

Abstract
Chordomas are an aggressive rare type of malignant bone tumors arising from the remnant of the notochord. Chordomas occur mainly in vertebral bones and account for 1–4% of malignant bone tumors. Management and treatment of chordomas are difficult as they are resistant to conventional chemotherapy; therefore, they are mainly treated with surgery and radiation therapy. In this study, we performed DNA methylation profiling of 26 chordomas and normal nucleus pulposus samples plus UCH-1 chordoma cell line using the Illumina Infinium HumanMethylation450 BeadChips. Combined bisulfite restriction analysis and bisulfite sequencing was used to confirm the methylation data. Gene expression was analyzed using RT-PCR before and after 5-aza-2’-deoxycytidine (5-azaDC) treatment of chordoma cell lines. Analysis of the HumanMethylation450 BeadChip data led to the identification of 8,819 loci (2.9%) that were significantly differentially methylated (>0.2 average β-value difference) between chordomas and nucleus pulposus samples (adjusted P < 0.05). Among these, 5,868 probes (66.5%) were hypomethylated, compared to 2,951 (33.5%) loci that were hypermethylated in chordomas compared to controls. From the 2,951 differentially hypermethylated probes, 33.3% were localized in the promoter region (982 probes) and, among these, 104 probes showed cancer-specific hypermethylation. Ingenuity Pathway Analysis indicates that the cancer-specific differentially methylated loci are involved in various networks including cancer disease, nervous system development and function, cell death and survival, cellular growth, cellular development, and proliferation. Furthermore, we identified a subset of probes that were differentially methylated between recurrent and non-recurrent chordomas. BeadChip methylation data was confirmed for these genes and gene expression was shown to be upregulated in methylated chordoma cell lines after treatment with 5-azaDC. Understanding epigenetic changes in chordomas may provide insights into chordoma tumorigenesis and development of epigenetic biomarkers.
Keywords: chordomas, DNA Methylation, HumanMethylation450 BeadChips, recurrent, non-recurrent
Introduction
Chordomas are rare, slow growing and locally aggressive notochordal tumors of adults arising from the clivus, sacrum, and coccyx. They account for 1–4% of all primary malignant bone tumors and show dual epithelial-mesenchymal differentiation. Chordomas are divided into three subtypes, conventional, chondroid and dedifferentiated. Conventional chordomas are the most common type and have distinct clinical and pathological features. These tumors primarily spread by local invasion but have the potential to spread to lungs, bone, and liver. Local recurrence rate is high and 5 y survival rate is approximately 60%.1,2 Due to the rarity of the samples and difficulty in obtaining appropriate control tissues, there is a paucity of genetic and, even more, epigenetic studies reported in the literature (reviewed in Yakkioui et al.3 and references within). Expression of brachyury (T) has been proposed as a diagnostic marker for chordomas and plays a critical role in chordoma biology.4,5 Only a limited number of chordoma cell lines have thus far been established; lack of in vitro and in vivo models has hindered the practical translation of the molecular findings.
DNA methylation is now a well-documented aspect of cancer development and hyper- or hypo-methylation of specific gene loci has been shown to be strongly associated with malignancy. For example, the hypermethylation of CpG islands in the promoter region of specific tumor suppressor genes, such as RASSF1A, in a wide range of cancers leads to silencing of the gene.6,7 Gene expression can be restored by treatment with DNA methyltransferase inhibitors such as decitabine (5-aza-2’-deoxycytidine). Decitabine use has been approved by the U.S. Food and Drug Administration for the treatment of myelodysplastic syndromes.
Genome-wide DNA methylation profiles using high-throughput platforms, such as methylation arrays, MeDIP, MIRA, and functional epigenomic approaches have been used for epigenetic profiling of many common cancers, including lung, breast, and colorectal cancer. While large scale epigenomic sequencing equivalent to whole genome sequencing is still too expensive to be used in the majority of laboratories for high-throughput analysis, DNA methylation arrays, such as those developed by Illumina, can be used for larger sample sets at a more reasonable cost.8
In this report, we have used the Infinium HumanMethylation450 BeadChips to compare the DNA methylation profile of chordomas and control tissues and between chordomas that undergo recurrence and those that do not recur, in order to decipher DNA methylation events associated with chordoma development and aggressiveness.
Results
HumanMethylation450 BeadChip array analysis
In order to explore genome-wide DNA methylation profiles of chordomas, we analyzed 26 chordomas plus UCH-1 chordoma cell line and 3 nucleus pulposus samples as controls using the Infinium HumanMethylation450 BeadChips. After array normalization, further criteria were used to remove certain probes, including probes located on X and Y chromosomes, polymorphic probes, and recently described cross-reactive probes (Figure S1). This resulted in retention of 301,576 probes for downstream analysis.
Unsupervised hierarchical clustering of the 1,000 most variable probes clearly split the samples into 2 major clusters, major cluster 1, containing all but one of the chordoma samples and showing higher level of methylation, and major cluster 2, consisting of the 3 control samples and one chordoma sample (Fig. 1).
Figure 1.
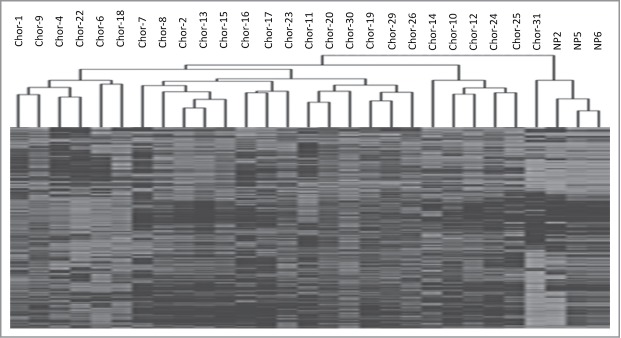
Unsupervised hierarchical cluster analysis of the 1000 most variable probes between chordoma samples and nucleus pulposus samples.
In order to analyze DNA methylation differences between chordomas and controls, we examined the average β-values between both groups for individual CpGs in the retained probe dataset. Probes with adjusted P-value of <0 .05 after FDR correction and >0.2 average β value difference (Δβ-value) between chordomas and controls were considered as differentially methylated loci. A total of 8,819 probes (2.9%) were significantly differentially methylated between chordomas and control samples (Table S1 and S2). Of these 8,819 differentially methylated loci, 5,868 (66.5%) were hypomethylated and 2,951 (33.5%) were hypermethylated (Supplementary Figure S2). We analyzed distribution of the differentially methylated probes based on their genomic context and the location of CpGs (Figs. 2A and 2B). From the 2,951 differentially hypermethylated probes, 33.3% (982 probes) were located in the promoter region (TSS200, TSS1500, 5’UTR, and 1st Exon), followed by 33% located in gene body, and 31% not linked to genes (Fig. 2C). In relation to CGI, 70% were in islands, shores or shelves, and the remaining (30%) were located in open seas (Fig. 2D). In contrast, the majority of hypomethylated loci were located in gene bodies (36.7%) and intragenic regions (36.5%), and 23% of the probes localized to promoter regions (Figure S3). In relation to CGI, the majority (66%) was located in open seas.
Figure 2.
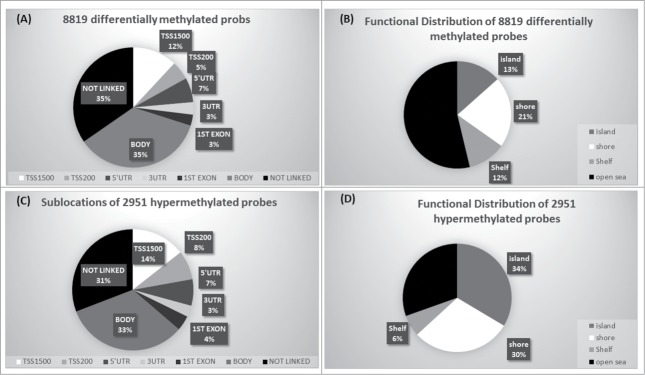
Genomic context of CpG methylation. (A) Differentially methylated probes in relation to gene. (B) Differentially methylated probes in relation to CGI context. (C) Sub-location of differentially hypermethylated probes in relation to gene. (D) Functional distribution of hypermethylated probes in relation to CGI.
Cancer-specific methylation profiles
In order to evaluate tumor specific methylation, probes were considered as hypermethylated if the average β-value was >0.5 in chordomas vs. average β-value <0.25 in control samples. This led to the identification of 364 cancer-specific hypermethylated probes (Table S3). Among these cancer-specific hypermethylated probes, 104 were located within the promoter region, and among these, 74 probes were located within CpG islands and adjacent regions (shore and shelves). The reverse criteria were applied to determine hypomethylated probes in chordoma vs. controls, 29 probes were found to be hypomethylated in chordoma (Table S4). Figure S2 illustrates the stepwise analysis for the Methylation450 BeadChip array to identify cancer-specific hypo- and hyper-methylated loci.
Recurrent vs. non-recurrent chordoma methylation
Interestingly, among our series of chordomas, 13 recurred, while the remaining (n=13) did not. Hypermethylation of gene promoter region CpG island loci is often associated with loss of gene expression and, therefore, likely to be biologically relevant. Hence, we decided to undertake the above analysis using only the 982 probes that were differentially hypermethylated (vs. controls) and were located in the promoter region. This led to the identification of 9 probes corresponding to 8 genes with >0.2 average β-value difference between the 2 groups of chordomas (recurrent vs. non-recurrent) and adjusted P-value of <0 .05 (FAM181B, KANK2, NPR3, PON3, RAB32, RAI1, SLC16A5 and ZNF397OS). These genes have higher average β-value in recurrent chordoma samples compared with non-recurrent samples (> 0.2 average β-value difference) (Table 1).
Table 1.
Differentially methylated genes between recurrent and non-recurrent chordoma samples
| UCSC_REF GENE_NAME | ID | Chromosome | CGI.Relation | Average β-value in recurrence | Average β-value in non-recurrence | Average β-value difference between recurrence and non-recurrence | P-value | Average in controls | UCSC_REF GENE_ACCESSION | Relation to Gene | UCSC_CPG_ISLANDS_NAME |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FAM181B | cg19320476 | chr11 | Island | 0.439897 | 0.235521 | 0.204376 | 0.022539 | 0.046153 | NM_175885 | 1stExon | chr11:82443435–82444976 |
| NPR3 | cg17630392 | chr5 | Island | 0.47289 | 0.261363 | 0.204376 | 0.000756 | 0.079265 | NM_000908 | 1stExon | chr5:32709910–32714450 |
| RAI1 | cg10140454 | chr17 | Island | 0.783044 | 0.549829 | 0.204376 | 0.00584 | 0.372009 | NM_030665 | 5'UTR | chr17:17625605–17628500 |
| SLC16A5 | cg09300114 | chr17 | Island | 0.700374 | 0.474643 | 0.204376 | 0.00878 | 0.127086 | NM_004695 | 5'UTR | chr17:73083866–73084495 |
| ZNF397OS;ZNF397OS | cg16657538 | chr18 | Island | 0.493249 | 0.215783 | 0.204376 | 0.002872 | 0.051907 | NM_001166012;NM_001112734 | 5'UTR;5'UTR | chr18:32847284–32848130 |
| KANK2;KANK2 | cg07888912 | chr19 | North Shore | 0.58871 | 0.344717 | 0.204376 | 0.000285 | 0.081067 | NM_001136191;NM_015493 | 5'UTR;TSS1500 | chr19:11307495–11308276 |
| RAB32 | cg23267550 | chr6 | North Shore | 0.566886 | 0.35041 | 0.204376 | 0.004754 | 0.216715 | NM_006834 | TSS1500 | chr6:146864481–146865426 |
| PON3 | cg08520743 | chr7 | Island | 0.564437 | 0.344416 | 0.204376 | 0.001623 | 0.052802 | NM_000940 | TSS200 | chr7:95025559–95026122 |
| PON3 | cg08828819 | chr7 | Island | 0.603982 | 0.393397 | 0.204376 | 0.005161 | 0.051629 | NM_000940 | TSS200 | chr7:95025559–95026122 |
Treatment of chordoma cell lines with 5-aza-2’-deoxycytidine and gene expression analysis
Chordoma cell lines (UCH-1 and MUG-Chor1) were obtained from the Chordoma Foundation and grown according to the recommended protocols and treated with 5-aza-2’-deoxycytidine (5-azaDC) as described previously (see Materials and Methods). A total of 18 genes were selected in order to analyze the expression of these genes in UCH-1 and MUG-Chor1 chordoma cell lines before and after treatment with the demethylating agent 5-azaDC using RT-PCR. Among these genes, 7 genes (COL11A2, NEFM, SCT, FOXP4, FAIM2, IFNGR2, and TUBB6) were from the differentially hypermethylated promoter region probe list, 3 genes were from the cancer-specific differentially methylated gene list, and 8 genes (FAM181B, KANK2, NPR3, PON3, RAB32, RAI1, SLC16A5, and ZNF397OS) were differentially methylated between recurrent and non-recurrent chordoma samples. RT-PCR analysis showed a good correlation with the array methylation results as these genes showed a high β-value (>0.6) for the UCH-1 cell line from the HumanMethylation450 BeadChips and they were upregulated or re-expressed after 5-azaDC treatment in UCH-1 cell line (Fig. 3A and Figure S4). Methylation status of the MUG-Chor1 cell line was determined using combined bisulfite restriction analysis (COBRA), since this cell line was not included in the HumanMethlation450 BeadChip array hybridization. The above results demonstrate that the hypermethylation of these genes is associated with loss of (or reduced) expression and the expression can be restored (or upregulated) after 5-azaDC treatment.
Figure 3.
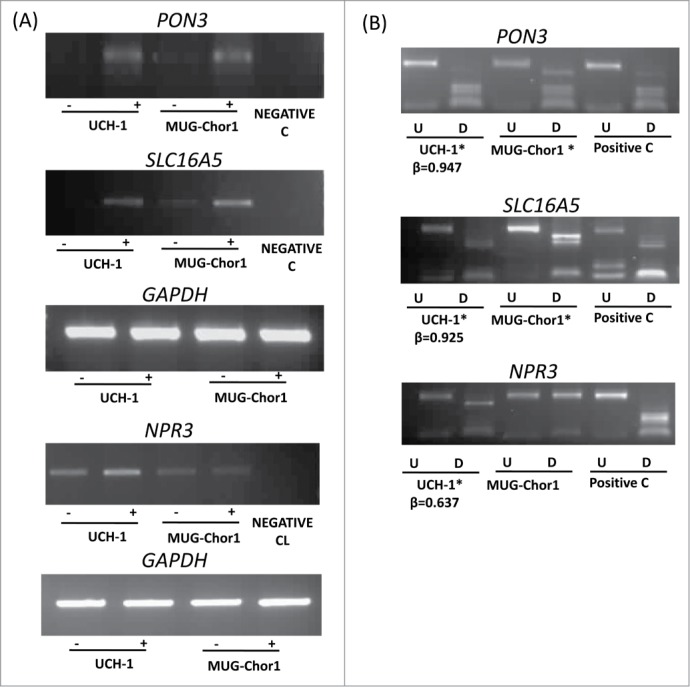
Expression and methylation analysis in ES cell lines. Expression and methylation results for 3 selected genes in chordoma cell lines (PON3, SLC16A5, and NPR3; GAPDH as control). (A) Expression analysis using RT-PCR in 2 chordoma cell lines are shown for each gene with (-) indicating the samples without 5-azaDC treatment and (+) indicating the samples after 5-azaDC treatment. (B) COBRA results for the selected genes in cell lines. Undigested (U) samples were loaded next to the BstU1 digested samples (D). Two chordoma cell lines are shown for each gene and the positive control (Positive C) is in vitro methylated DNA. (*) indicates that the cell line is methylated. β-values from the HumanMethylation450 BeadChip array for UCH-1 cell line is also shown under each digest.
Validation of the HumanMethylation450 BeadChip array
Confirmation of the HumanMethylation450 BeadChip data was carried out in chordoma cell lines and samples using COBRA. A total of 7 genes (FAM181B, NPR3, PON3, RAB32, RAI1, SLC16A5, and ZNF397OS) from the differentially methylated gene list between recurrent and non-recurrent chordoma samples were analyzed (Figs. 3B and 4A). In addition, 8 other genes from our 982 genes list of differentially hypermethylated probes in the promoter region were analyzed in chordoma samples and 4 of these genes were from the cancer-specific probe list (Figs. 4B and 4C). Our in house methylation analysis confirms that high β-values correspond to methylation and low β-values correspond to low or no methylation (Fig. 4). The methylation status was further confirmed by clone sequencing of bisulfite modified DNA for specific loci (Figure S5).
Figure 4.

COBRA analysis for selected genes in chordoma samples. (A) Results of COBRA analysis for 5 genes (RAB32, FAM181B, SLC16A5, NPR3, and PON3) selected from differentially methylated gene list between recurrent and non-recurrent chordoma samples. (B) Results of COBRA analysis for 2 genes (COL11A2 and TUBB6) from differentially methylated probe list. (C) Results of COBRA analysis for 2 genes (FGR and PSKH2) selected from cancer-specific hypermethylated probe list. COBRA results are shown for chordoma samples with in vitro methylated DNA as positive control (Positive C). The digested samples (D) are loaded next to the undigested samples (U). (*) indicates methylated samples. β-value from the HumanMethylation450 BeadChip array for each selected sample is shown.
Ingenuity Pathway Analysis
Ingenuity Pathway Analysis (IPA) was conducted for the 104 hypermethylated cancer-specific probes within the promoter region to identify the networks and interactions between hypermethylated genes in chordomas. IPA revealed 9 significant networks (Table S5). Network 1 contains 13 genes from the cancer-specific hypermethylated probes involved in cell-to-cell signaling and interaction, cellular function and maintenance, and tissue development. Network 2 contains 13 focus molecules from our list involved in cancer disease, nervous system development and function, cell death, and survival. Moreover, Network 3 has 12 genes that are hypermethylated in chordoma and this network is responsible for different functions, including cellular growth, cellular development, and proliferation. The fourth network contains 11 molcules from our list; these are associated with DNA replication, repair and recombination, cell cycle, cellular assembly, and organization (Figure S6). Each network from Networks 5–9 contains only one molecule from our hypermethylated list and they are involved in different biological functions and diseases including cancer.
Discussion
Current treatment options for chordomas are limited and surgical excision is the main curative route; however, excision is often difficult to perform due to the anatomic location of the tumor. Patients who relapse with local recurrence have a poor prognosis. Early detection is a key to better management of the disease and increasing number of molecular studies are being undertaken using chordoma tissues and the few chordoma cell lines that have been developed and are available to the scientific community.9,10 Patient-derived chordoma xenografts have also been recently developed for preclinical assessment of novel therapies and drug testing.11-14
Increasingly, DNA methylation has provided a wealth of information in establishing useful biomarkers for cancer diagnosis, prognosis and disease monitoring. In addition, epigenetic therapy is being actively pursued for haematological malignancies and more recently has been targeted towards solid tumors.15,16 Especially, DNA hypermethylation of CpG rich regions in the promoter region of tumor suppressor genes has been established as an alternative mechanism for silencing of tumor suppressor genes in many cancer types. RASSF1A and HIC1 tumor suppressor genes are among the most significantly hypermethylated genes in human cancer.17,18 In the present report, our aim was to determine methylation profiles of chordomas in relation to control tissue and to identify tumor specific hypermethylated gene loci in locally aggressive and non-aggressive chordomas.
We report one of the first major genome-wide DNA methylation analyses of chordomas, a rare bone tumor, in relation to nucleus pulposus control samples using the latest Illumina Infinium HumanMethylation450 BeadChips. We also provide a comparison between chordomas that undergo local recurrence and, hence, are more difficult to manage clinically and chordomas that do not recur. Chordomas are postulated to arise from the ‘notochordal remnant’ cells, which are derived from the embryonic notochord. Recent studies in mice have shown that embryonic notochord directly give rise to all cell types present in the nucleus pulposus.19 In the present study, we decided to use nucleus pulposus as the control tissue, since it is very difficult to get embryonic notochord cells.
We demonstrated that a large set of gene loci show differential methylation between chordomas and controls. In addition, a subset shows cancer-specific hypermethylation in chordomas and, hence, may be developed into diagnostic markers for early detection and early intervention for disease management. A recent study by Rinner et al.20 identified 20 genes (15 hyper- and 5 hypo-methylated) that showed significant differential methylation between chordomas and blood from healthy individuals. Four of the hypermethylated genes (RASSF1, IRF4, DLEC1, and HSD17B4) were also found in our significantly differentially hypermethylated gene set when comparing chordomas vs. nucleus pulposus samples, but were not among our cancer-specific hypermethylated gene set.
IPA analysis revealed that the cancer-specific hypermethylated gene loci fell into 9 significant networks, many of which are relevant to cancer development. Examples of genes associated with cancer include NFI; mutations in this gene cause neurofibromatosis type 1 disease associated with benign neurofibromas and malignant tumors of the central and peripheral nervous system. The protein product of NF1 gene, neurofibromin, is a multifunctional protein involved in many cellular signaling pathways, including RAS/MAPK and mTOR pathways, both of which are dysregulated in many cancer types.21 CTNNB1, Wnt/β-catenin, is a highly complex pathway and plays a vital role in bone development and homeostasis and is dysregulated in many human malignancies, including bone cancers and, especially, osteosarcomas.22
Although on a small sample set, we were able to identify a preliminary set of 9 probes (8 gene loci: FAM181B, KANK2, NPR3, PON3, RAB32, RAI1, SLC16A5, and ZNF397OS) that were differentially methylated between control samples and chordomas that recurred and tumors that did not recur. Eight of the 9 probes were unmethylated in the control samples (average β-value <0.25) and all 9 showed higher average β-values in chordomas that recurred compared to chordomas that did not recur and controls. Seven of the probes were located in gene promoter region CpG islands and 2 were located in CGI shores and, hence, likely to be associated with gene expression. Our data demonstrated that these genes were re-expressed and/or upregulated in methylated chordoma cell lines after treatment with 5-azaDC. These probes may represent markers for chordoma aggressiveness if confirmed in a larger number of samples. Among these loci is KANK2, which belongs to a family of ankyrin-repeat domain containing proteins (KANK1-KANK4). KANK1, the first identified member of this gene family, is a candidate tumor suppressor gene for kidney cancer and was shown to be involved in gene fusion in a case of myeloproliferative neoplasm.23 Northcott et al. have shown that immunohistochemistry analysis using a set of 4 antibodies (DKK1, SFRP1, NPR3, and KCNA1) can reliably divide medulloblastomas into 4 subgroups; furthermore, progression-free and overall survival of patients with NPR3 positive tumors was significantly reduced.24 PON3 belongs to the paraoxonase family of proteins (PON1, PON2, PON3). All 3 proteins prevent oxidative stress and have anti-inflammatory properties and are implicated in pathogenesis of several inflammatory diseases including cancer.25
In summary, we have identified a preliminary set of differential and cancer-specific methylated loci in chordomas that may be useful for future research into this rare cancer type.
Materials and Methods
Patient DNA and RNA samples
Chordoma samples (n=26) DNA and RNA was isolated as described previously by Gharani et al.26 The samples used in the study were anonymized. The study was ethically approved and followed institutional guidelines. The samples were obtained from a National Research Ethics Committee-approved tissue bank and patients gave informed consent. The study was conducted according to the principles expressed in the Helsinki Declaration of 1975. Among the 26 chordomas used in this study, 10 were from female and 16 were from male patients. The chordomas were conventional type and were located in the sacrum/pelvis. The age at diagnosis ranged from 37 to 74 y Thirteen patients developed a chordoma recurrence during the follow-up period and 6 had died of the disease, 6 were alive with disease, 11 had no evidence of disease and 2 had died of other causes. All were positive for brachyury. Nucleus pulposus (NP) specimens were collected from intervertebral disks harvested during discectomy of 3 patients affected by spine disorders, after informed consent and Institutional Review Board approval of Galeazzi orthopedic Institute, Milan, Italy.
Chordoma cell lines
Two chordoma cell lines (UCH-1 and MUG-Chor1)9,10 were kindly provided by the Chordoma Foundation. UCH-1 was maintained in IMDM (Invitrogen 12400)/RPMI 1,640 (Sigma R8758) medium (4 IMDM: 1 RPMI) with 10% FBS and 1% PenStrep at 37°C, 5% CO2. MUG-Chor1 was maintained in IMDM/RPMI (4:1) with 2 mM L-Glutamine, 1% ITS and 1% PenStrep at 37°C, 5% CO2. The cell lines were treated with 5-aza-2′-deoxycitidine (5-azaDC) over 5 d as described in Alholle et al.27
RT-PCR, bisulfite modification and COBRA
Superscript III cDNA synthesis kit (Invitrogen) was used to create cDNA from 1 μg RNA. RT-PCR was performed as described previously.27 Bisulfite modification of chordoma samples and cell lines was performed using Qiagen EpiTect kit (Qiagen) according to the manufacturer's instructions. COBRA analysis was performed as described previously in Alholle et al.27 Expression and Methylation primers can be provided upon request.
HumanMethylation450 BeadChips
High quality DNA (1 μg) from each sample was sent to Cambridge Genomic Services (Cambridge, UK). DNA bisulfite conversion and experimental processing of HumanMethylation450 BeadChips was carried out by Cambridge Genomic Services. The “.idat” files generated by the iScan scanner were processed using the RnBeads pipeline.28 The pipeline includes pre-processing with QC of the data to remove poor quality samples, normalization using the BMIQ normalization,29 and post filtering removing known problematic probes (containing 2 SNPs, probe below the detection threshold, etc.).
Statistical analysis and data processing
All the data processing and statistical analysis were conducted using Microsoft Excel 2013. Student t-test with FDR correction was performed to calculate the significance (P < 0.05 was considered significant).
Ingenuity Pathway Analysis
The Ingenuity Pathway Analysis (IPA) program was used to identify pathways, networks, and functional categories for genes of interest (http://www.ingenuity.com/products/ipa).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Chordoma cell lines were kindly provided by the Chordoma Foundation.
Funding
This work was supported by Bone Cancer Research Trust; Kuwait Medical Genetics Center (KMGC), Ministry of Health, Kuwait and the Royal orthopedic Hospital NHS Foundation trust, Birmingham, England. We would also like to acknowledge Ministero della Salute of Italy for financial support (RC-L2013 and RC-L2029).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Walcott BP, Nahed BV, Mohyeldin A, Coumans JV, Kahle KT, Ferreira MJ. Chordoma: current concepts, management, and future directions. Lancet Oncol 2012; 13:e69-76; PMID:22300861; http://dx.doi.org/ 10.1016/S1470-2045(11)70337-0 [DOI] [PubMed] [Google Scholar]
- 2. Williams BJ, Raper DM, Godbout E, Bourne TD, Prevedello DM, Kassam AB, Park DM. Diagnosis and treatment of chordoma. J Natl Compr Canc Netw 2013; 11:726-31; PMID:23744869 [DOI] [PubMed] [Google Scholar]
- 3. Yakkioui Y, van Overbeeke JJ, Santegoeds R, van Engeland M, Temel Y. Chordoma: the entity. Biochim Biophys Acta 2014; 1846:655-69; PMID:25193090 [DOI] [PubMed] [Google Scholar]
- 4. Vujovic S, Henderson S, Presneau N, Odell E, Jacques TS, Tirabosco R, Boshoff C, Flanagan AM. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol 2006; 209:157-65; PMID:16538613; http://dx.doi.org/ 10.1002/path.1969 [DOI] [PubMed] [Google Scholar]
- 5. Presneau N, Shalaby A, Ye H, Pillay N, Halai D, Idowu B, Tirabosco R, Whitwell D, Jacques TS, Kindblom LG, et al. . Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: a genetic and functional-based study. J Pathol 2011; 223:327-35; PMID:21171078; http://dx.doi.org/ 10.1002/path.2816 [DOI] [PubMed] [Google Scholar]
- 6. Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 2012; 13:679-92; PMID:22945394; http://dx.doi.org/ 10.1038/nrg3270 [DOI] [PubMed] [Google Scholar]
- 7. Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta 2009; 1796:114-28; PMID:19344752 [DOI] [PubMed] [Google Scholar]
- 8. Stirzaker C, Taberlay PC, Statham AL, Clark SJ. Mining cancer methylomes: prospects and challenges. Trends Genet 2014; 30:75-84; PMID:24368016; http://dx.doi.org/ 10.1016/j.tig.2013.11.004 [DOI] [PubMed] [Google Scholar]
- 9. Scheil S, Brüderlein S, Liehr T, Starke H, Herms J, Schulte M, Möller P. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer 2001; 32:203-11; PMID:11579460; http://dx.doi.org/ 10.1002/gcc.1184 [DOI] [PubMed] [Google Scholar]
- 10. Rinner B, Froehlich EV, Buerger K, Knausz H, Lohberger B, Scheipl S, Fischer C, Leithner A, Guelly C, Trajanoski S, Szuhai K, Liegl B. Establishment and detailed functional and molecular genetic characterisation of a novel sacral chordoma cell line, MUG-Chor1. Int J Oncol 2012; 40:443-51; PMID:22002331 [DOI] [PubMed] [Google Scholar]
- 11. Siu IM, Salmasi V, Orr BA, Zhao Q, Binder ZA, Tran C, Ishii M, Riggins GJ, Hann CL, Gallia GL. Establishment and characterization of a primary human chordoma xenograft model. J Neurosurg 2012;116:801-9; PMID:22283186; http://dx.doi.org/ 10.3171/2011.12.JNS111123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trucco MM, Awad O, Wilky BA, Goldstein SD, Huang R, Walker RL, Shah P, Katuri V, Gul N, Zhu YJ, et al. . A novel chordoma xenograft allows in vivo drug testing and reveals the importance of NF-κB signaling in chordoma biology. PLoS One 2013; 8:e79950; PMID:24223206; http://dx.doi.org/ 10.1371/journal.pone.0079950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karikari IO, Gilchrist CL, Jing L, Alcorta DA, Chen J, Richardson WJ, Gabr MA, Bell RD, Kelley MJ, Bagley CA, et al. . Molecular characterization of chordoma xenografts generated from a novel primary chordoma cell source and two chordoma cell lines. J Neurosurg Spine 2014; 21:386-93; PMID:24905390; http://dx.doi.org/ 10.3171/2014.4.SPINE13262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bozzi F, Manenti G, Conca E, Stacchiotti S, Messina A, Dagrada G, Gronchi A, Panizza P, Pierotti MA, Tamborini E, Pilotti S. Development of transplantable human chordoma xenograft for preclinical assessment of novel therapeutic strategies. Neuro Oncol 2014; 16:72-80; PMID:24366975; http://dx.doi.org/ 10.1093/neuonc/not152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest 2014; 124:56-63; PMID:24382390; http://dx.doi.org/ 10.1172/JCI69736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heyn H, Méndez-González J, Esteller M. Epigenetic profiling joins personalized cancer medicine. Expert Rev Mol Diagn 2013;13:473-9; PMID:23782254; http://dx.doi.org/ 10.1586/erm.13.36 [DOI] [PubMed] [Google Scholar]
- 17. Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 2000; 25:315-9; PMID:10888881; http://dx.doi.org/ 10.1038/77083 [DOI] [PubMed] [Google Scholar]
- 18. Wales MM, Biel MA, el Deiry W, Nelkin BD, Issa JP, Cavenee WK, Kuerbitz SJ, Baylin SB. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med 1995; 1:570-7; PMID:7585125; http://dx.doi.org/ 10.1038/nm0695-570 [DOI] [PubMed] [Google Scholar]
- 19. Choi KS, Cohn MJ, Harfe BD. Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: implications for disk degeneration and chordoma formation. Dev Dyn 2008; 237:3953-8; PMID:19035356; http://dx.doi.org/ 10.1002/dvdy.21805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rinner B, Weinhaeusel A, Lohberger B, Froehlich EV, Pulverer W, Fischer C, Meditz K, Scheipl S, Trajanoski S, Guelly C, Leithner A, Liegl B. Chordoma characterization of significant changes of the DNA methylation pattern. PLoS One 2013; 8:e56609; PMID:23533570; http://dx.doi.org/ 10.1371/journal.pone.0056609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sabova L, Kretova M, Luciakova K. New insights into the role of NF1 in cancer. Neoplasma 2013; 60:233-9; PMID:23373991; http://dx.doi.org/ 10.4149/neo_2013_031 [DOI] [PubMed] [Google Scholar]
- 22. Lin CH, Ji T, Chen CF, Hoang BH. Wnt signaling in osteosarcoma. Adv Exp Med Biol 2014; 804:33-45; PMID:24924167; http://dx.doi.org/ 10.1007/978-3-319-04843-7_2 [DOI] [PubMed] [Google Scholar]
- 23. Medves S, Duhoux FP, Ferrant A, Toffalini F, Ameye G, Libouton JM, Poirel HA, Demoulin JB. KANK1, a candidate tumor suppressor gene, is fused to PDGFRB in an imatinib-responsive myeloid neoplasm with severe thrombocythemia. Leukemia 2010; 24:1052-5; PMID:20164854; http://dx.doi.org/ 10.1038/leu.2010.13 [DOI] [PubMed] [Google Scholar]
- 24. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, et al. . Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 2011; 29:1408-14; PMID:20823417; http://dx.doi.org/ 10.1200/JCO.2009.27.4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Devarajan A, Shih D, Reddy ST. Inflammation, infection, cancer and all that…the role of paraoxonases. Adv Exp Med Biol 2014; 824:33-41; PMID:25038992; http://dx.doi.org/ 10.1007/978-3-319-07320-0_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gharanei S, Brini AT, Vaiyapuri S, Alholle A, Dallol A, Arrigoni E, Kishida T, Hiruma T, Avigad S, Grimer R, et al. . RASSF2 methylation is a strong prognostic marker in younger age patients with Ewing sarcoma. Epigenetics 2013; 8:893-8; PMID:23887284; http://dx.doi.org/ 10.4161/epi.25617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alholle A, Brini AT, Gharanei S, Vaiyapuri S, Arrigoni E, Dallol A, Gentle D, Kishida T, Hiruma T, Avigad S, et al. . Functional epigenetic approach identifies frequently methylated genes in Ewing sarcoma. Epigenetics 2013; 8:1198-204; PMID:24005033; http://dx.doi.org/ 10.4161/epi.26266 [DOI] [PubMed] [Google Scholar]
- 28. Assenov Y, Müller F, Lutsik P, Walter J, Lengauer T, Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods 2014; 11:1138-40; PMID: 25262207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013; 29:189-96; PMID:23175756; http://dx.doi.org/ 10.1093/bioinformatics/bts680 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.