

Abstract
To ensure duplication of the entire genome, eukaryotic DNA replication initiates from thousands of replication origins. The replication forks move through the chromatin until they encounter forks from neighboring origins. During replication fork termination forks converge, the replisomes disassemble and topoisomerase II resolves the daughter DNA molecules. If not resolved efficiently, terminating forks result in genomic instability through the formation of pathogenic structures. Our recent findings shed light onto the mechanism of replisome disassembly upon replication fork termination. We have shown that termination-specific polyubiquitylation of the replicative helicase component – Mcm7, leads to dissolution of the active helicase in a process dependent on the p97/VCP/Cdc48 segregase. The inhibition of terminating helicase disassembly resulted in a replication termination defect. In this extended view we present hypothetical models of replication fork termination and discuss remaining and emerging questions in the DNA replication termination field.
Keywords: DNA replication, Mcm2–7, p97 segregase, replication termination, replisome, replicative helicase, ubiquitin, Xenopus
Abbreviations
- CMG
Cdc45, Mcm2–7, GINS complex
- CRL
cullin-RING ligase
- DDR
DNA damage response
- D loop
displacement loop
- DSB
double strand break
- DUB
deubiquitylating enzyme
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum associated protein degradation
- GINS
Go-Ichi-Ni-San, complex made of Sld5, Psf1, Psf2, Psf3
- ICL
intra-strand crosslink
- MCM
Minichromosome maintenance
- OriC
chromosomal replication origin
- RING
really interesting gene
- R loop
RNA:DNA hybrid
- RPC
Replisome Progression Complex
- Ter
termination site
- Tus-Ter
terminus utilisation substance - termination
Introduction
The elucidation of the double helix structure by Watson and Crick in the 1950s led to the theory of semi-conservative replication: the double helix is unwound and new strands synthesized in a complementary manner, so that the new double strand consists of one new and one old strand.1,2 Ever since this discovery scientists have been trying to describe how this process works in detail. In eukaryotes, much of the work has focused on understanding the synthesis of DNA and, within the last 15 years, the initiation stage of DNA replication. However, very little is known as to how this process terminates. Resolution of structures created during termination of DNA replication is essential for accurate duplication of eukaryotic genomes since, if not resolved efficiently, terminating replication forks can result in genomic instability.3,4 Our recent work is the first indication of the mechanism that unloads the replication machinery at the termination of DNA replication forks.5-7
The replication machinery (replisome) consists of about 150 proteins and is built around a few key organizing centers. One of them is a replicative helicase, which is the protein complex that can unwind double stranded DNA and create the templates for DNA synthesis.8 In eukaryotic cells the core of this replicative helicase is formed by the hexameric Mcm2–7 (Minichromosome maintenance) complex. Hundreds of thousands of Mcm2–7 complexes are loaded onto DNA in the form of head-to-head double hexamers9-11 and 30–50 thousand of these are activated per human cell during DNA replication.12 Activation of the helicase requires the action of S-phase kinases, which recruit two additional factors, Cdc45 and GINS, to the Mcm2–7 hexamer leading to the formation of the CMG complex (Cdc45, Mcm2–7, GINS).13 This active helicase complex is built around a single Mcm2–7 hexamer and thus the two CMGs that are formed from an inactive double hexamer of Mcm2–7 establish bi-directional replication forks by unwinding DNA and moving away from each other.8,14,15 Only a small proportion of chromatin-loaded Mcm2–7 complexes are activated during S-phase, while the remaining serve as back-up “dormant origins”, which are activated only in the case of replication stress.16 The active helicase complexes form the tips of replication forks and travel through DNA, unwinding it until they meet forks coming from the opposite direction (originating from the neighboring origin). This convergence can happen anywhere within the genome and leads to the termination of the converging replication forks. During this process, DNA in between the forks needs to be fully replicated, the replisomes disassembled and the newly synthesized chromatids untangled, so that they can be divided in the forthcoming mitosis. How all these processes happen in eukaryotic cells is still poorly understood.
Our recent work focused on the process of the replisome disassembly at the termination of replication forks. As it is essential that DNA is replicated only once per cell cycle, S-phase kinases, apart from activating Mcm2–7 complexes, also inhibit their further loading onto DNA.17 This mechanism ensures that no Mcm2–7 complexes are loaded onto already replicated DNA, thus no re-replication occurs. It means, however, that if the active helicase is prematurely removed from the replication fork it cannot be reloaded due to inhibition of the Mcm2–7 loading mechanism in S-phase. Therefore, the presence of the active helicase is very precious at the fork and it is prevented from unnecessarily dissociating from DNA. Both the active and inactive helicases encircle DNA in a very stable way and cannot be removed from DNA with high salt buffers or detergent. Moreover, one of the main functions of the S-phase checkpoint is to ensure the stability of the active helicase and the replisome at the replication fork in case of DNA replication stress.18 Conversely, this means that a highly regulated mechanism is required to unload the active helicase once its role is fulfilled i.e. at the termination of DNA replication forks.
Our recent findings: dissolution of replisome at the terminating replication forks
Using the well-established Xenopus laevis egg extract system, which provides an excellent model for biochemical studies of DNA replication,19 we showed that blocking polyubiquitylation results in the prolonged association of the active helicase with replicating chromatin and a DNA replication termination defect. This accumulation was due to a defect in unloading of the active helicase at the terminating replication forks, and the accumulated terminating helicases remained in a complex of a size identical to the normal active helicase. This unloading defect was driven through ubiquitin chains linked through lysine 48 (K48), which are usually a marker for degradation of the modified substrate by the proteasome system. However, the inactivation of proteasomal activity, using a small drug inhibitor, could not recapitulate the replisome disassembly phenotype, suggesting that these K48 chains may play a signaling role.5
We observed that only one component of the active helicase was polyubiquitylated during S-phase on replicating chromatin: Mcm7, one of the subunits of the Mcm2–7 complex. Mcm7 was ubiquitylated with K48-linked ubiquitin chains but was not degraded on chromatin by proteasomal activity. Instead, the observed polyubiquitylation was followed by disassembly of the active helicase, and this was dependent on the p97/VCP/Cdc48 protein remodeller. Importantly, Mcm7 was polyubiquitylated only when replication forks were allowed to terminate. It was strongly inhibited when progression of the forks was blocked by inhibition of the DNA polymerases or when termination itself was perturbed with the Topoisomerase II (Topo II) inhibitor ICRF193. Both, the ubiquitylation of Mcm7 and disassembly of the active helicase, were dependent on activity of the cullin family of ubiquitin ligases, as both were blocked with MLN-4924, an inhibitor of cullin-activating neddylation. Altogether, our findings suggest an unloading mechanism whereby an unknown aspect of replication fork termination leads to cullin-dependent ubiquitylation of Mcm7 with a K48-linked ubiquitin chain. This, in turn is recognized by the p97 complex and remodeled causing the helicase to be removed from chromatin5 (Fig. 1).
Figure 1.
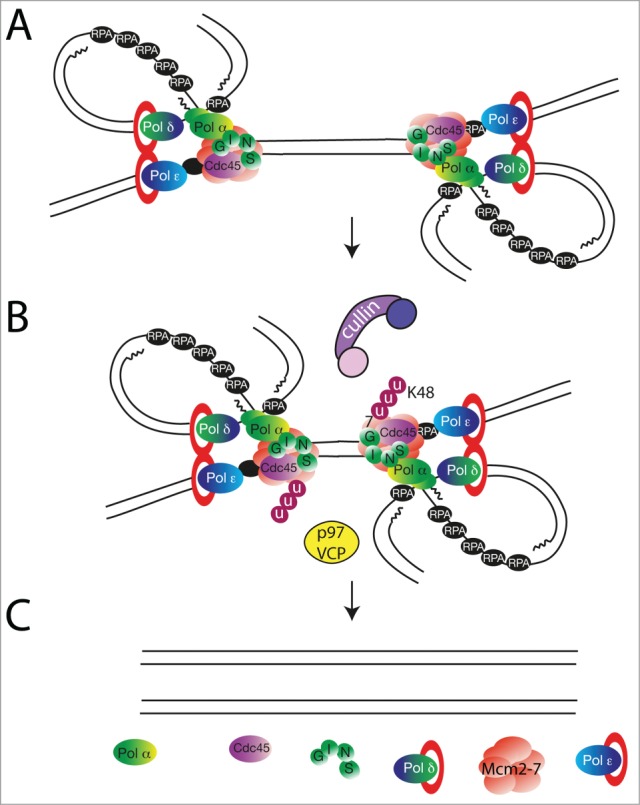
A speculative model of replisome dissolution at the termination of DNA replication forks based on data published in (5, 55). (A) Two replication forks from neighboring origins approach each other. (B) The Mcm7 subunit of the CMG complex becomes ubiquitylated with lysine-48-linked ubiquitin chains in a process dependent on cullin-type ubiquitin ligase. The ubiquitylated Mcm7 is recognized by protein segregase p97/VCP/Cdc48. (C) p97 activity is required to remodel the active helicase complex resulting in replisome disassembly and removal from chromatin.
Unloading of inactive and active Mcm2–7 complexes
Importantly, the ubiquitylation-driven disassembly of the helicase described above specifically affected the CMG (i.e., activated Mcm2–7) complexes. CMG complexes are formed from only 5–10% of all Mcm2–7 present on chromatin in Xenopus laevis egg extract, and we do not see a delay in the unloading of the inactive Mcm2–7 complexes.5 This suggests that the mechanisms involved in unloading these two types of Mcm2–7 complexes are different. This is not surprising as inactive Mcm2–7 complexes form double hexamers encircling double stranded DNA along which they can slide,9,20 while active helicase complexes contain a single Mcm2–7 hexamer, Cdc45, GINS and encircle the single strand of the leading strand at the fork: their movement along which unwinds DNA.13,15,21 CMG complexes are also surrounded by multiple regulatory proteins forming Replisome Progression Complexes (RPCs).8 It is currently unclear if the inactive Mcm2–7 complexes are unloaded as the progressing forks encounter them or if they slide in front of the progressing forks up to the point of termination of two neighboring forks (Fig. 2). In either case, they are removed from chromatin throughout S-phase as replication progresses, and for unloading depend on active replication.22
Figure 2.
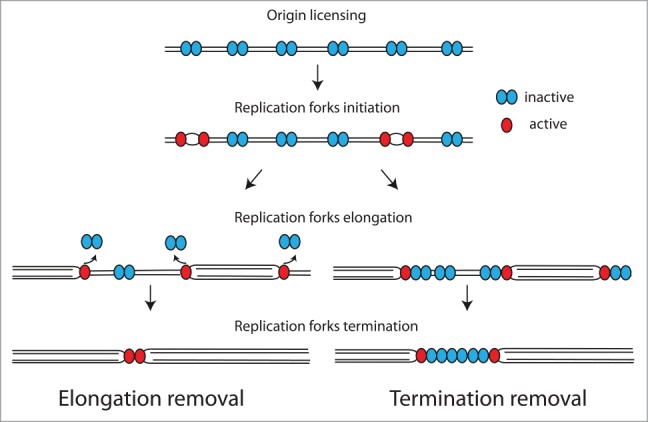
Two possible mechanisms for dormant origin removal from chromatin. Inactive Mcm2–7 complexes may be removed as active forks approach them (left panel, Elongation removal) or pushed in front of the progressing forks and removed at sites of replication fork termination (right panel, Termination removal).
It has been shown previously that MCM Binding Protein (Mcm-BP) and its interacting partner Usp7 can both stimulate the unloading of Mcm2–7 complexes from chromatin.23-25 Importantly, immunodepletion of Mcm-BP from Xenopus laevis egg extract does affect the bulk of Mcm2–7 complexes on chromatin, indicating its importance for unloading the majority of chromatin-loaded Mcm2–7 (the inactive Mcm2–7 double hexamers).24 Interestingly, this defect in removing inactive, dormant origins could also lead to a delay in termination by presenting a roadblock for terminating forks (Fig. 2, Termination removal model), although Mcm-BP depleted extract did not exhibit any defect in replicon fusing.24 In immortalized human cell lines, as well as affecting the chromatin levels of Mcm2–7, Mcm-BP downregulation resulted in increased cellular levels of Mcm2–7 complexes, replication initiation problems, replication stress, post-replicative cohesion defects, G2 checkpoint activation, mitotic problems and altered nuclear morphology.26-30 Usp7, a deubiquitylating enzyme (DUB), was also shown to affect the removal of Mcm2–7 complexes from chromatin in human cells, while also being able to interact with Mcm-BP.23 Similarly to Mcm-BP, Usp7 is reported to regulate many cell cycle and DNA repair processes: during S-phase Usp7 stabilizes the replisome component Claspin31 and epigenetic regulator DNA methyltransferase 1 DNMT1.32 During the DNA damage response, Usp7 deubiquitylates and stabilizes translesion polymerase Pol-eta,33 nucleotide excision repair factor XPC,34 checkpoint kinase Chk135 and p53 acetyltransferase Tip60.36 Finally, in mitosis, Usp7 regulates the level of the spindle assembly checkpoint component - Bub3,37 E3 ubiquitin ligase - CHFR and the cell cycle regulator kinase - Aurora A.38 With so many cellular functions reported for both Mcm-BP and Usp7 it remains to be determined if the Mcm2–7 unloading defect observed in human cells upon downregulation of Mcm-BP and Usp7 reflects indeed a direct role of these proteins in Mcm2–7 disassembly. Additionally, it was also suggested that CDK driven phosphorylation of Mcm4 can reduce the affinity of Mcm2–7 for chromatin.39 Altogether, the exact mechanism for the unloading of dormant Mcm2–7 complexes is still to be established.
Topological resolution of terminating replication forks
At present we know very little about the mechanism of resolution of DNA replication forks in eukaryotic cells, but it has been studied in more detail in bacteria. In the circular chromosomes of bacteria, replication typically begins at a single origin, oriC. The two resultant replication forks proceed in opposite directions and replication is terminated at a fixed site: the sequence-specific termination (Ter) site that is located directly opposite the replication origin.40,41 A specific terminator protein is bound at these sites, so that when the helicase collides with it, a “replication fork trap” is formed, which pauses the replicative machinery until the fork traveling from the opposite direction meets it.42 It is believed that this second approaching fork displaces the terminator protein and completes DNA unwinding.43 Additionally, RecQ helicase activity can resolve converging replication forks: thus it may replace the standard replicative helicase during the final stage of termination.44 The exact mechanism of disassembly of the replisomes remains unknown, but the existing evidence suggests that Pol I (which is normally involved in Okazaki fragment maturation) may replace Pol III to complete termination.45 The two entwined double stranded chromosomes are subsequently decatenated by Topoisomerase IV activity.46 Another DNA helicase, RecG, which is essential for DNA recombination and repair has also been shown as important for limiting re-replication, for example, at D and R loops near the termination site.47,48
In eukaryotic cells, which start replication from many thousands of origins, replication forks terminate when they converge at random sites thus, it is believed that specific DNA sequences are not generally required for this.49 Eukaryotic Ter sites have, however, previously been described and studied as examples of specific sites of replication forks termination. These Ter sites contain fork-pausing elements and help to coordinate fork fusion at these specific locations. In budding yeast, within Ter sites, the helicase Rrm3 is important for fork progression and Topo II facilitates fork fusion possibly by resolving precatenates behind the fork.3,50 Additionally, the fission yeast DNA helicase Pfh1 is known to promote merging of replication forks at termination by allowing fork progression past DNA-protein barriers present at barrier-specific termination sites.51 It is unclear however, whether these helicases play an important role at the majority of termination sites, which do not occur at Ter sites, but are spread randomly in the genome. Nevertheless, we do know that Topo II plays an essential role in the termination of replication forks irrespectively of where they occur. Topo II removes hemicatenates within the parental DNA in between the converging forks as well as precatenates of sister chromatids behind the converging forks. Expression of catalytically inactive Topo II leads to G2 arrest, with a failure to complete replication and persistence of hypercatenated DNA molecules.52 Moreover, an inhibitor of Topo II activity, ICRF-193, impairs the completion of DNA replication and leads to a termination defect in Xenopus laevis egg extract.53 Finally, we have shown that when replication termination is blocked using ICRF-193, Mcm7 ubiquitylation and active helicase disassembly are substantially reduced.5 To resolve topological stress, Topo II acts via an active strand passage mechanism: it creates a double strand break in a duplex DNA molecule and passes another duplex DNA molecule through this break.54 Topo II forms a covalent complex with DNA in a reversible reaction. This ensures that DNA ends are protected and do not start a DNA damage response (DDR) and also that they can be quickly and efficiently re-ligated without the need of a DDR.54
Based on our current knowledge of DNA replication and replication termination in bacteria and eukaryotic cells, there are at least 3 theoretical models of the termination mechanism (Fig. 3). Firstly, a direct collision between the two helicases may occur, after which they become disassembled (Fig. 3A – Simultaneous removal of converging replisomes). Additional helicases (like Rrm3) and polymerases may possibly be recruited to resolve the final sections of DNA.3 Secondly, after collision one helicase may first be disassembled before the remaining helicase can complete replication without the need for additional non-replisome factors (Fig. 3B – Sequential removal of converging replisomes). Thirdly, it is possible that the replicative helicases may cross each other at the termination site and slide onto double-stranded DNA before being removed. This may be possible as the eukaryotic helicase most likely encircles single-stranded DNA and travels along the leading strand15,21 (Fig. 3C – Passing replisomes). This last model could explain how dia2Δ S.cerevisiae cells can survive through chromosome segregation despite retaining their CMG complexes on DNA until the next cell cycle.55
Figure 3.
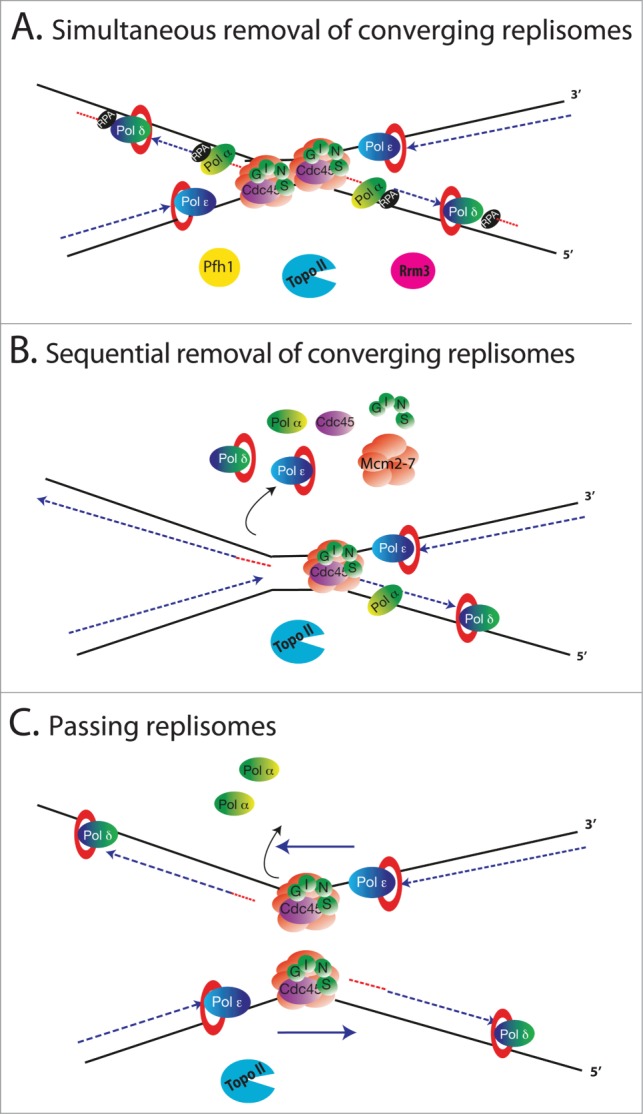
Speculative models of replication fork termination mechanisms. Two forks approach each other traveling in opposite directions with the CMG complexes unwinding DNA at the tips of the forks. Topo II helps to release the tension created by hemicatenates between approaching forks. (A) Simultaneous removal of converging replisomes. Two helicases collide with each other leading to disassembly of both replisomes and resolution of the terminating DNA with the potential help of additional factors and Topo II. (B) Sequential removal of converging replisomes. The convergence of two helicases leads to disassembly of one of the replisomes while the other finishes unwinding and replicating the remaining DNA. The sister chromatids are detangled by Topo II. (C) Passing replisomes. Two approaching forks pass each other at the termination site as each helicase is traveling only on the leading strand i.e., on the parental strand within the new sister chromatids. The CMG complex may be disassembled when it approaches the last Okazaki fragment of the lagging strand in front of it, start unwinding the last Okazaki fragment or start sliding on the double stranded DNA of the last Okazaki fragment. Again, Topo II detangles the new sister chromatids.
What ubiquitylates Mcm7 at replication forks termination?
The parallel study from Maric et al. showed that in the budding yeast, a cullin-RING family ubiquitin ligase SCFDia2 ubiquitylates Mcm7 at the terminated replisome.55 SCFDia2 is composed of the cullin scaffold protein Cdc53, RING protein Hrt1, adaptor protein Skp1 and substrate recognition F-box protein Dia2. In higher eukaryotes the identity of the ubiquitin ligase targeting Mcm7 is not yet known, as Dia2 does not have a sequence homolog in higher eukaryotes. Our study shows, however, that both Mcm7 ubiquitylation and replisome dissolution depend on activity of the cullin-family ubiquitin ligases in Xenopus egg extract. Taking into account that other elements of this novel mechanism are highly conserved throughout higher eukaryotes, it is likely that a cullin-RING type of ubiquitin ligase (CRL) directly ubiquitylates Mcm7 in higher eukaryotes. Higher eukaryotes express 7 different canonical cullins (Cul1, 2, 3, 4A, 4B, 5 and 7) that each form a multi-subunit ubiquitin ligase. A key feature of CRLs is the formation of a multi-subunit ligase with a common catalytic core but numerous substrate receptors, which can recruit different substrates.56 Altogether, there are over 600 possible combinations of cullin ligases and the challenge lies in finding the substrate receptor combination responsible for Mcm7 ubiquitylation. Cul1, Cul2 and Cul4 are all implicated in the regulation of eukaryotic replication and/or S-phase progression.57-59 Time will show which cullin/substrate receptor combination is responsible for Mcm7 ubiquitylation.
How is the terminating helicase selected for ubiquitylation?
While it is easy to see that inactive and active helicase complexes differ significantly in their structure, it is less clear how cells can specifically recognize the terminating active helicase for polyubiquitylation and disassembly over all other molecules of actively unwinding helicase. The key question worth considering is therefore what is different about the terminating helicase? It is likely that the replicative helicase undergoes a conformational change at termination (possibly followed by a post-translational modification), which acts as a signal to initiate the ubiquitylation of Mcm7. This conformational change or modification is likely to be stable as CMGs accumulated on G1 chromatin from a previous cell cycle (in dia2Δ cells) can readily be ubiquitylated and disassembled upon ligase re-expression.55 Taking into account the termination models outlined above (Fig. 3), the conformational change within the terminating helicase can take different forms. If one of the first two models is true, it may be a conformational change within the helicase induced by the torsion in DNA due to hemicatenates formed between the converging forks.60 Alternatively, the recognized termination structure may be two CMG complexes with the Mcm2–7 C-terminal helicase domains facing each other (opposite to the Mcm2–7 hexamers N-terminus to N-terminus orientation in inactive double hexamers). In the third model (Passing replisomes), the conformational change could be due to the helicase transferring onto the double stranded DNA upon passage of the termination site or the structure of 2 replisomes passing each other. This conformational change could then be recognized directly by the ubiquitin ligase leading to Mcm7 ubiquitylation and CMG disassembly, or by other post-translational modifying enzymes, creating the recognition signal for the ligase. Unfortunately, as the precise mechanism of resolution of the bacterial replisome at the termination of prokaryotic replication forks is not clear, we are unable to draw any analogies to help us distinguish between the above possibilities.
Another way to understand how terminating helicases differ from those unwinding DNA at the forks is to study the ubiquitin ligase targeting Mcm7 and to establish the signal recognized by it. Interestingly, Dia2 interacts with the replisome throughout S-phase and when replication fork progression is slowed down by hydroxyurea treatment.61 It therefore suggests that it is the ubiquitin ligase activity, but not localization, of SCFDia2 that is stimulated specifically at termination of replication forks. Interestingly, CRLs often target substrates upon their priming post-translational covalent modification such as phosphorylation (e.g. CDK inhibitor Sic1, Cyclin E and p27) or hydroxylation (e.g., the transcription factor, Hypoxia Inducible Factor 1α).56 Such a modification-dependent substrate choice can allow selective ubiquitylation of a specific substrate pool – i.e. specifically the helicase, which has no longer any function. It is unlikely, however, that this targeting modification could be applied just by cell cycle regulating kinases such as cyclin-dependent kinases CDKs or Cdc7 kinase, as termination can happen at any time throughout S-phase whenever two forks meet. We have also shown that forks accumulated in S-phase but prevented from physical termination do not support efficient Mcm7 ubiquitylation.5 To conclude, much remains to be learnt about the identity and regulation of the ligase responsible for replisome dissolution and the mechanism by which the terminated helicase is specifically recognized for dissolution.
Is Mcm7 ubiquitylation required for terminating replisome disassembly?
Our research raises another important question: is Mcm7 the only protein that needs to be ubiquitylated for replisome dissolution? At present we know that Mcm7 is the only CMG subunit that is found polyubiquitylated on chromatin during S-phase in both S.cerevisiae and X.laevis.5,55 To test whether it is the essential substrate we need to create a mutant of Mcm7 that is proficient in unwinding DNA but cannot be ubiquitylated. The human homolog of Mcm7 contains 32 lysine amino acids and at least 14 of them have been reported to be ubiquitylated in proteome-wide screens.62-65 Mutation of all of them is impossible as it would most likely result in a non-functional protein. It is also not known if any of these mapped sites are relevant for replication termination as unsynchronised cell populations were used for the proteome-wide screens. The mapped sites may therefore be involved in other cellular processes such as protein quality control, as other subunits of Mcm2–7 complex have also been reported as ubiquitylated substrates. Importantly, it has been shown that cullin complexes (especially Cul1 complexes) display a considerable plasticity in the selection of the lysine residue within the substrate.56 Therefore, a version of Mcm7 with mutated mapped ubiquitylation sites may still be ubiquitylated (on neighboring lysines). Altogether, this is a challenge that will require much further work to resolve.
p97 segregase and the fate of ubiquitylated CMG
Our findings on the final stage of replisome dissolution – its actual remodeling and removal from chromatin – also instigate a number of further questions that need to be answered. We have shown that p97/VCP/Cdc48 segregase activity is required for the disassembly of the CMG complexes at converging forks; most likely by applying its remodeling activity to helicase labeled with K48-linked ubiquitin chains on Mcm7.5 Maric et al. proposed a similar role for Cdc48, the yeast homolog of p97, in CMG disassembly,55 suggesting that this is an evolutionary conserved mechanism. p97 is a highly conserved homohexameric ring-shaped protein complex and a member of the family of AAA-ATPases.66,67 It functions as a ubiquitin-selective segregase, binding ubiquitylated substrates and extracting them from different cellular structures including protein complexes, membranes and chromatin.68,69 Although it was first described as a required factor in the process of endoplasmic reticulum-associated protein degradation (ERAD)70 and membrane fusion,71 in the last decade p97 has emerged as a central player in chromatin related processes such as DNA replication and repair, transcription, chromosome segregation and chromatin decondensation.72 This functional diversity of p97 can be explained by the fact that it is one of the most abundant proteins within the cell and that it can associate with a wide spectrum of “substrate-recruiting cofactors.”66,73,74 p97 segregase forms two major core-complexes by interacting with the well characterized cofactors p47 and the Ufd1-Npl4 heterodimer in a mutually exclusive manner.75 These core-complexes can then associate with extra cofactors that confer a higher degree of substrate specificity.76 Examples of these secondary cofactors are Ubx2, which links p97Ufd1-Npl4 to ERAD, or the recently described DVC1, which recruits p97Ufd1-Npl4 to stalled replication forks after DNA damage.77,78 Apart from interacting with this plethora of “substrate-recruiting cofactors”, p97 also associates with a broad spectrum of “substrate-processing cofactors”, including de-ubiquitylating enzymes (DUBs). The association of p97 with DUBs ultimately lead to de-ubiquitylation of the substrate and prevent its proteasomal degradation.79 Several DUBs have been found to interact with p97 including VCIP135 - a regulator of the p97-mediated Golgi and ER membrane fusion,80,81 YOD1 (and its yeast ortholog Otu1), USP13, ataxin3 and USP25 - linked to the regulation of ERAD79,82-85 and USP50 - which has been implicated in the regulation of the DNA damage checkpoint.84,86
The list of chromatin-bound ubiquitylated proteins that are processed by p97 has been largely expanded within the last decade. In general, these proteins become polyubiquitylated in a context-dependent manner, which induces their p97Ufd1-Npl4-mediated recognition and extraction from chromatin.72 In some instances p97Ufd1-Npl4 recruits the proteasome to ubiquitylated proteins to promote their degradation e.g., Cdt1 (after UV irradiation), SET8 and the RNA Polymerase II subunit Rpb1.87,88 Alternatively, it can also extract the substrate from chromatin and direct it for degradation by the proteasome e.g., Cdt1 during mitosis89 and proteins involved in the repair of DSBs.90,91 In other cases there is no evidence of proteasomal degradation e.g. mitotic kinase Aurora B,92 S.cerevisiae transcriptional repressor α293 and the Polycomb protein L3MBTL1.94
In our study there are still some open questions related to p97-dependent CMG unloading. Firstly, we do not know which “substrate-recruiting cofactors” associate with p97 during this process. It is likely that the heterodimer Ufd1-Npl4 is the major cofactor as for all other reported chromatin-associated p97 substrates. Moreover, p97Ufd1-Npl4 has been shown to be crucial for S-phase progression and normal DNA replication in C. elegans.95,96 Secondly, the fate of ubiquitylated Mcm7 after being removed from chromatin still remains unknown. Our data indicate that Mcm7 is not degraded by proteasome while still on chromatin.5 The ubiquitylated Mcm7 is therefore removed from chromatin by p97 and then either delivered to the proteasome for degradation or de-ubiquitylated by DUBs (either one associated with p97 or another member of the family). It remains to be determined which of the above is correct in order to fully understand the mechanism of active helicase unloading.
The relevance of faultless resolution of terminating replication forks
All of these new questions raised by our findings are of key importance for cell biology, aging and cancer development. Improper replication fork resolution, sections of unreplicated DNA and the resultant DNA breakages, or unloaded replication machinery, can all lead to genomic instability. In bacteria, inactivation of the termination Tus-Ter system in a plasmid replication model leads to rolling-circle replication and thus to plasmid instability.97 Furthermore, depletion of the helicase RecG results in major defects in chromosomal segregation due to a termination defect leading to the initiation of replication near the termination site.4 Given that in eukaryotic cells forks terminate at multiple sites, the potential effects are likely to be more severe. In budding yeast, a genome-wide correlation has been described between fragile sites and replication termination sequences, suggesting that the perturbed termination could lead to breakages at these sites.98 The chromosomes of top2 mutant cells exhibit breaks and remain entangled,3 while cells lacking Rrm3 or Pfh1 exhibit problems with chromosome segregation, including anaphase bridges and lagging chromosomes.51,99 Finally budding yeast dia2Δ cells exhibit highly increased levels of genomic instability, constitutive Rad53 activation and an increase in endogenous DNA repair foci.100 There are up to a hundred thousand replication forks established during every S-phase in human cells. If even a small proportion of these have problems, this has the potential to drive mutations and genomic instability.
It is also likely that the mechanism of disassembly of the active helicase discussed here is equally relevant in the case of disassembly of the replisome at sites of DNA damage. It is known, at least in the case of repair of intra-strand crosslinks (ICL), that the timely removal of the replisome plays a key role for the subsequent repair. A recent elegant study suggested that ubiquitylation and Brca1 are required for this replisome dissolution at ICLs, but the specific substrate of this ubiquitylation is not yet known.101 If polyubiquitylation of Mcm7 is indeed essential for efficient DNA damage response and repair, its significance in maintenance of genomic stability and cancer development will be even greater. Altogether, gaining a detailed understanding of the mechanism of replication fork termination and answering the questions raised here are therefore very important for developing our knowledge of the maintenance of genome integrity.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work has been supported by U.K. Medical Research Council Career Development Award MR/K007106/1 and Birmingham Fellows Fellowship to AG.
References
- 1. Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953. Apr 25; 171(4356):737-8; PMID:13054692; http://dx.doi.org/ 10.1038/171737a0 [DOI] [PubMed] [Google Scholar]
- 2. Watson JD, Crick FH. Genetical implications of the structure of deoxyribonucleic acid. Nature 1953. May 30; 171(4361):964-7; PMID:13063483; http://dx.doi.org/ 10.1038/171964b0 [DOI] [PubMed] [Google Scholar]
- 3. Fachinetti D, Bermejo R, Cocito A, Minardi S, Katou Y, Kanoh Y, Shirahige K, Azvolinsky A, Zakian VA, Foiani M. Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol Cell 2010. Aug 27; 39(4):595-605; PMID:20797631; http://dx.doi.org/ 10.1016/j.molcel.2010.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rudolph CJ, Upton AL, Stockum A, Nieduszynski CA, Lloyd RG. Avoiding chromosome pathology when replication forks collide. Nature 2013. Jul 28; 500(7464):608-11; PMID:23892781; http://dx.doi.org/ 10.1038/nature12312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moreno SP, Bailey R, Campion N, Herron S, Gambus A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 2014. Oct 24; 346(6208):477-81; PMID:25342805; http://dx.doi.org/ 10.1126/science.1253585 [DOI] [PubMed] [Google Scholar]
- 6. Bell SP. DNA Replication. Terminating the replisome. Science 2014. Oct 24; 346(6208):418-9; PMID:25342784; http://dx.doi.org/ 10.1126/science.1261245 [DOI] [PubMed] [Google Scholar]
- 7. Lengronne A, Pasero P. Closing the MCM cycle at replication termination sites. EMBO Rep 2014. Dec; 15(12):1226-7; PMID:25391904; http://dx.doi.org/ 10.15252/embr.201439774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, Labib K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol 2006. Apr; 8(4):358-66; PMID:16531994; http://dx.doi.org/ 10.1038/ncb1382 [DOI] [PubMed] [Google Scholar]
- 9. Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 2009. Nov 13; 139(4):719-30; PMID:19896182; http://dx.doi.org/ 10.1016/j.cell.2009.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc Natl Acad Sci U S A 2009. Dec 1; 106(48):20240-5; PMID:19910535; http://dx.doi.org/ 10.1073/pnas.0911500106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gambus A, Khoudoli GA, Jones RC, Blow JJ. MCM2-7 form double hexamers at licensed origins in Xenopus egg extract. J Biol Chem 2011. Apr 1; 286(13):11855-64; PMID:21282109; http://dx.doi.org/ 10.1074/jbc.M110.199521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mechali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat Rev Mol Cell Biol 2010. Oct; 11(10):728-38; PMID:20861881; http://dx.doi.org/ 10.1038/nrm2976 [DOI] [PubMed] [Google Scholar]
- 13. Ilves I, Petojevic T, Pesavento JJ, Botchan MR. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol Cell 2010. Jan 29; 37(2):247-58; PMID:20122406; http://dx.doi.org/ 10.1016/j.molcel.2009.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yardimci H, Loveland AB, Habuchi S, van Oijen AM, Walter JC. Uncoupling of sister replisomes during eukaryotic DNA replication. Mol Cell 2010. Dec 10; 40(5):834-40; PMID:21145490; http://dx.doi.org/ 10.1016/j.molcel.2010.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Costa A, Renault L, Swuec P, Petojevic T, Pesavento JJ, Ilves I, MacLellan-Gibson K, Fleck RA, Botchan MR, Berger JM. DNA binding polarity, dimerization, and ATPase ring remodeling in the CMG helicase of the eukaryotic replisome. Elife 2014; 3:e03273; PMID:25117490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McIntosh D, Blow JJ. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb Perspect Biol 2012; 4(10); PMID:22904560; http://dx.doi.org/ 10.1101/cshperspect.a012955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shreeram S, Blow JJ. The role of the replication licensing system in cell proliferation and cancer. Prog Cell Cycle Res 2003; 5:287-93; PMID:14593723 [PMC free article] [PubMed] [Google Scholar]
- 18. Hustedt N, Gasser SM, Shimada K. Replication checkpoint: tuning and coordination of replication forks in s phase. Genes (Basel) 2013; 4(3):388-434; PMID:24705211; http://dx.doi.org/ 10.3390/genes4030388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gillespie PJ, Gambus A, Blow JJ. Preparation and use of Xenopus egg extracts to study DNA replication and chromatin associated proteins. Methods 2012. Jun; 57(2):203-13; PMID:22521908; http://dx.doi.org/ 10.1016/j.ymeth.2012.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun J, Fernandez-Cid A, Riera A, Tognetti S, Yuan Z, Stillman B, Speck C, Li H. Structural and mechanistic insights into Mcm2-7 double-hexamer assembly and function. Genes Dev 2014. Oct 15; 28(20):2291-303; PMID:25319829; http://dx.doi.org/ 10.1101/gad.242313.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Schärer OD, Walter JC. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 2011. Sep 16; 146(6):931-41; PMID:21925316; http://dx.doi.org/ 10.1016/j.cell.2011.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuipers MA, Stasevich TJ, Sasaki T, Wilson KA, Hazelwood KL, McNally JG, Davidson MW, Gilbert DM. Highly stable loading of Mcm proteins onto chromatin in living cells requires replication to unload. J Cell Biol 2011. Jan 10; 192(1):29-41; PMID:21220507; http://dx.doi.org/ 10.1083/jcb.201007111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jagannathan M, Nguyen T, Gallo D, Luthra N, Brown GW, Saridakis V, Frappier L. A role for USP7 in DNA replication. Mol Cell Biol 2014. Jan; 34(1):132-45; PMID:24190967; http://dx.doi.org/ 10.1128/MCB.00639-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nishiyama A, Frappier L, Mechali M. MCM-BP regulates unloading of the MCM2-7 helicase in late S phase. Genes Dev 2011. Jan 15; 25(2):165-75; PMID:21196493; http://dx.doi.org/ 10.1101/gad.614411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kusunoki S, Ishimi Y. Interaction of human minichromosome maintenance protein-binding protein with minichromosome maintenance 2-7. Febs J 2013. Dec 2“ for ”FEBS J. 2014 Feb; 281(4):1057-67. doi: 10.1111/febs.12668 [DOI] [PubMed] [Google Scholar]
- 26. Jagannathan M, Sakwe AM, Nguyen T, Frappier L. The MCM-associated protein MCM-BP is important for human nuclear morphology. J Cell Sci 2012. Jan 1; 125(Pt 1):133-43; PMID:22250201; http://dx.doi.org/ 10.1242/jcs.089938 [DOI] [PubMed] [Google Scholar]
- 27. Ding L, Forsburg SL. Schizosaccharomyces pombe minichromosome maintenance-binding protein (MCM-BP) antagonizes MCM helicase. J Biol Chem 2011. Sep 23; 286(38):32918-30; PMID:21813639; http://dx.doi.org/ 10.1074/jbc.M111.282541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Santosa V, Martha S, Hirose N, Tanaka K. The fission yeast minichromosome maintenance (MCM)-binding protein (MCM-BP), Mcb1, regulates MCM function during prereplicative complex formation in DNA replication. J Biol Chem 2013. Mar 8; 288(10):6864-80; PMID:23322785; http://dx.doi.org/ 10.1074/jbc.M112.432393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takahashi N, Quimbaya M, Schubert V, Lammens T, Vandepoele K, Schubert I, Matsui M, Inzé D, Berx G, De Veylder L. The MCM-binding protein ETG1 aids sister chromatid cohesion required for postreplicative homologous recombination repair. PLoS Genet 2010. Jan; 6(1):e1000817; PMID:20090939; http://dx.doi.org/ 10.1371/journal.pgen.1000817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takahashi N, Lammens T, Boudolf V, Maes S, Yoshizumi T, De Jaeger G, Witters E, Inzé D, De Veylder L. The DNA replication checkpoint aids survival of plants deficient in the novel replisome factor ETG1. Embo J 2008. Jul 9; 27(13):1840-51; PMID:18528439; http://dx.doi.org/ 10.1038/emboj.2008.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Faustrup H, Bekker-Jensen S, Bartek J, Lukas J, Mailand N. USP7 counteracts SCFbetaTrCP- but not APCCdh1-mediated proteolysis of Claspin. J Cell Biol 2009. Jan 12; 184(1):13-9; PMID:19124652; http://dx.doi.org/ 10.1083/jcb.200807137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S, Kao HY, Xu Y, Willis J, Markowitz SD, et al. . DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci Signal 2010; 3(146):ra80; PMID:21045206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qian J, Pentz K, Zhu Q, Wang Q, He J, Srivastava AK, Wani AA. USP7 modulates UV-induced PCNA monoubiquitination by regulating DNA polymerase eta stability. Oncogene 2014. Dec 1; PMID:25435364; http://dx.doi.org/ 10.1038/onc.2014.394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. He J, Zhu Q, Wani G, Sharma N, Han C, Qian J, Pentz K, Wang QE, Wani AA. Ubiquitin-specific protease 7 regulates nucleotide excision repair through deubiquitinating XPC protein and preventing XPC protein from undergoing ultraviolet light-induced and VCP/p97 protein-regulated proteolysis. J Biol Chem 2014. Sep 26; 289(39):27278-89; PMID:25118285; http://dx.doi.org/ 10.1074/jbc.M114.589812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alonso-de Vega I, Martin Y, Smits VA. USP7 controls Chk1 protein stability by direct deubiquitination. Cell Cycle 2014; 13(24):3921-6; PMID:25483066; http://dx.doi.org/ 10.4161/15384101.2014.973324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dar A, Shibata E, Dutta A. Deubiquitination of Tip60 by USP7 determines the activity of the p53-dependent apoptotic pathway. Mol Cell Biol 2013. Aug; 33(16):3309-20; PMID:23775119; http://dx.doi.org/ 10.1128/MCB.00358-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giovinazzi S, Sirleto P, Aksenova V, Morozov VM, Zori R, Reinhold WC, Ishov AM. Usp7 protects genomic stability by regulating Bub3. Oncotarget 2014. Jun 15; 5(11):3728-42; PMID:25003721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Giovinazzi S, Morozov VM, Summers MK, Reinhold WC, Ishov AM. USP7 and Daxx regulate mitosis progression and taxane sensitivity by affecting stability of Aurora-A kinase. Cell Death Differ 2013. May; 20(5):721-31; PMID:23348568; http://dx.doi.org/ 10.1038/cdd.2012.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moritani M, Ishimi Y. Inhibition of DNA binding of MCM2-7 complex by phosphorylation with cyclin-dependent kinases. J Biochem 2013. Oct; 154(4):363-72; PMID:23864661; http://dx.doi.org/ 10.1093/jb/mvt062 [DOI] [PubMed] [Google Scholar]
- 40. Mott ML, Berger JM. DNA replication initiation: mechanisms and regulation in bacteria. Nat Rev Microbiol 2007. May; 5(5):343-54; PMID:17435790; http://dx.doi.org/ 10.1038/nrmicro1640 [DOI] [PubMed] [Google Scholar]
- 41. Louarn J, Patte J, Louarn JM. Evidence for a fixed termination site of chromosome replication in Escherichia coli K12. J Mol Biol 1977. Sep 25; 115(3):295-314; PMID:338909; http://dx.doi.org/ 10.1016/0022-2836(77)90156-5 [DOI] [PubMed] [Google Scholar]
- 42. Neylon C, Kralicek AV, Hill TM, Dixon NE. Replication termination in Escherichia coli: structure and antihelicase activity of the Tus-Ter complex. Microbiology and molecular biology reviews: MMBR 2005. Sep; 69(3):501-26; PMID:16148308; http://dx.doi.org/ 10.1128/MMBR.69.3.501-526.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Duggin IG, Wake RG, Bell SD, Hill TM. The replication fork trap and termination of chromosome replication. Mol Microbiol 2008. Dec; 70(6):1323-33; PMID:19019156; http://dx.doi.org/ 10.1111/j.1365-2958.2008.06500.x [DOI] [PubMed] [Google Scholar]
- 44. Suski C, Marians KJ. Resolution of converging replication forks by RecQ and topoisomerase III. Mol Cell 2008. Jun 20; 30(6):779-89; PMID:18570879; http://dx.doi.org/ 10.1016/j.molcel.2008.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Markovitz A. A new in vivo termination function for DNA polymerase I of Escherichia coli K12. Mol Microbiol 2005. Mar; 55(6):1867-82; PMID:15752206; http://dx.doi.org/ 10.1111/j.1365-2958.2005.04513.x [DOI] [PubMed] [Google Scholar]
- 46. Zechiedrich EL, Cozzarelli NR. Roles of topoisomerase IV and DNA gyrase in DNA unlinking during replication in Escherichia coli. Genes Dev 1995. Nov 15; 9(22):2859-69; PMID:7590259; http://dx.doi.org/ 10.1101/gad.9.22.2859 [DOI] [PubMed] [Google Scholar]
- 47. Rudolph CJ, Upton AL, Lloyd RG. Replication fork collisions cause pathological chromosomal amplification in cells lacking RecG DNA translocase. Mol Microbiol 2009. Nov; 74(4):940-55; PMID:19818016; http://dx.doi.org/ 10.1111/j.1365-2958.2009.06909.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rudolph CJ, Upton AL, Briggs GS, Lloyd RG. Is RecG a general guardian of the bacterial genome? DNA Repair (Amst) 2010. Mar 2; 9(3):210-23; PMID:20093100; http://dx.doi.org/ 10.1016/j.dnarep.2009.12.014 [DOI] [PubMed] [Google Scholar]
- 49. Santamaria D, Viguera E, Martinez-Robles ML, Hyrien O, Hernandez P, Krimer DB, Schvartzman JB. Bi-directional replication and random termination. Nucleic Acids Res 2000. May 15; 28(10):2099-107; PMID:10773078; http://dx.doi.org/ 10.1093/nar/28.10.2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lucas I, Germe T, Chevrier-Miller M, Hyrien O. Topoisomerase II can unlink replicating DNA by precatenane removal. Embo J 2001. Nov 15; 20(22):6509-19; PMID:11707421; http://dx.doi.org/ 10.1093/emboj/20.22.6509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Steinacher R, Osman F, Dalgaard JZ, Lorenz A, Whitby MC. The DNA helicase Pfh1 promotes fork merging at replication termination sites to ensure genome stability. Genes Dev 2012. Mar 15; 26(6):594-602; PMID:22426535; http://dx.doi.org/ 10.1101/gad.184663.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baxter J, Diffley JF. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell 2008. Jun 20; 30(6):790-802; PMID:18570880; http://dx.doi.org/ 10.1016/j.molcel.2008.04.019 [DOI] [PubMed] [Google Scholar]
- 53. Cuvier O, Stanojcic S, Lemaitre JM, Mechali M. A topoisomerase II-dependent mechanism for resetting replicons at the S-M-phase transition. Genes Dev 2008. Apr 1; 22(7):860-5; PMID:18381889; http://dx.doi.org/ 10.1101/gad.445108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 2009. May; 9(5):327-37; PMID:19377505; http://dx.doi.org/ 10.1038/nrc2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maric M, Maculins T, De Piccoli G, Labib K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 2014. Oct 24; 346(6208):1253596; PMID:25342810; http://dx.doi.org/ 10.1126/science.1253596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 2005. Jan; 6(1):9-20; PMID:15688063; http://dx.doi.org/ 10.1038/nrm1547 [DOI] [PubMed] [Google Scholar]
- 57. Craney A, Rape M. Dynamic regulation of ubiquitin-dependent cell cycle control. Curr Opin Cell Biol 2013. Dec; 25(6):704-10; PMID:23890701; http://dx.doi.org/ 10.1016/j.ceb.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 58. Feng H, Zhong W, Punkosdy G, Gu S, Zhou L, Seabolt EK, Kipreos ET. CUL-2 is required for the G1-to-S-phase transition and mitotic chromosome condensation in Caenorhabditis elegans. Nat Cell Biol 1999. Dec; 1(8):486-92; PMID:10587644; http://dx.doi.org/ 10.1038/70272 [DOI] [PubMed] [Google Scholar]
- 59. Cukras S, Morffy N, Ohn T, Kee Y. Inactivating UBE2M impacts the DNA damage response and genome integrity involving multiple cullin ligases. PLoS One 2014; 9(7):e101844; PMID:25025768; http://dx.doi.org/ 10.1371/journal.pone.0101844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vos SM, Tretter EM, Schmidt BH, Berger JM. All tangled up: how cells direct, manage and exploit topoisomerase function. Nat Rev Mol Cell Biol 2011. Dec; 12(12):827-41; PMID:22108601; http://dx.doi.org/ 10.1038/nrm3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Morohashi H, Maculins T, Labib K. The amino-terminal TPR domain of Dia2 tethers SCF(Dia2) to the replisome progression complex. Curr Biol 2009. Dec 1; 19(22):1943-9; PMID:19913425; http://dx.doi.org/ 10.1016/j.cub.2009.09.062 [DOI] [PubMed] [Google Scholar]
- 62. Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. . Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 2011. Oct 21; 44(2):325-40; PMID:21906983; http://dx.doi.org/ 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xu G, Paige JS, Jaffrey SR. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat Biotechnol 2010. Aug; 28(8):868-73; PMID:20639865; http://dx.doi.org/ 10.1038/nbt.1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics 2011. Oct; 10(10):M111.013284; PMID:21890473; http://dx.doi.org/ 10.1074/mcp.M111.013284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Emanuele MJ, Elia AE, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen YN, Rush J, Hsu PW, et al. . Global identification of modular cullin-RING ligase substrates. Cell 2011. Oct 14; 147(2):459-74; PMID:21963094; http://dx.doi.org/ 10.1016/j.cell.2011.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Peters JM, Walsh MJ, Franke WW. An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF. Embo J 1990. Jun; 9(6):1757-67; PMID:2140770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct 2006; 35:93-114; PMID:16689629; http://dx.doi.org/ 10.1146/annurev.biophys.35.040405.101933 [DOI] [PubMed] [Google Scholar]
- 68. Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 2014. Sep 15; 127(Pt 18):3877-83; PMID:25146396; http://dx.doi.org/ 10.1242/jcs.093831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, Milligan RA, Wilson-Kubalek EM. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat Struct Biol 2002. Dec; 9(12):950-7; PMID:12434150; http://dx.doi.org/ 10.1038/nsb872 [DOI] [PubMed] [Google Scholar]
- 70. Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol 2002. Feb; 4(2):134-9; PMID:11813000; http://dx.doi.org/ 10.1038/ncb746 [DOI] [PubMed] [Google Scholar]
- 71. Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G. p47 is a cofactor for p97-mediated membrane fusion. Nature 1997. Jul 3; 388(6637):75-8; PMID:9214505; http://dx.doi.org/ 10.1038/40411 [DOI] [PubMed] [Google Scholar]
- 72. Vaz B, Halder S, Ramadan K. Role of p97/VCP (Cdc48) in genome stability. Front Genet 2013; 4:60; PMID:23641252; http://dx.doi.org/ 10.3389/fgene.2013.00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zeiler M, Straube WL, Lundberg E, Uhlen M, Mann M. A Protein Epitope Signature Tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Mol Cell Proteomics 2012. Mar; 11(3):O111 009613; PMID:21964433; http://dx.doi.org/ 10.1074/mcp.O111.009613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jentsch S, Rumpf S. Cdc48 (p97): a "molecular gearbox" in the ubiquitin pathway? Trends Biochem Sci 2007. Jan; 32(1):6-11; PMID:17142044; http://dx.doi.org/ 10.1016/j.tibs.2006.11.005 [DOI] [PubMed] [Google Scholar]
- 75. Bruderer RM, Brasseur C, Meyer HH. The AAA ATPase p97/VCP interacts with its alternative co-factors, Ufd1-Npl4 and p47, through a common bipartite binding mechanism. J Biol Chem 2004. Nov 26; 279(48):49609-16; PMID:15371428; http://dx.doi.org/ 10.1074/jbc.M408695200 [DOI] [PubMed] [Google Scholar]
- 76. Hanzelmann P, Buchberger A, Schindelin H. Hierarchical binding of cofactors to the AAA ATPase p97. Structure 2011. Jun 8; 19(6):833-43; PMID:21645854; http://dx.doi.org/ 10.1016/j.str.2011.03.018 [DOI] [PubMed] [Google Scholar]
- 77. Schuberth C, Buchberger A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol 2005. Oct; 7(10):999-1006; PMID:16179952; http://dx.doi.org/ 10.1038/ncb1299 [DOI] [PubMed] [Google Scholar]
- 78. Davis EJ, Lachaud C, Appleton P, Macartney TJ, Nathke I, Rouse J. DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat Struct Mol Biol 2012. Nov; 19(11):1093-100; PMID:23042607; http://dx.doi.org/ 10.1038/nsmb.2394 [DOI] [PubMed] [Google Scholar]
- 79. Rumpf S, Jentsch S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol Cell 2006. Jan 20; 21(2):261-9; PMID:16427015; http://dx.doi.org/ 10.1016/j.molcel.2005.12.014 [DOI] [PubMed] [Google Scholar]
- 80. Uchiyama K, Jokitalo E, Kano F, Murata M, Zhang X, Canas B, Newman R, Rabouille C, Pappin D, Freemont P, et al. . VCIP135, a novel essential factor for p97/p47-mediated membrane fusion, is required for Golgi and ER assembly in vivo. J Cell Biol 2002. Dec 9; 159(5):855-66; PMID:12473691; http://dx.doi.org/ 10.1083/jcb.200208112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang X, Zhang H, Wang Y. Phosphorylation regulates VCIP135 function in Golgi membrane fusion during the cell cycle. J Cell Sci 2014. Jan 1; 127(Pt 1):172-81; PMID:24163436; http://dx.doi.org/ 10.1242/jcs.134668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ernst R, Mueller B, Ploegh HL, Schlieker C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell 2009. Oct 9; 36(1):28-38; PMID:19818707; http://dx.doi.org/ 10.1016/j.molcel.2009.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu Y, Soetandyo N, Lee JG, Liu L, Xu Y, Clemons WM, Jr., Ye Y. USP13 antagonizes gp78 to maintain functionality of a chaperone in ER-associated degradation. Elife 2014; 3:e01369; PMID:24424410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009. Jul 23; 138(2):389-403; PMID:19615732; http://dx.doi.org/ 10.1016/j.cell.2009.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Blount JR, Burr AA, Denuc A, Marfany G, Todi SV. Ubiquitin-specific protease 25 functions in Endoplasmic Reticulum-associated degradation. PLoS One 2012; 7(5):e36542; PMID:22590560; http://dx.doi.org/ 10.1371/journal.pone.0036542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Aressy B, Jullien D, Cazales M, Marcellin M, Bugler B, Burlet-Schiltz O, Ducommun B. A screen for deubiquitinating enzymes involved in the G(2)/M checkpoint identifies USP50 as a regulator of HSP90-dependent Wee1 stability. Cell Cycle 2010. Sep 15; 9(18):3815-22; PMID:20930503; http://dx.doi.org/ 10.4161/cc.9.18.13133 [DOI] [PubMed] [Google Scholar]
- 87. Raman M, Havens CG, Walter JC, Harper JW. A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol Cell 2011. Oct 7; 44(1):72-84; PMID:21981919; http://dx.doi.org/ 10.1016/j.molcel.2011.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Verma R, Oania R, Fang R, Smith GT, Deshaies RJ. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol Cell 2011. Jan 7; 41(1):82-92; PMID:21211725; http://dx.doi.org/ 10.1016/j.molcel.2010.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Franz A, Orth M, Pirson PA, Sonneville R, Blow JJ, Gartner A, Stemmann O, Hoppe T. CDC-48/p97 coordinates CDT-1 degradation with GINS chromatin dissociation to ensure faithful DNA replication. Mol Cell 2011. Oct 7; 44(1):85-96; PMID:21981920; http://dx.doi.org/ 10.1016/j.molcel.2011.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Meerang M, Ritz D, Paliwal S, Garajova Z, Bosshard M, Mailand N, Janscak P, Hübscher U, Meyer H, Ramadan K. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat Cell Biol 2011. Nov; 13(11):1376-82; PMID:22020440; http://dx.doi.org/ 10.1038/ncb2367 [DOI] [PubMed] [Google Scholar]
- 91. Ramadan K, Meerang M. Degradation-linked ubiquitin signal and proteasome are integral components of DNA double strand break repair: New perspectives for anti-cancer therapy. FEBS Lett 2011. Sep 16; 585(18):2868-75; PMID:21536036; http://dx.doi.org/ 10.1016/j.febslet.2011.04.046 [DOI] [PubMed] [Google Scholar]
- 92. Ramadan K, Bruderer R, Spiga FM, Popp O, Baur T, Gotta M, Meyer HH. Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 2007. Dec 20; 450(7173):1258-62; PMID:18097415; http://dx.doi.org/ 10.1038/nature06388 [DOI] [PubMed] [Google Scholar]
- 93. Wilcox AJ, Laney JD. A ubiquitin-selective AAA-ATPase mediates transcriptional switching by remodelling a repressor-promoter DNA complex. Nat Cell Biol 2009. Dec; 11(12):1481-6; PMID:19915556; http://dx.doi.org/ 10.1038/ncb1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, Dantuma NP. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat Struct Mol Biol 2011. Dec; 18(12):1345-50; PMID:22120668; http://dx.doi.org/ 10.1038/nsmb.2188 [DOI] [PubMed] [Google Scholar]
- 95. Mouysset J, Deichsel A, Moser S, Hoege C, Hyman AA, Gartner A, Hoppe T. Cell cycle progression requires the CDC-48UFD-1/NPL-4 complex for efficient DNA replication. Proc Natl Acad Sci U S A 2008. Sep 2; 105(35):12879-84; PMID:18728180; http://dx.doi.org/ 10.1073/pnas.0805944105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Deichsel A, Mouysset J, Hoppe T. The ubiquitin-selective chaperone CDC-48/p97, a new player in DNA replication. Cell Cycle 2009. Jan 15; 8(2):185-90; PMID:19158489; http://dx.doi.org/ 10.4161/cc.8.2.7356 [DOI] [PubMed] [Google Scholar]
- 97. Krabbe M, Zabielski J, Bernander R, Nordstrom K. Inactivation of the replication-termination system affects the replication mode and causes unstable maintenance of plasmid R1. Mol Microbiol 1997. May; 24(4):723-35; PMID:9194700; http://dx.doi.org/ 10.1046/j.1365-2958.1997.3791747.x [DOI] [PubMed] [Google Scholar]
- 98. Song W, Dominska M, Greenwell PW, Petes TD. Genome-wide high-resolution mapping of chromosome fragile sites in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 2014. May 27; 111(21):E2210-8; PMID:24799712; http://dx.doi.org/ 10.1073/pnas.1406847111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell 2003. Dec; 12(6):1525-36; PMID:14690605; http://dx.doi.org/ 10.1016/S1097-2765(03)00456-8 [DOI] [PubMed] [Google Scholar]
- 100. Blake D, Luke B, Kanellis P, Jorgensen P, Goh T, Penfold S, Breitkreutz BJ, Durocher D, Peter M, Tyers M. The F-box protein Dia2 overcomes replication impedance to promote genome stability in Saccharomyces cerevisiae. Genetics 2006. Dec; 174(4):1709-27; PMID:16751663; http://dx.doi.org/ 10.1534/genetics.106.057836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Long DT, Joukov V, Budzowska M, Walter JC. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol Cell 2014. Oct 2; 56(1):174-85; PMID:25219499; http://dx.doi.org/ 10.1016/j.molcel.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]