

ABSTRACT
Vibrio cholerae is an aquatic organism and facultative human pathogen that colonizes the small intestine. In the small intestine, V. cholerae is exposed to a variety of antimicrobial compounds, including bile. V. cholerae resistance to bile is multifactorial and includes alterations in the membrane permeability barrier that are mediated by ToxR, a membrane-associated transcription factor. ToxR has also been shown to be required for activation of the LysR family transcription factor leuO in response to cyclic dipeptides. LeuO has been implicated in the regulation of multiple V. cholerae phenotypes, including biofilm production and virulence. In this study, we investigated the effects of bile on leuO expression. We show that leuO transcription increased in response to bile and bile salts but not in response to other detergents. The bile-dependent increase in leuO expression was dependent on ToxR, which was found to bind directly to the leuO promoter. The periplasmic domain of ToxR was required for basal leuO expression and for the bile-dependent induction of both leuO and ompU transcription. V. cholerae mutants that did not express leuO exhibited increased bile susceptibility, suggesting that LeuO contributes to bile resistance. Our collective results demonstrate that ToxR activates leuO expression in response to bile and that LeuO is a component of the ToxR-dependent responses that contribute to bile resistance.
IMPORTANCE The success of Vibrio cholerae as a human pathogen is dependent upon its ability to rapidly adapt to changes in its growth environment. Growth in the human gastrointestinal tract requires the expression of genes that provide resistance to host antimicrobial compounds, including bile. In this work, we show for the first time that the LysR family regulator LeuO mediates responses in V. cholerae that contribute to bile resistance.
INTRODUCTION
Vibrio cholerae is a Gram-negative bacterial pathogen and the causal agent of the severe diarrheal disease cholera. V. cholerae exists naturally in aquatic reservoirs and is capable of colonizing the human small intestine. The transition of V. cholerae from the aquatic ecosystem to growth in the human gastrointestinal tract is mediated by transcriptional responses that are required for colonization and disease development. Many of the genes that contribute to intestinal colonization are under the control of the membrane-associated regulatory protein ToxR, which functions as one of the primary regulators in the ToxR regulon (reviewed in reference 1). The ToxR regulon is divided into two branches, a ToxT-dependent branch, which controls the expression of virulence factors, and a ToxT-independent branch, which reciprocally regulates the production of the outer membrane porins OmpU and OmpT. The ToxT-dependent branch of the ToxR regulon is a hierarchical regulatory cascade that regulates the expression of genes encoding the production of cholera toxin (CT) and the toxin-coregulated pilus (TCP) in response to environmental cues in the host.
ToxR is a membrane-associated regulatory protein that belongs to the winged-helix family of transcriptional regulators (2, 3). ToxR is a one-component signal-transducing protein that is composed of a periplasmic signaling domain and a cytoplasmic DNA binding domain that are linked by a single transmembrane-spanning domain (4). toxR is borne on an operon along with toxS, which is located downstream of toxR. ToxS is an inner membrane protein, which is thought to interact with ToxR to facilitate its transcriptional activity (5, 6). The ToxR periplasmic signaling domain is thought to sense and transduce environmental stimuli to affect the activity of the cytoplasmic DNA binding domain at its target genes. ToxR has been shown to respond to a variety of environmental stimuli, including acidity, nutrient availability, salinity, small molecules, and bile (4, 7–9).
It has been shown that ToxR plays an essential role in modulating adaptive responses that contribute to bile resistance. Bile is produced by the liver and secreted at high concentrations into the small intestine to aid in the digestion of lipids. Bile is composed primarily of bile salts but also contains significant amounts of phospholipids, cholesterol, protein, and bilirubin. While bile is important in digestion, bile also provides a barrier against intestinal colonization by restricting bacterial growth in the small intestine, presumably through its detergent-like effects on bacterial cell membranes (10). As such, enteric pathogens have evolved methods to overcome this barrier. This includes the modulation of outer membrane porin proteins to decrease the rate of diffusion of toxic molecules across the outer membrane and the expression of active efflux systems that remove bile salts from within the cell envelope (11–15). In V. cholerae, resistance to the antimicrobial effects of bile is due to the combined actions of multiple factors, including active efflux- and ToxR-regulated genes. This is evident from the observation that toxR mutant strains exhibit greatly increased susceptibility to bile and bile salts (16).
The elevated susceptibility of toxR mutant strains to bile salts has been linked to the expression of the ompU and ompT porins (16). OmpU and OmpT are general diffusion porins located in the outer membrane. Porins are responsible for allowing the diffusion of nutrients, metabolites, and signaling molecules into and out of the cell (17). OmpU and OmpT have been found to be regulated in response to environmental stimuli, including bile, osmolarity, organic acids, cyclic dipeptides, and amino acids (4, 7–9). The genes encoding OmpU and OmpT are reciprocally regulated by ToxR, which binds to conserved direct repeat elements that are located in the ompU and ompT promoters (18, 19). ToxR positively regulates ompU and negatively regulates ompT. OmpU is preferentially produced during growth in rich medium or in minimal medium containing certain amino acids, bile, or mucin (20). In nutrient-poor environments or in a toxR mutant strain, the porin profile is reversed, and OmpT becomes predominant, while OmpU is no longer produced. Consistently with the activation of ompU expression by bile salts, the production of OmpU is associated with bile salt resistance, while the production of OmpT is associated with bile salt susceptibility (21). This phenotype is presumably related to the fact that OmpU, in contrast to OmpT, is an anion-selective porin, which restricts the passage of negatively charged compounds (21, 22).
Previous studies in our laboratory suggested that in V. cholerae, ToxR activated leuO expression in response to cyclo(Phe-Pro) (cFP) (23). Increased leuO transcription was linked to downregulation of the ToxR regulon and attenuated CT and TCP production. These results suggested that leuO functioned downstream of ToxR to modulate gene expression in response to environmental cues. LeuO is a LysR family transcription factor that was first identified as a regulator of leucine biosynthetic genes in Salmonella enterica serovar Typhimurium (24). Subsequent studies have shown LeuO to be a global regulator of diverse and unrelated phenotypes in the Enterobacteriaceae. For example, in S. enterica, LeuO has been shown to regulate outer membrane proteins, virulence genes, transport genes, biofilm production, and quorum sensing (25). Likewise, LeuO has been shown to be involved in the regulation of genes involved in carbohydrate utilization, phage resistance, acid shock, temperature adaptation, and biofilm production in Escherichia coli (26). LeuO has also been associated with virulence gene regulation in Yersinia enterocolitica (27). The function of LeuO as a global regulator appears to be conserved in the Vibrionaceae, in which LeuO has been shown to contribute to biofilm production, cell wall degradation, and virulence gene regulation (23, 28–30).
Since bile acids are an important environmental cue during V. cholerae pathogenesis, and since ToxR regulates the expression of many of its target genes in response to bile salts, we tested the hypothesis that leuO expression was also modulated in response to bile via ToxR. The results of our experiments showed that leuO expression was activated upon exposure to bile salts by a mechanism that was dependent on ToxR. Multiple approaches were used to show that ToxR acted directly at the leuO promoter and that the ToxR periplasmic domain was required for basal leuO expression and the bile-dependent induction of leuO and ompU expression. Mutants that failed to express leuO exhibited reduced survival upon exposure to lethal concentrations of bile, indicating that LeuO contributed to bile resistance. Collectively, our results indicated that ToxR activated leuO and ompU expression in response to bile salts by a mechanism that was dependent on the ToxR periplasmic signaling domain and that LeuO contributed to the ToxR-mediated bile resistance.
MATERIALS AND METHODS
Strains, media, and chemicals.
The bacterial strains used in this study are listed in Table 1. E. coli strain EC100λpir was used for cloning and strain SM10λpir for plasmid mobilization. The V. cholerae strains used in this study were derivatives of O1 El Tor strain N16961 (31). V. cholerae strain N16961 ΔlacZ Smr (Smr, streptomycin resistant) was used as the wild-type (WT) control for all experiments. Bacterial strains were grown at 37°C in lysogeny broth (LB) or on LB agar. Modified T medium was prepared as previously described (20). Bacterial stocks were maintained in LB containing 25% glycerol at −80°C. Antibiotics were added to the growth medium at 100 μg/ml for carbenicillin (Cb) and streptomycin (Sm) and 25 μg/ml for chloramphenicol (Cm), as required. Arabinose was added to the growth medium to induce expression from the arabinose-regulated promoter in pBAD18 and pBAD33. Stock solutions of the detergents and bile (Oxgall; Difco) were made in water and filter sterilized before use.
TABLE 1.
Strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Relevant characteristics or sequence (5′ to 3′)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| EC100λpir | supE44 ΔlacU169 (ϕ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 (λpirR6K) | Epicentre |
| SM10λpir | thi-1 thr leu tonA lacY supE recA::RP4-2-Tc::mu Kmr (λpirR6K) | Lab collection |
| V. cholerae | ||
| JB58 | O1 El Tor strain N16961 ΔlacZ Smr | Lab collection |
| XBV222 | JB58 ΔleuO | 23 |
| DT733 | JB58 ΔtoxRS | 23 |
| SS4 | JB58 toxRΔppd | This study |
| Plasmids | ||
| pCM10 | luxCDABE reporter plasmid, Kmr | 51 |
| pJB906 | pCM10 containing leuO promoter | 23 |
| pTL61T | lacZ transcriptional reporter plasmid, Cbr | 52 |
| pXB266 | pTL61T containing leuO promoter region with two ToxR binding sites | |
| pVA258 | pTL61T containing leuO promoter region with one ToxR binding site | This study |
| pVA261 | pTL61T containing leuO promoter region with no ToxR binding sites | This study |
| pXB233 | pTL61T containing vexRAB promoter | 53 |
| pXB228 | pTL61T containing vexEF promoter | 54 |
| pXB229 | pTL61T containing vexGH promoter | 54 |
| pXB230 | pTL61T containing vexIJK promoter | 54 |
| pXB231 | pTL61T containing vexCD promoter | 54 |
| pXB232 | pTL61T containing vexLM promoter | 54 |
| pBAD18 | Arabinose-regulated expression plasmid, Cbr | 55 |
| pBAD33 | Arabinose-regulated expression plasmid, Cmr | 55 |
| pXB298 | pBAD18Km expressing leuO | 23 |
| pVA94 | pBAD18 expressing leuO | This study |
| pXB289 | pBAD18 expressing toxRS | 23 |
| pXB286 | pBAD18 expressing toxRΔppdS | 23 |
| pXB302 | pBAD33 expressing toxRS | This study |
| pDT1391 | pBAD33 expressing toxRΔppdS | This study |
| pWM91 | Suicide plasmid vector used for allelic exchange | 41 |
| pWM91::ΔtoxRppd | pWM91 containing a fragment of toxR harboring a deletion of periplasmic domain | This study |
| Oligonucleotides | ||
| PleuO1-F | CGCCCGGGAAATGCATTTTTATAGATTTTT | |
| PleuO2-F | CGCCCGGGAATCGTATTGATTATTAAGGCT | |
| PleuO-R | GGGGATCCGCGTCTTTTTTATCTAACATTTGCATGCCT | |
| toxRΔppd-F1 | GGGAGCTCGGTCCTCAAAAGAGATAT | |
| toxRΔppd-F2 | CTGCTCACTAACTAGGATCTTGCTAT | |
| toxRΔppd-R1 | AACCCGGGCATGCCGCTCAGTCAGG | |
| toxRΔppd-R2 | AGCAAGATCCTAGTTAGTGAGCAGTA | |
| 5′BIO | GCGGGAGTCGGCAGCG | |
| leuO-F-EMSA | GCGGGAGTCGGCAGCGGTTAAAACATTTTTGACGTGAATATTAGTG | |
| leuO-R-EMSA | GCGGGAGTCGGCAGCGCGTCACTAGCGATAAATATGCATAAATC | |
| ompU-F-EMSA | GCGGGAGTCGGCAGCGCAATTAGATTGCGTGCATTT | |
| ompU-R-EMSA | GCGGGAGTCGGCAGCGTTTTTTTACTCCCAAAGTTC | |
| vexR-F-EMSA | GCGGGAGTCGGCAGCGTGCAAAACAGGGGGTATTAG | |
| vexR-R-EMSA | GCGGGAGTCGGCAGCGGCCGTACACTATTTCAGACA | |
| leuO-qPCR-F | GACCACTTCGCCACAAATCACCA | |
| leuO-qPCR-R | CGTTGGATGGCGGAAAATGCG | |
| ompU-qPCR-F | ACACCGTATAGGCTGTCATTG | |
| ompU-qPCR-R | GTGCTGAAGCTCGCCTATCTC | |
| gyrA-qPCR-F | CAATGCCGGTACACTGGTACG | |
| gyrA-qPCR-R | AAGTACGGATCAGGGTCACG |
Kmr, kanamycin resistant; Cbr, carbenicillin resistant; Cmr, chloramphenicol resistant.
Plasmid and mutant construction.
The plasmids and oligonucleotides used in this study are listed in Table 1. Genomic DNA from N16961 ΔlacZ Smr was used as a PCR template for cloning. Plasmid reporters containing derivatives of the leuO promoter lacking one or both ToxR binding sites were constructed as follows. pVA258 (PleuO lacking the distal ToxR binding site) was generated by PCR using the PleuO1-F and PleuO-R oligonucleotide primers. The resulting amplicon was digested with BamHI and XmaI restriction endonucleases and ligated into similarly digested pTL61T. pVA261 (PleuO lacking both ToxR binding sites) was generated by PCR using the PleuO2-F and PleuO-R primers. The resulting PCR amplicon was digested with BamHI and XmaI restriction endonucleases and ligated into similarly digested pTL61T. The leuO expression vector pVA94 (pBAD18::leuO) was constructed by moving leuO from pXB298 as a NheI and XbaI restriction fragment into the same sites in pBAD18. pXB302 (pBAD33::toxRS) was made by moving the toxRS genes from pXB289 as a SacI and SphI restriction fragment into the same sites in pBAD33. pDT1391 (pBAD33::toxRΔppdS) (ppd, periplasmic domain) was made by moving the toxRΔppdS genes from pXB286 as a SacI and SmaI restriction fragment into the same sites in pBAD33. pWM91::ΔtoxRppd, which contains a 94-amino-acid C-terminal deletion of the ToxR periplasmic domain, was made by crossover PCR, as previously described (32, 33). Briefly, primer pairs toxRΔppd-F1/toxRΔppd-R2 and toxRΔppd-F2/toxRΔppd-R1 were used in separate PCRs with N16961 genomic DNA. The resulting ∼1-kb amplicons were collected and used as the template for second-round PCR amplification with the flanking toxRΔppd-F1/toxRΔppd-R1 PCR primers. The resulting ∼2-kb amplicon was then digested with SacI and SmaI restriction endonucleases before being ligated into similarly digested pWM91.
Deletion of the ToxR periplasmic domain in V. cholerae strain SS4 was accomplished as follows. pWM91::ΔtoxRppd was conjugated into V. cholerae strain JB58, and plasmid cointegrants were selected for Sm and Cb resistance. Sm- and Cb-resistant cointegrants were then plated onto LB agar plates containing 5% sucrose and no NaCl. Sucrose-resistant and Cb-sensitive colonies were then screened by PCR using the toxRΔppd-F1/toxRΔppd-R1 primers to confirm the deletion of the ToxR periplasmic domain. Verification of toxRΔppd in SS4 was accomplished by DNA sequencing of the toxR locus.
Reporter assays.
β-Galactosidase assays were performed as follows. V. cholerae strains carrying the indicated leuO-lacZ reporters were cultured overnight in LB at 37°C with shaking. The cultures were then diluted 1:100 into fresh LB and incubated at 37°C with shaking. Culture aliquots were collected in triplicate at mid-exponential phase (optical density at 600 nm [OD600], ∼0.5) to quantify β-galactosidase activity, as previously described (34). The E. coli two-plasmid reporter experiments were performed as follows. E. coli bearing an expression plasmid (pBAD33, pXB302, or pDT1391) and a lacZ reporter plasmid (pXB266, pVA258, or pVA261) was cultured overnight in LB with shaking at 37°C. The overnight cultures were then diluted 1:100 into fresh LB with or without 0.08% arabinose, and the cultures were incubated at 37°C with shaking. Culture aliquots were collected in triplicate at mid-exponential phase (OD600, ∼0.5) to quantify β-galactosidase activity, as previously described (34). All of the reporter experiments were performed independently at least three times. Expression from the lacZ reporter was calculated and is given in Miller units (MU).
The bioluminescence assays were performed as follows. V. cholerae strain JB58 containing pJB906 (leuO-lux) was cultured overnight in LB at 37°C with shaking. The overnight cultures were then diluted 1:100 into fresh LB and incubated at 37°C with shaking for 2 h. Aliquots (100 μl) of the culture were then diluted into 100 μl of LB plus the indicated substrates (i.e., dimethyl sulfoxide [DMSO], deoxycholate [DOC], or cFP) and distributed into triplicate wells of a white 96-well microtiter plate with a clear bottom (Corning). In these experiments, DOC was used at 0.0125%, cFP at 1 mM, and DMSO at 0.1%. The plates were then incubated at 37°C, and luminescence and the OD600 were measured at the time points indicated in Fig. 1 using a BioTek Synergy HT plate reader. The relative light units (RLU) for each sample were calculated by dividing the luminescence by the OD600. The reported results are the averages and standard deviations (SD) of the results from three independent experiments.
FIG 1.
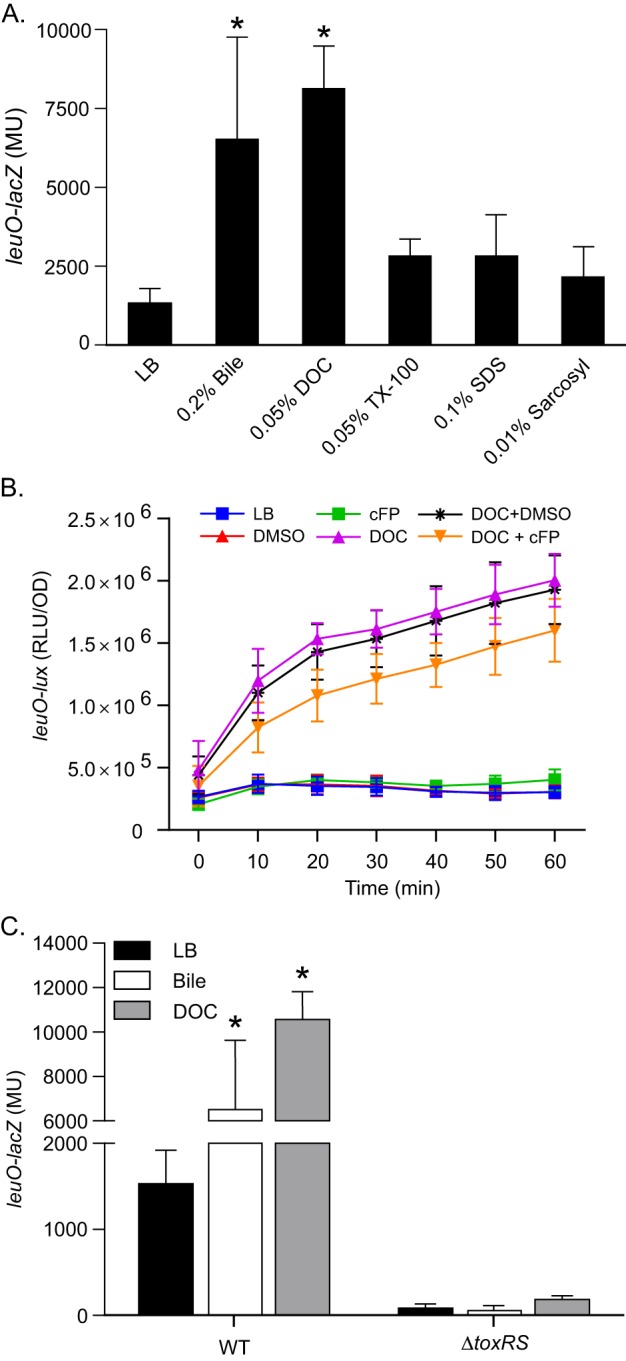
Induction of leuO transcription by bile and bile salts requires ToxR. (A) WT strain JB58 carrying the leuO-lacZ reporter plasmid pXB266 was grown in LB or LB supplemented with bile or the indicated detergents. Culture aliquots were collected at mid-logarithmic phase and assayed for leuO-lacZ expression by the β-galactosidase assay. (B) Expression of leuO-lux in the WT strain JB58::pJB906 (leuO-lux) during growth in LB containing the indicated compounds. DOC was used at 0.0125%, cFP at 1 mM, and DMSO at 0.1%. (C) WT strain JB58 and ΔtoxRS strain DT733 carrying pXB266 (leuO-lacZ) were grown in LB or LB plus 0.2% bile or 0.1% deoxycholate. Culture aliquots were collected at mid-logarithmic phase and assayed for leuO-lacZ expression by the β-galactosidase assay. The presented data are the means ± SD of the results from three independent experiments. Statistical significance in panel A was determined using one-way analysis of variance (ANOVA) with Dunnett's posttest, comparing each mean with that for the LB control; *, P < 0.01. Statistical significance in panel B was determined using two-way ANOVA with Tukey's posttest; *, P < 0.0001. DOC, deoxycholate; TX-100, Triton X-100; SDS, sodium dodecyl sulfate; MU, Miller units; DMSO, dimethyl sulfoxide; RLU, relative light units.
Bile killing assay.
Overnight cultures of the indicated strains were diluted 1:100 in LB with or without 0.1% arabinose (to induce expression from pBAD18) and incubated at 37°C with shaking for 4 h. Culture aliquots were then collected and washed once with phosphate-buffered saline (PBS) before being diluted in PBS to an OD600 of 0.5. Serial dilutions of each strain were then plated onto LB agar plates to determine the cell titer at time zero (CFUinput). Aliquots containing ∼106 CFU of each strain were then added to PBS containing 10% bile (toxR mutant strains) or 20% bile (toxR-positive strains); the bile concentrations were reduced for the toxR mutant strains due to their increased bile sensitivity. The cultures were then incubated statically at room temperature for 60 min, at which point the aliquots were collected, washed in PBS, and plated onto LB agar to quantify the viable cells (CFUoutput). The recovery ratio of each strain was then calculated as CFUoutput/CFUinput. The fold change in recovery was then determined by dividing the recovery ratio for the mutant strains by the recovery ratio for the WT. The fold change in recovery for the leuO-overexpressing strain was determined by dividing the recovery ratio for the ΔtoxRS::pBAD18-leuO mutant by the recovery ratio for the ΔtoxRS::pBAD18 mutant. The presented data are the means and standard deviations (SD) of the results from three independent experiments. Statistical significance was determined using Dunnett's multiple-comparison test relative to a recovery ratio of 1.0.
RNA extraction and quantitative real-time PCR.
RNA was extracted from V. cholerae cultures grown in modified T medium at 37°C with shaking to an OD600 of ∼0.3, at which point a mixture of the amino acids asparagine, arginine, glutamic acid, and serine (NRES) (50 mM) or bile (0.2%) was added to the cultures. The cultures were then incubated for an additional 15 min at 37°C with shaking before RNA was extracted using TRIzol, according to the manufacturer's instructions (Ambion). The resulting RNA was quantified using a NanoDrop spectrophotometer (Thermo) before being used to make cDNA with the Maxima first-strand cDNA synthesis kit (Life Technologies), according to the manufacturer's instructions. The resulting cDNA was then used with gene-specific primers (Table 1) and the SYBR green PCR master mix (Thermo) to quantify gene expression using a StepOnePlus real-time PCR system (Applied Biosystems). The relative gene expression levels were then calculated using the 2(−ΔΔCT) method (where CT is the threshold cycle) (35) using the A subunit of DNA gyrase (gyrA) as an internal control. The presented results are the means ± standard deviations of the results from three independent biological replicates, with each biological replicate being generated from at least two technical replicates.
Membrane isolation.
V. cholerae strain DT733 (ΔtoxRS) containing pXB289 (pBAD18::toxRS), pXB286 (pBAD18::toxRΔppdS), or pBAD18 was cultured in LB with shaking at 37°C to an OD600 of ∼1.0, at which point expression from the arabinose promoter was induced by the addition of arabinose to 0.2%. The cultures were then incubated with shaking at 37°C for an additional hour, and then the cells were harvested by centrifugation. The cell pellet was resuspended in 10 mM Tris-HCl (pH 8.0) in 0.75 M sucrose. Spheroplasts were then induced by the addition of 150 μg/ml lysozyme, followed by the addition of 2 volumes of buffer A (10 mM Tris-HCl [pH 8.0], 10 mM EDTA) (36, 37). The cells were then lysed by passage through a model M-11P Microfluidizer (Microfluidics). Particulate matter was removed from the cell lysate by centrifugation at 8,000 × g and 4°C for 10 min. The membrane-containing supernatant was then subjected to centrifugation in an SW-28 rotor (Beckman Coulter) at 24,000 rpm and 4°C for 2 h to pellet the membrane fraction. The membrane pellet was then suspended in 20% sucrose in buffer B (10 mM Tris-HCl [pH 8.0], 1 mM EDTA) and applied to a two-step 60% and 70% sucrose gradient. The sucrose gradients were then subjected to centrifugation in an SW-28 rotor at 23,000 rpm and 4°C for 18 h. The inner and outer membrane fractions were then decanted from the sucrose gradients and diluted with 2 volumes of cold buffer B before being centrifuged in a Ti55 rotor (Beckman Coulter) at 47,000 rpm and 4°C for 1 h. The resulting inner membrane pellets were resuspended in cold buffer B and frozen at −20°C until used. Protein concentrations were determined using the Pierce Coomassie Plus protein assay, according to the manufacturer's directions.
EMSA.
DNA probes for gel shift assays were generated by PCR using the primers listed in Table 1. The PCR primers for the electrophoretic mobility shift assays (EMSAs) were engineered to include a 5′ tail (5′-GCGGGAGTCGGCAGCG-3′), which facilitated biotinylation of the probes by a second PCR using the 5′BIO PCR primer, which hybridized to the 5′ tail. The 5′BIO PCR primer was purchased from the manufacturer (IDT) with a 5′ biotin label. The biotinylated probes were gel purified and quantified using a NanoDrop 2000 spectrophotometer (Thermo Scientific) before being used in the EMSAs. The DNA binding reactions were performed in a final volume of 10 μl of binding buffer (10 mM Tris-HCl [pH 7.4], 5 mM NaCl, 50 mM KCl, 1 mM EDTA, 50 μg/ml bovine serum albumin [BSA], 1.5 nM biotinylated probe, and 10 μg/ml sheared salmon sperm DNA) containing the indicated amounts of the V. cholerae inner membranes. The binding reaction mixtures were incubated at 30°C for 30 min before being subjected to electrophoresis at 150 V for 1 h on a nondenaturing 5% polyacrylamide Tris buffer-EDTA (TBE) gel that had been prerun with 5% thioglycolic acid for 1 h at 150 V in 1× TBE buffer. The resolved gels were electroblotted to positively charged nylon membranes in 0.5× TBE buffer at 380 mA for 1 h, before the nylon membrane was UV cross-linked at 120,000 μJ using a Stratalinker 1800 cross-linker (Stratagene). The biotinylated probes were then detected using the Pierce chemiluminescent nucleic acid detection module (Thermo Scientific) and visualized using a FluorChem E imaging system (ProteinSimple).
RESULTS
Bile salts induce expression of leuO.
In response to bile salts, ToxR has been shown to activate ompU and ctxAB expression while repressing ompT expression (16, 38). Given that ToxR activates the expression of at least some of its target genes in response to bile salts, we hypothesized that ToxR may also regulate leuO expression in response to bile salts. To test this hypothesis, we introduced a leuO-lacZ reporter plasmid, pXB266, into WT strain JB58. The resultant strain was then cultured in the presence of bile or the bile salt deoxycholate to the mid-logarithmic growth phase, when leuO expression was quantified by the β-galactosidase assay. The results showed an ∼5-fold increase in leuO expression in the presence of bile and an ∼6-fold increase in leuO expression in the presence of deoxycholate compared to that in the LB control (Fig. 1A). These results confirmed the hypothesis that leuO expression was upregulated in the presence of bile or deoxycholate.
Bile salts and other components of bile exhibit detergent-like properties that can affect the permeability barrier of the outer membrane and compromise the integrity of the cytoplasmic membrane (10). This alluded to the possibility that leuO was upregulated as a result of the deleterious effects from the detergent properties of bile/deoxycholate on the cell envelope. If this were true, we hypothesized that exposure of V. cholerae to other classes of membrane-active detergents should also result in leuO upregulation. We therefore tested the effects of two anionic detergents (SDS and sarcosyl) and one nonionic detergent (Triton X-100) on leuO-lacZ expression. The results showed that exposure of JB58(pXB266) to these three detergents did not significantly affect leuO expression (Fig. 1A). Taken together, these results suggested that leuO induction was specific for bile and bile salts and was probably not a result of the detergent-like properties of bile or deoxycholate.
The finding that bile salts induced leuO expression prompted us to investigate if cFP and bile salts functioned synergistically to regulate leuO expression. To test this, we quantified leuO expression in the WT strain JB58 containing pJB906 (leuO-lux) during growth in LB containing DOC, cFP, or DOC and cFP. As expected, the addition of DOC to the medium activated leuO expression (Fig. 1B). However, the addition of cFP to the medium did not significantly increase leuO expression compared to that in the DMSO control. cFP did activate leuO expression at high (nonphysiological) concentrations of cFP (data not shown). As cFP was shown to activate leuO expression under AKI growth conditions, this result suggests that cFP activity is dependent on the growth conditions (23). The addition of both DOC and cFP to the growth medium resulted in a small but reproducible decrease in leuO expression compared to that in cells grown in medium containing DOC and DMSO. Although the presence of DOC and cFP appeared to decrease leuO expression, the differences were not statistically significant. Based on these results, we concluded that cFP and DOC do not work synergistically to increase leuO expression during growth under standard laboratory conditions. It remains to be determined how cFP and DOC affect leuO expression in vivo.
Upregulation of leuO by bile is dependent on ToxR.
ToxR regulates OmpU and OmpT production in response to bile salts. This suggested the possibility that ToxR is responsible for the bile-dependent upregulation of leuO in response to bile and bile salts. To test this, we compared levels of leuO-lacZ expression in JB58(pXB266) and an isogenic ΔtoxRS mutant, DT733(pXB266), which had been cultured in the presence and absence of bile or deoxycholate, as described above. The results showed that leuO expression in JB58 was increased upon exposure to deoxycholate and bile relative to growth in LB alone (Fig. 1C), as shown above. In contrast, leuO expression in the ΔtoxRS mutant grown in LB decreased by ∼15-fold relative to that in JB58. This indicates that ToxR is a positive regulator of leuO, confirming previous findings (23). The deletion of toxRS also abolished the bile- and deoxycholate-dependent upregulation of leuO (Fig. 1C). Together, these data indicated that ToxR was required for basal leuO expression and the increased leuO expression in response to bile and bile salts. Further, given that none of the other tested detergents affected leuO expression, we speculate that the ToxR-dependent upregulation of leuO was specific for bile salts and was not due to other components of bile or to a general membrane stress response.
ToxR acts directly on the leuO promoter.
Previous studies showed that ToxR binds to direct repeat elements that represent a ToxR consensus binding sequence in the toxT, ompU, ompT, and ctxAB promoters to regulate their transcription (4, 18, 19, 39, 40). The gene encoding LeuO (VC2485) is located downstream from VC2486 in an apparent two-gene operon. Sequence analysis of the leuO promoter revealed the presence of two putative ToxR consensus binding sequences, suggesting that ToxR may directly regulate leuO expression (23). The distal ToxR binding site (i.e., site A in Fig. 2A) is located from −126 to −112 relative to the start codon for VC2486, while the proximal ToxR binding site (i.e., Fig. 2A, site B) is located on the complementary strand from −104 to −90 relative to the start codon for VC2486. To determine if both ToxR binding sites were required for leuO expression, derivatives of the leuO promoter lacking one or both ToxR consensus sequences were transcriptionally fused to the lacZ gene in pTL61T. All together, we generated three leuO-lacZ reporters: pXB266 (WT leuO promoter), pVA258 (deletion of the distal ToxR binding site), and pVA261 (no ToxR binding sites) (Fig. 2A).
FIG 2.

ToxR consensus binding sequences are required for ToxR activation of leuO transcription. (A) Schematic diagram of the leuO promoter in the indicated leuO-lacZ reporter plasmids. The locations of the two putative ToxR consensus binding sites are indicated by the letters A (distal) and B (proximal). The putative −10 and −35 promoter elements are indicated. pXB266 contains the native leuO promoter, which contains both ToxR consensus binding sites, pVA258 contains only the proximal ToxR binding site, and pVA261 lacks both ToxR binding sites but maintains the −35 and −10 basal promoter elements. (B) WT strain JB58, ΔtoxRS strain DT733, and toxRΔppd strain SS4 containing the indicated leuO-lacZ reporter plasmids were grown in LB to mid-logarithmic phase, at which point aliquots were collected and assayed for leuO-lacZ using the β-galactosidase assay. (C) WT strain JB58 containing the indicated leuO-lacZ reporter plasmids was grown in LB or LB plus 0.2% bile to mid-logarithmic phase, at which point aliquots were collected and assayed for leuO-lacZ expression using a β-galactosidase assay. (D) E. coli strains containing the indicated toxRS expression plasmid pBAD33::toxRS, pBAD33::toxRΔppdS, or pBAD33 and one of the leuO-lacZ reporter plasmids, pXB266, pVA258, or pVA261, were grown in LB containing 0.08% arabinose to mid-logarithmic phase, at which point aliquots were collected and assayed for leuO-lacZ expression using the β-galactosidase assay. Statistical significance in panel B was determined using two-way ANOVA with Dunnett's posttest, comparing the mean result for each plasmid to that for the control, pTL61T, in the designated strain. Statistical significance in panel C was determined using two-way ANOVA with Tukey's posttest, comparing the result for the overexpression plasmid to that of the pBAD33 control plasmid with the designated second plasmid. *, P < 0.05; **, P < 0.0001.
We introduced these three leuO-lacZ reporter plasmids into WT strain JB58 and the ΔtoxRS mutant strain DT733. The resulting strains were then grown to mid-logarithmic phase, when leuO-lacZ expression was quantified using β-galactosidase assays (Fig. 2B). Consistently with the above data, the results of these tests showed high levels of β-galactosidase production in JB58 containing the native leuO promoter (i.e., pXB266) and very little β-galactosidase production in the ΔtoxRS mutant containing the same promoter reporter. In contrast, very little β-galactosidase was produced in JB58 or the ΔtoxRS mutant containing the leuO reporters lacking one (pVA258) or both (pVA261) ToxR consensus binding sites. These results confirmed that ToxR was required for leuO expression and suggested that both sites A and B were required for basal leuO expression in V. cholerae.
We next tested to see if the addition of bile could bypass the requirement for both ToxR binding sites in the leuO promoter to induce leuO expression. We therefore cultured WT strain JB58 bearing the three leuO-lacZ reporter plasmids (Fig. 2A) in the presence and absence of bile and quantified leuO expression. The results showed that only the full-length leuO promoter (i.e., pXB266) supported the activation of leuO expression in the presence of bile (Fig. 2C). Very little β-galactosidase was produced in JB58 containing the leuO reporters lacking one (pVA258) or both (pVA261) ToxR consensus binding sites in the presence or absence of bile. These results indicated that both ToxR binding sites are required for the bile-dependent induction of leuO expression.
The presence of the ToxR binding sequences in the leuO promoter suggested that ToxR acts directly on the leuO promoter. If this is true, we hypothesized that ToxR expression in a heterologous host would result in activation of the leuO promoter. To test this, we expressed toxRS from the pBAD33 arabinose-inducible promoter in an E. coli host that contained each of the three leuO-lacZ reporters described above (i.e., pXB266, pVA258, and pVA261). We cultured the recombinant strains to mid-logarithmic phase in the presence of arabinose (to induce toxRS expression) before quantifying leuO-lacZ expression using β-galactosidase assays. The results showed that the expression of toxRS from pBAD33::toxRS resulted in high and equal levels of expression from the native leuO promoter (pXB266) and the promoter lacking the distal ToxR binding site (pVA258) (Fig. 2D). β-Galactosidase production was greatly diminished in the strain bearing the leuO promoter that lacked both ToxR consensus sequences (pVA261). The ∼18-fold increase in leuO-lacZ expression in the E. coli(pBAD33::toxRS) cultures bearing pXB266 and pVA258 relative to the strain bearing pVA261 suggests that ToxR directly binds to the ToxR consensus binding sites in the leuO promoter to facilitate activation of leuO transcription in E. coli. These results also imply that in E. coli, in contrast to V. cholerae, ToxR can bind to the proximal ToxR consensus site in pVA258 to activate transcription.
Activation of leuO requires the ToxR periplasmic domain.
The periplasmic domain of ToxR was shown to be important for the cFP-dependent activation of leuO expression (23) but dispensable for basal ompU expression and virulence factor production (2, 23). We therefore examined whether the ToxR periplasmic domain contributed to leuO expression during growth in LB. To test this, we generated a V. cholerae mutant (SS4) that produced a toxR allele in which we deleted the carboxy-terminal periplasmic domain (i.e., toxRΔppd). This mutant allele is localized to the membrane and was previously shown to be functional (2, 23).We then introduced pXB266, pVA258, and pVA261 into the toxRΔppd mutant SS4. The strains were then cultured as described above, and leuO-lacZ expression was quantified. The results showed that deletion of the ToxR periplasmic domain largely abolished leuO expression from all three leuO-lacZ reporters (Fig. 2B). This indicated that the ToxR periplasmic domain was important for leuO expression.
We also tested whether the ToxR periplasmic domain was necessary for the activation of leuO expression in E. coli. E. coli strains containing each of the leuO-lacZ reporter plasmids were transformed with plasmid pBAD33::toxRΔppdS (pXB286), and the resulting strains were cultured as described above before being assayed for leuO-lacZ expression. The results of these experiments mirrored the results obtained with E. coli expressing the WT toxRS allele. Overexpression of toxRΔppdS from pXB286 activated the WT leuO promoter in pXB266 and the leuO promoter lacking the distal ToxR binding site in pVA258 to similar levels (Fig. 2D). Further, the magnitude of activation was similar to that observed with the strain expressing the WT toxRS allele. This suggested that the toxRΔppd allele produces a functional protein that is able to bind to the ToxR consensus sequences in the leuO promoter and to activate leuO transcription. This suggests that the inability of ToxRΔppd to activate leuO expression in V. cholerae was not due to its inability to bind to DNA but rather due to other factors that are dependent on the presence of the ToxR periplasmic domain.
ToxR can directly bind the leuO promoter.
To further support the conclusion that ToxR bound directly to the leuO promoter, gel electrophoretic mobility shift assays were performed with the leuO promoter. The EMSAs were performed with crude membrane fractions that were isolated from V. cholerae strain DT733 (ΔtoxRS) containing pBAD18 or pBAD18 expressing toxRS or toxRΔppdS. As has been found with other gel shift assays using ToxR membrane fractions, the bound DNA probes do not enter the gel and are left in the wells of the gel; thus, the binding of ToxR to the labeled probe should be assessed by the disappearance of free probe and not by the presence of a shifted band (3). The results showed that the ToxRS-positive membrane fractions bound directly to the leuO promoter, which resulted in a decrease in the abundance of free leuO probe (Fig. 3A, lane 2). The decrease in free leuO probe was largely abolished in the presence of ToxRS-negative membrane fractions (lane 7). Taken together, these results indicated that ToxR can directly bind to the leuO promoter.
FIG 3.
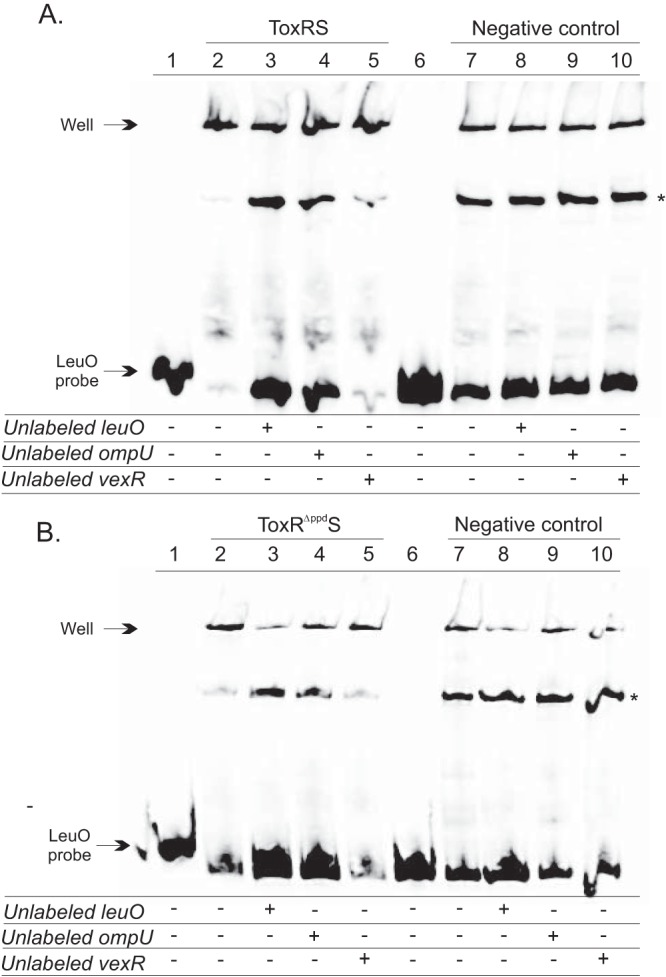
ToxR and ToxRΔppd interact with the leuO promoter. A biotinylated DNA fragment encompassing the leuO promoter was incubated with V. cholerae cytoplasmic membrane fractions isolated from strain DT733 (ΔtoxRS), which overproduces ToxRS or ToxRΔppdS from pBAD18, described in Materials and Methods. The negative-control membranes are ToxRS negative and were isolated from DT733 containing the empty vector control (i.e., pBAD18). The binding reaction mixtures contained either no membranes (lanes 1 and 6) or equal amounts of the indicated membranes (lanes 2 to 5 and 7 to 10). Unlabeled competitor DNA probes encompassing the leuO, ompU, or vexR promoter were added as indicated to the binding reaction mixture at a 10-fold excess relative to the level of the biotinylated leuO DNA probe. Equal aliquots of binding reaction mixtures were then electrophoresed on nondenaturing 5% acrylamide gels, and the position of the leuO probe was visualized by chemiluminescence detection, as described in Materials and Methods. (A) ToxR binding to the leuO promoter. The ToxR binding mixture contained 3 mg/ml ToxRS-positive membranes, while the control binding reaction mixtures contained an equivalent concentration of ToxRS-negative membranes. (B) ToxRΔppd binding to the leuO promoter. The ToxRΔppd binding mixture contained 1 mg/ml ToxRΔppdS-positive membranes, while the control binding reaction mixtures contained an equivalent concentration of ToxRS-negative membranes. The asterisks denote an unknown nonspecific mobility shift.
To determine whether ToxR binding to the leuO promoter was specific, we performed binding competition assays by including a 10-fold excess of unlabeled competitor DNA in the binding reaction mixtures. If ToxR bound specifically to the leuO promoter fragment, adding excess unlabeled leuO promoter would prevent a shift in the free probe. When excess leuO DNA was added in the assay, there was a reduced shift in the labeled leuO probe (Fig. 3A, lane 3). The leuO promoter was also competed with a known ToxR-specific promoter, the ompU promoter, which competed for leuO binding and prevented the shift in the free probe (lane 4). The addition of a nonspecific competitor encompassing the vexR promoter, which is not regulated by ToxR, did not alter the level of free leuO probe (lane 5), indicating that the observed shift was specific for the leuO promoter. These same binding conditions were also used for ToxRS-negative membranes (Fig. 3A, lanes 6 to 10). The results showed that although there was some decrease in free probe between the no-protein control and the ToxRS-negative membrane control, the decrease was much less than that observed with ToxRS-positive membranes and was unaffected by the addition of any of the unlabeled competitor probes. These results confirmed the specificity of the ToxR-positive membranes for the leuO promoter.
Gel shift assays were also performed to determine if deletion of the ToxR periplasmic domain altered ToxR binding to the leuO promoter. These experiments were performed as described above with membrane fractions from DT733 containing pBAD18 expressing toxRΔppdS. The results showed that the ToxRΔppdS membranes had shifts comparable to those of the ToxRS membranes. The ToxRΔppdS membrane fractions shifted the leuO promoter (Fig. 3B, lane 2). The leuO shift was competed by the addition of a 10-fold excess of unlabeled leuO or ompU promoter probes but not by the addition of the nonspecific vexR promoter probe (lanes 3 to 5). These same binding conditions were also used for ToxRΔppdS-negative membranes (Fig. 3B, lanes 6 to 10). The results showed a decrease in free probe between the no-protein control and the ToxRΔppdS-negative membrane control, but the decrease was much less than that observed with ToxRΔppdS-positive membranes and was unaffected by the addition of any of the unlabeled competitor probes; this confirmed the specificity of the ToxR-positive membranes for the leuO promoter. From these results, we concluded that the ToxR periplasmic domain was not required for ToxR binding to the leuO promoter.
ToxR periplasmic domain is important for responding to bile.
The role of the ToxR periplasmic domain in environmental sensing is poorly understood. ToxR inversely regulates ompU and ompT expression. OmpT is expressed during growth in minimal medium, while OmpU is expressed during growth in rich medium. The addition of bile salts or the amino acids asparagine, arginine, glutamic acid, and serine (NRES) to minimal medium results in ToxR-dependent porin switching that mimics growth in rich medium (i.e., expression of ompU and repression of ompT) (41). The mechanism by which ToxR activates ompU expression in minimal medium differs for bile salts and NRES (20). The addition of NRES to minimal medium results in toxR upregulation, which is sufficient to increase ompU expression. In contrast, bile salts activate ompU expression via a process that does not result in toxR upregulation and may involve transcriptional activation (20).
We took advantage of the ompT-ompU switching system described above to test the contribution of the ToxR periplasmic domain on leuO and ompU expression in response to bile. We cultured WT strain JB58, the ΔtoxRS strain DT733, and the toxRΔppd strain SS4 in T minimal medium or T medium containing bile or NRES and quantified leuO and ompU expression by quantitative real-time PCR (qRT-PCR). The results showed that leuO expression was induced ∼24-fold in response to bile and was unaffected by the addition of NRES to the WT strain (Fig. 4A). The addition of NRES or bile to the medium did not affect leuO expression in the toxRS or toxRΔppd mutant. In contrast to leuO, ompU expression increased in response to both NRES (13-fold) and bile (22-fold) relative to that in the control cultures (Fig. 4B). The expression of ompU was abolished under all conditions in the ΔtoxRS mutant. The expression of ompU in the toxRΔppd mutant following exposure to NRES increased to a level that was similar to that observed in the WT, further confirming that the toxRΔppd allele produced a functional protein in V. cholerae. The expression of ompU in the toxRΔppd mutant following exposure to bile resulted in a much lower level of ompU induction than that observed in the WT. Exposure to bile resulted in an ∼21-fold increase in ompU expression in the WT but only an ∼3-fold increase in the toxRΔppd mutant (Fig. 4B). Taken together, these results provide additional evidence that the ToxRΔppd protein is functional and that the periplasmic domain of ToxR is critical for the induction of ompU and leuO in response to bile. This suggests the possibility that bile or bile salts affect ToxR activity by a process that requires the periplasmic domain.
FIG 4.
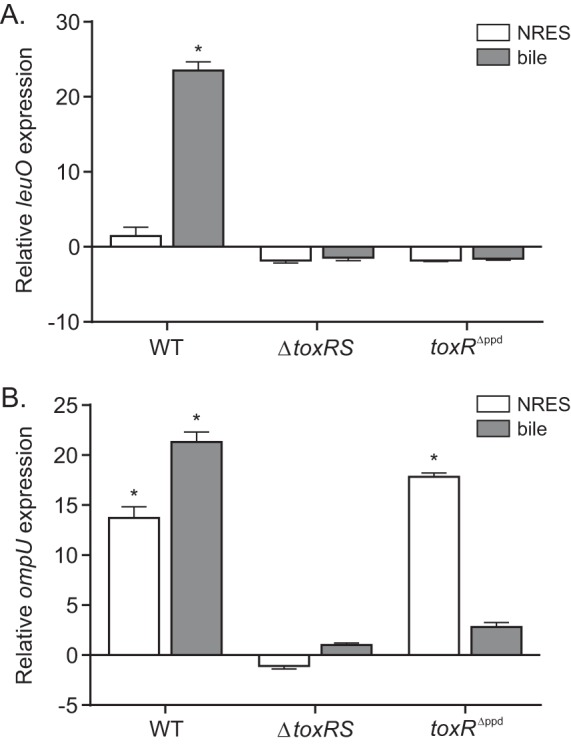
The ToxR periplasmic domain is required for leuO and ompU upregulation in response to bile. qRT-PCR was used to determine the effect of the amino acids NRES and bile on leuO (A) and ompU (B) expression during the growth of JB58 (WT), DT733 (ΔtoxRS), and SS4 (toxRΔppd) in modified T medium. The strains were cultured to an OD600 of ∼0.3 before being transferred to medium supplemented with either 50 mM NRES or 0.2% bile. The cultures were then grown for an additional 15 min before aliquots were collected for RNA isolation, as described in Materials and Methods. The presented data are the means ± SD of the results from three independent experiments. Statistical significance was determined using a one-sample t test comparing the sample mean to a hypothetical value of ±1. *, P < 0.01.
LeuO contributes to V. cholerae bile resistance.
Since ToxR functions in bile resistance (7, 16), we hypothesized that LeuO may also contribute to V. cholerae bile resistance. To test this, we performed bile killing assays. In these experiments, we quantified the survival of WT V. cholerae (JB58) and isogenic leuO, toxRS, and toxRΔppd deletion mutants upon exposure to a lethal concentration of bile for 1 h, as described in Materials and Methods. The results of these experiments showed an ∼4-fold decrease in the recovery of the leuO mutant strain XBV222 relative to the recovery of WT strain JB58 when exposed to 20% bile (Table 2). This suggested that the presence of leuO provided a survival advantage to V. cholerae in the presence of bile. The ΔtoxRS mutant strain DT733 and the toxRΔppd mutant SS4 were not recovered when exposed to 20% bile. This finding is consistent with the role of ToxR in bile resistance and is likely attributable to the combined dysregulation of ompU, ompT, and leuO expression in the ΔtoxRS mutant. We therefore performed the killing assays with the toxR mutant strains using 10% bile. When exposed to 10% bile, the ΔtoxRS mutant strain exhibited an ∼11-fold decrease in recovery relative to that of the WT, suggesting that the ΔtoxRS mutant was more sensitive to bile than the ΔleuO mutant. The toxRΔppd mutant exhibited an ∼7-fold decrease in recovery compared to that of the WT. This indicated that the toxRΔppd mutant exhibited greater susceptibility to bile than did a ΔleuO mutant. As ToxRΔppd has been shown to be sufficient for ompU expression under standard laboratory conditions, these results suggest the possibility that the periplasmic domain is required for the expression of other factors, in addition to leuO, that might contribute to bile resistance.
TABLE 2.
Bile killing assays
| Strain | Fold change in recovery (mean [SD]) for: |
|
|---|---|---|
| 10% bile | 20% bile | |
| ΔleuO mutant or WT | 1 | −4.5 (2.1)a |
| ΔtoxRS mutant or WT | −11.3 (3.7)a | NRb |
| toxRΔppd mutant or WT | −7.0 (1.4)a | NR |
| ΔtoxRS pBAD18::leuO or ΔtoxRS pBAD18 mutant | 5.2 (1.4)a | NR |
P < 0.01.
NR, not recovered.
The above data suggest that LeuO contributed to V. cholerae survival in the presence of bile. If this is true, the overexpression of leuO in a ΔtoxRS mutant should provide a survival advantage upon exposure to a lethal concentration of bile. We tested this by introducing pBAD18-leuO or pBAD18 into the ΔtoxRS mutant DT733. We chose a ΔtoxRS mutant for these experiments to negate leuO and ompU expression. We cultured the toxRS deletion strain bearing the pBAD18-leuO plasmid pVA94 or the empty vector control (pBAD18) to log phase in the presence of 0.1% arabinose to induce leuO expression. Aliquots of the induced cultures were then exposed to a lethal concentration of bile before being processed as described above. The results showed that leuO overexpression resulted in an ∼5-fold increase in cell recovery relative to the recovery of the empty vector control (Table 2). This indicated that LeuO contributed to V. cholerae survival in the presence of bile by a mechanism that is likely independent of ompU. Taken together, the results of these experiments confirm the importance of ToxR in bile resistance and support the conclusion that ToxR activation of leuO expression contributes to bile resistance.
LeuO does not contribute to bile resistance through regulation of the RND efflux pumps.
Our laboratory has previously shown that the V. cholerae resistance-nodulation-division (RND) family efflux systems are major contributors to V. cholerae bile resistance (11–15). We therefore tested whether the contribution of leuO to bile resistance was mediated by upregulation of any of the RND efflux systems. We quantified the expression of all six of the RND efflux pumps in the WT strain JB58 and the ΔleuO mutant strain XBV222 in response to bile using lacZ promoter reporter fusions. The strains were cultured in the absence or presence of bile to mid-logarithmic growth phase, at which point the expression of the individual RND efflux systems was quantified. The results showed that in the absence of bile, there were no significant differences between the expression of any of the RND efflux pumps in the ΔleuO mutant and that in the WT strain (Fig. 5). This indicated that LeuO did not contribute to the basal expression of any of the efflux pumps. When the same reporter strains were cultured in the presence of bile, as expected, vexRAB and vexCD expression levels were found to be significantly induced. However, there were no significant differences found in any of the RND efflux pump expression levels between the WT and leuO deletion strains in response to bile (Fig. 5). This indicated that LeuO does not regulate the expression of the RND efflux systems and is likely not working through the RND efflux pumps to contribute to bile resistance.
FIG 5.

Effect of leuO on the expression of the V. cholerae RND efflux systems. V. cholerae WT strain JB58 and ΔleuO mutant strain XBV222 carrying lacZ promoter fusion reporter plasmids for vexRAB-lacZ (pXB233) (A), vexCD-lacZ (pXB231) (B), vexEF-lacZ (pXB228) (C), vexGH-lacZ (pXB229) (D), vexIJK-lacZ (pXB230) (E), or vexLM-lacZ (pXB232) (F). All strains were cultured in LB in the presence or absence of 0.2% bile before being assayed for β-galactosidase activity, as described in Materials and Methods. The presented data are the means ± SD of the results from three independent experiments.
DISCUSSION
The ability of V. cholerae to respond to its environment is essential for its success as an enteric pathogen. This is critical upon entrance into the human host, where V. cholerae must express genes that are indispensable for colonization of the small intestine. Colonization and growth in the small intestine require the expression of virulence genes plus the expression of genes that combat antimicrobial agents present in the intestine. ToxR plays a critical role in this regard by regulating the expression of genes that are required for host adaptation. ToxR is thought to transduce in vivo signals to affect the expression of its target genes, but the mechanism by which this occurs is poorly understood.
In this study, we observed the upregulation of leuO expression in response to bile and the bile salt deoxycholate by a process that was dependent on ToxR (Fig. 1). Bile salts have detergent-like properties that make them bactericidal. The expression of leuO was not altered by other membrane-active detergents (e.g., SDS, Triton X-100, or sarcosyl), indicating that leuO induction was not the result of the general membrane stress response but instead was directly in response to bile and bile salts. The upregulation of leuO in response to bile suggested the possibility that leuO functions in bile resistance. Support for this conclusion was provided by the observation that leuO deletion resulted in increased bile susceptibility, while leuO overexpression resulted in increased bile resistance (Table 2). In light of recent studies using an infant mouse model showing that leuO is expressed in the intestine (23), we speculate that the findings observed here may extend to the host. These results were also similar to what has been reported for OmpU, a porin that is associated with bile resistance in V. cholerae and whose expression is also activated by ToxR in response to bile and deoxycholate (16).
While the contribution of leuO to bile resistance is clear, the mechanism by which leuO impacts bile resistance was not resolved. The ΔleuO mutant strain exhibited a bile susceptibility phenotype that was intermediate relative to that of the ΔtoxRS mutant (Table 2). This suggested that LeuO likely affected bile resistance by a mechanism that was distinct from ompU or ompT regulation. This conclusion was confirmed by the finding that leuO overexpression provided a survival advantage in a toxRS-negative strain exposed to bile (Table 2). Bile resistance results from the synergistic affects of reduced outer membrane permeability and active efflux (14). In V. cholerae, the RND efflux systems are major contributors to bile resistance, suggesting a potential mechanism by which LeuO might affect bile resistance. However, our results showed that there was no difference in the levels of expression of any of the RND efflux systems in a leuO mutant grown in the presence and absence of bile (Fig. 5), suggesting that leuO affects bile resistance by a mechanism that is independent of the RND efflux systems. There are a number of other potential mechanisms by which LeuO might impact bile resistance, including the expression of other transport systems, the production of other porins, alterations in cell physiology, and alterations to the cell envelope.
The expression of leuO was previously found to be dependent on ToxR (23). Sequence analysis of the leuO promoter revealed the presence of two putative ToxR consensus binding sites, both of which were required for basal-level leuO expression in V. cholerae (Fig. 2B). This finding was reminiscent of what was observed for ToxR activity at the ompU promoter, in which the most distal ToxR binding site was needed for full ompU activation in V. cholerae (18). In contrast, ToxR activated expression from the leuO promoter lacking the distal ToxR binding site (i.e., pVA258) in E. coli (Fig. 2D). The lack of expression from the same mutant promoter in V. cholerae suggested that other factors affect leuO expression in V. cholerae. We do not know what these factors are, but there are a number of potential explanations for this result. It is possible that other DNA binding proteins interact with the leuO promoter and impede ToxR binding at the proximal site. It is also possible that ToxR binds sequentially to the two ToxR binding sites in V. cholerae and has to bind to the distal site first, which then facilitates binding at the proximal site. This tandem fashion of binding is similar to that of other DNA binding domains of OmpR family proteins, which generally interact as dimers with direct repeat DNA sequences (40). This idea is also supported by cooperative binding studies comparing ToxR oligomerization and the regulation of target promoters containing multiple operator elements in E. coli and V. cholerae (42).
The periplasmic domain of ToxR has been implicated in the response to environmental signals, but how ToxR regulates its target genes in response to these signals is poorly understood. The finding that the ToxR periplasmic domain was required for the upregulation of both leuO and ompU in response to bile (Fig. 4) suggests the possibility that the periplasmic domain acts as a bile sensor, which can affect the activity of the cytoplasmic DNA binding domain at its target promoters. The mechanism by which the periplasmic domain senses bile is unclear. There are a number of potential mechanisms by which bile might affect ToxR activity. Bile might facilitate ToxR interaction with ToxS. ToxS has been shown to contribute to ToxR stability and to enhance its activity at target genes (4–6). Alternatively, bile might affect conformational changes in the ToxR periplasmic domain that affect DNA binding. Bile might also affect disulfide bond formation in the two cysteine residues located in the ToxR periplasmic domain. There is evidence that disulfide bond formation in the periplasmic domain of ToxR and ToxR-like proteins affects their activities. For example, disulfide bond formation in ToxR has been shown to contribute to ompU regulation in response to some growth conditions (43). ToxR and TcpP have also been shown to form homodimers and heterodimers (44, 45), and the bile salt taurocholate has been shown to induce intermolecular disulfide bond formation in the periplasmic domain of TcpP (46). Similarly, E. coli CadC has been shown to form disulfide bonds in response to pH, which result in the activation of cadBA transcription (47). Additional work will be required to differentiate between these potential mechanisms.
Previous studies showed that cFP activation of leuO resulted in the downregulation of the ToxR regulon (23). The data presented here show that leuO expression is also activated by bile and bile salts. While the fatty acid components of bile have been linked to downregulation of the ToxR regulon (48), bile salts have been shown to either be neutral or enhance virulence gene expression (38, 46). Thus, the role of LeuO in virulence gene regulation is a paradox. Although there are a number of potential explanations for the differential effects of LeuO on virulence, we suspect that bile salts and CDPs differentially affect the expression (or activities) of other proteins that contribute to virulence gene expression. For example, bile salts and cFP may differentially affect histone-like nucleoid-structuring protein (H-NS) or C-reactive protein (CRP), both of which have been shown to suppress the ToxR regulon and thus might contribute to the observed phenotype (49, 50). Studies to resolve the role of LeuO in virulence gene regulation are ongoing in our laboratory.
ACKNOWLEDGMENTS
This research was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) under award R01AI091845. M.F.H. was supported by NIH training grant T32AI049820.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Childers BM, Klose KE. 2007. Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol 2:335–344. doi: 10.2217/17460913.2.3.335. [DOI] [PubMed] [Google Scholar]
- 2.Crawford JA, Krukonis ES, DiRita VJ. 2003. Membrane localization of the ToxR winged-helix domain is required for TcpP-mediated virulence gene activation in Vibrio cholerae. Mol Microbiol 47:1459–1473. doi: 10.1046/j.1365-2958.2003.03398.x. [DOI] [PubMed] [Google Scholar]
- 3.Ottemann KM, DiRita VJ, Mekalanos JJ. 1992. ToxR proteins with substitutions in residues conserved with OmpR fail to activate transcription from the cholera toxin promoter. J Bacteriol 174:6807–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell 48:271–279. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 5.Pfau JD, Taylor RK. 1998. Mutations in toxR and toxS that separate transcriptional activation from DNA binding at the cholera toxin gene promoter. J Bacteriol 180:4724–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiRita VJ, Mekalanos JJ. 1991. Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell 64:29–37. doi: 10.1016/0092-8674(91)90206-E. [DOI] [PubMed] [Google Scholar]
- 7.Provenzano D, Klose KE. 2000. Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc Natl Acad Sci U S A 97:10220–10224. doi: 10.1073/pnas.170219997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park DK, Lee KE, Baek CH, Kim IH, Kwon JH, Lee WK, Lee KH, Kim BS, Choi SH, Kim KS. 2006. Cyclo(Phe-Pro) modulates the expression of ompU in Vibrio spp. J Bacteriol 188:2214–2221. doi: 10.1128/JB.188.6.2214-2221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merrell DS, Bailey C, Kaper JB, Camilli A. 2001. The ToxR-mediated organic acid tolerance response of Vibrio cholerae requires OmpU. J Bacteriol 183:2746–2754. doi: 10.1128/JB.183.9.2746-2754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Begley M, Gahan CG, Hill C. 2005. The interaction between bacteria and bile. FEMS Microbiol Rev 29:625–651. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Bina JE, Provenzano D, Wang C, Bina XR, Mekalanos JJ. 2006. Characterization of the Vibrio cholerae vexAB and vexCD efflux systems. Arch Microbiol 186:171–181. doi: 10.1007/s00203-006-0133-5. [DOI] [PubMed] [Google Scholar]
- 12.Bina XR, Provenzano D, Nguyen N, Bina JE. 2008. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect Immun 76:3595–3605. doi: 10.1128/IAI.01620-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerda-Maira FA, Ringelberg CS, Taylor RK. 2008. The bile response repressor BreR regulates expression of the Vibrio cholerae breAB efflux system operon. J Bacteriol 190:7441–7452. doi: 10.1128/JB.00584-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatterjee A, Chaudhuri S, Saha G, Gupta S, Chowdhury R. 2004. Effect of bile on the cell surface permeability barrier and efflux system of Vibrio cholerae. J Bacteriol 186:6809–6814. doi: 10.1128/JB.186.20.6809-6814.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor DL, Bina XR, Bina JE. 2012. Vibrio cholerae VexH encodes a multiple drug efflux pump that contributes to the production of cholera toxin and the toxin co-regulated pilus. PLoS One 7:e38208. doi: 10.1371/journal.pone.0038208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Provenzano D, Schuhmacher DA, Barker JL, Klose KE. 2000. The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infect Immun 68:1491–1497. doi: 10.1128/IAI.68.3.1491-1497.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crawford JA, Kaper JB, DiRita VJ. 1998. Analysis of ToxR-dependent transcription activation of ompU, the gene encoding a major envelope protein in Vibrio cholerae. Mol Microbiol 29:235–246. doi: 10.1046/j.1365-2958.1998.00925.x. [DOI] [PubMed] [Google Scholar]
- 19.Li CC, Crawford JA, DiRita VJ, Kaper JB. 2000. Molecular cloning and transcriptional regulation of ompT, a ToxR-repressed gene in Vibrio cholerae. Mol Microbiol 35:189–203. doi: 10.1046/j.1365-2958.2000.01699.x. [DOI] [PubMed] [Google Scholar]
- 20.Mey AR, Craig SA, Payne SM. 2012. Effects of amino acid supplementation on porin expression and ToxR levels in Vibrio cholerae. Infect Immun 80:518–528. doi: 10.1128/IAI.05851-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duret G, Delcour AH. 2006. Deoxycholic acid blocks Vibrio cholerae OmpT but not OmpU porin. J Biol Chem 281:19899–19905. doi: 10.1074/jbc.M602426200. [DOI] [PubMed] [Google Scholar]
- 22.Simonet VC, Baslé A, Klose KE, Delcour AH. 2003. The Vibrio cholerae porins OmpU and OmpT have distinct channel properties. J Biol Chem 278:17539–17545. doi: 10.1074/jbc.M301202200. [DOI] [PubMed] [Google Scholar]
- 23.Bina XR, Taylor DL, Vikram A, Ante VM, Bina JE. 2013. Vibrio cholerae ToxR downregulates virulence factor production in response to cyclo(Phe-Pro). mBio 4(5):e00366-13. doi: 10.1128/mBio.00366-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hertzberg KM, Gemmill R, Jones J, Calvo JM. 1980. Cloning of an EcoRI-generated fragment of the leucine operon of Salmonella Typhimurium. Gene 8:135–152. doi: 10.1016/0378-1119(80)90033-5. [DOI] [PubMed] [Google Scholar]
- 25.Hernández-Lucas I, Calva E. 2012. The coming of age of the LeuO regulator. Mol Microbiol 85:1026–1028. doi: 10.1111/j.1365-2958.2012.08175.x. [DOI] [PubMed] [Google Scholar]
- 26.Shimada T, Bridier A, Briandet R, Ishihama A. 2011. Novel roles of LeuO in transcription regulation of E. coli genome: antagonistic interplay with the universal silencer H-NS. Mol Microbiol 82:378–397. doi: 10.1111/j.1365-2958.2011.07818.x. [DOI] [PubMed] [Google Scholar]
- 27.Lawrenz MB, Miller VL. 2007. Comparative analysis of the regulation of rovA from the pathogenic yersiniae. J Bacteriol 189:5963–5975. doi: 10.1128/JB.00528-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moorthy S, Watnick PI. 2005. Identification of novel stage-specific genetic requirements through whole genome transcription profiling of Vibrio cholerae biofilm development. Mol Microbiol 57:1623–1635. doi: 10.1111/j.1365-2958.2005.04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitaker WB, Parent MA, Boyd A, Richards GP, Boyd EF. 2012. The Vibrio parahaemolyticus ToxRS regulator is required for stress tolerance and colonization in a novel orogastric streptomycin-induced adult murine model. Infect Immun 80:1834–1845. doi: 10.1128/IAI.06284-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JA, Park JH, Lee MA, Lee HJ, Park SJ, Kim KS, Choi SH, Lee KH. 2015. Stationary phase induction of vvpS expression by three transcription factors: repression by LeuO and activation by SmcR and CRP. Mol Microbiol 97:330–346. doi: 10.1111/mmi.13028. [DOI] [PubMed] [Google Scholar]
- 31.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imai Y, Matsushima Y, Sugimura T, Terada M. 1991. A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res 19:2785. doi: 10.1093/nar/19.10.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bina JE, Mekalanos JJ. 2001. Vibrio cholerae tolC is required for bile resistance and colonization. Infect Immun 69:4681–4685. doi: 10.1128/IAI.69.7.4681-4685.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 35.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Huntley JF, Robertson GT, Norgard MV. 2010. Method for the isolation of Francisella tularensis outer membranes. J Vis Exp 40:e2044. doi: 10.3791/2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osborn MJ, Gander JE, Parisi E, Carson J. 1972. Mechanism of assembly of the outer membrane of Salmonella Typhimurium. Isolation and characterization of cytoplasmic and outer membrane. J Biol Chem 247:3962–3972. [PubMed] [Google Scholar]
- 38.Hung DT, Mekalanos JJ. 2005. Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proc Natl Acad Sci U S A 102:3028–3033. doi: 10.1073/pnas.0409559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Higgins DE, DiRita VJ. 1994. Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol Microbiol 14:17–29. doi: 10.1111/j.1365-2958.1994.tb01263.x. [DOI] [PubMed] [Google Scholar]
- 40.Goss TJ, Morgan SJ, French EL, Krukonis ES. 2013. ToxR recognizes a direct repeat element in the toxT, ompU, ompT, and ctxA promoters of Vibrio cholerae to regulate transcription. Infect Immun 81:884–895. doi: 10.1128/IAI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol 170:2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dziejman M, Kolmar H, Fritz HJ, Mekalanos JJ. 1999. ToxR co-operative interactions are not modulated by environmental conditions or periplasmic domain conformation. Mol Microbiol 31:305–317. doi: 10.1046/j.1365-2958.1999.01173.x. [DOI] [PubMed] [Google Scholar]
- 43.Fengler VH, Boritsch EC, Tutz S, Seper A, Ebner H, Roier S, Schild S, Reidl J. 2012. Disulfide bond formation and ToxR activity in Vibrio cholerae. PLoS One 7:e47756. doi: 10.1371/journal.pone.0047756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ottemann KM, Mekalanos JJ. 1996. The ToxR protein of Vibrio cholerae forms homodimers and heterodimers. J Bacteriol 178:156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan F, Liu Z, Jabeen N, Birdwell LD, Zhu J, Kan B. 2014. Enhanced interaction of Vibrio cholerae virulence regulators TcpP and ToxR under oxygen-limiting conditions. Infect Immun 82:1676–1682. doi: 10.1128/IAI.01377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. 2013. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc Natl Acad Sci U S A 110:2348–2353. doi: 10.1073/pnas.1218039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tetsch L, Koller C, Dönhöfer A, Jung K. 2011. Detection and function of an intramolecular disulfide bond in the pH-responsive CadC of Escherichia coli. BMC Microbiol 11:74. doi: 10.1186/1471-2180-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chatterjee A, Dutta PK, Chowdhury R. 2007. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect Immun 75:1946–1953. doi: 10.1128/IAI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nye MB, Pfau JD, Skorupski K, Taylor RK. 2000. Vibrio cholerae H-NS silences virulence gene expression at multiple steps in the ToxR regulatory cascade. J Bacteriol 182:4295–4303. doi: 10.1128/JB.182.15.4295-4303.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skorupski K, Taylor RK. 1997. Cyclic AMP and its receptor protein negatively regulate the coordinate expression of cholera toxin and toxin-coregulated pilus in Vibrio cholerae. Proc Natl Acad Sci U S A 94:265–270. doi: 10.1073/pnas.94.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morin CE, Kaper JB. 2009. Use of stabilized luciferase-expressing plasmids to examine in vivo-induced promoters in the Vibrio cholerae vaccine strain CVD 103-HgR. FEMS Immunol Med Microbiol 57:69–79. doi: 10.1111/j.1574-695X.2009.00580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linn T, St. Pierre R. 1990. Improved vector system for constructing transcriptional fusions that ensures independent translation of lacZ. J Bacteriol 172:1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taylor DL, Ante VM, Bina XR, Howard MF, Bina JE. 2015. Substrate-dependent activation of the Vibrio cholerae vexAB RND efflux system requires vexR. PLoS One 10:e0117890. doi: 10.1371/journal.pone.0117890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor DL, Bina XR, Slamti L, Waldor MK, Bina JE. 2014. Reciprocal regulation of resistance-nodulation-division efflux systems and the Cpx two-component system in Vibrio cholerae. Infect Immun 82:2980–2991. doi: 10.1128/IAI.00025-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]