanine amidase, AmiA, from Bacteroides uniformis that hydrolyzes GlcNAc-1,6-anhydro-MurNAc-peptide, a major cell wall turnover product produced by lytic transglycosylases, into GlcNAc-1,6-anhydro-MurNAc and stem peptide (28). Thus, AmiA can produce stem peptide substrates for YkfC. In contrast, the substrate specificity of AmiA arises from internal insertions that remodel the binding pocket at nonprime sites (28). These results suggest that NlpC/P60 proteins may play a significant role in cell wall recycling.
Both AmiA and YkfC orthologs are commonly found in Bacteroides species. Their strict specificity toward only peptidoglycan degradation products and the sequential processing of the substrate suggest that they work together in a common enzymatic pathway. Here, we postulate the existence of such a pathway for cell wall recycling in B. thetaiotaomicron (Fig. 6B), a model bacterium that is used to study microbiome-host interactions (29) and which does not contain a recognizable MpaA ortholog. In this proposed pathway, GlcNAc-1,6-anhydro-MurNAc-tetrapeptide in the periplasm is cleaved by the C-terminal AmiA amidase domain of the putative BT1087 protein to generate GlcNAc-1,6-anhydro-MurNAc and tetrapeptide. The C-terminal d-Ala of the tetrapeptide would then be removed by the N-terminal α/β hydrolase domain of BT1087 to produce tripeptide, which would be further cleaved by the BtYkfC amidase (BT1314) to remove A2pm. The dipeptide l-Ala-d-Glu would then be subsequently converted by the recently characterized YkfB-like epimerase (BT1313) to l-Ala-l-Glu (30), which can then be imported into the cytoplasm by the putative oligopeptide permease, BT1086, for further processing. In comparison with the E. coli PG recycling pathway, although the intermediates are similar, none of the functionally equivalent enzymes are evolutionarily related, excepted for the epimerase YkfB, which suggests parallel evolution of these pathways. Further experimental evidence, such as in vivo assays in relevant bacteria, is needed to confirm or refute our hypotheses.
MATERIALS AND METHODS
Cloning and protein production.
Clones were generated using the polymerase incomplete primer extension (PIPE) cloning method (31). The genes encoding BoYkfC (GenBank accession number ZP_02063661.1) and BtYkfC (GenBank accession number NP_810227.1) were amplified by PCR from genomic DNA from Bacteroides ovatus or Bacteroides thetaiotaomicron using PfuTurbo DNA polymerase (Stratagene) and I-PIPE (insert) primers (BoYkfC forward primer, 5′ ctgtacttccagggcCAGGAAATACGTCCCATGCCTGCCG 3′; BoYkfC reverse primer, 5′ aattaagtcgcgttaTTGATAAAAGGGGTTCTTATTTGTGG 3′; BtYkfC forward primer, 5′ ctgtacttccagggcCAGGAAATACGCCCCATGCCTGCTG 3′; and BtYkfC reverse primer, 5′ aattaagtcgcgttaGTGATGTAGATAATAGAGGTTATGGTCG 3′ [target sequences are in uppercase letters]) that included sequences for the predicted 5′ and 3′ ends. The expression vector, pSpeedET, which encodes an amino-terminal tobacco etch virus (TEV) protease-cleavable expression and purification tag (MGSDKIHHHHHHENLYFQ/G), was PCR amplified with V-PIPE (vector) primers (forward primer, 5′ TAACGCGACTTAATTAACTCGTTTAAACGGTCTCCAGC 3′, and reverse primer, 5′ GCCCTGGAAGTACAGGTTTTCGTGATGATGATGATGATG 3′). V-PIPE and I-PIPE PCR products were mixed to anneal the amplified DNA fragments together. Escherichia coli GeneHogs (Invitrogen) competent cells were transformed with the I-PIPE/V-PIPE mixture and dispensed on selective LB agar plates. The cloning junctions were confirmed by DNA sequencing. Using the PIPE method, the gene segment encoding the predicted signal peptide (residues 1 to 21 based on SignalP [32]) was deleted from both the BoYkfC and the BtYkfC expression construct. Expression was performed in a selenomethionine-containing medium at 37°C. Selenomethionine was incorporated via inhibition of methionine biosynthesis (33), which does not require a methionine-auxotrophic strain. At the end of fermentation, lysozyme was added to the culture to a final concentration of 250 µg/ml, and the cells were harvested and frozen. After one freeze-thaw cycle, the cells were homogenized and sonicated in lysis buffer [40 mM Tris, 300 mM NaCl, 10 mM imidazole, 1 mM Tris(2-carboxyethyl)phosphine-HCl (TCEP), pH 8.0]. The remaining nucleic acids were digested with the addition of 0.4 mM magnesium sulfate and 1 µl of 250 U/µl Benzonase nuclease (Sigma) in the lysate. The lysate was clarified by centrifugation at 32,500 × g for 25 min. The soluble fraction was passed over nickel-chelating resin (GE Healthcare) preequilibrated with lysis buffer, the resin was washed with wash buffer (40 mM Tris, 300 mM NaCl, 40 mM imidazole, 10% [vol/vol] glycerol, 1 mM TCEP, pH 8.0), and the protein was eluted with elution buffer (20 mM Tris, 300 mM imidazole, 10% [vol/vol] glycerol, 150 mM NaCl, 1 mM TCEP, pH 8.0). The eluate was buffer exchanged with TEV buffer (20 mM Tris, 150 mM NaCl, 30 mM imidazole, 1 mM TCEP, pH 8.0) using a PD-10 column (GE Healthcare) and incubated with 1 mg of TEV protease per 15 mg of eluted protein for 2 h at room temperature and then overnight at 4°C. The protease-treated eluate was passed over nickel-chelating resin (GE Healthcare) preequilibrated with crystallization buffer (20 mM Tris, 150 mM NaCl, 30 mM imidazole, 1 mM TCEP, pH 8.0), and the resin was washed with the same buffer. The flowthrough and wash fractions were combined and concentrated to 21 mg/ml for BoYkfC and 14.1 mg/ml for BtYkfC by centrifugal ultrafiltration (Millipore) for biochemical assays and crystallization trials.
The DvLysin gene (GenBank accession number YP_010117.1; residues 32 to 464) was cloned using the PIPE cloning method (forward primer, 5′ ctgtacttccagggcTCCCGTCCTGCGACTCCGCCCGTCACTC 3′, and reverse primer, 5′ aattaagtcgcgttaTTGCGCCCCGCCGGGAAGGATGCTCATG 3′ [target sequences are in uppercase letters]) and expressed as described above, and the protein was purified as reported previously for BoYkfC (12). The purified protein was concentrated to 15.3 mg/ml.
Production of mutant proteins.
Mutagenesis was carried out by Genewiz, Inc. (La Jolla, CA), and all mutations were confirmed by DNA sequencing. The mutant proteins were expressed and purified as described above for the WT BoYkfC.
γ-d-Glu-meso-A2pm amidase activity assay.
The activities of DvLysin and YkfC proteins toward various PG-related compounds were tested in a 50 µl reaction mixture containing 50 mM Tris-HCl, pH 7.8, 2.5 mM MgCl2, 0.2 mM substrate, and pure enzyme. One microliter amounts of the undiluted protein stocks used for structure determination (i.e., 15 to 20 µg of protein) were used first in preliminary assays aiming at identifying substrates of these enzymes, and specific activities were then precisely determined using appropriate enzyme dilutions performed in 20 mM potassium phosphate buffer, pH 7.4. The mixtures were incubated for 30 min at 37°C, and reactions were stopped by flash freezing in liquid nitrogen. Substrate and reaction products were separated by high-performance liquid chromatography (HPLC) as follows. When peptides were used as the substrates, an ODS-Hypersil 3 µm particle-size C18 column (250 by 4.6 mm; Thermo Scientific) was utilized and elution was with 0.05% trifluoroacetic acid (TFA), supplemented with 5% methanol when required, at a flow rate of 0.6 ml/min. For the other substrates, a Nucleosyl 100 5 µm particle size C18 column (250 by 4.6 mm; Alltech France) was used and elution was with 50 mM sodium phosphate, pH 4.5, with or without the application of a linear gradient of methanol (MeOH; from 0 to 25%) between 0 and 40 min, at a flow rate of 0.6 ml/min. Peaks were detected by measuring the absorbance at 262 nm (nucleotide PG precursors) or 207 nm (other compounds). The retention times of substrates and products observed under these HPLC conditions are reported in Table S2 in the supplemental material. Compounds were identified based on their retention times compared to standards and on their amino acid and amino sugar composition as determined with a Hitachi model L8800 analyzer (ScienceTec, Les Ulis, France) after hydrolysis of samples in 6 M HCl at 95°C for 16 h. Some samples were also further characterized by matrix-assisted laser desorption ionization time of flight (MALDI-TOF) mass spectrometry.
Peptidoglycan precursors and muropeptides.
Peptidoglycan (PG) was purified from an E. coli Δlpp mutant strain that does not express the Lpp lipoprotein (34). GlcNAc-1,6-anhydro-MurNAc-l-Ala-γ-d-Glu-meso-A2pm-d-Ala (TCT) and its dimer (two cross-linked TCT monomers) were produced by digestion of PG with E. coli SltY lytic transglycosylase, and the nonanhydro GlcNAc-MurNAc-tetrapeptide and its dimer were generated by treatment of PG with mutanolysin (35). The different UDP-MurNAc peptides were prepared as described previously (36), and their MurNAc peptides derivatives were obtained by mild acid hydrolysis (0.1 M HCl at 100°C for 30 min) (37). Lactoyl-pentapeptide and free pentapeptide were produced by treating MurNAc-l-Ala-γ-d-Glu-meso-A2pm-d-Ala-d-Ala with 4 M ammonium hydroxide (35) and E. coli AmiD N-acetylmuramoyl-l-alanine amidase (38), respectively. The l-Lys-containing pentapeptide l-Ala-γ-d-Glu-l-Lys-d-Ala-d-Ala was obtained from Bachem (Bubendorf, Switzerland). The tetrapeptide l-Ala-γ-d-Glu-meso-A2pm-d-Ala and tripeptide l-Ala-γ-d-Glu-meso-A2pm were produced sequentially by treatments of pentapeptide with purified penicillin-binding protein 5 (PBP5) d,d-carboxypeptidase and LdcA l,d-carboxypeptidase, respectively (39). The same procedure was followed to generate the l-Lys-containing tetrapeptide and tripeptide. All of these compounds were purified by HPLC, and their composition was verified by amino acid and amino sugar content analysis and/or by MALDI-TOF mass spectrometry.
BoYkfC activity inhibition.
BoYkfC (2,500-fold dilution from 21 mg/ml stock) was preincubated for 5 min at 37°C with various reagents at a 60 µM final concentration in 20 mM potassium phosphate buffer, pH 7.4. Then, 2 µl aliquots of these mixtures were added to 50 µl standard assay reaction mixtures containing 0.1 mM of tetrapeptide as the substrate. After 30 min of incubation at 37°C, reactions were stopped and substrates and products were separated by HPLC as described above. Assays that used different preincubation times and/or concentrations of reagent were similarly performed.
Crystallization and diffraction screening.
All proteins studied here were crystallized using the nanodroplet vapor diffusion method (40) with standard JCSG crystallization protocols (17). Crystallization trials were performed with 200 nl protein solution mixed with 200 nl crystallization solution in a sitting-drop format and equilibrated against a 50 µl reservoir at 277 K. DvLysin crystals were grown in mother liquor consisting of 1.0 M LiCl, 20.0% polyethylene glycol (PEG) 6000, and 0.1 M MES pH 6.0. Glycerol was added to a final concentration of 20% (vol/vol) as a cryoprotectant. The crystallization reagent for BoYkfC consisted of 15% glycerol, 0.17 M sodium acetate, 25.5% PEG 4000, and 0.1 M Tris, pH 8.5, and for BtYkfC, it was 20% glycerol, 40 mM KH2PO4, and 16% PEG 8000 (crystal 1) or 15% glycerol, 8.5% isopropanol, 17% PEG 4000, 0.1 M HEPES, pH 7.5 (crystal 2). Initial screening for diffraction was carried out using the Stanford Automated Mounting system (SAM) (41) at the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA).
Data collection, structure solution, and refinement.
SAD or MAD data were collected at SSRL or Advanced Light Source (ALS) beamlines (see Table S1 in the supplemental material). Data processing and structure solution were carried out using automated structure determination protocols developed at JCSG (42). The data were processed using XDS (43). Each structure was determined independently using the SAD or MAD method, except for the second crystal of BtYkfC, which was determined by molecular replacement. The location of selenium sites, initial phasing, and identification of the space group were carried out using SHELXD (44). Phase refinement and initial model building were performed using autoSHARP (45) and ARP/wARP (46). Model completion and refinement were iteratively performed using COOT (47) and REFMAC5 (48) or BUSTER (49). The refinement included experimental phase restraints in the form of Hendrickson-Lattman coefficients, TLS (translation, libration, and screw-rotation) refinement with one TLS group per molecule in the ASU (asymmetric unit), and NCS (noncrystallographic symmetry) restraints. Structural comparison and clustering analysis were performed using Matt (50). Molecular graphics were prepared using PyMOL (Schrödinger LLC, USA), and electrostatic potential was calculated using Delphi (51).
Molecular modeling.
Initial conformations of various ligands in random orientations and their restraints were generated using JLigand (52) from the CCP4 package (53). Since small molecules at different subsites of DvLysin resembled various functional groups of the substrate, an initial pose of the substrate was obtained manually in COOT by fitting the parts of the substrate to the corresponding electron density of these fragments (see Fig. S5 in the supplemental material). Docking of substrates for other proteins was performed using Glide (Schrödinger LLC, USA), and the solutions were selected by visual inspection to satisfy known geometric and chemical restraints. All final poses were energy minimized to optimize local interactions.
Protein structure accession numbers.
The structure factors and atomic coordinates are deposited in the RCSB Protein Data Bank (http://www.rcsb.org) with PDB accession numbers 3M1U (DvLysin), 3NPF (BoYkfC), 3PVQ (BtYkfC crystal 1), and 4R0K (BtYkfC crystal 2).
SUPPLEMENTAL MATERIAL
Schematic representation of A2pm-type bacterial peptidoglycan. The sites cleaved by d-Glu-A2pm hydrolases are marked in red. Borrowing the terminology for peptidase (see reference 20), the residues of the substrate at two sides of the cleavage site (scissile bond) are numbered P3, P2, P1 (nonprime side) to P1′, P2′ (prime side), respectively, while the corresponding binding pocket of the protein is numbered S3, S2, S1, S1′, and S2′. Download
γ-d-Glu-A2pm amidase activity of DvLysin. GlcNAc-1,6-anhydro-MurNAc-tetrapeptide (TCT, a), TCT dimer (b), and peptidoglycan (c) were incubated with (continuous lines) or without (dotted lines) purified DvLysin, and reaction mixtures were analyzed by HPLC as described in the text. TCT and TCT dimer were cleaved into two products identified as GlcNAc-anhydro-MurNAc-l-Ala-d-Glu (TCT-dip) and either the dipeptide A2pm-d-Ala or a tetrapeptide consisting of two cross-linked A2pm-d-Ala, respectively, consistent with a γ-d-Glu-A2pm amidase activity of this enzyme. The latter A2pm- and d-Ala-containing peptides were also released following digestion of the PG polymer by the enzyme. Download
Active sites of DvLysin and YkfC orthologs (BoYkfC, BtYkfC, and BcYkfC). (A) Stereoview of the DvLysin active site. Protein residues (green, red, blue) and ligands (cyan, red, blue) are shown as sticks. Experimental density-modified (DM) map (prior to model building, light-blue meshes) for the ligands is contoured at 1.0 σ. Hydrogen bonds are shown as dashed lines. (B) Active site of BoYkfC (experimental DM map shown for ligands). (C) Active site of BtYkfC crystal 1 (experimental DM map for ligands). (D) Active site of BtYkfC crystal 2 (2Fo-Fc omit map). (E) Active site of BcYkfC in complex with l-Ala-d-Glu dipeptide (PDB accession number 3H41). Download
Inhibition studies of BoYkfC by thiol-reactive labeling reagents. (A) Inhibition of BoYkfC by various thiol reagents. (B) Effects of time and concentration of DTNB on inhibition. (C) Inhibition effect of DTNB can be reversed by 2-mercaptoethanol. Download
Modeling of substrate into the DvLysin active site. (A) Stereoview of the DvLysin active site (green) with the modeled substrate (shown as magenta colored thick sticks), superimposed with the dipeptide complex of BcYkfC (thin sticks) (PDB accession number 3H41). The region that binds the l-Ala-d-Glu dipeptide portion of the substrate in DvLysin adopts a conformation similar to that of BcYkfC (thin gray sticks) since the S2 and S1 subsites are highly conserved in the two proteins. (B) Stereoview of the DvLysin active site (green) and the modeled substrate, superimposed with S-acetonyl-cysteine-inhibited BoYkfC (thin sticks) (PDB accession number 3NPF) that may mimic the transition state. (C) The modeled substrate matches well with solvent moieties (their final 2Fo-Fc densities are shown as contours at 1 σ) that resemble parts of the substrate. For example, the sulfonic acid group of MES and the acetate resemble the carboxylate groups of the substrate, while glycerols occupy subsites for binding l-Ala (S2) or d-Ala (S2′). Download
Distal and RT loops of SH3b1 (pink) are involved in the formation of the substrate binding groove in SH3b-NlpC/P60 fusion proteins. The distal loop is located upstream from the S2 subsite, while the RT loop is located near the S1′ subsite. Each protein is colored by domain (magenta, SH3b1; orange, SH3b2; green, NlpC/P60; blue, c-clip). Modeled substrates are show as sticks. Download
Structure-based sequence alignment of CwlT NlpC/P60 domain (PDB accession number 4FDY), AvPCP (PDB accession number 2HBW), BoYkfC (PDB accession number 3NPF), BcYkfC (PDB accession number 3H41), and DvLysin (PDB accession number 3M1U). SH3b1, SH3b2, and NlpC/P60 domains are marked by magenta, orange, and green boxes, respectively. The catalytic triad (Cys-His-His) and a nearby Tyr are marked by red circles. Download
Structure comparison of SH3b1 and SH3b2 domains. (A) Stereoview of seven superimposed SH3b domains in AvPCP, BcYkfC, BoYkfC, and DvLysin. The equivalent residues are colored red (SH3b1) and green (SH3b2). β-Strands are labeled A, A1, A2, and B to E. (B) Clustering of SH3b domains by pairwise structural comparison. (C) Structure-based sequence alignment of SH3b domains. Residues that are strictly conserved only in SH3b1 are highlighted in red boxes, while highly conserved residues in both SH3b1 and SH3b2 are colored white on a blue background. Download
Crystallization, data collection, and refinement statistics.
HPLC retention times of compounds tested in this work.
ACKNOWLEDGMENTS
We thank the members of the JCSG high-throughput structural biology pipeline for their contribution to this work.
This work was supported by the NIH, National Institute of General Medical Sciences (NIGMS), Protein Structure Initiative (grant U54 GM094586), CNRS, and Université Paris-Sud.
Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including grant P41GM103393). Genomic DNAs from Desulfovibrio vulgaris Hildenborough (ATCC 29579D), Bacteroides ovatus (extracted from cells of ATCC 8483) and Bacteroides thetaiotaomicron VPI-5482 (ATCC 29148D) were obtained from the American Type Culture Collection (ATCC). I.A.W. is the principal investigator of the JCSG.
Q.X. and D.M.-L. conceived and designed the experiments. Q.X., D.M.-L., and X.W.L. performed the experiments. Q.X. and D.M.-L. analyzed the data. D.P., C.L.F., J.C.G., H.-J.C., L.J., and M.W.K. contributed reagents/materials/analysis tools. Q.X., D.M.-L., M.-A.E., A.M.D., and I.A.W. wrote the paper. A.G., S.A.L., M.-A.E., and A.M.D. supervised various sections of the JCSG structural genomics pipeline.
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Citation Xu Q, Mengin-Lecreulx D, Liu XW, Patin D, Farr CL, Grant JC, Chiu H-J, Jaroszewski L, Knuth MW, Godzik A, Lesley SA, Elsliger M-A, Deacon AM, Wilson IA. 2015. Insights into substrate specificity of NlpC/P60 cell wall hydrolases containing bacterial SH3 domains. mBio 6(5):e02327-14. doi:10.1128/mBio.02327-14.
Contributor Information
Thomas Bernhardt, Harvard Medical School.
Gerald B. Pier, Harvard Medical School.
REFERENCES
- 1.Vollmer W, Blanot D, de Pedro MA. 2008. Peptidoglycan structure and architecture. FEMS Microbiol Rev 32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 2.Vollmer W, Joris B, Charlier P, Foster S. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev 32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 3.Van Heijenoort J. 2011. Peptidoglycan hydrolases of Escherichia coli. Microbiol Mol Biol Rev 75:636–663. doi: 10.1128/MMBR.00022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reith J, Mayer C. 2011. Peptidoglycan turnover and recycling in Gram-positive bacteria. Appl Microbiol Biotechnol 92:1–11. doi: 10.1007/s00253-011-3486-x. [DOI] [PubMed] [Google Scholar]
- 5.Johnson JW, Fisher JF, Mobashery S. 2013. Bacterial cell-wall recycling. Ann N Y Acad Sci 1277:54–75. doi: 10.1111/j.1749-6632.2012.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodell EW. 1985. Recycling of murein by Escherichia coli. J Bacteriol 163:305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JT, Uehara T. 2008. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol Mol Biol Rev 72:211–227. doi: 10.1128/MMBR.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobs C, Huang LJ, Bartowsky E, Normark S, Park JT. 1994. Bacterial cell wall recycling provides cytosolic muropeptides as effectors for β-lactamase induction. EMBO J 13:4684–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anantharaman V, Aravind L. 2003. Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol 4:R11. doi: 10.1186/gb-2003-4-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aramini JM, Rossi P, Huang YJ, Zhao L, Jiang M, Maglaqui M, Xiao R, Locke J, Nair R, Rost B, Acton TB, Inouye M, Montelione GT. 2008. Solution NMR structure of the NlpC/P60 domain of lipoprotein Spr from Escherichia coli: structural evidence for a novel cysteine peptidase catalytic triad. Biochemistry 47:9715–9717. doi: 10.1021/bi8010779. [DOI] [PubMed] [Google Scholar]
- 11.Xu Q, Sudek S, McMullan D, Miller MD, Geierstanger B, Jones DH, Krishna SS, Spraggon G, Bursalay B, Abdubek P, Acosta C, Ambing E, Astakhova T, Axelrod HL, Carlton D, Caruthers J, Chiu HJ, Clayton T, Deller MC, Duan L, Elias Y, Elsliger MA, Feuerhelm J, Grzechnik SK, Hale J, Han GW, Haugen J, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Kumar A, Marciano D, Morse AT, Nigoghossian E, Okach L, Oommachen S, Paulsen J, Reyes R, Rife CL, Trout CV, van den Bedem H, Weekes D, White A, Wolf G, Zubieta C, Hodgson KO, Wooley J, Deacon AM, Godzik A, Lesley SA, Wilson IA. 2009. Structural basis of murein peptide specificity of a γ-d-glutamyl-l-diamino acid endopeptidase. Structure 17:303–313. doi: 10.1016/j.str.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Q, Abdubek P, Astakhova T, Axelrod HL, Bakolitsa C, Cai X, Carlton D, Chen C, Chiu H, Chiu M, Clayton T, Das D, Deller MC, Duan L, Ellrott K, Farr CL, Feuerhelm J, Grant JC, Grzechnik A, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Krishna SS, Kumar A, Lam WW, Marciano D, Miller MD, Morse AT, Nigoghossian E, Nopakun A, Okach L, Puckett C, Reyes R, Tien HJ, Trame CB, van den Bedem H, Weekes D, Wooten T, Yeh A, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wilson IA. 2010. Structure of the γ-d-glutamyl-l-diamino acid endopeptidase YkfC from Bacillus cereus in complex with l-Ala-γ-d-Glu: insights into substrate recognition by NlpC/P60 cysteine peptidases. Acta Crystallogr Sect F Struct Biol Cryst Commun 66:1354–1364. doi: 10.1107/S1744309110021214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Q, Chiu HJ, Farr CL, Jaroszewski L, Knuth MW, Miller MD, Lesley SA, Godzik A, Elsliger MA, Deacon AM, Wilson IA. 2014. Structures of a bifunctional cell wall hydrolase CwlT containing a novel bacterial lysozyme and an NlpC/P60 DL-endopeptidase. J Mol Biol 426:169–184. doi: 10.1016/j.jmb.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukushima T, Kitajima T, Yamaguchi H, Ouyang Q, Furuhata K, Yamamoto H, Shida T, Sekiguchi J. 2008. Identification and characterization of novel cell wall hydrolase CwlT: a two-domain autolysin exhibiting N-acetylmuramidase and DL-endopeptidase activities. J Biol Chem 283:11117–11125. doi: 10.1074/jbc.M706626200. [DOI] [PubMed] [Google Scholar]
- 15.Whisstock JC, Lesk AM. 1999. SH3 domains in prokaryotes. Trends Biochem Sci 24:132–133. doi: 10.1016/S0968-0004(99)01366-3. [DOI] [PubMed] [Google Scholar]
- 16.Eldholm V, Johnsborg O, Straume D, Ohnstad HS, Berg KH, Hermoso JA, Håvarstein LS. 2010. Pneumococcal CbpD is a murein hydrolase that requires a dual cell envelope binding specificity to kill target cells during fratricide. Mol Microbiol 76:905–917. doi: 10.1111/j.1365-2958.2010.07143.x. [DOI] [PubMed] [Google Scholar]
- 17.Lesley SA, Kuhn P, Godzik A, Deacon AM, Mathews I, Kreusch A, Spraggon G, Klock HE, McMullan D, Shin T, Vincent J, Robb A, Brinen LS, Miller MD, McPhillips TM, Miller MA, Scheibe D, Canaves JM, Guda C, Jaroszewski L, Selby TL, Elsliger MA, Wooley J, Taylor SS, Hodgson KO, Wilson IA, Schultz PG, Stevens RC. 2002. Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc Natl Acad Sci U S A 99:11664–11669. doi: 10.1073/pnas.142413399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wooley J, Wüthrich K, Wilson IA. 2010. The JCSG high-throughput structural biology pipeline. Acta Crystallogr Sect F Struct Biol Cryst Commun 66:1137–1142. doi: 10.1107/S1744309110038212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schechter I, Berger A. 1968. On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem Biophys Res Commun 32:898–902. doi: 10.1016/0006-291X(68)90326-4. [DOI] [PubMed] [Google Scholar]
- 21.Kippert F, Gerloff DL. 2009. Highly sensitive detection of individual HEAT and ARM repeats with HHpred and COACH. PLoS One 4:e7148. doi: 10.1371/journal.pone.0007148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pawson T. 1995. Protein modules and signalling networks. Nature 373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 23.Casares S, Ab E, Eshuis H, Lopez-Mayorga O, van Nuland NA, Conejero-Lara F. 2007. The high-resolution NMR structure of the R21A Spc-SH3:P41 complex: understanding the determinants of binding affinity by comparison with Abl-SH3. BMC Struct Biol 7:22. doi: 10.1186/1472-6807-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cloud-Hansen KA, Peterson SB, Stabb EV, Goldman WE, McFall-Ngai MJ, Handelsman J. 2006. Breaching the great wall: peptidoglycan and microbial interactions. Nat Rev Microbiol 4:710–716. doi: 10.1038/nrmicro1486. [DOI] [PubMed] [Google Scholar]
- 25.Lebeer S, Vanderleyden J, De Keersmaecker SC. 2010. Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat Rev Microbiol 8:171–184. doi: 10.1038/nrmicro2297. [DOI] [PubMed] [Google Scholar]
- 26.Uehara T, Park JT. 2003. Identification of MpaA, an amidase in Escherichia coli that hydrolyzes the γ-d-glutamyl-meso-diaminopimelate bond in murein peptides. J Bacteriol 185:679–682. doi: 10.1128/JB.185.2.679-682.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maqbool A, Hervé M, Mengin-Lecreulx D, Wilkinson AJ, Thomas GH. 2012. MpaA is a murein-tripeptide-specific zinc carboxypeptidase that functions as part of a catabolic pathway for peptidoglycan-derived peptides in γ-proteobacteria. Biochem J 448:329–341. doi: 10.1042/BJ20121164. [DOI] [PubMed] [Google Scholar]
- 28.Xu Q, Mengin-Lecreulx D, Patin D, Grant JC, Chiu HJ, Jaroszewski L, Knuth MW, Godzik A, Lesley SA, Elsliger MA, Deacon AM, Wilson IA. 2014. Structure-guided functional characterization of DUF1460 reveals a highly specific NlpC/P60 amidase family. Structure 22:1799–1809. doi: 10.1016/j.str.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. 2003. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 30.Lukk T, Sakai A, Kalyanaraman C, Brown SD, Imker HJ, Song L, Fedorov AA, Fedorov EV, Toro R, Hillerich B, Seidel R, Patskovsky Y, Vetting MW, Nair SK, Babbitt PC, Almo SC, Gerlt JA, Jacobson MP. 2012. Homology models guide discovery of diverse enzyme specificities among dipeptide epimerases in the enolase superfamily. Proc Natl Acad Sci U S A 109:4122–4127. doi: 10.1073/pnas.1112081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klock HE, Koesema EJ, Knuth MW, Lesley SA. 2008. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins 71:982–994. doi: 10.1002/prot.21786. [DOI] [PubMed] [Google Scholar]
- 32.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 33.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. 1993. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol 229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 34.Leulier F, Parquet C, Pili-Floury S, Ryu JH, Caroff M, Lee WJ, Mengin-Lecreulx D, Lemaitre B. 2003. The drosophila immune system detects bacteria through specific peptidoglycan recognition. Nat Immunol 4:478–484. doi: 10.1038/ni922. [DOI] [PubMed] [Google Scholar]
- 35.Stenbak CR, Ryu JH, Leulier F, Pili-Floury S, Parquet C, Hervé M, Chaput C, Boneca IG, Lee WJ, Lemaitre B, Mengin-Lecreulx D. 2004. Peptidoglycan molecular requirements allowing detection by the Drosophila immune deficiency pathway. J Immunol 173:7339–7348. doi: 10.4049/jimmunol.173.12.7339. [DOI] [PubMed] [Google Scholar]
- 36.Flouret B, Mengin-Lecreulx D, van Heijenoort J. 1981. Reverse-phase high-pressure liquid chromatography of uridine diphosphate N-acetylmuramyl peptide precursors of bacterial cell wall peptidoglycan. Anal Biochem 114:59–63. doi: 10.1016/0003-2697(81)90451-6. [DOI] [PubMed] [Google Scholar]
- 37.Hervé M, Boniface A, Gobec S, Blanot D, Mengin-Lecreulx D. 2007. Biochemical characterization and physiological properties of Escherichia coli UDP-N-acetylmuramate:l-alanyl-γ-d-glutamyl-meso-diaminopimelate ligase. J Bacteriol 189:3987–3995. doi: 10.1128/JB.00087-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pennartz A, Généreux C, Parquet C, Mengin-Lecreulx D, Joris B. 2009. Substrate-induced inactivation of the Escherichia coli AmiD N-acetylmuramoyl-l-alanine amidase highlights a new strategy to inhibit this class of enzyme. Antimicrob Agents Chemother 53:2991–2997. doi: 10.1128/AAC.01520-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das D, Hervé M, Elsliger MA, Kadam RU, Grant JC, Chiu HJ, Knuth MW, Klock HE, Miller MD, Godzik A, Lesley SA, Deacon AM, Mengin-Lecreulx D, Wilson IA. 2013. Structure and function of a novel LD-carboxypeptidase A involved in peptidoglycan recycling. J Bacteriol 195:5555–5566. doi: 10.1128/JB.00900-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santarsiero BD, Yegian DT, Lee CC, Spraggon G, Gu J, Scheibe D, Uber DC, Cornell EW, Nordmeyer RA, Kolbe WF, Jin J, Jones AL, Jaklevic JM, Schultz PG, Stevens RC. 2002. An approach to rapid protein crystallization using nanodroplets. J Appl Crystallogr 35:278–281. doi: 10.1107/S0021889802001474. [DOI] [Google Scholar]
- 41.Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. 2002. An automated system to mount cryo-cooled protein crystals on a synchrotron beamline, using compact sample cassettes and a small-scale robot. J Appl Crystallogr 35:720–726. doi: 10.1107/S0021889802016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van den Bedem H, Wolf G, Xu Q, Deacon AM. 2011. Distributed structure determination at the JCSG. Acta Crystallogr D Biol Crystallogr 67:368–375. doi: 10.1107/S0907444910039934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheldrick GM. 2008. A short history of SHELX. Acta Crystallogr A 64(Pt 1):112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 45.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. 2003. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr 59:2023–2030. [DOI] [PubMed] [Google Scholar]
- 46.Langer G, Cohen SX, Lamzin VS, Perrakis A. 2008. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. 2004. COOT: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G. 2004. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr 60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- 50.Menke M, Berger B, Cowen L. 2008. Matt: local flexibility aids protein multiple structure alignment. PLoS Comput Biol 4:e10. doi: 10.1371/journal.pcbi.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Honig B, Nicholls A. 1995. Classical electrostatics in biology and chemistry. Science 268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 52.Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. 2012. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D Biol Crystallogr 68:431–440. doi: 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic representation of A2pm-type bacterial peptidoglycan. The sites cleaved by d-Glu-A2pm hydrolases are marked in red. Borrowing the terminology for peptidase (see reference 20), the residues of the substrate at two sides of the cleavage site (scissile bond) are numbered P3, P2, P1 (nonprime side) to P1′, P2′ (prime side), respectively, while the corresponding binding pocket of the protein is numbered S3, S2, S1, S1′, and S2′. Download
γ-d-Glu-A2pm amidase activity of DvLysin. GlcNAc-1,6-anhydro-MurNAc-tetrapeptide (TCT, a), TCT dimer (b), and peptidoglycan (c) were incubated with (continuous lines) or without (dotted lines) purified DvLysin, and reaction mixtures were analyzed by HPLC as described in the text. TCT and TCT dimer were cleaved into two products identified as GlcNAc-anhydro-MurNAc-l-Ala-d-Glu (TCT-dip) and either the dipeptide A2pm-d-Ala or a tetrapeptide consisting of two cross-linked A2pm-d-Ala, respectively, consistent with a γ-d-Glu-A2pm amidase activity of this enzyme. The latter A2pm- and d-Ala-containing peptides were also released following digestion of the PG polymer by the enzyme. Download
Active sites of DvLysin and YkfC orthologs (BoYkfC, BtYkfC, and BcYkfC). (A) Stereoview of the DvLysin active site. Protein residues (green, red, blue) and ligands (cyan, red, blue) are shown as sticks. Experimental density-modified (DM) map (prior to model building, light-blue meshes) for the ligands is contoured at 1.0 σ. Hydrogen bonds are shown as dashed lines. (B) Active site of BoYkfC (experimental DM map shown for ligands). (C) Active site of BtYkfC crystal 1 (experimental DM map for ligands). (D) Active site of BtYkfC crystal 2 (2Fo-Fc omit map). (E) Active site of BcYkfC in complex with l-Ala-d-Glu dipeptide (PDB accession number 3H41). Download
Inhibition studies of BoYkfC by thiol-reactive labeling reagents. (A) Inhibition of BoYkfC by various thiol reagents. (B) Effects of time and concentration of DTNB on inhibition. (C) Inhibition effect of DTNB can be reversed by 2-mercaptoethanol. Download
Modeling of substrate into the DvLysin active site. (A) Stereoview of the DvLysin active site (green) with the modeled substrate (shown as magenta colored thick sticks), superimposed with the dipeptide complex of BcYkfC (thin sticks) (PDB accession number 3H41). The region that binds the l-Ala-d-Glu dipeptide portion of the substrate in DvLysin adopts a conformation similar to that of BcYkfC (thin gray sticks) since the S2 and S1 subsites are highly conserved in the two proteins. (B) Stereoview of the DvLysin active site (green) and the modeled substrate, superimposed with S-acetonyl-cysteine-inhibited BoYkfC (thin sticks) (PDB accession number 3NPF) that may mimic the transition state. (C) The modeled substrate matches well with solvent moieties (their final 2Fo-Fc densities are shown as contours at 1 σ) that resemble parts of the substrate. For example, the sulfonic acid group of MES and the acetate resemble the carboxylate groups of the substrate, while glycerols occupy subsites for binding l-Ala (S2) or d-Ala (S2′). Download
Distal and RT loops of SH3b1 (pink) are involved in the formation of the substrate binding groove in SH3b-NlpC/P60 fusion proteins. The distal loop is located upstream from the S2 subsite, while the RT loop is located near the S1′ subsite. Each protein is colored by domain (magenta, SH3b1; orange, SH3b2; green, NlpC/P60; blue, c-clip). Modeled substrates are show as sticks. Download
Structure-based sequence alignment of CwlT NlpC/P60 domain (PDB accession number 4FDY), AvPCP (PDB accession number 2HBW), BoYkfC (PDB accession number 3NPF), BcYkfC (PDB accession number 3H41), and DvLysin (PDB accession number 3M1U). SH3b1, SH3b2, and NlpC/P60 domains are marked by magenta, orange, and green boxes, respectively. The catalytic triad (Cys-His-His) and a nearby Tyr are marked by red circles. Download
Structure comparison of SH3b1 and SH3b2 domains. (A) Stereoview of seven superimposed SH3b domains in AvPCP, BcYkfC, BoYkfC, and DvLysin. The equivalent residues are colored red (SH3b1) and green (SH3b2). β-Strands are labeled A, A1, A2, and B to E. (B) Clustering of SH3b domains by pairwise structural comparison. (C) Structure-based sequence alignment of SH3b domains. Residues that are strictly conserved only in SH3b1 are highlighted in red boxes, while highly conserved residues in both SH3b1 and SH3b2 are colored white on a blue background. Download
Crystallization, data collection, and refinement statistics.
HPLC retention times of compounds tested in this work.