

Abstract
TET proteins have been found to play an important role in active demethylation at CpG sites in mammals. There are some reports implicating their functions in removal of DNA methylation imprint at the imprinted regions in the germline. However, it is not well established whether TET proteins can also be involved in demethylation of DNA methylation imprint in embryonic stem (ES) cells. Here we report that loss of TET proteins caused significant increase in DNA methylation at the Igf2-H19 imprinted region in ES cells. We also observed variable increase in DNA methylation at the Peg1 imprinted region in the ES clones devoid of TET proteins, in particular in the differentiated ES cells. By contrast, we did not observe significant increase of DNA methylation imprint at the Peg3, Snrpn and Dlk1-Dio3 imprinted regions in ES cells lacking TET proteins. Interestingly, loss of TET proteins did not result in significant increase of DNA methylation imprint at the Igf2-H19 and Peg1 imprinted regions in the embryoid bodies (EB). Therefore, TET proteins seem to be differentially involved in maintaining DNA methylation imprint at a subset of imprinted regions in ES cells and EBs.
Keywords: Genomic imprinting, TET, DNA methylation, ES cells, EB, COBRA analysis, Bisulphite sequencing
Introduction
Genomic imprinting is an epigenetic phenomenon characterized by parental origin-dependent expression of the imprinted genes (Barlow and Bartolomei, 2014; Bartolomei and Ferguson-Smith, 2011; Lawson et al., 2013; Li, 2013; Peters, 2014; Tomizawa and Sasaki, 2012). Roughly 150 imprinted genes have been identified in mouse and many of them are conserved in humans (Barlow and Bartolomei, 2014; Bartolomei and Ferguson-Smith, 2011; Kelsey and Bartolomei, 2012). A large number of the imprinted genes are clustered and co-regulated by a cis-acting imprinting control region called ICR (Ben-Porath and Cedar, 2000; Lewis and Reik, 2006; Robertson, 2005). ICRs are marked by germline-derived differential DNA methylation that is present at the CpG sites within the ICR of either paternal chromosome or maternal chromosome (Barlow and Bartolomei, 2014; Bartolomei and Ferguson-Smith, 2011; Ciccone et al., 2009). Allelic differential DNA methylation at the ICRs is maintained by DNA methyltransferase complexes to prevent its loss during cell divisions (Li et al., 1993; Li and Zhang, 2014; Moore et al., 2013; Reik et al., 2001; Tomizawa and Sasaki, 2012). Without DNA methyltransferase complexes, the newly synthesized DNA will lack DNA methylation at the CpG sites including those at the ICRs (Kaneda et al., 2004; Klose and Bird, 2006; Law and Jacobsen, 2010; Li and Zhang, 2014; Okano et al., 1999). ZFP57 and PGC7/Stella are two maternal-effect genes necessary for the maintenance of DNA methylation imprint at most imprinted regions examined (Li et al., 2008; Mackay et al., 2008; Nakamura et al., 2007; Payer et al., 2003; Quenneville et al., 2011; Strogantsev et al., 2015). Human and mouse ZFP57 proteins appear to play similar roles in genomic imprinting (Mackay et al., 2008; Takikawa et al., 2013b). Our previous studies have demonstrated that ZFP57 interacts with DNA methyltranferases via its cofactor KAP1/TRIM28 (Li et al., 2008; Zuo et al., 2012). Indeed, KAP1/TRIM28 also appears to be required for the maintenance of DNA methylation imprint (Messerschmidt et al., 2012; Zuo et al., 2012). Therefore, the DNA methyltransferase complexes containing ZFP57 and KAP1/TRIM28 play a major role in maintaining DNA methylation imprint (Li, 2010, 2013).
DNA methylation can be passively lost during DNA replication if it is not maintained by DNA methyltransferases (Chen and Riggs, 2011; Li and Zhang, 2014; Moore et al., 2013; Ooi et al., 2009). Besides passive loss due to DNA replication during cell divisions, DNA methylation can also be subject to active demethylation via DNA repair pathway (Walsh and Xu, 2006; Wu and Zhang, 2010). One recent exciting discovery in the epigenetics field is the presence of 5-hydroxymethylcytosine in mammalian DNA (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009). It is well-established that 5-methylcytosine can be catalytically converted to 5-hydroxymethylation by three mammalian TET proteins (Gu et al., 2011; Guo et al., 2014; Guo et al., 2011; He et al., 2011; Ito et al., 2011; Ko et al., 2015; Lee et al., 2014; Pastor et al., 2013; Tahiliani et al., 2009; Williams et al., 2012; Xu and Walsh, 2014). Interestingly, it is reported in some studies that DNA methylation imprint may be partially erased by TET proteins in the germline during the resetting of genomic imprinting (Dawlaty et al., 2013; Hackett et al., 2013; Ko et al., 2015; Nakamura et al., 2012; Piccolo et al., 2013; Yamaguchi et al., 2013). However, passive loss of DNA methylation through DNA replication may be more important in erasure of the original DNA methylation imprint in the germline (Kagiwada et al., 2013). Upon fertilization, both maternal and paternal pronuclear genomes undergo locus-specific passive and active demethylation in the zygote (Guo et al., 2014; Wang et al., 2014; Xu and Walsh, 2014). The patterns of differential DNA methylation at various ICRs are reformed in the zygote, and DNA methylation imprint is thought to be stably maintained in somatic cells after it is established in the germline (Barlow and Bartolomei, 2014; Bartolomei and Ferguson-Smith, 2011; Tilghman, 1999). Indeed, a recent whole-genome bisulphite sequencing analysis has confirmed this hypothesis although several imprinted regions may be subject to DNA demethylation during early embryogenesis (Wang et al., 2014). PGC7/Stella has been found to protect DNA methylation imprint from TET3-catalyzed 5mC oxidation in early mouse embryos (Nakamura et al., 2012). Despite these advances, it is not clear whether TET proteins could also play a role in maintaining genomic imprinting in ES cells. Here we provide evidence suggesting that loss of TET proteins in embryonic stem (ES) cells may affect the steady-state level of DNA methylation imprint at a subset of imprinted regions.
Materials and Methods
Undifferentiated ES cell culture for uES samples
For undifferentiated ES cells (Figure 1), ES clones were cultured with ES cell growth medium and plated on top of the irradiated SNL feeder cells that constitutively express leukemia inhibitory factor (LIF). The ES cell growth medium was made of DMEM medium plus 15% (v/v) fetal bovine serum (FBS). ES clones were passaged onto 6-well or 24-well plates once every 3–5 days. The uES genomic DNA samples were harvested from the undifferentiated ES cells grown on 6-well plates when they became confluent so that only a small portion of genomic DNA was derived from feeder cells. We estimated that less than 5% of the entire population of the cells might be SNL feeder cells and a vast majority of the cells used for DNA preparation should be ES cells.
Figure 1. The undifferentiated ES cell colonies and differentiated mature EBs of TET mutant ES clones displayed similar morphology to those of wild-type parental ES clone.
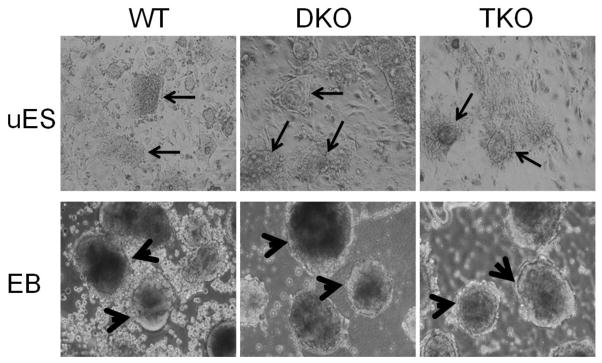
The undifferentiated ES cells and EBs derived from a TET DKO ES clone and a TET TKO ES clone were shown here, along with those derived from the wild-type parental ES clone. Arrows, undifferentiated ES cell colonies grown on top of the irradiated SNL feeder cells. Arrowheads, differentiated mature EBs cultured on non-adherent plates for 9 days.
Differentiated ES cell culture for dES samples
For dES samples, undifferentiated ES cells grown on top of the irradiated SNL feeder cells were plated onto gelatin-coated 6-well or 24-well plates without SNL feeder cells. The ES clones grown on gelatin-coated plates for one generation were passaged onto gelatin-coated 6-well or 24-well plates without SNL feeder cells for one more generation. The ES clones were mostly differentiated and free of almost all SNL feeder cells after two generations on gelatin-coated plates (Figure S1). These differentiated ES cells (dES) were harvested for preparation of dES genomic DNA samples when they became confluent on gelatin-coated plates.
Embryoid body (EB) culture for EB samples
About 1–2 million of the ES cells for these ES clones grown on top of the irradiated SNL feeder cells were plated onto the non-adherent 10-cm tissue culture dish plates coated with poly-hema (Sigma). ES cell growth medium without LIF was used for culturing EBs. Half of the medium for the EB culture was changed every 2–3 days by aspiration without removing the EBs in the dish plate. The mature EBs for each ES clone were harvested for genomic DNA preparation of EB samples after they had been cultured on non-adherent plates for 9 days (Figure 1).
Bisulphite mutagenesis
The uES, dES and EB genomic DNA samples for each ES clone were subjected to bisulphite treatment with the EZ DNA Methylation-Gold™ Kit (Zymo Research). The bisulphite-treated DNA was used for COBRA analysis of the imprinted regions and the non-imprinted IAP repeat regions (Figure S2).
Combined Bisulphite Restriction Analysis (COBRA)
COBRA was used for most analyses of DNA methylation levels at the imprinted regions and IAP repeats in this study (Eads and Laird, 2002; Xiong and Laird, 1997). After bisulphite mutagenesis, the purified mutagenized genomic DNA was subjected to PCR amplification with the primers covering a portion of the imprinting control region (ICR) for the imprinted regions or a portion of the non-imprinted IAP repeat regions (Takikawa et al., 2013a; Zuo et al., 2012). The resultant PCR product was used for restriction digestion for 2–3 hours with the restriction enzymes targeting the CpG sites within the amplified ICR or IAP regions (Figure S2). Then the digested PCR product was loaded to a gel for electrophoresis so that the undigested product indicative of unmethylated template DNA and digested product indicative of methylated template DNA were separated on the gel if the restriction enzyme sites for the unmethylated template DNA were lost after bisulphite mutagenesis (Figures 2–3). For each imprinted region, we performed triplicate COBRA analyses starting from the bisulphite-treated DNA samples for one restriction enzyme. The results for the triplicate COBRA are shown in Supplemental Figures S3–S7 with statistical analysis data included.
Figure 2. COBRA analysis of paternally inherited DNA methylation imprint at two imprinted regions.

PCR amplification was performed on bisulphite-treated DNA samples with the primers covering the paternally inherited imprinting control region of two imprinted regions (Figure S2). Then the PCR product was subjected to restriction enzyme digestion targeting the CpG sites. uES, undifferentiated ES cell samples derived from the ES cells grown on feeder cells. dES, differentiated ES cell samples derived from the ES cells passaged on gelatin-coated plates for two generations. EB, embryoid bodies formed after the ES cells grown on non-adherent 10-cm dish plates for 9 days. Lanes 1, wild-type (WT) ES clone. Lanes 2, TET DKO#1 ES clone. Lanes 3, TET DKO#2 ES clone. Lanes 4, TET TKO#1 ES clone. Lanes 5, TET TKO#2 ES clone. u, the restriction enzyme product of the unmethylated DNA. m, the restriction enzyme product of the methylated DNA.
A, COBRA analysis of the H19 DMR of the Igf2-H19 imprinted region, with a PCR product of 461 bp (Figure S2A). ClaI, RsaI, BstUI and TaqαI are four restriction enzymes used for digestion. The sizes of the restriction enzyme digestion product for the methylated DNA are: 350 bp and 111 bp for ClaI; 340 bp and 121 bp for RsaI; 379 bp and 82 bp for BstUI; 349 bp and 112 bp for TaqαI. B, COBRA analysis of the IG-DMR of the Dlk1-Dio3 imprinted region, with a PCR product of 384 bp (Figure S2B). HpyCH4IV and TaqαI are two restriction enzymes used for digestion. The sizes of the restriction enzyme digestion product for the methylated DNA are: 280 bp and 104 bp for ClaI; 267 bp and 117 bp for TaqαI.
Figure 3. COBRA analysis of maternally inherited DNA methylation imprint at three imprinted regions.

PCR amplification was performed on bisulphite-treated DNA samples with the primers covering the maternally inherited imprinting control region of three imprinted regions (Figure S2). Then the PCR product was subjected to restriction enzyme digestion targeting the CpG sites. uES, undifferentiated ES cell samples derived from the ES cells grown on feeder cells. dES, differentiated ES cell samples derived from the ES cells passaged on gelatin-coated plates for two generations. EB, embryoid bodies formed after the ES cells grown on non-adherent 10-cm dish plates for 9 days. Lanes 1, wild-type (WT) ES clone. Lanes 2, TET DKO#1 ES clone. Lanes 3, TET DKO#2 ES clone. Lanes 4, TET TKO#1 ES clone. Lanes 5, TET TKO#2 ES clone. u, the restriction enzyme product of the unmethylated DNA. m, the restriction enzyme product of the methylated DNA.
A, COBRA analysis of the Peg1 imprinted region, with a PCR product of 564 bp (Figure S2C). ClaI, RsaI and HpyCH4IV are three restriction enzymes used for digestion. The sizes of the restriction enzyme digestion product for the methylated DNA are: 341 bp and 223 bp for ClaI; 348 bp and 216 bp for RsaI; 367 bp and 197 bp for HpyCH4IV. B, COBRA analysis of the Peg3 imprinted region, with a PCR product of 375 bp (Figure S2D). BstUI and TaqαI are two restriction enzymes used for digestion. The sizes of the restriction enzyme digestion product for the methylated DNA are: 223 bp and 152 bp for BstUI; 248 bp and 127 bp for TaqαI. C, COBRA analysis of the Snrpn imprinted region, with a PCR product of 375 bp (Figure S2E). BstUI and HhaI are two restriction enzymes used for digestion. The sizes of the restriction enzyme digestion product for the methylated DNA are: 264 bp and 135 bp for BstUI; 265 bp and 136 bp for HhaI.
Bacterial colony bisulphite sequencing
Upon ligation, the purified bisulphite PCR product of the bisulphite-treated DNA samples was cloned into the pGEM-T vector system (Promega). After bacterial transformation, the bacterial colonies on the dish plates were sent for direct sequencing (Zuo et al., 2012). The sequence results for the imprinted regions were analyzed with the web-based bisulphite DNA sequence analysis program called QUMA (see the website: http://quma.cdb.riken.jp/).
Results
TET mutant ES clones were generated in the previous study (Hu et al., 2014). One wild-type parental ES clone, two TET DKO (Tet1−/−Tet2−/−) and two TET TKO (Tet1−/−Tet2−/−Tet3−/−) ES clones were cultured for the samples of the undifferentiated ES cells (uES) grown on SNL feeder cells (Figure 1), differentiated ES cells (dES) grown on gelatin-coated plates for two generations without LIF (Figure S1), and embryoid bodies (EB) after extended culture for 9 days on non-adherent tissue culture plates coated with poly-hema (Figure 1). These clones were named WT#1, DKO#1, DKO#2, TKO#1 and TKO#2, respectively. Genomic DNA samples were isolated from uES cells, dES cells and EBs for each ES clone. Then we subjected these 15 genomic DNA samples to COBRA analysis (Takikawa et al., 2013a; Zuo et al., 2012).
DNA methylation imprint at two paternally imprinted regions
COBRA analysis was performed at the region of H19 DMR and IG-DMR of Dlk1-Dio3 imprinted region, with a PCR product of 461 bp and 384 bp, respectively (Figures S2A and S2B). Compared with that of the wild-type parental ES cells (WT#1), H19 DMR appeared to be hypermethylated in the uES samples of two TET DKO and two TET TKO ES cell clones based on COBRA analyses of the genomic DNA samples with four different restriction enzymes recognizing the distinctive CpG sites within the H19 DMR (Figure 2A). When quantified by ImageJ, hypermethylation was observed at the H19 DMR in the uES samples of these TET DKO and TET TKO ES clones compared with that of WT#1 (Figure S3). Based on the triplicate COBRA analyses of the H19 DMR with ClaI, H19 DMR was significantly hypermethylated in the undifferentiated ES cells of four TET mutant ES clones in comparison to those of WT#1 (Figure S3A).
Compared with that of WT#1, the methylation level was not much different at the H19 DMR in the dES sample of TET DKO#2 (Figure 2 and Figure S3). By contrast, significant increase of methylation was observed at the H19 DMR in the dES samples of TET DKO#1 and two TET TKO ES clones in comparison to WT#1 (Figure 2 and Figure S3). For EBs, roughly similar levels of methylation were observed at the H19 DMR in WT#1, two TET DKO and two TET TKO ES clones (Figure 2 and Figure S3). These results were confirmed by statistical analyses for the COBRA of H19 DMR with ClaI (Figure S3A). Therefore, loss of TET proteins did not cause any significant increase of methylation at the H19 DMR in the TET mutant ES clones when they differentiated as EBs but it resulted in hypermethylation at the CpG sites of the H19 DMR when TET mutant ES clones were induced to differentiate by culturing on gelatin-coated plates for two generations.
By large, DNA methylation imprint at the IG-DMR of Dlk1-Dio3 imprinted region did not appear to be hypermethylated in the uES, dES and EB samples of two TET DKO and two TET TKO ES clones, compared with those of WT#1 (Figure 2B, Figure S4). This was confirmed by the statistical analysis of the triplicate COBRA of the IG-DMR by TaqαI (Figure S4A). Thus, DNA methylation imprint was relatively stable at the IG-DMR in the uES, dES and EB samples of the TET DKO and TKO ES clones, and loss of TET proteins did not apparently lead to hypermethylation at the IG-DMR of Dlk1-Dio3 imprinted region in either undifferentiated ES cells or their differentiated progeny.
DNA methylation imprint at three maternally imprinted regions
We also examined DNA methylation imprint by COBRA at three maternally imprinted regions (Peg1, Peg3 and Snrpn), with a PCR product of 564 bp, 375 bp and 401 bp, respectively (Figure S2 and Figure 3). Peg1 DMR appeared to be mildly hypermethylated, with variable degrees, in some samples derived from TET DKO and TET TKO ES clones in comparison to WT#1 (Figure 3A, Figure S5). Based on the results of triplicate COBRA of Peg1 DMR by HpyCH4IV, significant increase of methylation was observed at the Peg1 DMR in the uES samples of DKO#1 and TKO#2 compared with that of WT#1 (Figure S5C). Methylation appeared to be increased, although not statistically significant, at the Peg1 DMR in the uES samples of DKO#2 and TKO#1 in comparison to WT#1 (Figure S5C). This increase of methylation was more apparent in the dES samples. Peg1 DMR was significantly hypermethylated in the dES samples of DKO#1, TKO#1 and TKO#2 compared with that of WT#1 (Figure S5C). Methylation at the Peg1 DMR in the dES sample of DKO#2 also appeared to be increased in comparison to WT#1 although it was not statistically significant (Figure S5C). Methylation levels were more variable at the Peg1 DMR in the EB samples of TET DKO and TKO ES clones. Based on the triplicate COBRA of the Peg1 DMR by HpyCH4IV, methylation appeared to be increased in 3 out of 4 EB samples of TET DKO and TKO ES clones although this increase was not statistically significant (Figure S5C). Surprisingly, we did not observe similar increase of methylation at the Peg1 DMR in COBRA analyses by ClaI and RsaI (Figure 3A, Figure S5). Peg1 DMR appeared to be similarly methylated at the CpG site recognized by ClaI in almost all samples, whereas the CpG site of Peg1 DMR recognized by RsaI exhibited higher methylation in some TET TKO samples, in particular the dES samples of TET TKO ES clones (Figure 3A, Figure S5). It appears that loss of TET proteins caused variable increase of methylation at the Peg1 DMR with some CpG sites being more affected than others. Hypermethylation of Peg1 DMR was more apparent in the differentiated ES samples without all three TET proteins.
We also examined DNA methylation imprint at the Peg3 imprinted region (Figure S2D). Although it seems that BstUI digestion for Peg3 DMR was not as complete as TaqαI digestion, the results for these two restriction digestions of Peg3 DMR are largely consistent with each other (Figure 3B and Figure S6). Methylation appeared to be increased in the uES and dES samples of TET DKO and TKO ES clones in comparison to WT#1. However, it was not statistically significant based on triplicate COBRA analyses of Peg3 DMR by TaqαI (Figure S6B). Similar levels of DNA methylation were observed at the Peg3 DMR in the EB samples derived from WT#1, DKO and TKO ES clones (Figure 3B and Figure S6). We also performed bacterial colony bisulphite sequencing of the amplified bisulphite PCR product from the Peg3 DMR for these samples of WT#1, DKO#2 and TKO#2 ES clones (Figure 4). The levels of methylation at the Peg3 DMR inferred from bacterial colony bisulphite sequencing are generally consistent with those obtained in TaqαI digestion of these samples (compare Figure S6B with Figure 4). Relatively higher levels of methylation were obtained from the uES and dES samples but not from the EB samples of DKO#2 and TKO#2 ES clones in comparison to those of WT#1 according to the bisulphite sequencing results of Peg3 DMR (Figure 4). We also determined the sequencing results for only the unique clones based on the presence of unique methylated CpG sites or unique incompletely converted unmethylated cytosine (C) residues in the sequenced DNA molecules (Figure S8). Similarly, relatively higher levels of methylation were obtained from the uES and dES samples but not from the EB samples of DKO#2 and TKO#2 ES clones compared with WT#1 (Figure S8). Taken together, loss of TET proteins seemed to cause increased methylation at the Peg3 DMR in the uES and dES samples but not in the EB samples of TET mutant ES clones by COBRA and bisulphite sequencing, although it was not statistically significant based on triplicate COBRA analyses of Peg3 DMR by TaqαI.
Figure 4. Bisulphite sequencing results of the Peg3 DMR.

The bisulphite PCR product was amplified from the Peg3 DMR of the uES, dES and EB samples of the wild-type (WT), one TET DKO (DKO#2) and one TET TKO (TKO#2) ES clones after bisulphite mutagenesis, and then subjected to bacterial colony bisulphite sequencing. Filled circle, methylated CpG. Unfilled circle, unmethylated CpG. Cross (X), CpG site without a clear methylation status. Each row stands for a sequenced DNA template molecule from a single bacterial colony. The number underneath each sequencing diagram indicates the percentage of all methylated CpG sites over the total number of CpG sites of the sequenced bacterial colonies for each sample.
DNA methylation imprint at the Snrpn imprinted region was more variable in these samples based on COBRA (Figure 3C). Methylation appeared to be increased at Snrpn DMR in some samples of TET DKO and TKO ES clones, in particular the dES samples of TET TKO ES clones (Figure 3C and Figure S7). However, there was no significant increase in DNA methylation at Snrpn DMR in TET DKO and TKO ES clones based on triplicate COBRA analyses by HhaI (Figure S7B). We also performed bisulphite sequencing analysis of Snrpn DMR for the uES and dES samples of WT#1, DKO#2 and TKO#2 ES clones (Figure 5). The methylation levels at the Snrpn DMR inferred from bacterial colony bisulphite sequencing are in general agreement with those obtained in COBRA analysis (compare Figure S7 with Figure 5). Indeed, methylation at the Snrpn DMR was roughly similar in the uES and dES samples of DKO#2 and WT#1, whereas it was relatively increased in those of TKO#2 ES clone (Figure 5). We also determined the sequencing results for the Snrpn DMR of these samples based on only the unique clones containing either unique methylated CpG sites or unique incompletely converted unmethylated cytosine (C) residues (Figure S9). Compared with WT#1, Snrpn DMR was roughly similarly methylated in the uES of DKO#2 but relatively hypermethylated in the dES of DKO#2, whereas both the uES and dES samples of TKO#2 appeared to be relatively hypermethylated at Snrpn DMR (Figure S9). Taken together, Snrpn DMR appeared to be relatively hypermethylated in some uES and dES samples of TET mutant ES clones, in particular the dES samples of TET TKO ES clones, by COBRA and bisulphite sequencing. However, this increase was variable and not statistically significant based on triplicate COBRA analyses of Snrpn DMR by HhaI.
Figure 5. Bisulphite sequencing results of the Snrpn DMR.

The bisulphite PCR product was amplified from the Snrpn DMR of the uES and dES samples of the wild-type (WT), one TET DKO (DKO#2) and one TET TKO (TKO#2) ES clones after bisulphite mutagenesis, and then subjected to bacterial colony bisulphite sequencing. Filled circle, methylated CpG. Unfilled circle, unmethylated CpG. Cross (X), CpG site without a clear methylation status. Each row stands for a sequenced DNA template molecule from a single bacterial colony. The number underneath each sequencing diagram indicates the percentage of all methylated CpG sites over the total number of CpG sites of the sequenced bacterial colonies for each sample.
DNA methylation at the non-imprinted IAP repeats
In our previous study we found IAP repeat regions were almost fully methylated in undifferentiated ES cells (Zuo et al., 2012). We performed COBRA analysis for IAP repeats in these samples, with a PCR product of 257 bp (Figure S2F). Similarly, we found IAP repeats were fully methylated in the uES samples derived from WT#1, TET DKO and TET TKO ES clones (Figure 6). It remained highly methylated in the dES and EB samples derived from these ES clones, with slight loss of hypermethylation observed in WT#1 and TET TKO EB samples (Figure 6). It seems that loss of TET proteins did not have significant effect on the hypermethylation status at the IAP repeat regions in undifferentiated ES cells and their differentiated progeny.
Figure 6. COBRA analysis of non-imprinted IAP repeat regions.
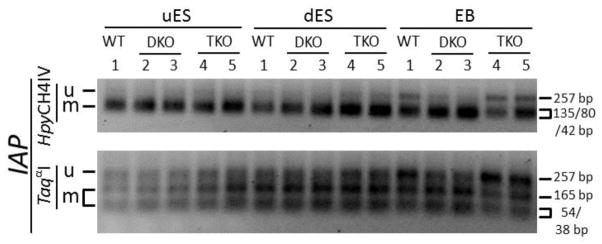
PCR amplification was performed on bisulphite-treated DNA samples with the primers covering the non-imprinted IAP repeat regions (Figure S2F). Then the PCR product was subjected to restriction enzyme digestion targeting the CpG sites. uES, undifferentiated ES cell samples derived from the ES cells grown on feeder cells. dES, differentiated ES cell samples derived from the ES cells passaged on gelatin-coated plates for two generations. EB, embryoid bodies formed after the ES cells grown on non-adherent 10-cm dish plates for 9 days. Lanes 1, wild-type (WT) ES clone. Lanes 2, TET DKO#1 ES clone. Lanes 3, TET DKO#2 ES clone. Lanes 4, TET TKO#1 ES clone. Lanes 5, TET TKO#2 ES clone. u, the restriction enzyme product of the unmethylated DNA. m, the restriction enzyme product of the methylated DNA.
Discussion
DNA methylation imprint is erased in the germline and re-established during gametogenesis (Barlow and Bartolomei, 2014; Bartolomei and Ferguson-Smith, 2011; Li, 2013). Upon fertilization, the patterns of differential DNA methylation at the ICRs are reformed in the zygote after germline-derived DNA methylation imprint is passed through the gametes. DNA methylation imprint is stably maintained during embryogenesis. It has been documented in a few recent studies that TET proteins may play a role in partial erasure of DNA methylation imprint at some imprinted regions in primordial germ cells (Dawlaty et al., 2013; Hackett et al., 2013; Ko et al., 2015; Nakamura et al., 2012; Yamaguchi et al., 2013). It was also reported in a recent study that passive demethylation caused by DNA replication during cell cycle in proliferating cells may be more important than active demethylation in erasure of the original DNA methylation imprint in the germline (Kagiwada et al., 2013). Our current study demonstrated that loss of TET proteins had significant effect on DNA methylation at the H19 DMR in undifferentiated ES cells and lesser so in differentiated ES cells. DNA methylation imprint was variably hypermethylated at the Peg1 DMR in undifferentiated ES cells without TET proteins, ranging from no effect to significant hypermethylation in TET DKO and TKO ES clones. Hypermethylation became more consistently apparent at the Peg1 DMR in the differentiated ES cells derived from TET mutant ES clones, in particular TET TKO ES clones. Hypermethylation was not observed at H19 DMR when the TET mutant ES clones were differentiated as EBs, and the increase of methylation at Peg1 DMR became insignificant in the EBs of TET mutant ES clones. Loss of TET proteins did not significantly affect DNA methylation at Peg3 DMR and Snrpn DMR although methylation appeared to be increased at these two imprinted regions in the undifferentiated and differentiated ES cells of TET mutant ES clones. By contrast, DNA methylation remained relatively stable at the IG-DMR of the Dlk1-Dio3 imprinted region in either undifferentiated ES cells or differentiated cells lacking TET proteins. Therefore, loss of TET proteins has variable effect on DNA methylation imprint at different imprinted regions with some being more affected than others.
A plausible interesting observation coming out from this study is that DNA methylation imprint at H19 DMR may be more sensitive to loss of TET proteins in undifferentiated ES cells than they are in differentiated cells, whereas DNA methylation imprint at Peg1 DMR may be more prone to hypermethylation in differentiated ES cells rather than in EBs. These effects may be due to the inherent differences in the stabilities of epigenetic modifications at these DMRs in undifferentiated ES cells, differentiated ES cells or EBs. DNA methylation imprint at H19 DMR may be more stably maintained in differentiated cells, whereas methylation at Peg1 DMR may be more stable in undifferentiated ES cells relative to the differentiated ES cells.
In general, the increase of methylation at the imprinted regions may be less apparent in EBs than in ES cells. This could be the result of genome-wide DNA methylation during ES cell differentiation, similar to the gain of de novo genome-wide DNA methylation after implantation in mouse embryos (Law and Jacobsen, 2010; Li and Zhang, 2014; Smith et al., 2012). This increase in the levels of de novo DNA methylation during differentiation may stabilize the DNA methylation imprint at the imprinted regions (Gopalakrishnan et al., 2008; Latos et al., 2009), rendering them less susceptible to loss of TET proteins in the differentiated cells. These hypotheses could be tested in a future study.
Hypermethylation was more consistently observed at the H19 and Peg1 DMRs in two TET TKO ES clones than in two TET DKO ES clones. This implies that three TET proteins may play a partially redundant role in DNA methylation imprint in ES cells although TET1 and TET2 may be responsible for most TET-mediated demethylation activities in ES cells. This observation is consistent with the expression patterns of these three TET proteins. Indeed, it is reported that notable expression of TET1 and TET2 is very much restricted to ES cells whereas TET3 is highly expressed in the adult tissues but only barely expressed in ES cells (Koh et al., 2011; Tsagaratou and Rao, 2013;Dawlaty et al., 2014).
Hypermethylation at the H19 DMR in the TET mutant ES clones (TET DKO or TKO ES clones) in our study is consistent with what was reported in Tet1−/−Tet2−/− DKO mutant embryos previously published by another group (Dawlaty et al., 2014). Peg1 DMR was found to be variably hypermethylated in the TET mutant ES clones. Similarly, methylation at the Peg1 (also called Mest) DMR was also reported to be variably mildly increased in a subset of Tet1−/−Tet2−/− DKO mutant embryos (Dawlaty et al., 2014). These results suggest that loss of TET proteins have similar effects on DNA methylation imprint in ES cells and mouse embryos, and TET mutant ES clones can be used as a model system to investigate the functions of TET proteins on genomic imprinting. This is consistent with some other previously published studies using ES cells as a model system for studying genomic imprinting (Kohama et al., 2012; Latos et al., 2009; Mann, 2001; Quenneville et al., 2011; Stelzer et al., 2015; Stelzer et al., 2014; Strogantsev et al., 2015; Takikawa et al., 2013b; Zuo et al., 2012). We will continue to employ these TET mutant ES clones to dissect the dynamic maintenance mechanisms of DNA methylation imprint in our future research.
Supplementary Material
Highlights.
Loss of TET proteins causes hypermethylation at a subset of imprinted regions.
TET proteins are involved in the steady-state DNA methylation imprint in ES cells.
TET proteins play partially redundant roles in the imprinted regions.
Imprinted regions respond differently to loss of TET proteins in ES cells.
Acknowledgments
The work in the author’s laboratory is currently supported by the grant from NIH (R01GM093335). XL and GX conceived the study. LL, SM, CR, YZ, FB, SN and XL performed the experiments. XL wrote the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barlow DP, Bartolomei MS. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I, Cedar H. Imprinting: focusing on the center. Curr Opin Genet Dev. 2000;10:550–554. doi: 10.1016/s0959-437x(00)00126-x. [DOI] [PubMed] [Google Scholar]
- Chen ZX, Riggs AD. DNA methylation and demethylation in mammals. J Biol Chem. 2011;286:18347–18353. doi: 10.1074/jbc.R110.205286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009;461:415–418. doi: 10.1038/nature08315. [DOI] [PubMed] [Google Scholar]
- Dawlaty MM, Breiling A, Le T, Barrasa MI, Raddatz G, Gao Q, Powell BE, Cheng AW, Faull KF, Lyko F, et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev Cell. 2014;29:102–111. doi: 10.1016/j.devcel.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng AW, Gao Q, Powell BE, Li Z, Xu M, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eads CA, Laird PW. Combined bisulfite restriction analysis (COBRA) Methods Mol Biol. 2002;200:71–85. doi: 10.1385/1-59259-182-5:071. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan S, Van Emburgh BO, Robertson KD. DNA methylation in development and human disease. Mutat Res. 2008;647:30–38. doi: 10.1016/j.mrfmmm.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- Guo F, Li X, Liang D, Li T, Zhu P, Guo H, Wu X, Wen L, Gu TP, Hu B, et al. Active and passive demethylation of male and female pronuclear DNA in the Mammalian zygote. Cell Stem Cell. 2014;15:447–458. doi: 10.1016/j.stem.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle. 2011;10:2662–2668. doi: 10.4161/cc.10.16.17093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, Surani MA. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Zhang L, Mao SQ, Li Z, Chen J, Zhang RR, Wu HP, Gao J, Guo F, Liu W, et al. Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell. 2014;14:512–522. doi: 10.1016/j.stem.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagiwada S, Kurimoto K, Hirota T, Yamaji M, Saitou M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013;32:340–353. doi: 10.1038/emboj.2012.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- Kelsey G, Bartolomei MS. Imprinted genes … and the number is? PLoS Genet. 2012;8:e1002601. doi: 10.1371/journal.pgen.1002601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, Rao A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev. 2015;263:6–21. doi: 10.1111/imr.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohama C, Kato H, Numata K, Hirose M, Takemasa T, Ogura A, Kiyosawa H. ES cell differentiation system recapitulates the establishment of imprinted gene expression in a cell-type-specific manner. Hum Mol Genet. 2012;21:1391–1401. doi: 10.1093/hmg/ddr577. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latos PA, Stricker SH, Steenpass L, Pauler FM, Huang R, Senergin BH, Regha K, Koerner MV, Warczok KE, Unger C, et al. An in vitro ES cell imprinting model shows that imprinted expression of the Igf2r gene arises from an allele-specific expression bias. Development. 2009;136:437–448. doi: 10.1242/dev.032060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson HA, Cheverud JM, Wolf JB. Genomic imprinting and parent-of-origin effects on complex traits. Nat Rev Genet. 2013;14:609–617. doi: 10.1038/nrg3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Hore TA, Reik W. Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell. 2014;14:710–719. doi: 10.1016/j.stem.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A, Reik W. How imprinting centres work. Cytogenet Genome Res. 2006;113:81–89. doi: 10.1159/000090818. [DOI] [PubMed] [Google Scholar]
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014;6:a019133. doi: 10.1101/cshperspect.a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Extending the maternal-zygotic effect with genomic imprinting. Mol Hum Reprod. 2010;16:695–703. doi: 10.1093/molehr/gaq028. [DOI] [PubMed] [Google Scholar]
- Li X. Genomic imprinting is a parental effect established in mammalian germ cells. Curr Top Dev Biol. 2013;102:35–59. doi: 10.1016/B978-0-12-416024-8.00002-7. [DOI] [PubMed] [Google Scholar]
- Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15:547–557. doi: 10.1016/j.devcel.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- Mann JR. Deriving and propagating mouse embryonic stem cell lines for studying genomic imprinting. Methods Mol Biol. 2001;181:21–39. doi: 10.1385/1-59259-211-2:21. [DOI] [PubMed] [Google Scholar]
- Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science. 2012;335:1499–1502. doi: 10.1126/science.1216154. [DOI] [PubMed] [Google Scholar]
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Arai Y, Umehara H, Masuhara M, Kimura T, Taniguchi H, Sekimoto T, Ikawa M, Yoneda Y, Okabe M, et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol. 2007;9:64–71. doi: 10.1038/ncb1519. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, Matoba S, Tachibana M, Ogura A, Shinkai Y, Nakano T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–419. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Ooi SK, O’Donnell AH, Bestor TH. Mammalian cytosine methylation at a glance. J Cell Sci. 2009;122:2787–2791. doi: 10.1242/jcs.015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–356. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payer B, Saitou M, Barton SC, Thresher R, Dixon JP, Zahn D, Colledge WH, Carlton MB, Nakano T, Surani MA. Stella is a maternal effect gene required for normal early development in mice. Curr Biol. 2003;13:2110–2117. doi: 10.1016/j.cub.2003.11.026. [DOI] [PubMed] [Google Scholar]
- Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet. 2014;15:517–530. doi: 10.1038/nrg3766. [DOI] [PubMed] [Google Scholar]
- Piccolo FM, Bagci H, Brown KE, Landeira D, Soza-Ried J, Feytout A, Mooijman D, Hajkova P, Leitch HG, Tada T, et al. Different roles for Tet1 and Tet2 proteins in reprogramming-mediated erasure of imprints induced by EGC fusion. Mol Cell. 2013;49:1023–1033. doi: 10.1016/j.molcel.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44:361–372. doi: 10.1016/j.molcel.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–344. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer Y, Bar S, Bartok O, Afik S, Ronen D, Kadener S, Benvenisty N. Differentiation of human parthenogenetic pluripotent stem cells reveals multiple tissue- and isoform-specific imprinted transcripts. Cell Rep. 2015;11:308–320. doi: 10.1016/j.celrep.2015.03.023. [DOI] [PubMed] [Google Scholar]
- Stelzer Y, Sagi I, Yanuka O, Eiges R, Benvenisty N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat Genet. 2014;46:551–557. doi: 10.1038/ng.2968. [DOI] [PubMed] [Google Scholar]
- Strogantsev R, Krueger F, Yamazawa K, Shi H, Gould P, Goldman-Roberts M, McEwen K, Sun B, Pedersen R, Ferguson-Smith AC. Allele-specific binding of ZFP57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol. 2015;16:112. doi: 10.1186/s13059-015-0672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikawa S, Ray C, Wang X, Shamis Y, Wu TY, Li X. Genomic imprinting is variably lost during reprogramming of mouse iPS cells. Stem Cell Res. 2013a;11:861–873. doi: 10.1016/j.scr.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikawa S, Wang X, Ray C, Vakulenko M, Bell FT, Li X. Human and mouse ZFP57 proteins are functionally interchangeable in maintaining genomic imprinting at multiple imprinted regions in mouse ES cells. Epigenetics. 2013b;8:1268–1279. doi: 10.4161/epi.26544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilghman SM. The sins of the fathers and mothers: genomic imprinting in mammalian development. Cell. 1999;96:185–193. doi: 10.1016/s0092-8674(00)80559-0. [DOI] [PubMed] [Google Scholar]
- Tomizawa S, Sasaki H. Genomic imprinting and its relevance to congenital disease, infertility, molar pregnancy and induced pluripotent stem cell. J Hum Genet. 2012;57:84–91. doi: 10.1038/jhg.2011.151. [DOI] [PubMed] [Google Scholar]
- Tsagaratou A, Rao A. TET proteins and 5-methylcytosine oxidation in the immune system. Cold Spring Harb Symp Quant Biol. 2013;78:1–10. doi: 10.1101/sqb.2013.78.020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CP, Xu GL. Cytosine methylation and DNA repair. Curr Top Microbiol Immunol. 2006;301:283–315. doi: 10.1007/3-540-31390-7_11. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang J, Duan J, Gao X, Zhu W, Lu X, Yang L, Li G, Ci W, Li W, et al. Programming and inheritance of parental DNA methylomes in mammals. Cell. 2014;157:979–991. doi: 10.1016/j.cell.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Christensen J, Helin K. DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 2012;13:28–35. doi: 10.1038/embor.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GL, Walsh CP. Enzymatic DNA oxidation: mechanisms and biological significance. BMB Rep. 2014;47:609–618. doi: 10.5483/BMBRep.2014.47.11.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Shen L, Liu Y, Sendler D, Zhang Y. Role of Tet1 in erasure of genomic imprinting. Nature. 2013;504:460–464. doi: 10.1038/nature12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, Bell FT, Iacovino M, Kyba M, Xu G, et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J Biol Chem. 2012;287:2107–2118. doi: 10.1074/jbc.M111.322644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.