

Summary
Lipids play central roles in physiology and disease, where their structural, metabolic, and signaling functions often arise from interactions with proteins. Here, we describe a set of lipid-based chemical proteomic probes and their global interaction map in mammalian cells. These interactions involve hundreds of proteins from diverse functional classes and frequently occur at sites of drug action. We determine the target profiles for several drugs across the lipidinteraction proteome, revealing that its ligandable content extends far beyond traditionally defined categories of druggable proteins. In further support of this finding, we describe a selective ligand for the lipid-binding protein nucleobindin-1 (NUCB1) and show that this compound perturbs the hydrolytic and oxidative metabolism of endocannabinoids in cells. The described chemical proteomic platform thus provides an integrated path to both discover and pharmacologically characterize a wide range of proteins that participate in lipid pathways in cells.
Graphical abstract
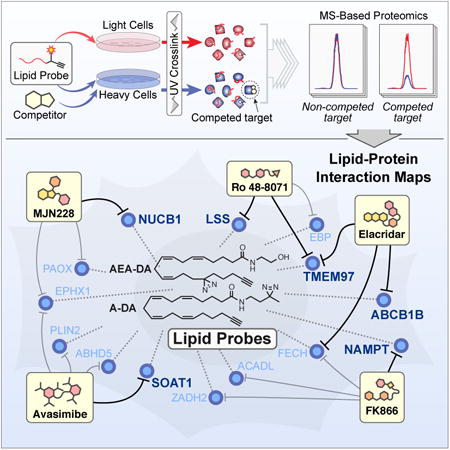
Small-molecule metabolites are central components of life, where their biological functions are often mediated and regulated by interactions with proteins. These metabolite-protein interactions include ligand-receptor, substrate-enzyme, and client-carrier relationships, many of which represent key nodes in biochemical networks that regulate cell physiology and disease. Eukaroytic and prokaryotic cells harbor numerous structurally distinct metabolites, and, among these natural products, lipids display a prominent capacity to interact with, and affect the functions of proteins (Muro et al., 2014). Sterol metabolites, for instance, interact with a broad set of enzymes, carriers, and receptors to regulate the composition and structure of cell membranes, as well as physiological processes, such as inflammation, metabolism, and blood pressure (Russell, 2009; Brown and Goldstein, 2009; Evans and Mangelsdorf, 2014).
Many fatty acid-derived lipids, including both phospholipids and neutral lipids, are also regulated by discrete enzymatic and transport pathways and transmit signals through an array of nuclear hormone receptors and G-protein-coupled receptors (GPCRs) (Evans and Hutchinson, 2010; Evans and Mangelsdorf, 2014). Lysophospholipids, for instance, have important roles in regulating immune and nervous system function (Mutoh et al., 2012; Shimizu, 2009), and their receptors have emerged as drug targets for diseases such as multiple sclerosis (Urbano et al., 2013). Oxidatively modified arachidonic acid (AA) metabolites, or eicosanoids, including prostaglandins and leukotrienes, serve as central mediators of pain and inflammation, cardiovascular function, and parturition (Harizi et al., 2008), inspiring the development of drugs that target proteins involved in eicosanoid production and signaling (Samad et al., 2002). Additional arachidonoyl metabolites include the endocannabinoids N-arachidonoyl ethanolamine (anandamide or AEA) and 2-arachidonoyl glycerol (2-AG), which are endogenous ligands for the cannabinoid CB1 and CB2 GPCRs (Mechoulam et al. 1998), and oxidized variants of these endocannabinoids – prostamides and prostaglandin esters, respectively – which represent an emerging class of inflammatory mediators (Rouzer & Marnett, 2011).
The established and emerging functions for lipids in mammalian biology, along with the substantial number of drug targets that are lipid-binding proteins, indicate that mapping the full complement of lipid-protein interactions in cells has the potential to uncover new modes of signaling that are amenable to pharmacological perturbation. Inspired by this concept, we describe herein a set of chemical proteomic probes to characterize the lipid-protein interaction landscape of mammalian cells and its sensitivity to drug action. We show that lipid probes can be used for 1) enrichment and identification of hundreds of lipid-binding proteins; 2) proteomewide in situ engagement assays to determine the targets and off-targets of drugs that impact lipid biology; and 3) high-throughput screening to identify small-molecule ligands for lipid-binding proteins. Using these methods, we provide evidence for the broad ligandability of the lipidinteraction proteome and exemplify this concept through development of selective ligands for a lipid-binding protein nucleobindin-1 (NUCB1) that perturb endocannabinoid and eicosanoid metabolism in cells.
Results
Chemical proteomic probes for mapping lipid-protein interactions
Chemical proteomic probes provide a versatile approach to globally map the cellular targets of both natural and unnatural small molecules in native biological systems (Lee and Bogyo, 2013; Simon et al., 2013; Su et al., 2013). Some probes rely on innate chemical reactivity with protein residues, whereas others exploit binding affinity and light-induced crosslinking reactions to capture proteins (Heal et al., 2011). The latter group typically possesses: 1) a photoreactive element that converts reversible small molecule-protein interactions into stable, covalent adducts upon ultraviolet (UV) light irradiation; 2) an alkyne, which serves as a sterically minimized surrogate reporter allowing late-stage conjugation to azide tags by copper-catalyzed azide-alkyne cycloaddition (CuAAC or “click”) chemistry (Rostovtsev et al., 2002); and 3) a binding element that directs the probe towards proteins that recognize specific structural features (Haberkant et al., 2013; Hulce et al., 2013; Li et al., 2013). With the goal of identifying proteins that interact with fatty acid-derived lipids in cells, we prepared a set of probes that contain a diazirine photoreactive group, an alkyne handle, and binding groups that resembled common fatty acids, including arachidonic (C20:4), oleic (C18:1), palmitic (C16:0), and stearic (C18:0) (Figure 1A).
Figure 1. Chemical proteomic probes for mapping lipid-binding proteins in cells.
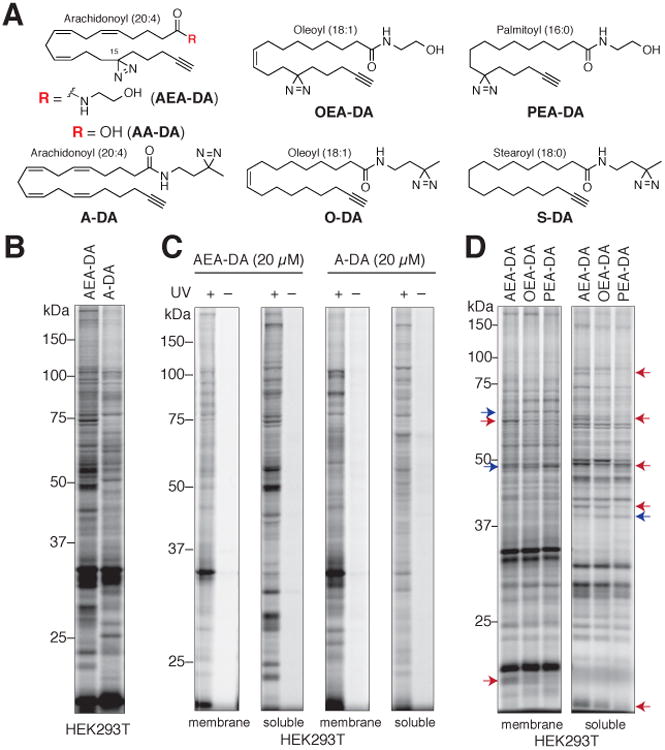
(A) Structures of lipid probes featuring arachidonoyl (AEA-DA, AA-DA and A-DA), oleoyl (OEA-DA and O-DA), palmitoyl (PEA-DA) and stearoyl (S-DA) acyl chains, as well as photoreactive (diazirine) and alkyne groups.
(B) AEA-DA and A-DA probes show overlapping, but distinct protein interaction profiles in HEK293T cells. Cells were treated with each probe (20 μM) for 30 min in situ before photocrosslinking and analysis of probe-modified proteins as described in Figure S1.
(C) Arachidonoyl probe labeling of membrane and soluble proteins depend on UV irradiation of cells.
(D) Comparative labeling profiles of lipid probes (20 μM, 30 min) in HEK293T cells. Red and blue arrows mark representative proteins preferentially labeled by arachidonoyl and oleoyl/palmitoyl probes, respectively. See Figure S1C for profiles of A-DA, O-DA and S-DA.
Within the arachidonoyl subset of probes, we synthesized both fatty acid- and fatty acid amide-based probes (AA-DA and AEA-DA, respectively) and evaluated their potential to bind and covalently modify (under UV-light exposure) proteins in human cells by gel-based profiling. HEK293T cells were treated with probe (AA-DA or AEA-DA; 20 μM, 30 min), irradiated with UV light (10 min, 4 °C), lysed, and the cell proteomes fractionated into membrane and soluble components by centrifugation prior to conjugation to a fluorescent reporter tag (Rh-N3) using CuAAC (Figure S1A). Analysis of probe targets by SDS-PAGE and in-gel fluorescence scanning revealed distinct protein labeling profiles for each probe (Figure S1B). The AA-DA probe showed almost exclusive labeling of membrane proteins, which we suspected was a consequence of rapid sequestration of this probe into membranes through its metabolic incorporation into phospho/neutral-lipids or into lipidated proteins, as has been noted for other fatty acid probes (Haberkant et al., 2013; Tate et al., 2014). In contrast, the AEA-DA probe showed substantial labeling of both soluble and membrane proteins in HEK293T cells (Figure S1B). The distinct labeling profile of the AEA-DA probe likely reflects the more limited capacity of the cell to metabolize this amidated probe, which must undergo enzyme-mediated hydrolysis prior to incorporation into other lipids or proteins. We therefore selected the fatty acid amide probes for chemical proteomic mapping of lipid-binding proteins in cells.
We also prepared a set of lipid probes that featured intact acyl chains and a diazirineamide head group (A-DA; O-DA, S-DA; Figure 1A). By including members with diazirines at distinct locations (acyl chain or head group), the lipid probe set thus had the potential, in our minds, to provide a more comprehensive portrait of interacting proteins. Consistent with this hypothesis, the AEA-DA and A-DA probes showed distinct protein labeling profiles in HEK293T cells (Figure 1B). Importantly, the protein-labeling events for both probes were UV light-dependent, confirming that they reflect reversible binding interactions between the probes and cellular proteins (Figure 1C). The polyunsaturated arachidonoyl probes (AEA-DA and A-DA) also showed more extensive proteomic labeling profiles compared to the monounsaturated (OEA-DA, O-DA) or saturated (PEA-DA, S-DA) lipid probes (Figure 1D and Figure S1C). Based on these results, and the diverse functional roles played by arachidonate-derived lipids in mammalian biology (Harizi et al., 2008; Rouzer and Marnett, 2011), we focused our initial MS-based proteomic studies on mapping the proteins that interact with the arachidonoyl lipid probes (AEA-DA and A-DA) in cells.
Landscape of lipid-binding proteins in cells
We identified the protein targets of the AEA-DA and A-DA probes using stable-isotope labeling by amino acids in cell culture (SILAC) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) following previously developed protocols (Hulce et al., 2013; Ong et al., 2002). Isotopically “light” cells served as a static control for each experiment and were treated with either AEA-DA or A-DA (20 μM, 30 min) before UV irradiation (Figure S2A). Isotopically “heavy” cells served as comparison groups and were treated with: 1) the same conditions as the “light” cells (probe-versus-probe control); 2) the same probe as the “light” cells, but were not crosslinked with UV light (probe-versus-No UV); or 3) the corresponding oleoyl (18:1, OEA-DA or O-DA) or fully-saturated (C16:0, PEA-DA; C18:0, S-DA) probes. Performing these SILAC experiments in both human (HEK293T) and mouse (Neuro2a) cell lines provided an extensive inventory of lipid probe targets—defined as proteins with at least three unique quantifiable peptides that were labeled by either A-DA or AEA-DA in a UV-dependent manner (SILAC ratio ≥ 3.0 in probe-versus-No UV experiments) and not enriched in probe-versus-probe control experiments (SILAC ratio < 2.0) (Figure 2A and Figure S2B).
Figure 2. Mapping protein targets of lipid probes by quantitative proteomics.

(A) Heavy/light SILAC ratio plots for total proteins identified in experiments comparing the labeling profiles of lipid probes (20 μM) versus a ‘No UV’ control (20 μM probe without UV irradiation) or the equivalent probe (both heavy and light cells treated with 20 μM of the same probe) in HEK293T cells. Dashed lines mark threshold ratio values (≥ 3-fold in No UV experiments) for designation of lipid probe targets (also see Figure S2).
(B) Venn diagram of shared and unique protein targets of AEA-DA and A-DA in HEK293T and Neuro2a cells.
(C-F) Analysis of lipid probe targets based on (C) presence (membrane) or absence (soluble) of known/predicted transmembrane domains; (D) known/predicted subcellular distribution; (E) involvement in specific biological processes; and (F) protein class distribution. Categories were assigned based on UniProt annotations.
(G) Diagram highlighting lipid probe targets (red) in major fatty acid metabolic pathways. SILAC ratios from probe-versus-No UV experiments are indicated in parentheses next to gene names (data shown are for the A-DA probe in HEK293T cells except for CPT1A, which was detected with the A-DA probe in Neuro2a cells). For instances where multiple isoforms of a given protein is enriched (i.e., ACSL and GPAT), the highest ratio across all isoforms is presented.
(H) Heat map showing the relative protein enrichment values for the AEA-DA probe compared to OEA-DA and PEA-DA probes, as well as compared to the AEA probe itself with (AEA-DA) or without UV irradiation (No UV) as controls, in HEK293T cells. See Figure S2D for a similar analysis of the A-DA probe series and Table S1 for complete list of lipid probe targets.
More than 1000 proteins in total were enriched from HEK293T and Neuro2a cells by the A-DA and AEA-DA probes. While each arachidonoyl probe shared several hundred common targets, a large set of probe-specific targets were also identified for both AEA-DA and A-DA (442 and 317, respectively; Figure 2B), emphasizing the value of employing both probes to maximize the capture of lipid-binding proteins. Of note, we found that the AEA-DA and A-DA probes targeted greater numbers of soluble and membrane proteins, respectively (Figure 2C), which was also manifested in the corresponding enrichment of these protein targets in cytoplasmic/nuclear and endoplasmic reticulum (ER) compartments (Figure 2D). These differences could reflect the higher lipophilicity (i.e., cLogP) of the A-DA probe, promoting its localization in membranes.
Categorizing the lipid probe targets in relation to their functions in biological pathways revealed enrichment of proteins involved in protein transport, lipid metabolism, and host-virus interactions (Figure 2E). Analysis of the probe targets through the Online Mendelian Inheritance in Man (OMIM) database further revealed links to numerous diseases, including metabolic disorders, cancer, and cardiovascular and neurological disease (Figure S2C). Proteins from a variety of functional classes were enriched by each lipid probes, with particularly high proportions of enzymes and transporters (Figure 2F). Prominent among these targets were enzymes and lipid carriers involved at key nodes of fatty acid uptake (SCARB1), transport (SLC25A20), biosynthesis (FASN, PNPLA2) and catabolism (ACADs, HADHA) (Figure 2G). Other known arachidonoyl lipid carrier proteins (e.g., FABP5) (Kaczocha et al., 2009) and metabolizing enzymes (e.g., PTGS1 or COX1) were also enriched. Of particular interest was the large number of poorly characterized proteins lacking prior links to lipid biology that were strongly enriched by at least one of the arachidonoyl probes (Table S1). Consistent with our gel-based profiles (Figure 1D and Figure S1C), we found that most lipid probe targets were preferentially enriched by the AEA-DA and A-DA probes compared to either monounsaturated (OEA-DA, O-DA) or saturated (PEA-DA, S-DA) probes (Figure 2H, S2D, S2E, and Table S1)
Validation and characterization of lipid probe-protein interactions
We next sought to validate representative probe-protein interactions and selected six probe targets, including both known (e.g., PTGR2) and unannotated (e.g., NUCB1, NENF) lipidinteracting proteins that displayed varying degrees of preferential labeling by the AEA-DA versus PEA-DA probes (Figure 3A and 3B). In situ probe treatment of HEK293T cells expressing Myc-tagged proteins confirmed, in each instance, the protein-lipid interaction (Figure 3C). NUCB1, NENF, and ZADH2 were each labeled by AEA-DA to a greater degree than PEA-DA, while ALDH1B1, VAT1, and PTGR2 exhibited similar extents of interaction with each probe, matching, in general, the lipid interaction profiles observed for endogenous forms of these proteins (compare Figure 3B and 3C).
Figure 3. Experimental validation of representative lipid probe targets.
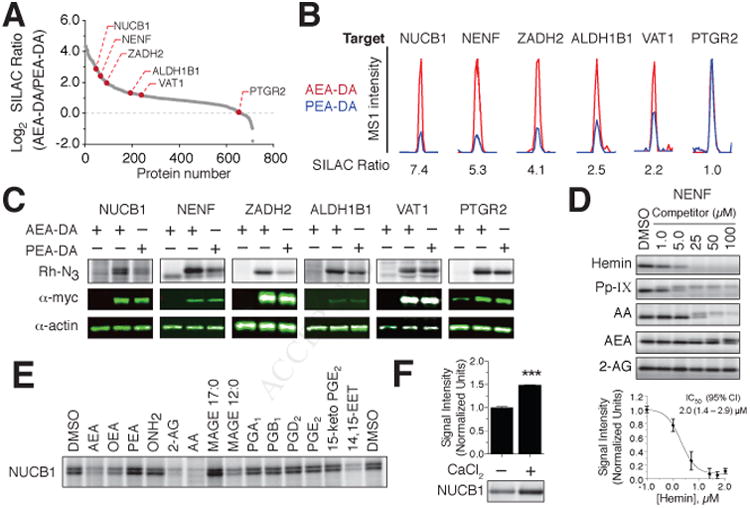
(A) SILAC ratio plot for AEA-DA versus PEA-DA (20 μM) probe labeling in HEK293T cells highlighting targets selected for experimental validation.
(B) Representative peptide MS1 chromatograms for selected targets showing relative labeling by AEA-DA and PEA-DA probes.
(C) Lipid probe labeling of myc-tagged recombinant proteins expressed by transient transfection in HEK293T cells. Top panels show in situ labeling profiles for the AEA-DA and PEA-DA probes with indicated targets (lane 1, mock-transfected cells; lanes 2 & 3, target-transfected cells; see Figure S3A for full gel profiles). Middle panels, anti-myc blotting. Lower panels, anti-actin blotting as a loading control.
(D) Upper panel shows in vitro competition profiles of AEA-DA probe labeling of NENF by hemin, protoporphyrin IX (Pp-IX), and the arachidonoyl lipids AA, AEA and 2-AG (1–100 μM) (experiments performed in NENF-transfected HEK293T lysates). Lower panel shows concentration-dependent inhibition of AEA-DA labeling of NENF by hemin (CI = 95% confidence interval) as determined from gel profiles. Data represent mean values ± SD from three independent experiments.
(E) In vitro competition profiles of NUCB1 labeling by the AEA-DA (5.0 μM) probe using variouslipids (20×) as competitors. Experiments were performed in lysates from NUCB1-transfectedHEK293T cells.
(F) Calcium-dependent enhancement of NUCB1 labeling by the AEA-DA probe. Data represent mean values ± SEM; n = 3/condition. ***P < 0.001 for untreated versus CaCl2 (100 μM)-treated samples.
NENF, also called neudesin, is a secreted protein from the cytochrome b5-like heme/steroid-binding family that promotes the survival of neurons (Kimura et al., 2008). NENF has been shown to bind hemin and protoporphyrin IX (Pp-IX) and these interactions can promote neurotrophic activity (Kimura et al., 2008); however, whether NENF can also bind steroids/lipids remains unknown. We found that hemin and Pp-IX inhibited in a concentrationdependent manner the labeling of recombinant NENF by the AEA-DA probe (Figure 3D), and the hemin-NENF interaction (IC50 = 2 μM), in particular, appeared selective, as other AEA-DA-modified proteins detected by SDS-PAGE were unaffected by hemin treatment (Figure S3B). AA and, to a lesser degree, AEA, but not 2-AG, also competitively blocked AEA-DA probe labeling of NENF (Figure 3D).
The nucleobindin proteins NUCB1 and NUCB2 are not known to bind small-molecule ligands, but NUCB1 has been shown to interact physically with the prostaglandin biosynthetic enzymes PTGS1 and PTGS2 enzymes and enhance PTGS2-mediated prostaglandin synthesis (Ballif et al., 1996; Leclerc et al., 2008). We found that AEA-DA probe labeling of recombinant NUCB1 was preferentially blocked by arachidonoyl lipids (AEA, AA and 2-AG) over saturated/shorter chain analogs [OEA, PEA, oleamide (ONH2)] and prostaglandins (Figure 3E). Nucleobindins also feature two EF-hand domains that undergo conformational changes upon binding calcium ions (de Alba and Tjandra, 2004). Using both purified, recombinant NUCB1 (Figure 3F) and NUCB1-transfected HEK293T cell lysates (Figure S3C), we found that CaCl2 (100 μM) significantly and selectively increased AEA-DA probe-labeling of NUCB1, whereas EDTA appeared to reduce this interaction.
In situ drug profiling with lipid probes
We found that the lipid-interaction proteome was substantially enriched in known drug targets (∼25%, or 280 proteins; Figure 4A and Table S2) compared to the total fraction of the human proteome represented in the DrugBank database (∼12%). The fraction of DrugBank proteins present among membrane and soluble probe targets were similar (Figure S4A; 18% and 29%, respectively) and included proteins from multiple functional classes, such as enzymes (e.g., LSS, PTGS1, SOAT1), transporters (e.g., ABCB1, ATP4A, VDAC1-3), and receptors (e.g., SCARB1, PGRMC1). The remaining lipid-probe targets not found in DrugBank included proteins that would be considered “ligandable” (e.g. enzymes, receptors, etc.), as well as a large number of proteins not predicted to interact with small molecules based on their ascribed biochemical activities or lack of functional characterization (Figure 4A and Table S2). These findings suggested that the lipid probes exhibit a preferential capacity to interact with known drug-binding proteins in cells and, by extension, might facilitate the discovery of many additional proteins with the potential to bind small-molecule ligands. Further, for ligandable proteins known or identified herein, we surmised that the lipid probes could provide a method to determine drug target engagement and the selectivity of these interactions in cells. We set out to test these concepts by first evaluating whether the lipid probes competed for drug-binding to DrugBank proteins in cells.
Figure 4. The lipid-interaction proteome is rich in drug targets.
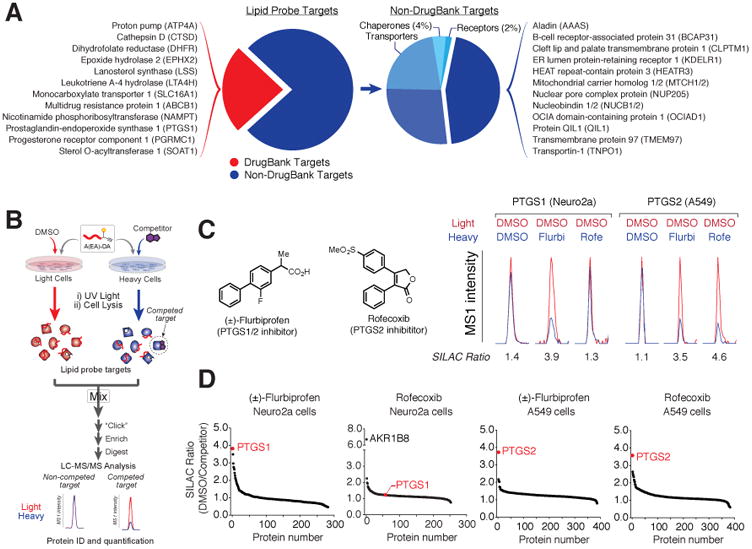
(A) Categorization of lipid probe targets based on distribution in DrugBank (left pie chart) and further analysis of non-DrugBank targets by protein classes considered ligandable (e.g., enzymes, receptors, transporters) or not (Others).
(B) Scheme for in situ competitive profiling of ligands using lipid probes. Isotopically light and heavy cells are treated with vehicle (DMSO) or competitor ligand, respectively, along with a lipid probe for 30 min. Cells are then UV-irradiated, lysed, and light and heavy lysates combined, enriched, and digested for LC-MS/MS analysis. Ligand targets are designated as proteins that show light/heavy ratios of ≥ 3.0.
(C) Chemical structures of the dual PTGS1/2-inhibitor (±)-flurbiprofen and PTGS2-selective inhibitor rofecoxib and representative peptide MS1 chromatograms for PTGS1 and PTGS2 in Neuro2a and A549 cells, respectively, showing that (±)-flurbiprofen (25 μM) competes A-DA (5 μM) labeling of both PTGS1 and PTGS2, whereas rofecoxib (25 μM) selectively competes PTGS2 labeling.
(D) SILAC ratio plots for in situ competition experiments of A-DA (5 μM) labeling by (±)-flurbiprofen (25 μM) and rofecoxib (25 μM) validating target engagement and selectivity across PTGS isoforms and other lipid probe targets.
The prostaglandin biosynthetic enzymes PTGS1 and PTGS2 were selected for initial analysis, as dual PTGS1/PTGS2 and selective PTGS2 inhibitors have been developed for treating inflammatory disorders. PTGS1 was detected as a lipid probe target in Neuro2A cells (Figure S4B), while PTGS2 was evaluated in phorbol-12-myristate-13-acetate (PMA)-stimulated A549 cells (Figure S4C and S4D). We generated drug-competition profiles with the lipid probes by co-treating heavy and light cells with the A-DA probe (5 μM) and either DMSO (light) or drug competitor (25 μM; heavy) for 30 min (Figure 4B). The cells were then irradiated with UV light, harvested and lysed, whereupon the heavy and light proteomes were mixed in equal proportions. Following CuAAC conjugation with biotin-N3, streptavidin enrichment, and onbead tryptic digestion, probe-labeled proteins were analyzed by LC-MS/MS. Drug-competed proteins were defined as those showing a substantial (≥ 3-fold) reduction in signal in drug-treated (heavy) versus DMSO-treated (light). The dual PTGS1/2 inhibitor (±)-flurbiprofen competitively blocked A-DA-labeling of both PTGS1 and PTGS2 in cells, whereas the selective PTGS2 inhibitor rofecoxib disrupted A-DA-labeling of PTGS2, but not PTGS1 (Figure 4C). Both drugs showed good selectivity for PTGS enzymes, which were among the most competed proteins across the hundreds of A-DA probe targets detected in Neuro2A and A549 cells (Figure 4D and Table S2). Some additional competed targets were also identified, including aldose reductase-related protein 2 (AKR1B8), which showed strong reductions in A-DA labeling in rofecoxib-, but not (±)-flurbiprofen-treated Neuro2A cells (Figure 4D and Table S2). AKR1B8 is a mouse ortholog of the human aldo-keto reductase AKR1B10, which is modified and inhibited by electrophilic prostaglandins (Diez-Dacal et al., 2011), providing further support that these enzymes specifically interact with arachidonoyl-related lipids and drugs.
We next expanded our analysis of drug action in cells to include several additional lipid probe targets with known ligands – sterol O-acyltransferase (SOAT1), nicotinamide phosphoribosyltransferase (NAMPT), lanosterol synthase (LSS), and multidrug resistance protein 1 (ABCB1) and their respective ligands – avasimibe, FK-866, Ro 48-8071, and elacridar (Figure 5A). Some of these targets were chosen because they are integral membrane proteins (LSS, SOAT1, ABCB1) and thus pose technical challenges for other drug-interaction profiling methods that measure ligand-induced changes in proteolytic (Lomenick et al., 2009) or thermal (Martinez Molina et al., 2013) stability. Each ligand was initially assayed at 25 μM in Neuro2a cells, as this concentration was predicted to fully engage the primary drug target and also facilitate a broader prospecting of the lipid-interaction proteome for other ligandable proteins. We tested one drug – Ro 48-8071 – across a broader concentration range (5 and 50 μM) to assess the potency of its interactions and to facilitate identification of additional drug-protein interactions. As before, we treated heavy and light cells with drug and DMSO, respectively, together with the arachidonoyl probes (5 μM). Following UV irradiation and proteomic analysis, we observed clear evidence of engagement with the primary established targets for each drug and, notably, little cross-reactivity with the targets of the other drugs tested (Figure 5B and Figure S5A). One exception was FK-866, which competed probe-labeling of both its established target NAMPT and ABCB1B (Figure 5B).
Figure 5. In situ drug profiling with lipid probes.

(A) Structures of compounds analyzed by competitive profiling with lipid probes.
(B) Heatmap showing SILAC ratios for primary targets of drugs (25 μM, except for Ro 48-8071, which was assayed at 5 μM) from competitive profiling experiments performed in Neuro2a and A549 cells. All drugs were profiled in both cell lines, and target engagement for SOAT1, NAMPT, and ABCB1B is shown for Neuro2a cells with the AEA-DA probe (5 μM) and, for LSS, in A549 cells with the A-DA probe (5 μM) (also see Table S3).
(C) Box-whisker plots of protein SILAC ratios from in situ competition experiments showing on-(red) and off- (blue) targets (ratios ≥ 3.0) for tested drugs.
(D) Heatmap of competed off-targets for tested drugs measured with the AEA-DA probe in Neuro2a cells.
(E) Plot of SILAC ratios from AEA-DA competition experiments with tested drugs (25 μM) versus the lipid competitor AEA (200 μM). For simplicity, only the highest drug competition SILAC ratio is plotted for each target.
(F) Categorization of drug-competed lipid probe targets based on their presence or absence in Drug Bank and by protein class (also see Table S3).
A greater survey of the lipid-interaction proteome revealed a unique set of additional targets for each drug (Figure 5C and Figure S5A), many of which were preferentially competed by one of the four tested drugs (Figure 5D). Clear concentration-dependent increases in the target landscape were observed for Ro 48-8071, with the principal target, LSS, being fully competed at 5 μM along with only two prominent off-targets (TMEM97 and EBP), whereas, at 50 μM, Ro 48-8071 suppressed probe labeling of many additional targets (Figure 5C and S5A). We also compared the drug competition profiles to that of the endogenous lipid transmitter AEA tested at 200 μM, which was found to be a suitable concentration for competitive profiling by gel-based analysis (Figure S5B). AEA competed several targets of the drugs FK-866 (e.g. PTGR2) and Ro 48-8071 (e.g. DHRS1), both of which possess lipid-like scaffolds, but not the targets of avasimibe or elacridar (Figure 5E).
Additional profiling of drugs in Neuro2a and A549 cells using both A-DA and AEA-DA probes to maximize coverage provided a rich set of competed targets (Table S3). Only ∼30% of the identified drug targets were listed in DrugBank, and, of the non-DrugBank targets, a substantial portion (∼40%) were uncharacterized proteins or proteins that belonged to classes that would traditionally be considered challenging to ligand based on their sparse representation in DrugBank (Figure 5F and Table S3). Some proteins interacted strongly with multiple drugs, such as ferrochelatase (FECH), which was recently found to bind kinase inhibitors in cells using thermal proteome profiling (Savitski et al., 2014). Arachidonoyl probe labeling of FECH, along with ABCB1B and TMEM97, was blocked by elacridar at concentrations as low as 0.5 μM (Figure S5D), indicating that these drug-protein interactions are high affinity events. We confirmed that both elacridar and Ro 48-8071 block AEA-DA probe labeling of recombinantly expressed TMEM97 in transfected HEK293T cells (Figure S5C). These data suggest that FECH and TMEM97 are highly ligandable proteins, as reflected by their capacity to interact with multiple small-molecule chemotypes in cells.
Discovery of selective ligands for the lipid-binding protein nucleobindin 1
While investigating the proteome-wide interactions of known drugs provides one path for discovering ligand-binding proteins, this approach is limited in throughput. We therefore asked whether the lipid probes could be adapted for the screening of larger compound libraries. As a proof-of-principle, we selected NUCB1, which, we hypothesized, based on its interactions with PTGS enzymes and preferential binding to arachidonoyl probes, to play a role in lipid metabolism in cells. We first synthesized a fluorescent arachidonoyl lipid probe (Fl-AEA; Figure 6A) and confirmed that it bound to recombinant, purified human NUCB1 (hNUCB1) protein to produce a substantial increase in fluorescence polarization (FluoPol) signal (Figure 6B). This FluoPol signal was significantly reduced by arachidonoyl, but not palmitoyl (Figure 6C) or other (Figure S6A) competitor lipids, recapitulating the selectivity observed by gel- and MS-based profiling with the photoreactive lipid probes (Figure 3E).
Figure 6. Adapting lipid probes for HTS to discover NUCB1 ligands.
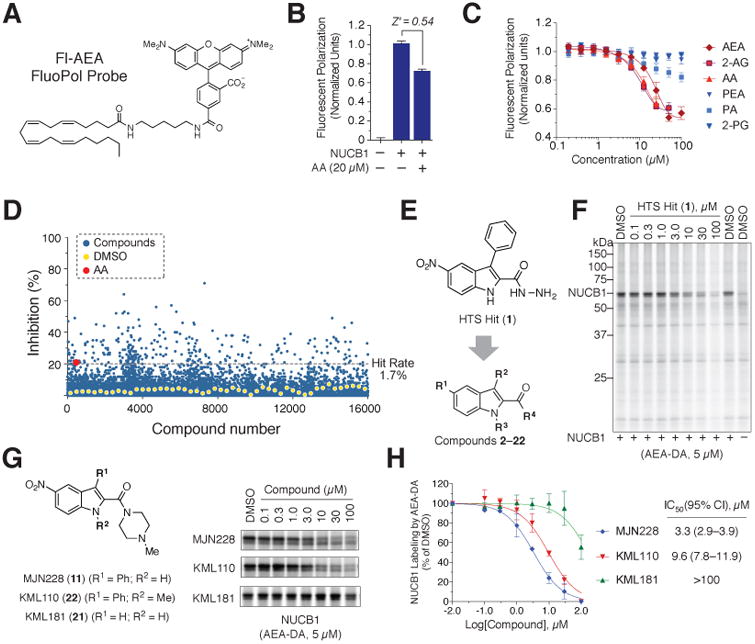
(A) Structure of Fl-AEA probe.
(B) Incubation of the Fl-AEA probe (0.5 μM) with recombinant human NUCB1 (1.0 μM) produced a strong FluoPol signal that was significantly suppressed by the competitive lipid AA (20 μM; Z′ = 0.54).
(C) Concentration-dependent suppression of the NUCB1-FluoPol signal by arachidonoyl lipids AEA, 2-AG and AA, but not palmitoyl lipids PEA, 2-palmitoyl glycerol (2-PG) or palmitic acid (PA). See Figure S6A for profiling of additional lipids.
(D) Screen of 16,000 compounds identified small molecules that inhibited the NUCB1-FluoPol signal by 20% or greater (dotted black line).
(E) Structure of confirmed HTS hit 1 and positions modified for medicinal chemistry optimization.See Figure S6B–D for summary of medicinal chemistry optimization of NUCB1 ligands.
(F) Concentration-dependent blockade of AEA-DA (5 μM) labeling of purified, recombinant NUCB1 (0.25 μM) doped into HEK293T lysates (0.75 mg/mL) by HTS hit 1 (0.1-100 μM).
(G, H) Structures and competitive profiling results (G) and IC50 curves and values (H) for NUCB1 ligands MJN228 and KML110 and the inactive control compound KML181.
Data in B, C and H represent mean values ± SD from at least three independent experiments.
The FluoPol assay was optimized (Z′ score of > 0.5 compared to assays performed with AA as a competitor ligand; Figure 6B) and used to screen 16,000 compounds from the Maybridge library at 10 μM in 384 well-plate format. Chemoinformatic analysis to remove frequent hit compounds and compounds with structural alerts yielded 100 compounds that produced a 20% or greater reduction in FluoPol signal (on par or greater than the reduction caused by AA; Figure 6D). These hits were assayed by gel-based competitive profiling with the AEA-DA probe against recombinant hNUCB1 doped into HEK293T cell lysates, and hydrazide 1 (Figure 6E) was identified as a strong competitor of NUCB1 labeling (Figure 6F and Figure S6B). Optimization of this compound (Figure S6) furnished N-methylpiperazine amide MJN228 (11; Figure 6G), which blocked AEA-DA probe labeling of NUCB1 with an IC50 value of 3.3 μM (Figure 6H) and did not appear to disrupt other arachidonoyl probe-protein interactions in HEK293T cell lysates (Figure S6E). A second NUCB1-active ligand was developed that contained a methyl substituent on the indole nitrogen (22, KML110), which only caused a slight reduction in potency (IC50 = 9.6 μM), as well as a structurally related inactive control compound KML181 that displayed markedly reduced potency for NUCB1 (IC50 > 100 μM; Figure 6G and 6H).
We next tested whether the ligands could bind to NUCB1 in cells. Treatment of Neuro2a cells with MJN228 or KML110 (25 μM) produced substantial (∼3-5-fold) reductions in lipid probe enrichment of NUCB1, while KML181 had no effect (Figure 7A). MJN228 inhibited lipid probe binding to NUCB1 at concentrations as low as 10 μM, with near-maximal inhibition observed at ∼25 μM (Figure S7A). A broader analysis of the lipid-interaction proteome revealed that NUCB1 was the most competed protein among the ∼400 AEA-DA probe targets detected in Neuro2a cells (Figure 7B). Finally, we sought to map the site of arachidonoyl probe (and MJN228) binding to NUCB1 in cells, which was accomplished by treating Neuro2A cells with the AEA-DA probe (50 μM) in the presence of DMSO or MJN228 (50 μM), followed by UV irradiation, CuAAC conjugation of AEA-DA-labeled proteins to isotopically light (DMSO-treated cells) and heavy (MJN228-treated cells) azide-biotin tags featuring a TEV protease-cleavable linker, and LC-MS/MS analysis using a previously described platform, termed isoTOP-ABPP, for mapping probe-modified peptides in proteomes (Speers and Cravatt, 2005). A single prominent AEA-DA-labeled peptide was identified for NUCB1 (aa 53-68) in DMSO-treated cells and the signals for this peptide were substantially (> 5-fold) reduced in MJN228-treated cells (Figure 7C). We confirmed this AEA-DA labeling site using recombinant, purified hNUCB1 and tandem MS analysis narrowed down the likely site of probe modification to His67 (Figure S7B and S7C). Interestingly, the MJN228-sensitive, AEA-DA-modified peptide resides within the previously mapped PTGS1/2-binding domain of NUCB1 (aa 1-123; Figure 7C) (Ballif et al., 1996), indicating that this region is responsible for both the lipid- and protein-protein interactions displayed by NUCB1.
Figure 7. Target engagement and lipid metabolism effects of NUCB1 ligands.
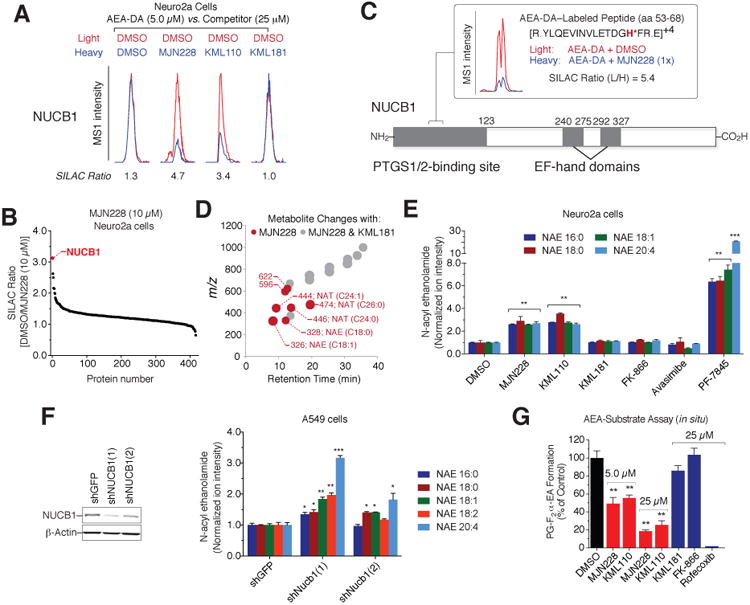
(A) Representative peptide MS1 chromatograms showing blockade of AEA-DA probe labeling of endogenous NUCB1 in Neuro2a cells by MJN228 and KML110, but not KML181.
(B) SILAC ratio plot for in situ competition experiment performed with MJN228 (10 μM) and the AEA-DA probe (5 μM).
(C) LC-MS/MS identification of a prominent MJN228-sensitive, AEA-DA-modified NUCB1 peptide (aa 53-68) in Neuro2a cells.
(D) Untargeted metabolite profiling reveals that Neuro2a cells treated with MJN228 (10 μM) show elevated fatty acid amides (NAEs and NATs) compared to cells treated with DMSO or KML181 (10 μM) (P < 0.0001, n = 5 per condition). See also Table S5.
(E) Targeted MRM measurements showing elevations in NAEs in Neuro2a cells treated withNUCB1 ligands MJN228 and KML110 (10 μM, 6 h), but not KML181, FK-866, or avasimibe. See Figure S7G for MRM measurements of NATs.
(F) Left, Western blot showing knockdown of NUCB1 in shNUCB1 A549 cell lines compared to a control cell line (shGFP). Right, both shNUCB1 cells show significant elevations in NAEs compared to the control shGFP cell line.
(G) NUCB1 ligands MJN228 and KML110 (5 and 25 μM), but not KML181 or FK-866 (25 μMeach), suppress the conversion of exogenous AEA (20 μM, 30 min) to PGF2α-EA in PMA-stimulated A549 cells. Rofecoxib (25 μM) also blocked PGF2 α-EA synthesis.
For (E-G), data represent mean values ± SEM; n = 3-4/group. *P < 0.05, **P < 0.01, ***P < 0.001 for DMSO-treated (E) or shGFP cells (F) versus compound-treated (E) or shNUCB1 (F) cells.
Deeper profiling of Neuro2a cells using the isoTOP-ABPP platform identified AEA-DA-modified peptides for an additional ∼150 proteins (Table S4), which accounted for ∼40% of the total AEA-DA targets mapped in this cell line. These findings demonstrate the potential for chemical proteomics to map not only lipid-binding proteins, but also the sites on these proteins that interact with lipids in cells.
NUCB1 ligands perturb multiple lipid pathways in cells
Having established that NUCB1 is a principal target of MJN228 in cells, we next investigated the metabolic consequences of this ligand-protein interaction by performing a lipidomic analysis of Neuro2a cells treated with DMSO, MJN228 (10 μM), or the inactive control probe KML181 (10 μM). Following a 6 h incubation with each compound, cells were harvested, lysed, and their lipids isolated by organic extraction and analyzed by untargeted LC-MS in both positive and negative ion modes. Using XCMS software (Smith et al., 2006) to quantitate differences between compound- and DMSO-treated samples, we identified a small group of metabolites that were significantly elevated in MJN228-treated Neuro2a cells (≥ 2 fold, P < 0.0001) but not in cells treated with KML181 (Figure 7D and Table S5). The chromatographic and tandem MS profiles of these lipids enabled their structural assignment as N-acyl ethanolamines (NAEs) and N-acyl taurines (NATs). NAEs and NATs are hydrolytically metabolized by the enzyme fatty acid amide hydrolase (FAAH) (Saghatelian et al., 2006); however, neither MJN228 nor KML110 showed substantial inhibitory activity against purified recombinant FAAH or endogenous FAAH in Neuro2a lysates (IC50 values > 100 μM, Figure S7D and S7E), suggesting that the compounds did not increase NAE or NAT levels through direct interactions with FAAH in cells. We had also hoped to directly assess ligand engagement of FAAH in Neuro2a cells using the lipid probes, but we were unable to detect substantial signals for this enzyme in our chemical proteomic data sets. The poor labeling of FAAH may be due to the lipid probes serving as substrates for this enzyme, as we observed an overall loss in probe labeling of proteins in cells that overexpress FAAH (Figure S7F).
We next used targeted metabolite analysis to evaluate the effects of NUCB1 ligands and control compounds on the fatty acid amide content of cells. Both NUCB1 ligands (MJN228 and KML110), as well as the FAAH inhibitor PF-7845 (Ahn et al., 2011) elevated the cellular concentrations of NAEs and NATs, including the endocannabinoid anandamide (C20:4 NAE, or AEA) and the TRPV4 ligand C20:4 NAT (Saghatelian et al., 2006) (Figure 7E and Figure S7G). Other arachidonoyl lipids, including AA and 2-AG, were either unaffected or marginally elevated by NUCB1 ligands (Figure S7H). Neither KML181, nor the additional control compounds avasimibe nor FK-866, which shared many off-targets with the NUCB1 ligands, but did not interact with NUCB1 itself (Figure S7I and Table S2), altered NAE/NAT content in cells (Figure 7E and Figure S7G). We also confirmed that the NUCB1 ligands elevated fatty acid amides in a human cell line (A549 cells; Figure S7J). Finally, we used RNA interference to stably lower the expression of NUCB1 in A549 cells using two distinct shRNA probes [shNUCB1(1) and shNUCB1(2)] (Figure 7F). Multiple NAEs, including AEA and OEA, were elevated in the shNUCB1-A549 cell lines, but not in the control shRNA (shGFP-A549) cell line (Figure 7F).
Our pharmacological and RNA interference data, taken together, indicate that NUCB1 plays a role in facilitating the metabolism of fatty acid amides, possibly by serving as an intracellular carrier to deliver these lipids to FAAH. Consistent with this model, treatment of cells with both a NUCB1 ligand and the FAAH inhibitor PF-7845 did not produce larger changes in NAEs than treatment with the PF-7845 alone (Figure S7K).
AEA is not only a substrate for FAAH, but also PTGS2, which converts this endocannabinoid into bioactive prostamides (Rouzer and Marnett, 2011). We therefore tested whether NUCB1 ligands might also perturb the oxidative metabolism of AEA by PTGS2. We first confirmed that NUCB1 ligands are not direct PTGS2 inhibitors (Figure S7L). We then treated PMA-stimulated A549 cells with NUCB1 ligands (MJN228 and KML110) and control compounds (KML181 and FK-866) followed by exogenous AEA (20 μM) and measured the formation of prostamides. Both NUCB1 ligands, but not the control compounds, produced a significant, concentration-dependent reduction in PGF2α-EA in A549 cells (Figure 7G). Since NUCB1 also bound AA in our biochemical assays (see Figure 3E and Figure 6B), we tested whether NUCB1 ligands affected prostaglandin production in A549 cells treated with PMA. The PMA-stimulated generation of PGE2 and, to a lesser degree, TXB2 was attenuated by NUCB1 ligands, but not the control compound KML181 (Figure S7M). The additional control compound FK-866 exhibited a curious profile, showing no effect on PGE2, but complete suppression of TXB2 (Figure S7M). It is not clear, however, whether FK-866 impairs TXB2 production through inhibiting its primary target NAMPT or another protein in A549 cells. Alterations in prostamides and prostaglandins were not observed in shNUCB1 cells, which could indicate that a more substantial reduction in NUCB1 expression than that achieved by RNA-interference is needed to perturb NUCB1-PTGS2 crosstalk in cells.
Discussion
Many life processes are regulated by the physical interactions between lipids and proteins, and these interactions constitute validated nodes for drug action to treat human diseases. Nonetheless, our understanding of the full scope of lipid-protein crosstalk in cells, as well as its accessibility to pharmacological perturbation, remains limited. Here, we have described a suite of chemical proteomic probes to inventory the landscape of lipid-binding proteins and map their ligandability in cells. We focused on fatty acid-based probes with limited potential for metabolic incorporation into more complex lipids, and, in this way, our studies complement previous work that has inventoried proteins that bind to other lipid classes (e.g., phospholipids, sterols) (Gallego et al., 2010; Gubbens et al., 2009; Haberkant et al., 2013; Hulce et al., 2013; Rowland et al., 2011). Key distinguishing features of our approach, however, include a comparative assessment of the protein interaction profiles of different structural lipids, revealing a preferential capacity for arachidonoyl lipids to interact with proteins, and the adaptation for competitive profiling to map drug activity across the lipid-interaction proteome. This latter advance not only offers a versatile method for determining drug-target (and off-target) engagement in cells, but also provides a glimpse into the broader ligandability of the lipid-interaction proteome. That many of the lipid- and drug-binding proteins discovered herein derive from classes not known to possess natural or synthetic ligands indicates the lipid-interaction proteome should constitute a rich source for future pharmacological inquiry.
Our studies with NUCB1 establish an experimental framework for efficiently progressing from the discovery of lipid-binding proteins to the development of selective ligands for the functional analysis of these proteins. Important to the success of these efforts was the adaptation of the lipid probes for high-throughput screening of a small-molecule library. We recognize that the described FluoPol assay may not be straightforward to apply to all of the lipid-interacting proteins discovered here — in particular, those that represent multi-pass transmembrane proteins, which are often difficult to purify and study in solution. Other interactions may reflect binding of lipids to protein complexes that require careful in vitro reconstitution for further analysis. Regardless, we are emboldened by the extent to which the lipid probes can be applied at each step in the experimental process of mapping lipid-regulatory pathways, including target discovery, ligand screening and optimization, and confirmation of selective target engagement for optimized ligands in cells.
Our findings also demonstrate how untargeted lipidomics of cells treated with selective (and control) ligands can provide initial insights into the biochemical functions of lipid-binding proteins like NUCB1, which may facilitate the intracellular transfer of NAEs/NATs for delivery to metabolic enzymes, such as FAAH and PTGS2. Other proteins have been found to contribute to NAE transport (Kaczocha et al., 2009; Oddi et al., 2009), including FABP5 (Kaczocha et al., 2009), which was also identified herein as a target of the arachidonoyl lipid probes. Notably, a similar mechanism exists for the biosynthesis of leukotrienes, where the non-enzymatic, auxiliary protein arachidonate 5-lipoxygenase activating protein (ALOX5AP or FLAP) facilitates transfer of AA to arachidonate 5-lipoxygenase (ALOX5) (Evans et al., 2008).
When considering the broader ligandability of the lipid-interaction proteome, it is noteworthy that the drugs assayed herein showed markedly distinct off-target profiles (Figure 5D and Table S3). This result indicates that the lipid-interaction proteome is rich and diverse in its ligand-binding content and the testing of additional, structurally distinct drugs should uncover even more ligand-protein interactions. We also observed that the off-targets for specific drugs, in some cases, share functionality. The three most potent targets of Ro 48-8071, for instance, were sequence-unrelated, membrane-bound enzymes (LSS, EBP) and proteins (TMEM97) involved in the metabolism and regulation cholesterol (Bartz et al., 2009; Laggner et al., 2005; Trapani et al., 2011), suggesting the potential to develop drugs that impact multiple nodes in lipid pathways.
We should also mention some of the technical limitations of mapping lipid- and ligand-binding proteins in cells using chemical proteomics. There is the potential for false-negative outcomes in the form of authentic lipid-binding proteins that fail to interact with the probes due to structural modifications imposed by the photocrosslinking and alkyne groups. As we have shown herein, these groups can be located at different positions on the lipid probes to overcome a negative impact on certain protein interactions. One also needs to be aware of the converse outcome in that we do not expect all lipid probe-enriched targets to reflect specific-binding interactions. Distinguishing specific from non-specific binding events can be facilitated by comparative profiling of structurally distinct lipid probes or competition experiments with exogenous ligands or drugs. It is also possible that some of the drug-dependent changes in lipid probe targets could reflect indirect effects on protein expression or function. We note, however, that more than 50% of the liganded proteins discovered herein showed evidence of interaction with more than one small-molecule competitor (Table S3). Since the tested ligands target distinct biochemical pathways, we interpret these data to indicate that most of the decreases in lipid probe labeling likely reflect primary interactions between competing ligands and proteins as opposed to secondary effects on protein function or expression.
In summary, the output of our studies is a global lipid-interaction map rich in both known and previously unannotated lipid-binding proteins, as well as the realization derived from competitive drug profiling experiments that these proteins constitute a fertile landscape for ligand development. We anticipate that the chemical proteomic methods described herein, by providing a fully integrated approach to discover both lipid-binding proteins and selective ligands to perturb the function of these proteins, will facilitate the characterization of lipid pathways that make important contributions to human health and disease.
Experimental Procedures
Materials
Internal standards for LC/MS analysis and competitors were purchased from Cayman Chemical Company, Sigma-Aldrich or Avanti Lipids. Lipid probes and NUCB1 ligands were synthesized according to methods outlined in the Supplemental Experimental Procedures. UV-mediated crosslinking was performed on a Stratagene, UV Stratalinker™ 1800 Crosslinker equipped with 365 nm light bulbs.
Live cell labeling with clickable photoaffinity probes
For gel-based profiling, cells were plated at a density of 2.5 × 106 cells/6cm plate and grown for 18-24 h prior to labeling. The indicated photoaffinity probe and, if applicable, competitors or vehicle were dissolved in fresh media (1.5 mL) and warmed to 37 °C. For competition experiments, serum-free media was used, whereas standard growth media containing 10% (v/v) fetal bovine serum (FBS) was used for probe-probe comparisons. The media from each 6-cm plate was then aspirated and the cells were washed with Dulbecco's Phosphate-Buffered Saline (DPBS) (2 × 3 mL) before adding media solutions containing probes and competitors. Cells were incubated at 37 °C for 30 min before the media was removed and the cells were directly exposed to 365 nm light for 10 min at 4 °C. Alternatively, for No UV control experiments, probe-treated cells were incubated at 4 °C for 10 min in ambient light.
For MS-based experiments, cell labeling was performed in a similar manner as described above. Modifications to this protocol included using isotopically light and heavy SILAC cells and increasing the cell count to increase protein yield. Specifically, SILAC cells were plated at a density of 4 × 106 cells/10cm plate and grown to near complete confluency prior to labeling. Additionally, probe and, if applicable, vehicles or competitors were dissolved together in SILAC media (4.0 mL) with or without dialyzed FBS (10%). Isotopically light cells were labeled with the arachidinoyl probe (AEA-DA or A-DA) and irradiated with UV for 10 min at 4 °C. ‘Heavy’ cells were subjected to variable conditions as specified in each experiment, including treatments with alternative lipid probes (OEA-DA, PEA-DA, O-DA or S-DA) or competitors.
Gel-based analysis of crosslinked proteins
Cell lysates prepared according to methods described in the Supplemental Experimental Procedures were diluted to 1.0 mg/mL (total protein concentration) and 50 μL of each proteome was transferred to separate wells in a 96-well plate and subjected a freshly prepared “click” reagent mixture containing TBTA (3.0 μL/sample, 1.7 mM in 4:1 DMSO:t-BuOH), CuSO4 (1.0 μL/sample, 50 mM in H2O), TCEP (1.0 μL/sample, 50 mM in DPBS and Rh-N3 (1.0 μL/sample, 1.25 mM in DMSO). After incubating for 1 h at room temperature, each reaction was quenched with 4× SDS loading buffer (17 μL), and proteins were immediately resolved using SDS-PAGE (10% acrylamide gel) and detected by in-gel fluorescent scanning on a Hitachi FMBIO-II flatbed fluorescence scanner.
Proteomic analysis by mass spectrometry and data analysis
Isotopically heavy and light proteomes derived from probe labeled cells were mixed in equal proportions and processed for CuAAC conjugation to biotin-N3, streptavidin enrichment and MS-analysis as described in the Supplemental Experimental Procedures. Proteomic samples were analyzed using a Thermo Orbitrap Velos mass spectrometer and the raw data was processed as described in detail in the Supplemental Experimental Procedures. SILAC results for identification of UV-dependent probe targets and comparison of structurally related lipid probes in HEK293T and Neuro2a represent data combined from 2–3 separate biological replicates. The soluble and membrane fractions from each biological replicate were analyzed separately to improve protein coverage. Median peptide SILAC ratios were then filtered to assure each protein ratio was derived from three or more unique and quantified peptides and that the combined quantified peptide ratios possessed a standard deviation of less than 10. SILAC ratios complying with these criteria were then averaged with ratios acquired from replicates and the alternate fraction (membrane or soluble) to provide a final value which is reported in Table S1. If no replicate values were detected, the SILAC ratio from this single occurrence was included only if each target was also quantified in probe-versus-probe experiments according to the above criteria. We reasoned that including data from instances where a target was identified in a single replicate was justified based on our analysis of probe-versus-No UV data where we found a >90% confirmation of UV-dependence (SILAC ratio ≥ 3.0) of targets identified across multiple replicates. Furthermore, many of these singly quantified targets were also identified in both cell lines (Neuro2a and HEK293T) further supporting their legitimacy as lipid probe targets. See Table S1 for a list of individual peptide sequences, charge states, and ratios detected for each protein in HEK293T cells. See Supplemental Experimental Procedures for details on mapping probe-modified peptides in purified NUCB1 and cell proteomes.
UV-dependent lipid probe targets were defined as proteins that complied with the following criteria: 1) the protein was identified and quantified (according to the above criteria) in both probe-versus-probe and probe-versus-No UV datasets; 2) the protein exhibited a mean SILAC ratio of ≥ 3.0 in probe-versus-No UV experiments; 3) the protein exhibited a mean SILAC ratio of < 2.0 and > 0.5 in probe-versus-probe. Only UV-dependent targets are shown in Table S1. For competition experiments, only proteins that qualified as UV-dependent targets according to the above criteria were included in the analysis (see Table S2 and S3). Furthermore, SILAC ratios for each competition experiment were only included if they were derived from two or more unique and quantified peptides.
Targeted and untargeted lipidomics
Cellular lipids were extracted and analyzed in a similar manner to previously described methods (Saghatelian et al., 2004). See Supplemental Experimental Procedures for details.
Supplementary Material
Acknowledgments
We thank G.M. Simon and M.M. Dix for assistance with proteomics (data analysis and technical assistance, respectively), L. Bar-Peled for assistance with shRNA studies, M.W. Buczynski and K.L. Hsu for guidance with LC-MS methods, and M.L. Matthews for assistance with recombinant protein expression and purification. This work was supported by the National Institutes of Health Grants CA132630 (B.F.C.) and DA032541 (M.J.N), and a predoctoral fellowship provided by NSS (PhD) A*STAR (K.M.L).
Footnotes
Author Contributions: M.J.N. and B.F.C. conceived of research and wrote the paper. M.J.N. synthesized lipid probes. M.J.N., K.M.L, A.B.C. F.P. and S.K. performed gel-based probe profiling experiments. M.J.N., K.M.L., B.E.C. and A.B.C. performed MS-based lipid probe profiling experiments and M.J.N., B.E.C. and T.-A.I. analyzed the MS data. B.E.C. designed, performed and analyzed data from site-of-labling experiments. J.O. assisted with recombinant NUCB1 expression and purification. A.B.C. developed the NUCB1 FluoPol assay and performed the HTS and hit validation with M.J.N. M.J.N. and K.M.L. performed medicinal chemistry for NUCB1 ligand optimization. M.J.N. and K.M.L. developed methods, performed experiments and analyzed data for lipidomic profiling experiments. S. J. B. and H. R. provided facilities for high-throughput screening.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, Young T, Wren P, Zhang Y, Swaney S, et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011;338:114–124. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballif BA, Mincek NV, Barratt JT, Wilson ML, Simmons DL. Interaction of cyclooxygenases with an apoptosis- and autoimmunity-associated protein. Proc Natl Acad Sci USA. 1996;93:5544–5549. doi: 10.1073/pnas.93.11.5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros RP, Gustafsson JA. Estrogen receptors and the metabolic network. Cell Metab. 2011;14:289–299. doi: 10.1016/j.cmet.2011.08.005. [DOI] [PubMed] [Google Scholar]
- Bartz F, Kern L, Erz D, Zhu M, Gilbert D, Meinhof T, Wirkner U, Erfle H, Muckenthaler M, Pepperkok R, et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab. 2009;10:63–75. doi: 10.1016/j.cmet.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J Lipid Res. 2009;50(Suppl 1):S15–27. doi: 10.1194/jlr.R800054-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol. 2012;13:213–224. doi: 10.1038/nrm3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alba E, Tjandra N. Structural studies on the Ca2+-binding domain of human nucleobindin (calnuc) Biochemistry. 2004;43:10039–10049. doi: 10.1021/bi049310a. [DOI] [PubMed] [Google Scholar]
- Diez-Dacal B, Gayarre J, Gharbi S, Timms JF, Coderch C, Gago F, Perez-Sala D. Identification of aldo-keto reductase AKR1B10 as a selective target for modification and inhibition by prostaglandin A(1): implications for antitumoral activity. Cancer Res. 2011;71:4161–4171. doi: 10.1158/0008-5472.CAN-10-3816. [DOI] [PubMed] [Google Scholar]
- Evans JF, Ferguson AD, Mosley RT, Hutchinson JH. What's all the FLAP about?: 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol Sci. 2008;29:72–78. doi: 10.1016/j.tips.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Evans JF, Hutchinson JH. Seeing the future of bioactive lipid drug targets. Nat Chem Biol. 2010;6:476–479. doi: 10.1038/nchembio.394. [DOI] [PubMed] [Google Scholar]
- Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis GA, Fayard E, Picard F, Auwerx J. Nuclear receptors and the control of metabolism. Annu Rev Physiol. 2003;65:261–311. doi: 10.1146/annurev.physiol.65.092101.142528. [DOI] [PubMed] [Google Scholar]
- Gallego O, Betts MJ, Gvozdenovic-Jeremic J, Maeda K, Matetzki C, Aguilar-Gurrieri C, Beltran-Alvarez P, Bonn S, Fernandez-Tornero C, Jensen LJ, et al. A systematic screen for protein-lipid interactions in Saccharomyces cerevisiae. Mol Syst Biol. 2010;6:430. doi: 10.1038/msb.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbens J, Ruijter E, de Fays LE, Damen JM, de Kruijff B, Slijper M, Rijkers DT, Liskamp RM, de Kroon AI. Photocrosslinking and click chemistry enable the specific detection of proteins interacting with phospholipids at the membrane interface. Chem Biol. 2009;16:3–14. doi: 10.1016/j.chembiol.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Haberkant P, Raijmakers R, Wildwater M, Sachsenheimer T, Brügger B, Maeda K, Houweling M, Gavin ACC, Schultz C, van Meer G, et al. In vivo profiling and visualization of cellular protein-lipid interactions using bifunctional fatty acids. Angew Chem Int Ed. 2013;52:4033–4038. doi: 10.1002/anie.201210178. [DOI] [PubMed] [Google Scholar]
- Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med. 2008;14:461–469. doi: 10.1016/j.molmed.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Heal W, Dang T, Tate E. Activity-based probes: discovering new biology and new drug targets. Chem Soc Rev. 2011;40:246–257. doi: 10.1039/c0cs00004c. [DOI] [PubMed] [Google Scholar]
- Hong C, Tontonoz P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr Opin Genet Dev. 2008;18:461–467. doi: 10.1016/j.gde.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods. 2013;10:259–264. doi: 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Glaser ST, Deutsch DG. Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci USA. 2009;106:6375–6380. doi: 10.1073/pnas.0901515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, Nakayama Y, Yamauchi H, Konishi M, Miyake A, Mori M, Ohta M, Itoh N, Fujimoto M. Neurotrophic activity of neudesin, a novel extracellular heme-binding protein, is dependent on the binding of heme to its cytochrome b5-like heme/steroid-binding domain. J Biol Chem. 2008;283:4323–4331. doi: 10.1074/jbc.M706679200. [DOI] [PubMed] [Google Scholar]
- Laggner C, Schieferer C, Fiechtner B, Poles G, Hoffmann RD, Glossmann H, Langer T, Moebius FF. Discovery of high-affinity ligands of sigma1 receptor, ERG2, and emopamil binding protein by pharmacophore modeling and virtual screening. J Med Chem. 2005;48:4754–4764. doi: 10.1021/jm049073+. [DOI] [PubMed] [Google Scholar]
- Leclerc P, Biarc J, St-Onge M, Gilbert C, Dussault AA, Laflamme C, Pouliot M. Nucleobindin co-localizes and associates with cyclooxygenase (COX)-2 in human neutrophils. PloS one. 2008;3:e2229. doi: 10.1371/journal.pone.0002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Bogyo M. Target deconvolution techniques in modern phenotypic profiling. Curr Opin Chem Biol. 2013;17:118–126. doi: 10.1016/j.cbpa.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hao P, Li L, Tan CY, Cheng X, Chen GY, Sze SK, Shen HM, Yao SQ. Design and synthesis of minimalist terminal alkyne-containing diazirine photo-crosslinkers and their incorporation into kinase inhibitors for cell- and tissue-based proteome profiling. Angew Chem Int Ed. 2013;52:8551–8556. doi: 10.1002/anie.201300683. [DOI] [PubMed] [Google Scholar]
- Lomenick B, Hao R, Jonai N, Chin R, Aghajan M, Warburton S, Wang J, Wu R, Gomez F, Loo J, et al. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci USA. 2009;106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson E, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Mechoulam R. Endocannabinoids. Eur J Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- Muro E, Atilla-Gokcumen GE, Eggert US. Lipids in cell biology: how can we understand them better? Mol Biol Cell. 2014;25:1819–1823. doi: 10.1091/mbc.E13-09-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh T, Rivera R, Chun J. Insights into the pharmacological relevance of lysophospholipid receptors. Br J Pharmacol. 2012;165:829–844. doi: 10.1111/j.1476-5381.2011.01622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddi S, Fezza F, Pasquariello N, D'Agostino A, Catanzaro G, De Simone C, Rapino C, Finazzi-Agro A, Maccarrone M. Molecular identification of albumin and Hsp70 as cytosolic anandamide-binding proteins. Chem Biol. 2009;16:624–632. doi: 10.1016/j.chembiol.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Ong SEE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective ‘ligation’ of azides and terminal alkynes. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Rouzer CA, Marnett LJ. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem Rev. 2011;111:5899–5921. doi: 10.1021/cr2002799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland MM, Bostic HE, Gong D, Speers AE, Lucas N, Cho W, Cravatt BF, Best MD. Phosphatidylinositol 3,4,5-trisphosphate activity probes for the labeling and proteomic characterization of protein binding partners. Biochemistry. 2011;50:11143–11161. doi: 10.1021/bi201636s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW. Fifty years of advances in bile acid synthesis and metabolism. J Lipid Res. 2009;50:120–125. doi: 10.1194/jlr.R800026-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghatelian A, McKinney MK, Bandell M, Patapoutian A, Cravatt BF. A FAAH-regulated class of N-acyl taurines that activates TRP ion channels. Biochemistry. 2006;45:9007–9015. doi: 10.1021/bi0608008. [DOI] [PubMed] [Google Scholar]
- Samad TA, Sapirstein A, Woolf CJ. Prostanoids and pain: unraveling mechanisms and revealing therapeutic targets. Trends Mol Med. 2002;8:390–396. doi: 10.1016/s1471-4914(02)02383-3. [DOI] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FBM, Franken H, Werner T, Savitski MF, Eberhard D, Molina DM, Jafari R, Dovega RB, Klaeger S, et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346 doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Shimizu T. Lipid Mediators in Health and Disease: Enzymes and Receptors as Therapeutic Targets for the Regulation of Immunity and Inflammation. Annu Rev Pharmacol. 2009;49:123–150. doi: 10.1146/annurev.pharmtox.011008.145616. [DOI] [PubMed] [Google Scholar]
- Simon G, Niphakis M, Cravatt B. Determining target engagement in living systems. Nat Chem Biol. 2013;9:200–205. doi: 10.1038/nchembio.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- Speers AE, Cravatt BF. A tandem orthogonal proteolysis strategy for high-content chemical proteomics. J Am Chem Soc. 2005;127:10018–10019. doi: 10.1021/ja0532842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Ge J, Zhu B, Zheng YG, Zhu Q, Yao SQ. Target identification of biologically active small molecules via in situ methods. Curr Opin Chem Biol. 2013;17:768–775. doi: 10.1016/j.cbpa.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Tate EW, Kalesh KA, Lanyon-Hogg T, Storck EM, Thinon E. Global profiling of protein lipidation using chemical proteomic technologies. Curr Opin Chem Biol. 2014;24C:48–57. doi: 10.1016/j.cbpa.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani L, Segatto M, Ascenzi P, Pallottini V. Potential role of nonstatin cholesterol lowering agents. IUBMB Life. 2011;63:964–971. doi: 10.1002/iub.522. [DOI] [PubMed] [Google Scholar]
- Urbano M, Guerrero M, Rosen H, Roberts E. Modulators of the Sphingosine 1-phosphate receptor 1. Bioorg Med Chem Lett. 2013;23:6377–6389. doi: 10.1016/j.bmcl.2013.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.