

Summary
Self-avoidance, a process preventing interactions of axons and dendrites from the same neuron during development, is mediated in vertebrates through the stochastic single-neuron expression of clustered protocadherin protein isoforms. Extracellular cadherin (EC) domains mediate isoform-specific homophilic binding between cells, conferring cell recognition through a poorly understood mechanism. Here, we report crystal structures for the EC1-EC3 domain regions from four protocadherin isoforms representing the α, β and γ subfamilies. All are rod-shaped and monomeric in solution. Biophysical measurements, cell aggregation assays, and computational docking reveal that trans binding between cells depends on the EC1-EC4 domains, which interact in an antiparallel orientation. We also show that the EC6 domains are required for the formation of cis-dimers. Overall, our results are consistent with a model in which protocadherin cis-dimers engage in a head-to-tail interaction between EC1-EC4 domains from apposed cell surfaces, possibly forming a zipper-like protein assembly thus providing a size-dependent self-recognition mechanism.
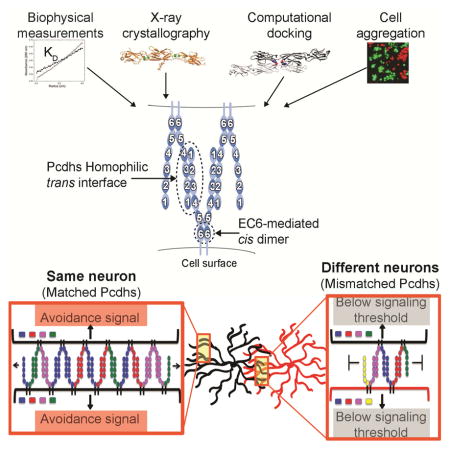
Graphical Abstract
Introduction
The human brain is comprised of approximately 10 billion neurons, each of which can connect with up to thousands of others. Neuronal self-avoidance is a process in which dendrites and axons originating from the same neuron repel one another, but can freely interact with neurites from other neurons. The combined properties of self-recognition and non-self discrimination require that contacting neurons display diverse cell surface identities that allow for discrimination between self and non-self (Hattori et al., 2009; Zipursky and Grueber, 2013; Zipursky and Sanes, 2010).
In Drosophila and other invertebrates self-avoidance is mediated by Dscam1 proteins; immunoglobulin superfamily members produced by alternative splicing of the DSCAM1 pre-mRNA. This cell-autonomous and stochastic alternative splicing can theoretically produce up to 19,008 Dscam1 isoforms with distinct ectodomains, each of which have highly specific homophilic trans binding specificity (Hattori et al., 2008; Miura et al., 2013; Schmucker et al., 2000; Wojtowicz et al., 2007). Distinct cell surface identities are generated in Drosophila by the stochastic expression of a small set of Dscam1 isoforms in each neuron (Miura et al., 2013). Homophilic interactions between identical sets of protein isoforms on the surface of neurites from the same neuron result in repulsion and neurite self-avoidance (Hattori et al., 2008). The expression of even a single Dscam1 isoform is sufficient for self-avoidance of neurites from the same neuron (Hughes et al., 2007; Matthews et al., 2007; Soba et al., 2007). However, robust non-self discrimination, which allows processes from different neurons to freely interact, requires thousands of distinct Dscam1 isoforms (Hattori et al., 2009).
Recent studies suggest that in vertebrate nervous systems neuronal self-avoidance functionality is provided, at least in part, by the clustered protocadherins (Pcdhs) (Chen and Maniatis, 2013; Zipursky and Grueber, 2013; Zipursky and Sanes, 2010). Mammalian Pcdhs are encoded in a contiguous genomic locus comprised of three adjacent gene clusters (Pcdh α, β and γ), each of which contains close to 60 “variable” exons (58 in mice, Figure 1A) (Wu and Maniatis, 1999). Only a few variable exons are stochastically chosen for expression in each cell by a mechanism involving alternative promoter choice (Ribich et al., 2006; Tasic et al., 2002). Each variable exon encodes an entire Pcdh ectodomain region consisting of six tandem extracellular cadherin (EC) domains, a single transmembrane region, and a short cytoplasmic region. In the α and γ gene clusters, a “constant” C-terminal cytoplasmic region encoding an intracellular domain (ICD) is joined to the variable ectodomain exon by pre-mRNA splicing. The β cluster does not contain such a constant region and therefore β-Pcdhs are lacking an ICD. The α and γ gene clusters also encode a small set of “C-type” Pcdhs, which are divergent from other members of their respective clusters, and appear to have distinct functions (Figure 1A) (Chen et al., 2012). Deletion of the Pcdhγ gene cluster in mice leads to the disruption of self-avoidance in retinal starburst amacrine cells and Purkinje cells with phenotypes similar to those described for Dscam1 deletion mutants in Drosophila (Lefebvre et al., 2012).
Figure 1. Crystal structures of four Pcdh EC1-EC3 isoforms.

A) The Pcdh genomic locus contains three adjacent clusters of variable exons. Each exon encodes an entire ectodomain comprising six EC domains, a transmembrane (TM) domain, and a short cytoplasmic region. Alpha and gamma clusters also contain three constant exons that encode a cluster-specific intracellular domain (ICD) which are joined by pre-mRNA splicing for alpha and gamma clusters. C-type Pcdh exons are shown in pink and light blue for the alpha and gamma clusters, respectively.
B) Crystal structures of EC1-EC3 regions from PcdhαC2, Pcdhβ1, PcdhγA8, and PcdhγC5 shown in ribbon representation. Ca2+ ions are drawn as green spheres. N-glycans and conserved O-mannose residues are drawn as sticks. The inter-domain calcium binding sites are arranged similarly to those observed in classical cadherins (expanded view). See also Figure S1 and Table S1.
C) Comparison of the PcdhγC5 and type I classical C-cadherin structures. The overall architecture of classical cadherin ectodomains have a curved shape with an approximate 90° angle between EC1 and EC5 (Boggon et al., 2002). In contrast, the architecture of Pcdh EC1-EC3 domain regions is characterized by an extended zigzagged conformation.
D) EC2-EC3 angles distinct from classical cadherins account for the extended zigzagged conformation of the Pcdh structures. EC1-EC3 domains are drawn as blue (PcdhγC5) and yellow (C-cadherin) ovals. Angles shown are between principal axes of inertia for adjacent domains.
Like invertebrate Dscam proteins, Pcdh isoforms engage in isoform-specific trans homophilic interactions (Schreiner and Weiner, 2010; Thu et al., 2014). It is remarkable that Pcdhs, with only 58 isoforms, can mediate neural self-recognition and non-self discrimination similar to Dscams, which have up to tens of thousands of distinct extracellular isoforms. Central to this capability is the observation that a single mismatched Pcdh isoform can interfere with recognition between cells that express an otherwise matching set of Pcdhs (Thu et al., 2014). Understanding the mechanism underlying this “interference” phenomenon is crucial, as it is likely to explain how only 58 Pcdh isoforms can provide sufficient functional diversity to enable self-recognition and non-self discrimination in the nervous system comparable to the much more diverse Drosophila Dscam gene.
Here we report crystal structures of Pcdh extracellular protein fragments comprising the previously mapped Pcdh specificity-determining EC1-EC3 domains for PcdhαC2, Pcdhβ1, PcdhγA8, and PcdhγC5 isoforms, thus providing examples from all three Pcdh gene clusters. Guided by these structures we used two orthogonal mutagenesis approaches – surface saturating arginine mutagenesis and bioinformatics-derived predictions – to map the isoform specificity-determining regions at the amino acid level using cell aggregation and biophysical experiments as readouts. The two approaches yielded consistent results, revealing an essential role for EC1 through EC4 in trans homophilic interactions and for EC6 in cis interactions. On the basis of these findings we propose a model for Pcdh mediated cell-cell recognition that is consistent with the remarkable ability of these cell surface proteins to provide diverse single-cell identities to vertebrate neurons.
Results
Structures of Pcdh EC1-EC3 region fragments from α, β and γ sub-families
We determined crystal structures of proteins composed of the three N-terminal EC domains of mouse PcdhαC2, Pcdhβ1, PcdhγA8, and PcdhγC5 to a resolution of 2.4 Å, 3.3 Å, 2.9 Å and 2.9 Å, respectively (Figure 1B, Table S1). We focused on protein fragments containing EC1-EC3, since the results of earlier cell aggregation experiments indicated that Pcdh isoform-specific recognition was mediated via the EC2-EC3 domains and that the EC1 domain is required for trans binding (Schreiner and Weiner, 2010).
The four structures show high overall similarity (Figures 1B and S1A). Each structure consists of three EC domains, each with the two-layer β-sheet fold observed in classical cadherins. Successive domains are connected by calcium-binding linkers, each of which coordinate three Ca2+ ions utilizing side chains in the same conserved motifs (Figure 1B). These motifs are also conserved within type-I and type-II classical cadherins with the exception of the EE motif (bottom of EC1 domain, Figure 1B), which is present only in type-II cadherins. In contrast with previous conclusions (Schreiner and Weiner, 2010), but consistent with the presence of Ca2+ at the inter-domain linkers and in common with classical cadherins, we have found that cell aggregation of Pcdhs is Ca2+ dependent (Figure S1B). Despite these similarities to classical cadherins, the Pcdh isoform structures are distinctive in several aspects. Most notably, the overall arrangement of the three EC domains in each structure is much straighter than the curved classical cadherin architecture (Figure 1C). This “straight-rod” architecture arises from an extended zigzagged conformation: an arrangement that is generated primarily by a very different EC2-EC3 angle than classical cadherins (>131° difference, Figure 1D).
In addition, mass spectrometry analyses showed that all four isoforms contain two sites of O-mannosylation at residues 194 and 196 (PcdhγC5 sequence numbering, Figures 1B and S1 panels G and H). These positions are conserved in sequence among most Pcdh isoforms (Fig. S1G) and among classical cadherins (Vester-Christensen et al., 2013), suggesting these O-glycans play important functional roles. O-mannosylation of cadherins and protocadherins were recently discovered (Vester-Christensen et al., 2013), and it was further shown that O-mannosylation of E-cadherin is essential for preimplantation development of the mouse embryo (Lommel et al., 2013).
The Pcdh structures show local Pcdh-specific embellishments on the EC domain fold. In particular Pcdh EC1 domains show a number of differences from vertebrate cadherin EC1 domains (Figure S1D), as was previously observed in NMR structures of Pcdhα4 and Pcdhβ14 EC1 domains (Morishita et al., 2006). The A-strand is shorter than that of classical cadherins and lacks the conserved Trp-2 residue, which anchors the strand-swap trans-binding interface of classical cadherins (Figures S1C and S1D; Posy et al., 2008). The EC1 EF loop region in each of the Pcdh structures contains a disulfide-constrained loop formed by a Pcdh-specific CX5C motif. The EC2 and EC3 domains of the Pcdh structures are each most similar to either the EC1 or EC2 domain from the atypical cadherin-23 (RMSD 1.5 and 1.2 Å). However, the D and E strands of Pcdh EC2 domains, and the CD loop region of EC3, are significantly longer than found in cadherin-23 or in classical cadherins (Figure S1E). There are also distinctive differences among the structures of the four Pcdh isoforms. The EC1 BC loop helix, C strand and CD loop regions display distinct conformations in all four structures (Figure S1F). In EC3 the two C-type structures (PcdhαC2 and PcdhγC5) have a longer FG loop than Pcdhβ1 and PcdhγA8, a feature conserved among α and C-type Pcdhs (Figure S1F).
Analysis of the molecular packing of the four Pcdh EC1-EC3 structures revealed different crystallographic contacts for each isoform, with no interfaces in common. Interfaces exhibiting typical protein-protein interface attributes were not identified in any of the crystal forms analyzed.
Analytical ultracentrifugation and cell aggregation assays define the multimeric structure of Pcdhs
We expressed and purified proteins from a C-terminal deletion series comprising EC1-EC6, EC1-EC5, EC1-EC4, and EC1-EC3, and a construct comprising domains EC2-EC6 where EC1 was deleted. Using AUC we assessed the oligomerization state of each of these ectodomain fragments in solution. With the exception of PcdhγA8, all EC1-EC3 Pcdh isoform fragments behaved as monomers (Table 1A). This finding was consistent with our crystal structures in which no apparent binding interfaces were detected. The PcdhγA8 EC1-EC3 fragment formed a disulfide-linked dimer through cysteine 283 in the EC3 domain (Figure S2A–B); however, this disulfide bond is likely artifactual since it is not detected in the larger PcdhγA8 isoform fragment (EC1-EC4) (Table 1A).
Table 1.
| Table 1A. Analytical ultracentrifugation analysis of clustered-protocadherins homodimerization | ||
|---|---|---|
| Protein | Oligomeric state | KD dimerization (μM) |
| α7EC1-EC3 | Monomer | NA |
| αC2EC1-EC3 | Non-specific dimer | 242 ± 0.1a |
| β1EC1-EC3 | Monomer | NA |
| γA8EC1-EC3 | Disulfide-linked dimer | NA |
| γC5EC1-EC3 | Monomer | NA |
| γC5EC1-EC3 extended N-term |
Monomer | NA |
|
| ||
| α7EC1-EC5 | Dimer | 2.9 ± 0.5 |
| αC2EC1-EC4 | Dimer | 20 ± 1.2 |
| αC2EC1-EC5 | Dimer | 5.9 ± 0.8 |
| γC5EC1-EC5 | Dimer | 100 ± 4.3 |
| γB6EC1-EC4 | Dimer | 29 ± 4.9 |
| γA8EC1-EC4 | Dimer | 30 ± 1.5 |
|
| ||
| γC5EC2-EC6 | Dimer | 18 ± 0.2 |
| γA8EC2-EC6 | Dimer | 23 ± 8.1 |
| αC2EC2-EC6 | Dimer | 8.9 ± 0.3 |
|
| ||
| αC2EC1-EC6 | Tetramer | 0.1b |
| γC5EC1-EC6 | Tetramer | 7.6b |
| γB6EC1-EC6 | Tetramer | 0.2 |
| Table 1B. Pcdhs form independent trans and cis interactions | ||
|---|---|---|
| Protein | Oligomeric state | KD dimerization (μM) |
| γA8EC1-EC4 | Dimer | 30 |
| γA8EC1-EC4 I116R | Monomer | NA |
|
| ||
| γC5EC2-EC6 | Dimer | 18 |
| γC5EC2-EC6 S116R | Dimer | 14 |
|
| ||
| αC2EC1-EC6 | Tetramer | 0.1b |
| αC2EC1-EC6 S118R | Tetramer | 1.8b |
| γC5EC1-EC6 | Tetramer | 7.6b |
| γC5EC1-EC6 S116R | Dimer | 5.7 |
n=2 Isodesmic Ki=359 μM, Ki/KD=1.48
KD of a tetramer was obtained by locking the Cis-interaction KD as obtained from EC 2–6 deletion constructs.
In contrast to monomeric EC1-EC3 fragments, EC1-EC4 or EC1-EC5 Pcdh fragments were observed to self-associate as dimers with dissociation constants (KD) in the micromolar range (2.9 – 100 μM) that varied significantly between isoforms (Table 1A). The EC1-deleted constructs comprising domains EC2-EC6 also formed homodimers in solution, with KD values in the low micromolar range (8.9 – 23 μM). Importantly, AUC measurements for complete ectodomains including EC1-EC6 could be fit only to a tetramer (dimer-of-dimers) model, indicating a crucial role for the EC6 domain in Pcdh association (Table 1A).
We expressed similarly truncated Pcdhs in K562 cells and assessed their ability to mediate cell aggregation. K562 cells provide a robust assay for Pcdh cell-cell recognition, as they do not express endogenous Pcdhs and do not spontaneously aggregate in liquid culture (Reiss et al., 2006; Schreiner and Weiner, 2010; Thu et al., 2014). Cells expressing the EC1-EC3 fragment, which was found to be monomeric in solution, failed to produce cell aggregates (Figure 2A). In contrast, with the exception of PcdhgC5 EC1-EC4 which forms a non-natural disulfide between monomers, cells expressing either EC1-EC4, EC1-EC5, or the complete ectodomain (EC1-EC6), showed extensive aggregation for all isoforms tested (Figure 2A). Consistent with previous studies (Schreiner and Weiner, 2010; Thu et al., 2014), cells expressing Pcdh EC2-EC6 fragments, which were shown above to homodimerize in solution, did not aggregate (Figure 2A). Detection of two independent dimers, one of which (generated by EC1-EC4 and EC1-EC5 fragments) correlates with cell-cell aggregation while the other (generated by EC2-EC6 fragments) does not (Figure 2A), strongly suggests that EC1-EC4 and EC1-EC5 fragments mediate trans interactions while the EC2-EC6 fragments mediate cis interactions involving the most membrane-proximal domain, EC6 (see also below). The observation that full-length ectodomains form apparent tetramers in AUC strongly suggests that this molecular species corresponds to a dimer-of-dimers formed by these two distinct interfaces, one mediating cis and the other trans interactions.
Figure 2. Elements of Pcdh cis and trans binding.

A) Correlating multimerization states of truncated Pcdh proteins with their cell-cell recognition properties. Cells transfected with Pcdh deletion series plasmid constructs were tested for aggregation. With the exception of EC2-EC6 Pcdh fragments and PcdhγC5 EC1-EC4, all deletion proteins that formed oligomers in solution also mediated cell aggregation. Full-length Pcdhα4 include the EC6 domain from PcdhγC3 so it could be delivered to cell surface.
B) Probing homophilic interaction interface by arginine-scanning mutagenesis. Residues mutated to arginine are drawn in space filling representation. In blue are mutations that did not disrupt recognition, in orange are mutations that weakened recognition and in red are mutations that abolished cell-cell recognition. Excluding residue 142, all the effective arginine mutants are located along one side of the molecule.
C) Cell aggregation experiments showing the mutations in part (B) that weakened or abolished interactions. See also Figure S2C.
D) In other Pcdh isoforms, residues analogous to the effective PcdhγC5 arginine mutants had similar effects on the cell-cell recognition in the majority of cases.
Structural elements of the trans-binding interface
Arginine-scanning mutagenesis
Selected non-basic surface residues of the PcdhγC5 EC1-EC3 domains revealed in the crystal structure were individually mutated to arginine, and the homophilic recognition function of these single-arginine-mutant proteins was assessed using the K562 cell aggregation assay. Selected basic surface residues were mutated to glutamic acid. As expected, the majority of single-point mutant proteins exhibited wild-type cell aggregation phenotypes (Figures S2C). In contrast, cells transfected with the arginine point mutant L87R in the EC1 domain, S116R and T142R in the EC2 domain, and M301R and E302R in the EC3 domain of PcdhγC5 showed no detectable aggregation (Figure 2B–C). Cells transfected with the EC2 S114R mutation showed diminished homophilic binding (Figure S2C). S114 and S116 are located in the AB loop connecting the A and B β-strands in EC2 while M301 and E302 are located in the FG loop of EC3. All are located on one side of the molecule and are very close to one another in space, thus defining a potentially continuous homophilic recognition interface with elements distributed over the EC2 and EC3 domains. Notably, L87 in EC1 faces in the same direction although T142 in EC2 does not.
To determine whether this binding region is unique to PcdhγC5, we produced mutants for isoforms from all three Pcdh gene clusters for residues structurally equivalent to PcdhγC5 positions 87, 116, and 301. Mutations equivalent to 301R abolished homophilic recognition for isoforms from all three gene clusters (Pcdhα7, PcdhαC2, Pcdhβ6, PcdhγA8, and PcdhγB6, Figure 2D). Homophilic recognition was abolished for mutations equivalent to 116R for isoforms from the α and γ gene cluster members (Pcdhα7, PcdhαC2, PcdhγA8), but not for the isoforms we tested from the β and γB cluster (Figure 2D). Finally, mutations equivalent to L87R abolished homophilic recognition for PcdhγA8 and diminished homophilic recognition for Pcdhα7. It is possible that homophilic recognition for the Pcdhβ6 and PcdhγB6 isoforms may not involve residues 87 in EC1 and 116 in EC2 or, alternatively, arginine mutants of these residues might not appropriately test their contribution to binding. Below we show that isoforms from the α and β gene clusters do in fact utilize interface residues in the EC2 AB loop region and others in close structural proximity to EC1 residue 87.
Domain shuffling to identify specificity-determining domains
Within each of the mouse gene clusters there exist pairs of Pcdh isoforms (Pcdhα7-Pcdhα8, Pcdhβ6-Pcdhβ8, and PcdhγA8-PcdhγA9) with greater than 80% pairwise sequence identity within their EC1-EC4 domain regions. Despite this high identity these pairs display strict homophilic specificities (Thu et al., 2014). In order to help identify the binding interface we produced chimeras in which EC domains were shuffled between the closely related isoforms. These proteins were tagged at the C-terminus with either of the fluorescent proteins mCherry or mVenus, and tested for binding specificity in the K562 cell assay. We confirmed that all three pairs bind strictly homophilically (Figure 3A1–4, 3B1–4, 3C1–4).
Figure 3. Pcdh trans binding depends on the four N-terminal domains EC1-EC4.

A–C) Domain-shuffled chimeras of closely related isoforms and their wild-type counterparts were assayed for binding specificity. Swapped specificity was noted for chimeras in which either the EC1-EC3 or EC2-EC4 domains were replaced with the corresponding domains of closely related isoforms. See also Figure S3.
D) Schematic representation of the domain-shuffled isoforms and their observed binding specificities to their wild-type isoform counterparts.
The results of cell aggregation experiments using different chimeric constructs are summarized in Figures 3 and S3. These results are presented in such a way that two closely related wild-type “parent” proteins appear at the left of each panel while each figure indicates whether a particular chimera co-aggregates with one or the other parent protein, or prefers to aggregate homophilically. Figure 3D summarizes the data presented in Figures 3A–C. All chimeric constructs containing EC1-EC3 domains from one isoform and EC4-EC6 domains from another co-aggregated with the wild-type “parent” isoform that contained the same EC1-EC3 domains (Figure 3A–C panel 6 and Figure S3B and D panel 13), whereas chimeric constructs with just EC2-EC3 shuffled, preferred to aggregate homophilically (Figure S3A–E panels 11 and 12).
Despite the fact that shuffling EC1-EC3 is sufficient to swap specificity in close pairs, our AUC and cell aggregation assay results (Table 1A and figure 2A) indicate that all four N-terminal domains (EC1-EC4) are required for trans homophilic recognition. We therefore generated a chimera of PcdhγA8 in which domains EC2-EC4 were replaced with the corresponding domains of the closely related PcdhγA9 isoform, while domains EC5-EC6 were replaced with the EC5-EC6 domains of the distant PcdhγB6 isoform, which would not be expected to interact in trans with PcdhγA8 or PcdhγA9. Cells expressing this chimera adhere to cells expressing PcdhγA9 indicating, consistent with AUC data, that the EC4 domain plays a role in determining homophilic binding specificity (Figure 3C panel 8). This conclusion is also supported by cell aggregation studies using chimeras where EC1 is derived from one parent and EC2-EC6 from another. In all cases, these chimeras co-aggregate with the parent containing the same EC2-EC6 domains (Figure S3A, C, and E panel 1 and S3B and D panel 2). Since domains EC5 and EC6 are not required for trans binding these results also implicate EC2-EC4 as sufficient to determine homophilic specificity.
The experiments reported in Figure S3 help define the minimal number of domains within the EC1-EC4 region that determine the binding properties of a chimera. The presence of a single domain is never enough to mediate co-aggregation with a parent isoform containing this domain (Figure S3A, C, and E panels 2, 4 and 6, S3B and S3D panels 1, 3, and 5) but, in some cases, a mismatched single domain is capable of disrupting binding to the parent isoforms (FigureS3C panel 5, S3D panel 6 and S3E panel 3). In a few cases, the presence of just two domains in common is sufficient to mediate co-aggregation with a parent even if the other four domains are different. This can be seen in: a chimera containing EC1 and EC3 from γA9 and EC2 and EC4-EC6 from γA8 which co-aggregates with wild-type γA9 (Figure S3C panel 10), and a chimera containing EC1 and EC2 from β8 and EC3-EC6 from β6 which co-aggregates with wild-type β8 (Figure S3E panel 8). Overall, these results are consistent with all four N-terminal domains, EC1-EC4, contributing to trans binding with the relative contributions of each domain to specificity varying from one isoform to another.
Rational design of point mutations to identify specificity-determining residues
Sequence alignment of specificity-determining EC3 domains shows that Pcdhα7 and Pcdhα8 differ in five amino acids whereas PcdhγA8 and PcdhγA9 differ in eight (Figure 4A). Notably, in both cases, three of these residues are located in the same structural element: the FG loop (Figures 4A, 5A, and 5C). In the case of PcdhγA8 and PcdhγA9 the three variable FG loop residues are highly conserved within their respective orthologs (Figure 4B). Together, these data strongly suggest that these three EC3 domain FG loop residues act as specificity determinants for α and γ Pcdh isoforms.
Figure 4. Candidate specificity determining residues.

A) Multiple sequence alignment of the three closely related Pcdh isoform pairs, along with PcdhγC5. Highlighted in gray are positions conserved in all Pcdh sequences. Sequence positions that differ between the closely related isoforms are shown in red; a subset of these residues determines binding specificity. Residues swapped between isoforms and assayed for binding properties are boxed. Secondary structure from PcdhγC5 is shown at the top of the alignment.
B) Multiple sequence alignment of the FG-loop region for PcdhγA8 and PcdhγA9 orthologs. Three of the residues that differ between mouse PcdhγA8 and PcdhγA9 are highly conserved in orthologs (highlighted in red), suggesting their functional importance.
Figure 5. Structural elements of the canonical Pcdh trans binding interface.

A–C) Assessing specificity-determining residues. Binding properties of wild-type isoforms (left side of each panel) or constructs with shuffled residues (top of each panel) were tested separately for each EC domain. Cases in which shuffled residues swapped specificities are indicated by an orange outline. Residues shuffled between closely related isoforms are shown in magenta on surface representations of the Pcdhα7, Pcdhβ6, and PcdhγA8 structures. Sequence alignments of shuffled regions are shown. See also Figure S4.
D) Correspondence between trans interface residues identified by arginine scanning and close-isoform pair analysis. Single arginine mutant residues that abolish or diminish homophilic binding, highlighted in red and orange respectively, are found in the same structural regions as the shuffled residues (see also Figure 2). Residues that swap binding specificity between closely related isoforms are shown in magenta on surface representations of the Pcdh-γC5 crystal structure.
To test this hypothesis experimentally, we swapped the three residues (Figure 5) between the EC3 domains of closely related isoforms and tested their binding specificities with their “parent” native isoforms. We produced chimeras with the three FG-loop residues of one isoform replaced with the corresponding residues of its close-pair isoform. These three-residue-swapped mutants were tested, along with their native “parents”, in the K562 cell aggregation assay. Cells expressing an isoform in which the three FG-loop residues were replaced with those from the close-pair isoform intermixed with cells expressing the wild-type isoform with residues identical to those at the shuffled positions (Figure 5A and 5C). In contrast, these cells segregated from cells expressing the wild-type isoform from which the EC3 domain originated (Figure S4). We conclude that the three variable residues of the EC3 FG loop are specificity-determining in the closely related α and γ isoforms.
A similar analysis was carried out for EC1 and EC2 domains with comparable results. As with the EC3 domains, we analyzed close isoform pairs (Figure 4A) and identified candidate specificity-determining residues located on the EC1 C strand and EC2 AB region (Figure 5). We validated these assignments by showing that shuffling residues between EC2 domain AB regions resulted in swapped specificities for close-pair isoforms from all three Pcdh gene clusters (Figures 5 & S4). Shuffling residues between EC1 domain C strand regions was sufficient to swap EC1 specificities from Pcdhβ6 to that of Pcdhβ8 or from Pcdhα7 to Pcdhα8. The contribution of this region in the Pcdhγ pair could not be determined since shuffling of residues in this region resulted in a protein that could not mediate cell aggregation (Figures S4D). We note that swapping EC1 specificities from Pcdhβ6 to Pcdhβ8 or EC2 specificities from Pcdhα7 to Pcdhα8 or from PcdhγA9 to PcdhγA8 required the alteration of only a single residue (residue R41N, L114P and S114N for β, α and γ respectively, Figure 5).
Rational and random mutagenesis identify the same functional binding surfaces
Figures 2 and 5 list specificity determining residues identified from arginine scanning and bioinformatics-based mutagenesis. The finding that two different approaches implicate the same structural regions in Pcdh homophilic binding, and that these regions are in common for isoforms from different Pcdh gene clusters, indicates that these regions – the EC1 C, and G strands, the EC2 AB loop and EC3 FG loop (Figure 5D) are likely to contribute to determining the binding specificities for other Pcdh isoforms as well. As shown above, EC4 contributes to the trans binding specificity in a similar way to that of EC1. However, we focused on the EC1-EC3 domains because this is the region for which we have atomic-level structures.
AUC experiments on mutant proteins confirm that Pcdh trans-interactions occur via EC1-EC4 domains, whereas cis interactions occur via the EC6 domain
We have provided evidence from both AUC and cell aggregation assays that the EC1-EC4 domains mediate Pcdh trans interactions, whereas the EC6 domain mediates an independent Pcdh cis interaction. To provide further evidence for these findings we expressed and purified various domain-truncated constructs of PcdhγA8-I116R, PcdhγC5-S116R, and PcdhαC2-S118R. Since an arginine at these positions ablates trans binding in cell aggregation assays these mutant constructs should only affect the Pcdh trans-association but not the cis-association in AUC experiments. As expected the EC1-EC4 fragment of I116R PcdhγA8 behaved differently from its wild-type counterpart and was monomeric in solution (Table 1B). In contrast, we found that similar to its wild-type counterpart, the EC2-EC6 fragment PcdhγC5-S116R behaved as a dimer with KD similar to wild-type EC2-EC6. This observation suggests that the EC2-EC6 protein dimerizes in cis through a region that is not involved in the trans interface (Table 1B). Finally, the complete ectodomain of PcdhαC2 containing an S118R mutation displayed tetramerization affinity, which was an order of magnitude lower than that of the wild-type protein. Similarly, the S116R mutant of PcdhγC5 EC1-EC6 did not form tetramers (as does its wild-type counterpart) but rather, similar to the EC2-EC6 fragment, self-associates as a dimer. Since trans binding has been ablated by this mutation, the observed dimer must correspond to association in cis (Table 1B).
The trans homophilic interface is formed via head-to-tail interactions of EC1-EC4 domains
Computational docking yields antiparallel orientations
We carried out modeling studies in an effort to elucidate the dimerization mode of Pcdhs. We limited our modeling to EC1-EC3 for which we have determined crystal structures and have identified specificity-determining residues. We used the M-zdock program (Pierce et al., 2005) to produce symmetric homodimeric models for the EC1-EC3 domain regions of PcdhαC2, Pcdhβ1, PcdhγA8, and PcdhγC5. We generated thousands of models for each crystal structure, and used the experimentally identified specificity determinant residues to filter the docked models; requiring models to include these residues at the binding interface. A second constraint required docking models to have a buried surface area at the binding interface of more than 1200 Å2 (600 Å2 per protomer). Applying these two conditions reduces the number of docked models from thousands to 149: 23, 40, 40 and 46 for PcdhγA8, Pcdhβ1, PcdhαC2, and PcdhγC5, respectively. We then structurally clustered the filtered docked homodimers with the expectation that there would be more docked structures near the native conformation.
Notably, the majority of the filtered docked homodimeric Pcdhs (62.5%) adopted a head-to-tail orientation of the two molecules in which the EC2 domain of one molecule interacts with the EC3 domain of its partner (Figures 6A and S5Ai–ii). Furthermore, most structures with this binding mode place the EC1 domain of one molecule adjacent to the expected position of the EC4 domain of its partner (Figure 6A). Only three of the docked and filtered complexes had a head-to-head orientation (two for PcdhγC5 and one for PcdhαC2, Figure 6Aiii) while filtered solutions for Pcdhβ1 and PcdhγA8 resulted solely in solutions with a head-to-tail orientation. We note that it is the application of the two constraints, one of which was experimentally derived, that results in this distribution of binding modes.
Figure 6. Molecular logic of Pcdh-mediated cell-cell recognition.

A) Shown in ribbon representation is the only orientation observed for docking of the four EC1-EC3 domains structures which position the EC2 AB loop in close proximity to the EC3 FG loop. EC2 AB loop residue 116 and FG loop residue 301 are drawn as space filling and colored red and blue respectively. The vast majority of the docked complexes were observed to interact in this mode. See also Figure S5A.
B) Cell aggregation assays on chimeric proteins that show EC1 interacts with EC4 and EC2 interacts with EC3. Schematic representation of the head-to-tail interaction between the domain-shuffled chimeras is shown above each panel. Mixed aggregates were formed where all interactions involve “matching” domains (panels 1–3). Separate aggregates were formed when there is a mismatch between EC1/EC4 (panel 4) or between EC2/EC3 (panel 5).
C) The EC2 domain AB region recognizes the EC3 domain FG loop. Cells expressing isoforms with single arginine mutants in the EC3 FG loop region, or with double mutations (aspartate at the AB region and arginine at the FG loop), were assayed for aggregation. The double-mutation rescued the non-adhesive phenotype, supporting the head-to-tail binding orientation shown in part (A).
D) Two possible models of Pcdh interaction. A discrete tetramer composed of a dimer of dimers is observed in analytical ultracentrifugation, but we suggest that a connected ribbon of molecules can form between cells via the trans and cis interactions.
E & F) A model for Pcdh mediated cell-cell recognition based on formation of a superstructure defined by promiscuous cis and specific trans interactions. Growth of the chain of molecules requires matching of all isoforms; a single mismatch can terminate chain extension. Dendrites of the same neuron will have the same isoform repertoire while dendrites of different neurons will differ. In this model, repulsion signaling is triggered, or achieves a sufficient level for response, only through the formation of an extended chain of Pcdhs.
G) For the case of 15 distinct Pcdh isoforms expressed per cell, Monte-Carlo simulations were used to estimate the average size of one-dimensional Pcdh assemblies between contacting cells. The average number of cis dimers that comprise such assemblies is shown on a logarithmic scale as a function of the number of mismatched isoforms. Two cases are shown: one for 15000 total Pcdh monomers (1000 per isoform, red), and one for 1500 total copies (100 per isoform). The model assumes that each cell contains a stable set of cis dimers formed from the random association of monomers present in each cell. See also Figure S5B.
Experimental validation of a head-to-tail orientation
The computational evidence for a head-to-tail dimer, taken together with our identification of EC1-EC4 as the specificity-determining region, suggests that EC1 interacts with EC4 and EC2 interacts with EC3. In order to validate this model we carried out cell aggregation assays on chimeras of the γA8 and γA9 Pcdh isoforms, which were designed to determine which domains physically interact. As shown in the schematic, diagrams in Figure 6B panels 1–3, head-to-tail binding would result in a dimer where all EC2/EC3 and EC1/EC4 interactions involve domains from the same wild type protein. In all three cases the chimeras form mixed aggregates thus providing strong evidence for our proposed model of the Pcdh-Pcdh interface. Note that if the monomers bound in a head-to-head orientation, some interacting domains would be derived from different wild type proteins so that mixed aggregates would not be expected to form.
Figure 6B panels 4 and 5 provide direct evidence that EC1 interacts with EC4 and EC2 interacts with EC3. Comparing panel 4 to panel 1, the only difference between the two is that there is a mismatch between EC4 and EC1 in panel 4. The two cell populations in panel 4 form separate aggregates indicating that this single mismatch is sufficient to ablate trans dimerization. An identical conclusion regarding EC2 and EC3 is reached by comparison of panel 5 to panel 2. Here again, a single-domain mismatch inhibits co-aggregation even though the remaining three domains are correctly matched.
To further validate the model of head-to-tail binding, we carried out mutagenesis experiments on specificity determining regions. Since, as shown above, for the α and γ close pairs the EC2 AB loop and the EC3 FG loop determine specificities we reasoned that the specificity-determining residues in the EC2 AB loop might interact with corresponding residues in the EC3 FG loop. Notably, the largest cluster of structurally-similar docked and filtered complexes is the only cluster that positions the EC2 AB loop near the EC3 FG loop and projected to position the EC1 near EC4 (Figures 6A and S5A). To test this model (Figure 6A), we relied on two observations. First, that arginine mutations of residue 301 in the EC3 FG loop region and residue 116 in the EC2 AB loop region (PcdhγC5 numbering) abrogate recognition in isoforms from different gene clusters (Figure 2B–D), and second, that docked models position residue 301 and residue 116 at close distance (less than 6Å, Figure 6A). Hypothesizing that residues 116 and 301 are near each other in the recognition complex, we attempted to rescue single-arginine mutants at residue 303 of PcdhαC2 or 298 of PcdhγA8 and Pcdhβ6 (analogous to PcdhγC5 301) by producing an aspartic acid mutation of PcdhαC2 residue 118, of PcdhγA8 residue 116 or of Pcdhβ6 residue 117 (analogous to PcdhγC5 116). The designed double-mutants could, in principle, form a salt bridge at the interface and thus might rescue recognition.
For all three isoforms (PcdhαC2, Pcdhβ6, and PcdhγA8), cells expressing the double arginine/aspartic-acid mutants tested positive for cell aggregation (Figure 6C), indicating that these two mutated residues (116 and 301), located respectively on domains EC2 and EC3, are in close proximity at the homophilic binding interface. This observation provides strong support for a head-to-tail binding mode where EC2 interacts with EC3 and where EC1 interacts with EC4. Moreover, since PcdhαC2, Pcdhβ6, and PcdhγA8 are not closely related, it is likely that the modeled interface represents the recognition interface for other Pcdhs as well.
Discussion
Counterintuitively, the phenomenon of neuronal self-avoidance is initiated by trans homophilic adhesive binding between Pcdhs. Presumably, repulsion is a consequence of the activation of downstream signals via the ICD, which is known to interact with signaling adaptors and kinases (Han et al., 2010; Schalm et al., 2010). This mechanism requires that different neurons express a sufficiently distinct set of Pcdh isoforms so that inappropriate “self”-recognition, and subsequent repulsion, will not occur. In the case of invertebrates, this is accomplished through the stochastic expression of about 10–50 different alternatively spliced Dscam isoforms in each cell (Hattori et al., 2008; Zipursky and Grueber, 2013; Zipursky and Sanes, 2010). With thousands of stochastically generated distinct Dscam isoforms, the probability that two different neurons express the same set of isoforms is extremely low (Miura et al., 2013). Considering the much smaller number of distinct Pcdh isoforms in vertebrates, isoform diversity alone cannot account for “non-self discrimination”.
As mentioned above, we have shown previously that an interference phenomenon plays a crucial role in Pcdh-based non-self discrimination (Thu et al., 2014). In this paper we present evidence from several independent sources of data that suggest that Pcdh cell-cell recognition is mediated by a mechanism that couples cis and trans interactions. Specifically, we propose that Pcdh isoforms form promiscuous EC6 dependent cis-dimers at the cell surface that associate specifically in trans via a stereotyped interface with elements in domains EC1-EC4. Below we summarize our findings and discuss their implications for the molecular mechanisms by which clustered Pcdhs mediate neuronal self-recognition and non-self discrimination.
Pcdh homophilic specificity is determined by a head-to-tail trans recognition interface
We found that Pcdh EC1-EC3 fragments do not associate in solution, nor do they mediate homophilic cell-cell recognition in cell aggregation assays. Rather, we showed both in AUC measurements and cell assays that stable trans dimerization requires all four of the N-terminal EC1-EC4 domains. Site-directed arginine scanning mutagenesis and rational mutagenesis based on analysis of sequence alignments allowed us to identify key structural elements in a trans interface that mediate cell-cell recognition between Pcdhs.
The identification of interfacial regions in EC2 and EC3 through computational modeling and mutagenesis experiments provided strong constraints that made it possible to demonstrate that Pcdh trans dimers adopt a head-to-tail orientation where EC2 interacts with EC3. This remarkable anti-parallel trans-interaction is in contrast to the parallel trans dimerization of classical cadherins. However, for classical cadherins the parallel binding mode is made possible by a significant intramolecular bend whereby the five EC domains form a highly curved structure so that interacting membrane-distal EC1 domains from apposed cells are parallel to one another. In contrast, since the EC1-EC3 domains in Pcdhs are straight rather than curved, binding in parallel would require a sharp bend between the three N-terminal and three C-terminal domains. Such a bend has been observed only in cadherins lacking inter-domain calcium binding sites (e.g. DN cadherin (Jin et al., 2012)), and the presence of complete calcium binding sites between all domains renders such significant bending highly unlikely in the case of Pcdhs.
Figure 6A shows the structure of an EC1-EC3 trans dimer obtained from our docking studies that satisfy all the constraints established by mutagenesis. The EC4 domain is represented as an ellipse in the diagram since its structure has not yet been determined. In addition to satisfying all the mutagenesis data used as constraints in the docking studies, independent evidence supporting the model include; 1) the set of five cell aggregation studies on γA8 and γA9 chimeras (Figure 6B) that show that EC1 interacts with EC4 and EC2 interacts with EC3; 2) the rescue experiments shown in Figure 6C that reveal that residue 116 in EC2 is in close proximity to residue 301 in EC3, as predicted by the head-to-tail model (Figure 6A).
The head-to-tail model shown in the figure provides a clear explanation of the binding affinity and cell aggregation data. In the model, the free energy of binding is distributed over all four domain-domain interfaces, and all must be present to generate sufficient affinity to produce a stable homodimer. This is evident from the observations that three domain constructs do not dimerize, and that interfacial mutations in only a single domain are sufficient to ablate binding. All EC1-EC3 ectodomain fragments studied here were monomeric and none revealed a likely trans interaction. With a head-to-tail orientation, deletion of only one domain in EC1-EC4 effectively removes half the interface, providing a likely explanation for the absence of native dimer interactions.
We note that the structural model itself is unlikely to be accurate in detail and will certainly be superseded once X-ray structures of all four interacting domains are available. The major significance of the model is the demonstration that Pcdhs dimerize in trans in a head-to-tail orientation with an extended interface formed from four inter-domain interfaces (two EC2/EC3 and two EC1/EC4). We note that the molecular dimerization logic of Pcdhs where different domains recognize one another through EC1/EC4 and EC2/EC3 trans interactions, is fundamentally different from that of Dscam1 where the dimerization interface is formed from three separate self-self interactions, Ig2/Ig2, Ig3/Ig3 and Ig7/Ig7.
Pcdhs form cis dimers mediated by EC6
We previously provided evidence for promiscuous Pcdh EC6/EC6 cis interactions. Specifically, any single carrier isoform (β, γ or C-type) can mediate cell-surface delivery of α isoforms, which are otherwise confined within the cell, through interactions involving the EC6 domain (Thu et al., 2014). In addition, the pairwise sequence identity between EC6 domains for all isoforms of Pcdhβ or Pcdhγ clusters averages over 90% (Thu et al., 2014), consistent with the idea of promiscuous interactions.
We show above that the EC6 domain mediates Pcdh cis dimerization even in the absence of trans interactions. Moreover, as shown in Table 1, the affinity of this interaction is comparable or even stronger than the trans interaction involving EC1-EC4. In general, cis interactions in the two dimensional environment of the plasma membrane would be significantly enhanced, and the effect is strongest for membrane proximal domains as there would be little entropy loss due to inter-domain flexibility upon binding (Wu et al., 2013; Wu et al., 2011). Indeed, even at low surface densities, molecules with substantial solution (3D) KDs, such as that of Pcdhs, will likely form dimers on cell surfaces. The promiscuity of the EC6 carrier function suggests that these dimers can form between essentially any two Pcdh isoforms, which in turn suggests that Pcdhs on cell surfaces exist as cis dimers formed by pairs of different isoforms from all three subfamilies as well the C-type isoforms.
Assembly termination by mismatched isoforms distinguishes self from non-self
We have shown above that full-length Pcdh ectodomains in solution form tetramers (a cis/trans dimer of dimers) mediated by head-to-tail trans interactions involving EC1-EC4, and a cis interaction involving EC6. A schematic of this molecular arrangement is shown in the left panel of Figure 6D. If Pcdhs on cell surfaces interacted in this manner, cellular recognition would be based on dimeric recognition units. However, as we have discussed in a previous study, dimeric recognition units are unlikely to provide sufficient diversity for neuronal non-self discrimination, and indeed all models based on multimeric recognition units encounter difficulties in accounting for both self-recognition and non-self discrimination (Thu et al., 2014). For this reason, we previously proposed an alternative recognition mechanism based on “junction-like” molecular assemblies at least partially reminiscent of those formed by classical cadherins.
As discussed above, each Pcdh molecule forms strong independent trans and cis interactions. This is in contrast to classical cadherins, in which each molecule forms relatively strong trans interactions and two weak asymmetrical cis interactions that become stronger on cell surfaces only once the trans interactions have been formed (Wu et al., 2011). In the case of classical cadherins, the combination of cis and trans interactions generates a two-dimensional lattice that corresponds to the extracellular structure of adherens junctions (Harrison et al., 2011). In contrast, the interactions defined here for Pcdhs suggest the formation of a one-dimensional zipper-like structure involving symmetrical cis and trans interactions. This structure is depicted in the right panel of Figure 6D, which shows how each bivalent Pcdh cis dimer could recognize two other dimers via independent trans interactions so as to form a connected ribbon of molecules that emanate from two apposed cell surfaces. We note that still undiscovered extracellular, trans-membrane or cytoplasmic interactions may ultimately reveal a more complex network of interactions than the one depicted in the figure. For example, the receptor tyrosine kinase Ret has been shown to associate with, and directly or indirectly phosphorylate Pcdhα and γ tyrosine residues in their ICD’s (Schalm et al., 2010). In any case, the existence of even a one-dimensional network would provide a mechanism for interference that does not encounter the problems based on models of isolated multimeric recognition units.
Figure 6E illustrates that cells with the same isoform composition would be able to form a large assembly upon contact. In contrast, cells with different isoform compositions would incorporate mismatches, preventing further growth of the lattice (Figure 6F). If downstream signaling leading to neurite repulsion depends on the size of the assembly, which in turn depends on isoform composition, the model offers a natural mechanism for Pcdh interference. Indeed, there is a striking dependency of the size of Pcdh assemblies on the number of mismatched Pcdh isoforms. Figure 6G plots the average size of such linear assemblies as a function of the number of mismatched isoforms between two contacting neurons. Assembly size is obtained from Monte-Carlo calculations based on a model that assumes that each cell contains a stable set of cis dimers formed from the random association of monomers present in each cell. When all isoforms are identical assembly size is limited solely by the number of copies of each isoform. Remarkably, the presence of even a single mismatched isoform is sufficient to reduce the average size of an assembly by at least two orders of magnitude. The results presented in Figure 6G thus suggest that a mechanism based on mismatched-isoform chain termination of a linear Pcdh-assembly could provide a binary definition of self and non-self.
While we recognize that this isoform mismatch chain-termination model is speculative, it is consistent with the presence of strong independent cis and trans interactions. Such signaling systems have been observed previously, including the one-dimensional network of CTLA-4/B7 immune receptors (Schwartz et al., 2001) where signaling has also been proposed to be based on large cell surface assemblies. Most importantly, the model provides a mechanism whereby 58 Pcdhs can generate the high level of diversity sufficient to allow for neuronal self-avoidance without encountering the problems for self-recognition, which is implicit in previous models that depend on discrete combinatorial multimeric recognition units.
Experimental procedures
Protein production and Crystallography
Proteins for crystallization or biophysical analysis were expressed in suspension-adapted HEK293 Freestyle cells (Invitrogen) and purified by nickel affinity and size exclusion chromatography. Pcdh crystals were grown by vapor diffusion in 1–2μl hanging drops, except the Pcdhβ1 EC1-3 crystals, which were grown in 0.2μl sitting drops. The PcdhγC5 EC1-3 P43212 crystal structure was solved using the MIRAS technique while all the other Pcdh crystal structures were solved by molecular replacement. See Extended Experimental Procedure for details.
Cell aggregation assays
Pcdh expression constructs were transfected into K562 cells by electroporation. The transfected cells were grown in culture for 24 hours. Cells were then allowed to aggregate for one to three hours on a rocker inside an incubator at 37°C. The cells were then fixed in 4% PFA for 10 minutes, washed in PBS, and cleared with 50% glycerol for imaging. See Extended Experimental Procedure for details.
Sedimentation equilibrium analytical ultracentrifugation
Proteins were diluted to an absorbance at 10mm path length and 280 nm of 0.65, 0.43 and 0.23 absorbance units. All samples were run at four speeds: 11000, 14000, 17000 and 20000 rpm (all EC 1-EC3 constructs) or 9000, 11000, 13000 and 15000 rpm (all EC1-EC4, EC1-EC5 and EC1-EC6 constructs), respectively. Measurements were carried out at 25°C, and detection was by UV at 280 nm.
Monte-Carlo simulations
A stochastic algorithm was used to estimate the average size of Pcdh-assemblies (number of linked cis dimers) formed between a pair of neurons each expressing 15 distinct isoforms with 0–15 common isoforms. It was assumed that a neuron expresses an equal number of copies of each of the 15 Pcdh isoforms, with either 1000 or 100 copies per isoform (i.e., 15,000 or 1,500 total Pcdh monomers respectively). 106 simulations were performed and in each simulation stable cis dimers were randomly and independently generated for the contacting neurons. Note that the distribution of cis dimers on both neurons will not in general be identical even for neurons with an identical set of monomers. A linear network was initiated by randomly choosing a dimer on one of the cells. In the next step, a cis dimer is chosen on the second cell where one of its monomer constituents matches one of the monomers in the dimer chosen on the first cell. This matching process is then repeated with the search for matching dimers alternating between the contacting neurons moving from one cell to the other as the chain extends in two directions. This extension process was repeated until there remained no matching dimers either due to a mismatch or to a depletion of dimers.
Supplementary Material
Acknowledgments
We thank Dr. David Hirsh for valuable comments on the manuscript. We thank Igor Kourinov, Surajit Banerjee, Narayanasami Sukumar at Advanced Photon Source (APS) for support with synchrotron data collection. This work was supported by the National Science Foundation grant to B.H. (MCB-1412472), National Institutes of Health grant to L.S. (R01GM062270), joint NIH grant to T.M. and L.S. (R01GM107571), NIH Training Programs to H.W. (T32GM008281), and Danish National Research Foundation (DNRF107) to A.H. and H.C..
Footnotes
Author Contributions
R.R., C.A.T., K.M.G., T.M., L.S., and B.H. designed research, analyzed data, and assembled and wrote the paper. R.R. carried out the computational analysis, C.A.T. carried out cell aggregation assays and analysis, F.B., S.M., H.N.W. and K.M.G. prepared and crystallized all proteins. H.N.W. and K.M.G. determined the crystal structures. C.A.T., S.M., H.N.W., and M.C. prepared Pcdh mutants. G.A. performed and analyzed the AUC experiments. A.H. and H.C. performed the glycosylation analysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- Chen WV, Alvarez FJ, Lefebvre JL, Friedman B, Nwakeze C, Geiman E, Smith C, Thu CA, Tapia JC, Tasic B, et al. Functional significance of isoform diversification in the protocadherin gamma gene cluster. Neuron. 2012;75:402–409. doi: 10.1016/j.neuron.2012.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WV, Maniatis T. Clustered protocadherins. Development. 2013;140:3297–3302. doi: 10.1242/dev.090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Lin C, Meng S, Wang X. Proteomics analysis reveals overlapping functions of clustered protocadherins. Mol Cell Proteomics. 2010;9:71–83. doi: 10.1074/mcp.M900343-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, et al. The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure. 2011;19:244–256. doi: 10.1016/j.str.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori D, Chen Y, Matthews BJ, Salwinski L, Sabatti C, Grueber WB, Zipursky SL. Robust discrimination between self and non-self neurites requires thousands of Dscam1 isoforms. Nature. 2009;461:644–648. doi: 10.1038/nature08431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori D, Millard SS, Wojtowicz WM, Zipursky SL. Dscam-mediated cell recognition regulates neural circuit formation. Annual review of cell and developmental biology. 2008;24:597–620. doi: 10.1146/annurev.cellbio.24.110707.175250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Bortnick R, Tsubouchi A, Baumer P, Kondo M, Uemura T, Schmucker D. Homophilic Dscam interactions control complex dendrite morphogenesis. Neuron. 2007;54:417–427. doi: 10.1016/j.neuron.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Walker MA, Felsovalyi K, Vendome J, Bahna F, Mannepalli S, Cosmanescu F, Ahlsen G, Honig B, Shapiro L. Crystal structures of Drosophila N-cadherin ectodomain regions reveal a widely used class of Ca(2)+-free interdomain linkers. Proc Natl Acad Sci U S A. 2012;109:E127–134. doi: 10.1073/pnas.1117538108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JL, Kostadinov D, Chen WV, Maniatis T, Sanes JR. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature. 2012;488:517–521. doi: 10.1038/nature11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommel M, Winterhalter PR, Willer T, Dahlhoff M, Schneider MR, Bartels MF, Renner-Muller I, Ruppert T, Wolf E, Strahl S. Protein O-mannosylation is crucial for E-cadherin-mediated cell adhesion. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:21024–21029. doi: 10.1073/pnas.1316753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BJ, Kim ME, Flanagan JJ, Hattori D, Clemens JC, Zipursky SL, Grueber WB. Dendrite self-avoidance is controlled by Dscam. Cell. 2007;129:593–604. doi: 10.1016/j.cell.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Miura SK, Martins A, Zhang KX, Graveley BR, Zipursky SL. Probabilistic splicing of Dscam1 establishes identity at the level of single neurons. Cell. 2013;155:1166–1177. doi: 10.1016/j.cell.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita H, Umitsu M, Murata Y, Shibata N, Udaka K, Higuchi Y, Akutsu H, Yamaguchi T, Yagi T, Ikegami T. Structure of the cadherin-related neuronal receptor/protocadherin-alpha first extracellular cadherin domain reveals diversity across cadherin families. The Journal of biological chemistry. 2006;281:33650–33663. doi: 10.1074/jbc.M603298200. [DOI] [PubMed] [Google Scholar]
- Pierce B, Tong W, Weng Z. M-ZDOCK: a grid-based approach for Cn symmetric multimer docking. Bioinformatics. 2005;21:1472–1478. doi: 10.1093/bioinformatics/bti229. [DOI] [PubMed] [Google Scholar]
- Posy S, Shapiro L, Honig B. Sequence and structural determinants of strand swapping in cadherin domains: do all cadherins bind through the same adhesive interface? Journal of molecular biology. 2008;378:954–968. doi: 10.1016/j.jmb.2008.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss K, Maretzky T, Haas IG, Schulte M, Ludwig A, Frank M, Saftig P. Regulated ADAM10-dependent ectodomain shedding of gamma-protocadherin C3 modulates cell-cell adhesion. The Journal of biological chemistry. 2006;281:21735–21744. doi: 10.1074/jbc.M602663200. [DOI] [PubMed] [Google Scholar]
- Ribich S, Tasic B, Maniatis T. Identification of long-range regulatory elements in the protocadherin-alpha gene cluster. Proc Natl Acad Sci U S A. 2006;103:19719–19724. doi: 10.1073/pnas.0609445104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalm SS, Ballif BA, Buchanan SM, Phillips GR, Maniatis T. Phosphorylation of protocadherin proteins by the receptor tyrosine kinase Ret. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13894–13899. doi: 10.1073/pnas.1007182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmucker D, Clemens JC, Shu H, Worby CA, Xiao J, Muda M, Dixon JE, Zipursky SL. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell. 2000;101:671–684. doi: 10.1016/s0092-8674(00)80878-8. [DOI] [PubMed] [Google Scholar]
- Schreiner D, Weiner JA. Combinatorial homophilic interaction between gamma-protocadherin multimers greatly expands the molecular diversity of cell adhesion. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14893–14898. doi: 10.1073/pnas.1004526107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature. 2001;410:604–608. doi: 10.1038/35069112. [DOI] [PubMed] [Google Scholar]
- Soba P, Zhu S, Emoto K, Younger S, Yang SJ, Yu HH, Lee T, Jan LY, Jan YN. Drosophila sensory neurons require Dscam for dendritic self-avoidance and proper dendritic field organization. Neuron. 2007;54:403–416. doi: 10.1016/j.neuron.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Nabholz CE, Baldwin KK, Kim Y, Rueckert EH, Ribich SA, Cramer P, Wu Q, Axel R, Maniatis T. Promoter choice determines splice site selection in protocadherin alpha and gamma pre-mRNA splicing. Mol Cell. 2002;10:21–33. doi: 10.1016/s1097-2765(02)00578-6. [DOI] [PubMed] [Google Scholar]
- Thu CA, Chen WV, Rubinstein R, Chevee M, Wolcott HN, Felsovalyi KO, Tapia JC, Shapiro L, Honig B, Maniatis T. Single-cell identity generated by combinatorial homophilic interactions between alpha, beta, and gamma protocadherins. Cell. 2014;158:1045–1059. doi: 10.1016/j.cell.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester-Christensen MB, Halim A, Joshi HJ, Steentoft C, Bennett EP, Levery SB, Vakhrushev SY, Clausen H. Mining the O-mannose glycoproteome reveals cadherins as major O-mannosylated glycoproteins. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:21018–21023. doi: 10.1073/pnas.1313446110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtowicz WM, Wu W, Andre I, Qian B, Baker D, Zipursky SL. A vast repertoire of Dscam binding specificities arises from modular interactions of variable Ig domains. Cell. 2007;130:1134–1145. doi: 10.1016/j.cell.2007.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Maniatis T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell. 1999;97:779–790. doi: 10.1016/s0092-8674(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Wu Y, Honig B, Ben-Shaul A. Theory and simulations of adhesion receptor dimerization on membrane surfaces. Biophysical journal. 2013;104:1221–1229. doi: 10.1016/j.bpj.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Vendome J, Shapiro L, Ben-Shaul A, Honig B. Transforming binding affinities from three dimensions to two with application to cadherin clustering. Nature. 2011;475:510–513. doi: 10.1038/nature10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipursky SL, Grueber WB. The molecular basis of self-avoidance. Annual review of neuroscience. 2013;36:547–568. doi: 10.1146/annurev-neuro-062111-150414. [DOI] [PubMed] [Google Scholar]
- Zipursky SL, Sanes JR. Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell. 2010;143:343–353. doi: 10.1016/j.cell.2010.10.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.