

Abstract
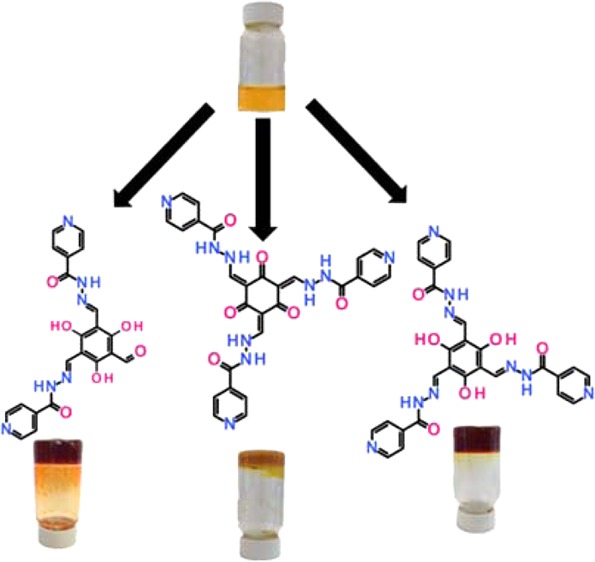
Simultaneous control of the kinetics and thermodynamics of two different types of covalent chemistry allows pathway selectivity in the formation of hydrogelating molecules from a complex reaction network. This can lead to a range of hydrogel materials with vastly different properties, starting from a set of simple starting compounds and reaction conditions. Chemical reaction between a trialdehyde and the tuberculosis drug isoniazid can form one, two, or three hydrazone connectivity products, meaning kinetic gelation pathways can be addressed. Simultaneously, thermodynamics control the formation of either a keto or an enol tautomer of the products, again resulting in vastly different materials. Overall, this shows that careful navigation of a reaction landscape using both kinetic and thermodynamic selectivity can be used to control material selection from a complex reaction network.
Both thermodynamic and kinetic parameters controlling self-assembly processes can be used to control pathway selection in assembling supramolecular materials. The resultant assemblies or materials can have vastly different properties, depending on the chosen self-assembly path.1 Thermodynamically, pushing an assembly down a certain pathway to a stable energy well within the assembly landscape can use solvent, temperature, or pH changes. Kinetically, the landscape can be navigated using the activation energies associated with certain assembly processes, to address metastable states over time or to kinetically trap a certain state. Materials generated from these complex pathways include crystal forms (polymorphism), viruses, protein networks, supramolecular polymers, and certain low-molecular-weight gelators (LMWGs).1 LMWGs represent the main materials of interest in this Communication, but the principles of the research may be applied to any supramolecular assembly process.2 LMWGs have been used and envisioned to have a number of applications, including drug delivery commercial products and as templates for crystal, particle, or cell growth, to name but a few.3 With this in mind, research groups from around the world have studied these materials for a number of years through the use of pre-synthesized LMW compounds which undergo supramolecular polymerization. More recently, exploitation of chemical reactivity to both form and deform the gelatinous materials and to sample the pathway complexity of the self-assembly, but to date have not used the chemical reaction pathways to control selectivity.4 These reported works have generally focused on single-step reactivity and have not researched multi-step reactivity or two or more types of reactivity in a single gelation system. Further work has begun to appear in which catalytic control over chemical reactivity is used to control the assembly processes of the gel components, the material properties and spatial distribution of the material.5 This has led to many elegant methodologies to tackle the pathway complexity of supramolecular assembly in which kinetic and thermodynamic materials are isolated. With this in mind we hypothesized that chemical reactivity could play an important role in the production of multiple materials by controlling pathway selection in complex reaction networks (instead of the supramolecular assembly landscape).4 To do this we introduced multiple-step reactivity (kinetic control) and pH-dependent tautomerization (thermodynamic control) to a reaction network capable of reversibly and irreversibly forming a range of potential hydrogelators from a set of simple starting chemicals (Figure 1). In the present case, this yielded three distinct gel materials from effectively the same chemical starting point (a mixed solution of the reactants).
Figure 1.

Pathway complexity reaction network diagram (left) showing the supramolecular assembly of three hydrogels from a single starting point (*) of dissolved core and periphery components. The multiple-step reactivity (horizontal arrows) between a core trialdehyde (Ie, orange triangle) and a peripheral hydrazide (II, black semicurve) samples kinetically the assembly landscape. Thermodynamically controlled tautomerization (hooked vertical arrows between orange (enol) and purple (keto) triangles) samples a different part of the conjectural gelation landscape. Self-assembly (vertical thick black arrows) from the chemical reactivity products is the result of the process conditions selecting the reaction product, giving three distinct gelatinous materials, Be (orange pathway), Ce (red pathway), and Ck (yellow pathway). Ae is the mono-substituted enol intermediate observed experimentally. Ie crystallizes out at low pH (<7) in water.
In situ covalent bond formation between the water-soluble trialdehyde 1,3,5-triformylphoroglucinol (Ie) and isoniazid (isonicotinic acid hydrazide, II) gave discotic compounds. This reactivity between hydrazides and aldehydes is well-known for its use in dynamic covalent chemistry.6 Hydrazone formation can be catalyzed by protons or hydroxyl anions as well as by certain amines, and although this catalysis can occur in our system, it has no effect on the kinetic or thermodynamic selectivity of the complex reaction pathway.7 The reaction pathway of Ie and II should give three possible enol hydrazone species, products Ae, Be, and Ce, and three possible keto hydrazone species, Ak, Bk, and Ck (Figure 1), referring to singly, doubly, and triply reacted sets of products, respectively. The triply reacted product is found in two tautomeric forms, enol (Ce) and keto (Ck), while Ae and Be are observed experimentally.
There are a number of ways in which to set gels formed from this reactive molecular system. Reaction conditions lead to three different gels, Ce, Ck, and Be gels (Figure 1). Gels are heat and time stable. The methodologies in brief for synthesizing the gels are as follow (see Supporting Information (SI) for details):
(1) Mix the core Ie with a number of equivalents of II at pH 8, and raise the pH to 9.5–12. This gives Ck gel, chemically the thermodynamically stable monomer. Equivalents of reactants, salts, or mode of changing the pH do not change the experimental outcome.
(2) Mix the core Ie with a number of equivalents of II at pH 8, and leave in solution for a set period of time (hours) before lowering the pH using glucono-δ-lactone (GdL).8 This gives Ce gel, a kinetically trapped monomer in reference to tautomerization, less stable than Ck.
(3) Mix the core Ie with a number of equivalents of II at pH 8, and immediately on mixing lower the pH using GdL at room temperature. The lower pH gives Be gel if the correct time variable within the reaction kinetics from reacting I and II through Ae to Be. This is the kinetic selectivity of an intermediate monomer in the stepped reaction sequence.
Gelation methodologies 1 and 2 form two different gels, Ck (high pH) and Ce (low pH), both triple-hydrazone compounds. The gels are distinctly different in terms of not only their rheological characteristics but also their color (Figures 1 and 2). The Ce gel is red whereas the Ck gel is yellow; the color is indicative of the tautomeric form and is thus chemically based. The Ce gel is more robust, having both higher G′ value and “yield stress” than the Ck gel (Figure 2 and SI, Figures S2–S7). The critical gelation concentrations (CGCs) for Ce and Ck are 0.2% and 0.5% by weight, respectively, also indicating a difference in the materials. Further rheological studies provided evidence on the connectivity between the supramolecular fibers of Ce and Ck. The effect on the rheology of increasing concentration of Ce revealed a good match with the cellular solid/SAFIN models for gels.4,5 The temporal changes during the kinetics of the gelation process also provide insights into the assembly process. The Avrami constants (also known as fractal dimensions) for Ce and Ck, 2.4 and 1.4, respectively, reveal a strong contrast in the assembly processes and connectivity, explaining the rheological differences. The self-assembled materials were found to be not only physically but also chemically different. Isolating Ce or Ck from these gels generated two distinct, analytically different tautomers (see SI for details of isolation through simple filtration and washes). Analytical data indicate that the tautomers isolated from the two pH ranges give Ce at low pH and Ck at high pH. This pH-dependent thermodynamic control selects the gelation pathway for Ce and Ck. The calculated reaction pathways and energy differences between the two tautomers, Ce and Ck, were determined to be relatively small. In the C3 geometry, Ck, as expected, is more stable.9Ce is kinetically trapped in a metastable state (see SI). Computational calculations indicate that Ck is 0.6 kcal/mol more stable than Ce and the reaction pathway has high-energy transition states (7–10 kcal/mol).
Figure 2.

Rheological frequency range sweeps of the gels showing the three different gel types, Ce, Ck, and Be, having three distinct rheological properties with G′ ≈ 11 000, 800, and 3000 Pa, respectively. Ce gels: black squares, ex situ gel; open squares, gel method 2. Ck gels: gray circles, ex situ gel; open circles, gelation and mixing at pH 10; black circles, gel method 1. Be gels: black triangles, gel method 3; open triangles, ex situ gel.
The setting of these gels is also reversible, indicating that the tautomerization is fully reversible (gelation reversibility is via the dissolved pH 8 species). To the best of our knowledge, this is the first example of reversibility of keto–enol tautomerization in this class of compounds in water.9 At pH 8 the fully soluble Ce species appears to be an anion in the form of either the mono-deprotonated or doubly deprotonated species as evidenced by MS and UV/vis spectroscopy (see SI). This indicates that the low-pH gelation trigger for Ce is the protonation of the anion, giving a very low-solubility neutral compound. The apparent pKa determined for Ce was ∼6.8(±0.1). The results also indicate that as the pH is increased the tautomer equilibrium between Ce and Ck is shifted (OH– induced, Figure S50). The pH change gives a compound which should have a very high pKa, Ck, which is neutral and once again has a low solubility and self-assembles into the gelatinous material.9,10 Chemical synthesis of Ce and gel setting of the dissolved Ce species at pH 8 yield the same gel as the in situ gelation method (i.e., an ex situ gelation method; see SI for details). This indicates that the gelation pathway is only dependent on the tautomerization covalent chemistry and not the hydrazone reactivity.
The importance of the pyridyl groups in the cross-linking of the supramolecular polymers is highlighted by the fact that the phenyl versions of Ce and Ck, De and Dk, form supramolecular polymers but do not cross-link to form a gel network (see SI). Crystallization of Ce and De and determination of the crystal structures reveal that both molecules are indeed the enol tautomer (the first such crystallographic determinations of enol forms of this class of molecules)9 and both show the propensity to stack one of top of the other due to their discotic shape (see SI for details of structures). The De tape motif is built from a discrete R2888 hydrogen-bonding pattern which involves the methanol solvent interrupting the well-known 1,3,5-benzenetriamide H-bonding patterns.9c,9d,9f,11,12 The Ce structure does not show this H-bonding as it crystallizes with DMSO, which acts as a strong H-bonding acceptor, again following the Etter rules.12 However, this does not prevent the molecule from forming a stacking assembly, showing that the dispersion forces and shape are sufficient to induce supramolecular polymerization. The pyridyl groups are not interacting directly with each other to cross-link the columns of Ce, but well-known H-bonding patterns with water may play an important role in cross-linking the hydrogel networks.13
Varying the ratios of Ie and II in the initial solution using methods 1 and 2 always gave Ck and Ce gels, respectively. Thus, Ck and Ce gel production is essentially independent of stoichiometry (see SI for details). When gelation method 3 was used, a different gel was formed, made up of the twice-reacted gelator Be rather than the thrice-reacted Ce materials, illustrating the kinetic selectivity within the reaction gelation landscape. The majority of cases using gelation method 3 resulted in the gels being orange in color and gave materials rheologically weaker than the Ce gels but stronger than the Ck gels (Figures 2 and S18–S20). Isolating the chemical component of these orange gels (see SI for details) revealed that the materials were exclusively made from Be. Rheological concentration studies and the Avrami constant (2.2) for Be gels show that the gels also follow the cellular solid theory of gels and are highly inter-connected gelatinous materials. All three gels are fibrous in nature, as seen in the SEM morphologies. PXRD stacking distances are 3.32–3.39 Å for all three types of gels, indicating recognizable molecular packing motifs, as confirmed by computational work and crystal structures.9c,9d,9f,11 We investigated trimers of structures Be, Ce, and Ck using B97D with a 6-311g(d) basis. Ce, Ck, and Be revealed their propensity to form supramolecular polymers but also the key difference in Be forming structurally different aggregates. These are all indicative of fiber formation through supramolecular polymerization resulting in gelation.2
The relation between the formation of the Be gel material and the kinetics of the reactions in different solution conditions was followed using MS and UV/vis spectroscopy to better understand the relationship between the chemical reactivity and the self-assembly of the gels (see SI for varied experimental settings). Figure 3 shows the reaction of Ie and II at pH 8 and indicates the immediate (seconds time scale) formation of Ae upon mixing Ie and II. Within seconds/minutes of mixing, Be is formed, almost completely depleting Ie and Ae. However, Ce is not observed in most cases until at least 5–10 min into the reaction. The rate of formation of Ce is dependent on the amount and type of catalyst present, i.e., pH. For example, Ce is formed completely within minutes at pH 10 (OH– catalyst) as noted by the quick gelation of Ck using gel method 2, compared to hours at pH 8. Ce always forms at high pH (>7) as Be is fully soluble at these pH vales and does not undergo tautomerization. At lower pH values (<7), a competition between formation of Ce from Be and the self-assembly of Be is established.
Figure 3.

Reaction pathway kinetics in solution for the formation of anion versions of Ae, Be, and Ce. Conversion of Ie (○) and II (at a ratio of 1:6, respectively) to Ce (■) via intermediates Ae (◆) and Be (△) at a constant pH 8 in water. Potential “gelling zones” based on the occurrence for Be and Ce are shown as light gray (first 10 min) and dark gray zones (after several hours), respectively. Solid lines are used as a guide for the eye. II is not shown for clarity. (a) Analysis of the entire reaction; (b) inset showing the first 10 min of the reaction.
The reaction kinetics give clear evidence on why the methodology for gelation (method 3 compared to 2) yields two distinct materials. Adding GdL to the gelation mixture via method 3 results in a pH drop to <6.6 (apparent pKa for Be is 6.6(±0.1)), indicating that the metastable Be is kinetically trapped through self-assembly. The explanation for this kinetic trapping is the mass-transfer limitation of Be from the solid-state network of the gel, resulting in limited concentration and low reactivity in solution (see SI for details). Indeed, increasing the pH of a gel via method 3 to <6.6 to resolubilize the compounds causes Be to react further to generate Ce in solution (without adding further II). Again lowering the pH below the apparent pKa of Ce results in a Ce gel. This gel cannot be converted back to a Be gel without extensive chemical isolation (i.e., theoretical breaking of covalent bonds between Ie and II and isolation of individual species). Isolated Be is indeed capable of forming gels through the pH-triggered mechanism as indicated by the rheology, morphology, and appearance of the gels made from dissolving isolated pure Be at pH 8 and lowering the pH. The CGCs of Be from both methodologies were identical at 0.3% by weight. Unlike Ce, when Be is added to a high-pH water solution, no gelation occurs and a clear, colorless solution results, indicative of a soluble deprotonated species.
In conclusion, by coupling two chemical reactivities (hydrazone bond formation and tautomerization) to the self-assembly of supramolecular gels, we have shown that engineered reaction pathways within a gelation landscape can be created. We have described the intended production of a number of gel materials starting from one initial solution mixture, depending on kinetic and thermodynamic control over reactions. We hope this connectivity hypothesis between chemical reactivity and self-assembly will lead to new pathway complexity studies in a variety of research fields where multi-step reactivity can be introduced.
Acknowledgments
Heriot-Watt University (G.O.L. and J.S.F.), the Royal Society of Edinburgh/Scottish Government Fellowship (G.O.L.), and the Scottish Funding Council Exchange Scheme (G.O.L.) are thanked for funding. G.O.L. and H.M. thank the Scottish Crucible for the project award “Custom Bubbles”. M.J.P. and N.A. thank the European Research Council (ERC FP7/2007-2013/ERC Grant No. 258990). J.M.Z. acknowledges the support of the Leverhulme Trust (RPG-165) and EPSRC U.K. (EP/J006602/1).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b06988.
The authors declare no competing financial interest.
Supplementary Material
References
- a Korevaar P. A.; George S. J.; Markvoort A. J.; Smulders M. M. J.; Hilbers P. A. J.; Schenning A. P. H. J.; De Greef T. F. A.; Meijer E. W. Nature 2012, 481, 492. 10.1038/nature10720. [DOI] [PubMed] [Google Scholar]; b Korevaar P. A.; Newcomb C. J.; Meijer E. W.; Stupp S. I. J. Am. Chem. Soc. 2014, 136, 8540. 10.1021/ja503882s. [DOI] [PubMed] [Google Scholar]; c Raeburn J.; Zamith Cardoso A.; Adams D. J. Chem. Soc. Rev. 2013, 42, 5143. 10.1039/c3cs60030k. [DOI] [PubMed] [Google Scholar]; d Cardoso A. Z.; Alvarez Alvarez A. E.; Cattoz B. N.; Griffiths P. C.; King S. M.; Frith W. J.; Adams D. J. Faraday Discuss. 2013, 166, 101. 10.1039/c3fd00104k. [DOI] [PubMed] [Google Scholar]; e Aggeli A.; Nyrkova I. A.; Bell M.; Harding R.; Carrick L.; McLeish T. C.; Semenov A. N.; Boden N. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 11857. 10.1073/pnas.191250198. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Van Bommel K. J. C.; Stuart M. C. A.; Feringa B. L.; van Esch J. Org. Biomol. Chem. 2005, 3, 2917. 10.1039/b507157g. [DOI] [PubMed] [Google Scholar]; g Qiu H.; Hudson Z. M.; Winnik M. A.; Manners I. Science 2015, 347, 1329. 10.1126/science.1261816. [DOI] [PubMed] [Google Scholar]; h Korevaar P. A.; de Greef T. F. A.; Meijer E. W. Chem. Mater. 2014, 26, 576. 10.1021/cm4021172. [DOI] [Google Scholar]; i Baskakov I. V.; Legname G.; Baldwin M. A.; Prusiner S. B.; Cohen F. E. J. Biol. Chem. 2002, 277, 21140. 10.1074/jbc.M111402200. [DOI] [PubMed] [Google Scholar]; j Misra N.; Lees D.; Zhang T.; Schwartz R. Comput. Math. Methods Med. 2008, 9, 277. 10.1080/17486700802168379. [DOI] [Google Scholar]; k Kumar M.; Brocorens P.; Tonnelé C.; Beljonne D.; Surin M.; George S. J. Nat. Commun. 2014, 5, 5793. 10.1038/ncomms6793. [DOI] [PubMed] [Google Scholar]; l Tidhar Y.; Weissman H.; Wolf S. G.; Gulino A.; Rybtchinski B. Chem.-Eur. J. 2011, 17, 6068. 10.1002/chem.201003419. [DOI] [PubMed] [Google Scholar]
- a Estroff L. A.; Hamilton A. D. Chem. Rev. 2004, 104, 1201. 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]; b Weiss R. G. J. Am. Chem. Soc. 2014, 136, 7519. 10.1021/ja503363v. [DOI] [PubMed] [Google Scholar]
- Hirst A. R.; Escuder B.; Miravet J. F.; Smith D. K. Angew. Chem., Int. Ed. 2008, 47, 8002. 10.1002/anie.200800022. [DOI] [PubMed] [Google Scholar]
- a Sreenivasachary N.; Lehn J.-M. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 5938. 10.1073/pnas.0501663102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ossipov D. A.; Yang X.; Varghese O.; Kootala S.; Hilborn J. Chem. Commun. 2010, 46, 8368. 10.1039/c0cc03055d. [DOI] [PubMed] [Google Scholar]; c Lloyd G. O.; Steed J. W. Nat. Chem. 2009, 1, 437. 10.1038/nchem.283. [DOI] [PubMed] [Google Scholar]; d Yang Z.; Gu H.; Fu D.; Gao P.; Lam J. K.; Xu B. Adv. Mater. 2004, 16, 1440. 10.1002/adma.200400340. [DOI] [Google Scholar]; e Colomb-Delsuc M.; Mattia E.; Sadownik J. W.; Otto S. Nat. Commun. 2015, 6, 7427. 10.1038/ncomms8427. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Li J.; Carnall J. M. A.; Stuart M. C. A.; Otto S. Angew. Chem., Int. Ed. 2011, 50, 8384. 10.1002/anie.201103297. [DOI] [PubMed] [Google Scholar]; g Sadownik J. W.; Ulijn R. V. Chem. Commun. 2010, 46, 3481. 10.1039/c001982h. [DOI] [PubMed] [Google Scholar]
- a Hirst A. R.; Roy S.; Arora M.; Das A. K.; Hodson N.; Murray P.; Marshall S.; Javid N.; Sefcik J.; Boekhoven J.; van Esch J. H.; Santabarbara S.; Hunt N. T.; Ulijn R. V. Nat. Chem. 2010, 2, 1089. 10.1038/nchem.861. [DOI] [PubMed] [Google Scholar]; b Boekhoven J.; Poolman J. M.; Maity C.; Li F.; van der Mee L.; Minkenberg C. B.; Mendes E.; van Esch J. H.; Eelkema R. Nat. Chem. 2013, 5, 433. 10.1038/nchem.1617. [DOI] [PubMed] [Google Scholar]; c Shi J.; Du X.; Huang Y.; Zhou J.; Yuan D.; Wu D.; Zhang Y.; Haburcak R.; Epstein I. R.; Xu B. J. Am. Chem. Soc. 2015, 137, 26. 10.1021/ja5100417. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Maity C.; Hendriksen W. E.; van Esch J. H.; Eelkema R. Angew. Chem., Int. Ed. 2015, 54, 998. 10.1002/anie.201409198. [DOI] [PubMed] [Google Scholar]; e Olive A. G. L.; Abdullah N. H.; Ziemecka I.; Mendes E.; Eelkema R.; van Esch J. H. Angew. Chem., Int. Ed. 2014, 53, 4132. 10.1002/anie.201310776. [DOI] [PubMed] [Google Scholar]; f Nalluri S. K. M.; Berdugo C.; Javid N.; Frederix P. W. J. M.; Ulijn R. V. Angew. Chem., Int. Ed. 2014, 53, 5882. 10.1002/anie.201311158. [DOI] [PubMed] [Google Scholar]
- a Smith M. M.; Edwards W.; Smith D. K. Chem. Sci. 2013, 4, 671. 10.1039/C2SC21547K. [DOI] [Google Scholar]; b Bhat V. T.; Caniard A. M.; Luksch T.; Brenk R.; Campopiano D. J.; Greaney M. F. Nat. Chem. 2010, 2, 490. 10.1038/nchem.658. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dirksen A.; Dirksen S.; Hackeng T. M.; Dawson P. E. J. Am. Chem. Soc. 2006, 128, 15602. 10.1021/ja067189k. [DOI] [PubMed] [Google Scholar]; d Levrand B.; Ruff Y.; Lehn J.-M.; Herrmann A. Chem. Commun. 2006, No. 282965. 10.1039/b602312f. [DOI] [PubMed] [Google Scholar]; e Kool E. T.; Park D.-H.; Crisalli P. J. Am. Chem. Soc. 2013, 135, 17663. 10.1021/ja407407h. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Belowich M. E.; Stoddart J. F. Chem. Soc. Rev. 2012, 41, 2003. 10.1039/c2cs15305j. [DOI] [PubMed] [Google Scholar]
- a Ramström O.; Lohmann S.; Bunyapaiboonsri T.; Lehn J.-M. Chem.-Eur. J. 2004, 10, 1711. 10.1002/chem.200305551. [DOI] [PubMed] [Google Scholar]; b Crisalli P.; Kool E. T. Org. Lett. 2013, 15, 1646. 10.1021/ol400427x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Crisalli P.; Kool E. T. J. Org. Chem. 2013, 78, 1184. 10.1021/jo302746p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bhat V. T.; Caniard A. M.; Luksch T.; Brenk R.; Campopiano D. J.; Greaney M. F. Nat. Chem. 2010, 2, 490. 10.1038/nchem.658. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Dirksen A.; Dirksen S.; Hackeng T. M.; Dawson P. E. J. Am. Chem. Soc. 2006, 128, 15602. 10.1021/ja067189k. [DOI] [PubMed] [Google Scholar]
- Adams D. J.; Butler M. F.; Frith W. J.; Kirkland M.; Mullen L.; Sanderson P. Soft Matter 2009, 5, 1856. 10.1039/b901556f. [DOI] [Google Scholar]
- a Yelamaggad C. V.; Achalkumar A. S.; Shankar Rao D. S.; Prasad S. K. J. Am. Chem. Soc. 2004, 126, 6506. 10.1021/ja0495967. [DOI] [PubMed] [Google Scholar]; b Chong J. H.; Sauer M.; Patrick B. O.; MacLachlan M. J. Org. Lett. 2003, 5, 3823. 10.1021/ol0352714. [DOI] [PubMed] [Google Scholar]; c Kandambeth S.; Mallick A.; Lukose B.; Mane M. V.; Heine T.; Banerjee R. J. Am. Chem. Soc. 2012, 134, 19524. 10.1021/ja308278w. [DOI] [PubMed] [Google Scholar]; d Plaul D.; Plass W. Inorg. Chim. Acta 2011, 374, 341. 10.1016/j.ica.2011.03.018. [DOI] [Google Scholar]; e Riddle J. A.; Lathrop S. P.; Bollinger J. C.; Lee D. J. Am. Chem. Soc. 2006, 128, 10986. 10.1021/ja061307m. [DOI] [PubMed] [Google Scholar]; f Jędrzejewska H.; Wierzbicki M.; Cmoch P.; Rissanen K.; Szumna A. Angew. Chem., Int. Ed. 2014, 53, 13760. 10.1002/anie.201407802. [DOI] [PubMed] [Google Scholar]
- Carey A. R. E.; Fukata G.; O’Ferrall R. A. M.; Murphy M. G. J. Chem. Soc., Perkin Trans. 2 1985, 11, 1711. 10.1039/p29850001711. [DOI] [Google Scholar]
- Howe R. C. T.; Smalley A. P.; Guttenplan A. P. M.; Doggett M. W. R.; Eddleston M. D.; Tan J. C.; Lloyd G. O. Chem. Commun. 2013, 49, 4268. 10.1039/C2CC37428E. [DOI] [PubMed] [Google Scholar]
- Etter M. C.; Urbanczyk-Lipkowska Z.; Zia-Ebrahimi M.; Panunto T. W. J. Am. Chem. Soc. 1990, 112, 8415. 10.1021/ja00179a028. [DOI] [Google Scholar]
- Byrne P.; Turner D. R.; Lloyd G. O.; Clarke N.; Steed J. W. Cryst. Growth Des. 2008, 8, 3335. 10.1021/cg800247f. [DOI] [Google Scholar]
- a Palatinus L.; Chapuis G. J. Appl. Crystallogr. 2007, 40, 786. 10.1107/S0021889807029238. [DOI] [Google Scholar]; b Parois P.; Cooper R. I.; Thompson A. L. Chem. Cent. J. 2015, 9, 30. 10.1186/s13065-015-0105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Flack H. D. Acta Crystallogr. 1983, A39, 876. 10.1107/S0108767383001762. [DOI] [Google Scholar]; d Cooper R. I.; Thompson A. L.; Watkin D. J. J. Appl. Crystallogr. 2010, 43, 1100. 10.1107/S0021889810025598. [DOI] [Google Scholar]; e Thompson A. L.; Watkin D. J. J. Appl. Crystallogr. 2011, 44, 1017. 10.1107/S0021889811034066. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.