

Abstract
Next-generation sequencing technologies have revolutionarily advanced sequence-based research with the advantages of high-throughput, high-sensitivity, and high-speed. RNA-seq is now being used widely for uncovering multiple facets of transcriptome to facilitate the biological applications. However, the large-scale data analyses associated with RNA-seq harbors challenges. In this study, we present a detailed overview of the applications of this technology and the challenges that need to be addressed, including data preprocessing, differential gene expression analysis, alternative splicing analysis, variants detection and allele-specific expression, pathway analysis, co-expression network analysis, and applications combining various experimental procedures beyond the achievements that have been made. Specifically, we discuss essential principles of computational methods that are required to meet the key challenges of the RNA-seq data analyses, development of various bioinformatics tools, challenges associated with the RNA-seq applications, and examples that represent the advances made so far in the characterization of the transcriptome.
Keywords: RNA-seq, data preprocessing, differential gene expression, alternative splicing, variants detection, pathway analysis, co-expression network, systems biology
Introduction
High-throughput sequencing technologies are being widely applied in biomedical research. Since the initial application, it has expedited tremendous advances in the characterization and quantification of genomes, epigenomes, and transcriptomes over the last few years. Next-generation sequencing (NGS) technology is free from many of the confines dictated by previous technologies, such as the bias due to the probe selection in array technology, cross-hybridization background, and signal saturation-induced detection dynamic range limitation.1,2 Moreover, this high-throughput technology produces large and complex datasets at single nucleotide resolution, and the cost is continuously dropping so that it offers the possibility of investigating the molecular biology genome widely in a far more precise and comprehensive manner as has been previously achieved.
RNA-seq is the set of experimental procedure that generates cDNA sequences derived from the entire RNA molecules, followed by library construction and massively parallel deep sequencing. Gene expression is known to be time-, cell-type-, and stimulus-dependent, and many loci are only expressed under very specific conditions. In fact, the genome-sequencing project has revealed numerous open reading frames encoding “hypothetical” genes, for which expression patterns are not established yet.3,4 RNA-seq allows quantifying the abundance level or relative changes of each transcript during defined developmental stages or under specific treatment conditions. Also, RNA-seq allows for analysis of the transcriptome in a rather unbiased way, with single base pair resolution, a tremendous dynamic detection range (>8,000 fold), and low background signals.5 In contrast to hybridization-based technologies, it is not limited to the interrogation of selected probes on an array and can be also applied in species, for which the whole reference genome is not assembled yet.
RNA-seq is not only a tool for quantitative assessment of RNA but can also be exploratory. Only until recently, it was appreciated that 85% of the human genome can be transcribed, albeit only 3% of the genome encodes protein-coding genes.6 Thus, RNA-seq has been instrumental to catalog the diversity of novel transcript species including long non-coding RNA, miRNA, siRNA, and other small RNA classes (eg, snRNA and piRNA) involved in regulation of RNA stability, protein translation, or the modulation of chromatin states.7,8 For instance, RNA-seq has been used to discover enhancer RNA, a class of short transcript directly transcribed from the enhancer region, which contributes to our knowledge of epigenetic gene regulation.9,10 In addition, RNA-seq can give information about transcriptional start sites, revealing alternative promoter usage, information about mRNA isoforms derived from alternative splicing, and premature transcription termination at the 3′ end, which is critical from mRNA stability.11–15 Most recently, RNA-seq was used to study biological problems including precisely locating regulatory elements.16,17 RNA-seq information can also identify allele-specific expression, disease-associated single nucleotide polymorphisms (SNP), and gene fusions contributing to our understanding about disease causal variants in cancer.18–21 Furthermore, RNA-seq can provide information about the transcription of endogenous retrotransposons and other parasitic repeat elements that may influence the transcription of neighboring genes or may result in somatic mosaicism in the brain.22 Finally, single-cell RNA-seq analysis has been widely applied to study the cellular heterogeneity and diversity in stem cell biology and neuroscience.23–25
While RNA-seq technology is considered unbiased, it is important to note that the preparation and fragmentation of RNA and the library construction (which includes size selection) can be biased.5 This bias may be undesired or unfavorable; for example, the use of oligo (dT) primers in the first strand synthesis enriches poly (A) mRNA, which is useful to study expression of most protein coding genes, but misses on canonical histones,26 some histone variants, and subclasses of non-coding RNA.27 Strand-specific sequencing retains the orientation of the original RNA transcript, which may be critical to identify antisense or non-coding RNA.28
The interpretation of the NGS datasets requires sophisticated and powerful computational programs. The RNA-seq data generation is an ever-evolving process, which includes development in sequencing technology, experiment design, and algorithm development. Accompanied with this, computational tools with varying performances are emerging constantly. A wealth of mature tools exists to meet the basic requirements of RNA-seq data analysis, for instance, the quality assessment and reads mapping. Meanwhile, challenges remain that require comprehensive solutions, such as differential gene expression analysis, as well as the detection of fusion genes. Instead of describing each software, we outline in this study the available tools to perform the analysis of data preprocessing, differentially gene expression (DGE), alternative splicing, variants detection and allele-specific expression, pathway analysis, co-expression network analysis and highlight the essential principles of computational methods in the RNA-seq data analysis, describe the challenges associated with the RNA-seq application, and discuss examples that represent the most advances in the transcriptome characterization.
RNA-seq Workflow
An overview of a typical RNA-seq workflow is outlined in Figure 1. Three main sections are presented: the Experimental Biology, the Computational Biology, and the Systems Biology. The experimental part includes the methods’ choice of RNA collection, first strand synthesis, and library construction, resulting in millions of short reads from the NGS sequencer. Multiple platforms (Table 1) have been applied for the RNA-seq including sequencing-by-synthesis approach Illumina GA IIx and HiSeq,29,30 Applied Biosystems SOLiD,31 Roche 454 Life Science,32 semi-conductor technology-driven Ion torrent Personal Genome Machine,33 single molecule real-time PCR machine PacBio,34 and nanopore technology-driven portable device MinION, and PromethION (http://allseq.com/blog/minion-and-promethion-oxford-nanopore-s-present-and-future).
Figure 1.
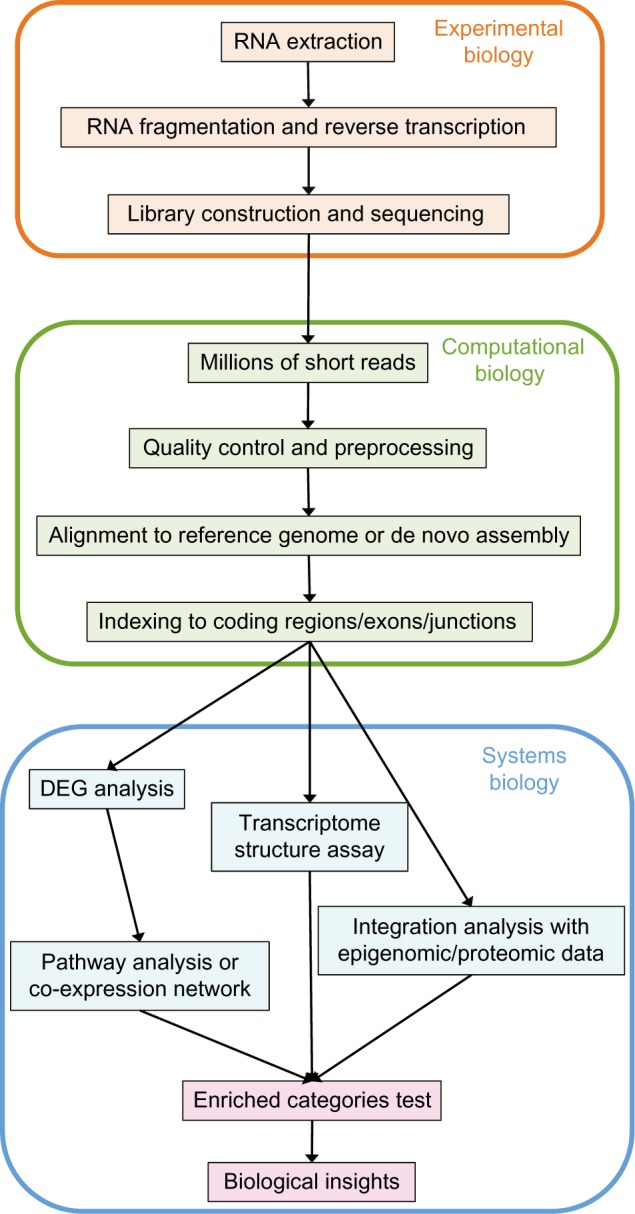
Overview of the typical RNA-seq pipeline. Three main sections are presented: The Experimental Biology, The Computational Biology and The Systems Biology. The pipeline starts from the experimental preparation and come with the work flow to the sequencing and analysis steps as the arrows point from step to step.
Table 1.
Overiew of technical specifications of next generation sequencing platforms.*
| PLATFORM | ILLUMINA GAIIx | ILLMINA HiSeq 2000 | ILLUMINA MiSeq V2 | SOLiD–5500xl | 454 GS FLX+ | ION TORRENT PGM | PacBio RS |
|---|---|---|---|---|---|---|---|
| Chemistry principle | Sequence-by-synthesize | Sequence-by-synthesize | Sequence-by-synthesize | Ligation and two base coding | Pyro-sequencing | Proton detection | Real-time sequencing |
| Instrument price | $256 K | $654 K | $128 K | $251 | $450 K | $80 K (System price including PGM, server, OneTouch and OneTouch ES.) | $695 K |
| Sequence yield per run | 30Gb | 600Gb | 1.5–2Gb | 150Gb | 0.7Gb | 50 Mb on 314 chip, 400 Mb on 316 chip, 1.5Gb on 318 chip | 100 Mb |
| Sequence cost per GB | $148 | $45 | $502 | $67.00 | $50 | $800 (318 chip) | $2,000 |
| Reagent cost per run** | $17,575 | $23,470 | $1,070 | $10,503 | $4,842 | $349 on 314 chip, $549 on 316 chip, $749 on 318 chip | ≥$300 |
| Reagent cost per MB | $0.19 | >$0.04 | $0.14 | <$0.07 | $7 | $5 on 314 chip, $1.2 on 316 chip, $0.6 on 318 chip | $2–17 |
| Run time | 10 days | 11 days | 27 hours*** | 7 Days for SE 14 Days for PE | 20 hours | 2–5 hours | 2 hours |
| Observed raw error rate | 0.76% | 0.26% | 0.80% | <0.1% | 1% | ∼1% | ∼10% |
| Read length | Up to 150 bases | Up to 150 bases | Up to 150 bases | 85 bases | 700 base | ∼200 bases | 3000 bases, up to 15000 bases |
| Read type | PE | PE | PE | PE | SR | PE | SR |
| Insert size | Up to 700 bases | Up to 700 bases | Up to 700 bases | 300 bases | Up to 40 kb | Up to 250 bases | Up to 10 kb |
| Typical DNA amount requirement | 50–1000 ng | 50–1000 ng | 50–1000 ng | 400–4000 ng | 25–1000 ng | 100–1000 ng | ∼1 µg |
| Computation resources | $222 cluste | $222 cluste | Desktop/cloud | $35 cluster | $5 (desktop) | $16.5 (desktop) | $65 cluster |
| Data file sizes (GB)*** | 600 | <600 | 1 | 148 | 40 images, 8 sff | 0.1 sff, 0.2 fastq on 314 chip, 5 sff, 1 fastq on 316 chip, 10 sff, 2.5 fastq on 318 chip | 2 (basecalls, QV, kinetics) |
Notes:
Information based on company sources alone, data update to 2013–2014.
Cost only count the cost per run, does not include general purpose and library preparation equipment, annual maintenance agreements and extra sevices.
New compressed binary data format saves base and quality-value data in a 1byte: 1base ratio.
RNA preparation methods may vary for different kinds of sequencing platforms, RNA subtypes, and sequencing purposes. However, sample quality is always the determinant of acquiring qualified data and deriving biological insights from unbiased analysis. Poly-A-selection of sufficient mRNA is well used in a variety of whole-transcriptome analysis including gene expression, alternative splicing, and variations detection.35,36 While for single cell sequencing, molecular labeling, and random sequencing, the labeled molecules on Illumina platform can achieve remarkable mRNA capture efficiency.36 With more RNA-seq applications in clinical samples, formalin-fixed paraffin-embedded (FFPE) tissue samples became invaluable recourse for transcriptomic studies. Ribo rRNA depletion is the preferred method for archival and long-aged FFPE samples.37
The raw reads served as starting material of the second part, the computational biology. First, technical and biological contaminations were removed from preprocessing steps, followed by mapping the qualified reads to the genome or transcriptome. The mapped reads for each sample were subsequently indexed into gene-level, exon-level, or transcript-level to assess the abundance of each category depending on the experimental purpose. The summarized data were then assessed by statistical models of differentially expression gene list and alternative splicing events, or regulatory mechanisms were evaluated via integration analysis with other datasets such as epigenomic or proteomic data. Finally, pathway or network level analyses were implemented to gain biological insight through the systems biology approaches.
Data Preprocessing
Quality assessment
Since RNA-seq is a complicated, multiple-step process involving sample preparation, fragmentation, purification, amplification, and sequencing, it is not straightforward to identify and quantify all RNA species from the reads sequenced. Hence, quality assessment is the first step of the bioinformatics pipeline of RNA-seq, and also, it is important as a step before analysis. Often, it is necessary to filter data, removing (trimming) low-quality sequences or bases adaptors, contaminations, or overrepresented sequences to ensure a coherent final result. An array of tools are available for this purpose with reads quality visualized graphically such as FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc), HTQC,38 as listed in Table 2. Recently, more flexible and efficient preprocessing tools were developed: Trimmomatic was developed to remove adapters and scan every read with a 4-base sliding window and trim the lower-scored bases and low-quality N bases to enhance the quality of reads39 and HTSeq to depict the base calling and evaluate the base quality at position-based way as well as the overall read features.40
Table 2.
Selected list of packages and tools for RNA-seq data analysis.
| ANALYSIS STEP | PACKAGE | DESCRIPTION AND COMMENTS | REFERENCES |
|---|---|---|---|
| Quanlity assessment and preprocessing | FastQC | A sequencing quality evaluator, easy to use, reports with reads quality visualized graphically. | http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc |
| HTQC | A toolkit including statistics tool for illumina high-throughput sequencing data, and filtration tools for sequence quality, length, tail quality. Depict the base calling and evaluate the base quality at position based way and the overall read features. | 38 | |
| Trimmomatic | Trimmomatic performs a variety of useful trimming tasks for illumina paired-end and single ended data. Remove PCR primers, adpater sequences, scan every read with a 4-base sliding window and trimming the lower-scored bases and low quality N bases to enhance the reads qualityflexible, can handle paired end data. | 39 | |
| BBMap | Short read aligner for DNA and RNA-seq data. Capable of handling arbitrarily large genomes with millions of scaffolds. Handles Illumina, PacBio, 454, and other reads; very high sensitivity and tolerant of errors and numerous large indels. Very fast. BBMerge included which can merge paired reads based on overlap to create longer reads and creates an insert-size histogram. | http://sourceforge.net/projects/bbmap/ | |
| FLASH | A rapid and cost-effective method for large-scale assembly of TALENs. combines paired-end reads that overlapped and converts them to single long reads. | 41 | |
| RSeQC | RSeQC package provides powerful modules that can comprehensively evaluate RNA-seq data after the preprocessing procedure. Some basic modules quickly inspect sequence quality, nucleotide composition bias, PCR bias and GC bias, while RNA-seq specific modules evaluate sequencing saturation, mapped reads distribution, coverage uniformity, strand specificity, etc. | 42 | |
| Mapping | ELAND | The first short read aligner but not the fastest any more. Eland substantially influences many aligners in this category and still outperforms many followers. Eland itself works for 32 bp single-end reads only. Additional Perl scripts in GAPipeline extend its ability. | http://support.illumina.com/help/SequencingAnalysisWorkflow/Content/Vault/Informatics/Sequencing_Analysis/CASAVA/swSEQ_mCA_ReferenceFiles.htm |
| SOAP | A program for efficient gapped and ungapped alignment of short oligonucleotides onto reference sequences. SOAP is compatible with numerous applications, including single-read or pair-end resequencing, small RNA discovery and mRNA tag sequence mapping. SOAP is a command-driven program, which supports multithreaded parallel computing, and has a batch module for multiple query sets. | 45 | |
| SOAP2 | An updated version of SOAP software for short reads alignment. Super fast and accurate alignment for huge amounts of short reads, includes a single individual genotype caller (SOAPsnp, SOAPsnv, SOAPindel) | 46 | |
| MAQ | A program to align short reads and to call variants. Features includes PET mapping, quality aware, gapped alignment for PET, mapping quality, adapter trimming, partial occurrences counting, and SNP caller. | 47 | |
| Bowtie | An ultrafast, memory-efficient short read aligner. Bowtie indexes the genome with a Burrows-Wheeler index to keep its memory footprint small. Useful unspliced aligners. | 48 | |
| BWA | A software package for mapping low-divergent sequences against a large reference genome. It consists of three algorithms: BWA-backtrack, BWA-SW and BWA-MEM, which are suitable for reads length from 70 bp to 1Mb. | 49 | |
| ZOOM | A framework that is able to map the Illumina/Solexa reads of 15x coverage of a human genome to the reference human genome in one CPU-day, allowing two mismatches, at full sensitivity. | 50 | |
| STAR | An ultrafast universal RNA-seq aligner which utilizes sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure. STAR has a potential for accurately aligning long (several kilobases) reads that are emerging from the third-generation sequencing technologies. | 51 | |
| BLAT | 54,55 | ||
| HTSeq | A Python framework to work with high-throughput sequencing data, able to perform sequencing quality evaluation, reads counting. It is flexible that customize the needs by writing scripts or just use the stand alone scripts. | 40 | |
| Easy RNASeq | A bioconductor package for processing RNA-Seq data, which perform count summarization per feature of interest and count normalization. | 64 | |
| Geno micRanges | A bioconductor package defines general purpose containers for storing genomic intervals. Specialized containers for representing and manipulating short alignments against a reference genome are defined in the GenomicAlignments package. | 65 | |
| Feature-Counts | An R package suitable for counting reads generated from either RNA or genomic DNA sequencing. It implements highly efficient chromosome hashing and feature blocking techniques so considerably faster than existing methods and requires far less computer memory. | 66 | |
| Expression quantification | Alexa-seq | A comprehensive package that include a database for alignment, gene expression euantification, extract isoform features and visualize the results. | 12 |
| Cufflinks | Transcriptome assembly and differential expression analysis for RNA-Seq. It also can perform Isoform Quantification, Maximum likelihood estimation of relative isoform expression. | 7,83,84 | |
| RSEM | A package for quantifying gene and isoform abundances from single-end or paired-end RNA-Seq data. RSEM outputs abundance estimates, 95% credibility intervals, and visualization files and can also simulate RNA-Seq data. In contrast to other existing tools, the software does not require a reference genome. Thus, in combination with a de novo transcriptome assembler, RSEM enables accurate transcript quantification for species without sequenced genomes. | 79 | |
| Differential expression | Cuffdiff | A robust and accurate tool for differential analysis of RNA-Seq experiments. isoform level analysis, Uses isoform levels in analysis. | 7,83,84 |
| DESeq | An R package to analyse count data from high-throughput sequencing assays such as RNA-Seq and test for differential expression. It uses multi-factors analysis, Poisson GLM. | 31 | |
| DESeq2 | A method for differential analysis of count data, using shrinkage estimation for dispersions and fold changes to improve stability and interpretability of estimates. This enables a more quantitative analysis focused on the strength rather than the mere presence of differential expression. | 85 | |
| EdgeR | A bioconductor software package for examining differential expression of replicated count data. An overdispersed Poisson model is used to account for both biological and technical variability. Empirical Bayes methods are used to moderate the degree of overdispersion across transcripts, improving the reliability of inference. The methodology can be used even with the most minimal levels of replication, provided at least one phenotype or experimental condition is replicated. The software may have other applications beyond sequencing data, such as proteome peptide count data. | 82 | |
| PoissonSeq | A method for normalization, testing, and false discovery rate estimation for RNA-sequencing data based on poisson log-linear model. | 86 | |
| Limmavoom | Limma is data analysis R package based on linear models and differential expression for microarray data. voom function in the limma package offers a way to transform count data into Gaussian distributed data so that significance can be tested statistically. | 87, 88, 89 | |
| MISO | A probabilistic framework that quantitates the expression level of alternatively spliced genes from RNA-Seq data, and identifies differentially regulated isoforms or exons across samples. | 105 | |
| Altenative splicing | TopHat | A widely used, fast splice junction mapper for RNA-Seq reads. It aligns RNA-Seq reads to mammalian-sized genomes using the ultra high-throughput short read aligner Bowtie, and then analyzes the mapping results to identify splice junctions between exons. | 56, 57 |
| MapSplice | An algorithm for mapping RNA-seq data to reference genome for splice junction discovery. It utilizes the exon-first methods, supports both single-end and pair-end reads with high memory efficiency and accuracy. | 59 | |
| SpliceMap | A de novo splice junction discovery and alignment tool. It offers high sensitivity and accuracy and support for arbitrary RNA-seq read lengths. | 106 | |
| SplitSeek | A program for de novo prediction of splice junctions in RNA-seq data. It utilizes the exon-first method. | 107 | |
| GEM mapper | A fast, accurate and versatile alignment by filtration. It can leverage string matching by filtration to search the alignment space more efficiently, simultaneously delivering precision and speed. | 58 | |
| SpliceR | An easy-to-use tool that extends the usability of RNA-seq and assembly technologies by allowing greater depth of annotation of RNA-seq data. | 108 | |
| Splicing-Compass | A method and software to predict genes that are differentially spliced between two different conditions using RNA-seq data. | 109 | |
| GliMMPS | A robust statistical method for detecting splicing quantitative trait loci (sQTLs) from RNA-seq data. | 110 | |
| MATS | A computational tool to detect differential alternative splicing events from RNA-Seq data. The statistical model of MATS calculates the P-value and false discovery rate that the difference in the isoform ratio of a gene between two conditions exceeds a given user-defined threshold. From the RNA-Seq data, MATS can automatically detect and analyze alternative splicing events corresponding to all major types of alternative splicing patterns. MATS handles replicate RNA-Seq data from both paired and unpaired study design. | 111 | |
| rMATS | A statistical model and computer program designed for detection of differential alternative splicing from replicate RNA-Seq data. rMATS uses a hierarchical model to simultaneously account for sampling uncertainty in individual replicates and variability among replicates. | 112 | |
| Varients detection | GATK | Package for aligned NGS data analysis, which includes a SNP and genotype caller (Unifed Genotyper), SNP filtering (Variant Filtration) and SNP quality recalibration (Variant Recalibrator). | http://gatkforums.broadinstitute.org/discussion/3891/calling-variants-in-rnaseq). |
| ANNOVAR | An efficient software tool to functionally annotate genetic variants (gene-based, region-based or filter-based) detected from diverse genomes. | 115 | |
| SNPiR | A highly accurate approach termed SNPiR to identify SNPs in RNA-seq data. | 116 | |
| SNiPlay3 | A web-based application for exploration and large scale analyses of genomic variations. | 117 | |
| Pathway analysis | GSEA | A knowledge-based approach for interpreting genome-wide expression profiles. It determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (eg, phenotypes). | 130 |
| GSVA | A non-parametric, unsupervised method for estimating variation of gene set enrichment through the samples of a expression data set. GSVA performs a change in coordinate systems, transforming the data from a gene by sample matrix to a gene-set by sample matrix, thereby allowing the evaluation of pathway enrichment for each sample. | 131 | |
| SeqGSEA | The package generally provides methods for gene set enrichment analysis of high-throughput RNA-Seq data by integrating differential expression and splicing. It uses negative binomial distribution to model read count data, which accounts for sequencing biases and biological variation. Based on permutation tests, statistical significance can also be achieved regarding each gene’s differential expression and splicing, respectively. | 132 | |
| GAGE | An evaluation of the very latest large-scale genome assembly algorithms. | 133 | |
| SPIA | An R package that uses the information form a list of differentially expressed genes and their log fold changes together with signaling pathways topology, in order to identify the pathways most relevant to the condition under the study. | 135 | |
| TAPPA | A java-based tool, for identification of phenotype-associated genetic pathways utilizing the pathway topological measures. | 136 | |
| DEAP | A tool capitalizes on information about biological pathways to identify important regulatory patterns from differential expression data. It makes significant improvements over existing approaches by including information about pathway structure and discovering the most differentially expressed portion of the pathway. | 137 | |
| GSAA SeqSP | A toolset for gene set association analysis of RNA-Seq count data. GSAASeqSP identify pathways/gene sets significantly associated with a disease or a phenotype by analyzing genome-wide patterns of gene expression variation measured by RNA-Seq technology. | 134 | |
| Co-expression network | GSCA | An open source software package to help researchers use massive amounts of publicly available gene expression data (PED) to make discoveries. Users can interactively visualize and explore gene and gene set activities in 25,000+ consistently normalized human and mouse gene expression samples representing diverse biological contexts. | 146 |
| DICER | A method for detecting differentially co-expressed gene sets using a novel probabilistic score for differential correlation. DICER goes beyond standard differential co-expression and detects pairs of modules showing differential co-expression. | 151 | |
| WGCNA | A powerful method to extract co-expressed groups of genes from large microarray data sets and has been successfully applied to RNA-seq data. It is suggested to remove genes whose read counts are consistently low and normalize the data with a variance-stabilizing transformation before calculating pairwise similarity of expression pattern. | 152 |
Before alignment to the reference genome, RNA-seq data can be further preprocessed to meet expectations in the next sequencing mapping steps. There are multiple tools available for this purpose, for example, BBMerge from BBMap package (http://sourceforge.net/projects/bbmap/) merges paired reads based on overlap to create longer reads and creates an insert-size histogram. FLASH41 combines paired-end reads that overlapped and converts them to single long reads. It is also a good practice to assess the RNA-seq data quality after the preprocessing procedure, and there are packages, for example, RSeQC package, to comprehensively evaluate the reads that will go to analysis.42
Reads mapping
Once high-quality data are obtained from preprocessing, the next step is to map the short reads to the reference genome or to assemble them into contigs and align them to the reference genome. This procedure refers to the classic bioinformatics problem of discovering the most reliable original sources of a large scale of short DNA sequences from the genome in a speed- and memory-efficient manner.43,44 There are many popular bioinformatics programs that can be used for this purpose, including ELAND (http://support.illumina.com/help/SequencingAnalysisWorkflow/Content/Vault/Informatics/Sequencing_Analysis/CASAVA/swSEQ_mCA_ReferenceFiles.htm), SOAP,45 SOAP2,46 MAQ,47 Bowtie,48 BWA,49 ZOOM,50 STAR,51 etc. Comparable analyses on real data have been done to assess most mapping tools.52 These programs are typically suitable for reads that are not located at the poly (A) tails or exon-intron splicing junctions. Poly (A) tails can be easily identified by the presence of multiple As or Ts, and a partial junction library that contains the known junction sequence has been compiled to allow the alignment of difficult mapping reads.23,53 From a different point of view, the reads that locate at the exon–intron boundaries are helps with the determination of the alternative splicing pattern, where advent RNA-seq promotes the development of new generation of slice-alignment software such as BLAT,54,55 TopHat,56,57 GEM,58 and MapSplice.59
Another problem in reads mapping is that of polymorphisms, which occur when sequence reads align to multiple locations of the genome. Polymorphisms are especially common for the large and complex transcriptomes. For lower repetitive reads, one can employ the solution of assigning the reads to multiple locations proportionally based on the neighboring unique reads.31,53 However, for the short reads that have a very high copy number and repetitive sequences, polymorphism is still a great challenge. A longer read sequencer such as the Roche 454 or PacBio sequence analyzer might be required. Alternatively, there are bioinformatics solutions to extend the short pair-end reads into 200–500 bp fragments before deciding upon the multiple-aligned reads.60–62
Reads counting
RNA-seq reads number that map to a gene is the measurement of the gene’s expression level. After mapping the reads to the reference genome, counting the reads number that mapped to gene body will facilitate the next steps. Library preparation methods, such as whether the protocol is strand-specific, whether first read is on the same strand or opposite strands, are determinant factors for the counting of reads. One example of tools and packages for read counting from bam file is the multicov command in bedtools that takes a feature file (GFF) and read counts in certain regions, such as all exons of a gene.63 By default, it counts reads on both strands within interested regions. But it can work in a strand specific manner if necessary. HTseq is a specialized utility for counting reads although speed lifting is necessary in the future.40 However, it allows us to look for more fine-grained controls on read counting by setting different parameters. This is very useful, especially when a read overlaps more than one gene and we want to use customized strategy. Note that HTseq-counts assume that the RNA-seq data is strand-specific; it will only count those reads that were mapped to the strand that the feature is on. R packages include easyRNASeq, summarizeOverlaps and featureCounts for reads counting. easyRNASeq hides the complex interplay of the required packages and thus can be easily used. summarizeOverlaps, which is a function in the Genomics-Ranges package in Bioconductor, and featureCounts, which have implemented, highly efficient chromsome hashing and feature blocking methods, are suitable for RNA-seq or genomic DNA sequencing data. Different tools and their different related parameters generate different reads’ numbers, and thus affect downstream analysis because they use different strategies to assign reads to features.
In addition, the gene model that hypothesizes the structure of transcripts produced by a gene also affects the analysis. Among multiple genome annotation databases, RefGene, Ensembl, and the UCSC annotation databases are the most popular ones. The choice of genome annotation directly affects gene expression estimation. Recently, Zhao and Zhang systematically characterized the impact of genome annotation datasets choice on read mapping and transcriptome quantification.67 They found that the impact of a gene model on mapping of nonjunction reads is different from junction reads. The percentage of correct mapped nonjunction reads was much higher than that of the junction reads for all gene models. Surprisingly, although there are 21,958 common genes among RefGene, Ensembl, and UCSC annotation, only 16.3% of genes obtained identical quantification results. Approximately 28.1% of genes’ expression levels differed by ≥5% when using different annotation, and of those, the relative expression levels for 9.3% of genes differed by ≥50%. This study revealed that the different gene definition of gene models frequently result in inconsistency in gene quantification.
Normalization
After getting the read counts, data normalization is one of the most crucial steps of data processing, and this process must be carefully considered, as it is essential to ensure accurate inference of gene expression and subsequent analyses thereof. There are multiple facets of the RNA-seq data to be taken into account including transcript size, GC-content, sequencing depth sequencing error rate, insert size, etc.68,69 Multiple normalization methods should be compared for the specific bias elimination of a dataset, which can be done by comparing their corresponding estimated performance parameters using measurement error models.69,70 Plenty of comparative analysis or integrative analysis concluded the best approach in different types of RNA-seq data analysis. For instance, quantile normalization can improve the mRNA-seq data quality including those from low amounts of RNA.71,72 R package EDASeq using within-lane normalization procedures followed by between-lane normalization can reduce GC-content bias.73 Lowess normalization and quantile normalization worked well in microRNA-seq data normalization.74 Further advancement of RNA-seq application calls for the development of effective statistical and computational methods for RNA-seq data normalization.
There are other bioinformatics challenges for the RNA-seq reads mapping, for example, reducing the errors in image analysis and base calling to enhance sequencing accuracy; removing low-quality reads; and the development of applicable approaches to store, retrieve, and process large datasets in a time- and energy-efficient manner.
Differential Gene Expression
The transcriptome is the complete set of transcripts in a cell or cell population, and transcriptome analysis provides information about the identity and quantity of all RNA molecules. An important application of RNA-seq is the comparison of transcriptomes across different developmental stages, across a disease state compared to normal cells, or specific experimental stimuli compared to physiologic conditions. This type of analysis requires identification of genes along with their isoforms and precise assessment of their abundance comparing two or multiple samples. It is essential for interpreting the functional elements of the genome and uncovering the molecular constitution providing important insights in the biological mechanisms of development and diseases.
After the step of preprocessing RNA-seq reads, it is an important question to reveal how the transcripts level differs across samples, known as DGE analysis. Numerous statistical methods have been developed that use read coverage to quantify transcript abundance since the microarray era.75,76 The RPKM (reads per kilo base per million mapped reads) is widely used method to account for expression and normalized read counts with respect to overall mapped read number and gene length.32,53 However, beside the read coverage, there are other factors that determine the estimated transcript abundance including sequencing depth, gene length, and isoforms abundance.72,77,78 Since the RPKM method handles all the RNA-seq reads almost equally, for example, without concern for isoforms, it has been criticized. RNA-Seq by Expectation Maximization (RSEM) is a newly developed software tool, which gives accurate estimates for gene and isoform expression levels and can be used even for species without a reference genome assembly.79
Most algorithms to date for differential gene expression analysis apply simple count-based probability distributions (eg, Poisson distribution) followed by Fisher’s exact test without accounting for biological variability among samples.32,53,80 While the technical variability of RNA-seq is extremely low compared with microarray data,32 the biological variability could be significantly reduced by analyzing several replicates through a permutation-derived methods.75 Serial analysis of gene expression has been developed for biological variability assessment, in which larger scale datasets are used so that an additional dispersion parameter can be estimated based on an extended Poisson distribution, allowing extensive molecular characterization capability.81,82
However, for most applications, a large number of replica may be too costly, and many developed methods have overcome the problem by modeling biological variability and measuring the significance with limited number of samples, applying pairwise or multiple group comparisons.75 Several programs offer well-done solution for this purpose and have been applied in numerous studies for biomedical and clinical research. Examples of these programs are Cuffdiff from the Cufflinks package,7,83,84 DESeq,31 DESeq2,85 and EdgeR.82 Since RNA-seq read counts are integer numbers that range from zero to millions and are highly skewed, many kinds of transformation algorithms have been applied to the counts so that the numbers can be fit to statistic distribution models for differential expression detection. For instance, Li et al developed PoissonSeq, a Poisson log-linear model for differential gene expression assay.86 Approaches developed for microarray data analysis based on continuous distribution have been improved for RNA-seq counts. Excellent example is the voom function in the limma package, which offers a way to transform count data into Gaussian distributed data so that significance can be tested statistically.87–89 An extensive comparison to evaluate the performances of several DGE packages has been recently reported.90,91 However, to the best of our knowledge, there is no one-size-fits-all strategy. Also, space for refinement of existing pipelines exists to develop effective strategies for the following questions: how to uniform the reads coverage along the genome with the nucleotide composition variation; how to detect the “within-sample” variations without simply assuming that the underlying conditions or treatments affect all individual gene equally; how to improve current methods to detect differences in gene isoform preferences and abundance level in varying conditions; and how to account for the different probability in read coverage in long genes versus short genes since we can gain great sequencing depth nowadays.
Alternative Splicing
The biological complexity and genomic diversity are determined, to a large degree, by the alternative splicing events.92 Alternative splicing shapes the control of numerous pivotal cellular processes, and abnormal splicing events are involved in 15%–50% of disease-causing mutations in human.93 Compared with constitutive splicing, alternative splicing refers to the differential inclusion/exclusion of exons in the processed RNA product after splicing of a precursor RNA segment.94 It is a crucial step in controlling the expression of ~95% of all multiexon genes, and an increasing number of diseases are found to be associated with the “wrong” splice sites usage, while the overall transcript abundance does not change.95,96 Spliceosomes, composited of intricate structures with RNA–RNA, protein–protein, and RNA–protein interactions, carry out the splicing reaction.97 Splicing mechanism studies on model genes have deduced many regulatory principles including the role of negative intrinsic sites binding and positive enhancement of splicing sites selection in the formation of spliceosome assembly.94
However, given the variety of cis-acting elements and trans-acting factors involved in splicing, either cooperatively or in a competing manner, the “code” for controlling alternative splicing needs still further deciphering using high-throughput approaches.98 RNA-seq technology allows us to estimate alternative splicing events on genome-wide scales and in an unbiased manner. Deep surveying of alternative splicing by RNA-seq revealed unprecedented wealth of splice junctions and RNA-binding motifs and provides more reliable measurements compared with microarray technology.99–101 Furthermore, alternative splicing is tissue-specific, with hundreds of context-sensitive RNA features and tissue-dependent splicing regulatory elements, which generate thousands of combinations of alternative splicing events.102,103 In-depth of RNA-sequencing analysis yield a digital inventory of gene and mRNA isoform expression with tissue specificity and high sensitivity of single cells and provides a framework of understanding alternative splicing pattern on genome-wide scales.15,104
With the rapid accumulation of RNA-seq data, many methods and tools have been developed to infer alternative splicing events. These tools generally focus on either gapped alignment of short reads or de novo assembly and characterization of transcript models. Examples of these methods are MISO for identification and regulation of isoforms from CLIP-seq data and105 SpliceMap,106 SplitSeek,107 spliceR,108 and SplicingCompass109 for detection of splice junctions and exon usage from pair-end RNA-seq. GLiMMPS provides a useful tool for elucidating the genetic variation of alternative splicing in humans and model organisms.110 MATS is developed from a statistical method and used for detecting differential alternative splicing events from RNA-seq data.111 rMATS is a statistical method for robust and flexible detection of genome-wide differential alternative splicing from paired or unpaired replicates.112 ALEXA-Seq assesses the differential and alternative expression of the mRNA isoforms after cataloging transcripts.12 An integrative analysis approach constructed an exon co-splicing network based on distances combined with matrix correlations and found that the co-splicing network was distinct and complementary to the co-expression network, although they both possess scale-free properties.113,114
The field of alternative splicing analysis using RNA-seq data is still in its infancy and would benefit from new strategies. An extensive evaluation and comparison of the existing methods would be desirable, and to date, there is no general consensus regarding which method performs best under given conditions. We are expecting to see the novel, exploring methods to be developed in this flourishing field in the near future.
Variants Detection and Allele-Specific Expression
The main applications of RNA-seq analysis are novel gene identification, expression, and splicing analysis. However, RNA-seq data is also a useful by-product of sequence-based mutation analysis, though there are many limitations, such as highly differential coverage between different genes. Among many variants calling and annotation methods such as ANNOVAR,115 SNPiR,116 and SNiPlay3,117 the best practical workflow provided by GATK may be still the best pipeline to identify mutations from RNA-seq data, although it is still far from perfect and under heavy development (http://gatkforums.broadinstitute.org/discussion/3891/calling-variants-in-rnaseq). In GATK pipeline, the sequence reads are first mapped to the reference using STAR aligner (2-pass protocol) to produce a file in SAM/BAM format sorted by a coordinate. After marking and removing duplicates, GATK splits reads with N operators in the CIGAR strings into component reads and trims to remove any overhangs into splice junctions to reduce the occurrence of artifacts. The remaining steps are similar to DNA-seq variants calling, such as local alignment and haplotype variant call.
Heterozygous SNP, which means two different alleles in the same position in the DNA, may lead to the following: one of two alleles is highly transcribed into mRNA and another is lowly transcribed or even not transcribed at all. This is called as allele-specific expression (ASE). Both genetic and epigenetic determinants govern transcriptional activity at the different alleles of a gene in a non-haploid genome, and impairment of this highly regulated process can lead to disease.118,119 Whole genome DNA sequencing (WGS) allows identification of single nucleotide mutations or polymorphisms in the entire human genome. The expression state of the heterozygous loci can be investigated in the matched RNA-Seq and WGS sample from the same individual, and ASE activity can be identified to uncover the instances of allele silencing.120 Though conceptual simple, there is still a challenge to identify ASE due to many problems, such as reads bias and lack of sophisticated statistical model.121 Recently, Mayba et al developed a pipeline, MBASED to ASE detection, through aggregating information across multiple single nucleotide variation loci to obtain a gene-level ASE.122 More sophisticated softwares are needed for ASE identification.
Beyond the Differentially Expressed Gene Lists
Creating lists of the differentially expressed genes is only the starting point of gaining biological insights into experimental systems, developmental stages, or specific disease scenarios. To understand the biologic context of differentially expressed genes, many advanced analyses have been working on gene ontology,123,124 gene sets,125 network inference, and knowledge databases.126,127
Pathway Analysis
The interpretation of gene expression data is based on the function of individual genes as well as their role in pathways since genes work connectively in all biological processes. In addition, for some genes, a small expression change may be not significant at a single gene level, but minor changes of several genes may be relevant in a pathway and may have dramatic biological consequences. Thus, differentially expressed biological pathways provide better explanatory results than a long list of seemingly unrelated genes.128
One traditional analysis works with a gene list of interest, identified with genomics methods or curated by biologists, and applies statistical methods, such as the Fisher Exact Test, on contingency tables to test for enrichment of each annotated gene set.129 Such approaches can be applied to the differentially expressed gene list identified with RNA-seq data directly. Another class of analysis ranks all expressed genes according to metrics of expression difference and then uses Kolmogorov–Smirnov like tests to obtain enrichment significance. Gene set enrichment analysis (GSEA) is one such highly effective method that has been widely used in studying functional enrichment between two biological groups.130
Many studies have adapted pathway analysis tools from microarray data analysis and developed new tools applicable to RNA-seq data. For example, a non-parametric competitive GSA approach named Gene Set Variation Analysis has been developed to fit RNA-seq data characteristics. Such analyses have given highly correlated results between microarrays and RNA-Seq sample sets of lymphoblastoids cell lines that have been profiled using both technologies.131 SeqGSEA uses count data modeling with negative binomial distributions to score differential expression and then executes gene set enrichment analysis to achieve biological insights. In real applications, SeqGSEA detects more biologically meaningful gene sets without biases toward longer or more highly expressed genes.132 GAGE is another method for pathway analysis that is applicable to both microarray and RNA-seq data. It is unaffected by sample sizes, experimental designs, assay platforms, or other types of heterogeneity.133 GSAASeqSP offers a variety of statistical procedures by adapting and combining multiple gene-level and gene set-level statistics for RNA-seq count-based data. Such statistics include Weighted_KS, L2Norm, Mean, WeightedSigRatio, SigRatio, GeometricMean, TruncatedProduct, FisherMethod, MinP, and RankSum.134 GSAASeqSP is a powerful platform for investigating molecular differential activity within biological pathways.
The limitations of the gene set analysis methods developed for microarrays in the context of RNA-seq data have been comprehensively investigated.128 Several frequently used RNA-seq normalization strategies were studied to examine the performance of multivariate tests. Data transformations were also investigated in an attempt to extend other approaches beyond microarray data analysis. It was found that the use of log counts when normalized for sequence depth is a good strategy for data transformation prior pathway analysis.
Previously, pathway analysis methods had been developed based on algorithms considering pathways as simple gene lists and ignoring pathway structure. Recently, methods have been developed that incorporate various aspects of pathway topology. For example, SPIA captures pathway topology through its scoring system, in which the positions and the interactions of the genes in the pathway are considered.135 Accordingly, interacting differentially expressed gene pairs are preferentially weighted over two non-interacting genes. Similarly, TAPPA is a scoring method in which higher weights are automatically assigned to hub genes and interacting gene pairs.136 DEAP identifies the most differentially expressed path to provide a refined focus for further biological exploration.137 Accordingly, biological pathways are represented by directed graphs, where nodes are biological compounds and the edges represent catalytic or inhibitory regulatory.
Applying methods developed for microarray data analysis without considering specific data features of RNA-seq data may lead to biases. For example, long or highly expressed transcripts are more likely to be detected as differentially expressed than are the short and/or lowly expressed ones. By developing new statistical framework, the new problem of gene length bias and total reads number bias from RNA-seq could be well corrected. One good example is the GOseq package for gene ontology analysis. It considered the read counts bias by estimating the probability weighting function and used resampling strategy beyond the differentially expressed gene expression so that it can highlight GO categories more consistent with the known biology.138 Development of good methods to correct the biases in pathway analysis brought by GC content, dinucleotide distribution, and other factors is challenging.139
Although many pathway databases are available, high-resolution annotation of such knowledge bases is still lacking. For example, >90% of the human genome is alternatively spliced and transcripts from the same gene may have distinct, even opposing functions. However, current knowledge bases only are curated at gene level. It is essential to also include knowledge about pathway-specific transcript activity. In addition, high-quality annotations for genes are still needed, although there are enormous numbers of annotations available in the public domain.140 We expect to see more sophisticated data mining and machine learning algorithms applicable to RNA-seq data, especially those methods considering the gene in the context of its pathway.
Co-expression network analysis
Co-expression network analysis is an important complement to DGE analysis. A gene co-expression network is represented as an undirected graph, in which each node corresponds to a gene, and two nodes are linked if there is a significant co-expression relationship between them. Because co-expressed genes are often functionally related, controlled by the same set of transcriptional factors, or work together within same pathway, building co-expression networks can help to extract meaningful biological modules that are tightly associated within a specific biological process.141
The co-expression network has been extensively studied since microarray era and such data have been examined using RNA-seq data with the emergence of NGS technology. Comparison studies between RNA-seq co-expression networks and microarray data-derived networks revealed that correlations from RNA-seq data are much higher due to the reason that RNA-seq data is of greater sensitivity and larger dynamic range. Although both co-expression networks show scale-free properties, there is low overlap between hub-like genes. This phenomenon can be explained by low correlation between microarray and RNA-seq data, especially for high- and low-transcript abundances.142
Both sample size and reads’ depth affect the quality of RNA-seq-derived co-expression networks.143 Larger sample sizes and greater read depth can increase the functional connectivity of the networks. The minimal suggested experimental criteria to obtain performance on par with microarrays are at least 20 samples with total number of reads greater than 10 million per sample. Meta-analysis across multiple data sets is a good solution to increase the relatively poor performance of individual co-expression networks. Aggregation across different experiments can improve performance significantly beyond that attained by even the largest individual co-expression networks in one experiment. However, thousands of samples from different conditions are necessary to obtain the “gold standard” co-expression networks.
The high quality of co-expression network by large meta-analysis promises the power of a functional genomics tool to biologists and clinicians. GeneFriends project team has constructed co-expression maps for human and mouse with RNA-seq datasets of 4,000 and 2,500 samples from different experiments, respectively.144 This information can be used statistically, such as using a guilt by association approach to predict gene function, identifying and prioritizing novel candidate genes involved in biological processes. COXPRESdb is another database of RNAseq-based gene co-expression networks.145 The co-expressed gene list in COXPRESdb provides a comparable view of orthologous genes among several species (human, mouse, rat, chicken, fly, zebra fish, nematode, monkey, dog, and yeast) and the numbers of common edges for all pairs of species.
Besides building gene co-expression networks under defined conditions, finding co-expression modules in one condition and then testing if these modules show different co-expression in other conditions can assist in understanding the regulatory change under disease conditions. Gene set co-expression analysis was proposed to test differential co-expression of known pathways through testing the changes in co-expression over all gene pairs in the pathway.146 Based on theoretical analysis, a small highly co-expressed subnetwork was found to be a good indicator of disease onset or other biological process. This finding has been validated with real data and confirmed that this small set of genes clustered within a strongly correlated subnetwork is able to provide the significant warning signal just before onset of disease.147 The current approach of building dynamic network biomarker is based on population data. It might be interesting to build a co-expression of network with time series data from same subject with self-correlation or synchronization,148,149 such that we can use it to predict disease onset for diagnosis and personalized medicine.
Interestingly, in biological systems, antagonistic and self-reinforcing co-expressed modules have been found in system stability and adaptability.150 Algorithm have been designed to model this phenomenon, for instance, DICER, in which the expression profiles of genes within each module of the pair are correlated across all samples and the correlation between the two modules differ dramatically between the disease and normal samples.151 Weighted gene co-expression network analysis is a powerful method to extract co-expressed groups of genes from large microarray data sets and has been successfully applied to RNA-seq data. It is suggested to remove genes whose read counts are consistently low and normalize the data with a variance-stabilizing transformation before calculating pairwise similarity of expression pattern. It can perform various aspects of weighted correction of co-expression network analysis including network construction, module detection, gene selection, calculations of topological properties, data simulation, visualization, and interfacing with external software.152
As more and more RNA-seq data become publically available, there is a great need to develop new algorithms to formulate both the global and local characteristics of co-expression networks, especially those dynamic changes associated with biological processes. Much work still remains for the development of RNA-seq co-expression methodologies. So far, there have been few published statistical studies that have examined metrics for similarity of expression profiles with RNA-seq data. Si et al designed an algorithm to cluster genes by measuring the differential expression patterns across treatments using model-based statistical methods according to either Poisson or NB models for RNA-seq data, using the mean expression level as reference, bypassing treating RNA-seq data directly.153 The co-expression networks built with different expression measurements, such as those using raw counts, RPKM, or variance-stabilizing transformation have low overlap. Therefore, the development of new metrics for co-expression network establishment is urgently needed.142
Centrality and network flow have been successful for the identification of important genes and modules from co-expression networks. However, we lack a good way to formulate the network structure. Some network metric, such as percolation, is too simple to grasp network characteristics efficiently due to the dynamic nature of the biological processes. The lack of a model that represents the dynamic change of co-expression network at different time points limits our ability to observe biological system changes at a network level.150
Systems Biology
High-throughput sequencing technologies are now routinely being applied to a wide range of topics in biology and medicine, allowing scientists to address important questions and reveal difficult discoveries that were impossible before. Advances in genome sequencing and data analysis are of critical roles, while the procedure for how to prepare samples selectively and how to generate qualified data requires sophisticated experimental design, which is essential part of systems biology (Fig. 2).
Figure 2.
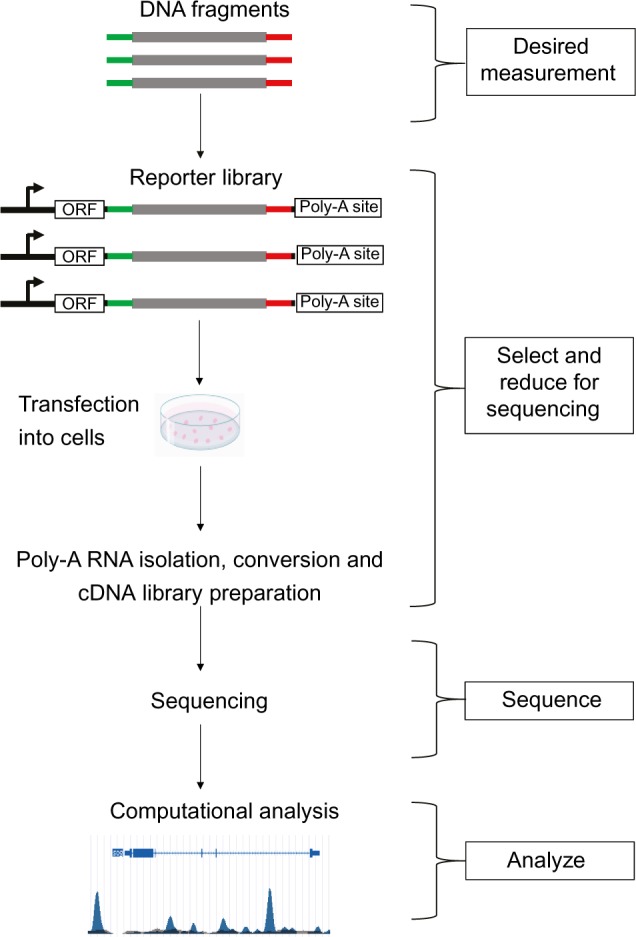
The STARR-seq pipeline and the corresponding ‘systems biology’ steps. The sonicated genomic DNA are PCR amplified and placed downstream of a minimal promoter in reporter vectors. The desired measurement are embedded in the genome. The reporter library is transfected into the cultured cell lines and Poly-A RNAs are isolated from the pool of total RNA. These steps are selectively to enrich the targets interested. After RNA-seq is performed, the reads are mapped to the reference genome and their enrichment over input are measured to reflect enhancer activity. The steps of systems biology including mathematics and computational biology analysis will help with the interpretation.
Concerning gene expression analysis, the integration datasets from diverse platforms in this Next-Generation Genomics era, including genomics, epigenomics, and proteomics with transcriptomics, is critical in the effort to understand complex biological systems. A wide scope of integrating analysis projects were well defined for a more complete picture of gene regulation such as the Roadmap Epigenomics Project, the ENCODE Project, and The Cancer Genome Atlas.154 RNA-seq has been used in combination with transcription factor (TF) binding,155,156 histone modification,157,158 DNA methylation,159,160 genotyping data,161,162 and RNA interference.163 In this study, we summarize two excellent examples to illustrate RNA-seq application in the frame of systems biology.
STARR-seq: whole genome functional readout of enhancers
Enhancers are functional non-coding DNA sequences that can recruit TFs, physically interact with promoters, and regulate the timing and tissue specificity of gene expression.164–169 Despite their important roles during development, in response to stimuli and various diseases, a genome-wide approach to identify functional enhancer regions is still lacking. Current high-throughput enhancer detection methods can be grouped into three categories: (1) identification of open chromatins, including deep sequencing of DNase hypersensitive sites (DHS-seq)170 and formaldehyde-assisted isolation of regulatory elements sequencing (FAIRE-seq)171; (2) chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) on enhancer-associated histone modifications (H3K4me1, H2K27ac, H3K18ac, etc.)172–175; and (3) ChIP-seq on TFs or cofactors (p300, CEBPB, etc.).176 However, the mapping of open chromatins and histone modifications usually lacks sufficient resolution and specificity to detect precise enhancer locations, and the binding of some specific TFs or cofactors can hardly cover all the active enhancers. Moreover, none of these methods can provide a quantitative measurement of enhancers’ activities. The traditional quantitative reporter assays, on the other hand, cannot be scaled up to a high-throughput genome-wide manner.177,178
To address this question, Arnold and colleagues developed a method, named self-transcribing active regulatory region sequencing (STARR-seq),17 which quantitatively measures the activity of enhancers in the whole genome. They sheared Drosophila melanogaster genomic DNA and selected ~600 bp fragments. These random fragments were PCR amplified and placed downstream of a minimal promoter in reporter vectors (Fig. 2). The reporter library contains 11.3 million candidate fragments, which covered 96% of the non-repetitive genome by 10-folds. In these constructs, if candidate DNA fragments are enhancers, they will have an opportunity to activate their own transcription. Furthermore, by transfecting the reporter library into Drosophila cell lines, isolating polyadenylated RNA, and performing RNA-seq, the authors were able to quantitatively estimate the enhancers’ strength based on the amount of their transcription.
Computational analyses include mapping the STARR-seq data to the genome and examining their enrichment over input. From this, the authors identified 5,499 enhancers in Drosophila S2 cells and validated 77 in addition to 65 negative controls by luciferase assays. As a result, 81% of the predicted enhancers and 14% of negative controls showed enhancer activity. There was a strong linear correlation (r = 0.83) between the levels of luciferase activity and their STARR-seq transcription readouts, indicating STARR-seq is a reliable quantitative measurement of enhancers’ strength.
STARR-seq is a high-throughput application of the traditional enhancer reporter assay that directly and quantitatively assesses enhancers’ activities in a genome-wide manner. It complements existing enhancer detection methods based mainly on chromatin features. One of the limitations of STARR-seq, as the authors pointed out, is that it only assesses the potential enhancer ability of DNA sequences irrespective of the endogenous genomic context, such as DNA accessibility and histone modification.
Structural genome (re)annotation
The task of defining the complete set of transcripts is complicated because of the fact that transcriptomes are of high dynamic entities, which change in response to both of the intracellular signals and extracellular environment. In addition, expression level, allele expression, and alternative splicing events are involved in increasing the complexity of transcriptome defining with regard to the development stages, growth condition, or disease status.
Genomic studies including gene expression by microarray and chromatin feature assays by tiling array are based on genome annotations. However, the genome annotation is continuously being updated and even the current annotation is incomplete indicating that the previous studies might have missed important information or they are not precise enough to uncover the biological insight. Accumulating studies using RNA-seq to reveal the genome and transcriptome annotation structurally have been generating a more complete and more precise map to facilitate our understanding of the gene transcription. We highlight in this study examples that finely annotated transcriptional landscapes in a major invasive fungal pathogen with combined elegant experiment design and RNA-seq following comprehensive data analysis.
Candida species is a major invasive fungal pathogen of humans, responsible for diseases ranging from superficial skin infections to deep-seated systematic candidiasis with high mortality rates, for which progression and severity are determined by the host immune system.179 The disease caused by Candida albicans largely depends on the feature to change its transcription landscape thus switch its morphologies in response to different host niches or environmental stimuli.180,181 Because of the clinical significance, based on the feature of change transcriptome upon environmental clue, Beuno with colleagues generated RNA-seq data from in vitro-cultured C. albicans with diverse growth conditions including hyphae-inducing condition, high/low oxidative stress/pH condition, nitrosative stress, and cell wall damage-inducing condition.182 From a total of 177 million mapped reads, they have remarkably refined the primary genome annotations by determining transcripts position, identifying new genes and new introns, and determining expression levels under each growth condition and condition-specific expression of novel transcripts. With similar experimental design strategy, Linde et al depicted an even detailed transcriptional map by annotating protein coding genes and non-coding genes, intron and UTR in another Candida species Candida glabrata under pH and nitrosative stress.183 Comparison genomics also fueled this study to determine species-specific and condition-specific adaptions are regulated by individual genetic repertories and conserved orthologs on transcriptional level.184
Outlook/Perspective
In this review, we have outlined major applications of the RNA-seq in biomedical research, highlighted the computational approach in data preprocessing, differential gene expression, alternative splicing, pathway analysis, and co-expression network, and presented examples to show how this technology can be applied in systems biology field to advance our understanding in genomic level. Since it is potent in investigating the transcriptome in a highly quantitative manner at single nucleotide resolution, complex disease diagnosis, and precision medicine, the rapidly accumulating genome sequence data allow researchers to address fundamental biological questions that were not even asked just a few years ago. Although many progresses have been made since the initial application of this technology, there are still more applications possible if further refinement is provided for each of the topics.
Single RNA-seq
RNA-seq in single cells has provided a new powerful approach to study complex biological processes, for instance, promoting advances in cancer studies starting from qualitative microscopic images to quantitative genomic datasets in recent year.185 Single-cell genome and exome sequencing fueled the investigation of fundamental questions including resolving solid tumor heterogeneity, identifying stem cells, tracking cell lineages and population consumption, measuring mutation rates, and detecting fusion gene events.19,186–188 Although single-cell sequencing can provide far more accurate measurement, however, the challenges of the single-cell sequencing in cancer cells exist in the sequencing and data analysis steps beyond the cancer cell isolation. First, the two copies of DNA strands as the input material results in technical errors including insufficient coverage, difficulties in mutation calling, and false-positive error in heterogeneity characterization. Multiple datasets from different single-cell sequencing encompass even higher requirements for the post-sequencing comparison analysis. In the near future, we expect to see that the single-cell sequencing will be applied in much more new issues of cancer genomics study such as differentiate extensive biological complexity or extensive technical errors, rare cancer diagnosis, and early development stage tumor discovery.
Dual RNA-seq
Pathogen–host interactions study including the immune response of eukaryotic cells is another important battlefield, where RNA-seq plays a critical role. Transcriptomic analysis has predominantly focused on either the host or the pathogen, which requires the RNA molecule separation from the host or the pathogen at specific time point, prior to the high-throughput sequencing era.189 Deeper understandings of the interaction process, identification of new virulence factors, immune response mechanism, and development of therapeutic approach will require the simultaneous analysis of interaction partners because the battle leads to a constantly changing environment and complex gene expression patterns. A “dual RNA-seq” approach allows to monitor the genes from both host and pathogen without RNA separation throughout the infection process.190 It enables the study of dynamic response and interspecies gene regulatory networks in both the interaction partners from initial contact through to invasion and the final persistence of the pathogen or clearance by the host immune system with high level of accuracy and depth. Dual RNA-seq attempt studies are in widespread areas such as molecular and cellular biology,191,192 public health,191 immune response in disease,193,194 and bacteria and plant interactions.195–197 As a discovery-from-data approach, computational process and storage of the high magnitude data are of great challenge recently, although project-specific packages have been developed.198–200 Computational modeling and algorithm design beyond the existing ones will facilitate greatly for answering emerging questions by ever-developing applications from NGS to nanopore sequencing and single-cell sequencing.
As the biological complexity, the challenges of development of computational methods also exist in multiple dimensions. We have to consider the particular situation and design experiment accordingly, and no single method or pipeline is optimal under all circumstances even in the same fields. In addition, with rapid accumulation of data in public repositories, new challenges arise from the urgent need to effectively integrate many different RNA-seq datasets, as well as different levels omics data to study the biological complexity and ultimately facilitate the precision and personalized medicine.
Acknowledgments
We thank the NIH Fellows Editorial Board and Dr. Cuncong Zhong for suggestions on the manuscript. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Footnotes
ACADEMIC EDITOR: J.T. Efird, Associate Editor
PEER REVIEW: Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1186 words, excluding any confidential comments to the academic editor.
FUNDING: The Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and National Heart Lung Blood Institute, NIH supported this work. WZ is sponsored by NIH grant ES014811 funded to Dr. Trey Ideker. BZ is supported by NIH grants GM049369, HG004659, and GM052872 funded to Dr. Xiangdong Fu. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Wrote the first draft of the manuscript: YH, SG, WZ. Contributed to the writing of the manuscript: YH, SG, KM, WZ. Agree with manuscript results and conclusions: YH, SG, KM, WZ, BZ. Jointly developed the structure and arguments for the paper: YH, SG. Made critical revisions and approved final version: YH, SG, WZ, BZ. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Okoniewski MJ, Miller CJ. Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations. BMC Bioinformatics. 2006;7:276. doi: 10.1186/1471-2105-7-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Royce TE, Rozowsky JS, Gerstein MB. Toward a universal microarray: prediction of gene expression through nearest-neighbor probe sequence identification. Nucleic Acids Res. 2007;35(15):e99. doi: 10.1093/nar/gkm549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolker E, Makarova KS, Shabalina S, et al. Identification and functional analysis of ‘hypothetical’ genes expressed in Haemophilus influenzae. Nucleic Acids Res. 2004;32(8):2353–61. doi: 10.1093/nar/gkh555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galperin MY, Koonin EV. From complete genome sequence to ‘complete’ understanding? Trends Biotechnol. 2010;28(8):398–406. doi: 10.1016/j.tibtech.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z, Gerstein M, Snyder M. RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9(6):e1003569. doi: 10.1371/journal.pgen.1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robertson G, Schein J, Chiu R, et al. De novo assembly and analysis of RNA-seq data. Nat Methods. 2010;7(11):909–12. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- 9.Andersson R, Gebhard C, Miguel-Escalada I, et al. FANTOM Consortium An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–61. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim TK, Hemberg M, Gray JM, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465(7295):182–7. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camarena L, Bruno V, Euskirchen G, Poggio S, Snyder M. Molecular mechanisms of ethanol-induced pathogenesis revealed by RNA-sequencing. PLoS Pathog. 2010;6(4):e1000834. doi: 10.1371/journal.ppat.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffith M, Griffith OL, Mwenifumbo J, et al. Alternative expression analysis by RNA sequencing. Nat Methods. 2010;7(10):843–7. doi: 10.1038/nmeth.1503. [DOI] [PubMed] [Google Scholar]
- 13.Picardi E, Horner DS, Chiara M, Schiavon R, Valle G, Pesole G. Large-scale detection and analysis of RNA editing in grape mtDNA by RNA deep-sequencing. Nucleic Acids Res. 2010;38(14):4755–67. doi: 10.1093/nar/gkq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilhelm BT, Briau M, Austin P, et al. RNA-seq analysis of 2 closely related leukemia clones that differ in their self-renewal capacity. Blood. 2011;117(2):e27–38. doi: 10.1182/blood-2010-07-293332. [DOI] [PubMed] [Google Scholar]
- 15.Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Han D, Han Y, et al. Ab initio identification of transcription start sites in the Rhesus macaque genome by histone modification and RNA-Seq. Nucleic Acids Res. 2011;39(4):1408–18. doi: 10.1093/nar/gkq956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013;339(6123):1074–7. doi: 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]
- 18.Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458(7234):97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berger MF, Levin JZ, Vijayendran K, et al. Integrative analysis of the melanoma transcriptome. Genome Res. 2010;20(4):413–27. doi: 10.1101/gr.103697.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Supper J, Gugenmus C, Wollnik J, et al. Detecting and visualizing gene fusions. Methods. 2013;59(1):S24–8. doi: 10.1016/j.ymeth.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Conde L, Bracci PM, Richardson R, Montgomery SB, Skibola CF. Integrating GWAS and expression data for functional characterization of disease-associated SNPs: an application to follicular lymphoma. Am J Hum Genet. 2013;92(1):126–30. doi: 10.1016/j.ajhg.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erwin JA, Marchetto MC, Gage FH. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci. 2014;15(8):497–506. doi: 10.1038/nrn3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilhelm BT, Marguerat S, Watt S, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453(7199):1239–43. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 24.Saliba AE, Westermann AJ, Gorski SA, Vogel J. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014;42(14):8845–60. doi: 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson NK, Kent DG, Buettner F, et al. Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell. 2015;16(6):712–24. doi: 10.1016/j.stem.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marzluff WF, Wagner EJ, Duronio RJ. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Genet. 2008;9(11):843–54. doi: 10.1038/nrg2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011;12(2):R16. doi: 10.1186/gb-2011-12-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parkhomchuk D, Borodina T, Amstislavskiy V, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37(18):e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagalakshmi U, Wang Z, Waern K, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320(5881):1344–9. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, Li Y, Li S, et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012;2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cloonan N, Forrest AR, Kolle G, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5(7):613–9. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- 32.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18(9):1509–17. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–52. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 34.Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323(5910):133–8. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 35.Tariq MA, Kim HJ, Jejelowo O, Pourmand N. Whole-transcriptome RNAseq analysis from minute amount of total RNA. Nucleic Acids Res. 2011;39(18):e120. doi: 10.1093/nar/gkr547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carrara M, Lum J, Cordero F, et al. Alternative splicing detection workflow needs a careful combination of sample prep and bioinformatics analysis. BMC Bioinformatics. 2015;16(suppl 9):S2. doi: 10.1186/1471-2105-16-S9-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webster AF, Zumbo P, Fostel J, et al. Mining the archives: a cross-platform analysis of gene expression profiles in archival formalin-fixed paraffin-embedded (FFPE) tissue. Toxicol Sci. 2015 doi: 10.1093/toxsci/kfv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang X, Liu D, Liu F, et al. HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics. 2013;14:33. doi: 10.1186/1471-2105-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anders S, Pyl PT, Huber W. HTSeq – a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28(16):2184–5. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 43.Trapnell C, Salzberg SL. How to map billions of short reads onto genomes. Nat Biotechnol. 2009;27(5):455–7. doi: 10.1038/nbt0509-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flicek P, Birney E. Sense from sequence reads: methods for alignment and assembly. Nat Methods. 2009;6(11 suppl):S6–12. doi: 10.1038/nmeth.1376. [DOI] [PubMed] [Google Scholar]
- 45.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24(5):713–4. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 46.Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–7. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 47.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18(11):1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin H, Zhang Z, Zhang MQ, Ma B, Li M. ZOOM! Zillions of oligos mapped. Bioinformatics. 2008;24(21):2431–7. doi: 10.1093/bioinformatics/btn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bao S, Jiang R, Kwan W, Wang B, Ma X, Song YQ. Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet. 2011;56(6):406–14. doi: 10.1038/jhg.2011.43. [DOI] [PubMed] [Google Scholar]
- 53.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 54.Kent WJ. BLAT – the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fonseca NA, Rung J, Brazma A, Marioni JC. Tools for mapping high-throughput sequencing data. Bioinformatics. 2012;28(24):3169–77. doi: 10.1093/bioinformatics/bts605. [DOI] [PubMed] [Google Scholar]
- 56.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marco-Sola S, Sammeth M, Guigo R, Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat Methods. 2012;9(12):1185–8. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 59.Wang K, Singh D, Zeng Z, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38(18):e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holt RA, Jones SJ. The new paradigm of flow cell sequencing. Genome Res. 2008;18(6):839–46. doi: 10.1101/gr.073262.107. [DOI] [PubMed] [Google Scholar]
- 61.Hillier LW, Marth GT, Quinlan AR, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods. 2008;5(2):183–8. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- 62.Campbell PJ, Stephens PJ, Pleasance ED, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40(6):722–9. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Delhomme N, Padioleau I, Furlong EE, Steinmetz LM. easyRNASeq: a bioconductor package for processing RNA-seq data. Bioinformatics. 2012;28(19):2532–3. doi: 10.1093/bioinformatics/bts477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lawrence M, Huber W, Pagès H, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9(8):e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 67.Zhao S, Zhang B. A comprehensive evaluation of ensembl, RefSeq, and UCSC annotations in the context of RNA-seq read mapping and gene quantification. BMC Genomics. 2015;16:97. doi: 10.1186/s12864-015-1308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li S, ġabaj PP, Zumbo P, et al. Detecting and correcting systematic variation in large-scale RNA sequencing data. Nat Biotechnol. 2014;32(9):888–95. doi: 10.1038/nbt.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Filloux C, Cédric M, Romain P, et al. An integrative method to normalize RNA-seq data. BMC Bioinformatics. 2014;15:188. doi: 10.1186/1471-2105-15-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun Z, Zhu Y. Systematic comparison of RNA-Seq normalization methods using measurement error models. Bioinformatics. 2012;28(20):2584–91. doi: 10.1093/bioinformatics/bts497. [DOI] [PubMed] [Google Scholar]
- 71.Ager-Wick E, Henkel CV, Haug TM, Weltzien FA. Using normalization to resolve RNA-Seq biases caused by amplification from minimal input. Physiol Genomics. 2014;46(21):808–20. doi: 10.1152/physiolgenomics.00196.2013. [DOI] [PubMed] [Google Scholar]
- 72.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-seq data. BMC Bioinformatics. 2011;12:480. doi: 10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garmire LX, Subramaniam S. Evaluation of normalization methods in mammalian microRNA-Seq data. RNA. 2012;18(6):1279–88. doi: 10.1261/rna.030916.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grant GR, Manduchi E, Stoeckert CJ., Jr Analysis and management of microarray gene expression data. Curr Protoc Mol Biol. 2007 doi: 10.1002/0471142727.mb1906s77. Chapter 19:Unit19.6. [DOI] [PubMed] [Google Scholar]
- 77.Oshlack A, Wakefield MJ. Transcript length bias in RNA-seq data confounds systems biology. Biol Direct. 2009;4:14. doi: 10.1186/1745-6150-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang X, Wu Z, Zhang X. Isoform abundance inference provides a more accurate estimation of gene expression levels in RNA-seq. J Bioinform Comput Biol. 2010;8(suppl 1):177–92. doi: 10.1142/s0219720010005178. [DOI] [PubMed] [Google Scholar]
- 79.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiang H, Wong WH. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009;25(8):1026–32. doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sengoelge G, Winnicki W, Kupczok A, et al. A SAGE based approach to human glomerular endothelium: defining the transcriptome, finding a novel molecule and highlighting endothelial diversity. BMC Genomics. 2014;15:725. doi: 10.1186/1471-2164-15-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li J, Witten DM, Johnstone IM, Tibshirani R. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostatistics. 2012;13(3):523–38. doi: 10.1093/biostatistics/kxr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ritchie ME, Silver J, Oshlack A, et al. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007;23(20):2700–7. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- 90.Soneson C, Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics. 2013;14:91. doi: 10.1186/1471-2105-14-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kvam VM, Liu P, Si Y. A comparison of statistical methods for detecting differentially expressed genes from RNA-seq data. Am J Bot. 2012;99(2):248–56. doi: 10.3732/ajb.1100340. [DOI] [PubMed] [Google Scholar]
- 92.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17(2):100–7. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 93.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8(10):749–61. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 94.Fu XD, Ares M., Jr Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15(10):689–701. doi: 10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 96.Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136(4):701–18. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 97.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10(11):741–54. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pandit S, Zhou Y, Shiue L, et al. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013;50(2):223–35. doi: 10.1016/j.molcel.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413–5. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 100.Ray D, Kazan H, Cook KB, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499(7457):172–7. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sultan M, Schulz MH, Richard H, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321(5891):956–60. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 102.Reddy AS, Rogers MF, Richardson DN, Hamilton M, Ben-Hur A. Deciphering the plant splicing code: experimental and computational approaches for predicting alternative splicing and splicing regulatory elements. Front Plant Sci. 2012;3:18. doi: 10.3389/fpls.2012.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Barash Y, Calarco JA, Gao W, et al. Deciphering the splicing code. Nature. 2010;465(7294):53–9. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 104.Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 105.Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7(12):1009–15. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38(14):4570–8. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ameur A, Wetterbom A, Feuk L, Gyllensten U. Global and unbiased detection of splice junctions from RNA-seq data. Genome Biol. 2010;11(3):R34. doi: 10.1186/gb-2010-11-3-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vitting-Seerup K, Porse BT, Sandelin A, Waage J. spliceR: an R package for classification of alternative splicing and prediction of coding potential from RNA-seq data. BMC Bioinformatics. 2014;15:81. doi: 10.1186/1471-2105-15-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aschoff M, Hotz-Wagenblatt A, Glatting KH, Fischer M, Eils R, Konig R. SplicingCompass: differential splicing detection using RNA-seq data. Bioinformatics. 2013;29(9):1141–8. doi: 10.1093/bioinformatics/btt101. [DOI] [PubMed] [Google Scholar]
- 110.Zhao K, Lu ZX, Park JW, Zhou Q, Xing Y. GLiMMPS: robust statistical model for regulatory variation of alternative splicing using RNA-seq data. Genome Biol. 2013;14(7):R74. doi: 10.1186/gb-2013-14-7-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shen S, Park JW, Huang J, et al. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012;40(8):e61. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shen S, Park JW, Lu ZX, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A. 2014;111(51):E5593–601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li W, Dai C, Kang S, Zhou XJ. Integrative analysis of many RNA-seq datasets to study alternative splicing. Methods. 2014;67(3):313–24. doi: 10.1016/j.ymeth.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Iancu OD, Colville A, Darakjian P, Hitzemann R. Chapter four – coexpression and cosplicing network approaches for the study of mammalian brain transcriptomes. In: Robert H, Shannon M, editors. International Review of Neurobiology. Vol. 116. Waltham: Academic Press; 2014. pp. 73–93. [DOI] [PubMed] [Google Scholar]
- 115.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet. 2013;93(4):641–51. doi: 10.1016/j.ajhg.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dereeper A, Homa F, Andres G, et al. SNiPlay3: a web-based application for exploration and large scale analyses of genomic variations. Nucleic Acids Res. 2015;43(W1):W295–300. doi: 10.1093/nar/gkv351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chuang LC, Kao CF, Shih WL, Kuo PH. Pathway analysis using information from allele-specific gene methylation in genome-wide association studies for bipolar disorder. PLoS One. 2013;8(1):e53092. doi: 10.1371/journal.pone.0053092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pastinen T. Genome-wide allele-specific analysis: insights into regulatory variation. Nat Rev Genet. 2010;11(8):533–8. doi: 10.1038/nrg2815. [DOI] [PubMed] [Google Scholar]
- 120.Costa V, Aprile M, Esposito R, Ciccodicola A. RNA-Seq and human complex diseases: recent accomplishments and future perspectives. Eur J Hum Genet. 2013;21(2):134–42. doi: 10.1038/ejhg.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Degner JF, Marioni JC, Pai AA, et al. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009;25(24):3207–12. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mayba O, Gilbert HN, Liu J, et al. MBASED: allele-specific expression detection in cancer tissues and cell lines. Genome Biol. 2014;15(8):405. doi: 10.1186/s13059-014-0405-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dennis G, Jr, Sherman BT, Hosack DA, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):3. [PubMed] [Google Scholar]
- 124.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27(1):29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Du J, Yuan Z, Ma Z, Song J, Xie X, Chen Y. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol Biosyst. 2014;10(9):2441–7. doi: 10.1039/c4mb00287c. [DOI] [PubMed] [Google Scholar]
- 128.Rahmatallah Y, Emmert-Streib F, Glazko G. Comparative evaluation of gene set analysis approaches for RNA-Seq data. BMC Bioinformatics. 2014;15:397. doi: 10.1186/s12859-014-0397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huang D, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:47. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 130.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1[alpha]-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 131.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 2013;14(1):7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang X, Cairns M. Gene set enrichment analysis of RNA-Seq data: integrating differential expression and splicing. BMC Bioinformatics. 2013;14(suppl 5):S16. doi: 10.1186/1471-2105-14-S5-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Luo W, Friedman M, Shedden K, Hankenson K, Woolf P. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009;10(1):161. doi: 10.1186/1471-2105-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xiong Q, Mukherjee S, Furey TS. GSAASeqSP: a toolset for gene set association analysis of RNA-seq data. Sci Rep. 2014;4:6347. doi: 10.1038/srep06347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tarca AL, Draghici S, Khatri P, et al. A novel signaling pathway impact analysis. Bioinformatics. 2009;25(1):75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gao S, Wang X. TAPPA: topological analysis of pathway phenotype association. Bioinformatics. 2007;23(22):3100–2. doi: 10.1093/bioinformatics/btm460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Haynes WA, Higdon R, Stanberry L, Collins D, Kolker E. Differential expression analysis for pathways. PLoS Comput Biol. 2013;9(3):e1002967. doi: 10.1371/journal.pcbi.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Young M, Wakefield M, Smyth G, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zheng W, Chung LM, Zhao H. Bias detection and correction in RNA-sequencing data. BMC Bioinformatics. 2011;12:290. doi: 10.1186/1471-2105-12-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012;8(2):e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302(5643):249–55. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 142.Giorgi FM, Fabbro CD, Licausi F. Comparative study of RNA-seq- and microarray-derived coexpression networks in Arabidopsis thaliana. Bioinformatics. 2013;29(6):717–24. doi: 10.1093/bioinformatics/btt053. [DOI] [PubMed] [Google Scholar]
- 143.Ballouz S, Verleyen W, Gillis J. Guidance for RNA-seq co-expression network construction and analysis: safety in numbers. Bioinformatics. 2015;31(13):2123–30. doi: 10.1093/bioinformatics/btv118. [DOI] [PubMed] [Google Scholar]
- 144.van Dam S, Craig T, de Magalhães JP. GeneFriends: a human RNA-seq-based gene and transcript co-expression database. Nucleic Acids Res. 2015;43(D1):D1124–32. doi: 10.1093/nar/gku1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Obayashi T, Okamura Y, Ito S, Tadaka S, Motoike IN, Kinoshita K. COX-PRESdb: a database of comparative gene coexpression networks of eleven species for mammals. Nucleic Acids Res. 2013;41(D1):D1014–20. doi: 10.1093/nar/gks1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Choi Y, Kendziorski C. Statistical methods for gene set co-expression analysis. Bioinformatics. 2009;25(21):2780–6. doi: 10.1093/bioinformatics/btp502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen L, Liu R, Liu Z-P, Li M, Aihara K. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci Rep. 2012;6:2. doi: 10.1038/srep00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bar-Joseph Z, Gitter A, Simon I. Studying and modelling dynamic biological processes using time-series gene expression data. Nat Rev Genet. 2012;13(8):552–64. doi: 10.1038/nrg3244. [DOI] [PubMed] [Google Scholar]
- 149.Gao S, Wang X. Identification of highly synchronized subnetworks from gene expression data. BMC Bioinformatics. 2013;14(suppl 9):S5. doi: 10.1186/1471-2105-14-S9-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yosef N, Shalek AK, Gaublomme JT, et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496(7446):461–8. doi: 10.1038/nature11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Amar D, Safer H, Shamir R. Dissection of regulatory networks that are altered in disease via differential co-expression. PLoS Comput Biol. 2013;9(3):e1002955. doi: 10.1371/journal.pcbi.1002955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Si Y, Liu P, Li P, Brutnell TP. Model-based clustering for RNA-seq data. Bioinformatics. 2013;30(2):197–205. doi: 10.1093/bioinformatics/btt632. [DOI] [PubMed] [Google Scholar]
- 154.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;11(7):476–86. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wei G, Abraham BJ, Yagi R, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. 2011;35(2):299–311. doi: 10.1016/j.immuni.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ouyang Z, Zhou Q, Wong WH. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc Natl Acad Sci U S A. 2009;106(51):21521–6. doi: 10.1073/pnas.0904863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Han Y, Han D, Yan Z, et al. Stress-associated H3K4 methylation accumulates during postnatal development and aging of Rhesus macaque brain. Aging Cell. 2012;11(6):1055–64. doi: 10.1111/acel.12007. [DOI] [PubMed] [Google Scholar]
- 158.Wei G, Hu G, Cui K, Zhao K. Genome-wide mapping of nucleosome occupancy, histone modifications, and gene expression using next-generation sequencing technology. Methods Enzymol. 2012;513:297–313. doi: 10.1016/B978-0-12-391938-0.00013-6. [DOI] [PubMed] [Google Scholar]
- 159.Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yu W, McIntosh C, Lister R, et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014;24(10):1613–23. doi: 10.1101/gr.172015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464(7289):773–7. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pickrell JK, Marioni JC, Pai AA, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464(7289):768–72. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Solana J, Kao D, Mihaylova Y, et al. Defining the molecular profile of planarian pluripotent stem cells using a combinatorial RNAseq, RNA interference and irradiation approach. Genome Biol. 2012;13(3):R19. doi: 10.1186/gb-2012-13-3-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424(6945):147–51. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 165.Levine M. Transcriptional enhancers in animal development and evolution. Curr Biol. 2010;20(17):R754–63. doi: 10.1016/j.cub.2010.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Levine M, Cattoglio C, Tjian R. Looping back to leap forward: transcription enters a new era. Cell. 2014;157(1):13–25. doi: 10.1016/j.cell.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Buecker C, Wysocka J. Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. 2012;28(6):276–84. doi: 10.1016/j.tig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49(5):825–37. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144(3):327–39. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Boyle AP, Davis S, Shulha HP, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132(2):311–22. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gaulton KJ, Nammo T, Pasquali L, et al. A map of open chromatin in human pancreatic islets. Nat Genet. 2010;42(3):255–9. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 173.Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–12. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–83. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bonn S, Zinzen RP, Girardot C, et al. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet. 2012;44(2):148–56. doi: 10.1038/ng.1064. [DOI] [PubMed] [Google Scholar]
- 176.Visel A, Blow MJ, Li Z, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457(7231):854–8. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Melnikov A, Murugan A, Zhang X, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30(3):271–7. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Patwardhan RP, Hiatt JB, Witten DM, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30(3):265–70. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Klepser ME. Candida resistance and its clinical relevance. Pharmacotherapy. 2006;26(6 pt 2):68S–75S. doi: 10.1592/phco.26.6part2.68S. [DOI] [PubMed] [Google Scholar]
- 180.Biswas S, Van Dijck P, Datta A. Environmental sensing and signal transduction pathways regulating morphopathogenic determinants of Candida albicans. Microbiol Mol Biol Rev. 2007;71(2):348–76. doi: 10.1128/MMBR.00009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cutler JE. Putative virulence factors of Candida albicans. Annu Rev Microbiol. 1991;45:187–218. doi: 10.1146/annurev.mi.45.100191.001155. [DOI] [PubMed] [Google Scholar]
- 182.Bruno VM, Wang Z, Marjani SL, et al. Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 2010;20(10):1451–8. doi: 10.1101/gr.109553.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Linde J, Duggan S, Weber M, et al. Defining the transcriptomic landscape of Candida glabrata by RNA-Seq. Nucleic Acids Res. 2015;43(3):1392–406. doi: 10.1093/nar/gku1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Grumaz C, Lorenz S, Stevens P, et al. Species and condition specific adaptation of the transcriptional landscapes in Candida albicans and Candida dubliniensis. BMC Genomics. 2013;14:212. doi: 10.1186/1471-2164-14-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Navin NE. Cancer genomics: one cell at a time. Genome Biol. 2014;15(8):452. doi: 10.1186/s13059-014-0452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Van Loo P, Campbell PJ. ABSOLUTE cancer genomics. Nat Biotechnol. 2012;30(7):620–1. doi: 10.1038/nbt.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sirbu A, Kerr G, Crane M, Ruskin HJ. RNA-seq vs dual- and single-channel microarray data: sensitivity analysis for differential expression and clustering. PLoS One. 2012;7(12):e50986. doi: 10.1371/journal.pone.0050986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol. 2012;10(9):618–30. doi: 10.1038/nrmicro2852. [DOI] [PubMed] [Google Scholar]
- 191.Dodsworth BT, Flynn R, Cowley SA. The current state of naive human pluripotency. Stem Cells. 2015 doi: 10.1002/stem.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Das A, Chai JC, Kim SH, et al. Dual RNA sequencing reveals the expression of unique transcriptomic signatures in lipopolysaccharide-induced BV-2 microglial cells. PLoS One. 2015;10(3):e0121117. doi: 10.1371/journal.pone.0121117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Pittman KJ, Aliota MT, Knoll LJ. Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics. 2014;15:806. doi: 10.1186/1471-2164-15-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Choi YJ, Aliota MT, Mayhew GF, Erickson SM, Christensen BM. Dual RNA-seq of parasite and host reveals gene expression dynamics during filarial worm-mosquito interactions. PLoS Negl Trop Dis. 2014;8(5):e2905. doi: 10.1371/journal.pntd.0002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lu M, Zhang PJ, Li CH, Lv ZM, Zhang WW, Jin CH. miRNA-133 augments coelomocyte phagocytosis in bacteria-challenged Apostichopus japonicus via targeting the TLR component of IRAK-1 in vitro and in vivo. Sci Rep. 2015;5:12608. doi: 10.1038/srep12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Camilios-Neto D, Bonato P, Wassem R, et al. Dual RNA-seq transcriptional analysis of wheat roots colonized by Azospirillum brasilense reveals up-regulation of nutrient acquisition and cell cycle genes. BMC Genomics. 2014;15:378. doi: 10.1186/1471-2164-15-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lange M, Eisenhauer N, Sierra CA, et al. Plant diversity increases soil microbial activity and soil carbon storage. Nat Commun. 2015;6:6707. doi: 10.1038/ncomms7707. [DOI] [PubMed] [Google Scholar]
- 198.Schulze S, Henkel SG, Driesch D, Guthke R, Linde J. Computational prediction of molecular pathogen-host interactions based on dual transcriptome data. Front Microbiol. 2015;6:65. doi: 10.3389/fmicb.2015.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Torres-García W, Zheng S, Sivachenko A, et al. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics. 2014;30(15):2224–6. doi: 10.1093/bioinformatics/btu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Xu G, Strong MJ, Lacey MR, Baribault C, Flemington EK, Taylor CM. RNA CoMPASS: a dual approach for pathogen and host transcriptome analysis of RNA-seq datasets. PLoS One. 2014;9(2):e89445. doi: 10.1371/journal.pone.0089445. [DOI] [PMC free article] [PubMed] [Google Scholar]