

Abstract
Reactome (http://www.reactome.org) is an open source, expert-authored, peer-reviewed, manually curated database of reactions, pathways and biological processes. We provide an intuitive web-based user interface to pathway knowledge and a suite of data analysis tools. The Pathway Browser is a Systems Biology Graphical Notation (SBGN)-like visualization system that supports manual navigation of pathways by zooming, scrolling and event highlighting, and that exploits PSI Common Query Interface (PSIQUIC) web services to overlay pathways with molecular interaction data from the Reactome Functional Interaction (FI) Network and interaction databases such as IntAct, ChEMBL, and BioGRID. Pathway and Expression Analysis tools employ web services to provide ID mapping, pathway assignment and over-representation analysis of user-supplied datasets. By applying Ensembl Compara to curated human proteins and reactions, Reactome generates pathway inferences for 20 other species. The Species Comparison tool provides a summary of results for each of these species as a table showing numbers of orthologous proteins found by pathway from which users can navigate to inferred details for specific proteins and reactions. Reactome’s diverse pathway knowledge and suite of data analysis tools provide a platform for data mining, modeling and the analysis of large-scale proteomics datasets.
Keywords: Pathway database, Pathway visualization, Pathway analysis, BioMart, Data integration
Introduction
A major challenge for researchers and bioinformaticians is the integration of experimental and computational proteomics results with information relating to specific biological pathways. How are lists of protein-coding genes with somatic mutations identified in a survey of tumors, or lists of proteins whose expression level is changed in response to an experimental stress or a clinical disease to be mined effectively for insights into causes of disease and their physiological mechanisms? Biological pathway databases can help meet this need by facilitating the capture of the relationships between genes, proteins and small molecules in a computable data model. They provide information on biological reactions at the molecular level, and indicate how these reactions can be grouped to provide a specification of a higher-order process such as apoptosis. Unlike printed textbooks, pathway databases have the freedom to expand in breadth and depth, to be queried interactively, to adapt their visual display to the needs of individual research communities, and to connect to other internet resources.
There are several distinctive approaches to constructing a useful pathway database. One distinguishing feature is the domain of the database. Some databases focus on the transformation of small molecules (intermediary metabolism) while others place more emphasis on signal transduction and higher-order biological processes. Another distinguishing feature is the level of curation. Some pathway databases are fully curated, where each pathway is authored and reviewed by an expert and include numerous comments, literature citations, and data such as enzyme activators and inhibitors. The other extreme is an automatic scheme in which pathways are inferred by computational processes with little or no literature curation occurs.
One of the earliest and best-known databases of biological pathways is The Kyoto Encyclopedia of Genes and Genomes, or KEGG [1, 2]. KEGG’s focuses on intermediary metabolism rather than higher-level pathways, for a broad range of species. The BioCarta project (www.biocarta.com) is a human-specific pathways database that focuses on higher-order processes such as signaling. At heart the of BioCarta pathway data is a series of colorful high-resolution diagrams oriented towards education. MetaCyc is a richly curated database of pathways involved in primary and secondary metabolism that supports data mining and other software-driven applications [3]. Built on the principles of Wikipedia, the WikiPathways is an open, community-curated biological pathway database that aggregates material from the individual databases mentioned here [4]. NCI-PID is a collection of Reactome, BioCarta pathway data, and curated biomolecular interactions and signaling pathways related to cancer [5]. Science Signaling’s Database of Cell Signaling, is an expert-authored and peer-reviewed curated database of signaling pathways [6]. NCBI BioSystems functions as a repository of pathway data integrated with gene, protein, associated literature, and chemical data present within the Entrez database system [7].
Gathering, integrating, visualizing and analyzing pathway data with other types of biological data is a challenging undertaking. Pathway databases gather and exchange data in different files formats and database dumps. However, two standard pathway data formats have reduced the complexity of data exchange and allow databases to cooperate more effectively. Systems Biology Markup Language (SBML) is an XML format language for the exchange of computational models of biological pathways and processes [8]. Visualization and model simulation tools such as CellDesigner [9] and COPASI [10] are compatible with SBML files. Biological Pathway Exchange (BioPAX) uses a Web Ontology Language (OWL) to support the exchange of biomolecular and genetic interactions, gene regulation networks and metabolic and signaling pathway data. Tools such as Cytoscape [11] and Chisio BioPAX Editor [12] enable visualization and manipulation of BioPAX files.
The Reactome database of human pathways, reactions and biological processes [13-16] employs a reductionist data model, which attempts to represent all of biology as reactions that convert input physical entities into output physical entities. The input and output entities of a reaction can be proteins, nucleic acids, chemical compounds, or complexes of these entities. Every reaction and entity in Reactome is associated with a species and is assigned to a cellular location. Some reactions may span more than one compartment. For example, the reaction representing the P2Y11 receptor binding adenosine nucleotides (ATP) would have plasma membrane and extracellular components [17]. Each reaction is supported by experimental evidence represented by links to the appropriate literature references. Reactions are then grouped into ordered causal chains to form pathways. Pathways can contain reactions, pathways, or both. Pathways in turn have been grouped into approximately 160 canonical pathways each of which corresponds to a substantial, tightly-connected domain of human biology like carbohydrate metabolism [18], solute transport regulatory pathways, GPCR signal transduction [19], cell cycle regulation, and innate immunity [20].
Reactome curators work with collaborating faculty-level biologists to create pathways and reactions from published primary research article and reviews. Together they work through a domain of biology to create a human- and computer-accessible description, linking all of the genes, proteins, literature citations, and controlled vocabulary data together. Pathways, reactions, protein and small molecule entities are cross referenced with accession numbers and identifiers to a number of well-established databases, including NCBI Entrez Gene, Ensembl and UniProt databases, UCSC and HapMap Genome Browsers, KEGG Compound, PubChem Substance and ChEBI [1, 2, 21-29]. Physical entities and events are further linked to ‘Molecular Function’, ‘Biological Process’ and ‘Cellular Component’ ontology terms found in the Gene Ontology (GO) vocabularies [30, 31] and literature citation linked to PubMed [32]. Post-translational modifications are represented in Reactome with terms from PSI-MOD [33]. In the other direction, incoming links connect UniProt, ChEBI, Ensembl, Entrez Gene, WormBase, and the GO Consortium back to Reactome [21, 24, 26, 27, 30, 31, 34]. For example, an incoming link from a UniProt protein entry to Reactome links pages that describe the function the protein plays in one or more biological processes, the complexes it participates in, its position in the pathway diagrams, and the literature citations that back these assertions.
The tutorial that follows will illustrate how browsing, searching, analyzing and visualizing Reactome pathway data is useful in interpreting proteomics datasets. Please note that this information is based on Reactome in early 2011. The contents of the database and some of the web pages may have changed slightly since this tutorial was written.
Navigating the Reactome Website
The main user-entry point to Reactome is the website, located at http://www.reactome.org (Fig. 1). This intuitive home page, divided into three main sections, provides access to the database and the suite of pathway analysis and data mining tools. The navigation bar at the top of the page provides access to background information describing Reactome (‘About’), a list of pathways (‘Content’), the user guides and a description of the data model (‘Documentation’), additional data analysis tools (‘Tool’), software and datasets in MySQL, BioPAX, SBML, PSI-MITAB formats (‘Download’). The buttons on the left-hand side of the home page provides access to some of the popular data analysis tools and downloadable datasets. A simple search tool allows the user to query the contents of the Reactome database. The main text section provides information ‘About Reactome’, an example pathway (‘Pathway of the Month’), access to web tutorials and up-to-the-minute Reactome news.
Figure 1.

The Reactome website (www.reactome.org) homepage. A navigation bar and the side panel to the left provide access to the pathway data and pathway analysis tools.
Browsing Reactome Pathway Diagrams
The visualization of full pathway data in a consistent and navigable format is vital to support the pathway-based analysis of complex proteomics data sets. Clicking on the ‘Pathway Browser’ button on the left side of the home page will open a webpage displaying all the pathways in the Reactome database. The Reactome Pathway Browser (Fig. 2A) uses pre-computed tiles for fast zooming and scrolling, a custom Javascript for navigation and molecular overlays (described later), and the guiding principles of the Systems Biology Graphical Notation (SBGN) to provide an interactive and dynamic framework for pathway visualization and data analysis [35].
Figure 2.

The Reactome Pathway Browser and Molecular Interaction Overlays. (A) The main features of the Pathway Browser are the ‘Search” bar at the top, the ‘Pathways” panel on the left, the ‘Visualization” panel on the right and a ‘Details” panel. A description of any reaction or pathway in the pathway diagrams can be displayed below the diagram in the ‘Details” panel by selecting the event within the diagram. Right-clicking the mouse whilst the cursor is over a physical entity displays the context sensitive menus. (B) Protein-protein interactions are displayed in the Pathway Browser for SHC1 [cytosol] and SHP2 [cytosol] physical entities. (C) Selecting ‘Other Pathways” displays additional Reactome pathways that contain the highlighted physical entity. Navigating to the additional pathway is achieved by clicking the pathway name. (D) Selecting ‘Participating Molecules” displays additional components of the macromolecular complex. Navigating to the component is achieved by clicking the entity name.
At the top of the browser page is the ‘Search and Analyze’ panel that consists of a search text box to query the elements of the pathway diagram and the ‘Analyze, Annotate & Upload’ button that controls the interactive tools associated with pathway diagrams. The ‘Pathways’ panel, on the left side, organizes all the canonical pathways in a hierarchy. The sub-pathways and reactions within each canonical pathway can be displayed or hidden by clicking on the plus (+) symbol to the left of the pathway name. Navigating to pathways is achieved by clicking on the pathway name in the pathway hierarchy on the left. This displays the corresponding pathway diagram or diagram section in the ‘Visualization’ panel to the right. The Google map-like tools in the upper left corner of the ‘Visualization’ panel enable zooming and scrolling across the Pathway diagram.
When a pathway in the hierarchy is selected, it is highlighted in bright green in the hierarchy and its parent terms are green-highlighted. In the diagram on the right, green squares highlight the nodes of all reactions that are components of that pathway. Scrolling over any sub-pathway or reaction in the hierarchy of the selected pathway will highlight that event with a green square on each reaction node. Highlighted reactions are also visible in the thumbnail diagram, which can be used to navigate quickly to the region of interest in the main diagram. Canonical pathway diagrams, such as ‘Cell Cycle, Mitotic’ may contain sub-pathways that may have their own diagrams. These are represented as boxes with green boundaries in the diagram. This process of navigating downward in the event hierarchy, either by choosing sub-processes of the current one in the hierarchy on the left or by choosing a pathway box or reaction node in the diagram window on the right can be continued until a single reaction is highlighted. Moving the cursor over a reaction edge or physical entity node of the pathway will cause its name to appear in a popup window.
Underneath the ‘Visualization’ panel is the ‘Details’ panel that provides a description for the pathway, reaction or physical entity. Pathway Descriptions provide a text summary giving an overview of the pathway, the GO biological process term for the pathway and the GO cellular compartment term for its location in the cell, as well as published literature references linked to PubMed. If the pathway is not supported by direct experimental data but has been inferred from a pathway in another species, this is noted with the phrase ‘This event is deduced on the basis of event(s)’ and a link to the reference pathway.
Clicking a reaction box will present the reaction description in the ‘Details panel’ with information about the reaction, including the input and output physical entities, the catalyst, the precise component within a catalyst complex (or domain within a simple catalyst) that enables the reaction to occur. A description of any molecule represented in the pathway diagrams can be displayed below the diagram by selecting the physical entity node within the diagram. Physical entities within the ‘Details’ panel are seamlessly linked to other external bioinformatics resources.
Context-sensitive menus accessible from the ‘Visualization’ panel view of a reaction provide additional functionality whilst navigating the Pathway browser (Fig. 2C, 2D). The exact features of the context sensitive menus are determined as the user right-clicks on a physical entity: i) a list of the other pathways in Reactome in which the selected entity participates; ii) a display of the physical entities that contribute to the complex; and iii) a list of interactors of the entity (described later). The menu bar at the bottom of the ‘Details’ panel provides download options to retrieve static pathway diagram files and BioPAX level 2 and 3, SBML and Protégé formats. These batch data dumps allow researchers to download lists of all proteins that participate in a pathway or subpathway, and support data exchange, analysis and modeling [8, 36-38].
Integrating Molecular Interactions onto Reactome Pathway Diagrams
Reactome datasets are a high quality resource for a pathway-based data analysis. However, the usage of Reactome as a platform for high-throughput data analysis is limited by a low coverage of human proteins. To increase protein coverage and associated functional annotations, we have integrated molecular interaction and network data into the Reactome pathway diagrams. The molecular interaction overlay allows the display of proteins and chemicals interacting with proteins in a Reactome pathway.
As mentioned before, selecting ‘Display Interactors’ from the context sensitive menus will display the individual protein interactors (Fig. 2B). The ‘Analyze, Annotate and Upload’ feature located at the top left corner of the Pathway Browser is used to overlay all interactors for all pathway proteins. Mousing the cursor over a protein interactor displays a popup window with the gene name and Uniprot accession number of the protein. The molecular interaction overlay employs PSI Common Query Interface (PSIQUIC) web services to import binary interaction data from individual interaction databases into Reactome pathway diagrams (http://code.google.com/p/psicquic). The default interaction database is IntAct. Other PSIQUIC data sources available for overlay in this way include APID, BioGRID, ChEMBL, DIP, InnateDB, IntAct, iRefIndex, MatrixDB, MINT, MPIDB, Reactome, Reactome-FIs (functional interactions) and STRING [25, 27, 39-57]. Two the data sets, ‘Reactome’ and ‘Reactome-FIs’, were generated by the Reactome group. ‘Reactome’ represents interaction data derived from Reactome reactions and complexes. ‘Reactome-FIs’ contains approximately 201K functional interactions encompassing over 10,000 human proteins (46% of SwissProt entries for human). It combines curated interactions from Reactome and other pathway databases, including Panther [58], KEGG, NCI-PID, CellMap (http://cancer.cellmap.org/cellmap/), interaction datasets and interactions derived from co-expression data, protein domain-domain interactions, text mining, and GO annotations [53].
The nodes and edges of the overlaid network are interactive, providing links to relevant data sources. For example, clicking on a protein interactor opens a new web page displaying the Uniprot entry for the selected protein. Selecting an edge will open a new web page displaying the interaction entry in the current interaction database. If a new database is selected from the ‘Analyze, Annotate and Upload’ button while interactors are displayed for a set of pathway proteins, those proteins will be submitted to the new database and the display will automatically updated. As well as querying databases for interactions it is also possible to upload user-defined interactions, in the PSI-MITAB format, that will be overlaid onto the pathway. Launching ‘Submit a new PSIQUIC Service’ will access a PSICQUIC service not listed in the PSICQUIC registry. Interactions can be coloured based on the confidence level that reflects the amount of experimental data available. All the interactions displayed in the pathway diagram can be viewed as a list in the ‘Table of Interactors for Pathway’ of the ‘Analyze, Annotate and Upload’ feature. When displayed this table lists the proteins in the pathway along with their interactors from the currently selected interaction database. A full list of interactors for each pathway protein can be downloaded in the PSI-MITAB format.
Analysis of proteomics data sets using Reactome tools
Protein-protein interaction detection methodologies such as yeast two-hybrid (Uetz et al., 2000), phage-display [59], protein microarray [60] and affinity chromatography followed by mass spectrometry (MS) [61] have been used to create large interaction datasets. Biomolecular interaction databases such as DIP [62], BIND [55], BioGRID [56], IntAct [43] and MINT contain interaction datasets for yeast [61, 63, 64], bacteria [65-68], fruit fly [69], worm [70] and human [71-73]. Several MS-based technologies including MALDI-TOF-MS [74], LC-MS [75] and SELDI-TOF-MS [76] have been used to study the proteome. Mass spectrometry has also been instrumental in the discovery and characterization of protein post-translational modifications [77] and biomarkers [78]. Numerous protein fragment databases facilitate the identification of peptides within MS or tandem MS profiles, such as Mascot [79] and XTandem [80]. The PRIDE (PRoteomics IDEntifications) database integrates protein databases, literature citations and post-translational modification data to promote proteomics data analysis [81]. Nevertheless, with high-throughput proteomic technologies it has become increasingly important to have analysis tools that can integrate and visualize thousands of data points in the context of the pathway diagrams. Reactome facilitates detailed computational analysis of proteomics data through the capture of published knowledge about reactions, pathways and biological processes and providing a series of bioinformatics tools that integrate the results with the pathway visualization system.
Querying Reactome
Most users will probably find the Simple search tool, accessible on all the webpages, sufficient for querying the Reactome database and website (Fig. 3A). Users can submit a word, database identifier or phrase and retrieve a list of corresponding database records. For example, a simple query for the protein name TP53 will yield 524 hits in different data categories (Pathways, Reactions, Proteins and Others). The ‘Others’ category represents literature references, complexes, inhibitions, activations or anything else not covered by the first three categories. A subset of the results can be displayed if some of these categories are not required. Simply deselect the boxes that are not required and click the Show button to refresh the search results page. Each of the results returned is clickable and will link to the appropriate Reactome page when clicked. Should it be necessary to restrict the search to a specific species this can be achieved through the second Species drop-down menu of the search results page.
Figure 3.

Simple and Advanced Search. (A) Results for a simple query for ‘TPI1’ protein name. (B) The query form for the Advanced search. The search modes, include i) ‘with EXACT PHRASE ONLY’: returns hits that contain the exact query phrase; ii) ‘matching REGULAR EXPRESSION’: treats the query phrase as a PERL regular expression and returns hits that contain the query words, even as a substring; iii) ‘with ALL of the words’: returns hits that contain all of the query words in any order; iv) with ANY of the words: returns hits to any word in the query phrase; v) ‘with the EXACT PHRASE’: returns only those hits that exactly MATCH the query phrase; vi) ‘!=’: returns hits that do NOT MATCH query phrase; vii) ‘with no value’: returns hits for which the selected field of a given class has no value; and viii) ‘with any value’: returns hits for which the selected field of a given Class has any value (not zero or blank).
Working example of Reactome Simple Search: retrieve all Reactome instances that involve the TPI1 enzyme
Go to the Reactome homepage (http://www.reactome.org).
Enter ‘TPI1’ in the search box and click the ‘Search’ button. In a few seconds, a list of Reactome reactions, pathways and entities should appear in the search results tab.
Click ‘Protein: UniProt:P60174 TPI (Homo sapiens)’ to connect with the TPI1 protein summary page.
Returning to the Search page, click ‘Pathway: Gluconeogenesis (Homo sapiens)’ to open the Gluconeogenesis pathway diagram in the Pathway Browser.
The Advanced (Extended) search will provide more customizable, complex and logical queries that can be accessed via the Tools menu located in the main menu bar on all webpages. This Extended search method allows specific schema-based queries for particular types of Reactome data (Fig. 3B). Specifically, this option searches for records (instances) in the database by multiple field (attribute) values. Queries are combined together with boolean AND operators. For example, a query to retrieve all reactions which consume GDP and produce GTP would be prepared by choosing class ‘Reaction’, then selecting field name input and entering GDP into the search box, then picking field name output on the next row and entering GTP.
Working example of Reactome Advanced Search: find all plasma membrane-associated complexes whose name includes the word EGFR
Go to the Reactome homepage (http://www.reactome.org).
Under the ‘Tools’ in the Navigation bar, select ‘Advanced Search’.
Select ‘Complex’ under the ‘Restrict search to class’ drop-down menu.
Select ‘name’ under the first row ‘Field name’, select ‘with the EXACT PHRASE’ from the next drop-down menu and type ‘EGFR’ into the final text box.
Select ‘species’ under the second row ‘Field name’, select ‘with the EXACT PHRASE’ from the next drop-down menu and type ‘Homo sapiens’ into the final text box.
Select ‘compartment’ under the third row ‘Field name’, select ‘with the EXACT PHRASE’ from the next drop-down menu and type ‘Plasma membrane’ into the final text box.
Click the ‘Search’ button to retrieve from the advanced query, human complexes that contain EGFR and are located in the plasma membrane.
Pathway Analysis
The Pathway Analysis tool analyzes user-supplied lists of genes, proteins and small molecules and provides ID mapping, pathway assignment and overrepresentation analysis. Clicking the ‘Pathway Analysis’ button on the Reactome homepage launches a data entry page that allows the user to input a list of gene, protein or small molecule identifiers. Several identifier types and accession numbers are currently supported, including UniProt, GenBank/EMBL/DDBJ, RefPep, RefSeq, EntrezGene, OMIM, InterPro, Affymetrix, Agilent, Illumina and Ensembl. The data entry page supports both typing and pasting identifiers into the text area provided, or uploading a text file of identifiers from the user’s computer. Two pathway analyses can be performed. By default, the simpler of these analyses will be selected, ‘ID mapping and pathway assignment’. This analysis takes a set of accession numbers or identifiers and maps them to Reactome pathways. The results are presented in a sortable table that can be downloaded as a spreadsheet or as a comma-separated or tab-delimited file, for further analysis (Fig. 4A).
Figure 4.

Pathway Analysis. (A) Results table for the “ID mapping and pathway assignment”. The sortable table contains one row for each Reactome pathway and four columns: the user-supplied ID, the corresponding UniProt ID, the species name, and names of pathways in which this ID can be found. The names/IDs in the last two columns are clickable links that take the user to a diagram of the named pathway. (B) The results for the ‘overrepresentation analysis” are presented as a list of clickable links of enriched events. The warmer the colour, the higher the level of overrepresentation in the given pathway. Clicking on the “+” next to the pathway name gives access to the user-supplied identifiers that are found in the pathway, along with the corresponding UniProt IDs.
A more complex pathway analysis tool is ‘Overrepresentation analysis’. This tool determines which events (pathways and/or reactions) are statistically enriched in a set of genes or proteins as specified by a submitted list of identifiers (Fig. 4B). The results of the overrepresentation analysis are provided as a colour-coded interactive list of events. Each event is coloured according to the probability (from a hypergeometric test) of seeing a given number or more proteins in this event by chance. The top-level events are ordered according to the lowest p-value of their components. The warmer the colour, the higher the level of overrepresentation is for a given pathway. Selecting an event name will link to the Pathway Browser and clicking on the plus (+) next to the pathway name provides access to the protein identifiers from the submitted list that are found in the pathway, along with the corresponding UniProt IDs. The results are also provided as a table of statistically over-represented events as an ordered list that can be downloaded.
Working example of Reactome Pathway Analysis: annotate a list of UniProt identifiers with Reactome reaction and pathway data and identify statistically overrepresented events
Go to the Reactome homepage (http://www.reactome.org).
Click ‘Pathway Analysis’ in the sidebar.
Click the ‘Example’ button on the ‘Pathway Analysis’ page and then click ‘Analyse’. This will demonstrate the ‘ID mapping and pathway assignment’ feature. After a few seconds, a table of results entitled ‘Pathway Assignment’ will appear.
In the ‘UniProt’ column, click on the UniProt ID: O00139 link to open the reference UniProt protein record in a new page.
Return to the ‘Pathway Assignment’ table and click the upside-down triangle (on the left) of the ‘ID’ column header to sort the table based upon the UniProt IDs; UniProt ID: Q9Y6Y9 should now be in the top row.
In the ‘Pathway names’ column, click on ‘Toll Receptor Cascades’; the Pathway Browser should open in a new page.
Return to the results table. At the top of the table, you should see a download bar. Select the file format of your choice and click the ‘Download’ button to a file.
Repeat Steps 1-2 but select ‘Overrepresentation analysis’ before clicking ‘Analyse’. This will demonstrate the ‘Overrepresentation analysis’ feature. After a few seconds, a colour-coded interactive list of events will appear.
Click the plus (+) before the event name to reveal the ‘Matching identifiers’ list of the identifiers and associated proteins that contributed to the over-representation score.
Scroll down the page to the ‘Statistically over-represented events as an ordered list’ section to view the same results in a tabular form.
Click ‘Results in a tab-delimited text file” to download the results data.
Scroll down the page to the ‘Mapping from submitted identifiers to Reactions’ section to view the same results in as a list of reactions for each protein.
Expression Analysis
Proteomic researchers are producing vast quantities of structural and functional data of proteins through large-scale experiments that assess the abundance of proteins, post-translational modifications and protein-protein interactions. The Expression analysis tool will help with the biological interpretation of these different data types. Clicking the ‘Expression Analysis’ button on the Reactome homepage opens a form that allows entry of a user-specified list of identifiers and numerical values. As with the pathway analysis, the expression analysis tool will accept the same protein accession numbers and identifiers that are associated with the popular commercial proteomics platforms. However, the expression analysis tool will also accept numerical values (e.g. abundance, fold change or statistical value) and show how abundance levels affect events (reactions and pathways) in the cell. Once the data is submitted for analysis, the expression results will be presented as a sortable tabular format that can be downloaded as a comma- and tab-separated formats or a spreadsheet (Fig. 5A). A View button embedded in the results table will launch the Pathway Browser and displays the relevant pathway diagram (Fig 5B). The physical entities in the pathway diagram are colour-coded according to the submitted numerical values. The colour scale automatically adjusts to fit the range represented in the dataset, with red for the highest values to dark blue for the lowest values and the submitted identifier and value are overlaid onto the physical entities. Grey boxes are proteins or small molecules with no associated values in the input data. Black entities represent complexes that have values for at least one of the proteins. The ‘Experiment Browser’, at the bottom of the coloured pathway diagram, allows the user to step through different time points or visualize changes in abundance levels across multiple samples.
Figure 5.

Expression Analysis. The results table for the Expression Analysis. (A) The sortable table contains one row for each Reactome pathway and six columns: Name of the pathway, Species of presented results, Total number of proteins in pathway, Number of proteins in the user-supplied data that fall into the pathway, Graphical representation of the ratio of these 2 values, and a “View” button that creates a pathway diagram. (B) The pathway browser displaying the coloured physical entities that correspond to expression values of the experimental data. The nodes in this diagram are colour-coded: gray, no match; black, a (multicomponent) complex entity; and other colours represent expression levels. If the numerical data are a time series, the grey bar at the bottom of the coloured pathway diagram allows the user to step through time points and visualize changes in expression levels with time of the individual genes involved in the pathway.
Working example of Reactome Expression Analysis: visualize thousands of data points from an expression dataset in the context of Reactome pathway diagrams
Go to the Reactome homepage (http://www.reactome.org).
Click ‘Expression Analysis’ in the sidebar.
Click the ‘Example’ button on the ‘Upload expression data’ page and then click ‘Analyse’. After a few seconds, a table of results entitled ‘Expression per Pathway’ will appear.
Click on the arrows of the ‘% in data’ column to reorder based upon the highest percentage hits from the dataset at the top.
Click the ‘View’ button for ‘Intrinsic Pathway for Apoptosis’ to open the Pathway Browser in a new window. Be sure your browser is configured to see pop-ups for Reactome.
In the top left-hand corner of the diagram, there is an icon with 4 different sizes of blue circle, which allows you to chose your zoom level and scroll across the pathway diagram. Click on the second highest circle to zoom out and use the arrows to scroll about the pathway diagram.
Mouse over one of the black coloured physical entities (complexes) to show the name of the complex.
Right click on the same complex entity and select ‘Display Participating Molecules’. A popup box should appear, with a grid of coloured squares inside it, representing expression levels for the complex components.
At the base of the diagram, you will see a bar containing the text ‘Experiment: 10h_control’ and two arrows. Click on the forward arrow five times. Colours of some of the entities will change reflecting changes in their abundance over the course of the study.
Type ‘Smac’ into the Search box in the ‘Search and Analyze’ panel. You will see a demonstration of the auto complete feature of the pathway search. Select ‘SMAC [cytosol]’ and click ‘Search map’. In the ‘Search results’ panel to the left, click the ‘SMAC [cytosol] query link. This will open a Pathway hierarchy for the ‘Apoptosis’ pathway in the left panel and center the pathway diagram, and highlight the ‘SMAC’ physical entity of the ‘Intrinsic Pathway for Apoptosis’ sub-pathway diagram with a green box.
Mouse over this entity. The ‘SMAC[cytosol]’ popup window will appear, displaying the data point identifier and expression value.
Zoom back in to the highest zoom level, navigate to ‘SMAC[cytosol]’ and right click to show the context sensitive menu. Select ‘Display Interactors’. A halo of interacting proteins will appear around the physical entity.
Click on the line connecting ‘SMAC[cytosol]’ to ‘RNF85’ to open a new page with the IntAct interaction for these two proteins?
Click on the ‘RNF85’ node to open a new page with the UniProt protein page.
Comparative Analysis of Biological Pathways
Organism-based comparative analysis of biological pathways yields information on their evolution, on disease, on biotechnogical applications, and on pharmacological targets. Reactome provides the opportunity to view predicted pathways for twenty evolutionary divergent model organisms, including Arabidopsis thaliana, Bos taurus, Caenorhadbitis elegans, Canis familiaris, Danio renio, Dictyostelium discoideum, Drosophila melanogaster, Escherichia coli, Mus musculus, Saccharomyces cerevisiae, and Rattus norvegius. These species were chosen because of the fullness of their genome sequences and annotations, and because they embody more than four billion years of evolution and span the major branches of life. Twelve of the twenty non-human species also belong to the GO Reference Genome annotation project [30]. Protein homology data obtained from Ensembl Compara [82] is used to support orthology-based inference of reactions for which high-quality whole-genome sequence data are available. Selecting the species of interest in the ‘Switch species’ dropdown menu in the upper left corner of Pathway Browser will view model organism pathway diagrams. (Fig. 2A)
The Species Comparison tool allows users to compare the predicted pathways with those of Homo sapiens to find reactions and pathways common to both your selected species and human. This tool is launched by pressing the ‘Species Comparison’ button on the sidebar, on the left-hand side of the home page. Having selected a non-human species, the results of the species comparison are presented as a sortable HTML table that can be downloaded, as a spreadsheet, or as a comma-separated or tab-delimited file, for further analysis (Fig. 6A). A ‘View’ button embedded in the results table will launch the Pathway Browser and displays the comparative pathway diagram (Fig. 6B). The physical entities in the pathway diagram are colour-coded: 1) yellow indicates the protein’s ortholog is present in the comparison species; 2) blue indicates that the protein is only known in human and that no ortholog could be found in the comparison species; 3) grey indicates that inference was not possible, e.g. for small molecules; and 4) black indicates the entity is a complex.
Figure 6.

Species Comparison Tool. (A) Results for the comparison of human and mouse pathways. Each row in the table is a pathway; the columns are pathway name, model organism name, number of proteins in the human pathway, number of orthologous proteins in the inferred model organism pathway, a graphical representation of the ratio of these two values, and a “View” button that creates a pathway diagram. ‘Sort” buttons at the top of each column allow the table to be re-ordered according to cell contents in that column. (B) The pathway browser displaying the comparison of the human and mouse DNA Damage Bypass pathways. Physical entities in the pathway diagram are colour-coded: gray, no match; black, a complex (multicomponent) entity; and yellow, the protein’s ortholog is present in human.
Working example of Reactome Species Comparison Tool: compare the murine predicted pathways with those of Homo sapiens to identify common reactions and pathways
Go to the Reactome homepage (http://www.reactome.org).
Click Species Comparison’ in the sidebar.
Select species ‘Mus musculus’ from the drop-down menu and click the ‘Apply’ button. After a few seconds, a table of results entitled ‘Species Comparison’ will appear.
Click at the head of the column labeled ‘% in other species’. The table rows should reorder so that the pathways with the greatest overlap between mouse and human are at the top.
Click at the head of the column labeled ‘Pathway name’. The table rows should revert to being ordered alphabetically according to pathway name.
Scroll down the results page and click the ‘View’ button for the pathway ‘Metabolism of amino acids and derivatives’ to open the Pathway Browser in a new window. Be sure your browser is configured to see pop-ups for Reactome.
In the top left-hand corner of the diagram, there is an icon with 4 different sizes of blue circle, which allows the user to choose a zoom level and scroll across the pathway diagram. Click on the second lowest circle to zoom out and use the arrows to scroll about the pathway diagram. About two dozen yellow coloured entities are visible. These are pathway entities conserved between both species. The two blue coloured entities represent proteins that are only found in Homo sapiens.
Mouse over one of the black coloured entities (complexes); right click and select ‘Display Participating Molecules’. A popup should appear, with a grid of coloured squares inside it, representing complex components common to both species.
Using Reactome BioMart for Data Integration
BioMart [83] is a query-orientated data mining tool that can be used for rapid bulk querying, data integration and downloading of Reactome data. BioMart can link queries together, so that the results contain information from more than one database. For example, it is possible to find the ENSEMBL IDs associated with the genes in selected Reactome pathways by linking a Reactome query to an ENSEMBL query. The Reactome BioMart can be accessed via the Tools menu located in the main navigation bar on all webpages (Fig. 7). Simple or complex queries can be created through the BioMart interface. Firstly, the Reactome preformatted queries can be accessed at the top of the page and secondly, the Regular BioMart query interface that is below the canned query selector.
Figure 7.
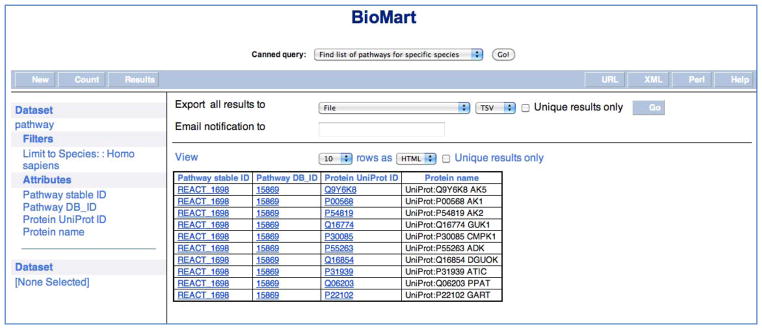
BioMart. Results of a BioMart query to find all human pathways in Reactome and to retrieve the UniProt annotations for the physical entities of the pathways.
The small set of preformatted or canned queries can be used without needing to understand the details of the BioMart query interface. A canned query selector allows users to choose from one of the currently available queries, to find: i) a list of pathways for specific species; ii) a list of reactions for specific pathway; iii) a list of proteins for specific pathways; iv) a list of complexes for specific proteins; v) a list of pathways for specific genes; vi) a list of genes for specific pathways; and vii) a list of reactions for specific genes. The results are presented in a regular BioMart results page (Fig. 8) and can be exported as HTML, tab-separated values or as an Excel spreadsheet.
The regular BioMart query interface provides users with opportunity to define their own queries. Users have control over both how the data is ‘filtered’, to limit the records that are integrated and also the ‘attributes’, corresponding to columns of data that are included in the results. There are two ways that proteomic researchers might want to use Reactome BioMart. Selecting the ‘database’ and ‘dataset’ initiate the regular query. In addition to the Reactome database, there are a number of other databases available, currently UniProt, ENSEMBL and PRIDE. Reactome provides four datasets, ‘complex’, ‘interaction’, ‘pathway’ and ‘reaction’ that are accessible to the BioMart query. For example, select the ‘pathway’ dataset if you would like to find all pathways associated with a given UniProt ID. The next step is to select the ‘Filters’ to restrict the query, e.g. ‘Limit to Species’ - Homo sapiens. If you do not use the species filter, then the results will contain information from all species known to Reactome. The ‘Attributes’ selected will specifically define what data is displayed in the results.
The second ‘Dataset’ link in the left hand panel is used to choose another dataset, providing the opportunity to integrate Reactome data with a dataset from another database. For example, if you want to find the evidence that supports the existence of the protein, associated with a set of pathways, select ‘pathway’ as the first dataset, then select ‘UNIPROT (EBI UK)] UNIPROT’ as the second. In the second dataset, click on ‘Attributes’, expand the ‘Protein attributes’ category by clicking the ‘+’ symbol in the right panel and select ‘Protein existence’ to include this attribute in the final results display.
Application programming interfaces (API) provide more flexible and interactive connections for automated data exchange between local programs and pathway databases. Reactome has a Perl-based API providing access to BioMart datasets. Perl and JAVA APIs and SOAP also give programmatic access to Reactome’s MySQL database. Describing their functionality is beyond the scope of this tutorial, but documentation is available via the Reactome website download page (http://www.reactome.org/download/index.html).
Working example of Reactome BioMart: query and extract Reactome protein and pathway annotations
Go to the Reactome homepage (http://www.reactome.org).
Under the ‘Tools’ in the Navigation bar, select ‘BioMart: query, link’.
Select ‘Find list of pathways for specific proteins’ from the ‘Canned query’ drop-down menu and click the ‘GO’ button. This will perform a preformatted BioMart query.
Click the ‘Show example’ button on the BioMart page and then click ‘Run query’. After a few seconds, a table of results will appear. Clicking the ‘Protein UniProt IDs’ and ‘Pathway ID’s in the table will connect to the UniProt database and Reactome pathway diagrams, respectively.
Click ‘New’ button towards the top of the page to reset the query submission page.
Choose the database ‘REACTOME’ from the ‘CHOOSE DATABASE’ drop-down menu of the regular BioMart query section (below the canned query). A new selector should appear, saying ‘CHOOSE DATASET’.
Click on the ‘CHOOSE DATASET’ selector. There should be 4 datasets: ‘complex’, ‘interaction’ ‘pathway’ and ‘reaction’. Select ‘reaction’.
Click on ‘Filters’ on the left hand side of the page, and then click on the right hand side of the page to select the ‘Homo sapiens’ filter from the ‘Limit to Species:’ drop-down menu.
Click on ‘Attributes’ on the left hand side of the page, and then on the right hand side of the page select the attributes ‘Reaction name’, ‘Protein UniProt ID’ and ‘Protein name.
Click on the ‘Results’ button towards the top of the page.
Click on a ‘Reaction DB_ID’ link to visualize the corresponding Reactome reaction in the Pathway Browser.
Go back to the BioMart results table, and then click the ‘Go’ button above the results table. This will download the BioMart results as a tab-separated value (TSV) file.
Challenges and Future Directions for Reactome
Pathway databases such as Reactome have made important contributions and advances in recent years in the way of data visualization and analysis. However, there are still some challenges outstanding. Our current curation practices allow Reactome to capture, to a high degree of accuracy, pathway annotations encompassing many areas of normal and developmental biology. However, one major caveat of manually curated databases is the low coverage of physical entities. Reactome curators will continue to systematically annotate proteins. We intend to extend our annotations to new signaling pathways, biological processes. Reactome already contains annotations a few tissue-specific processes derived from annotations of generic processes. An area of focus for future curation is pathways associated with pathological and infectious disease. Furthermore, pathway annotations in Reactome have concentrated on the properties and functions of proteins. However, a substantial part of the human genome is transcribed into noncoding RNAs, and these entities contribute to the regulation of signaling and other biological processes [84-86].
Pathway databases like Reactome must maintain and increase their commitment to collaboration and integrating biochemical, biological, biophysical and chemical information data exchange formats. Reactome has been exchanging data with a number of databases, including NCI-PID and is currently working with WikiPathways to create a specific data exchange framework. We would be encouraged to see the linkages to between databases and Reactome, and the integration of Reactome pathway data into other bioinformatics resources. Reactome does not currently store information on enzyme kinetics or protein binding affinities. Information on enzyme kinetics is highly dependent on experimental conditions, which would need to be described in a systematic way in order to allow for one-to-one comparisons. Reactome can provide systems biologists with a reaction graph into which kinetic data from other sources could be integrated. There is a need for much more quantitative data, such as reaction kinetics, entity stoichiometry, molecule concentrations, and other cell- or tissue-specific data. Reactome will continue to support SBML and BioPAX data structures as these formats support these additional attributes.
The integration of pathway and interaction data has been a key element of the Reactome redevelopment. There is only one drug interaction database (ChEMBL) that currently provides a PSIQUIC web service; the rest are all protein-protein interaction datasets. Overlaying protein-small molecule data from resources such as PubChem, or proprietary sources may enable identification of novel lead compounds. Reactome will need to maintain the molecular interaction interface as these web services are deployed.
The Reactome data model, curation software tools, data visualization and analysis have focused on pathways and reactions associated with human biology. We have previously worked with model organism groups, notably rice, Arabidopsis thaliana [87], fruit fly (http://fly.reactome.org) and other plants (Gramene) and M. tuberculosis, to build pathway databases on the Reactome model. One future goal is to create other manually curated model organism pathway databases.
We have developed a new intuitive web interface to visualize and analyze pathway data, and promote integrated research on pathways. Further development will centre on evaluating the usability and functionality and refining it appropriately. New analysis tools could be developed to improve the visualization of expression data, integrate data from others ‘omics’ databases, such as expression, protein localization or transcription factors data, and clinical resources. Reactome group will continue to develop and distribute open software and standard operating procedures for the management of pathway information in order to encourage standardization and reuse.
Supplementary Material
Acknowledgments
Development of the Reactome website, data model and data analysis tools described in this tutorial are a result of concerted work of the Reactome curators and developers. We are also grateful to the many scientists who collaborated with us to build the Reactome pathway content. This work was supported by grants from the National Human Genome Research Institute at the National Institutes of Health [grant number P41 HG003751], and the European Union 6th Framework Programme ‘ENFIN” [grant number LSHG-CT-2005-518254].
Footnotes
Conflict of interest
Authors declare no conflict of interests.
References
- 1.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogata H, Goto S, Fujibuchi W, Kanehisa M. Computation with the KEGG pathway database. Biosystems. 1998;47:119–128. doi: 10.1016/s0303-2647(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 3.Karp PD, Riley M, Paley SM, Pellegrini-Toole A. The MetaCyc Database. Nucleic Acids Res. 2002;30:59–61. doi: 10.1093/nar/30.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pico AR, Kelder T, van Iersel MP, Hanspers K, et al. WikiPathways: pathway editing for the people. PLoS Biol. 2008;6:e184. doi: 10.1371/journal.pbio.0060184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaefer CF, Anthony K, Krupa S, Buchoff J, et al. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37:D674–679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gough NR, Ray LB. Mapping cellular signaling. Sci STKE. 2002;135:1632–1633. doi: 10.1126/stke.2002.135.eg8. [DOI] [PubMed] [Google Scholar]
- 7.Geer LY, Marchler-Bauer A, Geer RC, Han L, et al. The NCBI BioSystems database. Nucleic Acids Res. 2010;38:D492–496. doi: 10.1093/nar/gkp858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hucka M, Finney A, Sauro HM, Bolouri H, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 9.Funahashi A, Tanimura N, Morohashi M, Kitano H. CellDesigner: a process diagram editor for gene-regulatory and biochemical networks. BioSilico. 2003;1:159–162. [Google Scholar]
- 10.Hoops S, Sahle S, Gauges R, Lee C, et al. COPASI--a COmplex PAthway SImulator. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- 11.Shannon P, Markiel A, Ozier O, Baliga NS, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babur O, Dogrusoz U, Demir E, Sander C. ChiBE: interactive visualization and manipulation of BioPAX pathway models. Bioinformatics. 2010;26:429–431. doi: 10.1093/bioinformatics/btp665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Croft D, O’Kelly G, Wu G, Haw R, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joshi-Tope G, Gillespie M, Vastrik I, D’Eustachio P, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matthews L, Gopinath G, Gillespie M, Caudy M, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37:D619–622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vastrik I, D’Eustachio P, Schmidt E, Gopinath G, et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. 2007;8:R39. doi: 10.1186/gb-2007-8-3-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qi AD, Kennedy C, Harden TK, Nicholas RA. Differential coupling of the human P2Y(11) receptor to phospholipase C and adenylyl cyclase. Br J Pharmacol. 2001;132:318–326. doi: 10.1038/sj.bjp.0703788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dall’olio GM, Jassal B, Montanucci L, Gagneux P, et al. The annotation of the Asparagine N-linked Glycosylation pathway in the Reactome Database. Glycobiology. 2011 doi: 10.1093/glycob/cwq215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jassal B, Jupe S, Caudy M, Birney E, et al. The systematic annotation of the three main GPCR families in Reactome. Database (Oxford) 2010 doi: 10.1093/database/baq018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillespie M, Shamovsky V, D’Eustachio P. Human and chicken TLR pathways: manual curation and computer-based orthology analysis. Mamm Genome. 2010 doi: 10.1007/s00335-010-9296-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39:D52–57. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q, Cheng T, Wang Y, Bryant SH. PubChem as a public resource for drug discovery. Drug Discov Today. 2010;15:1052–1057. doi: 10.1016/j.drudis.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flicek P, Aken BL, Ballester B, Beal K, et al. Ensembl’s 10th year. Nucleic Acids Res. 2010;38:D557–562. doi: 10.1093/nar/gkp972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Overington J. ChEMBL. An interview with John Overington, team leader, chemogenomics at the European Bioinformatics Institute Outstation of the European Molecular Biology Laboratory (EMBL-EBI). Interview by Wendy A. Warr. J Comput Aided Mol Des. 2009;23:195–198. doi: 10.1007/s10822-009-9260-9. [DOI] [PubMed] [Google Scholar]
- 26.Jain E, Bairoch A, Duvaud S, Phan I, et al. Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics. 2009;10:136. doi: 10.1186/1471-2105-10-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Degtyarenko K, de Matos P, Ennis M, Hastings J, et al. ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008;36:D344–350. doi: 10.1093/nar/gkm791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frazer KA, Ballinger DG, Cox DR, Hinds DA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kent WJ, Sugnet CW, Furey TS, Roskin KM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The Genome Ontology Consortium. The Gene Ontology’s Reference Genome Project: a unified framework for functional annotation across species. PLoS Comput Biol. 2009;5:e1000431. doi: 10.1371/journal.pcbi.1000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The Genome Ontology Consortium. The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res. 2010;38:D331–335. doi: 10.1093/nar/gkp1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McEntyre J, Lipman D. PubMed: bridging the information gap. CMAJ. 2001;164:1317–1319. [PMC free article] [PubMed] [Google Scholar]
- 33.Montecchi-Palazzi L, Beavis R, Binz PA, Chalkley RJ, et al. The PSI-MOD community standard for representation of protein modification data. Nat Biotechnol. 2008;26:864–866. doi: 10.1038/nbt0808-864. [DOI] [PubMed] [Google Scholar]
- 34.Harris TW, Antoshechkin I, Bieri T, Blasiar D, et al. WormBase: a comprehensive resource for nematode research. Nucleic Acids Res. 2010;38:D463–467. doi: 10.1093/nar/gkp952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Novere N, Hucka M, Mi H, Moodie S, et al. The Systems Biology Graphical Notation. Nat Biotechnol. 2009;27:735–741. doi: 10.1038/nbt.1558. [DOI] [PubMed] [Google Scholar]
- 36.Demir E, Cary MP, Paley S, Fukuda K, et al. The BioPAX community standard for pathway data sharing. Nat Biotechnol. 2010;28:935–942. doi: 10.1038/nbt.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luciano JS. PAX of mind for pathway researchers. Drug Discov Today. 2005;10:937–942. doi: 10.1016/S1359-6446(05)03501-4. [DOI] [PubMed] [Google Scholar]
- 38.Noy NF, Crubezy M, Fergerson RW, Knublauch H, et al. Protege-2000: an open-source ontology-development and knowledge-acquisition environment. AMIA Annu Symp Proc. 2003:953. [PMC free article] [PubMed] [Google Scholar]
- 39.Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000;28:3442–3444. doi: 10.1093/nar/28.18.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xenarios I, Rice DW, Salwinski L, Baron MK, et al. DIP: the database of interacting proteins. Nucleic Acids Res. 2000;28:289–291. doi: 10.1093/nar/28.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bader GD, Donaldson I, Wolting C, Ouellette BF, et al. BIND--The Biomolecular Interaction Network Database. Nucleic Acids Res. 2001;29:242–245. doi: 10.1093/nar/29.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, et al. MINT: a Molecular INTeraction database. FEBS Lett. 2002;513:135–140. doi: 10.1016/s0014-5793(01)03293-8. [DOI] [PubMed] [Google Scholar]
- 43.Hermjakob H, Montecchi-Palazzi L, Lewington C, Mudali S, et al. IntAct: an open source molecular interaction database. Nucleic Acids Res. 2004;32:D452–455. doi: 10.1093/nar/gkh052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prieto C, De Las Rivas J. APID: Agile Protein Interaction DataAnalyzer. Nucleic Acids Res. 2006;34:W298–302. doi: 10.1093/nar/gkl128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stark C, Breitkreutz BJ, Reguly T, Boucher L, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goll J, Rajagopala SV, Shiau SC, Wu H, et al. MPIDB: the microbial protein interaction database. Bioinformatics. 2008;24:1743–1744. doi: 10.1093/bioinformatics/btn285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lynn DJ, Winsor GL, Chan C, Richard N, et al. InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol Syst Biol. 2008;4:218. doi: 10.1038/msb.2008.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Michaut M, Kerrien S, Montecchi-Palazzi L, Chauvat F, et al. InteroPORC: automated inference of highly conserved protein interaction networks. Bioinformatics. 2008;24:1625–1631. doi: 10.1093/bioinformatics/btn249. [DOI] [PubMed] [Google Scholar]
- 49.Razick S, Magklaras G, Donaldson IM. iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformatics. 2008;9:405. doi: 10.1186/1471-2105-9-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chautard E, Ballut L, Thierry-Mieg N, Ricard-Blum S. MatrixDB, a database focused on extracellular protein-protein and protein-carbohydrate interactions. Bioinformatics. 2009;25:690–691. doi: 10.1093/bioinformatics/btp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aranda B, Achuthan P, Alam-Faruque Y, Armean I, et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ceol A, Chatr Aryamontri A, Licata L, Peluso D, et al. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010;38:D532–539. doi: 10.1093/nar/gkp983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010;11:R53. doi: 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chautard E, Fatoux-Ardore M, Ballut L, Thierry-Mieg N, Ricard-Blum S. MatrixDB, the extracellular matrix interaction database. Nucleic Acids Res. 2011;39:D235–240. doi: 10.1093/nar/gkq830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Isserlin R, El-Badrawi RA, Bader GD. The Biomolecular Interaction Network Database in PSI-MI 2.5. Database (Oxford) 2011 doi: 10.1093/database/baq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mi H, Thomas P. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol Biol. 2009;563:123–140. doi: 10.1007/978-1-60761-175-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crameri R, Kodzius R. The powerful combination of phage surface display of cDNA libraries and high throughput screening. Comb Chem High Throughput Screen. 2001;4:145–155. doi: 10.2174/1386207013331237. [DOI] [PubMed] [Google Scholar]
- 60.Zhu H, Bilgin M, Bangham R, Hall D, et al. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 61.Ho Y, Gruhler A, Heilbut A, Bader GD, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 62.Salwinski L, Miller CS, Smith AJ, Pettit FK, et al. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krogan NJ, Cagney G, Yu H, Zhong G, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 64.Uetz P, Giot L, Cagney G, Mansfield TA, et al. A comprehensive analysis of protein- protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 65.Arifuzzaman M, Maeda M, Itoh A, Nishikata K, et al. Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 2006;16:686–691. doi: 10.1101/gr.4527806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Butland G, Peregrin-Alvarez JM, Li J, Yang W, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 67.Parrish JR, Yu J, Liu G, Hines JA, et al. A proteome-wide protein interaction map for Campylobacter jejuni. Genome Biol. 2007;8:R130. doi: 10.1186/gb-2007-8-7-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rain JC, Selig L, De Reuse H, Battaglia V, et al. The protein-protein interaction map of Helicobacter pylori. Nature. 2001;409:211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- 69.Giot L, Bader JS, Brouwer C, Chaudhuri A, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 70.Li S, Armstrong CM, Bertin N, Ge H, et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ewing RM, Chu P, Elisma F, Li H, et al. Large-scale mapping of human protein- protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 73.Stelzl U, Worm U, Lalowski M, Haenig C, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 74.Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, et al. Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci U S A. 1996;93:14440–14445. doi: 10.1073/pnas.93.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCormack AL, Schieltz DM, Goode B, Yang S, et al. Direct analysis and identification of proteins in mixtures by LC/MS/MS and database searching at the low-femtomole level. Anal Chem. 1997;69:767–776. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 76.GW, Cazares LH, Leung SM, Nasim S, et al. Proteinchip(R) surface enhanced laser desorption/ionization (SELDI) mass spectrometry: a novel protein biochip technology for detection of prostate cancer biomarkers in complex protein mixtures. Prostate Cancer Prostatic Dis. 1999;2:264–276. doi: 10.1038/sj.pcan.4500384. [DOI] [PubMed] [Google Scholar]
- 77.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20:301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 78.Zhao Y, Lee WN, Xiao GG. Quantitative proteomics and biomarker discovery in human cancer. Expert Rev Proteomics. 2009;6:115–118. doi: 10.1586/epr.09.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 80.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 81.Vizcaino JA, Reisinger F, Cote R, Martens L. PRIDE and “Database on Demand” as valuable tools for computational proteomics. Methods Mol Biol. 2011;696:93–105. doi: 10.1007/978-1-60761-987-1_6. [DOI] [PubMed] [Google Scholar]
- 82.Vilella AJ, Severin J, Ureta-Vidal A, Heng L, et al. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Durinck S, Moreau Y, Kasprzyk A, Davis S, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 84.Rinn JL, Kertesz M, Wang JK, Squazzo SL, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guttman M, Amit I, Garber M, French C, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Loewer S, Cabili MN, Guttman M, Loh YH, et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010;42:1113–1117. doi: 10.1038/ng.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsesmetzis N, Couchman M, Higgins J, Smith A, et al. Arabidopsis reactome: a foundation knowledgebase for plant systems biology. Plant Cell. 2008;20:1426–1436. doi: 10.1105/tpc.108.057976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.