

ABSTRACT
During pneumonic plague, the bacterium Yersinia pestis elicits the development of inflammatory lung lesions that continue to expand throughout infection. This lesion development and persistence are poorly understood. Here, we examine spatially distinct regions of lung lesions using laser capture microdissection and transcriptome sequencing (RNA-seq) analysis to identify transcriptional differences between lesion microenvironments. We show that cellular pathways involved in leukocyte migration and apoptosis are downregulated in the center of lung lesions compared to the periphery. Probing for the bacterial factor(s) important for the alteration in neutrophil survival, we show both in vitro and in vivo that Y. pestis increases neutrophil survival in a manner that is dependent on the type III secretion system effector YopM. This research explores the complexity of spatially distinct host-microbe interactions and emphasizes the importance of cell relevance in assays in order to fully understand Y. pestis virulence.
IMPORTANCE
Yersinia pestis is a high-priority pathogen and continues to cause outbreaks worldwide. The ability of Y. pestis to be transmitted via respiratory droplets and its history of weaponization has led to its classification as a select agent most likely to be used as a biological weapon. Unrestricted bacterial growth during the initial preinflammatory phase primes patients to be infectious once disease symptoms begin in the proinflammatory phase, and the rapid disease progression can lead to death before Y. pestis infection can be diagnosed and treated. Using in vivo analyses and focusing on relevant cell types during pneumonic plague infection, we can identify host pathways that may be manipulated to extend the treatment window for pneumonic plague patients.
INTRODUCTION
Neutrophils are an important and necessary component of the innate immune system. During infection, neutrophils are rapidly called to the site of insult and can quickly activate a myriad of antimicrobial functions in an attempt to control invading pathogens (1–4). This rapid antimicrobial response is facilitated by neutrophil priming from signals initiated by the innate immune system upon detection of a pathogen (5–7). Upon direct interaction with a secondary stimulus, such as invading microbes, there is enhancement of the respiratory burst, cytoskeletal rearrangements that retain neutrophils in the lung and facilitate phagocytosis, regulation of neutrophil surface antigens (7–9), and release of antimicrobial peptides/proteins from granules (10–12). Inadvertent acute lung injury can occur if the recruited neutrophils are not efficiently released from the pulmonary compartment back into circulation or if appropriate neutrophil turnover and removal do not occur (6, 7, 12). After mounting an antimicrobial response, neutrophils typically undergo necrosis, apoptosis, or neutrophil extracellular trap formation (NETosis), all of which ultimately lead to macrophage infiltration and clearance of dead or dying neutrophils as well as any remaining bacteria (9, 12–18). Pathogenic microbes, however, have evolved ways to evade activated neutrophils (10, 19, 20), resulting in increased disease severity and mortality (20, 21). Pathogens have been shown to utilize various mechanisms to alter neutrophil function, including inhibiting neutrophil chemotaxis (22–26), preventing phagocytosis (27–31), altering degranulation (32, 33), and, importantly for this work, inhibiting neutrophil apoptosis (34–38).
Yersinia pestis, the causative agent of plague, is a high-priority pathogen that poses a severe threat to both human and animal health and continues to cause modern day outbreaks worldwide (39, 40). The ability of Y. pestis to be transmitted via respiratory droplets and its history of weaponization have led to its classification as a Tier 1 select agent. Inhalation of the bacterium leads to the most deadly form of plague disease, known as primary pneumonic plague. If antibiotic treatment is not administered within 24 h after the onset of symptoms, the disease approaches 100% mortality (40).
Despite the recruitment of neutrophils to control Y. pestis in the lungs, bacterial numbers continue to increase throughout the duration of infection (41). The increase in bacterial burden coincides with continued neutrophil recruitment, accumulation in the lungs, and subsequent targeting of neutrophils by the Y. pestis type III secretion system (T3SS) (42, 43). The massive neutrophilic infiltration during pneumonic plague results in the formation of histopathologically distinct lung lesions that arise during the later (proinflammatory) phase of disease. These lesions expand in the lungs as neutrophils continue to accumulate until death of the host, with no evidence of bacterial or neutrophil clearance (41). The continued influx of neutrophils and their apparent prolonged survival are contrary to typical neutrophil function but have also been observed in neutrophils of cystic fibrosis patients (44). The continuous neutrophil influx during pneumonic plague results in alveolar destruction within lung lesions and culminates in a severe and deadly necrotizing pneumonia (45).
Yersinia pestis is known to inhibit the host immune response via various virulence mechanisms (10, 46–49). However, it has just recently been appreciated that the functions of specific T3SS effectors may vary, depending on the cell type being targeted (50). Neutrophils are the most prominent immune infiltrate during pneumonic plague, are selectively targeted by Y. pestis, and are necessary for development of lung lesions (42). Early recruitment of neutrophils is also linked to decreased bacterial burden and increased survival of Y. pestis-infected mice (51). It is necessary to interrogate this inflammatory environment in the context of neutrophils to understand how interactions with Y. pestis determine pneumonic plague presentation and progression.
In this work, fully virulent Y. pestis was employed along with the novel use of laser capture microdissection (LCM) and transcriptome sequencing (RNA-seq) technology to evaluate host transcription within the expanding neutrophil-rich lung lesions that arise during pneumonic plague. Yersinia pseudotuberculosis has been shown to regulate genes spatially within infected tissue (52), and we hypothesized that spatially distinct transcriptional modulation would also be observable in Y. pestis lung lesions. We combined global transcriptional analysis in defined lesion microenvironments with in vitro and in vivo characterization of neutrophils that have interacted with Y. pestis. Together, these studies provide evidence that Y. pestis promotes neutrophil survival and inhibits apoptosis in a novel T3SS effector-dependent manner. This work is the first application of LCM to evaluate the effects of a bacterial pathogen on a host microenvironment during disease-mediated injury. By determining how Y. pestis alters the neutrophil life span and induces unique transcriptional responses within sites of infection, we will better understand the mechanisms by which neutrophils typically work in a broad range of diseases in which neutrophil infiltration becomes harmful to the host.
RESULTS
Characterizing host gene regulation in spatially defined regions of pneumonic plague lung lesions.
Pulmonary infection of mice with Y. pestis results in the formation of distinct inflammatory lesions in the lung during the proinflammatory phase of disease. The lesions consist primarily of Y. pestis bacteria and densely packed pockets of neutrophils, and these inflamed sites expand throughout the duration of infection, ultimately covering entire lobes of the lung (41). At 48 h postinoculation (hpi) with Y. pestis strain CO92, we observed that a typical 5-µm hematoxylin and eosin (H&E)-stained lung section sampled centrally through all 5 lung lobes contained up to 5 distinct foci of inflammation per lobe. Individual inflammatory foci were as large as 3 mm in diameter, with inflammation occupying 5 to 10% of the total examined tissue plane (Fig. 1A). The inflammatory infiltrate was composed predominantly of neutrophils, with fewer macrophages within alveolar air spaces. Interspersed amid the inflammatory infiltrate were abundant extracellular bacterial organisms and variable amounts of fibrin, hemorrhage, edema, and cellular debris. The majority of large and small bronchi remained unaffected, even those located immediately adjacent to inflammatory foci. Occasional dense aggregates of extracellular bacteria formed small colonies, particularly at the periphery of lesions (Fig. 1C). These observations suggested that we could learn more quantitative information from these lesions by spatially dissecting them to analyze unique microenvironments.
FIG 1 .

Lung lesion histology and laser capture microdissection of lesions. Representative H&E-stained sections of a Y. pestis-infected lung at 48 hpi are shown at different magnifications (A to C) Scale bars equal 500, 50, and 20 µm, respectively. (C) The green arrows indicate dense aggregates of Y. pestis. (D and E) Representative images of a lung section before (D) and after (E) laser capture microdissection. The scale bar equals 300 µm. The lung lesion (outlined in black) was identified, and the periphery and center of the lesions were spatially defined (outlined in white). Laser cutting and pulse catapulting into a preservative resin selectively removed white-outlined lesion sections.
To further characterize these inflammatory lung lesions, we focused on analyzing transcriptional responses in neutrophils to understand the lack of turnover and clearance of this key cell type. The T3SS of Y. pestis has potent anti-inflammatory effects on targeted mammalian cells (53). As a result of their direct contact with Y. pestis, we predicted that those neutrophils found within the initiating focus of the lung lesions, the lesion “center,” would have a different transcriptional profile than those found in the lesion periphery, where we assumed the most recent wave of migrating neutrophils would be found. To test this, we devised an approach to identify transcriptionally regulated host pathways in neutrophils found within these spatially distinct regions. Briefly, we infected mice with 104 cells of Yersinia pestis CO92, a fully virulent strain. At 48 hpi, we treated lungs with an RNA preservative and then used LCM on fixed and frozen sections to collect tissue from the center and periphery of the lesions (Fig. 1D and E).
Total RNA was isolated from these captured microenvironments, RNA-seq libraries were prepared, and samples were sequenced to define the host transcriptional responses. Additionally, we harvested total RNA from magnetically activated cell sorting (MACS)-isolated neutrophils (polymorphonuclear leukocytes) residing in the bone marrow (BM-PMNs) of Y. pestis-infected and mock-infected mice at 48 hpi for comparison. Using Student’s t test, 976 genes were identified that were differentially expressed between the periphery and center of the lung lesions (P < 0.05). This analysis was simultaneously performed on the infected and mock-infected BM-PMN samples. This comparison identified 3,198 genes that were differentially expressed in BM-PMNs from infected mice compared to those from mock-infected mice. Importantly, transcriptionally altered BM-PMN genes represent neutrophil-specific responses to Y. pestis infection at 48 hpi and are a heterologous tissue match to the lung lesion samples collected at the same time point during infection. Matching genes in the lesion periphery/center comparison to those genes found in the BM-PMN comparison allowed us to narrow the lung lesion gene set to those genes regulated specifically in neutrophils rather than other cell types in the lung. The initial list of 976 genes was reduced to 224 genes that were differentially expressed in both the BM-PMN and lung lesion comparisons, with a significant enrichment value of P = 0.02 (Fig. 2; see Table S1 in the supplemental material). This confirms that there are transcriptional alterations in neutrophil genes between the lesion periphery and center and establishes that Y. pestis causes a sustained change in neutrophil transcription at distinct locations in lung lesions. Interestingly, when the four conditions (infected BM-PMNs, mock-infected BM-PMNs, lesion periphery, and lesion center) were clustered across samples, the differential expression patterns of the 224 genes showed that (i) the BM-PMNs from mock-infected mice and lesion center conditions clustered together, and (ii) the BM-PMNs from Y. pestis-infected mice and lesion periphery conditions clustered together (Fig. 2). This suggests that the neutrophils in the lesion center had a transcriptional pattern more similar to unstimulated bone marrow neutrophils, despite residing in a highly inflammatory microenvironment in the infected lungs.
FIG 2 .
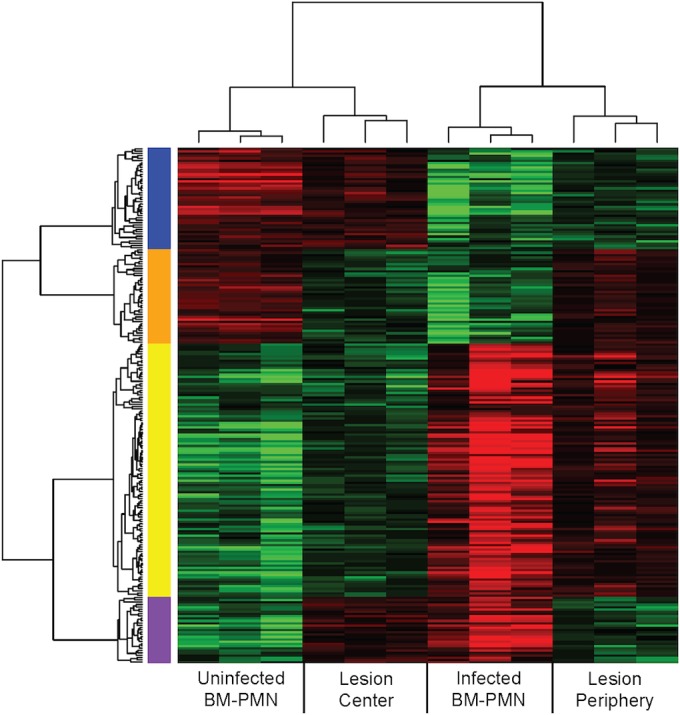
Clustered genes showing differential transcription. A heat map shows 224 genes differentially expressed in two comparisons (BM-PMNs from mock-infected versus infected mice and lesion periphery versus center). The red and green colors represent an increase or decrease, respectively, relative to the median value of each of the two initial comparisons. The x-axis clustering shows that all triplicates clustered together and that mock-infected BM-PMN and lesion center samples clustered more closely, as did infected BM-PMN and lesion periphery samples. Each gene shows significant differences (P < 0.05 by Student’s t test) in each of the two initial comparisons. The blue, orange, yellow, and purple bars represent the four possible outcomes of gene regulation comparisons: e.g., the blue bar shows genes that were decreased in infected compared to mock-infected BM-PMNs and increased in the lesion center compared to the periphery.
Gene set analysis implications for lesion development and neutrophil fate.
Using the Ingenuity Pathway Analysis (IPA) program (Qiagen, Redwood City, CA) and the Molecular Signatures Database (54), we compiled various gene sets (see Table S2 in the supplemental material) to analyze pathways for differential gene expression patterns between the lesion periphery and center. Density curves were generated based on the ratio of RNA expression estimates between the lesion periphery and lesion center (when there were more than three reads in both conditions). A Wilcoxon rank-sum test was performed comparing the log ratio distribution from the individual gene sets to the entire transcriptome, with the goal of identifying bias in specific gene sets.
Based on the log ratio of lesion periphery compared to the center, there was a statistically significant shift for genes in “leukocyte migration” pathways compared to the entire transcriptome (Fig. 3B). This shift indicates relatively lower expression in the center of lesions: i.e., the lesion centers show an overall downregulation of genes in this list. In contrast, “general cell migration” genes showed no shift in the lesion periphery/center ratio compared to the entire transcriptome (Fig. 3A; see Table S2). These transcriptional comparisons in the “leukocyte migration” pathways suggest that newly infiltrating neutrophils are localized at the periphery of the expanding lesions, while the neutrophils at the center were likely recruited much earlier. The histopathological evidence in the lungs also supports this scenario. Centrally within inflammatory foci, neutrophils are densely packed, completely filling alveolar spaces and leading to loss of the alveolar septa. Peripherally there are fewer, loosely packed neutrophils with some remaining clear air space.
FIG 3 .
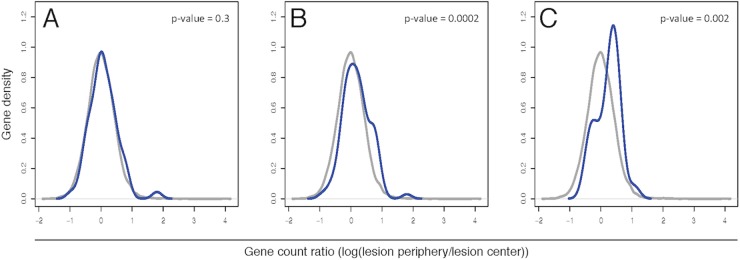
Density curves of defined gene sets compared to the entire transcriptome. Gene sets were compiled to represent the general cell migration (A), leukocyte migration (B), and apoptosis (C) pathways. Using the log ratio of RNA-seq reads in the lesion periphery compared to the lesion center, log ratios of each of these compiled gene sets (blue lines) were contrasted against the entire transcriptome (gray lines) using a Wilcoxon rank-sum test. The y-axis values are arbitrary units as calculated by Gaussian kernel density estimates. Only genes with >3 reads from the RNA-seq analysis were graphed.
We additionally evaluated whether neutrophil apoptosis/survival was being altered in the center of lung lesions, since this could explain continued neutrophil accumulation due to lack of turnover in the lung. A set of genes representing various apoptosis pathways was compiled (see Table S2 in the supplemental material) and showed a statistically significant shift in the lesion periphery/center ratio compared to the entire transcriptome, indicating a downregulation of genes in the apoptosis pathways in the lesion center (Fig. 3C).
Yersinia pestis effects on human neutrophil survival in vitro.
Based on pathway analysis of the LCM/RNA-seq data, we wanted to verify that Y. pestis alters neutrophil death, leading to increased survival, which may contribute to continued lesion expansion. We compared the survival of uninoculated primary human neutrophils to those of both Y. pestis- and nonpathogenic Escherichia coli-inoculated neutrophils in vitro. At 24 hpi, neutrophils were harvested for analysis of apoptotic and dying cells by flow cytometry. An aliquot of cells was also fixed at 0 hpi to retain the initial starting concentration of neutrophils in each assay. After 24 hpi, the population of remaining “healthy” (annexin V negative [annexin V−], propidium iodide negative [PI−]) neutrophils ranged from 40 to 60% of the initial seeding population, depending on the donor. Inoculation with nonpathogenic E. coli resulted in survival of only 20 to 30% of the initial population. In contrast, inoculation with Y. pestis resulted in 60 to 80% survival of the initial population (Fig. 4A), a significant increase in surviving healthy neutrophils compared to uninoculated or E. coli-inoculated neutrophils.
FIG 4 .

The type III secretion system effector YopM is necessary for enhanced neutrophil survival. Primary human neutrophils were incubated in buffered RPMI at 37°C and 5% CO2, with or without bacterial inoculation. After 24 h, the remaining “healthy neutrophils” were assayed by flow cytometry (CD16+, CD66b+, annexin V−, 7-aminoactinomycin D [7-AAD]/PI−). (A) After incubation with wild-type Y. pestis, there were significantly more healthy neutrophils than both uninoculated cells and nonpathogenic E. coli-inoculated cells. (B) After incubation with a Δpla mutant, there was no difference in healthy neutrophils compared to inoculation with wild-type Y. pestis. However, inoculation with a pCD1− (T3SS-negative) mutant caused a significant decrease in healthy neutrophils. (C) Comparing inoculations with six individual T3SS effector mutants, only the ΔyopM mutant significantly decreased the percentage of healthy neutrophils compared to inoculation with wild-type Y. pestis. (D) Complementation of the ΔyopM strain restored neutrophil survival to the level seen with wild-type Y. pestis. Results are from representative experiments performed in triplicate. Data are represented as means ± standard errors of the means (SEM). Asterisks represent statistical significance based on Tukey’s multiple comparison tests from an ordinary one-way analysis of variance (ANOVA) (**, P < 0.01; ***, P < 0.001).
Role of the type III secretion system in increased survival of human neutrophils.
After determining that Y. pestis significantly increased the survival of neutrophils, we tested if this could be attributed to a known virulence determinant of Y. pestis. We inoculated human neutrophils in vitro with Y. pestis strains lacking the pigmentation locus (pgm-negative) (data not shown), the plasminogen activator protease (Δpla), and the plasmid encoding the T3SS (pCD1−). We observed that neutrophils inoculated with a pgm-negative or Δpla mutant showed similar survival to those inoculated with wild-type Y. pestis, while infection with a pCD1− mutant significantly decreased neutrophil survival (Fig. 4B), indicating that the phenotype is dependent on the T3SS. We next tested mutants of the six primary T3SS effectors (ΔyopE, ΔyopH, ΔyopJ, ΔyopM, ΔyopO, and ΔyopT) in the neutrophil assay and observed that only infection with the ΔyopM mutant showed a significant decrease in neutrophil survival compared to inoculation with wild-type Y. pestis (Fig. 4C). We also observed that a ΔyopM/yopM complemented strain reproduced the phenotype of the wild-type strain (Fig. 4D) and validated our conclusion that YopM is important in modulating neutrophil survival.
A novel function for YopM in neutrophil survival.
YopM is known to have two intracellular functions: (i) binding ribosomal S6 kinase (RSK) and protein kinase N1 (PKN) (27), which induces prolonged activation of RSK (55), and (ii) inhibiting caspase-1 activity and inflammasome formation that leads to pyroptotic cell death (56, 57). To determine which function of YopM might be responsible for enhancing neutrophil survival, we performed a neutrophil infection assay in the presence of inhibitors of these two functions. To test the effect of continued YopM-induced RSK activity on neutrophil survival, we used the RSK inhibitor BI-D1870 (58). Addition of increasing doses of BI-D1870 to neutrophils inoculated with wild-type Y. pestis showed no decrease in neutrophil survival (Fig. 5A), indicating that the prolonged kinase activity of RSK induced by YopM had no effect on neutrophil survival. We validated the functionality of BI-D1870 by monitoring the phosphorylation of the S6 protein (59) at the highest tested dose of this RSK inhibitor. Even after 24 h, neutrophils treated with BI-D1870 showed decreased phosphorylation of S6 (see Fig. S1 in the supplemental material). We next tested the effect of inhibiting caspase-1 during inoculation with a ΔyopM mutant, using the caspase-1 inhibitor Z-YVAD-FMK. In the presence of this inhibitor, neutrophil survival was not enhanced after inoculation with the ΔyopM mutant compared to survival of uninoculated neutrophils. We did observe an overall increase in neutrophil survival in samples treated with Z-YVAD-FMK, though this was significantly lower than the survival seen during infection with wild-type Y. pestis. This could indicate that there is a certain amount of caspase-1-dependent neutrophil death, but this is not the mechanism through which YopM is acting (Fig. 5B). Additionally, no significant increase in survival was seen when neutrophils inoculated with wild-type Y. pestis were treated with this caspase-1 inhibitor (data not shown). This could alternatively indicate that the increase in survival seen under the other conditions is due to the potential cross reactivity of Z-YVAD-FMK with caspases-4 and -5 and may be more related to apoptosis than pyroptosis. These findings suggest a novel function of YopM specifically related to neutrophil survival that does not involve its known functions of RSK and PKN binding or caspase-1 inhibition.
FIG 5 .
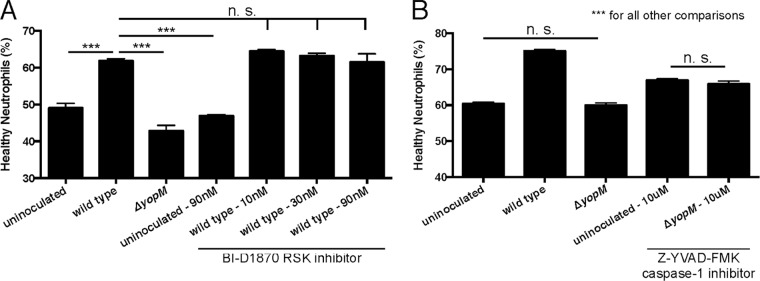
Inhibition of known functions of YopM does not alter neutrophil survival. Primary human neutrophils were incubated with or without bacterial inoculation and drug treatment. After 24 h, the remaining “healthy neutrophils” were assayed by flow cytometry (CD16+, CD66b+, annexin V−, 7-AAD/PI−). (A) Treatment with the RSK inhibitor BI-D1870 had no effect on the percentage of healthy neutrophils in culture for either uninoculated or wild-type Y. pestis-inoculated samples. (B) Treatment with the caspase-1 inhibitor Z-YVAD-FMK showed a significant increase in the percentage of healthy neutrophils compared to untreated samples. However, there was also still a significant difference in the percentage of healthy neutrophils remaining in uninoculated and ΔyopM mutant-inoculated samples compared to wild-type Y. pestis-inoculated samples. Results are from representative experiments performed in triplicate. Data are represented as means ± SEM. n.s., not significant. Asterisks represent statistical significance based on Tukey’s multiple comparison tests from an ordinary one-way ANOVA (***, P < 0.001).
Role of YopM in the inhibition of apoptosis in lung lesions.
Since YopM is important for prolonging neutrophil survival in vitro, we tested if YopM is involved in sustaining neutrophil health within inflammatory lung lesions in vivo. Lungs of mice infected with wild-type or ΔyopM Y. pestis were collected at 48 hpi and sectioned for H&E staining. Neither the size nor total number of inflammatory lesions was significantly altered in the ΔyopM-infected lungs compared to wild-type infection. The bacterial burdens in the lungs were similar between the two infections by CFU count assays (Fig. 6A). However, while the overall pattern of pulmonary inflammation was similar between the two infections (Fig. 6B and C), the centers of the largest inflammatory foci in the ΔyopM mutant-infected lungs displayed more prominent necrosis of the alveolar septa. We also observed a striking difference in lung lesion cellular appearance between wild type and ΔyopM mutant-infected mice (Fig. 6D and E). In the ΔyopM mutant-infected tissues, increased numbers of neutrophils were degranulated and had faded nuclei. Additionally, amid viable inflammatory cells, there were individual amphophilic cell bodies (3 to 5 µm in diameter, round to slightly misshapen) that lacked a discernible nucleus and often displayed faint stippling (Fig. 6E, blue arrows). Correspondingly, we observed increased DNA damage via fluorescent terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining in lung lesions of ΔyopM mutant-infected mice compared to wild-type Y. pestis-infected mice (Fig. 7). This TUNEL staining is indicative of apoptotic cell death and correlates with the presence of the amphophilic cell bodies. These in vivo observations confirm the importance of YopM-induced neutrophil survival seen in vitro. Furthermore, these results are consistent with the RNA-seq analysis suggesting that the cells in the center of lesions have a decreased capacity to undergo apoptosis in a wild-type Y. pestis infection, leading to the distinctive lung lesions seen in pneumonic plague.
FIG 6 .

YopM-dependent effects on bacterial burden and histopathology during pulmonary infection with Y. pestis. (A) At 48 hpi, there is no difference in lung bacterial burdens in mice infected with wild-type or ΔyopM mutant Y. pestis as quantified by CFU. (B and C) On microscopic examination of H&E-stained tissues, there is no difference in the size or number of lung lesions in mice infected with wild-type or ΔyopM mutant Y. pestis. (D and E) At higher magnification, however, large numbers of anucleate cell bodies can be seen throughout the lung lesions in mice infected with the ΔyopM mutant (representative cells marked with blue arrows) that are not present during an infection with wild-type Y. pestis. Scale bars in images represent 200 µm (B and C) or 20 µm (D and E). CFU data are represented as geometric means ± 95% confidence intervals (CI).
FIG 7 .

TUNEL staining of infected mouse lung sections. Shown are representative composite images of lung lesions (48 hpi) stained for damaged DNA using TUNEL (green) and total DNA using DAPI (blue). TUNEL staining, generally indicative of apoptosis, was primarily restricted to lung lesions during Y. pestis infection. More apoptosis was evident in the lung lesions of mice infected with the ΔyopM mutant than in those infected with wild-type Y. pestis.
DISCUSSION
Despite previous research on interactions between neutrophils and Y. pestis in vivo (42, 43, 60–62), there is little information to explain the architecture and rapid expansion of neutrophil-rich lesions that are the pathological hallmark of pneumonic plague. We hypothesized that Y. pestis directly alters the ability of neutrophils to progress through their cellular life cycle, leading to a failure of clearance from the lungs. This lack of clearance could then lead to development of the continually expanding inflammatory lesions, causing significant pulmonary damage. In this work, we have described a novel approach to examining bacterial pathogenesis, using laser capture microdissection (LCM) to dissect unique spatial phenotypes of tissue damage during infection in vivo that cannot be studied in vitro.
LCM was first described in 1996 (63) and is sophisticated enough to allow for DNA, RNA, and protein extraction at the single-cell level (64). Its use in the field of microbial pathogenesis, however, has rarely been exploited and has mainly been limited to plant pathogens (65–69) or in vitro studies (70). In the work presented here, we show that RNA-seq analysis after LCM is a viable technique to study host-pathogen interactions during infection. Furthermore, we can glean information about disease progression by spatially dissecting regions of inflammation and damage in vivo that could not be obtained through in vitro studies alone. In the future, it will be beneficial to be able to perform dual RNA-seq to obtain both the host and bacterial transcriptomes simultaneously.
The formation of lung lesions during pneumonic plague disease is well documented in several animal models (41, 71–77). In this study, we present the first analysis of how these lung lesions develop and persist during pneumonic plague. Further, we probe the transcriptomes of host cells in the periphery and center of these lesions in an effort to better understand this complex microenvironment. The resulting data indicated that within the lung lesions, there are spatially defined differences in transcriptional gene expression. Due to the high concentration of Y. pestis-targeted neutrophils within the lesions (42, 43), we were most interested in dissecting out neutrophil-specific transcriptional responses. When performing LCM, nonneutrophil RNA would also be isolated and complicate the neutrophil-specific analysis we were seeking. To abrogate this, we isolated BM-PMNs from mock-infected and Y. pestis-infected mice at 48 hpi to generate a list of genes we could identify as neutrophil-regulated responses to Y. pestis infection. By this time during infection, Y. pestis has disseminated into the bloodstream (41) and can even be detected in the bone marrow (78). We anticipated that any BM-PMN genes responsive to Y. pestis infection might also be identified in neutrophils in the lesion comparison. Analyzing the lesion periphery and lesion center gave us an unbiased list of differentially expressed genes between these two regions in any cell type present. Matching differentially expressed genes from the lesion periphery/center comparison to the same genes in the BM-PMN comparison resulted in a set of 224 genes we identified as likely neutrophil-specific transcriptional responses to Y. pestis infection. Using the BM-PMN list gave us confidence that the 224 genes identified in both comparisons are indicating a significant alteration in neutrophil gene expression between the lesion periphery and center, as opposed to contaminating monocytic infiltrate or lung epithelium.
In addition to analyzing differential expression between regions of lung lesions, we wanted to test the hypothesis that lesions develop from small initiating foci and continue expanding outward throughout the course of infection. Alternatively, neutrophils could be infiltrating randomly into the lesion space. To examine this, the RNA-seq data were analyzed based on defined lists of genes involved in general cell migration and leukocyte-specific migration compared to the entire transcriptome. The density distribution of the ratios of genes in each list was analyzed comparing the lesion periphery to the lesion center. This distribution was then compared to the same distribution from the entire transcriptome. This statistical comparison helped evaluate if transcription of genes in each of the lists was being skewed compared to the entire transcriptome. We observed a statistically significant shift in the log ratio in the leukocyte migration gene set, indicating decreased transcription in the center of lung lesions; in contrast, no shift was observed for genes involved in general cell migration. This implies that neutrophils in the center of lesions were recruited earlier during infection, and these cells subsequently decreased transcription of genes such as those encoding cytokine and cell-cell adhesion receptors that are necessary for chemotaxis and lung infiltration (see Table S2 in the supplemental material).
We also postulated that the lesions continue to expand over time as a result of Y. pestis-mediated alteration of neutrophil apoptosis and survival. The apoptotic pathway gene list was analyzed as described above. A significant shift in the log ratio distribution of the apoptosis gene set suggested that these pathways were downregulated in the center of lesions compared to the periphery. This list of apoptosis-related genes contains both pro- and antiapoptotic genes that need to be analyzed to determine which category of genes was being affected. Analysis of the RNA-seq data at the pathway level with the Ingenuity Pathways Analysis “apoptosis signaling” pathway (Qiagen) allowed for visualization of how the altered genes were affecting apoptosis (pro- or anti-). We could also observe how small changes in expression of pathway-related genes may escalate into greater effects downstream in the pathway. Individual genes in pathways did not always show discrete statistically significant differences (P < 0.05) between areas of the lesion but suggested overall changes when compiled with other gene alterations. For example, caspases-8 and -9 both had “nonsignificant” P values (<0.5 but >0.05). Looking more closely at where these caspases intersect in apoptosis signaling pathways, one can see how a transcriptional decrease in caspase-8 can prevent the activation of BID to tBID, ultimately preventing the release of proteins allowing for downstream caspase activation. Both caspases-8 and -9 are important for direct cleavage of caspase-3, the converging point of both the extrinsic and intrinsic apoptosis pathways (79). The decrease in transcription of both of these initiator caspases is an additional downstream hindrance to apoptosis (see Fig. S2 in the supplemental material).
We next sought to confirm that apoptosis is inhibited during Y. pestis infection in vitro and in vivo and also identify the bacterial factor or factors that are responsible for this phenotype. We therefore inoculated primary human neutrophils with fully virulent Y. pestis, as well as a repertoire of mutant strains lacking key virulence-related genes for a more detailed analysis. Prior to inoculation, Y. pestis was grown at 37°C and was not preopsonized to most closely mimic conditions during pneumonic plague disease (41). It has previously been observed that the pCD1 virulence plasmid of Y. pestis is necessary to inhibit neutrophil production of reactive oxygen species and decrease neutrophil apoptosis (50). Although we initially began evaluating apoptosis and death of neutrophils, we found that, despite an obvious loss of viable neutrophils, annexin V staining was not consistently different by 24 hpi. While neutrophils express phosphatidylserine as a marker of apoptosis, they continue to progress through apoptosis and are no longer detectable by flow cytometry at later time points. Thus, examination of a singular time point only visualizes cells that are currently apoptotic and fails to capture those that have already gone through apoptosis. We therefore changed our readout to detect surviving “healthy neutrophils” by subtracting the apoptotic neutrophils (annexin V+), dead neutrophils (PI+), and neutrophils that became undetectable by flow cytometry due to turnover (subtracting neutrophils at 24 hpi from total starting neutrophils) from the starting number of neutrophils. We have shown with this assay that Y. pestis leads to increased survival of primary human neutrophils and that the T3SS effector YopM is necessary for this phenotype. This is the first direct evidence that a single Y. pestis gene is able to extend the survival of neutrophils.
After testing for the known YopM functions of RSK activation and caspase-1 inhibition, we were unable to confirm that either of these functions had any effect on neutrophil survival in vitro. We hypothesize that, similar to YopJ/P (50), YopM has a unique function in neutrophils that differs from what is observed in macrophages. Among the six T3SS effectors that are encoded on the pCD1 plasmid, only YopM has no known enzymatic function. Rather, YopM has been shown to have protein-protein binding functions (80). It was initially shown to bind α-thrombin (81) and subsequently α1-antitrypsin (82), although neither of these binding functions is thought to contribute to Y. pestis virulence (81). YopM was later shown to bind p90 ribosomal S6 kinase (RSK) and protein kinase N1 (PKN) isoforms (27). The C-terminal end of YopM binds phosphorylated RSK (83), inhibiting dephosphorylation and prolonging its kinase activity (55). YopM binding to RSK and PKN also facilitates activated RSK to phosphorylate PKN and activate its kinase activity (27). The RSK and PKN binding domains of YopM are required for interleukin-10 (IL-10) production during a Y. pseudotuberculosis intravenous infection (84) and result in attenuation of virulence (85). More recently it has been shown that YopM can also bind both active caspase-1 (56) and IQGAP1 (57), depending on the isoform of YopM being tested. Both of these binding activities lead to an inhibition of inflammasome assembly and impede pyroptosis in macrophages. Other studies have shown the effects of YopM on bubonic plague and systemic infection through cytokine analysis and organ-specific cell composition of lesions (86–89), but this is the first study to suggest a function of YopM during pneumonic plague as well as its ability to promote neutrophil survival.
The influence of YopM on apoptosis of neutrophils has been previously studied in systemic infection by a pgm-negative strain of Y. pestis and appears to be organ specific (89). Our work focuses on neutrophils and pathology in the lung during infection with a fully virulent Y. pestis strain. We demonstrate the importance of YopM for neutrophil survival in vitro as well as for neutrophil integrity and reduction of apoptosis in vivo, as demonstrated by lesion histopathology and TUNEL staining. Linking the in vitro phenotype of increased neutrophil survival to apoptosis in vivo further validates the LCM/RNA-seq conclusion that the apoptosis pathway is inhibited in the center of lung lesions compared to the periphery. Along with observing cells that lacked a discernible nucleus, we detected increased, diffuse TUNEL staining after infection with the ΔyopM mutant strain. This physiology and diffuse damaged DNA staining could suggest the formation of neutrophil extracellular traps (NETs) within the lung lesions (90). Further investigation of the formation of NETs after infection with a ΔyopM mutant strain of Y. pestis could elucidate the specific effect that YopM is having on neutrophils.
In summary, we introduced a unique application of LCM technology to examine directly the spatially distinct microenvironments resulting from host-pathogen interactions during primary pneumonic plague. As a result, we were able to show that Y. pestis modulates neutrophil survival in vivo in a YopM-dependent manner, interfering with progression through apoptosis. Based on the healthy appearance of the neutrophils by histology and TUNEL analysis during Y. pestis infection, it is possible that YopM is preventing the degranulation of neutrophils or inhibiting the initiation of NET formation. Hindering either of these functions would ultimately result in neutrophils that are not working efficiently to clear the infection. The active process of Y. pestis inhibiting neutrophil function consequently leads to a lack of neutrophil death and immune cell-mediated bacterial clearance, contributing to lesion formation in the lung. Further definition and characterization of mediators of the dramatic pulmonary inflammation that occurs during pneumonic plague will help in understanding the lethality of this disease and may contribute to our understanding of severe pulmonary infection with other respiratory pathogens.
MATERIALS AND METHODS
All reagents were obtained from Sigma-Aldrich unless otherwise noted.
Ethics statement.
The use of live vertebrate animals was performed in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, the Amended Animal Welfare Act of 1985, and the regulations of the United States Department of Agriculture (USDA), All animal studies were approved by the University of North Carolina at Chapel Hill Office of Animal Care and Use, protocols 12-028.0 and 15-022.0.
Bacterial strains and culture conditions.
The fully virulent Yersinia pestis strain CO92 and the plasmid-cured strain (pCD1−) were obtained from the U.S. Army, Ft. Detrick, MD. The presence or absence of pCD1 and the pgm locus was confirmed by PCR for each strain before use. The construction of the Δpla and ΔyopH Y. pestis strains has been described previously (10, 20). Yop deletion strains (ΔyopE, -J, -M, -O, and -T) were constructed using a lambda red recombination system as described previously (20). The yopM gene was complemented back into its native site on the pCD1 plasmid using the pSR47S recombination method as previously described (22, 24). The Escherichia coli strain pir116 was used in the in vitro assays.
Strains were grown on brain heart infusion (BHI) agar (Difco Laboratories) at 26°C for 2 days. For infections, liquid cultures of Y. pestis CO92 were grown in BHI broth for 6 to 12 h at 26°C. The cultures were then diluted to an optical density at 620 nm (OD620) of 0.05 to 0.1 in BHI supplemented with 2.5 mM CaCl2 and grown for 12 to 16 h at 37°C with constant shaking.
Animals and infections.
Six- to 8-week-old female C57BL/6J mice were obtained from Jackson Laboratories. Mice were provided with food and water ad libitum and maintained at 25°C and 15% humidity with alternating 12-h periods of light and dark. For animal infections, groups of 3 to 10 mice were lightly anesthetized with ketamine/xylazine and inoculated intranasally with a lethal dose of 104 CFU suspended in 20 µl phosphate-buffered saline (PBS). The number of actual CFU inoculated was determined by plating serial dilutions of the inoculum on BHI. Moribund animals were euthanized with an overdose of sodium pentobarbital. For determination of bacterial burden, lungs were removed at the indicated times and homogenized in PBS using a Dremel tissue homogenizer. Serial dilutions of each organ homogenate were plated on BHI agar and reported as CFU per lung.
Isolation of primary human neutrophils and in vitro assays.
Human blood samples were obtained anonymously from consenting donors through the UNC Center for AIDS Research Virology Core Laboratory by common venipuncture according to IRB protocols 96-0859 and 08-0328. Blood was mixed with equal parts of a 3% dextran in 0.9% saline solution at room temperature for erythrocyte sedimentation. The top serum layer was removed and centrifuged at 250 × g at 4°C for 10 min, and the supernatant was removed. The pellet was resuspended in 0.9% saline and underlaid with Ficoll-Paque Plus (GE Healthcare). The gradient was centrifuged at 400 × g at 26°C for 40 min, and both gradient layers were removed. To remove the remaining erythrocytes, the pellet was resuspended in 0.2% saline for 1 min. An equal volume of 1.6% saline was added, and the suspension was centrifuged at 250 × g at 4°C for 6 min. This erythrocyte wash was repeated, and the final pellet was resuspended in cold PBS. Neutrophils were enumerated using a hemocytometer.
For in vitro assays, 12-well cell culture plates (Costar) were preincubated with fetal bovine serum (FBS) for 1 h at 37°C. FBS was removed, and the wells were washed three times with PBS. Wells were plated with ~106 freshly isolated neutrophils suspended in modified RPMI medium (RPMI 1640 without phenol red and HEPES and with l-glutamine [Gibco] buffered to pH 7.2 with 0.25 mM HEPES), and each well was supplemented with 5% FBS (HyClone), 2.0 mM l-glutamine (CellGro), and 2.5 mM CaCl2. The cells were then inoculated with various strains of Y. pestis or treated with chemical inhibitors for 24 h at 37°C with 5% CO2. The chemical inhibitors BI-D1870 (Cayman Chemical Co.) and Z-YVAD-FKM (BioVision) were used at various concentrations to inhibit the kinase activity of RSK isoforms and inhibit the active site of caspase-1, respectively.
Tissue preparation, laser capture microdissection, and RNA isolation.
All solutions are either RNase free or have been treated with diethylpyrocarbonate (DEPC). Lungs were inflated via tracheal cannulation with RNAlater (Ambion) and incubated for 30 min. Lungs were then fully inflated with 4% fresh paraformaldehyde and incubated for 2 h for fixation and removal from the biosafety level 3 (BSL3) laboratory. Lungs were placed in phosphate buffer (pH 7.4) with 30% sucrose and 20% OCT compound (Tissue-Tek) for 3 h with intermittent inverting. Lungs were removed, covered in OCT for 10 min, frozen on dry ice, and stored at −80°C.
Lungs were sectioned into 20-µm slices using a Leica-1850 cryostat and adhered to 1.0-mm PEN membrane slides (Zeiss). To remove OCT medium from sections, slides were dipped in 100% ethanol and allowed to dry, incubated in DEPC-treated water for 3 to 5 min, and then dipped into increasing concentrations of ethanol (70, 95, and 100%) and allowed to fully dry.
Slides were then loaded onto a Zeiss Palm laser capture microdissection (LCM) system, and a 4× objective was used to observe lung lesions. Opaque adhesive cap Eppendorf tubes were loaded and positioned directly over the slides. The desired sections of each lung lesion were traced, cut by a laser, and laser pulse catapulted into the resin preservative in the top of the adhesive cap tubes. The tubes containing isolated lesion pieces were stored at −80°C prior to RNA isolation. Total RNA was isolated from the LCM-extracted lesion pieces using an RNeasy FFPE (formalin-fixed, paraffin-embedded) kit (Qiagen) as per the manufacturer’s instructions, with the exception of a 3-h incubation at 56°C after the addition of proteinase K.
For bone marrow neutrophil isolation, femurs of C57BL/6J mice were collected and flushed with cold PBS to extract marrow. Marrow cells were disaggregated by repetitive passes through an 18-gauge needle. Bone marrow neutrophils (BM-PMNs) were then isolated using a MACS neutrophil isolation kit (Miltenyi Biotec) per the manufacturer’s instructions. The final volume of BM-PMNs was resuspended in TRIzol reagent (Ambion), and total RNA was isolated per the manufacturer’s instructions.
RNA-seq library preparation and analysis.
Total RNA was sent for library preparation and sequencing at the High-Throughput Sequencing Facility at the University of North Carolina at Chapel Hill. Briefly, a Qubit RNA assay kit for use with a Qubit fluorometer was used to quantify the total RNA concentration of each sample. Both eukaryotic RNA and prokaryotic rRNA were then removed from the samples using an Epidemiology Ribo-Zero Magnetic Gold kit (Epicentre). Complementary DNA was generated using a SMARTer Universal low-input RNA kit (Clontech) followed by RNA-seq library sequencing template preparation using a Clontech low-input library prep kit (Clontech). The library was prepared and run on a MiSeq sequencer (Illumina) to test the quality of the library. After library verification, samples were diluted and bar coded for paired end amplification and clustered (TruSeq SBS kit v2 [200 cycles] and TruSeq PE Cluster kit v2 [Illumina]) using a cBot (Illumina) for running on a HiSeq 2000 system (Illumina). Quality control steps throughout the RNAseq library preparation were performed using a DNA 12-K analysis kit (Experion) or a LabChipGX HT DNA LabChip kit (Caliper). All MiSeq and HiSeq kits used the v2 chemistry.
Purity-filtered reads were aligned to the Mus musculus reference genome (mm9) using MapSplice (91). The alignment profile was summarized by Picard Tools v1.64 (Broad Institute). Aligned reads were sorted and indexed using SAMtools (92), translated to transcriptome coordinates, and filtered for indels, large inserts, and zero mapping quality using UBU v1.0. Transcript abundance estimates for each sample were performed using an expectation-maximization algorithm implemented in RSEM (93) based on the UCSC knownGene definitions. Raw counts were normalized to the upper quartile (94). Log-transformed normalized gene expression estimates were assessed for differential expression using Student’s t test, and these results were filtered to highlight genes achieving P values of <0.05 in both comparisons (BM-PMN and lung lesions). Cluster analysis was performed to assess the patterns of differential expression (hierarchical clustering based on complete linkage and a Pearson correlation) for several defined gene sets testing for differential activity between the conditions. The gene set test performed a Wilcoxon rank-sum test of the log fold change estimates between genes in a set and those in the entire transcriptome where there were greater than three gene reads. Gene set results were visualized using density plots in R (version 3.1.1).
Harvesting and staining of neutrophils for flow cytometry.
Neutrophils were resuspended in wells by repetitive pipetting, collected into Eppendorf tubes, and spun for 2 min at 1,000 × g. Cells were washed in 2% FBS in PBS (flow buffer) and pelleted by spinning 2 min at 1,000 × g. Cells were resuspended in antibody staining solution (flow buffer plus the desired conjugated antibodies at a 1:200 concentration: CD11b-phyoerythrin [clone M1/70.15; Invitrogen] and Ly6G-phycoerythrin-Cy7 [clone 1A8; BD Bioscience]) on ice for 30 min. Cells were then stained for the apoptotic marker phosphatidylserine using annexin V-fluorescein isothiocyanate (FITC) and for cell death using propidium iodide (PI) (annexin V apoptosis detection kit FITC; Affymetrix) as per the manufacturer’s instructions. A Guava easyCyte 5HT flow cytometer (EMD Millipore) was used to detect cell staining in a 96-well plate format. Flow cytometry results were analyzed using InCyte software (EMD Millipore) and analyzed in Microsoft Excel, and graphs were generated using GraphPad Prism.
Western blotting.
Pellets of primary human neutrophils were lysed in equal amounts of loading buffer and heated at 80°C prior to equal volumes of lysate being resolved on 10% SDS-PAGE gels. Proteins were transferred onto nitrocellulose membrane and blocked with 5% nonfat dry milk in 1× Tris-buffered saline (TBS)–0.1% Tween 20. Membranes were incubated with primary antibody overnight at 4°C. The following day, membranes were incubated with secondary antibody conjugated to horseradish peroxidase (HRP) for 1 h at room temperature. Membranes were incubated and developed using Pico West chemiluminescent reagent (Thermo). The primary antibodies used were phospho-S6 (S235/S236) (Cell Signaling) and actin (Santa Cruz), and the secondary antibodies used were HRP-conjugated anti-rabbit (Cell Signaling) and anti-goat (Santa Cruz).
HRP-conjugated anti-rabbit, anti-mouse, anti-rat (Cell Signaling), and anti-goat (Santa Cruz) secondary antibodies were used. Normal rabbit IgG (Santa Cruz; sc-2027) and protein A beads (Santa Cruz; sc-2003) were used for preclearing immunoprecipitation samples.
Histopathology and TUNEL staining.
Groups of three mice were inoculated intranasally as described above, and lungs were inflated with 10% neutral buffered formalin via tracheal cannulation and then removed and incubated in 10% formalin for a minimum of 24 h. Lungs were immersed in 70% EtOH, and embedded in paraffin. Five-micrometer lung sections were adhered to glass slides and stained with hematoxylin and eosin for examination, and a coverslip was added.
A TUNEL Apo-Green detection kit (Biotool) was used to detect damaged DNA indicative of apoptosis. Stained slides were mounted with SlowFade Gold with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen), and coverslips were added prior to imaging with an Olympus BX60 fluorescence microscope and iVision software v4.0.0 (BioVision Technologies). All images were imported into Adobe Photoshop to merge color channels. Input levels were then uniformly adjusted for images from the same staining experiment for publication purposes.
Microarray data accession number.
The raw RNA-seq data are available in the NCBI repository under series record no. GSE70819.
SUPPLEMENTAL MATERIAL
BI-D1870 continues to inhibit RSK activity after 24 h in isolated human neutrophils. Primary human neutrophils were incubated with or without bacterial inoculation and RSK inhibitor (BI-D1870). After 24 h, neutrophils were harvested and lysed for Western blot analysis. Treatment with BI-D1870 caused a decrease in the phosphorylation of S6 in both uninoculated and wild-type Y. pestis-inoculated samples, even after 24 h. Download
Ingenuity Pathway Analysis (IPA) “apoptosis signaling” pathway overlaid with relevant genes from density curve analysis. Genes from the “apoptosis signaling” pathway from IPA were included in the “apoptosis genes” list used in the density curve analysis. The density curve analysis identified the “apoptosis genes” list as being transcriptionally decreased in the center of lesions so individual genes with a P value of <0.5 were overlaid onto the IPA “apoptosis signaling” pathway (green) to visually represent small transcriptional changes throughout the pathway in the center of lesions. Red lines represent direct interactions that would be affected by decreased transcription of identified genes in cells. Orange lines represent secondary effects of the identified genes being downregulated in cells. Download
List of 224 genes differentially regulated in both the infected versus uninfected BM-PMN and lesion periphery versus center comparisons.
Gene composition of the lists used for density curve analysis.
ACKNOWLEDGMENTS
We thank Robert Bagnell at the UNC Microscopy Services Laboratory Core for technical assistance with laser capture microdissection. We thank Nicole Maponga for coordinating human blood draws through the UNC Center for AIDS Research Virology Core Laboratory, as well as Alicia Brandt for coordinating RNA-seq library preparation and analysis at the UNC High-Throughput Sequencing Facility. Additionally we thank Carolyn Suitt and the Center for Gastrointestinal Biology and Disease Histology Core for sectioning and staining of lung sections.
This work was supported by National Institutes of Health grants AI099698 and AI057157 to William E. Goldman. Lung histology sectioning and staining were supported by the Center for Gastrointestinal Biology and Disease Histology Core, supported by National Institutes of Health grant P30 DK 034987. The funding agency had no role in study design, data collection, or interpretation of this work and was not involved in the decision to submit this work for publication.
Footnotes
Citation Stasulli NM, Eichelberger KR, Price PA, Pechous RD, Montgomery SA, Parker JS, Goldman WE. 2015. Spatially distinct neutrophil responses within the inflammatory lesions of pneumonic plague. mBio 6(5):e01530-15. doi:10.1128/mBio.01530-15.
REFERENCES
- 1.Hinnebusch BJ, Perry RD, Schwan TG. 1996. Role of the Yersinia pestis hemin storage (hms) locus in the transmission of plague by fleas. Science 273:367–370. doi: 10.1126/science.273.5273.367. [DOI] [PubMed] [Google Scholar]
- 2.Craig A, Mai J, Cai S, Jeyaseelan S. 2009. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun 77:568–575. doi: 10.1128/IAI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fetherston JD, Kirillina O, Bobrov AG, Paulley JT, Perry RD. 2010. The yersiniabactin transport system is critical for the pathogenesis of bubonic and pneumonic plague. Infect Immun 78:2045–2052. doi: 10.1128/IAI.01236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Segal AW. 2005. How neutrophils kill microbes. Annu Rev Immunol 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quenee LE, Hermanas TM, Ciletti N, Louvel H, Miller NC, Elli D, Blaylock B, Mitchell A, Schroeder J, Krausz T, Kanabrocki J, Schneewind O. 2012. Hereditary hemochromatosis restores the virulence of plague vaccine strains. J Infect Dis 206:1050–1058. doi: 10.1093/infdis/jis433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. 2010. Neutrophil kinetics in health and disease. Trends Immunol 31:318–324. doi: 10.1016/j.it.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh NRP, Johnson A, Peters AM, Babar J, Chilvers ER, Summers C. 2012. Acute lung injury results from failure of neutrophil de-priming: a new hypothesis. Eur J Clin Invest 42:1342–1349. doi: 10.1111/j.1365-2362.2012.02720.x. [DOI] [PubMed] [Google Scholar]
- 8.Wren BW. 2003. The yersiniae—a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol 1:55–64. doi: 10.1038/nrmicro730. [DOI] [PubMed] [Google Scholar]
- 9.Sodeinde OA, Sample AK, Brubaker RR, Goguen JD. 1988. Plasminogen activator/coagulase gene of Yersinia pestis is responsible for degradation of plasmid-encoded outer membrane proteins. Infect Immun 56:2749–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lathem WW, Price PA, Miller VL, Goldman WE. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509–513. doi: 10.1126/science.1137195. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi SD, Voyich JM, DeLeo FR. 2003. Regulation of the neutrophil-mediated inflammatory response to infection. Microbes Infect 5:1337–1344. doi: 10.1016/j.micinf.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 12.Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin Microbiol Rev 10:35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 14.Caulfield AJ, Lathem WW. 2012. Substrates of the plasminogen activator protease of Yersinia pestis, p 253–260. In Advances in experimental medicine and biology. Springer Verlag, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silva MT. 2011. Macrophage phagocytosis of neutrophils at inflammatory/infectious foci: a cooperative mechanism in the control of infection and infectious inflammation. J Leukoc Biol 89:675–683. doi: 10.1189/jlb.0910536. [DOI] [PubMed] [Google Scholar]
- 16.Caulfield A, Walker M, Gielda L, Lathem W. 2014. The Pla protease of Yersinia pestis degrades Fas ligand to manipulate host cell death and inflammation. Cell Host Microbe 15:424–434. doi: 10.1016/j.chom.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Kebir D, Filep JG. 2013. Modulation of neutrophil apoptosis and the resolution of inflammation through β2 integrins. Front Immunol 4:60. doi: 10.3389/fimmu.2013.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo HR, Loison F. 2008. Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol 83:288–295. doi: 10.1002/ajh.21078. [DOI] [PubMed] [Google Scholar]
- 19.Urban CF, Lourido S, Zychlinsky A. 2006. How do microbes evade neutrophil killing? Cell Microbiol 8:1687–1696. doi: 10.1111/j.1462-5822.2006.00792.x. [DOI] [PubMed] [Google Scholar]
- 20.Price PA, Jin J, Goldman WE. 2012. Pulmonary infection by Yersinia pestis rapidly establishes a permissive environment for microbial proliferation. Proc Natl Acad Sci U S A 109:3083–3088. doi: 10.1073/pnas.1112729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balamayooran G, Batra S, Fessler MB, Happel KI, Jeyaseelan S. 2010. Mechanisms of neutrophil accumulation in the lungs against bacteria. Am J Respir Cell Mol Biol 43:5–16. doi: 10.1165/rcmb.2009-0047TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker KA, Miller VL. 2004. Regulation of the Ysa type III secretion system of Yersinia enterocolitica by YsaE/SycB and YsrS/YsrR. J Bacteriol 186:4056–4066. doi: 10.1128/JB.186.13.4056-4066.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Dyke TE, Bartholomew E, Genco RJ, Slots J, Levine MJ. 1982. Inhibition of neutrophil chemotaxis by soluble bacterial products. J Periodontol 53:502–508. doi: 10.1902/jop.1982.53.8.502. [DOI] [PubMed] [Google Scholar]
- 24.Cathelyn JS, Crosby SD, Lathem WW, Goldman WE, Miller VL. 2006. RovA, a global regulator of Yersinia pestis, specifically required for bubonic plague. Proc Natl Acad Sci U S A 103:13514–13519. doi: 10.1073/pnas.0603456103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Haas CJC, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, Poppelier MJ, Van Kessel KP, van Strijp JA. 2004. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med 199:687–695. doi: 10.1084/jem.20031636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bignold LP, Rogers SD, Siaw TM, Bahnisch J. 1991. Inhibition of chemotaxis of neutrophil leukocytes to interleukin-8 by endotoxins of various bacteria. Infect Immun 59:4255–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDonald C, Vacratsis PO, Bliska JB, Dixon JE. 2003. The Yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. J Biol Chem 278:18514–18523. doi: 10.1074/jbc.M301226200. [DOI] [PubMed] [Google Scholar]
- 28.Visser LG, Annema A, van Furth R. 1995. Role of Yops in inhibition of phagocytosis and killing of opsonized Yersinia enterocolitica by human granulocytes. Infect Immun 63:2570–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grosdent N, Maridonneau-Parini I, Sory M-, Cornelis GR. 2002. Role of Yops and adhesins in resistance of Yersinia enterocolitica to phagocytosis. Infect Immun 70:4165–4176. doi: 10.1128/IAI.70.8.4165-4176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spinner JL, Carmody AB, Jarrett CO, Hinnebusch BJ. 2013. Role of Yersinia pestis toxin complex family proteins in resistance to phagocytosis by polymorphonuclear leukocytes. Infect Immun 81:4041–4052. doi: 10.1128/IAI.00648-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spinner JL, Cundiff JA, Kobayashi SD. 2008. Yersinia pestis type III secretion system-dependent inhibition of human polymorphonuclear leukocyte function. Infect Immun 76:3754–3760. doi: 10.1128/IAI.00385-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arnett E, Vadia S, Nackerman CC, Oghumu S, Satoskar AR, McLeish KR, Uriarte SM, Seveau S. 2014. The pore-forming toxin listeriolysin O is degraded by neutrophil metalloproteinase-8 and fails to mediate Listeria monocytogenes intracellular survival in neutrophils. J Immunol 192:234–244. doi: 10.4049/jimmunol.1301302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertram TA, Canning PC, Roth JA. 1986. Preferential inhibition of primary granule release from bovine neutrophils by a Brucella abortus extract. Infect Immun 52:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi K-, Park JT, Dumler JS. 2005. Anaplasma phagocytophilum delay of neutrophil apoptosis through the p38 mitogen-activated protein kinase signal pathway. Infect Immun 73:8209–8218. doi: 10.1128/IAI.73.12.8209-8218.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ge Y, Rikihisa Y. 2006. Anaplasma phagocytophilum delays spontaneous human neutrophil apoptosis by modulation of multiple apoptotic pathways. Cell Microbiol 8:1406–1416. doi: 10.1111/j.1462-5822.2006.00720.x. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz JT, Bandyopadhyay S, Kobayashi SD, McCracken J, Whitney AR, DeLeo FR, Allen LH. 2013. Francisella tularensis alters human neutrophil gene expression: insights into the molecular basis of delayed neutrophil apoptosis. J Innate Immun 5:124–136. doi: 10.1159/000342430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwartz JT, Barker JH, Kaufman J, Fayram DC, McCracken JM, Allen L-H. 2012. Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J Immunol 188:3351–3363. doi: 10.4049/jimmunol.1102863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erttmann SF, Gekara NO, Fällman M. 2014. Bacteria induce prolonged PMN survival via a phosphatidylcholine-specific phospholipase C- and protein kinase C-dependent mechanism. PLoS One 9:0087859. doi: 10.1371/journal.pone.0087859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stenseth NC, Atshabar BB, Begon M, Belmain SR, Bertherat E, Carniel E, Gage KL, Leirs H, Rahalison L. 2008. Plague: past, present, and future. PLoS Med 5:e3. doi: 10.1371/journal.pmed.0050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Koerner JF, Layton M, McDade J, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Schoch-Spana M, Tonat K, Working Group on Civilian Biodefense . 2000. Plague as a biological weapon—medical and public health management. JAMA 283:2281–2290. [DOI] [PubMed] [Google Scholar]
- 41.Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A 102:17786–17791. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pechous RD, Sivaraman V, Price PA, Stasulli NM, Goldman WE. 2013. Early host cell targets of Yersinia pestis during primary pneumonic plague. PLoS Pathog 9:e1003679. doi: 10.1371/journal.ppat.1003679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O. 2005. Plague bacteria target immune cells during infection. Science 309:1739–1741. doi: 10.1126/science.1114580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tirouvanziam R, Gernez Y, Conrad CK, Moss RB, Schrijver I, Dunn CE, Davies ZA, Herzenberg LA, Herzenberg LA. 2008. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc Natl Acad Sci U S A 105:4335–4339. doi: 10.1073/pnas.0712386105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finegold MJ, Petery JJ, Berendt RF, Adams HR. 1968. Studies on pathogenesis of plague—blood coagulation and tissue responses of Macaca mulatta following exposure to aerosols of Pasteurella pestis. Am J Pathol 53:99. [PMC free article] [PubMed] [Google Scholar]
- 46.Du Y, Rosqvist R, Forsberg A. 2002. Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect Immun 70:1453–1460. doi: 10.1128/IAI.70.3.1453-1460.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trosky JE, Liverman ADB, Orth K. 2008. Yersinia outer proteins: Yops. Cell Microbiol 10:557–565. doi: 10.1111/j.1462-5822.2007.01109.x. [DOI] [PubMed] [Google Scholar]
- 48.Burrows TW, Jackson S. 1956. The pigmentation of Pasteurella pestis on a defined medium containing haemin. Br J Exp Pathol 37:570–576. [PMC free article] [PubMed] [Google Scholar]
- 49.Ben-Gurion R, Shafferman A. 1981. Essential virulence determinants of different Yersinia species are carried on a common plasmid. Plasmid 5:183–187. doi: 10.1016/0147-619X(81)90019-6. [DOI] [PubMed] [Google Scholar]
- 50.Spinner JL, Seo KS, O’Loughlin JL, Cundiff JA, Minnich SA, Bohach GA, Kobayashi SD. 2010. Neutrophils are resistant to Yersinia YopJ/P-induced apoptosis and are protected from ROS-mediated cell death by the type III secretion system. PLoS One 5:e9279. doi: 10.1371/journal.pone.0009279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vagima Y, Zauberman A, Levy Y, Gur D, Tidhar A, Aftalion M, Shafferman A, Mamroud E. 2015. Circumventing Y. pestis virulence by early recruitment of neutrophils to the lungs during pneumonic plague. PLoS Pathog 11:e1004893. doi: 10.1371/journal.ppat.1004893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davis K, Mohammadi S, Isberg R. 2015. Community behavior and spatial regulation within a bacterial microcolony in deep tissue sites serves to protect against host attack. Cell Host Microbe 17:21–31. doi: 10.1016/j.chom.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aepfelbacher M, Trasak C, Ruckdeschel K. 2007. Effector functions of pathogenic Yersinia species. Thromb Haemost 98:521–529. doi: 10.1160/TH07-03-0173. [DOI] [PubMed] [Google Scholar]
- 54.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hentschke M, Berneking L, Belmar Campos C, Buck F, Ruckdeschel K, Aepfelbacher M. 2010. Yersinia virulence factor YopM induces sustained RSK activation by interfering with dephosphorylation. PLoS One 5:13165. doi: 10.1371/journal.pone.0013165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.LaRock C, Cookson B. 2012. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe 12:799–805. doi: 10.1016/j.chom.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chung LK, Philip NH, Schmidt VA, Koller A, Strowig T, Flavell RA, Brodsky IE, Bliska JB. 2014. IQGAP1 is important for activation of caspase-1 in macrophages and is targeted by Yersinia pestis type III effector YopM. mBio 5:e01402-14. doi: 10.1128/mBio.01402-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sapkota G, Cummings L, Newell F, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, Cohen P, Alessi D. 2007. Bi-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J 401:29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. 2007. RAS/ERK signaling promotes site-specific ribosomal protein s6 phosphorylation via RSK and stimulates Cap-dependent translation. J Biol Chem 282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laws TR, Davey MS, Titball RW, Lukaszewski R. 2010. Neutrophils are important in early control of lung infection by Yersinia pestis. Microbes Infect 12:331–335. doi: 10.1016/j.micinf.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 61.Shannon JG, Hasenkrug AM, Dorward DW, Nair V, Carmody AB, Hinnebusch BJ. 2013. Yersinia pestis subverts the dermal neutrophil response in a mouse model of bubonic plague. mBio 4:e00170-13. doi: 10.1128/mBio.00170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lukaszewski RA, Kenny DJ, Taylor R, Rees DGC, Hartley MG, Oyston PCF. 2005. Pathogenesis of Yersinia pestis infection in BALB/c mice: effects on host macrophages and neutrophils. Infect Immun 73:7142–7150. doi: 10.1128/IAI.73.11.7142-7150.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA, Liotta LA. 1996. Laser capture microdissection. Science 274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 64.Suarez-Quian CA, Goldstein SR, Pohida T, Smith PD, Peterson JI, Wellner E, Ghany M, Bonner RF. 1999. Laser capture microdissection of single cells from complex tissues. Biotechniques 26:328–335. [DOI] [PubMed] [Google Scholar]
- 65.Klink VP, Alkharouf N, MacDonald M, Matthews B. 2005. Laser capture microdissection (LCM) and expression analyses of Glycine max (soybean) syncytium containing root regions formed by the plant pathogen Heterodera glycines (soybean cyst nematode). Plant Mol Biol 59:965–979. doi: 10.1007/s11103-005-2416-7. [DOI] [PubMed] [Google Scholar]
- 66.Klink VP, Overall CC, Alkharouf NW, MacDonald MH, Matthews BF. 2007. Laser capture microdissection (LCM) and comparative microarray expression analysis of syncytial cells isolated from incompatible and compatible soybean (Glycine max) roots infected by the soybean cyst nematode (Heterodera glycines). Planta 226:1389–1409. doi: 10.1007/s00425-007-0578-z. [DOI] [PubMed] [Google Scholar]
- 67.Klink VP, Matthews BF. 2008. The use of laser capture microdissection to study the infection of Glycine max (soybean) by Heterodera glycines (soybean cyst nematode). Plant Signal Behav 3:105–107. doi: 10.4161/psb.3.2.4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balestrini R, Gómez-Ariza J, Lanfranco L, Bonfante P. 2007. Laser microdissection reveals that transcripts for five plant and one fungal phosphate transporter genes are contemporaneously present in arbusculated cells. Mol Plant Microbe Interact 20:1055–1062. doi: 10.1094/MPMI-20-9-1055. [DOI] [PubMed] [Google Scholar]
- 69.Berruti A, Borriello R, Lumini E, Scariot V, Bianciotto V, Balestrini R. 2013. Application of laser microdissection to identify the mycorrhizal fungi that establish arbuscules inside root cells. Front Plant Sci 4:135. doi: 10.3389/fpls.2013.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schulte LN, Eulalio A, Mollenkopf H, Reinhardt R, Vogel J. 2011. Analysis of the host microRNA response to Salmonella uncovers the control of major cytokines by the let-7 family. EMBO J 30:1977–1989. doi: 10.1038/emboj.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anderson DM, Ciletti NA, Lee-Lewis H, Elli D, Segal J, DeBord KL, Overheim KA, Tretiakova M, Brubaker RR, Schneewind O. 2009. Pneumonic plague pathogenesis and immunity in Brown Norway rats. Am J Pathol 174:910–921. doi: 10.2353/ajpath.2009.071168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fellows P, Lin W, Detrisac C, Hu S-, Rajendran N, Gingras B, Holland L, Price J, Bolanowski M, House RV. 2012. Establishment of a Swiss Webster mouse model of pneumonic plague to meet essential data elements under the animal rule. Clin Vaccine Immunol 19:468–476. doi: 10.1128/CVI.05591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van Andel R, Sherwood R, Gennings C, Lyons CR, Hutt J, Gigliotti A, Barr E. 2008. Clinical and pathologic features of cynomolgus macaques (Macaca fascicularis) infected with aerosolized Yersinia pestis. Comp Med 58:68–75. [PMC free article] [PubMed] [Google Scholar]
- 74.Agar SL, Sha J, Foltz SM, Erova TE, Walberg KG, Baze WB, Suarez G, Peterson JW, Chopra AK. 2009. Characterization of the rat pneumonic plague model: infection kinetics following aerosolization of Yersinia pestis CO92. Microbes Infect 11:205–214. doi: 10.1016/j.micinf.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 75.Koster F, Perlin DS, Park S, Brasel T, Gigliotti A, Barr E, Myers L, Layton RC, Sherwood R, Lyons CR. 2010. Milestones in progression of primary pneumonic plague in cynomolgus macaques. Infect Immun 78:2946–2955. doi: 10.1128/IAI.01296-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Layton RC, Brasel T, Gigliotti A, Barr E, Storch S, Myers L, Hobbs C, Koster F. 2011. Primary pneumonic plague in the African Green monkey as a model for treatment efficacy evaluation. J Med Primatol 40:6–17. doi: 10.1111/j.1600-0684.2010.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Adamovicz JJ, Worsham PL. 2006. Biodefense research methodology and animal models, 2nd ed, p 107–135. Taylor & Francis, Abingdon, Oxford, United Kingdom. [Google Scholar]
- 78.Vagima Y, Levy Y, Gur D, Tidhar A, Aftalion M, Abramovich H, Zahavy E, Zauberman A, Flashner Y, Shafferman A, Mamroud E. 2012. Early sensing of Yersinia pestis airway infection by bone marrow cells. Front Cell Infect Microbiol 2:143. doi: 10.3389/fcimb.2012.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McCracken JM, Allen L-AH. 2014. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death 7:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Viboud GI, Bliska JB. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Microbiology 59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 81.Leung KY, Reisner BS, Straley SC. 1990. YopM inhibits platelet aggregation and is necessary for virulence of Yersinia pestis in mice. Infect Immun 58:3262–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heusipp G, Spekker K, Brast S, Fälker S, Schmidt MA. 2006. YopM of Yersinia enterocolitica specifically interacts with alpha1-antitrypsin without affecting the anti-protease activity. Microbiology 152:1327–1335. doi: 10.1099/mic.0.28697-0. [DOI] [PubMed] [Google Scholar]
- 83.McCoy MW, Marre ML, Lesser CF, Mecsas J. 2010. The C-terminal tail of Yersinia pseudotuberculosis YopM is critical for interacting with RSK1 and for virulence. Infect Immun 78:2584–2598. doi: 10.1128/IAI.00141-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McPhee JB, Mena P, Bliska JB. 2010. Delineation of regions of the Yersinia YopM protein required for interaction with the RSK1 and PRK2 host kinases and their requirement for interleukin-10 production and virulence. Infect Immun 78:3529–3539. doi: 10.1128/IAI.00269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McPhee JB, Mena P, Zhang Y, Bliska JB. 2012. Interleukin-10 induction is an important virulence function of the Yersinia pseudotuberculosis type III effector YopM. Infect Immun 80:2519–2527. doi: 10.1128/IAI.06364-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kerschen EJ, Cohen DA, Kaplan AM, Straley SC. 2004. The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect Immun 72:4589–4602. doi: 10.1128/IAI.72.8.4589-4602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ye Z, Kerschen EJ, Cohen DA, Kaplan AM, van Rooijen N, Straley SC. 2009. Gr1+ cells control growth of YopM-negative Yersinia pestis during systemic plague. Infect Immun 77:3791–3806. doi: 10.1128/IAI.00284-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ye Z, Uittenbogaard AM, Cohen DA, Kaplan AM, Ambati J, Straley SC. 2011. Distinct CCR2(+) Gr1(+) cells control growth of the Yersinia pestis delta yopM mutant in liver and spleen during systemic plague. Infect Immun 79:674–687. doi: 10.1128/IAI.00808-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ye Z, Gorman AA, Uittenbogaard AM, Myers-Morales T, Kaplan AM, Cohen DA, Straley SC. 2014. Caspase-3 mediates the pathogenic effect of Yersinia pestis YopM in liver of C57BL/6 mice and contributes to YopM’s function in spleen. PLoS One 9:110956. doi: 10.1371/journal.pone.0110956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury Chevance A, Zhang K, Conly J, Kubes P. 2012. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18:1386–1393. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, MacLeod JN, Chiang DY, Prins JF, Liu J. 2010. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bullard JH, Purdom E, Hansen KD, Dudoit S. 2010. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
BI-D1870 continues to inhibit RSK activity after 24 h in isolated human neutrophils. Primary human neutrophils were incubated with or without bacterial inoculation and RSK inhibitor (BI-D1870). After 24 h, neutrophils were harvested and lysed for Western blot analysis. Treatment with BI-D1870 caused a decrease in the phosphorylation of S6 in both uninoculated and wild-type Y. pestis-inoculated samples, even after 24 h. Download
Ingenuity Pathway Analysis (IPA) “apoptosis signaling” pathway overlaid with relevant genes from density curve analysis. Genes from the “apoptosis signaling” pathway from IPA were included in the “apoptosis genes” list used in the density curve analysis. The density curve analysis identified the “apoptosis genes” list as being transcriptionally decreased in the center of lesions so individual genes with a P value of <0.5 were overlaid onto the IPA “apoptosis signaling” pathway (green) to visually represent small transcriptional changes throughout the pathway in the center of lesions. Red lines represent direct interactions that would be affected by decreased transcription of identified genes in cells. Orange lines represent secondary effects of the identified genes being downregulated in cells. Download
List of 224 genes differentially regulated in both the infected versus uninfected BM-PMN and lesion periphery versus center comparisons.
Gene composition of the lists used for density curve analysis.