

Abstract
Nuclear factor-erythroid 2 p45-related factor 2 (Nrf2) regulates the basal and stress-inducible expression of a battery of genes encoding key components of the glutathione-based and thioredoxin-based anti-oxidant systems, as well as aldo-keto reductase, glutathione S-transferase, and NAD(P)H:quinone oxi-doreductase-1 drug-metabolizing isoenzymes along with multidrug-resistance-associated efflux pumps. It therefore plays a pivotal role in both intrinsic resistance and cellular adaptation to reactive oxygen species (ROS) and xenobiotics. Activation of Nrf2 can, however, serve as a double-edged sword because some of the genes it induces may contribute to chemical carcinogenesis by promoting futile redox cycling of polycyclic aromatic hydrocarbon metabolites or confer resistance to chemotherapeutic drugs by increasing the expression of efflux pumps, suggesting its cytoprotective effects will vary in a context-specific fashion. In addition to cytoprotection, Nrf2 also controls genes involved in intermediary metabolism, positively regulating those involved in NADPH generation, purine biosynthesis, and the β-oxidation of fatty acids, while suppressing those involved in lipogenesis and gluconeogenesis. Nrf2 is subject to regulation at multiple levels. Its ability to orchestrate adaptation to oxidants and electrophiles is due principally to stress-stimulated modification of thiols within one of its repressors, the Kelch-like ECH-associated protein 1 (Keap1), which is present in the cullin-3 RING ubiquitin ligase (CRL) complex CRLKeap1. Thus modification of Cys residues in Keap1 blocks CRLKeap1 activity, allowing newly translated Nrf2 to accumulate rapidly and induce its target genes. The ability of Keap1 to repress Nrf2 can be attenuated by p62/sequestosome-1 in a mechanistic target of rapamycin complex 1 (mTORC1)-depen-dent manner, thereby allowing refeeding after fasting to increase Nrf2-target gene expression. In parallel with repression by Keap1, Nrf2 is also repressed by β-transducin repeat-containing protein (β-TrCP), present in the Skp1–cullin-1–F-box protein (SCF) ubiquitin ligase complex SCFβ-TrCP. The ability of SCFβ-TrCP to suppress Nrf2 activity is itself enhanced by prior phosphorylation of the transcription factor by glycogen synthase kinase-3 (GSK-3) through formation of a DSGIS-containing phosphodegron. However, formation of the phosphodegron in Nrf2 by GSK-3 is inhibited by stimuli that activate protein kinase B (PKB)/Akt. In particular, PKB/Akt activity can be increased by phosphoinositide 3-kinase and mTORC2, thereby providing an explanation of why antioxidant-responsive element-driven genes are induced by growth factors and nutrients. Thus Nrf2 activity is tightly controlled via CRLKeap1 and SCFβ-TrCP by oxidative stress and energy-based signals, allowing it to mediate adaptive responses that restore redox homeostasis and modulate intermediary metabolism. Based on the fact that Nrf2 influences multiple biochemical pathways in both positive and negative ways, it is likely its dose–response curve, in terms of susceptibility to certain degenerative disease, is U-shaped. Specifically, too little Nrf2 activity will lead to loss of cytoprotection, diminished antioxidant capacity, and lowered β-oxidation of fatty acids, while conversely also exhibiting heightened sensitivity to ROS-based signaling that involves receptor tyrosine kinases and apoptosis signal-regulating kinase-1. By contrast, too much Nrf2 activity disturbs the homeostatic balance in favor of reduction, and so may have deleterious consequences including overproduction of reduced glutathione and NADPH, the blunting of ROS-based signal transduction, epithelial cell hyperplasia, and failure of certain cell types to differentiate correctly. We discuss the basis of a putative U-shaped Nrf2 dose–response curve in terms of potentially competing processes relevant to different stages of tumorigenesis.
Keywords: Nrf2, Keap1, β-TrCP, GSK-3, Reactive oxygen species, Inflammation, Nutrient supply, Lipid metabolism, Glutathione, Thioredoxin, Free radicals, aldo-keto reductase (AKR)
1. Introduction
1.1. Oxidative stress
Aerobic metabolism of a single molecule of glucose leads to the formation of 36 molecules of ATP, whereas anaerobic metabolism yields only 2 molecules of ATP. However, the increased yield in energy produced by aerobic metabolism comes at a cost; specifically, incomplete oxidation of O2 results in the formation of potentially damaging reactive oxygen species (ROS) such as the superoxide anion radical (O2●−), hydrogen peroxide (H2O2), and the hydroxyl radical (HO●). ROS can damage proteins, lipids, and DNA, and when the burden of ROS in the cell exceeds its antioxidant capability a state of oxidative stress is considered to exist [1].
Endogenous cellular ROS are generated as by-products of the actions of organelles, e.g., peroxisomes and mitochondria, and by the action of enzymes involved in electron transfer such as the cytochrome P450 (CYP) isoenzymes [2, 3]. Mitochondria are the principle producers of ATP within the cell, generating high yields of energy by oxidative phosphorylation using the electron transport chain located in the inner mitochondrial membrane. During the process of electron transport, molecular oxygen is reduced to H2O by cytochrome c oxidase. In some cases, reduction of O2 is incomplete and results in the generation of ROS.
Although ROS are generally regarded as harmful, it is worth noting that some enzymes, such as the NADPH oxidase (NOX) family, produce ROS in a controlled fashion that is necessary for cell signaling [4]. The production of ROS by membrane-associated oxidases is required for correct receptor tyrosine kinase signaling upon the binding of ligands such as insulin, epidermal growth factor (EGF), and platelet-derived growth factor (PDGF) to their cognate receptors [5–8]. The thiol (–SH) moiety in the side chain of the amino acid cysteine is a nucleophilic functional group that is susceptible to oxidation, particularly by ROS. Oxidation of the –SH group in the side chain of cysteine by ROS can result in creation of intramolecular and intermolecular disulfide bridges and the formation of sulfenic acid (–S–OH), sulfinic acid (–SO2H), and sulfonic acid (–SO3H), all of which can alter the function or activity of proteins, enabling ROS to participate in signal transduction. Therefore, in the case of insulin signaling, protein-tyrosine phosphatase (PTP) 1B is transiently inactivated by a controlled burst of NOX-generated ROS and this is required for optimal tyrosine phosphorylation [9]. Also, phosphatase and tensin homolog (PTEN) is oxidized in response to insulin, EGF, and PDGF and is similarly required for correct signaling [10–12]. Proteomics has suggested many PTP isoenzymes are subject to reversible oxidation [13].
Modification of protein thiols by ROS accounts for the mechanism by which oxidative stress activates the apoptosis signal-regulating kinase (ASK1; also called mitogen-activated protein kinase kinase kinase 5)–c-Jun N-terminal kinase (JNK) pathway and the ASK1-p38 mitogen-activated protein kinase (p38MAPK) pathway; in this case, oxidation of Cys residues in thioredoxin (TXN), resulting in formation of an intramolecular disulfide bridge, allows ASK1 to dissociate from TXN and activate downstream kinases [14, 15]. In a manner similar to that of the ASK1–JNK and ASK1–p38MAPK pathways, a TXN-related protein has been reported to regulate the Wnt-β-catenin signaling pathway via redox chemistry [16].
Exogenous causes of oxidative stress include exposure to ultraviolet irradiation, exposure to harmful environmental pollutants or carcinogens, and cigarette smoke [17]. Exposure to xenobiotics that are quinone-based or give rise to quinones or quinoneimines is also a cause of oxidative stress through one-electron and two-electron redox cycling [18]. Certain xenobiotics are electrophilic in nature or are metabolized to such agents, e.g., Michael acceptors, enabling them to modify proteins. The metabolism of xenobiotics can thus generate oxidative and electrophilic stress, which can in turn induce cellular defense mechanisms against these stressors. Oxidative stress is thought to contribute to many degenerative pathologies, including cancer, chronic inflammation, diabetes mellitus (DM), Alzheimer disease, and Parkinson disease [19, 20]. To reduce the risk of cellular damage by oxidative stress, cells employ intricate defense mechanisms.
1.2. Cellular defense against reactive oxygen species
Cells have evolved complex biochemical processes to limit the impact of ROS on essential cellular components, including enzymes that allow them to inactivate such species. Enzymes such as superoxide dismutase and catalase function in the reduction of O2●− to H2O2 and the conversion of two molecules of H2O2 to two H2O molecules and O2, respectively, thus limiting ROS levels in the cell and reducing the risk of oxidative damage to macromolecules [21].
Antioxidants play a key role in cellular defense against ROS and can act either directly or indirectly. Direct antioxidants are small molecules that possess redox-active properties, capable of scavenging ROS or reactive nitrogen species. They are depleted or modified in the process and must be replenished or restored after their interaction with ROS. Indirect antioxidants may or may not be redox active and exert their biological effects through induction of cytoprotective genes that recycle and/or regenerate direct antioxidants. As a result of gene induction, indirect antioxidants tend to have longer lasting effects than direct antioxidants because the proteins involved typically have longer half-lives than small molecules [22].
An additional defense mechanism against ROS involves the elimination of potentially harmful xenobiotics via the combined actions of drug-metabolizing enzymes and drug-efflux pumps. Thus the induction of proteins involved in metabolism and disposition of xenobiotics by indirect antioxidants aids this process by removing molecules that can generate ROS by redox cycling or by removing electrophiles that deplete endogenous antioxidants.
1.2.1. Direct antioxidants
Some antioxidants, such as glutathione, are synthesized de novo in the cell (Fig. 1A). Others, such as ascorbate and tocopherol, are absorbed through the diet.
Fig. 1.
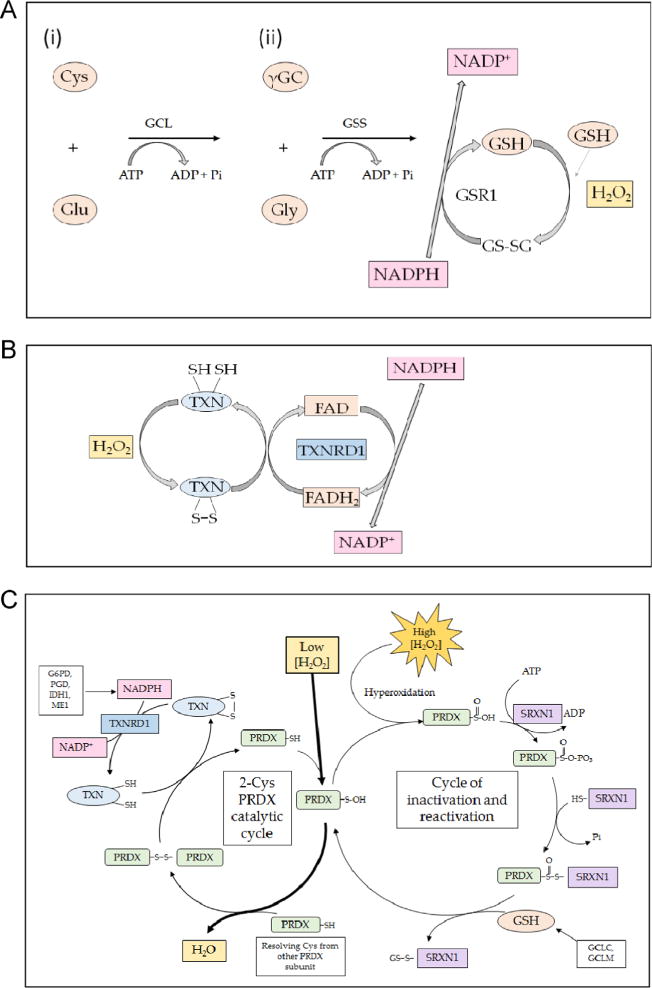
Mechanism of action of endogenous small protein antioxidants. (A) The biosynthesis of glutathione involves two steps. First (i), glutamate-cysteine ligase (GCL) conjugates cysteine (Cys) with glutamate (Glu), in a rate-limiting reaction that requires ATP, to produce γ-glutamyl-cysteine (γGC). Second (ii), glutathione synthetase (GSS) attaches glycine (Gly) to the C-terminal cysteine of γGC to produce the tripeptide glutathione (GSH). In turn, GSH may be oxidized by ROS (shown as H2O2), generating a disulfide bridge between two glutathione molecules, resulting in the formation of GSSG. The GSSG can be reduced back to two GSH molecules by the action of GSR1, an enzyme that utilizes NADPH as the electron donor. (B) The small protein dithiol TXN is oxidized, producing an intermolecular disulfide that is reduced by the flavoprotein TXNRD1, using NADPH as the electron donor. (C) The typical 2-Cys PRDX isoenzymes 1, 2, 3, and 4 reduce H2O2 through a catalytic cycle (shown on the left-hand side) that involves oxidation of the – SH group in an active-site Cys residue to sulfenic acid (–S–OH) in one subunit of the dimeric proteins. The oxidized thiol then forms an intermolecular disulfide bridge with a Cys residue in the other subunit before it is reduced by TXN; the resulting oxidized TXN is reduced by TXNRD1 in an NADPH-dependent manner. During reduction of H2O2, the active-site Cys in a small fraction of PRDX is hyperoxidized to sulfinic acid (–SO2H) (shown on the right-hand side). Overoxidation of the peroxidatic Cys to sulfinic acid inactivates PRDX, but it can be reactivated by SRXN1 through a mechanism that involves a transient covalent linkage between the two proteins, followed by a thiol-mediated reduction that is likely to involve GSH.
Glutathione is a tripeptide antioxidant, which is present in millimolar concentrations in cells [23]. It directly scavenges ROS within the cell, with two molecules of reduced glutathione (GSH) each donating an electron to generate oxidized glutathione (GSSG) that contains a disulfide bridge. The glutathione peroxidase (GPX) enzyme family also uses GSH to reduce H2O2 [24]. The oxidized GSSG is reduced by glutathione reductase (GSR1) in an NADPH-dependent manner to regenerate two GSH molecules. Under normal conditions, the amount of GSH is between 10- and 100-fold higher than the concentration of GSSG. Therefore the ratio of GSH: GSSG is an important indicator of cellular redox status. Although critical for the regeneration of two GSH molecules from GSSG, genetic knockout of GSR1 is not lethal, with the mutant animal continuing to thrive under unstressed conditions [25], presumably owing to redundancy between GSH and other antioxidant systems.
Another example of an endogenous direct antioxidant is provided by TXN [26]. The TXN system consists of small 12-kDa ubiquitously expressed dithiol proteins and their selenoprotein partner thioredoxin reductase (TXNRD) isoenzymes; TXNRD catalyzes the cycling of TXN proteins from the oxidized to the reduced state in an NADPH-dependent manner (Fig. 1B). There are two distinct pools of TXN activity in the cell, cytosolic and mitochondrial. The cytosolic pool makes use of the TXN1 and TXNRD1 isoforms of the proteins, whereas mitochondria use TXN2 and TXNRD2 [27]. Like the GSH-based system, the TXN system is capable of directly scavenging ROS and can also make use of reversible oxidation of its key conserved cysteine residues (C-X-X-C) to reduce disulfide bridges in oxidized proteins. TXNRD plays an essential role in reducing oxidized TXN. In mice, complete knockout of Txnrd1 is embryonic lethal [28]. However, hepatocyte-specific knockout of Txnrd1 yields viable mice and causes constitutive overexpression of GSH-associated enzymes and sulfiredoxin 1 (Srxn1), presumably as a means of compensating for the lack of reducing power available in the cell [29]. Interestingly, the TXN and GSH systems are themselves jointly redundant, as mice with hepato-cyte-specific dual disruption of Txnrd1 and Gsr1 are also viable. Under unstressed conditions these double-knockout mice can sustain hepatic redox homeostasis by using methionine as the sole source of disulfide reducing power and sulfur amino acids [30].
In addition to the GSH and TXN systems, cells utilize the per-oxiredoxin (PRDX) family as cellular antioxidants [31, 32]. Like catalase and superoxide dismutase, PRDX family members (isoenzymes 1–4) are capable of scavenging H2O2 directly using their peroxidatic Cys residue (CP) as an electron source and forming a disulfide bridge with their other reactive Cys residue, known as the resolving Cys (CR) [33]. The resulting intermolecular disulfide (CP–CR) is reduced by TXN, which is in turn reduced by TXNRD isoenzymes in an NADPH-dependent manner (summarized in Fig. 1C). In the case of overoxidation of the peroxidatic Cys to sulfinic acid, PRDX isoenzymes 1–4 employ SRXN1 and GSH to reactivate its Cys residues [34, 35]. The fact that TXN1, TXNRD1, TXNRD6, and SRXN1 are all regulated by nuclear factor-erythroid 2 p45-related factor 2 (Nrf2; with the gene usually designated NFE2L2), as are enzymes involved in the regeneration of NADPH (glucose 6-phosphate 1-dehydrogenase (G6PD), 6-phosphogluconate (PGD), isocitrate dehydrogenase 1 (IDH1) and malic enzyme 1 (ME1)) and those involved in the synthesis of GSH (glutamate–cysteine ligase catalytic (GCLC) and glutamate–cysteine ligase modifier (GCLM) subunits) [36–41] suggests that the Nrf2 transcription factor makes a substantial contribution to the efficiency of reduction of H2O2 by PRDX.
Some direct antioxidants are ingested in the diet. For example, ascorbate (vitamin C), found abundantly in citrus fruits, and to-copherols (e.g., vitamin E), found in plant oils, and napthoquinoids (e.g., vitamin K), found in leafy green vegetables, are direct-acting antioxidants that are chiefly not synthesized de novo in mammals, but are acquired through dietary intake.
1.2.2. Indirect antioxidants
Redox-dependent systems that recognize indirect antioxidants have evolved to allow cells to adapt to oxidative stress by increasing their ability to neutralize ROS and prevent cumulative damage. This class of antioxidant acts by inducing cytoprotective genes involved in the rate of synthesis and regeneration of endogenous direct antioxidants and by influencing the metabolism and elimination of xenobiotics [42].
Indirect antioxidants can be found in the human diet. Groups of natural and synthetic molecules such as polyphenols and isothiocyanates induce genes encoding GSH biosynthetic enzymes [43, 44], drug-metabolizing enzymes such as NAD(P)H:quinone oxidoreductase-1 (NQO1) [45], glutathione S-transferase (GST) isoenzymes [46, 47], and aldo-keto reductase (AKR) isoenzymes [48], which will be discussed in detail later. As the name suggests, the polyphenol family of compounds encompasses a diverse set of molecules containing multiple phenol structures. They can be divided into many subgroups, including flavones, isoflavones, lignans, stilbenoids, tannins, and curcuminoids, some examples of which are shown in Fig. 2. Polyphenols can act as direct anti-oxidants by scavenging ROS. Some polyphenols also contain an α, β-unsaturated carbonyl group that serves as an electrophile, enabling them to act as indirect antioxidants by increasing Nrf2 activity, leading to induction of its target genes. Also, glucosinolates that are present in cruciferous vegetables are degraded to yield isothiocyanates that contain the electrophilic –N=C=S functional group, as well as epithionitriles, both of which increase Nrf2 activity, resulting in induction of its target genes [49, 50].
Fig. 2.
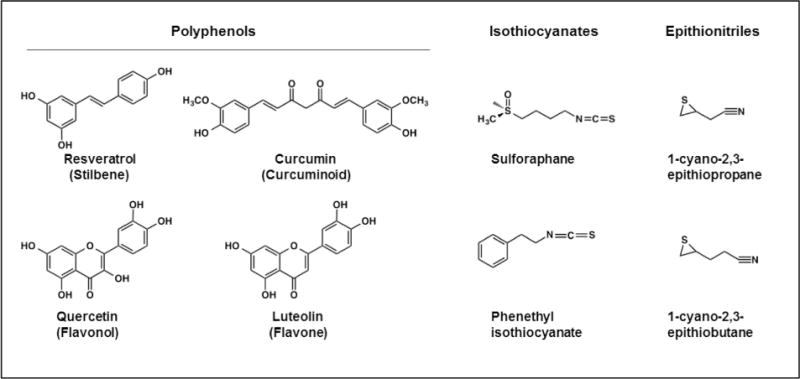
Chemical structures of dietary antioxidant compounds. Polyphenol family members that exhibit indirect antioxidant activity include resveratrol, a stilbenoid found in red wine; quercetin, a flavonol found in red and yellow onions; curcumin, a curcuminoid found in turmeric and mustard; and luteolin, a flavone found in celery, parsley, and thyme. Glucosinolate breakdown products that have indirect antioxidant properties include the isothiocyanates sulforaphane and phenethyl isothiocyanate and the epithionitriles 1-cyano-2,3-epithiopropane and 1-cyano-3,4-epithiobutane: glucosinolates that give rise to these compounds are found in cruciferous vegetables, such as broccoli.
1.3. Metabolism of xenobiotics and environmental toxins
Foreign chemicals, including drugs and environmental toxicants, represent another source of stress for cells and it is important that they be effectively inactivated [51]. Drug metabolism occurs in three phases, which act to make lipophilic xenobiotics more water soluble, to render them less harmful, and finally to export them from the cell [52].
Phase I drug metabolism involves functionalization of the parent xenobiotic/endobiotic, such as introduction of a hydroxyl group, so that phase II conjugation reactions, e.g., addition of a sulfonate group, can proceed to make the xenobiotic more water soluble. Paradoxically, the reactions that take place in phase I often serve to make compounds more reactive and can increase the toxicity of the agent [53]. Functionalization involves the introduction of new polar groups by oxidation, reduction, hydration, and hydrolysis reactions or by the exposure of polar groups by the removal of alkyl groups. This can be catalyzed by a variety of phase I drug-metabolizing enzymes such as CYP, alcohol dehydrogenases, short-chain dehydrogenase reductase isoenzymes, AKR isoenzymes, and flavin-containing monooxygenases [54].
Phase II metabolism involves the conjugation of the functionalized xenobiotic/endobiotic with GSH, catalyzed by GST isoenzymes [46]; glucuronic acid, catalyzed by UDP-glucuronosyl-transferases (UGT); or an SO3 group, catalyzed by sulfotransferases (SULT) [55]. Once metabolites have been conjugated appropriately, they are usually less reactive and are more readily excretable. It is the balance of phase I and phase II reactions that determines whether this leads to toxication or detoxication of a xenobiotic in a particular organ or cell type.
Phase III of drug metabolism involves the active transport of conjugated metabolites across the cell membrane by transport proteins, such as the ATP-binding cassette family of transporters, including multidrug resistance [56] and multidrug-resistance-associated protein (MRP) [57], where they can be excreted via the biliary or renal routes.
2. The Nrf2 antioxidant defense system
Nrf2 is a transcription factor that controls both the basal expression of genes under unstressed homeostatic conditions and the inducible expression of genes upon redox perturbation [58–61]. In total, it regulates directly a battery of approximately 250 genes involved in a wide variety of cellular functions ranging from cytoprotection against endogenous and environmental stressors to lipid and carbohydrate metabolism [62]. It also controls other genes indirectly by virtue of the fact that its targets include other transcription factors [62].
2.1. Discovery of Nrf2
Nrf2 was first identified by researchers in the laboratory of Yuet Wai Kan using a λgt11 cDNA expression library to screen for proteins that bind to an extended activator protein 1 (AP-1) sequence (i.e., 5′-TGAC/GTCA-3′) [63]. The 2.2-kb cDNA isolated by this strategy was predicted to encode a ubiquitously expressed 66-kDa protein that contained toward its C-terminus a cap’n’collar basic-region leucine zipper (CNC-bZIP) domain. It was recognized as the third member of the mammalian CNC-bZIP family of proteins and named Nrf2.
2.2. Structure of Nrf2
Nrf2 is a modular protein that in the mouse and rat comprises 597 amino acids and in the human consists of 605 amino acids. On the basis of sequence identity shared between human Nrf2 and the orthologous chicken protein, which when first cloned by Ma-sayuki Yamamoto and colleagues was called erythroid cell-derived protein with CNC homology (ECH), the transcription factor can be divided into a number of distinct regions referred to as Nrf2–ECH homology (Neh) domains [64]. To date, seven Neh domains have been reported, and the structure of human Nrf2 is presented in Fig. 3. The individual Neh domains have different functions.
Fig. 3.
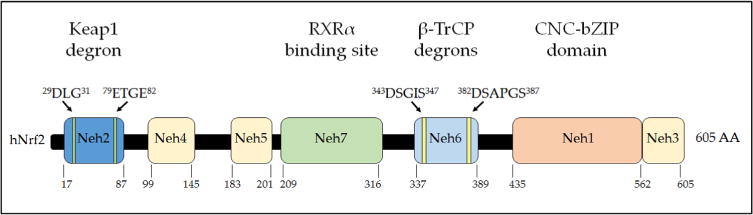
Domain structure of human Nrf2. The relative positions of the Neh domains of transcription factor Nrf2 are shown. The degrons responsible for targeting of Nrf2 protein by Keap1 and β-TrCP for proteasomal degradation are indicated above the domains, and the region of Nrf2 through which it is inhibited by RXRα is indicated. The numbering of amino acids is based on the human sequence and is shown below the cartoon.
The Neh1 domain in Nrf2 comprises the conserved CNC-bZIP region, a feature that was first described as a DNA-binding peptide sequence in Drosophila melanogaster[65] and is essential for its activity as a transcription factor and heterodimerization with other bZIP proteins.
The N-terminal Neh2 domain negatively controls the activity of Nrf2 and is crucial for Kelch-like ECH-associated protein 1 (Keap1)-mediated repression of the transcription factor [66]. It contains two highly conserved peptide sequences to which Keap1 binds; these are the high-affinity ETGE motif and the lower-affinity DLG motif [67–70].
The C-terminal Neh3 domain is involved in transcriptional activation of Nrf2. Removal of 16 amino acids from the C-terminus of the protein inactivates the CNC-bZIP factor, indicating a role for the Neh3 domain in the transactivation of target genes. This region has also been shown to interact with the chromodomain helicase DNA-binding protein 6, which is consistent with the idea that it is involved in transcriptional activation [71].
Neh4 and Neh5 have been shown to represent Nrf2 transactivation domains, which act cooperatively in binding the coactivator CREB binding protein, thereby synergistically increasing the rate of gene transcription [72].
The Neh6 domain negatively controls Nrf2 and is responsible for Keap1-independent regulation of Nrf2 [73]. It contains two conserved peptide motifs, DSGIS and DSAPGS, which are recognized by β-transducin repeat-containing protein (β-TrCP). The DSGIS motif includes a glycogen synthase kinase-3 (GSK-3) phosphorylation site that when modified by GSK-3 increases the ability of β-TrCP to repress Nrf2 [74–76]. This is referred to as the DSGIS-containing phosphodegron.
Neh7 is the most recently described domain. It includes a region (yet to be mapped) that can engage in a direct protein–protein interaction between Nrf2 and the DNA-binding domain of retinoid X receptor α (RXRα), which causes suppression of Nrf2 activity by preventing recruitment of coactivators to the Neh4 and Neh5 domains [77]. The RXRα ligand bexarotene can inhibit the expression of Nrf2-target genes presumably by promoting interaction between RXRα and the Neh7 domain of Nrf2 [78]. The retinoic acid receptor α (RARα) ligand all-trans-retinoic acid is also a potent inhibitor of Nrf2 [79] but it has not been established if RARα binds to the Neh7 domain of Nrf2.
2.3. Members of the cap’n’collar family
Among the CNC-bZIP family of transcription factors, Nrf2 shows the highest transactivation activity [72]. Other members of the mammalian CNC-bZIP family include the founding member, NF-E2 p45, and also Nrf1 and Nrf3 (Fig. 4). Expression of NF-E2 p45 is limited to hematopoietic tissue [80], whereas Nrf1 and Nrf2 expression is ubiquitous. The NF-E2 p45 and Nrf2 transcription factors are soluble proteins, whereas upon translation Nrf1 and Nrf3 are initially anchored in the endoplasmic reticulum (ER) as glycosylated proteins [81–83]. NF-E2 p45 is involved in the production of platelets, and its genetic knockout is lethal in mice because of internal bleeding [84]. Knockout of Nrf1 is embryonic lethal, suggesting it fulfills an essential role that cannot be compensated for by other family members [85]. Despite Nrf1 having a critical function in development, its physiological roles are not yet fully understood, though it does influence uptake of cystine, via the Slc7a11 cystine/glutamate exchange transporter, and lipid metabolism [86, 87] while contributing positively to the basal and in-ducible expression of proteasome subunits [88]. Knockout of Nrf2 in the mouse does not exhibit an obvious phenotype [89], other than decolorization of teeth due to iron transport impairment [90]. The remaining family member, Nrf3, is expressed in a number of tissues, including placenta, B cells, and monocytes, but it is not ubiquitously expressed. Mice lacking Nrf3 develop normally and display no obvious phenotypic differences to the wild-type animal [91–93]. Interestingly, Nrf3 is inducible [94, 95] and may on occasion compensate for the absence of other CNC-bZIP transcription factors.
Fig. 4.
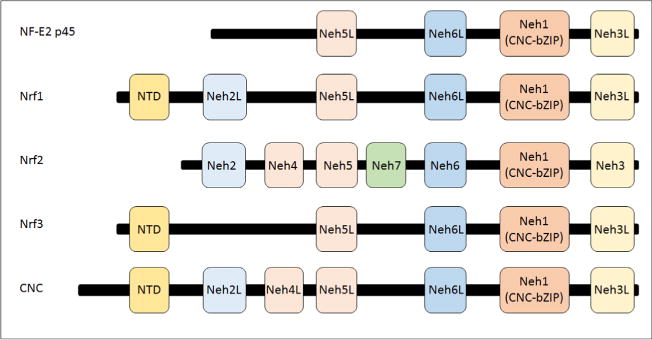
Structure of CNC-bZIP family members. By definition, all family members contain both CNC and bZIP sequences, which together comprise the Neh1 domain. The Neh2 or the Neh2-like (Neh2L) domain is present in mammalian Nrf1 and Nrf2 as well as the Drosophila CNC protein. The Neh3, Neh5, and Neh6 domains are common to all family members. The Neh4 domain is represented only in Nrf2 and CNC. The Neh7 domain has to date been identified only in Nrf2. An N-terminal domain (NTD) is found in Nrf1, Nrf3, and CNC, which directs them to the endoplasmic reticulum.
2.4. Cooperation between Nrf2 and small Maf proteins
Nrf2 binds DNA as an obligatory heterodimer with small musculoaponeurotic fibrosarcoma (Maf) proteins [96, 97]. Three small Maf proteins, MafF, MafG, and MafK, have been described. Bach proteins also dimerize with small Maf (sMaf) proteins, leading to competitive binding of the Nrf2–sMaf dimers and the Bach–sMaf dimers [98, 99]. The level of sMaf protein must be carefully regulated in the cell, as too little impairs activation of CNC and Bach factors, but similarly too much will result in homodimer formation, causing fewer sMaf proteins to be available for dimerization with CNC and Bach proteins [98]. Compound knockout fibroblasts that lack all three MafF, MafG, and MafK proteins have been found to possess a severely impaired anti-oxidant response, indicating that sMaf proteins are essential for the recruitment of Nrf2 to its cognate binding sites in the promoter of target genes [100, 101].
2.5. The antioxidant response element
The antioxidant response element (ARE) is the cis-acting DNA sequence to which small Nrf2–sMaf heterodimers are recruited. Its name originates from the fact that it was first found in the promoter regions of genes that can be induced by synthetic phenolic antioxidants, with the metabolite of butylated hydroxyanisole (BHA), tert-butyl hydroquinone (tBHQ), being one of the prototypic inducing agents [102]. In the early 1990s, researchers in the laboratory of Cecil Pickett and that of Violet Daniel first described the ARE. The research group of Cecil Pickett reported an ARE in the promoter of the rat GSTA2 subunit gene as a novel cis-element that was responsive to tBHQ and β-naphthoflavone (β-NF), but not to the classic arylhydrocarbon receptor (AhR) ligand dioxin [103]. Thereafter they demonstrated that the element was required for induction of gene expression by metabolizable planar aromatic compounds, such as 3-methylcholanthrene, and diphenols that undergo redox cycling, such as catechol and hydroquinone [104]. At around the same time, an essentially identical cis-element was reported in the mouse Gsta1 subunit gene promoter by the research group of Violet Daniel and named the electrophile response element (EpRE) because it responded to the soft electrophiles trans-4-phenyl-3-buten-2-one and dimethyl fumarate, as well as to tBHQ and β-NF [105]; the EpRE was formally defined by Friling et al. [106]. The term ARE is now more commonly used than EpRE, and in this review we use the former term. Following the characterization of the ARE, Paul Talalay and co-workers demonstrated that the phytochemical sulforaphane (SFN), an isothiocyanate derived from the glucosinolate glucoraphanin that is abundant in cruciferous vegetables [49], can induce gene expression though this cis-element, as did 1,2-dithiole-3-thione [107].
Shortly after the description of a consensus sequence for the ARE in rodent GST genes, closely related sequences were found in the regulatory regions of the rat and human NQO1 genes, which can be induced by tBHQ and β-NF [108, 109]. These findings suggested that the ARE plays a role in genetic programs involved in cellular responses to oxidative stress [104, 110]. Since its discovery, the core ARE sequence has been described frequently as 5′-A/GTGAC/GNNNGCA/G-3′, where “N” signifies apparently redundant residues. In some cases, the flanking sequence of the ARE and also the nucleotides designated “N” influence its function, suggesting that the element is somewhat plastic in nature (see Fig. 5) [110–112].
Fig. 5.
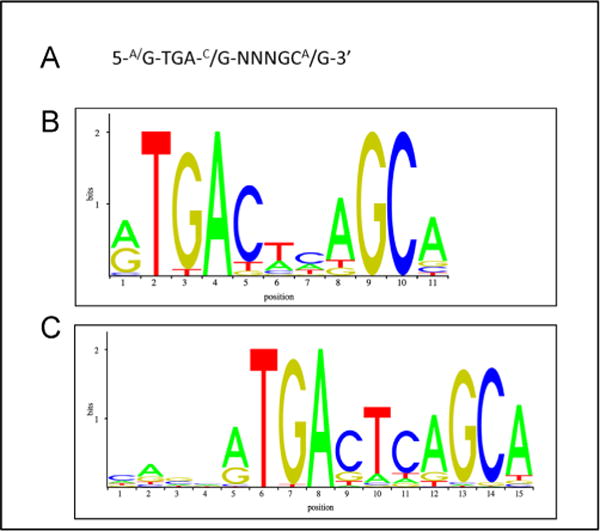
The antioxidant response element. The characteristics of ARE sequences across human and rodent genes, and Nrf2-binding profiles, have been assessed using bioinformatics. (A) The ARE consensus sequence as usually reported in the literature on the basis of mutation analyses of gene reporter plasmids based primarily on rat, mouse, and human GST and NQO1 genes. (B and C) The positional matrices for the human and murine ARE sequences, Jasper IDs MA0150.1 and MA0150.2, respectively, which have been generated by the frequency at which these sites have been found experimentally to be occupied by Nrf2.
2.6. The identity of Nrf2-target genes
Masayuki Yamamoto and colleagues played a major role in linking the in vivo regulation of ARE-driven genes with Nrf2 by demonstrating loss of induction of various Gst subunits and Nqo1 by BHA in the livers and small intestine of Nrf2−/− mice [96]. While historically Nrf2 has largely been studied in the context of its ability to regulate genes involved in GSH synthesis, redox regulation, and drug metabolism, it has also more recently been recognized to play a role in the regulation of genes that contribute to NADPH generation, lipid metabolism, glucose/glycogen metabolism, and H2S production [113–116]. Studies of Nrf2−/− mice have allowed researchers to generate lists of genes and proteins regulated by the CNC-bZIP transcription factor. Table 1 provides some examples of genes that have been shown to be regulated, both positively and negatively, by Nrf2 in mouse and in human cell lines [36–41, 58, 115–125].
Table 1.
Examples of genes regulated by Nrf2 and their biological functions.
| Primary role | Gene | Protein | Function | Cellular processes | References |
|---|---|---|---|---|---|
| Redox homeostasis | GCLC | Glutamate–cysteine ligase catalytic subunit | Catalytic subunit of the enzyme responsible for the rate-limiting step in synthesis of the cellular antioxidant glutathione | Maintenance of cellular redox homeostasis | [36, 37, 40, 41] |
| Redox homeostasis | GLCM | Glutamate–cysteine ligase modifier subunit | Modifier subunit of the enzyme responsible for the rate-limiting step in synthesis of the cellular antioxidant glutathione | Maintenance of cellular redox homeostasis | [36–38, 40, 123] |
| Redox homeostasis | GPX2 | Glutathione peroxidase 2 | Detoxification of H2O2 and an important cellular antioxidant | Maintenance of cellular redox homeostasis | [126, 127] |
| Redox homeostasis | PRDX1 | Peroxiredoxin 1 | Reduces peroxides, regulates cellular concentrations of H2O2 | Maintenance of cellular redox homeostasis | [36–38, 41, 117] |
| Redox homeostasis | SRXN1 | Sulfiredoxin | Contributes to the thioredoxin-based antioxidant system that reduces oxidized protein thiols; reduces sulfinic acid in proteins | Thiol maintenance, maintenance of cellular redox homeostasis | [36, 37, 40, 41] |
| Redox homeostasis | TXN1 | Thioredoxin 1 | Reversible oxidation of active center allows participation in dithiol–disulfide exchange reactions; reduces sulfenic acid in proteins | Thiol maintenance, maintenance of cellular redox homeostasis | [36, 37, 39–41, 117] |
| Detoxification | ABCB6 | ATP-binding cassette B6 (MDR/TAP) | Mitochondrial transporter; transports heme and porphyrin in an ATP-dependent manner, crucial for heme metabolism | Phase III drug metabolism, heme metabolism | [37, 41] |
| Detoxification | ABCC2 | ATP-binding cassette C2 (MRP2) | Biliary transport/hepatic excretion; involved in the extrusion of certain anti-cancer drugs and implicated in multidrug resistance | Phase III drug metabolism | [36, 38, 39, 41, 120] |
| Detoxification | AKR1B10 | Aldo-keto reductase 1B10 | Converts retinal to retinol; reduces aromatic and aliphatic aldehydes | Phase I drug metabolism and retinal metabolism | [36, 38–40] |
| Detoxification | AKR1C1 | Aldo-keto reductase 1C1 | Converts 4-hydroxy-2-nonenal to 1,2-dihydroxy nonene; inactivates progesterone | Phase I drug metabolism | [36, 39, 40, 128] |
| Detoxification | AKR1C3 | Aldo-keto reductase 1C3 | Type 5 17β-hydroxysteroid dehydrogenase and prostaglandin F2α synthase | Phase I drug metabolism | [129–131] |
| Detoxification | CES1G | Carboxyl esterase 1G | Catalyzes the trans-esterification of xenobiotics, hydrolysis of long-chain fatty acid esters | Phase I drug metabolism, fatty acid oxidation, fatty acid degradation | [123] |
| Detoxification | CES1H | Carboxyl esterase 1H | Catalyzes the trans-esterification of xenobiotics, hydrolysis of long-chain fatty acid esters | Phase I drug metabolism, fatty acid oxidation, fatty acid degradation | [123] |
| Detoxification | GSTA1 | Glutathione S-transferase A1 | Detoxification and metabolism of electrophilic compounds; metabolism of bilirubin and certain anti-cancer drugs; also displays glutathione peroxidase activity | Phase II drug metabolism, cytoprotection | [38, 41, 58] |
| Detoxification | GSTM1 | Glutathione S-transferase M1 | Detoxification and metabolism of electrophilic compounds | Phase II drug metabolism | [38, 41, 58] |
| Detoxification | NQO1 | NAD(P)H quinone oxidoreductase 1 | Reduces quinones to hydroquinones and prevents the one-electron reduction of quinones that would otherwise produce free radicals | Phase I drug metabolism | [36, 37, 40] |
| Heme metabolism | FECH | Ferrochelatase | Catalyzes the insertion of Fe2+ into protoporphyrin IX during heme synthesis; localized in mitochondria | Heme metabolism | [37, 40] |
| Heme metabolism | HMOX1 | Heme oxygenase 1 | Cleaves heme to produce biliverdin during heme catabolism | Heme metabolism | [36–41] |
| Lipid metabolism | AWAT1 | Acyl-CoA wax alcohol acyltransferase 1 | Catalyzes the production of wax esters from long-chain alcohols and acyl-CoA-derived fatty acids; enriched in skin | Lipid metabolism | [123] |
| Lipid metabolism | FABP1 ↓ | Fatty acid binding protein 1 | Binds long-chain fatty acids, their CoA derivatives, and bile acids in the cytoplasm for intracellular transport | Lipid uptake and intracellular transport | [116] |
| Lipid metabolism | LIPH | Lipase H | Membrane-bound triglyceride lipase that hydrolyzes phosphatidic acid to produce 2-acyl lysophosphatidic acid, which is a potent bioactive lipid mediator | Platelet aggregation, smooth muscle contraction, cell proliferation and motility | [123] |
| Lipid metabolism | PPARγ | Peroxisome proliferator-activated receptor γ | Transcription factor that orchestrates lipid metabolism; key regulator of adipocyte differentiation and glucose homeostasis | Lipid mobilization, β-oxidation of fatty acids, adipocyte differentiation, glucose metabolism | [37, 116] |
| Lipolysis | ACOT7 | Acyl-CoA thioesterase 7 | Catalyzes the hydrolysis of palmitoyl-CoA and other long-chain fatty acids to form free fatty acid and CoA | Fatty acid oxidation, fatty acid degradation | [115] |
| Lipolysis | ACOX2 | Acyl-CoA oxidase 2 | Catalyzes peroxisomal degradation of long branched-chain fatty acids and bile acid intermediates | Fatty acid oxidation, fatty acid degradation | [123] |
| Lipogenesis | ACLY ↓ | ATP citrate lyase | Catalyzes the production of acetyl-CoA and oxaloacetate from CoA and citrate using ATP | Lipogenesis, cholesterol synthesis, gluconeogenesis | [115, 116, 118] |
| Lipogenesis | FASN ↓ | Fatty acid synthase | Generation of long-chain fatty acids such as palmitate from malonyl-CoA and acetyl-CoA using NADPH | Lipogenesis | [115, 116, 118] |
| Lipogenesis | SCD1 ↓ | Stearoyl-CoA desaturase 1 | Catalyzes the introduction of a double bond into stearoyl-CoA to create the mono-unsaturated fatty acid oleic acid | Lipogenesis | [115, 122] |
| Lipogenesis | SREBF1 ↓ | Sterol-regulatory element binding transcription factor 1 | Transcription factor that controls expression of the LDL receptor, and genes involved in glucose metabolism and lipid synthesis | Lipogenesis, glucose metabolism | [116] |
| Glucose metabolism | FGF21 ↓ | Fibroblast growth factor 21 | Stimulates glucose uptake in adipocytes, involved in regulation of insulin | Glucose uptake and clearance, insulin signaling | [132] |
| Cholesterol synthesis | HMGCS1 ↓ | 3-Hydroxy-3-methylglutaryl-CoA synthase 1 | Catalyzes the condensation of acetyl-CoA and acetoacetyl-CoA to produce HMG-CoA, the substrate for HMG-CoA reductase | Cholesterol synthesis | [115, 116] |
| NADPH generation | G6PD | Glucose-6-phosphate dehydrogenase | Generates NADPH in the pentose phosphate pathway; maintains cellular glutathione redox status | Pentose phosphate pathway, NADPH generation, maintenance of cellular redox homeostasis | [36, 38–41, 115] |
| NADPH generation | IDH1 | Isocitrate dehydrogenase 1 | Catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate, using NADP+ as a cofactor, outside the context of the citric acid cycle in the cytoplasm or in peroxisomes | NADPH generation | [38, 41, 115] |
| Pentose synthesis | TALDO1 | Transaldolase 1 | Involved in production of ribose 5′-phosphate required for nucleic acid synthesis | Pentose phosphate pathway | [36, 37, 39, 115] |
| Pentose synthesis | TKT | Transketolase | Channels excess sugars from the pentose phosphate pathway to glycolysis by the creation of glyceraldehyde 3-phosphate | Pentose phosphate pathway, glycolysis | [36] |
| Scavenger receptor | CD36 | CD36 molecule/fatty acid translocase | Major platelet glycoprotein that binds long-chain fatty acids and functions in the transport and regulation of transport of fatty acids | Fatty acid transport, adhesion | [133] |
| Autophagy | p62/SQSTM1 | Sequestosome 1 | Required for formation and autophagic degradation of polyubiquitin-containing bodies; used as a scaffold protein | Autophagy, inflammation | [134] |
| Tissue regeneration | NOTCH1 | Notch 1 | Transmembrane protein containing multiple epidermal growth factor-like repeats that is involved in signaling processes during development and tissue regeneration | Cell fate determination, developmental signaling | [135] |
| Regulation of xenobiotic response | AHR | Aryl hydrocarbon receptor | Transcription factor that binds planar aromatic compounds and upregulates genes involved in xenobiotic metabolism, including CYP family members | Xenobiotic metabolism | [124] |
| E3 ligase substrate adaptor | Keap1 | Kelch-like ECH associated protein 1 | E3 ubiquitin ligase substrate adaptor that targets proteins for degradation by the 26S proteasome; known negative regulator of Nrf2 | Targeting for ubiquitination, Nrf2 repression, regulation of antioxidant response | [37] |
| Heterodimeric binding partner | MAFG | Musculoaponeurotic fibrosarcoma G | Transcriptional regulator; forms heterodimers with a number of transcription factors, including Nrf2, allowing their activation; also forms homodimers restricting the activation of obligate heterodimeric partner molecules | Transcriptional activation, regulation of anti-oxidant response | [37, 40, 41, 123] |
| Regulation of antioxidant response | NFE2L2 | NF-E2 p45-like 2 | Transcription factor that regulates genes involved in the oxidative stress response; maintains cellular redox homeostasis and detoxification and contributes to lipid and carbohydrate metabolism | Drug metabolism, xenobiotic metabolism, maintenance of cellular redox homeostasis | [119] |
| Regulation of lipid metabolism | PPARα ↓ | Peroxisome proliferator-activated receptor α | Transcription factor, key regulator of lipid metabolism; activator of Nrf2 | Lipid mobilization, fatty acid oxidation, lipogenesis, gluconeogenesis, ketogenesis | [116, 125] |
| Transcriptional regulator | RXRα | Retinoid X receptor α | Transcriptional regulator; mediates the biological effect of retinoids by forming homo- or heterodimers and binds to target gene sequences; known negative regulator of Nrf2; known binding partner and activator of PPARα | Transcriptional activation, Nrf2 repression, regulation of antioxidant response, PPARα activation | [37, 41] |
A selection of Nrf2-target genes taken from the literature is shown. Genes that are in bold (with an adjacent down arrow) are those that have been reported to be downregulated by Nrf2.
In the human, it is notable that AKR isoenzymes are among the most highly induced mRNAs/proteins upon activation of Nrf2 and that GST mRNAs/proteins are not dominantly regulated by the CNC-bZIP transcription factor. Specifically, in human HaCaT keratinocytes, MCF10A mammary cells, IMR-32 neuroblastoma cells, and U937 lymphoma cells, treatment with SFN or tBHQ, or knockdown of the Nrf2 repressor Keap1, has been reported to result in substantial induction of AKR1B10, AKR1C1, AKR1C2, and AKR1C3, which is as least as great as that observed for NQO1 [36, 39, 40, 136]. The nomenclature used to describe AKR is species specific, and thus the identity of genes in the mouse that are equivalent to human AKR1B10, AKR1C1, AKR1C2, and AKR1C3 has been the source of some confusion in the literature. In the mouse, microarray experiments have indicated that hepatic Akr1a1 is induced by 1,2-dithiole-3-thione in an Nrf2-dependent manner [59] and chromatin immunoprecipitation (ChIP)-seq analyses of cell lysates from Hepa1c1c7 hepatoma cells treated with diethyl maleate has revealed Akr1b8 to be proximal to an Nrf2 ChIP-seq site [38]. Moreover, members of the Akr1b and Akr1c family are overexpressed in a mouse lung KrasG12D-driven tumor model [137]. A list of the human, rat, and murine AKR isoenzymes that contain an ARE sequence(s) in their respective promoter regions is provided in Table 2. It is difficult to assign murine orthologs for the human enzymes: the laboratory of Christopher Bunce has conducted a detailed characterization of the murine Akr1c enzymes and concluded from tissue expression profiling and analysis of enzyme function that none are equivalent to human AKR1C family members [138]. However, it is clear that murine Akr1a4 represents aldehyde reductase and that murine Akr1b3 represents aldose reductase. Because human AKR1B10, AKR1C1, AKR1C2, and AKR1C3 are among the most inducible human genes to be regulated by Nrf2, it is important to consider the catalytic properties of the enzymes they encode (see later).
Table 2.
Human, rat, and murine AKR genes with ARE sequences in their promoter regions
| Species | Gene name | Nonsystematic name | No. of AREs based on Nrf2 consensus sequence | Positional matrix human LD <6 | Positional matrix mouse LD <6 |
|---|---|---|---|---|---|
| Human | AKR1A1 | Aldehyde reductase; dihydrodiol dehydrogenase (DD) 3 | 6 | 5 | 0 |
| Human | AKR1B1 | Aldose reductase | 1 | 3 | 1 |
| Human | AKR1B10 | Retinaldehyde reductase; small-intestine-like aldose reductase | 4 | 2 | 0 |
| Human | AKR1B15 | 3-Keto-acyl CoA reductase; 17β-hydroxysteroid dehydrogenase (HSD) | 4 | 5 | 0 |
| Human | AKR1C1 | 3(20α)-HSD; DD1 | 10 | 16 | 5 |
| Human | AKR1C2 | Type 3 3α-HSD; DD2 | 15 | 24 | 7 |
| Human | AKR1C3 | Type 5 17β-HSD; prostaglandin F synthase; DDX | 4 | 6 | 2 |
| Human | AKR1C4 | Type 1 3α-HSD; DD4; chlordecone reductase | 2 | 2 | 0 |
| Human | AKR1D1 | Steroid 5β-reductase | 11 | 27 | 0 |
| Human | AKR1E2 | 1,5-Anhydro-D-fructose reductase | 6 | 3 | 0 |
| Human | AKR6A5 | Shaker channel β-subunit (Kvb2) | 26 | 8 | 1 |
| Human | AKR7A2 | Aflatoxin aldehyde reductase; succinic semialdehyde reductase | 9 | 10 | 0 |
| Human | AKR7A3 | Aflatoxin aldehyde reductase | 4 | 4 | 0 |
| Rat | AKR1A3 | Aldehyde reductase | 1 | 3 | 0 |
| Rat | AKR1B4 | Aldose reductase | 3 | 2 | 1 |
| Rat | AKR1B13 | Aldose reductase-like | 3 | 3 | 1 |
| Rat | AKR1B14 | Aldose reductase-related protein | 2 | 2 | 0 |
| Rat | AKR1C9 | 3α-HSD, DD | 4 | 4 | 0 |
| Rat | AKR1C17 | Rat AKR D | 2 | 5 | 1 |
| Rat | AKR1D2 | Steroid 5β-reductase | 1 | 2 | 0 |
| Rat | AKR6A2 | Shaker channel β-subunit (Kvb2) | 11 | 14 | 0 |
| Rat | AKR7A1 | Aflatoxin aldehyde reductase | 4 | 2 | 0 |
| Rat | AKR7A4 | Aflatoxin aldehyde reductase | 3 | 4 | 0 |
| Mouse | AKR1A4 | Aldehyde reductase | 2 | 2 | 0 |
| Mouse | AKR1B3 | Aldose reductase | 0 | 3 | 0 |
| Mouse | AKR1B7 | Aldose reductase-related protein | 3 | 0 | 0 |
| Mouse | AKR1B8 | Fibroblast growth factor-induced protein | 0 | 5 | 2 |
| Mouse | AKR1C6 | Estradiol 17β-HSD; prostaglandin F synthase | 0 | 6 | 0 |
| Mouse | AKR1C12 | 3α/β-HSD | 0 | 8 | 1 |
| Mouse | AKR1C13 | Interleukin-3-regulated AKR | 0 | 6 | 1 |
| Mouse | AKR1C14 | 3α/β-HSD | 2 | 4 | 1 |
| Mouse | AKR1C18 | 20α-HSD | 1 | 3 | 0 |
| Mouse | AKR1C19 | 3(20α)-HSD | 4 | 6 | 0 |
| Mouse | AKR1C21 | DD | 2 | 1 | 0 |
| Mouse | AKR1E1 | Mouse liver keto reductase | 1 | 1 | 0 |
| Mouse | AKR6A4 | Shaker channel β-subunit (Kvb2) | 11 | 13 | 0 |
| Mouse | AKR6A8 | Shaker channel β-subunit (Kvb2) | 23 | 30 | 2 |
| Mouse | AKR6A14 | Shaker channel β-subunit (Kvb2) | 4 | 7 | 0 |
| Mouse | AKR7A5 | Aflatoxin aldehyde reductase: succinic semialdehyde reductase | 2 | 2 | 0 |
The species, gene, and original nonsystemic name of enzymes are shown. Identification of putative ARE sequences within AKR genes was performed using the Tess PWM search software, the source code for which is available at http://www.cbil.upenn.edu/downloads. This software implements a search algorithm of the target gene sequence using a positional/partial weight matrix to describe the ARE. PWMs encode a ranked representation of base values accommodating some level of flexibility for certain positions in the sequence while mandating that other positions match exactly [139]. From left to right, the ARE sequences included in our search included the sequence 5′-G-TGAC-NNN-GC-3′, the matrices assigned the Jasper IDs MA0150.1 (human Nrf2, NFE2L2) and MA0150.2 (mouse Nrf2, Nfe2l2). Gene sequences for each of the human, rat, and mouse AKR genes were downloaded from the Ensembl database. In the search, we included 7000 flanking bases upstream and downstream from each gene. For further details visit the AKR Web site at http://www.med.upenn.edu/akr/.
2.7. Loss ofNrf2 changes cell physiology under unstressed homeostatic conditions
Because Nrf2 is widely regarded to be an oxidative stress-activated transcription factor, the reason it contributes to the expression of ARE-driven genes under normal physiological conditions is not immediately obvious. Evidence from knockout mice indicates that Nrf2 exhibits modest activity even under normal homeostatic unstressed conditions [58, 60, 102], and it seems likely this reflects low-level antagonism of repression of the transcription factor by Keap1 and/or β-TrCP. It is possible that Keap1 activity is slightly impaired under basal conditions, by ROS and thiol-reactive endogenous electrophilic metabolites (e.g., acrolein, 15-deoxy-Δ1214-prostaglandin J2, dopa-O-quinone, fumarate, 4-hydroxynonenal, and maleylacetoacetate), as well as by dietary factors. Alternatively, it is feasible that other proteins that bind Keap1, such as p62/sequestosome-1 (SQSTM1), diminish the ability of Keap1 to target Nrf2 for proteasomal degradation. A further possibility is that under normal physiological conditions GSK-3 activity is inhibited to a sufficient extent to cause sub-optimal phosphorylation of the DSGIS-containing phosphodegron in the Neh6 domain, and this blunts β-TrCP-mediated turnover of Nrf2. Presumably low-level antagonism of the inhibitory actions of Keap1, β-TrCP, or GSK-3 occurs under normal physiological processes and this is sufficient to allow a portion of Nrf2 to escape repression under unstressed conditions.
2.7.1. Redox homeostasis and cell proliferation are compromised in the absence of Nrf2
A cardinal feature of cells lacking Nrf2 is that their antioxidant capacity is compromised, and this is the justification for it being cast in the role of a master regulator of redox homeostasis. Mouse embryonic fibroblast (MEF) cells and macrophages from Nrf2−/− mice possess only about 25% of the intracellular levels of GSH of wild-type MEF cells [140, 141], and this is associated with reduced expression of antioxidant genes such as Gclc, Gclm, Prdx1 (also called MSP23), Slc7a11, Txn1, and Txnrd1[140, 142, 143]. In livers of Nrf2-null mice, the level of GSH is not decreased to such a great extent as it is in MEF cells, with it being estimated to be 70–80% of that in wild-type livers [144, 145], but in the mutant mice the amount of GSH secreted by the liver into bile is almost half that of the wild type [146]. In alveolar type II cells from Nrf2-null mice, the level of GSH has been shown to be about 50% of that in their wild-type counterparts [147].
It is likely that the decrease in GSH in tissues of Nrf2-null mice, relative to those of wild-type mice, is sufficient to compromise their ability to scavenge ROS. Consistent with this hypothesis, a number of research groups have reported that ROS levels are between 1.6- and 4.0-fold higher in Nrf2-null cells than in the equivalent wild-type cells. Thus Nrf2−/− MEF cells contain approximately 3.5-fold higher O2●− levels and about 2.3-fold higher total ROS levels than Nrf2+/+ MEF cells [148, 149]. Similarly, primary cardiomyocytes from Nrf2−/− mice contain 2.5-fold higher ROS levels than wild-type cardiomyocytes [150], bone marrow-derived macrophages from Nrf2−/− mice have about 4.0-fold higher ROS levels than wild-type bone marrow-derived macrophages [151], and dendritic cells from Nrf2−/− mice have 1.6-fold higher ROS levels than wild-type dendritic cells [152]. In addition to the loss of antioxidant capacity, it seems that the extra ROS burden in Nrf2-null cells may arise from increased production of ROS by certain NOX enzymes. Support for this idea comes from the observation that mRNA for Nox4 is increased in mouse Nrf2−/− endothelial cells compared with Nrf2+/+ endothelial cells [153] and mRNA for Nox2 and Nox4 is increased in mouse Nrf2−/− brain hippocampal glioneuronal cultured cells compared with the wild type [154]. Together, these results suggest that in the mouse, Nrf2 may repress the expression of Nox2 and Nox4 genes in a cell-specific fashion.
Accumulating data indicate that Nrf2-null cells have lower GSH levels and a higher burden of ROS than equivalent wild-type cells. In primary cultures of alveolar epithelial cells from Nrf2−/− mice, the higher levels of ROS result in activation of the ataxia telangiectasia mutated DNA damage-response pathway and impaired cell cycle progression, with the mutant cells exhibiting G2/M-phase arrest and fewer cells in S-phase [155]. The G2/M arrest in Nrf2−/− alveolar epithelial cells has been reported to be associated with reduced phosphorylation of retinoblastoma protein (at Ser-807/Ser-811) and inactivation and mislocalization of cyclin B1 and cyclin-dependent kinase (CDK) 1, both of which are involved in the mitosis-promoting factor complex. Interestingly, treatment of Nrf2−/− alveolar cells with GSH restored phosphorylation of retinoblastoma protein, corrected localization of the mitosis-promoting factors cyclin B1 and CDK1, and normalized cell cycle progression [155]. These results indicate that loss of Nrf2 may have complex effects on signal transduction pathways and that its influence on GSH homeostasis is possibly of principal significance.
2.7.2. Cell differentiation is altered in the absence of Nrf2
A number of experimental models have been employed to demonstrate that Nrf2 is involved in maintenance of the stem cell phenotype. In Drosophila intestinal stem cells, loss of the CncC bZIP-CNC transcription factor has been reported to increase ROS levels and their proliferation rates, suggesting that CncC is required to keep the intestinal stem cells in a state of proliferative quiescence and to prevent them from entering the cell cycle [156]. In mouse hematopoietic stem cells, loss of Nrf2 has been shown to lead to an expansion of the progenitor pool of myeloid and lymphoid lineages, again suggesting that Nrf2 supports stem cell renewal and proliferative quiescence [157]. Interestingly, it has also been found that Nrf2 supports the homing of hematopoietic cells to bone marrow because it positively regulates CXCR4 [157]. Nrf2 has additionally been implicated in the self-renewal of human airway basal stem cells, but in this case the flux of ROS levels appeared to be the critical factor, rather than the absolute levels, and the Notch1 signaling pathway was implicated in helping establish dynamic changes in ROS levels [158].
It is becoming apparent that Nrf2 may be specifically down-regulated in certain cell types during development and differentiation, possibly because high levels of expression of the ARE-gene battery would compromise certain ROS-based signaling pathways. Thus, during development in the mouse, Nrf2 is repressed by epigenetic inactivation of its gene promoter [159]. Moreover, forced overexpression of Nrf2 in young neurons impairs dendritic outgrowth and their development, which is thought to involve antagonism of JNK and Wnt pathways.
2.7.3. The unfolded protein response is perturbed in the absence of Nrf2
The unfolded protein response (UPR), which is triggered by ER stress, appears to be perturbed in the livers of Nrf2−/− mice under basal conditions. Specifically, initiation of the UPR is carried out by the three ER-resident stress sensors, namely, activating transcription factor 6, inositol-requiring enzyme 1 (IRE1), and protein kinase RNA-like ER kinase (PERK). When stimulated, these sensors initiate activation of complex pathways that include induction of C/EBP homologous protein (CHOP), splicing of mRNA for X-box-binding protein 1 (XBP1), called XBP1s, and phosphorylation of eukaryotic initiation factor 2α (eIF2α) [160, 161]. In the case of Nrf2−/− mice, it has been concluded that the UPR is at least partially triggered under basal conditions on the basis that Chop and Xbp1s are elevated and the phosphorylation of eIF2α is increased in livers of mutant mice fed a normal chow diet [162]. Moreover, Chop has also been reported to be upregulated in Nrf2−/− MEFs under basal conditions [163].
It is not clear why ER stress is apparent in livers and fibroblasts of mutant mice. One possibility is that diminished expression of Slc7a11 in Nrf2-null mice limits the supply of intracellular cystine, which is reduced to cysteine for protein synthesis [164], and that lower levels of cysteine may compromise the correct folding of membrane proteins and secretory proteins within the ER Another possibility is that the diminished availability of GSH, or a relative inability to reduce GSSG, compromises the ability of cells to fold proteins into their appropriate native tertiary structures. In this case, correct oxidative protein folding within the ER requires GSH in two distinct but interrelated ways that involve the concerted actions of the oxidoreductases ER oxidoreductin-1 (ERO1) and protein disulfide isomerase (PDI); both ERO1 and PDI family members contain multiple Cys residues that influence their activities by forming intramolecular disulfide bridges [165–167]. First, GSH is required for the activity of ERO1, which is a flavoprotein responsible for oxidizing Cys residues in PDI by coupling reduction of O2 to H2O2 with the introduction of disulfide bridges into substrate proteins [168], because it itself contains regulatory disulfides that require to be reduced for it to exhibit oxidoreductase activity [169]. Second, GSH is required for the reductive proofreading activity of PDI in which non-native disulfides in proteins are corrected by isomerization and reduction to form correctly folded protein [170]. The redox state of the Cys residues in PDI is poised in a semioxidized state by a feedback regulatory mechanism with those in ERO1, such that when the major portion of the PDI protein population is reduced (following substrate oxidation) the regulatory disulfides in ERO1 are reduced by PDI and allow a further cycle of protein oxidation to be initiated. Through the actions of ERO1 and PDI, GSH is continuously oxidized to GSSG [170, 171]. The significance of the contribution of GSH to this process is emphasized by the finding that treatment of cells with the GSH synthesis inhibitor buthionine sulfoxamine increases disulfide bond formation but does not result in correct protein folding [172]. It is also noteworthy that treatment of mice with BHA alleviates ER stress [173] but it is not known if the protective effects of BHA are dependent on Nrf2.
2.7.4. Mitochondrial function is disturbed in the absence of Nrf2
In addition to loss of cellular antioxidant capacity and modest activation of ER stress, knockout of Nrf2 diminishes mitochondrial bioenergetics and activity. Thus in MEF cells and neurons from Nrf2−/− mice, loss of the transcription factor decreases the mitochondrial membrane potential, ATP production, and respiration [149], and this is consistent with the observation that the mitochondrial oxidation of long-chain and short-chain fatty acids is diminished in Nrf2−/− mice [174].
2.8. Loss of Nrf2 compromises intrinsic resistance to xenobiotics, proinflammatory agents, and tissue regeneration, but increases ROS-dependent signaling and insulin sensitivity
The influence that basal Nrf2 activity has on redox homeostasis and the expression of drug-metabolizing enzymes and drug transporters influences significantly the intrinsic interactions of cells with toxins and stressors present in the environment.
2.8.1. Cells are less capable of withstanding toxic xenobiotics in the absence of Nrf2
Nrf2-null mice are sensitive to a range of stressors, including hyperoxia, acetaminophen, benzo[a]pyrene, butylated hydroxytoluene, diesel exhaust fumes, 7,12-dimethylbenz[a]anthracene, nitrosamine, and tobacco smoke [175–177]. Furthermore, Nrf2−/− MEF cells have been demonstrated to be substantially more sensitive to a range of isothiocyanates, α,β-unsaturated carbonyls, aryl halides, epoxides, peroxides, quinones, metal salts, and DNA-damaging agents than wild-type fibroblasts [140, 178]. In addition to a deficiency in antioxidant enzymes, the Nrf2−/− MEF cells also express lower levels of Alpha-, Mu-, and Pi-class Gst isoenzymes and Nqo1 [140]. Among the compounds examined, the increased sensitivity of Nrf2−/− fibroblasts to the peroxide cumene hydroperoxide and the nitrogen mustard chlorambucil is principally attributable to the lower levels of GSH, whereas this is not the case for the redox cycling agent menadione [140]. Presumably the greater sensitivity of Nrf2−/− compared to Nrf2+/+ MEF cells to menadione is due to the lower expression of Nqo1 in the mutant fibroblasts, as it is well known to be regulated by Nrf2 and to convert harmful quinones to less reactive hydroquinones [179, 180]. It is likely the hydroquinones produced by Nqo1 are themselves glucuronidated by 5′-diphosphate-glucuronosyl transferase before elimination from the cell via MRP [181].
The notion that because Nrf2−/− mice are less able to detoxify harmful xenobiotics they are more sensitive than wild-type mice to chemically induced carcinogenesis seems to be broadly valid. There is, however, an interesting caveat to this interpretation because evidence suggests Nrf2 may support the later stages of lung carcinogenesis. Specifically, it has been reported that 4 weeks after intraperitoneal injection of urethane, Nrf2−/− mice produce approximately sevenfold more microscopic nodules in their lungs than wild-type mice, and that 8 weeks after urethane treatment all lungs of Nrf2−/− mice develop surface tumors, whereas only half of the wild-type lungs do so [182]. Remarkably, the larger number of lesions in the lungs of Nrf2-null mice treated with urethane, compared with lungs of wild-type mice, in the early stages of carcinogenesis is reversed at later stages of the disease. Thus it has been observed that between 16 and 24 weeks after urethane treatment, the lungs of Nrf2−/− mice contain significantly fewer tumors than their wild-type counterparts. Moreover, the tumors in the mutant mice were less malignant than those in wild-type mice [182]. Taken together, these findings suggest that Nrf2 does indeed prevent initiation of urethane-induced lung carcinogenesis, but that in the later promotion and progression stages of the disease it supports the more malignant oncogenic pathway(s) that is driven by mutant Kras. This interpretation is based on the finding that Kras mutations were observed in only 1 in 13 of the lung tumors examined from urethane-treated Nrf2−/− mice but were observed in 15 of 15 lung tumors obtained from the urethane-treated wild-type mice [182]. Moreover, it was also discovered that although very few Nrf2-null tumors expressed mutant Kras, they did over-express the tumor cell survival gene osteopontin (also called secreted phosphoprotein-1) and so may have been directed to an alternative Kras-independent oncogenic pathway through mechanisms that are poorly understood [182].
2.8.2. Inflammatory responses are exacerbated in the absence of Nrf2
In addition to being susceptible to acute chemical toxicity, Nrf2−/− mice are more susceptible to the proinflammatory effects of allergens, lipopolysaccharide, and a high-fat diet [162, 183, 184]. A contributing factor to the increased sensitivity of Nrf2−/− mice to inflammation may be loss of peroxisome proliferator-activated receptor γ (Pparγ) because it is known that Nrf2 positively regulates Pparγ [185] and that Pparγ exerts strong anti-inflammatory effects [186]. The increased sensitivity of Nrf2−/− mice to inflammation is consistent with the existence of a multitiered hierarchical oxidative stress defense in which low and localized production of ROS is used in signalling to control proliferation and differentiation. However, modest increases in ROS levels above normal stimulate Nrf2-orchestrated adaptation to relatively low levels of oxidative stress, whereas higher increases in ROS stimulate nuclear factor-κB (NF-κB) along with AP-1 to provide a second tier of defense against more severe oxidative stress [187, 188]. Exposure of HeLa, HEK293E, and MDA-MB-435 cells to H2O2 has been shown to activate the NF-κB pathway through signaling via the nonreceptor proto-oncogene tyrosine-protein kinase c-Src, Abelson murine leukemia viral oncogene homolog 1 (c-Abl), protein kinase C (PKC) δ, and protein kinase D (PKD) [189–191], which are involved in the induction of the inflammatory response and the production of proinflammatory cytokines. Presumably the diminished antioxidant capacity of Nrf2−/− cells ensures they have higher ROS levels than wild-type cells, and this allows c-Src, c-Abl, PKCδ, and PKD to be more readily activated and NF-κB more readily stimulated. It is also possible that the increased sensitivity of Nrf2−/− mice to proinflammatory stimuli is a consequence of an increased susceptibility to ER stress and may occur via induction of TXN-interacting protein, downstream of the PERK and IRE1 pathways, to cause increased production of interleukin-1β by the NLRP3 inflammasome [192, 193].
When wounded, Nrf2−/− mice have been shown to display delayed healing and prolonged inflammation in the affected area, with expression of proinflammatory cytokines altered such that although their expression levels are lower than those of wild-type mice their production continues for a significantly longer time period [94]. During carrageenan-induced acute inflammation, it has been proposed that accumulation of 15-deoxy-Δ12,14-prostaglandin J2 in peritoneal macrophages is responsible for activation of Nrf2 through inhibition of Keap1 [194]. Mice lacking Nrf2 have been reported to exhibit a delayed response to carrageenan stimulation followed by persistent inflammation. Taken together with the findings of Sabine Werner and co-workers [94], these data suggest a possible role for Nrf2 in the resolution of inflammation as well as regulation of the magnitude of the inflammatory response.
2.8.3. Liver regeneration is compromised in the absence of Nrf2
Three days after a two-thirds partial hepatectomy, the regeneration of liver in Nrf2−/− mice is significantly lower than in Nrf2+/+ mice as a consequence of blunted Notch1 signaling [135]. Tom Kensler and colleagues have provided compelling evidence that Notch1 contributes to the diminished capacity to regenerate liver in Nrf2-null mice based on the fact that the proximal region of the Notch1 promoter contains an ARE sequence (5′-CTGAGCGGGCG-3′) and that Nrf2−/− MEF cells exposed to Notch1 signaling ligands display reduced expression of Notch1, p21, Hes1, Herp1, and Nrarp compared with wild-type fibroblasts treated in a similar manner [135]. Moreover, the relative inability of Nrf2-null mice to regenerate liver after a partial hepatectomy could be rescued by hepatic overexpression of the Notch1 intracellular domain [135].
2.8.4. ROS-dependent signal transduction is increased in the absence of Nrf2
Diminished antioxidant status and the increased production of ROS that accompanies loss of Nrf2 are likely to alter significantly many cellular signaling pathways. It is, for example, reasonable to suppose that signal transduction instigated by growth factors and cytokines, which entail H2O2 as a second messenger [15, 195], are likely to be more readily activated in Nrf2-null cells than in equivalent wild-type cells. Stimulation of receptor tyrosine kinases, such as the insulin receptor, EGF receptor (EGFR), and PDGF receptor (PDGFR), by their ligands results in localized production of O2●− by NOX isoenzymes, which serves as a second messenger to activate downstream kinases via the reversible inactivation of protein tyrosine phosphatase (PTP) enzymes including PTP1B and SHP-2 (i.e., Src-homology 2 domain-containing phosphatase 2) and dual-specificity phosphatases MAPK phosphatase (MKP) isoenzymes and PTEN [6, 8–11, 196–198]. Reactivation of oxidized PTP enzymes is achieved through reduction by TXN1 [199], and we therefore envisage that in Nrf2−/− cells the combined effects of higher ROS levels coupled with diminished TXN1 activity (as a consequence of lower expression of TXNRD1 and SRXN1) will heighten signaling through the insulin, EGFR, and PDGFR pathways. Support for the notion that ROS-dependent signaling through the insulin receptor is increased in Nrf2−/− mice exists insofar as the mutant mouse displays increased insulin sensitivity and increased glucose disposal when fed a high-fat diet [162] (see below).
The non-receptor tyrosine kinase c-Abl, which regulates many cell processes including stimulation of cell survival in response to growth factors and cell death in response to DNA damage [200, 201], is activated by oxidative stress [202, 203]. Interestingly, c-Abl is inhibited by direct physical interaction with PRDX1 [204], and oxidative stress results in dissociation of c-Abl from PRDX1 [205]. In view of the ability of ROS to activate c-Abl and PRDX1 to inhibit c-Abl, it seems probable that the tyrosine kinase will assume increased activity in Nrf2-null cells. Moreover, c-Abl activity has been reported to lead to proteasomal degradation of catalase and also to activation of GPX1 [206, 207], and these modifications of antioxidant enzyme activity may attenuate oxidative stress responses in Nrf2-null cells.
The ASK1–JNK and ASK1–p38MAPK pathways seem to be more readily activated in Nrf2-null cells than in wild-type cells. A cartoon illustrating how loss of Nrf2 might result in increased sensitivity to ASK1 signal transduction through increased production of ROS by NOX2 and NOX4 enzymes and diminished redox buffering by GSH and TXN is presented in Fig. 6. Consistent with the role of ROS in activating ASK1, the ASK1–JNK and the ASK1–p38MAPK pathways have been found to be activated more readily upon treatment with the redox-cycling agent paraquat in Nrf2-null fibroblasts than in wild-type fibroblasts, and this was shown to be associated with activation of cell death triggered by oxidative stress [143]. The increased sensitivity of these two ASK1-dependent pathways in Nrf2-null fibroblasts is consistent with the mutant cells possessing heightened activity of ROS-sensitive signal transduction pathways. Critically, ASK1 is directly inhibited by TXN1 in a redox-dependent manner through protein–protein interactions [208]. In unstressed cells, individual ASK1 subunits exist in a homo-oligomer complex through interactions in its C-terminal coiled-coil region with other ASK1 subunits (residues 1239–1295 of mouse ASK1) [209]. Under such circumstances, TXN1 binds to the N-terminal noncatalytic region of ASK1 (i.e., residues 46–277 of mouse ASK1) when the Cys residues in its redox-active31WCGPC35 motif are reduced, and the binding of TXN1 to ASK1 keeps the kinase in an inactive state by preventing its N-terminal coiled-coil regions from associating with other ASK1 subunits through homophilic interactions [210]. However, oxidation of TXN1 results in an intramolecular disulfide bridge being formed between Cys-32 and Cys-35 and a conformational change in the dithiol oxidoreductase that results in its dissociation from ASK1 and allows the kinase to recruit tumor necrosis factor receptor-associated factor 2 (TRAF2) and TRAF6 to regions adjacent to those to which TXN1 binds (i.e., residues 384–655 of mouse ASK1), thereby forming a high-molecular-mass complex [211]. In turn, the recruitment of TRAF2 and TRAF6 to ASK1 promotes the N-terminal homophilic interaction between ASK1 proteins and facilitates trans-autophosphorylation (of Thr-845 in mouse ASK1) and activation of the kinase in its catalytic center (i.e., residues 687–945) [210]. Thus in Nrf2-null cells the relative lack of TXNRD1 and SRXN1 will result in lower levels of reduced TXN1 being available to repress ASK1, and consequently ASK1 is more readily activated by ROS.
Fig. 6.
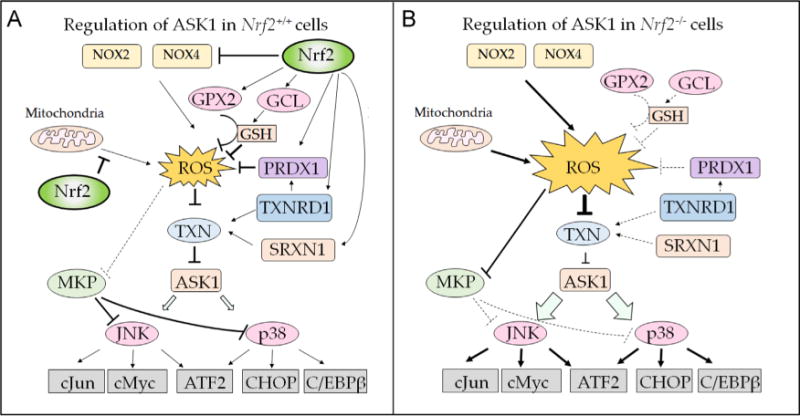
Model to explain the influence of Nrf2 on ROS-dependent ASK1 signaling. Intracellular levels of ROS represent a major regulator of ASK1 activity, as the kinase is repressed by reduced TXN but not by oxidized TXN. Transcription factor Nrf2 is predicted to modulate the sensitivity of ASK1 to ROS-dependent activation by its ability to increase GSH-based and TXN-based antioxidant systems, its ability to suppress production of ROS by modulating mitochondrial function, and possibly also its ability to repress the expression of NOX2 and NOX4. (A) In wild-type cells, Nrf2 negatively controls expression of NOX2 and NOX4, which produce ROS (top), with inhibition depicted by a blunt-headed arrow. Moreover, Nrf2 supports inactivation of ROS by increasing the expression of GCL (upper right-hand side), which catalyzes the rate-limiting step in GSH synthesis; by increasing the expression of GPX2, which reduces H2O2 using GSH as a cofactor; and by increasing the expression of PRDX1, which reduces H2O2 in a TXN-dependent manner (all are shown as arrows). Nrf2 maintains TXN in a reduced state by increasing expression of TXNRD1 and SRXN1 (middle and lower right-hand side), and TXN, TXNRD1, and SRXN1 contribute to the reduction of H2O2 by PRDX1 (Fig. 1). Last, by improving the efficiency of oxidative phosphorylation, Nrf2 limits production of ROS by mitochondria (left-hand side). (B) In Nrf2-null cells, production of ROS by NOX2 and NOX4 is increased (top), as is the production by mitochondria (left-hand side). In addition, the GSH-based antioxidant and TXN-based antioxidant systems are diminished through decreased expression of GPX2, GCL, PRDX1, TXNRD1, and SRXN1 that results from loss of Nrf2 (right-hand side). The increased production of ROS and the diminished antioxidant capacity in Nrf2-null cells combine to cause the redox status of TXN to be shifted toward oxidation, and therefore its inhibition of ASK1 will be less effective. As a consequence, the downstream p38MAPK and JNK kinases are more readily activated in Nrf2-null cells. Moreover, the increase in ROS in Nrf2-null cells will increase inhibition of MKP enzymes, and this will decrease the ability of the phosphatases to inhibit JNK and p38MAPK.
In addition to regulation of JNK and p38MAPK by ASK1, the kinases are also controlled by the MKP family of phosphatases [196]. In Nrf2-null cells, JNK and p38MAPK may be activated, at least in part, by the increase in ROS causing inhibition of MKP isoenzymes. For example, MKP3 can be reversibly inactivated by treatment with H2O2 via formation of sulfenic acid at Cys-293 and reactivated by dithiothreitol [196]. As MKPs inhibit JNK and p38MAPK, it is likely their inactivation by high levels of ROS in Nrf2-null cells will facilitate MAPK signaling by stimuli such as tumor necrosis factor α that can trigger apoptosis.
The activity of various serine/threonine PKC isoenzymes is increased by oxidative stress [212]. Upon treatment with ROS, the PKCα, β, δ, and ε isoforms are recruited to plasma membranes where they are activated by diacylglycerol, whereas PKCζ translocates to the nucleus [213]. Activation of PKC isoenzymes by ROS may entail phosphorylation of certain tyrosine residues that lie adjacent to the regulatory domain by non-receptor-type kinases [214], possibly by c-Abl [190], but it may also occur via oxidation of Cys residues in its regulatory or catalytic domains [215]. PKC isoenzymes serve many roles, including immune cell signaling, cell polarity, and cell migration [216, 217]. An important feature of PKC enzymes is that they bind and phosphorylate the cytoplasmic p47phox regulatory subunit within NOX enzyme complexes and contribute to the propagation of ROS signaling by stimulating formation of O2●− by NOX [218–220], which in turn stimulates downstream signaling cascades, resulting in activation of NF-κB and AP-1. Last, it is interesting to note that in addition to the ability of PKC enzymes to amplify ROS signaling and increase NF-κB and AP-1 activity, it has also been reported that PKCδ activates Nrf2 by phosphorylating Ser-40 in the Neh2 domain of the CNC-bZIP transcription factor [221–223]; it remains unclear, however, whether phosphorylation of Nrf2 at Ser-40 alters its activity, its subcellular localization, and/or its stability. Nevertheless, based on the role played by ROS and tyrosine kinases in activating PKC, and the possibility that activation of Nrf2 by PKCδ forms a negative feedback loop, it seems likely that these kinases are activated in Nrf2−/− mice, and the increase in inflammatory response that is observed in cells lacking Nrf2 is consistent with this hypothesis.
2.8.5. Insulin sensitivity is increased in the absence of Nrf2
Many research groups have studied the influence that Nrf2 has on DM because of its prevalence and the role played by oxidative stress in progression of the disease [224]. It should, however, be mentioned at the outset that ROS have both positive and negative effects on insulin signaling [225]. Whereas it was initially anticipated that Nrf2−/− mice would be susceptible to DM, most studies have indicated this is not the case. Comparison of blood glucose from nonfasted Nrf2-null and wild-type mice fed on a regular chow diet have been reported to be closely similar, as were overnight-fasted blood glucose levels [144, 226, 227], and intraperitoneal glucose tolerance tests of fasted Nrf2−/− and Nrf2+/+ mice on a regular chow diet revealed similar levels of glucose disposal in the two genotypes. However, in mice fed a high-fat diet (usually 40–60% of total calories from fat) the absence of Nrf2 has been reported to improve glucose homeostasis [132, 226, 227]. This improvement in glucose disposal may be due partly to increased expression of fibroblast growth factor 21, which is repressed by Nrf2 [132, 227]. It also seems to entail an increase in insulin signaling, because after the intraperitoneal injection of insulin, protein kinase B/Akt (henceforth referred to as Akt) displayed greater phosphorylation at Ser-473 in the livers and skeletal muscle of Nrf2−/− mice than it did in the same organs of wild-type mice [162]. The heightened insulin sensitivity in Nrf2-null mice is consistent with the PTP1B phosphatase being more readily inactivated by higher levels of ROS and lower TXN1 activity than in wild-type mice. Moreover, the increase in insulin sensitivity in Nrf2−/− mice is reminiscent of that observed in Gpx1−/− mice, in which the improved sensitivity was accompanied in the muscle by enhanced insulin-induced ROS production, phosphoinositide 3-kinase (PI3K)-Akt signaling, and PTEN oxidation and could be reversed by treatment with N-acetylcysteine [228]. It should be noted that improved glucose homeostasis and increased insulin sensitivity in Nrf2−/− mice has not been reported by all researchers, and in one of the early studies it was reported that the mutant mice show lower plasma insulin levels and an impaired glucose tolerance test [229].
3. Molecular regulation of Nrf2
Considering the fact that Nrf2 is a stress-activated transcription factor and that it regulates a wide variety of biological processes, it seems likely that alterations in the biochemical pathways that Nrf2 regulates will negatively feed back on its activity in a manner that allows physiological functions to be maintained. Specifically, Nrf2 broadly regulates pathways involved in combatting oxidative stress as well as metabolic pathways leading to the production of NADPH and ATP [61, 149]. Given the multiple stimuli that activate Nrf2 and the diverse pathways it controls, it is important to understand how Nrf2 activity is in turn regulated by oxidative stressors and by metabolic stimuli that require NADPH and ATP production. It is therefore possibly not surprising that Nrf2 is subject to complex regulatory mechanisms at both the transcriptional and the posttranslational levels.
3.1. Transcriptional regulation of the NFE2L2 gene
Two major pathways exist by which xenobiotics induce gene induction. The first involves regulation by the AhR which when occupied by ligand, e.g., 2,3,7,8-tetrachlorodibenzodioxin, binds to the xenobiotic response element (XRE; 5′-TA/TGCGTGA/C-3′) in the promoter regions of inducible genes, e.g., CYP1A1 and CYP1B1 [230, 231]. The second involves activation of Nrf2 so that it is recruited to ARE sequences in the promoters of target genes. The gene encoding Nrf2, NFE2L2, contains an XRE and two XRE-like sequences, which can recruit the AhR, enabling NFE2L2 to be transcriptionally activated by polycyclic aromatic hydrocarbons (PAHs) [232, 233]. Nrf2 has also been shown to play a role in the regulation of AhR indicating that a feedback loop exists between the two xenobiotic-sensing transcription factors [124]. In addition, planar PAH o-quinones produced by human AKR1C enzymes, which are regulated by Nrf2, have been found to bind to the AhR leading to gene activation, thereby demonstrating a level of cross talk involving metabolites generated by Nrf2-regulated genes [234, 235]. Upstream of the transcription start site (TSS), the NFE2L2 promoter contains two ARE-like sequences that allow the transcription factor to increase modestly its own expression [119]. Moreover, downstream from the TSS, the mouse Nfe2l2 gene contains a 12-O-tetradecanoylphorbol-13-acetate-response element that allows it to be transcriptionally activated by oncogenic KrasG12D via c-Jun and c-Fos [137, 236]. The NFE2L2 gene also contains an NF-κB binding site downstream from the TSS, which allows it to be induced by inflammatory stimuli [237].
The abundance of Nrf2 mRNA has also been shown to be influenced by the levels of peroxisome proliferator-activated receptor α (PPARα) during fasting conditions [238]. Taken together with the finding that Nrf2 appears to negatively regulate the level of PPARα mRNA expression [116], it is reasonable to suppose that dynamic cross talk exists between Nrf2 and PPARa, which will be discussed in detail below.
3.2. Posttranslational regulation ofNrf2
In common with other stress-responsive transcription factors, Nrf2 is largely controlled at the level of protein stability. Thus Nrf2 protein is constantly targeted for degradation by the 26S proteasome by the actions of several E3 ubiquitin ligases, including cullin–RING ubiquitin ligase, called CRL, and S-phase kinase-associated protein 1 (Skp1), cullin-1 (Cul1), F-box protein E3 ubiquitin ligase, called SCF. The rapid turnover of Nrf2 provides a readily available pool of newly translated protein to allow almost immediate accumulation of the factor when redox stressors and metabolic stimuli inhibit its ubiquitylation and proteasomal degradation. This process therefore ensures an efficient mechanism of adaptation to stress and metabolic perturbations, provided the agents and stimuli that cause cessation of degradation of Nrf2 are coupled with the function of Nrf2-target genes. It remains unclear whether the translocation of Nrf2 from the cytoplasm to the nucleus is regulated. Rather, it appears most likely that Nrf2 translocates to the nucleus spontaneously through the nuclear localization signal in its Neh1 domain.
3.2.1. Repression of Nrf2 by Keap1
The best studied negative regulator of Nrf2 is Keap1, a dimeric protein that is located principally in the cytoplasm [239]. It was initially thought that Keap1 inhibits Nrf2 by tethering the transcription factor in the cytoplasm and preventing its translocation to the nucleus [240] before it was recognized that Keap1 serves to target Nrf2 for proteasomal degradation [70, 241]. It is now recognized that Keap1 is a substrate adaptor protein that enables the Cul3 RING-box 1 (RBX1) E3 ubiquitin ligase complex (called CRLKeap1) to ubiquitylate Nrf2 under normal homeostatic conditions [242–244]. The importance of Keap1 in the repression of Nrf2 has been demonstrated clearly by its knockout in the mouse [245] and its knockdown in human cell lines [246].
Keap1 comprises five domains, depicted in Fig. 7, which are as follows: an N-terminal region (NTR); a conserved N-terminal broad complex, tram-track, bric-à-brac (BTB) protein-protein interaction domain; an intervening region (IVR); a C-terminal double-glycine repeat (DGR) domain (that consists of six subdomains, each of which contains a distinctive Gly-Gly motif, and is sometimes called the Kelch-repeat domain), and a C-terminal region (CTR) [247, 248]. The BTB provides a protein-protein interaction domain to which Cul3 is recruited, whereas the DGR along with the CTR forms a six-bladed β-propeller structure that interacts with the Neh2 domain of Nrf2 [243, 247–249].
Fig. 7.
Domain structure of human Keap1. The Keap1 protein can be divided into five domains, the N- and C-terminal NTR and CTR sequences and the BTB, IVR, and DGR sequences. The BTB domain is responsible for dimerization and also the recruitment of cullin-3 to the CRLKeap1 complex. The DGR domain along with the CTR form the β-propeller structure to which Nrf2 binds, with the DLG motif in the Neh2 domain of Nrf2 docking onto one Keap1 subunit and the ETGE motif docking onto the other Keap1 subunit. Cysteine residues that are functionally important or unusual are highlighted: Cys-151 is crucial for the ability of tBHQ and SFN to inhibit the substrate adaptor activity of Keap1 [250], and it forms a transient disulfide bridge with Cys-151 in the other Keap1 subunit [251]; Cys-273 and Cys-288 recognize alkenals and cyclopentanone prostaglandins [252, 253]; Cys-434 recognizes 8-nitro-cGMP [254]; Cys-226 and Cys-613 recognize metals such as Zn2+, Cd2+, As3+, and Se4+[253], and they form a transient disulfide bridge upon exposure to H2O2 [113, 251], which is represented by a horizontal two-headed arrow. Mutation of Cys-23 to tyrosine has been shown to impair the ability of Keap1 to ubiquitylate Nrf2 in breast cancer cells [255, 256]. The three amino acids at the C-terminus of human, mouse, and rat Keap1 comprises a distinctive CTC motif but its functional significance is obscure.
Inactivation of Keap1 can be stimulated by a structurally diverse range of chemicals that possess the ability to modify cysteine residues, including tBHQ, SFN, curcumin, 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid-imidazolide (CDDO-Im), tricyclic bis-(cyanoenone)-31 (TBE-31), and diethyl maleate [250–256], as shown in Figs. 2 and 8. Compared to most proteins that possess, on average, a total cysteine content of ~2%, Keap1 is a relatively cysteine-rich protein with a total cysteine content of ~4% [257]. Mammalian Keap1 proteins contain multiple conserved and highly reactive cysteine residues, some of which have the pKa value of their thiol group lower than that of a free thiol and therefore exist as a thiolate anion (i.e., S−), by virtue of the fact that they are situated in close proximity to polar and basic amino acids [253, 258, 259]. The chemical inducers of ARE-driven gene expression modify reactive cysteine residues in Keap1. By employing the differential ability of each reactive Cys residue to interact with a distinct spectrum of electrophiles and oxidants, Keap1 is equipped with at least four discrete stress sensors that enable it to be inactivated by a wide range of agents [141, 252–254, 260]. Interestingly, the stress sensors in Keap1 evolved separately [253].
Fig. 8.
Inducers of Nrf2-target genes. Many small molecules that induce ARE-driven gene expression are indirect antioxidants or share structural similarity with indirect antioxidants. Examples shown include the metabolite of butylated hydroxyanisole, tert-butyl hydroquinone (tBHQ), the diethyl ester of fumaric acid and the related compound diethyl maleate, the synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazolide (CDDO-Im), the acetylenic tricyclic bis (cyanoenone) designated TBE-31, and 15-deoxy-Δ12,14-prostaglandin J2.
Two conserved Keap1 binding sites exist in the Neh2 domain of Nrf2 that have been referred to as the high-affinity ETGE motif and the low-affinity DLG motif, though it is now recognized that the DLG motif encompasses at least 10 amino acids [261]. Mutation of either the ETGE or the DLG motif largely abolishes Keap1-mediated Nrf2 degradation [67–69]. The presence of the high- and low-affinity sites, along with seven conserved Lys residues located between DLG and ETGE, has led to the development of the “hinge and latch” model of binding between Keap1 and Nrf2 [262], also called the “two-site tethering” model [69]. The model proposes that Keap1 binds Nrf2 initially through the high-affinity ETGE motif, followed by docking of the low-affinity DLG motif onto the β-propeller of the adjacent Keap1 molecule. Under conditions of oxidative stress or electrophilic insult, modification of the cysteine residues in Keap1 impairs its ability to present Nrf2 for ubiquitylation, resulting in failure of Keap1 to target the CNC-bZIP factor for proteasomal degradation and thereby allowing accumulation of newly translated Nrf2 [242–244, 263]. More recently, it has been shown in single cells using fluorescence lifetime imaging microscopy that Keap1-inactivating compounds such as SFN cause the substrate adaptor to bind Nrf2 more tightly via the ETGE and DLG motifs [264]. This suggests that the conformational change of Keap1 that occurs in response to oxidative insult alters the orientation of components within the RBX1–Cul3–Keap1 complex. The altered conformation prevents ubiquitylation of bound Nrf2, thus trapping the complex and preventing sequestration of newly synthesized Nrf2. As a consequence of the stalling of Nrf2 ubiquitylation, and the resulting “logjam,” newly translated Nrf2 bypasses the CRLKeap1 complex and accumulates in the nucleus (see Fig. 9). Importantly, there is no evidence that stressors such as SFN enable CRLKeap1 to release nonubiquitylated Nrf2 once the stressor has been eliminated from the cell [69, 264–266].
Fig. 9.
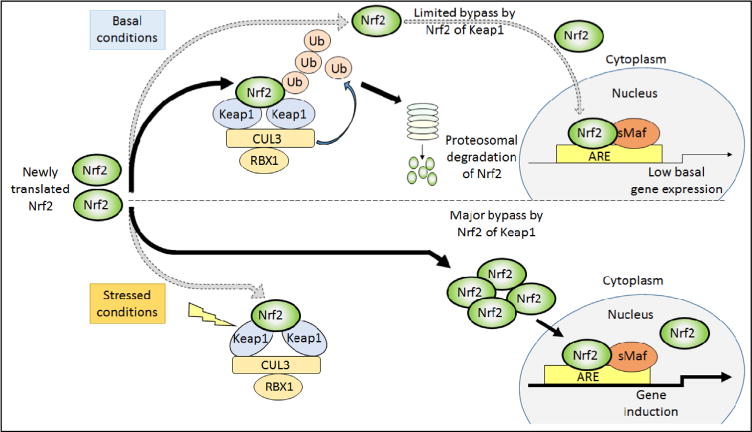
Regulation of Nrf2 by Keap1 under basal and stressed conditions. Under unstressed conditions, newly synthesized Nrf2 protein is short lived, being sequestered in the cytoplasm by Keap1 and targeted immediately for proteasomal degradation by CRLKeap1. Under such circumstances, Nrf2 is rapidly ubiquitylated and degraded, leaving the Keap1 homodimer free to sequester and process newly translated Nrf2 for degradation. A small fraction of the Nrf2 escapes degradation and translocates to the nucleus, maintaining a low basal level of ARE-driven gene expression even under normal homeostatic circumstances. Under stressed conditions, modification of Keap1 causes a conformational change that prevents the CRLKeap1 protein complex from ubiquitylating Nrf2. In this situation, Keap1 is unable to turn over bound Nrf2, and as “free-Keap1” cannot be regenerated, it is unable to bind newly translated Nrf2. Thus under stressed conditions, newly synthesized Nrf2 evades Keap1 capture and translocates directly to the nucleus where it heterodimerizes with sMaf proteins, and together they bind ARE sequences to transactivate target genes.
3.2.2. Electrophiles trigger several stress sensors in Keap1
Several of the reactive cysteines in Keap1 act as independent sensors for a variety of soft electrophiles [267]. Modification of these sensor Cys residues by electrophiles leads to the stabilization of Nrf2 and induction of ARE-driven gene expression. Within the IVR domain of Keap1, Cys-273 and Cys-288 are essential for the turnover of Nrf2 in vitro and in vivo, with mutation of either of these residues resulting in the loss of Nrf2 ubiquitination by Keap1 [250, 268]. These residues have been shown to recognize cyclopentanone prostaglandins such as 15-deoxy-Δ12,14-prostaglandin J2 (by Cys-273) and alkenals such as acrolein and 4-hydroxynonenol (by Cys-288) [252, 253]. Within the BTB domain of Keap1, Cys-151 is critical for the detection of electrophiles such as tBHQ, SFN, and nitric oxide (NO), and modification of Cys-151 by these agents results in the activation of Nrf2 through inhibition of Keap1 substrate adaptor activity [253]. Mutation of Cys-151 to serine or alanine results in loss of Nrf2 induction in response to certain stimuli [253, 268]. Furthermore, replacement of Lys-131, Arg-135, and Lys-150, which surround Cys-151, to Met residues has been shown to reduce the ability of tBHQ, SFN, and NO to induce ARE-driven gene expression, which is consistent with the hypothesis that these basic residues increase the reactivity of Cys-151 through the formation of a thiolate anion [253]. Within the DGR of Keap1, Cys-434 represents a further electrophile cysteine sensor in Keap1, but in this case it is triggered by 8-nitroguanosine-3′,5′-cyclic monophosphate (8-nitro-cGMP), which is generated after NO production, causing S-guanylation of Keap1 and induction of Nrf2-target genes [254].
3.2.3. Reactive metals trigger a unique stress sensor in Keap1
Nrf2 activity is increased by a number of reactive metal(loid)s through their interaction with Keap1. Specifically, Zn2+, Cd2+, Se4+, and As3+ all interact noncovalently with Keap1 through Cys-226 and Cys-613. Together, these two residues have been designated the Zn2+ sensor because zinc is the most likely endogenous trigger [253]. Evidence from using nonreducing SDS–polyacrylamide gel electrophoresis suggests that the Cys-226 and Cys-613 residues in Keap1 also sense ROS because upon exposure to H2O2 they can form a transient intramolecular disulfide bridge that is associated with brief stabilization of Nrf2 protein [251]. Furthermore, H2S also triggers the Cys-226/Cys-613 sensor, and this may involve formation of H2O2 as an intermediate that in turn stimulates formation of the Cys-226–Cys-613 intramolecular disulfide bridge as it can be modulated by forced expression of catalase [113].
3.2.4. Suppression of Nrf2 by TXNRD1 may entail reduction of Cys-based stress sensors in Keap1
Examination of rodents fed a Se-deficient diet or conditional knockout mice lacking the tRNASec gene has revealed that the resulting loss in expression of selenoproteins elicits a compensatory increase in expression of cytoprotective proteins [269–273]. Many of the genes that are upregulated upon loss of selenoproteins, such as AKR7A1, GCLC, GST, HMOX1, and NQO1, are regulated by Nrf2. Among the selenoproteins that may modulate Nrf2 activity, TXNRD1 seems to be of particular significance because liver-specific knockout of Txnrd1 results in upregulation of the ARE-gene battery [29, 274]. The mechanism(s) by which TXNRD1 inhibits Nrf2 activity is uncertain. However, the dimeric Keap1 protein has been shown to form an intermolecular disulfide bridge between Cys-151 and Cys-151 in different Keap1 subunits, and the formation of an intramolecular disulfide bridge between Cys-226 and Cys-613 within the same subunit [113, 251]. Most interestingly, Toledano and colleagues discovered that use of buthionine sulfoximine (BSO) to inhibit GSH synthesis (diminished to approx. 20%) had no effect on either the Cys-151 or the Cys-226/Cys-613 sensors, and knockdown of TXNRD1 resulted in just modest stabilization of the Cys-151–Cys-151 and Cys-226–Cys-613 disulfide bridges [251]. However, the combined use of both BSO and TXNRD1 knockdown in the presence of H2O2 produced substantial stabilization of both disulfide bridges and a profound increase in Nrf2 protein [251]. These results suggest that both the GSH and the TXN systems antagonize triggering of the Cys-151 and Cys-226/Cys-613 sensors by ROS. They also imply that the overexpression of ARE-driven genes observed in Se-deficient animals is due to exacerbation of oxidative stress resulting from an inability to remove H2O2 by GPX and PRDX, the latter of which requires TXN and TXNRD (see Fig. 1C), as well as failure to efficiently reduce the inter- and intramolecular disulfide bridges formed in Keap1 by increased ROS levels. It is notable that as TXNRD1 is regulated by Nrf2, its induction after inactivation of Keap1 through disulfide bond formation allows rapid reactivation of Keap1, thereby providing a negative feedback loop.
3.2.5. Activation of Nrf2 by endogenous protein inducers that compete for binding to Keap1
Keap1 acts as a CRL substrate adaptor for proteins other than Nrf2. The autophagy cargo receptor and signaling adaptor protein p62/SQSTM1 can act as an activator of Nrf2 by competing with Nrf2 for binding to Keap1. Immediately adjacent to its light chain 3-interacting region, p62/SQSTM1 contains an evolutionarily conserved Keap1-interacting region that contains an STGE binding motif. The STGE motif is closely related to the Nrf2 ETGE motif, allowing for competitive binding by p62/SQSTM1 and Nrf2 to the DGR domain (also called the Kelch-repeat domain) of Keap1 [134, 275, 276]. An ARE has been identified in the p62/SQSTM1 gene promoter, indicating a mechanism by which Nrf2 can modestly upregulate its own expression [134]. Importantly, Nrf2 can be regulated by p62/SQSTM1 under physiological conditions, i.e., refeeding with a high-carbohydrate diet after fasting, because Ser-351 in the STGE motif can be phosphorylated by mechanistic target of rapamycin complex 1 (mTORC1). Phosphorylation of Ser-351 in the STGE motif of p62/SQSTM1 leads to a marked increase in its affinity for Keap1, which causes autophagosome-mediated degradation of Keap1 and consequently an increase in the expression of Nrf2-target genes [277]. Fasting causes an increase in 5′-AMP-activated protein kinase (AMPK) activity and inhibition of mTORC1, which presumably decreases the phosphorylation of Ser-351 in the STGE motif of p62/SQSTM1 and minimizes autophagosome-mediated degradation of Keap1. High-carbohydrate refeeding after fasting increases the degradation of Keap1 through the combined actions of p62/SQSTM1 and sestrin-1 and sestrin-2, resulting in the induction of Nrf2-target genes [278]. It remains to be established if the sestrin-1/2-dependent degradation of Keap1 upon refeeding fasted mice with a high-carbohydrate diet involves phosphorylation of the STGE motif in p62/Sqstm1 by mTORC1, but it seems likely. Interestingly, treatment with As3+ markedly increases induction of Nrf2-target genes in a p62/SQSTM1-dependent manner by sequestration of Keap1 into autophagosomes but it is not known whether this involves phosphorylation of the STGE motif in p62/SQSTM1 [279].
It seems probable that once modified by electrophiles, Keap1 is eliminated from the cell via p62/SQSTM1-mediated autophagy [280]. In such circumstances, Keap1 repression of Nrf2 is restored by Nrf2-mediated induction of Keap1 [281].
Phosphoglycerate mutase 5 (PGAM5) may also be degraded in a Keap1-dependent fashion [282], as may inhibitor of κB kinase β (IKKβ) [283, 284]. In the former case, PGAM5, a serine–threonine phosphatase known to activate ASK1 [285], contains a conserved N-terminal ESGE motif, which binds to Keap1 in a manner similar to that of the Nrf2 ETGE motif. Moreover, PGAM5 has been reported to be associated with mitochondria and therefore its interaction with Keap1 may be regulated by mitochondrial signaling processes [286]. In the latter case, IKKβ, a regulator of the NF-κB pathway [287, 288], contains a conserved ETGE motif. It seems likely that both PGAM5 and IKKβ can increase the amount of free Nrf2 by competing directly with it for binding sites on Keap1.
3.3. Repression of Nrf2 by β-TrCP
The negative regulation of Nrf2 by β-TrCP is a more recent discovery than its repression by Keap1, and therefore it is less well characterized. Keap1-independent degradation was first noted when it was recognized that Nrf2 mutant proteins lacking the Neh2 domain were not particularly stable [73]. Examination of Nrf2 deletion mutants that individually lacked the Neh4, Neh5, or Neh6 domains revealed that loss of the last region significantly increased the half-life of the mutant protein, ascribing a degron role to a region of Nrf2 that previously had no known function. The Neh6 domain was shown to contain two highly conserved regions, deletion of either of which increased the half-life of Nrf2 protein. Within these conserved regions, sequences that resemble the β-TrCP substrate recognition motif, DSGϕXS, were identified (ϕ represents a hydrophobic residue, and X is any amino acid). Although neither of the two recognition motifs, DSGIS or DSAPGS (Fig. 10A), conforms to the consensus β-TrCP binding site, both have been shown to recruit β-TrCP [74–76].
Fig. 10.
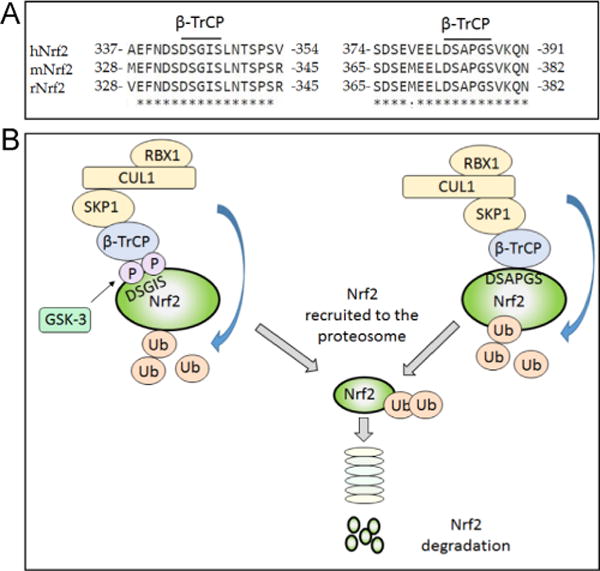
Degradation of Nrf2 by β-TrCP-mediated ubiquitylation. In addition to ubiquitylation of Nrf2 by CRLKeap1, Nrf2 is also targeted for proteasomal degradation by SCFβ-TrCP through sequences in its Neh6 domain. (A) Sequence alignment of human, mouse, and rat Nrf2 (hNrf2, mNrf2, and rNrf2, respectively) using Clustal Omega shows that the two β-TrCP binding sites, and the surrounding residues in the Neh6 domain of Nrf2, are highly conserved across species. (B) The SCF complex mediates ubiquitylation of Nrf2 via the substrate receptor β-TrCP. Through its WD40 domain, β-TrCP binds to the DSGIS and DSAPGS peptide sequences in Nrf2. Phosphorylation of at least one of the Ser residues in the DSGIS motif by GSK-3 leads to increased activity of the degron.
The substrate receptor β-TrCP is an F-box-containing protein, which uses its C-terminal WD40 protein–protein interaction domain to bind substrates and its F-box motif to bind to the SCF E3 ubiquitin ligase complex [289]. The members of the SCF family of E3 ubiquitin ligases each consist of the adapter protein Skp1, the scaffold protein Cul1, the RING box protein RBX1 (also called ROC1 or HRT1), and an F-box substrate receptor protein that varies depending on the substrate to be ubiquitinated [290, 291]. The E3 ligase complex formed by β-TrCP is called SCFβ-TrCP. It is noteworthy that ubiquitylation of Nrf2 by SCFβ-TrCP does not require both the DSGIS and the DSAPGS motifs to be present simultaneously in the Neh6 domain; each can function independent of the other, and this distinguishes these degrons from the DLG and ETGE motifs in the Neh2 domain, which are both required by CRLKeap1 for ubiquitylation of Nrf2. Two different isoforms of β-TrCP exist, called β-TrCP1 and β-TrCP2, encoded by BTRC and FBXW11 [292].
3.3.1. β-TrCP-mediated degradation of Nrf2 is regulated by GSK-3
A GSK-3 phosphorylation site exists within the atypical DSGIS β-TrCP-binding site in the Neh6 domain of Nrf2. GSK-3 is a key regulator of glycogen metabolism that inhibits glycogen synthase, as well as being a regulator of insulin signaling, cell fate specification in embryo development, and cell cycle and apoptosis (reviewed in [293, 294]). In its active form, GSK-3 requires its substrate to first be phosphorylated by a “priming” kinase at an amino acid that typically resides four residues to the C-terminal side of the GSK-3 site [295–297]. To date, no specific kinase has been identified that primes Nrf2 for GSK-3 phosphorylation. Unlike most other kinases, GSK-3 is active in unstressed cells and is regulated by inhibitory phosphorylation. Two GSK-3 isoforms exist, GSK-3α and GSK-3β, both of which are inhibited by growth factors via Akt phosphorylation of their N-terminal Ser-21 and Ser-9 residues, respectively; inhibition of GSK-3 upon phosphorylation occurs because the modified N-terminal region acts as a pseudo-substrate and competes for binding with primed substrates [296].
It has been shown previously that GSK-3 phosphorylates many proteins that SCFβ-TrCP ubiquitylates and targets for degradation, including β-catenin; as such, the substrate is said to contain a phosphodegron [298]. Antonio Cuadrado and colleagues first showed that GSK-3β inhibits Nrf2 activity, but identification of the sites in Nrf2 that are phosphorylated by GSK-3 was achieved several years later using two-dimensional electrophoresis and mass spectrometry [75, 76, 299, 300]. One of the two β-TrCP recognition sites, the DSGIS motif, contains a functional GSK-3 phosphorylation site, which increases the activity of the phosphodegron (Fig. 10B), whereas the DSAPGS motif does not appear to be phosphorylated by GSK-3 [74].
Repression of Nrf2 by GSK-3 can be suppressed, and ARE-driven genes induced, by agents that increase PI3K activity, because stimulation of PI3K activates Akt, and this in turn inhibits GSK-3. We propose that phosphorylation of GSK-3α/β at Ser-21/9 decreases formation of the DSGIS-containing phosphodegron, and thus inhibitory phosphorylation of GSK-3 results in loss of repression of Nrf2 by β-TrCP [74]. Support for this hypothesis comes from the study of PTEN mutant cells, in which PI3K-Akt signaling is greatly increased, as is Nrf2 activity [299]. Interestingly, the Parkinson-associated protein PARK7/DJ-1 has been reported to stimulate Nrf2 activity [301], and loss of PARK7/DJ-1 appears to diminish Nrf2 activity [302]. Moreover, PARK7/DJ-1 is capable of activating Akt [303, 304], suggesting that the mechanism by which PARK7/DJ-1 increases Nrf2-target gene expression most likely involves inhibition of formation of the DSGIS-containing phosphodegron and possibly not blocking of the interaction between Nrf2 and Keap1, as was suggested originally.
N-terminal inhibitory phosphorylation of GSK-3 can also be catalyzed by p70 S6 kinase (p70S6K), p90 ribosomal S6 kinase (p90RSK), and PKC [305]. We therefore speculate that Nrf2 may also be activated by stimulation of the mechanistic target of rapamycin (mTOR)–p70S6K, extracellular signal-regulated kinase (ERK)–p90RSK, and PKC signaling pathways [306]. Recently, Nrf2 has been reported to be regulated by WNT-3A in a β-catenin and Keap1-independent manner through its interaction with GSK-3 and AXIN1 [307]. Specifically, a physical association between Nrf2 and AXIN1 facilitates phosphorylation of the transcription factor by GSK-3, and subsequent ubiquitylation by SCFβ-TrCP, but this is inhibited by WNT-3A. Mice with a hepatocyte-specific deletion of Axin1 have been reported to show upregulation of Nrf2 and its target genes, attesting to the importance of canonical WNT signaling in regulation of Nrf2 in the liver.
3.3.2. Repression of Nrf2 by GSK-3 via Src kinases
It should also be noted that repression of Nrf2 by GSK-3 may occur independently of SCFβ-TrCP. In this alternative scenario it has been proposed that GSK-3β represses Nrf2 via activation of tyrosine kinases. Thus, GSK-3β phosphorylates Fyn at Tyr-213, and in turn the activated Fyn accumulates in the nucleus where it phosphorylates mouse and rat Nrf2 on Tyr-568, and human Nrf2 on Tyr-576, which results in nuclear export and degradation of the transcription factor [308, 309].
3.4. Regulation of Nrf2 by AMPK and mTOR in response to nutrient supply
The fact that Nrf2 serves to decrease gluconeogenesis and to increase mitochondrial activity and the β-oxidation of fatty acids [149, 174] suggest that its activity is regulated by cellular energy and nutrition, but the details of how this may occur are not fully understood. Under conditions of glucose deprivation and low energy availability, AMPK is activated by its upstream kinases, liver kinase B-1 (LKB-1) and Akt [310]. AMPK is a serine/threonine-specific kinase that responds to alterations in levels of adenylate in the cell; a low-ATP/high-AMP ratio, such as that observed during fasting, activates AMPK [311, 312].
3.4.1. Role of mTOR as a stress sensor
Evidence suggests that mTOR controls Nrf2 because mTOR engages in signaling upstream of both Keap1 and β-TrCP (see Fig. 11). The mTOR protein is a ubiquitously expressed serine–threonine protein kinase, and a member of the PI3K-related kinase family. It acts as a cellular energy sensor by integrating signals from hormones, energy levels, growth factors, and nutrients into the regulation of protein translation, autophagy, and lipid metabolism [313]. The active site of mTOR contains multiple phosphorylation sites, which are targeted by kinases that lie both upstream and downstream of mTOR in the signaling pathway, thereby allowing fine-tuning of its activity [314, 315].
Fig. 11.
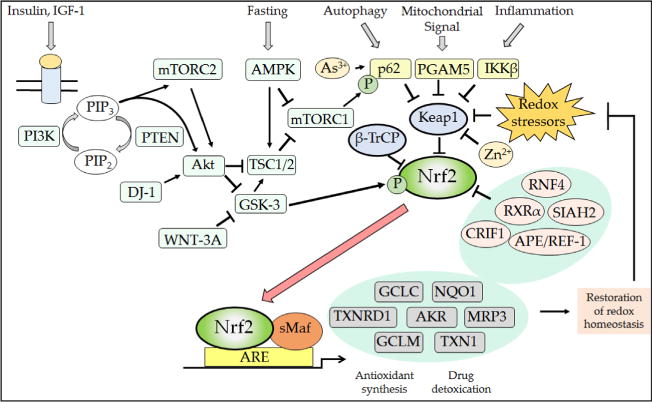
Posttranslational mechanisms that regulate Nrf2. The Nrf2 transcription factor is repressed under normal homeostatic conditions by the dual actions of β-TrCP and Keap1, thereby ensuring that the expression of antioxidant proteins and detoxication systems is restricted when the cell is not exposed to stress. However, numerous signaling processes allow derepression of Nrf2 by β-TrCP and Keap1, and this ensures the induction of ARE-driven genes to enable the elimination of electrophiles and oxidative stressors as well as metabolic adaptation to nutrients and growth cues. Thus, from the top left, the binding of insulin and growth factors to their cognate receptors will activate PI3K, which in turn increases Akt and mTORC2 activity while decreasing that of mTORC1 (not shown, but see Fig. 12). Increases in the PI3K–Akt–mTORC2 signaling system will inhibit GSK-3 activity, causing a decrease in the rate of β-TrCP-mediated Nrf2 turnover through reduced formation of the DSGIS-containing phosphodegron. In addition to insulin and growth factor signaling, the activity of mTORC1 is also decreased under conditions of low nutrient availability by the activation of AMPK. As shown top center, the activity of mTORC1 increases competitive inhibition of Keap1 by p62/SQSTM1 because it phosphorylates the STGE motif in the autophagy cargo receptor, which increases the affinity of p62/SQSTM1 for Keap1 and stimulates elimination of Keap1 by autophagy. Thus, during fasting, phosphorylation by mTORC1 of the STGE motif in p62/SQSTM1 is diminished and Keap1 is able to repress Nrf2 more effectively. As shown top right, signaling from autophagy, mitochondria, and inflammation pathways will diminish Keap1-mediated degradation of Nrf2 via competitive binding by p62/SQSTM1, PGAM5, and IKKβ to Keap1. Electrophiles and ROS also antagonize Keap1 substrate adaptor activity by modifying its sensor Cys-151, Cys-273, Cys-288, Cys-226/Cys-613, and Cys-434 residues. When small-molecule inducers inactivate Keap1, the conformation and/or orientation of the Keap1 dimer relative to other proteins within the Cul3–RBX1 complex is altered, producing a stalling of ligase activity that traps bound Nrf2 within CRLKeap1. As a consequence, newly translated Nrf2 evades degradation by CRLKeap1. This free fraction of Nrf2 accumulates and translocates to the nucleus where it heterodimerizes with sMaf proteins before binding to the ARE sequences in the promoters of target genes, causing induction of cytoprotective proteins. The increased expression of ARE-driven genes results in the synthesis and recycling of antioxidants, detoxification of harmful agents, and excretion of drugs. The physiological consequence of the induction of ARE-driven genes is the restoration of cellular redox homeostasis, through a negative feedback loop that ultimately allows Keap1 repression of Nrf2 to be reinstated through a variety of mechanisms that may include reactivation of oxidized Keap1 by TXNRD1, the induction of p62/SQSTM1 leading to increased degradation of electrophile-modified Keap1 by autophagy, and increased synthesis of Keap1.
3.4.2. Composition of mTOR-containing complexes
The ability of mTOR to regulate cell function is achieved through the existence of two structurally and functionally distinct complexes, mTORC1 and mTORC2, depicted in Fig. 12. First, mTORC1 comprises mTOR, regulatory-associated protein of mTOR (RAPTOR), mammalian lethal SEC13 protein 8 (mLST8), proline-rich Akt substrate 40 (PRAS40), and DEP domain-containing mTOR-interacting protein (DEPTOR). The mTORC1 complex is activated by amino acids (especially leucine), growth factors, nutrients, and oxidative stress, and its activation is associated with augmented cell survival because it results in increased protein synthesis, initiation of lipid biosynthesis, and diminished autophagy [316]. Second, mTORC2 consists of mTOR, rapamycin-in-sensitive companion of mTOR (RICTOR), mLST8, stress-activated protein kinase-interacting protein 1 (SIN1), protein observed with RICTOR-1 (PROTOR-1), and DEPTOR. The mTORC2 complex affects cell survival through signaling via the Akt pathway and cytoskeletal dynamics through interactions with PKC.
Fig. 12.
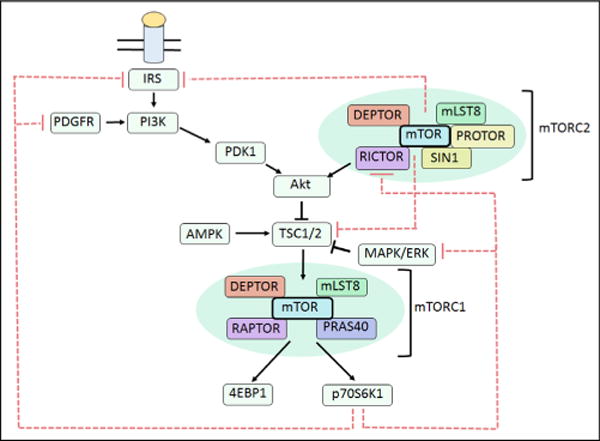
Complexity of the direct and indirect effects of mTOR on signaling pathways. Metabolic signaling through insulin receptor substrate-1 (IRS) leads to activation of PI3K and subsequent recruitment of phosphoinositide-dependent kinase-1 (PDK1) to the cell membrane. Here, PDK1 phosphorylates Akt at Thr-308, which in turn triggers phosphorylation of Akt at Ser-473 by mTORC2. Upon its activation, Akt becomes capable of inhibiting the TSC1/2 complex, leading to the stimulation of mTORC1 and its downstream targets 4EBP1 and p70S6K1. The resulting stimulation of p70S6K1 activity can then cause negative feedback of the mTOR signaling cascade at several points of the pathway. Thus, increased p70S6K1 activity can lead to phosphorylation of IRS, leading to degradation of the receptor. Active p70S6K1 also negatively regulates Akt signaling through inhibition of PDGFR and ERK/MAPK signaling. The two mTOR complexes are also able to regulate each other in an inter-complex feedback loop, as activation of p70S6K1 stimulates phosphorylation of the RICTOR subunit of mTORC2, causing a decrease in Akt signaling thorough impaired phosphorylation of Ser-473. In addition, mTORC2, via p70S6K1, also inhibits IRS, leading to a reduction in Akt signaling.
3.4.3. Direct regulation of Nrf2 by mTORC1
As described above, mTORC1 can increase Nrf2 protein levels and induce its target genes because it phosphorylates Ser-351 in p62/SQSTM1, thereby enabling p62/SQSTM1 to bind to Keap1 and repress CRLKeap1 activity by promoting Keap1 degradation via autophagy [277]. It is likely that AMPK also influences Nrf2 activity because of its influence on mTORC1 activity, but in this case it involves an interaction with tuberous sclerosis complex 1 (TSC1) and TSC2, proteins that together form a physical complex. The TSC1/2 complex is frequently activated under adverse physiological conditions such as hypoxia, nutrient starvation, and oxidative stress: during low oxygen availability, hypoxia-inducible factor 1-α is activated, which in turn activates TSC1/2; during nutrient deprivation, LKB-1 is activated, leading to activation of AMPK, which then phosphorylates TSC1/2 directly, leading to attenuation of mTORC1 signaling and a switch in metabolism from lipogenesis to lipolysis. In the case of DNA damage, the TSC1/2 complex is activated via AMPK through interactions with the tumor suppressor protein p53 [310]. The catalytic activity of the TSC1/2 complex is governed solely by the TSC2 protein, which exhibits GTPase-activating protein activity toward Rheb, a member of the Ras family of GTPases. Rheb lies upstream of mTORC1, and once activated, it stimulates mTORC1, leading to subsequent phosphorylation of the translational regulators p70S6K and eukaryotic translation initiation factor 4E binding protein 1 (4EBP1). Therefore, AMPK regulates cell growth and survival through mTORC1 signaling [317].
Under energy starvation conditions, GSK-3 phosphorylates TSC1/2, leading to inhibition of the complex. As discussed above, GSK-3 has a unique requirement for a priming kinase, and in the case of mTORC1 signaling, phosphorylation of TSC1/2 by AMPK could provide the priming event required for subsequent phosphorylation of TSC1/2 by GSK-3 [294]. This suggests that AMPK and GSK-3 cooperatively inhibit mTORC1, insofar as the ability of GSK-3 to inhibit mTORC1 is enhanced when AMPK primes TSC1/2 for phosphorylation [317]. Taken together, the above data suggest that under fasting conditions, when AMPK is stimulated, mTORC1 activity will be low and p62/SQSTM1 will not be phosphorylated. Such conditions will allow CRLKeap1 to maximally repress Nrf2 provided normal intracellular redox is maintained.
3.4.4. Direct regulation of Nrf2 by mTORC2
It seems likely that mTORC2 also influences Nrf2 activity, but in this case it arises through its ability to control PI3K–Akt signaling. Growth factors and insulin activate PI3K via receptor tyrosine kinases leading to increased production of phosphatidylinositol (3,4,5)-trisphosphate (PIP3). Once formed, PIP3 binds both phosphoinositide-dependent protein kinase 1 (PDK1) and Akt, bringing them into close proximity in the cytoplasm. The juxtapositioning of PDK1 and Akt initiates the first of two phosphorylation events required for full Akt activation: an initial weak phosphorylation on Thr-308 mediated by PDK1, which then triggers a second stronger phosphorylation event at Ser-473 of Akt that is carried out by mTORC2 [318, 319]. The resulting full activation of Akt results in inhibition of GSK-3 and therefore loss of repression of Nrf2 by β-TrCP.
3.4.5. Indirect regulation of Nrf2 by mTORC1 and mTORC2 via feedback loops
In addition to the direct phosphorylation of Akt by mTORC2, a series of complex regulatory loops exist between mTORC1, mTORC2 and Akt that are likely to influence Nrf2 activity. Activation of mTORC1 by an overabundance of nutrients leads to activation of p70S6K and phosphorylation of the insulin receptor substrate-1 (IRS-1), which results in the subsequent degradation of IRS-1. Activation of p70S6K via mTORC1 also negatively regulates PI3K–Akt through inhibition of PDGF receptor (PDGFR) and ERK–MAPK signaling [320]. An intercomplex feedback loop is also present between mTORC1 and mTORC2. In this case, activation of p70S6K through mTORC1 signaling leads to phosphorylation of RICTOR at Thr-1135 in the mTORC2 complex, leading to impaired phosphorylation of Akt at Ser-473 and a subsequent reduction in Akt signaling [321]. Nutrient levels have also been linked to the mTORC2–Akt negative feedback loop as supplementation with amino acids leads to enhanced phosphorylation of TSC1/2 at Ser-2448. Activation of mTORC1 and mTORC2 signaling due to high glucose levels leads to increased phosphorylation of several Ser residues in IRS-2 that results in its degradation and subsequent inhibition of apoptotic death in B cells [313]. In turn, activation of Akt by phosphorylation of Ser-473 leads to inhibition of GSK-3 activity, decreased formation of the DSGIS-containing phosphodegron in Nrf2, which leads to decreased turnover of Nrf2 by SCFβ-TrCP and increased expression of ARE-driven genes.
3.5. Repression of Nrf2 during endoplasmic reticulum stress
Cross talk exists between the oxidative stress and the ER stress pathways in the context of liver cirrhosis produced experimentally by administration of CCl4. In this instance, upregulation of the ER stress response downregulates the Nrf2 signaling pathway [78]. Thus during liver cirrhosis, Nrf2 interacts directly with the ERAD-associated E3 ubiquitin-associated ligase synovial apoptosis inhibitor 1 (also called synoviolin or HRD1), causing ubiquitylation and degradation of the CNC-bZIP transcription factor. This prevents Nrf2 from orchestrating antioxidant responses and countering the high levels of ROS produced during cirrhosis. Interestingly, during ER stress HRD1 is transcriptionally activated byXBP1, which lies downstream of IRE1 in the UPR [160]. HRD1 therefore seems to be a potential new target for ameliorating cirrhosis-induced liver oxidative stress and preventing progression of the disease.
3.6. Other proteins involved in the regulation of Nrf2
Other proteins have emerged as repressors of Nrf2 activity. CR6-interacting factor 1 interacts with Nrf2 in a redox-independent fashion, promoting the ubiquitylation of Nrf2 by binding at both the C- and the N-terminal Neh3 and Neh2 domains of the transcription factor [322].
The protein apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/REF-1) negatively regulates Nrf2 through its redox activity, with inhibition of APE1/REF-1 causing significant increases in the levels of Nrf2 and selected target genes HMOX1, GCLC, and GCLM in pancreatic cancer cell lines without causing the generation of ROS [323].
During hypoxia, Nrf2 levels are suppressed and seven in absentia homolog 2 (SIAH2) levels increase. It has been proposed that SIAH2 suppresses Nrf2 under conditions of hypoxia through a Keap1-independent means as experiments using ectopic expression of wild-type and mutant forms of Nrf2 have shown that loss of the Keap1-binding domain did not influence hypoxic levels of the CNC-bZIP transcription factor, whereas knockdown of SIAH2 prevented the hypoxic suppression of Nrf2 [324].
The small ubiquitin-like modifier (SUMO)-specific RING finger protein 4 (RNF4) has also been linked to the regulation of Nrf2, with SUMOylated Nrf2 being ubiquitinated by RNF4 in the nucleus, which leads to degradation of the transcription factor [325].
4. Biochemical consequences of Nrf2 upregulation on redox and detoxification
Based on its essential role in redox homeostasis it is likely the dose-response curve for the pharmacological activation of Nrf2 is U-shaped, at least with regard to the risk of several degenerative diseases [326, 327]. Thus in the “normal” physiological homeostatic range, Nrf2 is required to maintain the ROS/antioxidant balance. By contrast, at the low end of the dose-response curve when Nrf2 is unable to maintain antioxidant capacity, toxicity may arise because of the inability to eliminate ROS; indeed, elevation of intracellular ROS above a critical threshold activates Nrf2 to induce Kruppel-like factor 9, which in turn suppresses the expression of TXNRD2 and exacerbates oxidative stress [328]. At the high end of the dose–response curve, when elevated Nrf2 activity results in ROS levels being quenched, the consequences include a relative overproduction of GSH and NADPH, epithelial cell hyperplasia, and drug resistance. As described above, from studies of Nrf2-null mice and cells, low levels of the CNC-bZIP transcription factor are likely to lead to diminished cell proliferation [155], alterations in cell differentiation [156–158], perturbation of the UPR [162, 163], increased sensitivity to the toxic effects of xenobiotics [140, 143, 175], increased sensitivity to inflammation [144, 162, 183–185], increased sensitivity to hepatic steatosis [144, 162], and a compromised ability to regenerate the liver [135], but conversely an increased sensitivity to insulin [132, 162, 227]. Many of these phenotypes can be attributed to the loss of redox homeostasis through decreased capacity of the GSH-dependent and TXN-dependent antioxidant systems to suppress ROS levels and maintain thiol groups in proteins in their appropriate redox state [140–152]. As a consequence of diminished antioxidant capacity, the activity of some of the protein phosphatases, such as PTP1B, SHP-2, PTEN, and MKP, that attenuate the progression of ROS-dependent signal transduction by being subject to reversible oxidation by H2O2 [15, 195] is likely to be compromised. Moreover, the loss of TXN activity will allow ASK1 to be more readily activated [208–211]. Thus signaling through receptor tyrosine kinases, ASK1–JNK, ASK1-p38MAPK, and Wnt-β-catenin is likely to be more active in cells that have low Nrf2 levels than in cells with high Nrf2 activity. Evidence suggests high Nrf2 levels are likely to increase the GSH and TXN antioxidant systems and blunt the sensitivity of ROS-dependent signaling pathways, including sensitivity to inflammation and apoptosis. To a greater or lesser extent, these changes will contribute to drug tolerance, inhibition of inflammatory reactions, increase in cell proliferation, and changes in cell differentiation that are observed when Nrf2 is overexpressed [121, 140, 159, 185].
It is important to distinguish between the relatively modest and transient upregulation of Nrf2 achieved pharmacologically by chemopreventive agents and the supraphysiological upregulation of Nrf2 that occurs by genetic knockout of Keap1, as the continuous upregulation of Nrf2 resulting from genetic loss-of-function mutations in Keap1, or gain-of-function mutations in NFE2L2, is much more likely than pharmacological agents to produce deleterious effects. During hyperactivation of Nrf2, an overabundance of GSH and NADPH can produce “reductive stress” that is associated with protein misfolding and aggregation and cardiac hypertrophy [329–332]. Also, excessive levels of Nrf2 may increase expression of the oxidized-LDL scavenger receptor CD36, which under certain conditions leads to the formation of foam cells and an increased risk of atherosclerosis [133, 333].
The influence that activation of Nrf2 has on cell function has been examined using pharmacological agents and genetic manipulation as a means to upregulate the transcription factor. Chemical inducers that have been used for this purpose include tBHQ SFN, the dithiole-thione oltipraz, the triterpenoid CDDO-Im, and the acetylenic bis(cyanoenone) TBE-31 [59,61,116,334–336]. Genetic activation of Nrf2 as a means of studying the function of the transcription factor has been achieved by global knockout or tissue-specific disruption of the Keap1 gene in the mouse [116, 337] or by knockdown of Keap1 in human cell lines [36, 40, 246]. In addition, knockdown of Nrf2 in human cells that harbor somatic mutations in Keap1, and thus possess constitutively high levels of the transcription factor, has also been employed as a means of studying the pathways it controls [121, 338, 339].
4.1. Effect of Nrf2 upregulation on intracellular antioxidant status
An overwhelming body of evidence indicates that upregulation of Nrf2 increases intracellular antioxidant capacity. Paul Talalay and colleagues found that treatment of human ARPE-19 adult retinal pigment epithelial cells and HaCaT keratinocytes with nontoxic doses of SFN produced an approximate twofold increase in GSH levels that was sustained for several days and provided prolonged protection against oxidants and electrophiles, such as menadione, tert-butylhydroperoxide, 4-hydroxynonenal, and peroxynitrite [42]. Similar effects were observed in Nrf2+/+ MEFs treated with SFN [140] and HaCaT cells after knockdown of Keap1 [40], and in both instances it was associated with induction of GCLC and GCLM. Treatment of human K562 erythroleukemia cells with tBHQ and human K-1034 RPE cells with SFN has been reported to induce TXN [340, 341], and the increase in TXN protects mice against light-induced retinal damage [341]. Importantly, the increase in GSH and TXN upon activation of Nrf2 is associated with upregulation of reductases, GSR1, TXNRD1, and SRXN1 [35, 342, 343], which along with the increase in expression of enzymes involved in NADPH regeneration ensures these thiol-based antioxidant systems are kept reduced [61].
The induction of antioxidant genes by thiol-reactive electrophiles is thought to represent an adaptive response insofar as their induction augments GSH and TXN antioxidant systems, enhances the capacity to metabolize xenobiotics, and increases drug efflux transporters. Thus, induction of antioxidant and detoxication genes represents a major means by which chemoprotective agents provide inducible resistance against subsequent exposure to a toxic dose of a xenobiotic by a type of “preconditioning” or “drug-priming” mechanism [140], which may be an aspect of habituation. This seems to be the same principle by which cancer-chemopreventive “blocking agents” inhibit chemical carcinogenesis [344].
4.2. Effect of Nrf2 upregulation on drug metabolism and drug transport
Nrf2 regulates xenobiotic metabolizing enzymes in a species-specific manner. It is well known that NQO1 is highly inducible in many rodent and human tissues and cell lines in which it increases reduction of quinones to hydroquinones, which can in turn be glucuronidated by UGTs or sulfonated by SULTs and eliminated from the cell by MRPs, thereby preventing redox cycling [179, 181]. By contrast with the protection provided against quinones, NQO1 is capable of activating the carcinogenic nitro-aromatics 3-nitrobenzanthrone and aristolochic acid I [345], suggesting it can occasionally adversely affect health.
Class Alpha, Mu, Pi, and Theta GSTs are induced in the liver and gastrointestinal tract of rats and mice by cancer-chemopreventive agents such as butylated hydroxyanisole, ethoxyquin, coumarin, SFN, 4-methyl-5-[2-pyrazinyl]-1,2-dithiole-3-thione (oltipraz), CDDO-Im, and TBE-31, but are not obviously inducible in the human [111]. The consequences of GST induction can be profound. For example, the rat is highly sensitive to hepatocarcinogenesis caused by the mycotoxin aflatoxin B1 (AFB1), but induction of the GST A5-5 isoenzyme (originally called Yc2Yc2) in rat liver by the above chemopreventive agents results in increased detoxification of the ultimate carcinogen, AFB1-8,9-epoxide, reduction in DNA adduct formation, and protection against tumorigenesis [346–351]. Although workers in the laboratory of Masayuki Yamamoto have created an Nrf2-knockout rat, it has not yet been investigated whether Nrf2 mediates induction of the GSTA5 gene by chemopreventive agents in rat liver, but it appears probable. In addition to induction of GST in the rat, the AFB1-dialdehyde-metabolizing AKR7A1 reductase (originally cloned by Elizabeth Ellis and called AFAR [352,353]), which protects against cytotoxicity, is also highly inducible by cancer-chemopreventive agents in rat liver [352–355]. Although it is not known if induction of AKR7A1 is mediated by Nrf2 its gene contains four ARE sequences in its promoter (Table 2). As a cautionary note, induction of class Theta GST T1-1 isoenzyme in rat liver increases activation of the dihaloalkane carcinogens dichloromethane and dibromomethane [356], and therefore the consequences of GST induction depend on the likelihood of exposure to particular environmental toxins.
By contrast with the rat, mice are intrinsically resistant to AFB1, and mouse liver contains high basal levels of a class Alpha Gst a3-3 enzyme (originally called YcYc or Ya3Ya3) that catalyzes the conjugation of GSH with AFB1-8,9-epoxide and appears to be responsible for the intrinsic resistance of the mouse to AFB1 hepatocarcinogenesis [357–360]. Knockout of the Gsta3 subunit renders mice sensitive to AFB1 toxicity, and this cannot be rescued by treatment of Gsta3−/− mice with chemopreventive agents that activate Nrf2 [360], despite the fact that many other class Alpha, Mu, and Pi Gst subunits are inducible in mouse liver [59, 61, 96, 361, 362]. Examples of the dependency of chemopreventive agents on Nrf2 to inhibit chemical carcinogenesis in the mouse are provided by the protection conferred by oltipraz against benzo[a] pyrene-initiated stomach cancer, through induction of Gst and Nqo1, and protection conferred by oltipraz against N-nitrosobutyl (4-hydroxybutyl)amine bladder carcinogenesis, through induction of Ugt1a [363–365]. Nrf2 also mediates the chemopreventive effects of SFN against 7,12-dimethylbenz[a]anthracene-induced skin tumorigenesis [176], but the enzymes responsible have not been identified.
Whereas many Gst genes are regulated in mice by chemopreventive agents in an Nrf2–Keap1-dependent manner, this is not the case in humans [246], in which Nrf2 is more commonly associated with the induction of AKR1A1, AKR1B10, AKR1C1, AKR1C2, and AKR1C3 genes. Among these inducible human oxidoreductases, AKR1A1 is implicated in the reduction of doxorubicin to doxorubicinol [366], whereas AKR1C1 and AKR1C2 are involved in the metabolism of the steroid hormone replacement therapeutic tibolone [367, 368] and the oral contraceptive northeynodrel [369]. These findings suggest that activation of Nrf2 in humans may lead to more rapid clearance of certain therapeutic drugs. In humans, the AKR1B10, AKR1C1, AKR1C2, and AKR1C3 genes are upregulated by the Keap1–Nrf2 pathway as part of the redundancy built in to protect against the deleterious consequences of lipid peroxidation and its breakdown products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal [128, 370]. In the case of AKR isoenzymes, the bifunctionality of the electrophile is inactivated when the aldehyde is reduced by AKR1C enzymes to a primary alcohol to form 1,4-dihydroxynonene [128]. The AKR1C isoenzymes are also capable of activating PAH trans-dihydrodiol carcinogens including 7,12-dimethylbenz[a]anthracene-3,4-dihydrodiol to form a ketol, which can spontaneously form a catechol that can in turn redox cycle to an ortho-quinone [371, 372]. Finally, it is noteworthy that although AKR7A1 can be induced by BHA, ethoxyquin, oltipraz, and coumarin in rat liver [348, 352], the related human AKR7A2 and ARK7A3 genes do not appear to be inducible even though they contain ARE sequences in their gene promoters (Table 2).
MRP transporters are part of the mammalian phase III drug detoxication system, responsible for the elimination of xenobiotics. In mice, induction of Mrp2, Mrp3, and Mrp4 by BHA and oltipraz is dependent on Nrf2 [120]. It remains unclear, however, whether Nrf2 regulates these transporters in rat and human [373], and further work is required to address this issue.
5. Biochemical consequences of Nrf2 upregulation on metabolic processes
As discussed earlier, Nrf2 activity is influenced by signaling though the mTOR pathways, suggesting a role for the transcription factor outside its traditional role in the oxidative stress response and detoxification and, in so doing, implicating the transcription factor in the regulation of metabolic processes such as carbohydrate and lipid metabolism and the utilization and generation of NADPH. The links between Nrf2 and metabolism are relatively recent discoveries, first noted by the research groups of Thomas Kensler, Curtis Klaassen, and Masayuki Yamamoto [115, 116, 121]. Although the role of Nrf2 in these processes is slowly being clarified, the body of literature available on the subject is relatively small, with much remaining to be discovered.
5.1. Effect of Nrf2 upregulation on carbohydrate metabolism
Carbohydrates not only constitute an important energy source for cells, but their structural diversity allows them to serve as key structural components involved in defining the function of the cell and in cell-cell interactions [374]. The metabolism of carbohydrates is controlled at many levels by the availability of certain key nutrients and through feedback from other metabolic processes. The links between Nrf2 and the regulation of carbohydrate metabolism are relatively recent discoveries [116], which are not yet fully understood.
5.1.1. The role of Nrf2 upregulation in glucose metabolism and the pentose phosphate pathway
Availability of glucose dictates the pathway by which it is processed. During times of energy requirement, glucose is catabolized to produce two molecules of pyruvate, which is directed into the citric acid cycle for oxidative phosphorylation. Under conditions of high glucose availability, glucose is directed to anabolic reactions such as the pentose phosphate pathway (PPP) and the generation of glycogen [374]. Under sustained PI3K-Akt signaling, Nrf2 redirects glucose into the anabolic PPP with knockdown of the transcription factor leading to the increased accumulation of glycolysis intermediate metabolites in the cell [121]. In cancer cells with constitutively active Nrf2, knockdown of the transcription factor causes downregulation of all enzymes in the PPP, i.e., G6PD, PGD, transketolase, and transaldolase 1 [121, 375]. In agreement, pharmacological (by SFN) or genetic (by Keap1 knockdown) upregulation of Nrf2 in human nontumorigenic breast epithelial MCF10A cells causes increases in the PPP enzymes [36]. The influence of Nrf2 on the PPP can also be seen in its effect on nucleotide synthesis; Nrf2 is important for efficient synthesis of purine, but not pyrimidine, nucleotides from glucose [121]. Indeed, Nrf2 regulates the expression of phosphoribosyl pyrophosphate amidotransferase, which catalyzes entry into the de novo purine nucleotide biosynthetic pathway, as well as the mitochondrial methylenetetrahydrofolate dehydrogenase 2, which provides glycine and formate, the sources of one-carbon units for purine biosynthesis [121]. The upregulation of anabolic pathways by Nrf2 drives in vitro and in vivo cell proliferation and is thought to contribute to accelerated tumor growth [121, 375].
5.1.2. The role of Nrf2 in glutamine metabolism
Levels of the nonessential amino acid glutamine also play a role in the regulation of cellular energy homeostasis. Glutamine can be converted via a two-step process to α-ketoglutarate, which is a citric acid cycle intermediate. As with glucose, Nrf2 redirects glutamine into anabolic pathways under sustained PI3K–Akt signaling [121]. Knockdown of Nrf2 results in increased levels of glutamine and glutamate, with decreased production of GSH presumably contributing to this increase.
5.2. Influence of Nrf2 upregulation on NADPH generation and consumption
The reducing agent NADPH is a cofactor used in anabolic reactions, such as lipid and nucleic acid synthesis, as well as acting as a source of reducing equivalents in many reactions. Nrf2 has been linked to the regulation of generation and consumption of NADPH. The main enzymes involved in NADPH production, ME1, IDH1, G6PD, and PGD, are all regulated in part by Nrf2 [61, 115, 121, 375, 376], with low levels of the cofactor seen in Nrf2−/− mice and correspondingly high levels in mice with hepatocyte-specific Keap1 knockout [115]. Additionally, the levels of NADPH and the NADPH/NADP+ ratio are lower in Nrf2−/− than in wild-type MEF cells, and the NADPH levels decrease upon Nrf2 knockdown in cancer cell lines in which Nrf2 is constitutively active [375]. This decreased availability of reducing power in Nrf2−/− mice will not only negatively affect lipogenesis and nucleic acid synthesis, but will also contribute to reduced antioxidant availability in the cell, as NADPH acts as a reducing factor for both the GSH and the TXN systems.
5.3. Impact of Nrf2 on lipid metabolism
Tom Kensler and colleagues first noticed that genetic or pharmacological upregulation of Nrf2 caused changes in expression of genes involved in lipid metabolism [116]. Subsequently, various groups have reported that Nrf2−/− mice succumb rapidly to fatty liver (i.e., steatosis) when placed on a methionine- and choline-deficient diet [144, 377, 378]. In the wake of these observations, Keith Blackwell and Sean Curran and co-workers noted that genetic upregulation of Nrf2 in murine Keap1−/− lung tissue resulted in induction of genes involved in fatty acid oxidation and a decrease in expression of genes involved in fatty acid synthesis [123], which is consistent with the observation that Nrf2−/− mice are susceptible to steatosis. The increase in fatty acid oxidation upon genetic upregulation of Nrf2 is interesting because it accords with an earlier study in which expression of the oxidized-LDL scavenger receptor CD36 in macrophages was reported to be controlled by Nrf2 [133], suggesting that the increase in fatty acid oxidation is coupled with increased uptake of lipids.
Hepatic lipid metabolism is largely regulated by the nuclear receptor PPARa, which controls the expression of genes involved in lipid transport, β-oxidation of fatty acids, ketogenesis, lipogenesis, lipid mobilization, and cholesterol metabolism, some examples of which are shown in Table 3[379]. PPARα is a transcription factor that is activated under conditions of low energy availability, which is critical in the longer-term adaptation to fasting conditions. By contrast, the expression of Pparα has been reported to be decreased upon upregulation of Nrf2 [116]. Microarray studies involving 24-h-fasted Pparα−/− mice revealed a highly significant downregulation of Nrf2 mRNA levels compared to wild-type mice, whereas no change in Nrf2 mRNA was seen in Pparα-null mice that had been fed [238]. Taken together, these data suggest a potential feedback loop exists between the transcription factors Nrf2 and Pparα during fasting conditions, the mechanism of which is not yet fully understood. As outlined in Fig. 13 the activation of Nrf2 during fasting could be a direct transcriptional effect of Pparα activation, an indirect consequence of the production of ROS resulting from β-oxidation of fatty acids that results in increased Nrf2 protein stability, a combination of the two mechanisms, or something completely unrelated. Both Yates et al. [116] and Sanderson et al. [238] reported changes in gene expression of Nrf2 and Pparα at the level of mRNA, suggesting that the putative feedback could be a direct transcriptional mechanism, but this does not rule out a contribution from indirect means.
Table 3.
Examples of PPARα target genes and the biological processes in which they are involved
| Gene | Protein | Function | Cellular processes |
|---|---|---|---|
| ACACA | Acetyl-CoA carboxylase α | Catalyzes the rate-limiting step in fatty acid synthesis; carboxylation of acetyl-CoA to malonyl-CoA | Lipogenesis |
| GPAM | Glycerol-3-phosphate acyltransferase, mitochondrial | Catalyzes the first step in synthesis of glycerolipids; plays a role in the level of available cellular triglycerides | Lipogenesis, glycerolipid synthesis |
| SLC27A4 | Solute carrier family 27a4 | Translocates long-chain fatty acids across the plasma membrane | Lipid transport |
| ABCD2 | ATP-binding cassette D2 | Peroxisomal import of fatty acids and fatty acyl-CoA | Lipid transport, peroxisomal β-oxidation |
| ACOX1 | Acyl-CoA oxidase 1, palmitoyl | Catalyzes first desaturation step in β-oxidation of fatty acids; donates electrons to O2 leading to production of H2O2 | Peroxisomal β-oxidation |
| CPT1A | Carnitine palmitoyltransferase 1A | Transport of long-chain fatty acids across the mitochondrial membrane | Mitochondrial β-oxidation |
| CPT2 | Carnitine palmitoyltransferase 2 | Mitochondrial oxidation of long-chain fatty acids | Mitochondrial β-oxidation |
| FXR | Farnesoid X-activated receptor | Bile acid receptor that regulates the expression of genes involved in bile acid transport and synthesis | Bile acid transport, bile acid synthesis |
| LXR | Liver X receptor | Obligate heterodimeric binding partner of RXRs; involved in cholesterol homeostasis and innate immunity | Cholesterol homeostasis, innate immunity, RXR activation |
Modified from [379].
Fig. 13.
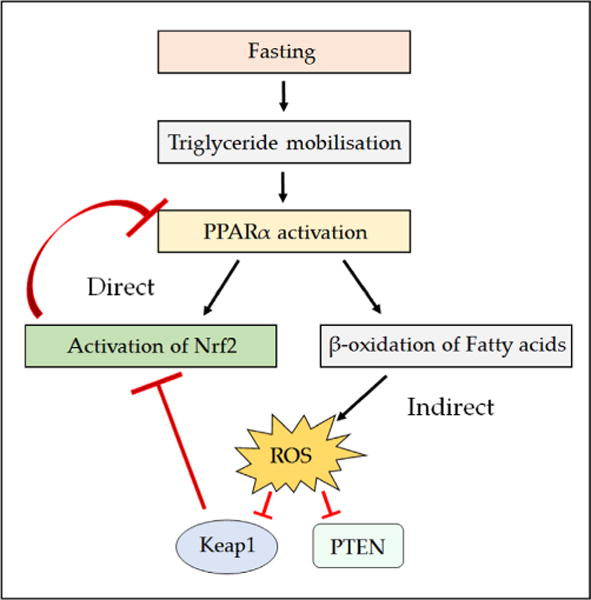
Potential role of PPARα in the activation of Nrf2 in response to fasting. During fasting, triglyceride stores are mobilized and broken down to glycerol and free fatty acids (FFA). Once formed, the FFA act as ligands for PPARα, activating it and causing transcription of genes involved in their β-oxidation. Upon activation, PPARα could induce the NFE2L2 gene encoding Nrf2, which could in turn suppress PPARα mRNA levels. Another possibility is that the β-oxidation of fatty acids, which occurs in a PPARα-dependent manner, causes the production of ROS and lipid peroxides that indirectly cause activation of Nrf2 by antagonizing Keap1 and/or β-TrCP. It is also possible that a combination of both mechanisms is responsible for the activation of Nrf2 seen during fasting.
Previous work on the potential role of Nrf2 in lipid metabolism by researchers in the laboratory of Curtis Klaassen examined the impact that loss of Nrf2 has on the effects of 24-h fasting in mice [145]. They found that during acute fasting, whole body energy utilization, hepatic glycogen content, and gluconeogenesis were not significantly influenced by Nrf2 deficiency. Levels of oxidative stress were shown to be higher in the Nrf2−/− mice owing to their inability to induce antioxidant genes, although it is worth noting that the wild-type fasted mice also failed to show significant induction of Nrf2 and its target genes after 24 h fasting. Examination of the effects of acute fasting on gene expression in Keap1+/− mice, with genetically upregulated Nrf2 [380], largely agreed with the findings of Zhang et al. [145], although they also noted a small but significant induction of Nqo1 in wild-type fasted mice. These data pose more questions about the exact role of genetic and pharmacological upregulation of Nrf2 on lipid metabolism, with further work needed to define the contribution made by the transcription factor.
The role played by Nrf2 in the regulation of metabolic processes is, at present, still unclear with much remaining to be elucidated about the contribution of the transcription factor to metabolism in vivo. Despite the limited availability of data on these topics, it has become clear that the contribution made to the regulation of many metabolic processes by Nrf2 is significant and that the field would benefit from further study in this area.
6. The varied contributions made by Nrf2 to degenerative disease
6.1. Nrf2 plays apparently contradictory roles in cancer
Nrf2 plays an important, but somewhat ambiguous, role in the initiation, promotion, and progression of cancer. Too little Nrf2 activity clearly results in loss of cytoprotection and therefore increased risk of chemical carcinogenesis. However, Nrf2-null mice do not spontaneously develop cancer. The Nrf2−/− mice succumb to carcinogenesis only if they are exposed to environmental carcinogens such as benzo[a]pyrene, 7,12-dimethylbenz[a]anthracene, nitrosamines, or tobacco smoke [175]. The only exception to the general rule that Nrf2−/− mice are more sensitive than Nrf2+/+ mice to chemical carcinogenesis is provided by urethane and the incidence of lung cancer, in that wild-type mice suffer a higher tumor burden than mutant mice [381]; however, even in this situation, after urethane treatment, the Nrf2−/− mice develop far more early preneoplastic lesions than the wild-type mice, and it seems that postinitiation events prevent these early lesions from developing into tumors [182]. A large body of literature indicates that Nrf2 mediates the beneficial effects of many chemopreventive agents in rodents when administered before exposure to chemical carcinogens (defined as “blocking” agents [344]). Set against the general role of Nrf2 as an inhibitor of carcinogenesis, it is possibly counterintuitive that somatic mutations resulting in its constitutive activation are a prominent feature of many cancers, a characteristic that is often referred to as the “dark side” of Nrf2 [382] and implies that the transcription factor is capable of supporting tumorigenesis [383, 384]. This conclusion should, however, be tempered by the discovery that the long-lived naked mole rat, which enjoys an extremely low cancer incidence, has higher Nrf2 activity than shorter-lived rodents [385, 386]. The dichotomy that Nrf2 mediates chemoprevention but is frequently upregulated in tumors is consistent with the notion that the dose-response curve for Nrf2 and cancer susceptibility is U-shaped [326, 327]. This nonlinear curve infers that upregulation of Nrf2 may be necessary for the promotion and progression of carcinogenesis in certain circumstances and is probably likely to depend on the context in terms of the carcinogenic stimulus involved, the somatic mutations that have arisen, and the microenvironment of the tumor.
6.1.1. Contribution of Nrf2 to cancer chemoprevention
Carcinogenesis is initiated by a series of DNA mutations, often caused by electrophiles or ROS insults, which lead to loss of normal growth control. Upregulation of enzymes involved in detoxification would be expected to reduce greatly the risk of chemical carcinogenesis, with the proviso that most detoxication enzymes will on occasions catalyze toxication reactions (for examples, see [345, 356, 371, 372]). In the vast majority of cases it has been found that chemopreventive agents that activate Nrf2 and induce phase II drug-metabolizing enzymes diminish tumorigenesis, especially when protection is compared between wild-type and Nrf2-knockout mice [387]. It is well established that chemical activators of Nrf2 such as oltipraz, SFN, and TBE-31 reduce the likelihood of cancers developing when administered before challenge with carcinogens [95, 102, 180, 388]. In addition to decreasing the likelihood of tumorigenesis, activation of Nrf2 by pharmacological (SFN) or genetic (Keap1 knockdown) means has been shown to reduce the number of harmful DNA-damaging effects of estrogens in MCF-10A cells [389].
The fact that phenolic antioxidants, such as BHA, are capable of inhibiting the initiation of chemical carcinogenesis was one of the discoveries that helped establish the cancer chemoprevention field [390–392]. It was, however, later reported that BHA and other phenolic antioxidants may promote tumorigenesis in the forestomach of some rodent species (reviewed in [393, 394]). Specifically, among F344 rats in which cancer had been initiated by injection of N-methyl-N-nitrosourea over a period of 4 weeks, subsequent feeding of a diet containing 2% BHA over the next 32 weeks resulted in 92 and 88% of animals developing forestomach papillomas and carcinomas, respectively, whereas among rats that had similarly received N-methyl-N-nitrosourea but were subsequently placed on a regular chow diet, only 36% developed forestomach papillomas and none had carcinomas [395]. BHA has also been reported to promote N-methyl-N-nitrosourea-initiated adenoma of the thyroid in F344 rats, but in this case the incidence increased from 0 to 16% [395]. It should be noted that similar findings have not to our knowledge been reported in the mouse. The significance for human health of the findings that BHA can promote forestomach cancer and thyroid cancer in the rat is uncertain because humans do not possess an anatomical equivalent to the rodent forestomach and because the dose of BHA used in the study was extremely high. Most importantly in the context of this article, it is not known if the ability of BHA to promote tumorigenesis is mediated by Nrf2.
In view of the ability of high doses of BHA to promote forestomach cancer in the rat, coupled with the fact that Nrf2 is a cell survival factor, it is reasonable to question whether activation of the transcription factor by chemopreventive agents once carcinogenesis has been initiated might serve to accelerate tumorigenesis rather than inhibit it. Most importantly, the available evidence does not support such a hypothesis. Thus in experiments using UV-irradiation (35% UVA and 65% UVB) to initiate skin carcinogenesis in SKH-1 mice over a period of 20 weeks, the subsequent topical application of an SFN-enriched broccoli sprout extract (containing 0.3 μmol SFN or 1.0 μmol SFN) for 5 days/week over 11 weeks was found to reduce significantly the tumor burden in a dose-dependent fashion [396]. In chemically initiated lung cancer in A/J mice, using two doses of vinyl carbamate given 1 week apart, the subsequent administration of the oleanane triterpenoids CDDO-ethyl amide (80 mg/kg in the diet) and CDDO-methyl ester (800 mg/kg in the diet) for 15 weeks, starting 1 week after the second injection of vinyl carbamate, modestly decreased the number of tumors and markedly inhibited their growth [397]. The vinyl carbamate lung carcinogenesis experiment has been extended more recently using the potent Nrf2 activator CDDO-Im and the weak Nrf2 activator dimethyl fumarate, and like CDDO-ethyl amide and CDDO-methyl ester, CDDO-Im was found to inhibit lung carcinogenesis but, unexpectedly, the weak Nrf2 activator dimethyl fumarate was found to increase lung carcinogenesis [398]. In the mouse breast cancer-associated gene 1 (BRCA1) genetic model of mammary carcinogenesis, using BrcaCo/Co::MMTV-Cre ::p53+/− mice, administration of CDDO-Me (50 mg/kg diet), starting at 12 weeks of age and continuing for approximately 25 weeks, inhibited constitutive phosphorylation of ErbB2, induced G0/G1 arrest, and delayed tumor development in the mice by 5.2 weeks [399]. Using the LSL-KrasG12D/+::LSL-Trp53R127H/+ ::Pdx-1-Cre genetic model of pancreatic cancer, administration of CDDO-Me (60 mg/kg in diet) after 4 weeks of age increased survival by about 3 weeks [400]. Collectively, the above findings indicate that in a variety of mouse cancer models the administration of potent Nrf2 activators, such as SFN and oleanane triterpenoids, after carcinogenesis has been initiated decreases tumor burden. However, it remains to be established why the weak Nrf2 activator dimethyl fumarate increases lung tumorigenesis and whether it involves the CNC-bZIP transcription factor.
6.1.2. Upregulation of Nrf2 in tumors
Nrf2 seems to be commonly upregulated in cancer. It was first reported that cell lines and tumors from patients with non-small-cell lung carcinoma contain somatic mutations that inactivate Keap1, thereby causing constitutive Nrf2 activation [249, 401]. Since this discovery, other somatic mutations that are believed to activate Nrf2 have been reported in lung, breast, gall-bladder, kidney, bladder, esophagus, and skin cancers [182, 249, 255, 383, 402–405]. In addition to somatic mutations in Keap1, “hot-spot” mutations in the NFE2L2 gene in regions encoding the ETGE and DLG motifs in Nrf2 that bind to Keap1 have also been reported [383, 406–410]. Recent profiling by The Cancer Genome Atlas (TCGA) research network and other consortia has documented the high frequency of mutations in these genes, with Nrf2 being estimated to be upregulated in 25–35% of lung cancers [384, 411, 412], 8% of bladder carcinomas [413], and 5% of head and neck squamous cell carcinomas [414]. Most remarkably, TCGA found that somatic mutations in Keap1 are the second most common genetic lesion in lung adenoma carcinoma and the fourth most common in lung squamous cell carcinoma, whereas gain-of-function DLG and ETGE hot-spot mutations in NFE2L2 are the sixth most common in lung squamous cell carcinoma [415]. In addition to mutations in Keap1 and NFE2L2, Nrf2 is likely to be upregulated in human KRAS mutant tumors (Fig. 14), because in mouse cancer models mutant KrasG12D drives transcriptional upregulation of the Nfe2l2 gene by both Jun–Fos and Myc [137, 236]. Moreover, Nrf2 is upregulated as a consequence of PTEN mutations [299, 416], presumably through its ability to activate the PI3K–Akt pathway and consequently inhibit GSK-3 [74].
Fig. 14.
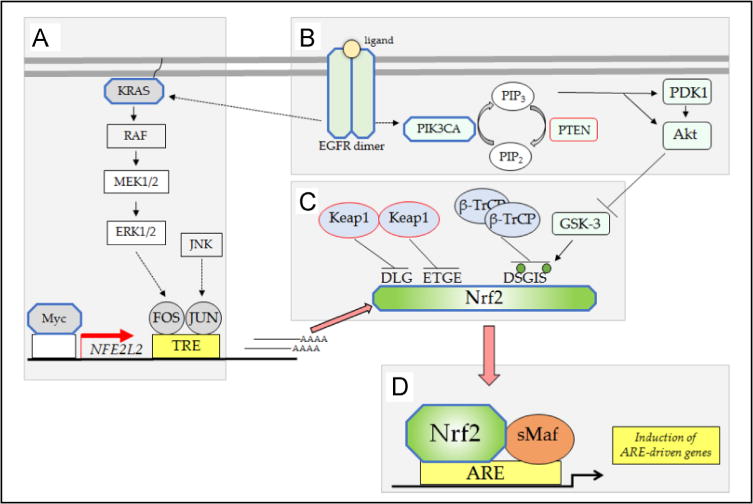
Somatic mutations that upregulate Nrf2. Mutations that result in gain of function are depicted in octagonal shapes outlined in blue, and those that result in loss of function are outlined in red. (A) Transcription of the NFE2L2 gene is increased by mutant KRAS and Myc. (B) Inhibition of GSK-3 and failure to form the DSGIS phosphodegron is likely to occur from mutations in EGFR, PIK3CA and PTEN. (C) Mutations in Keap1 or mutations in the DLG or ETGE motifs in Nrf2 result in failure of CRLKeap1 to ubiquitylate and repress Nrf2. (D) Accumulation of Nrf2 in tumors induces ARE genes, leading to increases in ROS scavenging, increases in metabolic detoxication, and efflux of endogenous electrophiles.
Other mutations that increase Nrf2 activity include fumarate hydratase (FH)-inactivating mutations, which are found in hereditary type-2 papillary renal cell carcinoma (PRCC2). These mutations cause the accumulation of fumarate, leading to the activation of ARE-driven gene expression, with loss of FH producing a gene expression signature that significantly overlaps with that observed after Keap1 knockdown [417]. Notably, addition of dimethyl fumarate was shown to cause direct stabilization of Nrf1 and Nrf2, whereas knockdown of the CNC-bZIP factors resulted in loss of fumarate-stimulated ARE-driven gene expression. Complementary reconstitution of cells with functional FH resulted in restoration of basal Nrf2 levels and decreased fumarate concentration [417, 418]. The mechanism by which fumarate affects Nrf2 upregulation is via succination of Keap1 Cys residues, which abrogates its ability to bind and ubiquitylate Nrf2 [418]. Sporadic PRCC2 has also been shown to be linked to sustained activation of Nrf2 through mutations of CUL3 and NFE2L2, which render it insensitive to Keap1-mediated repression, indicating a central role for the dysregulation of Nrf2 in PRCC2 [408].
6.1.3. Consequence of Nrf2 upregulation during tumorigenesis
The above data suggest that although the upregulation of Nrf2 in cancer seems paradoxical, it is not an inconsequential observation or artifact—the mutations seen are specifically targeted to activate and cause accumulation of Nrf2, either by increasing stability of the transcription factor (by inhibiting its primary negative regulator Keap1 or suppressing repression by β-TrCP) or by increasing transcription of NFE2L2. From this, it is apparent that in vivo regulation of Nrf2 is a careful balancing act. The evidence suggests acute activation of Nrf2 allows chemopreventive agents to confer beneficial cytoprotective effects on normal cells; however, if Nrf2 activation is high and sustained for a prolonged period it may in certain tissues and in certain contexts promote carcinogenesis [326].
Two principal hypotheses have been advanced to account for the fact that Nrf2 is upregulated in cancer. First, it has been proposed that Nrf2 upregulation increases the antioxidant capacity of tumors and thus prevents activation of apoptosis or senescence caused by high levels of ROS produced by certain oncogenes [236]. As mentioned previously, Nrf2 might influence oncogenic pathways once cancer has been initiated by supporting the survival of cells harboring particular oncogenes, such as KrasG12D[182]. Key to this proposal is the fact that upregulation of Nrf2 should prevent excessive levels of ROS from stimulating apoptosis, possibly via ASK1 signaling. Support for this hypothesis comes from the finding that knockout of the Nrf2-target gene Gclm in mice decreases GSH levels to just 20% with respect to wild-type mice and this is sufficient to prevent malignant transformation in murine tumorigenesis models, including mammary cancer produced in MMTV-PyMT mice, lymphomas produced in Pten+/− mice, and sarcomas induced by Cre recombinase in KrasLSL–G12D/+::Trp53fl/fl mice [419]. Second, Nrf2 upregulation increases cell proliferation and this is accompanied by hyperphosphorylation of retinoblastoma protein, possibly brought about by increased activity of the cyclin D1/CDK4/CDK6 complex and the cyclin E/CDK2 complex that has been speculated to be a consequence of repression of p21 expression [420]; this contrasts with the G2/M cell cycle arrest arising from hypophosphorylation of retinoblastoma protein and mislocalization of cyclin B1 and CDK1 in Nrf2-null cells [155]. It has been proposed that the increase in cell proliferation observed upon Nrf2 upregulation is supported by higher synthesis of NADPH and the redirection of glucose metabolism toward NADPH generation and anabolic pathways that are required for synthesis of macromolecules [121, 375]. Increased Nrf2 activity might also increase the supply of ATP, required for the synthesis of macromolecules, by improving mitochondrial function [149, 174], and this would be expected to contribute to the ability of cells to proliferate rapidly.
In addition to the contribution that Nrf2 might make to promotion of tumorigenesis by suppressing apoptosis and increasing cell proliferation, it is also possible that it contributes to cancer by increasing the likelihood of certain xenobiotics producing mutations. Specifically, in humans, upregulation of Nrf2 induces AKR and NQO1 enzymes that are capable of activating certain carcinogens. Thus human AKR1A1, AKR1C1, AKR1C2, and AKR1C3 are all capable of metabolically activating PAH carcinogens by their dihydrodiol dehydrogenase activity, through which they catalyze the NADP+-dependent oxidation of PAH-trans-dihydrodiols to create redox-active and electrophilic PAH ortho-quinones [421, 422]. Not only are the ortho-quinones capable of reacting with macromolecules, but they also undergo one- and two-electron enzymatic reductions, thereby creating ROS. Thus AKR1A1 and AKR1C family members can facilitate futile redox cycling of PAH ortho-quinones (Fig. 15A). Once formed, the PAH ortho-quinones can stimulate Nrf2-mediated gene induction and further exacerbate PAH activation [234, 421, 422]. This sequence of events has been demonstrated in human lung cells and shown to lead to an increase in the mutagenic lesion 8-oxo-dG [423]. Furthermore, AKR1C1 and AKR1C2 also reduce tobacco-specific nitrosamines such as nicotine-derived nitrosamine ketone (NNK) to 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol ((S)-NNAL), which is the stereoisomer that cannot be eliminated by glucuronidation [424–426]. In the absence of glucuronidation, S-NNAL represents a pool from which α-hydroxylation leading to the formation of DNA adducts can still occur (Fig. 15B). In a similar manner, human NQO1 has been identified as the major nitroreductase involved in the metabolic activation of nitroarene carcinogens, such as 3-nitobenzanthrone, present in diesel exhaust, to form DNA adducts [427, 428]. Metabolic activation also leads to the generation of ROS, suggesting that nitroarenes may stimulate their own genotoxicity by inducing NQO1 (Fig. 15C). Additionally, AKR1B10, AKR1C1, and AKR1C2 have been identified as part of a gene battery that is induced by cigarette smoke in lung epithelial and buccal cells and reduced in smokers who quit, suggesting that these genes are induced as part of a stress response to inhaled toxicants, but that an undesirable consequence of its activation could be a contribution to cancer initiation [429]. Unlike AKR1C1 and AKR1C2, AKR1B10 is a retinal reductase and converts retinal to retinol [430], thereby depriving the retinoic acid receptor of its ligand and preventing cellular differentiation. AKR1B10 can reduce NNK, the antiemetic 5-HT3 receptor agonist dolasetron, and the anticancer drug daunorubicin [431].
Fig. 15.
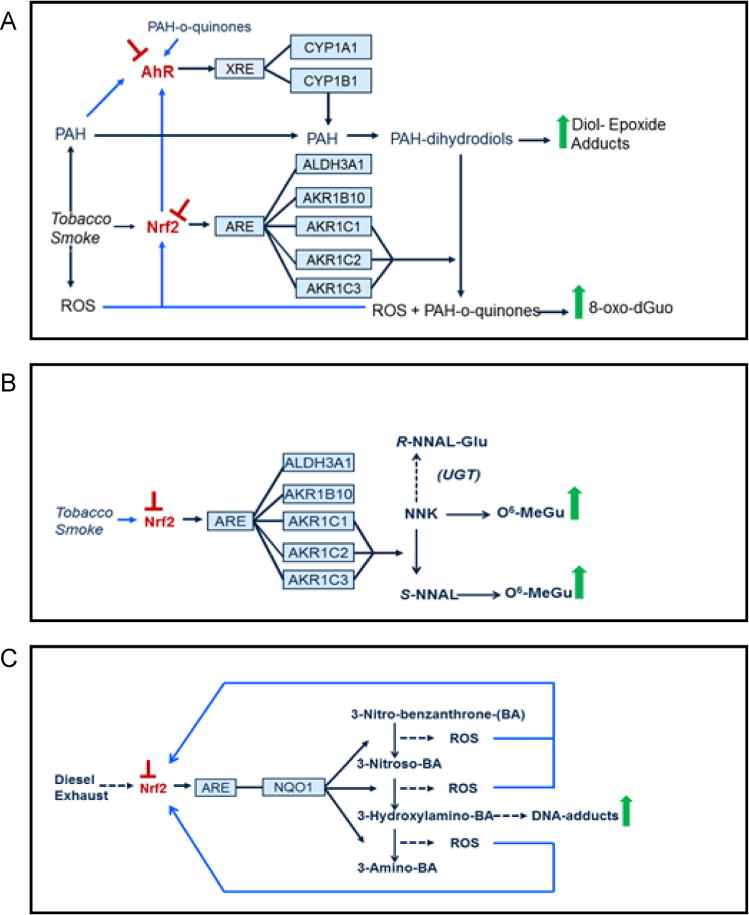
Metabolic activation of carcinogens by inducible human reductases encoded by genes that are regulated via ARE sequences. AKR1C family members and NQO1 are capable of activating carcinogens. The AhR and Nrf2 transcription factors are depicted in red letters (toward the left-hand side) with an arrow pointing to their cognate cis-elements through which they regulate their target genes; a blunted red arrow above the transcription factors indicates circumstances when it might be desirable to inhibit their activity. To the right of the enzymes (depicted in blue boxes, shown slightly left of center), reactions representing the activation of carcinogens are shown linked to individual isoenzymes, and the consequences of carcinogen activation on DNA adduct formation are presented on the far right-hand side. The blue lines leading from carcinogen metabolites and ROS toward AhR and Nrf2 refer to pathways that result in transcription factor activation. The individual pathways are described below. (A) PAHs, e.g., tobacco and environmental carcinogens, are activated by AKR1C isoenzymes through NADP+-dependent oxidation of PAH-trans-dihydrodiols to their PAH ortho-quinones. In turn, PAH ortho-quinones are unstable and spontaneously generate ROS. Furthermore, PAH ortho-quinones are ligands for the AhR, and both PAH ortho-quinones and ROS induce Nrf2-target gene expression, which further exacerbates activation of PAHs. (B) NNK is a tobacco carcinogen that is converted by AKR1C isoenzymes to S-NNAL, which cannot be glucuronidated and as a consequence provides an additional source of DNA adducts. The induction of Nrf2-target gene expression by tobacco smoke further exacerbates the conversion of NNK to S-NNAL. (C) Nitroarenes, a carcinogenic component of diesel exhaust, undergo an NQO1-catalyzed six-electron reduction to yield aminoarenes, resulting in DNA adducts and ROS formation. The generation of ROS can induce Nrf2-target gene expression, which further exacerbates nitroarene activation. It is likely that Nrf2 is activated by diesel exhaust exposure.
In the clinical setting, Nrf2 upregulation is likely to contribute to tumor cell survival. Thus, acquired resistance to chemotherapeutic drugs is a major obstacle that must be overcome during the treatment of cancer. Unfortunately, a significant number of chemotherapeutic agents are carcinogenic or mutagenic, resulting in a higher frequency of mutations in the tumor, which leads to drug resistance [432]. Many tumors with acquired drug resistance have high levels of GSH and TXN that are accompanied by overexpression of TXNRD1, GST, AKR, and NQO1 drug-metabolizing enzymes as well as several multidrug efflux pumps [433–437]. As the synthesis of GSH and the expression of some of these proteins are, at least in part, regulated by Nrf2, it seems probable that upregulation of the transcription factor may confer acquired drug resistance on tumors. Indeed, it has recently been reported by Donna Zhang and colleagues that in a KrasG12D-driven lung cancer model, cotreatment with the Nrf2 inhibitor brusatol increased the efficacy of cisplatin treatment and in so doing reduced tumor burden and increased survival [137].
In addition to the mechanisms proposed above by which induction of Nrf2-target genes might accelerate the promotion and progression of cancer, a convincing case could be argued that upregulation of Nrf2 during tumorigenesis provides cancer cells with additional diversity, possibly by allowing them to be reprogrammed toward pluripotency, which might increase their ability to evolve further. According to this view, the upregulation of Nrf2 would allow cancer cells to escape from being locked into proscribed developmental programs, in the sense of being obliged to follow a particular predetermined “epigenetic landscape,” as originally hypothesized by Waddington [438–440]. The idea that upregulation of Nrf2 could allow reprogramming of initiated cells is consistent with the evidence that the transcription factor is expressed at high levels in stem cells [156] and also that it controls self-renewal and pluripotency in human embryonic stem cells [441].
In summary, the above findings suggest that the ascending slope in the proposed U-shaped susceptibility to cancer curve that occurs at high levels of Nrf2 activity might arise as a consequence of the CNC-bZIP transcription factor’s contribution to inhibition of apoptosis, increase in cell proliferation (in part by directing metabolism toward NADPH generation and improving ATP synthesis for the synthesis of macromolecules), and activation of PAH and nitrosamine chemical carcinogens, as well as its possible influence on dedifferentiation that might allow developmental reprogramming during oncogenesis.
6.2. The interplay between Nrf2 and diabetes mellitus
DM is a metabolic disease characterized by chronic hyperglycemia and the development of associated complications, particularly in the microvasculature owing to changes in myogenic tone, leading to injury of the retina, renal glomerulus, and peripheral nerves [442, 443]. Obesity is a common risk factor for development of DM. The role Nrf2 plays in obesogenesis is likely to attenuate the initiation of diabetes; pharmacological activation of Nrf2 has been shown to inhibit weight gain and increase energy expenditure in wild-type but not Nrf2−/− mice fed on a high-fat diet [444]. Similarly, genetic activation of Nrf2 by knockdown of Keap1 in leptin-deficient obese (Lepob/ob) mice also results in a decrease in weight gain, food intake, and lipogenesis relative to wild-type animals [445]. The initial stages in the development of DM, as well as the pathogenesis of the disease itself, are thought to involve excessive production of ROS and reactive nitrogen species due to sustained hyperglycemia, which will in turn exacerbate redox dysfunction [446]. Nrf2 is thought to marshal cellular defenses against these oxidative insults, with loss of Nrf2 causing mitochondrial dysfunction, further ROS generation, insulin resistance, and diabetic complications [443, 446, 447].
Nrf2 contributes to the regulation of blood glucose content, with Nrf2-null mice shown to have lower basal insulin levels and longer periods of hyperglycemia than their wild-type counterparts, whereas mice with genetically or pharmacologically activated Nrf2 have lower blood glucose, improved insulin secretion, and improved insulin sensitivity [114, 229]. The increased levels of glucose-derived ROS also act as signaling molecules for glucose-stimulated insulin secretion (GSIS) in pancreatic β-cells. Nrf2 plays a paradoxical role in the regulation of GSIS in that its activation protects β-cells from injury by oxidative stress but in turn blunts the stimulatory signal, resulting in reduced GSIS [448]; this paradoxical role is seen clearly under conditions of metabolic stress. Lepob/ob::Keap1-knockdown mice display prolonged hyperglycemia and induced insulin resistance [445, 449], but on a high fat-diet Nrf2−/− mice have been shown to display improved insulin sensitivity relative to their wild-type counterparts [162]. The impaired insulin signaling in Lepob/ob::Keap1-knockdown mice may arise from the quenching of ROS-based signal transduction required for insulin signaling [9], by upregulation of GSH, and/or because of increased PTP1B activity [199], as a consequence of upregulation of the TXN1-TXNRD1-SRXN1 pathway.
In addition to its context-dependent effects on insulin resistance, Nrf2 has also been shown to protect against microvascular complications resulting from diabetes; in streptozotocin-induced diabetic models, Nrf2−/− mice have been found to experience more profound nephropathy, retinopathy, and cardiomyopathy as a result of oxidative stress than their wild-type counterparts [450–452]. Conversely, pharmacological activation of Nrf2 by SFN or cinnamic aldehyde is protective in these models [453–456]. A recent study has reported increased promoter DNA methylation, mRNA, and protein levels of Keap1, and corresponding decreased Nrf2 levels, in myocardial biopsies of diabetic patients [457]. Taken together, these data suggest a protective role for Nrf2 in the development and progression of DM and indicate that Nrf2-activating agents may be of value in the treatment of complications caused by the disease [453, 458].
The global changes in the levels of and sensitivity to insulin will themselves cause changes to Nrf2 signaling during DM. Insulin signaling has been shown to decrease the level of translation taking place in the cell through interactions with the mTOR pathway, which feeds into the regulation of Nrf2 protein stability (Fig. 11). Stimulation of mTOR through insulin signaling has been shown in Caenorhabditis elegans to lead to the induction of SKN-1 and DAF-16 (the mammalian orthologs of which are Nrf2 and FOXO, respectively) target genes that protect cells by diminishing metabolic stress [459]. Both mTORC1 and mTORC2 have been shown to be dysregulated during the development and progression of DM through independent signaling mechanisms; mTORC1 plays a role in the initiation of DM with inhibition of the transcriptional regulator p70S6K, an mTORC1 target, leading to glucose intolerance, loss of insulin sensitivity to glucose secretion, and hypoinsulinemia [460]. In the case of mTORC2, existing DM causes disruption to its signaling pathways. High glucose levels seen in DM can cause a loss of sirtuin-1 signaling, resulting in decreased RICTOR expression, leading to attenuated mTORC2 signaling, which in turn has downstream consequences on signaling through Akt-GSK-3 causing changes in Nrf2 protein stability.
6.3. The role of Nrf2 in neurodegenerative disease
Nrf2 status has been linked to many kinds of neurodegenerative disorders, such as Parkinson disease, multiple sclerosis, Huntington disease, Alzheimer disease, and amyotrophic lateral sclerosis (reviewed in [461, 462]). The common themes associated with these conditions are inflammation and oxidative stress; it has been shown previously that loss of Nrf2 increases sensitivity to oxidative insults and proinflammatory stimuli, indicating that Nrf2 is likely to play a significant role in many neurodegenerative conditions [60, 183, 463]. In addition to this, activators of Nrf2 such as carnosic acid have been shown to have in vivo neuroprotective function [464].
Parkinson disease (PD) is a condition characterized by deposition of Lewy bodies within the brain and progressive loss of dopaminergic neurons in the substantia nigra, leading to movement-related issues such as tremor and rigidity of movements [465]. Approximately 5–10% of Parkinson cases show a monogenic component with mutations in PARK1, PARK4, PARK5, PARK8, PARK11, and PARK13 associated with an autosomal dominant form, whereas mutations in PARK2, PARK6, and PARK7/DJ-1 are associated with an autosomal recessive form [466]. Another gene that is mutated in hereditary early-onset parkinsonism is PTEN-induced putative kinase 1 (PINK1) [467] encoding the mitochondrial serine/threonine-protein kinase, which, together with PARKIN, plays a role in mitochondrial function and quality control [468]. The remaining 90–95% of cases are considered sporadic; however, oxidative stress is believed to play a role in the development and progression of the disease. The beneficial effects of Nrf2 in PD have been shown in Drosophila, with upregulation of the CNC-bZIP factor or downregulation of one of its negative regulators, Keap1, partially restoring locomotor function lost in the PD model [469]. In parkinsonian mouse models induced by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 6-hydroxydopamine, compared to wild-type, Nrf2−/− mice showed greater loss of dopaminergic neurons in the substantia nigra [470–472] and more severe astrogliosis and microgliosis [471, 473]; comparatively, pharmacological (e.g., by 1,2-dithiole-3-thione, SFN, triterpenoids, or licochalcone E) or genetic (by astrocytic Nrf2 overexpression or Keap1 knockdown) upregulation of Nrf2 in wild-type, but not in Nrf2−/− mice, showed a protective effect against MPTP-induced neuronal damage [470, 474–483]. In addition, overexpression of Nrf2 in astrocytes has been shown to be sufficient to protect mice against MPTP-induced toxicity, suggesting that modulation of the Nrf2 pathway is a promising drug target for treatment of PD [470]. Interestingly, SFN restores the compromised mitochondrial membrane potential in PINK1-deficient cells and protects them against dopamine toxicity [484], suggesting that the role of Nrf2 in mitochondrial function [149, 174] may constitute part of the protective mechanism of pharmacological Nrf2 activators in Parkinson disease. The link between Nrf2 and Parkinson disease is also seen in humans, with evidence to suggest that variation in the Nrf2 haplotype is associated with changes in Parkinson disease risk and age of onset [485, 486].
Multiple sclerosis (MS) is a chronic autoimmune and inflammatory disease that causes damage to white matter and the spinal cord. The exact cause of MS is not known; however, it is widely accepted that dysregulation of immune cells causes damage to oligodendrocytes and axons. The overactivation of immune cells results in secondary damage through oxidative stress. In a mouse model of MS, it has been shown that Nrf2−/− mice experience an earlier onset and faster progression of the disease, in addition to greater demyelination of neurons and more robust activation of immune cells within the brain, than Nrf2+/+ mice [487]. Administration of the Nrf2 inducer dimethyl fumarate in MS models showed reduced levels of oxidative stress and preservation of nerve fiber myelination, which is lost in Nrf2−/− mice [488, 489]. The anti-inflammatory effects exhibited by Nrf2 may also be involved in its protective role in MS. To date, dimethyl fumarate that is used to treat MS is the only Nrf2 activator used in clinical practice.
7. Concluding comments and perspective
In this article we have provided an overview of the processes such as redox status, cell differentiation, ER stress, mitochondrial function, drug metabolism, inflammation, tissue regeneration, and signal transduction that are altered in Nrf2-null mice and cells. It is curious how little is understood about the processes that Nrf2 controls under normal basal conditions, possibly because Nrf2−/− mice have most frequently been used as controls in experiments designed to establish the contributions made by the transcription factor to combatting oxidative stress. It does, however, now seem clear that Nrf2−/− cells have their own phenotype, at least with regard to heightened activity of some signal transduction pathways. An important gap in current knowledge is a lack of understanding of tissue-specific effects of Nrf2, and this is of particular importance in metabolic and inflammatory disease. Shyam Biswal and colleagues have reported the generation of an Nrf2-floxed mouse [490] that will allow the tissue-specific roles of the transcription factor to be investigated.
Whereas humans lacking Nrf2 have not been reported, polymorphisms exist that entail interindividual variations in the promoter of the human NFE2L2 gene that influence its expression [491, 492]. Therefore it will be important in the future to explore whether humans that are “Nrf2-low expressors” exhibit alterations in the pathways we have described. It should, for example, be possible to subject human volunteers to physiological and metabolic challenge and examine whether the NFE2L2 genotype influences the stress and signaling pathways that are perturbed in Nrf2-null mice.
Herein we have described the mechanisms by which Nrf2 is regulated by CRLKeap1 and SCFβ-TrCP with a view to providing a better understanding of how redox status, growth factors, nutrient availability, and energy levels modulate the activity of the transcription factor and thus induction of its target genes. It is, however, unclear how these two systems are integrated and whether there is cross talk between them. Further experiments are required to determine whether CRLKeap1 and SCFβ-TrCP repress Nrf2 in different organs, cell types, or subcellular compartments, and whether either principally controls the magnitude or the duration of Nrf2 activation. It is also not known whether the β-TrCP1 and β-TrCP2 isoforms regulate Nrf2 differently, nor is it clear why Nrf2 contains two binding sites for β-TrCP1/2. Evidence exists that certain xenobiotics activate Nrf2 by antagonizing GSK-3 [299, 306], but further work is required to establish the spectrum of chemicals that work through this mechanism and how they compare with those that activate Nrf2 by inhibiting Keap1. Moreover, a complex signal transduction network exists upstream of GSK-3, including Akt, PI3K, and mTORC2, which enables growth factors and nutrients to increase Nrf2 activity. Additional studies are required to establish the extent to which xenobiotics and endobiotics control Nrf2 through this network.
We have also described the ambiguous role played by Nrf2 in cancer insofar that although in rodents it mediates the benefits of cancer-chemopreventive agents against many chemical carcinogens, it also seems to support KrasG12D-driven carcinogenesis in mouse tumor models and it is constitutively upregulated in many human tumors through somatic loss-of-function mutations in Keap1 and PTEN and gain-of-function mutations in NFE2L2 and KRAS. Future research should possibly be directed toward determining a better understanding of how constitutive Nrf2 upregulation supports tumorigenesis by suppressing proapoptotic signaling pathways, by mediating overexpression of AKR isoenzymes that promote redox cycling of PAH metabolites, and by providing an environment that facilitates the establishment of oncogenic pathways that are particularly malignant. Knowledge of the genes regulated by Nrf2 that are responsible for supporting tumorigenesis will allow alternative chemotherapeutic strategies to be devised.
Acknowledgments
This paper is dedicated to the memory of our friend and colleague Dr. Elizabeth M. Ellis (1964–2015), who pioneered research into the inducible aflatoxin-metabolizing aldo-keto reductases that are regulated by Nrf2. She is fondly remembered as an enthusiastic, accomplished, and meticulous scientist who was undeterred by technical challenges and was an outstanding teacher of graduate students; she is sorely missed. We thank Professor Stephen M. Keyse, Dr. Sudhir Chowdhry, and Dr. Ritu Sharma for illuminating discussions and for their critical comments. This article was supported in part by Cancer Research UK Grant C4909/A13786 (awarded to J.D.H.), MRC Project Grant MR/J001465/1 (awarded to J.D.H.), MRC ITTP Ph.D. Studentship Ref. 1313992 (awarded to J.D.H.), BBSRC Industrial CASE Ph.D. Studentship BB/L01405X/1 (awarded to S.T.D. and J.D.H.), BBSRC Project Grant BB/L01923X/1 (awarded to A.D.K.), and P30-ES013508 (awarded to T. M.P.).
References
- 1.Halliwell B. Biochemistry of oxidative stress. Biochem Soc Trans. 2007;35:1147–1150. doi: 10.1042/BST0351147. [DOI] [PubMed] [Google Scholar]
- 2.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 6.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, et al. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 7.Kang SW. Two axes in platelet-derived growth factor signaling: tyrosine phosphorylation and reactive oxygen species. Cell Mol Life Sci. 2007;64:533–541. doi: 10.1007/s00018-007-6437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Truong TH, Carroll KS. Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry (Moscow) 2012;51:9954–9965. doi: 10.1021/bi301441e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meng TC, Buckley DA, Galic S, Tiganis T, Tonks NK. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 10.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci USA. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boivin B, Zhang S, Arbiser JL, Zhang Z-Y, Tonks NK. A modified cysteinyl-labeling assay reveals reversible oxidation of protein tyrosine phosphatases in angiomyolipoma cells. Proc Natl Acad Sci USA. 2008;105:9959–9964. doi: 10.1073/pnas.0804336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karisch R, Fernandez M, Taylor P, Virtanen C, St-Germain JR, Jin LL, et al. Global proteomic assessment of the classical protein-tyrosine phosphatome and “redoxome”. Cell. 2011;146:826–840. doi: 10.1016/j.cell.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seki E, Brenner DA, Karin M. A Liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-β-catenin signalling through Dishevelled. Nat Cell Biol. 2006;8:501–508. doi: 10.1038/ncb1405. [DOI] [PubMed] [Google Scholar]
- 17.Rendic S, Guengerich FP. Summary of information on the effects of ionizing and non-ionizing radiation on cytochrome P450 and other drug metabolizing enzymes and transporters. Curr Drug Metab. 2012;13:787–814. doi: 10.2174/138920012800840356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 19.Gupta SC, Hevia D, Patchva S, Park B, Koh W, Aggarwal BB. Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signaling. 2012;16:1295–1322. doi: 10.1089/ars.2011.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med. 2013;62:90–101. doi: 10.1016/j.freeradbiomed.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauer G. Targeting extracellular ROS signaling of tumor cells. Anticancer Res. 2014;34:1467–1482. [PubMed] [Google Scholar]
- 22.Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008;52:S128–S138. doi: 10.1002/mnfr.200700195. [DOI] [PubMed] [Google Scholar]
- 23.Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 24.Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 25.Rogers LK, Tamura T, Rogers BJ, Welty SE, Hansen TN, Smith CV. Analyses of glutathione reductase hypomorphic mice indicate a genetic knockout. Toxicol Sci. 2004;82:367–373. doi: 10.1093/toxsci/kfh268. [DOI] [PubMed] [Google Scholar]
- 26.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 27.Bodenstein J, Follmann H. Characterization of two thioredoxins in pig heart including a new mitochondrial protein. Z Naturforsch, C J Biosci. 1991;46:270–279. doi: 10.1515/znc-1991-3-418. [DOI] [PubMed] [Google Scholar]
- 28.Bondareva AA, Capecchi MR, Iverson SV, Li Y, Lopez NI, Lucas O, et al. Effects of thioredoxin reductase-1 deletion on embryogenesis and transcriptome. Free Radic Biol Med. 2007;43:911–923. doi: 10.1016/j.freeradbiomed.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suvorova ES, Lucas O, Weisend CM, Rollins MF, Merrill GF, Capecchi MR, et al. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One. 2009;4:e6158. doi: 10.1371/journal.pone.0006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eriksson S, Prigge JR, Talago EA, Arnér ESJ, Schmidt EE. Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nat Commun. 2015;6:6479. doi: 10.1038/ncomms7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanschmann EM, Godoy JR, Berndt C, Hudemann C, Lillig CH. Thioredoxins, glutaredoxins, and peroxiredoxins–molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid Redox Signaling. 2013;19:1539–1605. doi: 10.1089/ars.2012.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perkins A, Poole LB, Karplus PA. Tuning of peroxiredoxin catalysis for various physiological roles. Biochemistry. 2014;53:7693–7705. doi: 10.1021/bi5013222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H2O2: regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signaling. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 34.Jeong W, Bae SH, Toledano MB, Rhee SG. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic Biol Med. 2012;53:447–456. doi: 10.1016/j.freeradbiomed.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 35.Soriano FX, Léveillé F, Papadia S, Higgins LG, Varley J, Baxter P, et al. Induction of sulfiredoxin expression and reduction of peroxiredoxin hyperoxidation by the neuroprotective Nrf2 activator 3H-1,2-dithiole-3-thione. J Neurochem. 2008;107:533–543. doi: 10.1111/j.1471-4159.2008.05648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agyeman AS, Chaerkady R, Shaw PG, Davidson NE, Visvanathan K, Pandey A, et al. Transcriptomic and proteomic profiling of Keap1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res Treat. 2012;132:175–187. doi: 10.1007/s10549-011-1536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40:7416–7429. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirotsu Y, Katsuoka F, Funayama R, Nagashima T, Nishida Y, Nakayama K, et al. Nrf2–MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012;40:10228–10239. doi: 10.1093/nar/gks827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung KA, Choi B, Nam CW, Song M, Kim ST, Lee JY, et al. Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol Lett. 2013;218:39–49. doi: 10.1016/j.toxlet.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 40.MacLeod AK, McMahon M, Plummer SM, Higgins LG, Penning TM, Igarashi K, et al. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the Keap1–NRF2 pathway, and not the BACH1–NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis. 2009;30:1571–1580. doi: 10.1093/carcin/bgp176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38:5718–5734. doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao X, Dinkova-Kostova AT, Talalay P. Powerful and prolonged protection of human retinal pigment epithelial cells, keratinocytes, and mouse leukemia cells against oxidative damage: the indirect antioxidant effects of sulforaphane. Proc Natl Acad Sci USA. 2001;98:15221–15226. doi: 10.1073/pnas.261572998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dahl EL, Mulcahy RT. Cell-type specific differences in glutamate cysteine ligase transcriptional regulation demonstrate independent subunit control. Toxicol Sci. 2001;61:265–272. doi: 10.1093/toxsci/61.2.265. [DOI] [PubMed] [Google Scholar]
- 44.Bonnesen C, Eggleston IM, Hayes JD. Dietary indoles and isothiocyanates that are generated from cruciferous vegetables can both stimulate apoptosis and confer protection against DNA damage in human colon cell lines. Cancer Res. 2001;61:6120–6130. [PubMed] [Google Scholar]
- 45.Dinkova-Kostova AT, Fahey JW, Talalay P. Chemical structures of inducers of nicotinamide quinone oxidoreductase 1 (NQO1) Methods Enzymol. 2004;382:423–448. doi: 10.1016/S0076-6879(04)82023-8. [DOI] [PubMed] [Google Scholar]
- 46.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 47.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 48.Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol. 2007;47:263–292. doi: 10.1146/annurev.pharmtox.47.120505.105337. [DOI] [PubMed] [Google Scholar]
- 49.Fahey JW, Zalcmann AT, Talalay P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry. 2001;56:5–51. doi: 10.1016/s0031-9422(00)00316-2. [DOI] [PubMed] [Google Scholar]
- 50.Kelleher MO, McMahon M, Eggleston IM, Dixon MJ, Taguchi K, Yamamoto M, et al. 1-Cyano-2,3-epithiopropane is a novel plant-derived chemopreventive agent which induces cytoprotective genes that afford resistance against the genotoxic α, β-unsaturated aldehyde acrolein. Carcinogenesis. 2009;30:1754–1762. doi: 10.1093/carcin/bgp182. [DOI] [PubMed] [Google Scholar]
- 51.Osburn WO, Kensler TW. Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat Res. 2008;659:31–39. doi: 10.1016/j.mrrev.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koshiba S, An R, Saito H, Wakabayashi K, Tamura A, Ishikawa T. Human ABC transporters ABCG2 (BCRP) and ABCG4. Xenobiotica. 2008;38:863–888. doi: 10.1080/00498250801986944. [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez FJ. Role of cytochromes P450 in chemical toxicity and oxidative stress: studies with CYP2E1. Mutat Res. 2005;569:101–110. doi: 10.1016/j.mrfmmm.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 54.Gibson GG, Skett P. Introduction to Drug Metabolism. 3rd. Nelson Thornes; Cheltenham: 2001. [Google Scholar]
- 55.Chen Y, Tang Y, Guo C, Wang J, Boral D, Nie D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem Pharmacol. 2012;83:1112–1126. doi: 10.1016/j.bcp.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borowski E, Bontemps-Gracz MM, Piwkowska A. Strategies for overcoming ABC-transporters-mediated multidrug resistance (MDR) of tumor cells. Acta Biochim Pol. 2005;52:609–627. [PubMed] [Google Scholar]
- 57.Deeley RG, Westlake C, Cole SPC. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev. 2006;86:849–899. doi: 10.1152/physrev.00035.2005. [DOI] [PubMed] [Google Scholar]
- 58.Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J. 2002;365:405–416. doi: 10.1042/BJ20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway: identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 60.McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, et al. The Cap “n”Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–3307. [PubMed] [Google Scholar]
- 61.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 62.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 63.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol. 1995;15:4184–4193. doi: 10.1128/mcb.15.8.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mohler J, Vani K, Leung S, Epstein A. Segmentally restricted, cephalic expression of a leucine zipper gene during Drosophila embryogenesis. Mech Dev. 1991;34:3–9. doi: 10.1016/0925-4773(91)90086-l. [DOI] [PubMed] [Google Scholar]
- 66.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katoh Y, Iida K, Kang MI, Kobayashi A, Mizukami M, Tong KI, et al. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys. 2005;433:342–350. doi: 10.1016/j.abb.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 68.Kobayashi M, Itoh K, Suzuki T, Osanai H, Nishikawa K, Katoh Y, et al. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells. 2002;7:807–820. doi: 10.1046/j.1365-2443.2002.00561.x. [DOI] [PubMed] [Google Scholar]
- 69.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem. 2006;281:24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 70.McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 71.Nioi P, Nguyen T, Sherratt PJ, Pickett CB. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol. 2005;25:10895–10906. doi: 10.1128/MCB.25.24.10895-10906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6:857–868. doi: 10.1046/j.1365-2443.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- 73.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem. 2004;279:31556–31567. doi: 10.1074/jbc.M403061200. [DOI] [PubMed] [Google Scholar]
- 74.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32:3765–3781. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol Cell Biol. 2012;32:3486–3499. doi: 10.1128/MCB.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013;73:3097–3108. doi: 10.1158/0008-5472.CAN-12-3386. [DOI] [PubMed] [Google Scholar]
- 78.Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708–722. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang XJ, Hayes JD, Henderson CJ, Wolf CR. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor α. Proc Natl Acad Sci USA. 2007;104:19589–19594. doi: 10.1073/pnas.0709483104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mignotte V, Wall L, deBoer E, Grosveld F, Romeo PH. Two tissue-specific factors bind the erythroid promoter of the human porphobilinogen deaminase gene. Nucleic Acids Res. 1989;17:37–54. doi: 10.1093/nar/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang W, Chan JY. Nrf1 Is targeted to the endoplasmic reticulum membrane by an N-terminal transmembrane domain: inhibition of nuclear translocation and transacting function. J Biol Chem. 2006;281:19676–19687. doi: 10.1074/jbc.M602802200. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Y, Crouch DH, Yamamoto M, Hayes JD. Negative regulation of the Nrf1 transcription factor by its N-terminal domain is independent of Keap1: Nrf1, but not Nrf2, is targeted to the endoplasmic reticulum. Biochem J. 2006;399:373–385. doi: 10.1042/BJ20060725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Y, Kobayashi A, Yamamoto M, Hayes JD. The Nrf3 transcription factor is a membrane-bound glycoprotein targeted to the endoplasmic reticulum through its N-terminal homology box 1 sequence. J Biol Chem. 2009;284:3195–3210. doi: 10.1074/jbc.M805337200. [DOI] [PubMed] [Google Scholar]
- 84.Shivdasani RA, Rosenblatt MF, Zucker-Franklin D, Jackson CW, Hunt P, Saris CJM, et al. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell. 1995;81:695–704. doi: 10.1016/0092-8674(95)90531-6. [DOI] [PubMed] [Google Scholar]
- 85.Chan JY, Kwong M, Lu R, Chang J, Wang B, Yen TS, et al. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998;17:1779–1787. doi: 10.1093/emboj/17.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsujita T, Peirce V, Baird L, Matsuyama Y, Takaku M, Walsh SV, et al. Transcription factor Nrf1 negatively regulates the cystine/glutamate transporter and lipid-metabolizing enzymes. Mol Cell Biol. 2014;34:3800–3816. doi: 10.1128/MCB.00110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu Z, Chen L, Leung L, Yen TSB, Lee C, Chan JY. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc Natl Acad Sci USA. 2005;102:4120–4125. doi: 10.1073/pnas.0500660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee CS, Ho DV, Chan JY. Nuclear factor-erythroid 2-related factor 1 regulates expression of proteasome genes in hepatocytes and protects against endoplasmic reticulum stress and steatosis in mice. FEBS J. 2013;280:3609–3620. doi: 10.1111/febs.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yanagawa T, Itoh K, Uwayama J, Shibata Y, Yamaguchi A, Sano T, et al. Nrf2 deficiency causes tooth decolourization due to iron transport disorder in enamel organ. Genes Cells. 2004;9:641–651. doi: 10.1111/j.1356-9597.2004.00753.x. [DOI] [PubMed] [Google Scholar]
- 91.Chevillard G, Blank V. NFE2L3 (NRF3): the Cinderella of the Cap“n”Collar transcription factors. Cell Mol Life Sci. 2011;68:3337–3348. doi: 10.1007/s00018-011-0747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Derjuga A, Gourley TS, Holm TM, Heng HHQ, Shivdasani RA, Ahmed R, et al. Complexity of CNC transcription factors as revealed by gene targeting of the Nrf3 locus. Mol Cell Biol. 2004;24:3286–3294. doi: 10.1128/MCB.24.8.3286-3294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kobayashi A, Ito E, Toki T, Kogame K, Takahashi S, Igarashi K, et al. Molecular cloning and functional characterization of a new Cap’n’ collar family transcription factor Nrf3. J Biol Chem. 1999;274:6443–6452. doi: 10.1074/jbc.274.10.6443. [DOI] [PubMed] [Google Scholar]
- 94.Braun S, Hanselmann C, Gassmann MG, auf dem Keller U, Born-Berclaz C, Chan K, et al. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol Cell Biol. 2002;22:5492–5505. doi: 10.1128/MCB.22.15.5492-5505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Knatko EV, Ibbotson SH, Zhang Y, Higgins M, Fahey JW, Talalay P, et al. Nrf2 activation protects against solar-simulated ultraviolet radiation in mice and humans. Cancer Prev Res. 2015;8:475–486. doi: 10.1158/1940-6207.CAPR-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 97.Toki T, Itoh J, Kitazawa J, Arai K, Hatakeyama K, Akasaka J, et al. Human small Maf proteins form heterodimers with CNC family transcription factors and recognize the NF-E2 motif. Oncogene. 1997;14:1901–1910. doi: 10.1038/sj.onc.1201024. [DOI] [PubMed] [Google Scholar]
- 98.Motohashi H, O’Connor T, Katsuoka F, Engel JD, Yamamoto M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294:1–12. doi: 10.1016/s0378-1119(02)00788-6. [DOI] [PubMed] [Google Scholar]
- 99.Oyake T, Itoh K, Motohashi H, Hayashi N, Hoshino H, Nishizawa M, et al. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol Cell Biol. 1996;16:6083–6095. doi: 10.1128/mcb.16.11.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol. 2005;25:8044–8051. doi: 10.1128/MCB.25.18.8044-8051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Blank V. Small Maf proteins in mammalian gene control: mere dimerization partners or dynamic transcriptional regulators? J Mol Biol. 2008;376:913–925. doi: 10.1016/j.jmb.2007.11.074. [DOI] [PubMed] [Google Scholar]
- 102.Hayes JD, Chanas SA, Henderson CJ, McMahon M, Sun C, Moffat GJ, et al. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem Soc Trans. 2000;28:33–41. doi: 10.1042/bst0280033. [DOI] [PubMed] [Google Scholar]
- 103.Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene: characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem. 1990;265:14648–14653. [PubMed] [Google Scholar]
- 104.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element: activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 105.Friling RS, Bensimon A, Tichauer Y, Daniel V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc Natl Acad Sci USA. 1990;87:6258–6262. doi: 10.1073/pnas.87.16.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Friling RS, Bergelson S, Daniel V. Two adjacent AP-1-like binding sites form the electrophile-responsive element of the murine glutathione S-transferase Ya subunit gene. Proc Natl Acad Sci USA. 1992;89:668–672. doi: 10.1073/pnas.89.2.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci USA. 1993;90:2965–2969. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Favreau LV, Pickett CB. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene: identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J Biol Chem. 1991;266:4556–4561. [PubMed] [Google Scholar]
- 109.Jaiswal AK. Human NAD(P)H:quinone oxidoreductase (NQO1) gene structure and induction by dioxin. Biochemistry. 1991;30:10647–10653. doi: 10.1021/bi00108a007. [DOI] [PubMed] [Google Scholar]
- 110.Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD (P)H: quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J. 2003;374:337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1–Nrf2 pathway. Antioxid Redox Signaling. 2010;13:1713–1748. doi: 10.1089/ars.2010.3221. [DOI] [PubMed] [Google Scholar]
- 112.Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci USA. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hourihan JM, Kenna JG, Hayes JD. The gasotransmitter hydrogen sulfide induces Nrf2-target genes by inactivating the Keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between Cys-226 and Cys-613. Antioxid Redox Signaling. 2013;19:465–481. doi: 10.1089/ars.2012.4944. [DOI] [PubMed] [Google Scholar]
- 114.Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, et al. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol. 2013;33:2996–3010. doi: 10.1128/MCB.00225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu KC, Cui JY, Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci. 2011;123:590–600. doi: 10.1093/toxsci/kfr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yates MS, Tran QT, Dolan PM, Osburn WO, Shin S, McCulloch CC, et al. Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoidtreated mice. Carcinogenesis. 2009;30:1024–1031. doi: 10.1093/carcin/bgp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hawkes HJK, Karlenius TC, Tonissen KF. Regulation of the human thioredoxin gene promoter and its key substrates: a study of functional and putative regulatory elements. Biochim Biophys Acta. 2014;1840:303–314. doi: 10.1016/j.bbagen.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 118.Kitteringham NR, Abdullah A, Walsh J, Randle L, Jenkins RE, Sison R, et al. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J Proteomics. 2010;73:1612–1631. doi: 10.1016/j.jprot.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol. 2002;22:2883–2892. doi: 10.1128/MCB.22.9.2883-2892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maher JM, Dieter MZ, Aleksunes LM, Slitt AL, Guo G, Tanaka Y, et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology. 2007;46:1597–1610. doi: 10.1002/hep.21831. [DOI] [PubMed] [Google Scholar]
- 121.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 122.Okada K, Warabi E, Sugimoto H, Horie M, Gotoh N, Tokushige K, et al. Deletion of Nrf2 leads to rapid progression of steatohepatitis in mice fed atherogenic plus high-fat diet. J Gastroenterol. 2012;48:620–632. doi: 10.1007/s00535-012-0659-z. [DOI] [PubMed] [Google Scholar]
- 123.Paek J, Lo JY, Narasimhan SD, Nguyen TN, Glover-Cutter K, Robida-Stubbs S, et al. Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 2012;16:526–537. doi: 10.1016/j.cmet.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, et al. NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol. 2007;27:7188–7197. doi: 10.1128/MCB.00915-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tanaka Y, Ikeda T, Yamamoto K, Ogawa H, Kamisako T. Dysregulated expression of fatty acid oxidation enzymes and iron-regulatory genes in livers of Nrf2-null mice. J Gastroenterol Hepatol. 2012;27:1711–1717. doi: 10.1111/j.1440-1746.2012.07180.x. [DOI] [PubMed] [Google Scholar]
- 126.Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohé R, et al. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol. 2006;35:639–650. doi: 10.1165/rcmb.2005-0325OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Banning A, Deubel S, Kluth D, Zhou Z, Brigelius-Flohé R. The GI-GPx gene is a target for Nrf2. Mol Cell Biol. 2005;25:4914–4923. doi: 10.1128/MCB.25.12.4914-4923.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Burczynski ME, Sridhar GR, Palackal NT, Penning TM. The reactive oxygen species- and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the α,β-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene. J Biol Chem. 2001;276:2890–2897. doi: 10.1074/jbc.M006655200. [DOI] [PubMed] [Google Scholar]
- 129.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, et al. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2 3 α-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3α/17β-HSD activity and cellular distribution. Mol Endocrinol. 1997;11:1971–1984. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- 131.Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Ninomiya M, et al. Identification of a principal mRNA species for human 3α-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem (Tokyo) 1998;124:940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 132.Chartoumpekis DV, Ziros PG, Psyrogiannis AI, Papavassiliou AG, Kyriazopoulou VE, Sykiotis GP, et al. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes. 2011;60:2465–2473. doi: 10.2337/db11-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004;94:609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 134.Jain A, Lamark T, Sjottem E, Bowitz Larsen K, Atesoh Awuh J, Overvatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wakabayashi N, Shin S, Slocum SL, Agoston ES, Wakabayashi J, Kwak MK, et al. Regulation of notch1 signaling by nrf2: implications for tissue regeneration. Sci Signaling. 2010;3:ra52. doi: 10.1126/scisignal.2000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li J, Lee JM, Johnson JA. Microarray analysis reveals an antioxidant responsive element-driven gene set involved in conferring protection from an oxidative stress-induced apoptosis in IMR-32 cells. J Biol Chem. 2002;277:388–394. doi: 10.1074/jbc.M109380200. [DOI] [PubMed] [Google Scholar]
- 137.Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK, et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014;74:7430–7441. doi: 10.1158/0008-5472.CAN-14-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Veliça P, Davies NJ, Rocha PP, Schrewe H, Ride JP, Bunce CM. Lack of functional and expression homology between human and mouse aldo-keto reductase 1C enzymes: implications for modelling human cancers. Mol Cancer. 2009;8:121. doi: 10.1186/1476-4598-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 140.Higgins LG, Kelleher MO, Eggleston IM, Itoh K, Yamamoto M, Hayes JD. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol Appl Pharmacol. 2009;237:267–280. doi: 10.1016/j.taap.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 141.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 143.Niso-Santano M, González-Polo RA, Bravo-San Pedro JM, Gómez-Sánchez R, Lastres-Becker I, Ortiz-Ortiz MA, et al. Activation of apoptosis signal-regulating kinase 1 is a key factor in paraquat-induced cell death: modulation by the Nrf2/Trx axis. Free Radic Biol Med. 2010;48:1370–1381. doi: 10.1016/j.freeradbiomed.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 144.Chowdhry S, Nazmy MH, Meakin PJ, Dinkova-Kostova AT, Walsh SV, Tsujita T, et al. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic Biol Med. 2010;48:357–371. doi: 10.1016/j.freeradbiomed.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 145.Zhang YKJ, Wu KC, Klaassen CD. Genetic activation of Nrf2 protects against fasting-induced oxidative stress in livers of mice. PLoS One. 2013;8:e59122. doi: 10.1371/journal.pone.0059122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Weerachayaphorn J, Mennone A, Soroka CJ, Harry K, Hagey LR, Kensler TW, et al. Nuclear factor-E2-related factor 2 is a major determinant of bile acid homeostasis in the liver and intestine. Am J Physiol Gastrointest Liver Physiol. 2012;302:G925–G936. doi: 10.1152/ajpgi.00263.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Reddy NM, Kleeberger SR, Cho HY, Yamamoto M, Kensler TW, Biswal S, et al. Deficiency in Nrf2-GSH signaling impairs type II cell growth and enhances sensitivity to oxidants. Am J Respir Cell Mol Biol. 2007;37:3–8. doi: 10.1165/rcmb.2007-0004RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.McDonald JT, Kim K, Norris AJ, Vlashi E, Phillips TM, Lagadec C, et al. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010;70:8886–8895. doi: 10.1158/0008-5472.CAN-10-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Holmström KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open. 2013;2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.He X, Kan H, Cai L, Ma Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol. 2009;46:47–58. doi: 10.1016/j.yjmcc.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 151.Hyeon S, Lee H, Yang Y, Jeong W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic Biol Med. 2013;65:789–799. doi: 10.1016/j.freeradbiomed.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 152.Yeang HXA, Hamdam JM, Al-Huseini LMA, Sethu S, Djouhri L, Walsh J, et al. Loss of transcription factor nuclear factor-erythroid 2 (NF-E2) p45-related factor-2 (Nrf2) leads to dysregulation of immune functions, redox homeostasis, and intracellular signaling in dendritic cells. J Biol Chem. 2012;287:10556–10564. doi: 10.1074/jbc.M111.322420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Goettsch C, Goettsch W, Brux M, Haschke C, Brunssen C, Muller G, et al. Arterial flow reduces oxidative stress via an antioxidant response element and Oct-1 binding site within the NADPH oxidase 4 promoter in endothelial cells. Basic Res Cardiol. 2011;106:551–561. doi: 10.1007/s00395-011-0170-3. [DOI] [PubMed] [Google Scholar]
- 154.Kovac S, Angelova PR, Holmström KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. 2015;1850:794–801. doi: 10.1016/j.bbagen.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Reddy NM, Kleeberger SR, Bream JH, Fallon PG, Kensler TW, Yamamoto M, et al. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene. 2008;27:5821–5832. doi: 10.1038/onc.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011;8:188–199. doi: 10.1016/j.stem.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tsai JJ, Dudakov JA, Takahashi K, Shieh JH, Velardi E, Holland AM, et al. Nrf2 regulates haematopoietic stem cell function. Nat Cell Biol. 2013;15:309–316. doi: 10.1038/ncb2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Paul MK, Bisht B, Darmawan DO, Chiou R, Ha VL, Wallace WD, et al. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent Notch signaling. Cell Stem Cell. 2014;15:199–214. doi: 10.1016/j.stem.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bell KFS, Al-Mubarak B, Martel MA, McKay S, Wheelan N, Hasel P, et al. Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat Commun. 2015;6:7066. doi: 10.1038/ncomms8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 161.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 162.Meakin PJ, Chowdhry S, Sharma RS, Ashford FB, Walsh SV, McCrimmon RJ, et al. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes, but not with insulin resistance. Mol Cell Biol. 2014;34:3305–3320. doi: 10.1128/MCB.00677-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279:20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 164.Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- 165.Sevier CS, Qu H, Heldman N, Gross E, Fass D, Kaiser CA. Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell. 2007;129:333–344. doi: 10.1016/j.cell.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 166.Appenzeller-Herzog C, Ellgaard L. In vivo reduction-oxidation state of protein disulfide isomerase: the two active sites independently occur in the reduced and oxidized forms. Antioxid Redox Signaling. 2008;10:55–64. doi: 10.1089/ars.2007.1837. [DOI] [PubMed] [Google Scholar]
- 167.Baker KM, Chakravarthi S, Langton KP, Sheppard AM, Lu H, Bulleid NJ. Low reduction potential of Ero1alpha regulatory disulphides ensures tight control of substrate oxidation. EMBO J. 2008;27:2988–2997. doi: 10.1038/emboj.2008.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sevier CS, Kaiser CA. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta. 2008;1783:549–556. doi: 10.1016/j.bbamcr.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 169.Kim S, Sideris DP, Sevier CS, Kaiser CA. Balanced Ero1 activation and in-activation establishes ER redox homeostasis. J Cell Biol. 2012;196:713–725. doi: 10.1083/jcb.201110090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kojer K, Riemer J. Balancing oxidative protein folding: The influences of reducing pathways on disulfide bond formation. Biochim Biophys Acta BBA -Proteins Proteomics. 2014;1844:1383–1390. doi: 10.1016/j.bbapap.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 171.Delaunay-Moisan A, Appenzeller-Herzog C. The antioxidant machinery of the endoplasmic reticulum: protection and signaling. Free Radic Biol Med. 2015;83:341–351. doi: 10.1016/j.freeradbiomed.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 172.Chakravarthi S, Bulleid NJ. Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J Biol Chem. 2004;279:39872–39879. doi: 10.1074/jbc.M406912200. [DOI] [PubMed] [Google Scholar]
- 173.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, et al. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci USA. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ludtmann MHR, Angelova PR, Zhang Y, Abramov AY, Dinkova-Kostova AT. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem J. 2014;457:415–424. doi: 10.1042/BJ20130863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 176.Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 2006;66:8293–8296. doi: 10.1158/0008-5472.CAN-06-0300. [DOI] [PubMed] [Google Scholar]
- 177.Becks L, Prince M, Burson H, Christophe C, Broadway M, Itoh K, et al. Aggressive mammary carcinoma progression in Nrf2 knockout mice treated with 7,12-dimethylbenz[a]anthracene. BMC, Cancer. 2010;10:540. doi: 10.1186/1471-2407-10-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Higgins LG, Hayes JD. Mechanisms of induction of cytosolic and microsomal glutathione transferase (GST) genes by xenobiotics and pro-inflammatory agents. Drug Metab Rev. 2011;43:92–137. doi: 10.3109/03602532.2011.567391. [DOI] [PubMed] [Google Scholar]
- 179.Dinkova-Kostova AT, Talalay P. Persuasive evidence that quinone reductase type 1 (DT diaphorase) protects cells against the toxicity of electrophiles and reactive forms of oxygen. Free Radic Biol Med. 2000;29:231–240. doi: 10.1016/s0891-5849(00)00300-2. [DOI] [PubMed] [Google Scholar]
- 180.Nioi P, Hayes JD. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helixloop-helix transcription factors. Mutat Res. 2004;555:149–171. doi: 10.1016/j.mrfmmm.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 181.Miura T, Shinkai Y, Jiang HY, Iwamoto N, Sumi D, Taguchi K, et al. Initial response and cellular protection through the Keap1/Nrf2 system during the exposure of primary mouse hepatocytes to 1,2-naphthoquinone. Chem Res Toxicol. 2011;24:559–567. doi: 10.1021/tx100427p. [DOI] [PubMed] [Google Scholar]
- 182.Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158–4168. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 183.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Thimmulappa RK. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cho HY, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, et al. Nrf2-regulated PPARγ expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med. 2010;182:170–182. doi: 10.1164/rccm.200907-1047OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 187.Nel A, Xia T, Mädler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- 188.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Storz P, Döppler H, Toker A. Protein kinase Cδ selectively regulates protein kinase D-dependent activation of NF-κB in oxidative stress signaling. Mol Cell Biol. 2004;24:2614–2626. doi: 10.1128/MCB.24.7.2614-2626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Storz P, Toker A. Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J. 2003;22:109–120. doi: 10.1093/emboj/cdg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Waldron RT, Rozengurt E. Oxidative stress induces protein kinase D activation in intact cells: involvement of Src and dependence on protein kinase C. J, Biol Chem. 2000;275:17114–17121. doi: 10.1074/jbc.M908959199. [DOI] [PubMed] [Google Scholar]
- 192.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y, et al. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Δ12,14-prostaglandin J2. Mol Cell Biol. 2004;24:36–45. doi: 10.1128/MCB.24.1.36-45.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci. 2011;32:82–89. doi: 10.1016/j.tips.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 196.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 197.Kwon J, Shatynski KE, Chen H, Morand S, de Deken X, Miot F, et al. The nonphagocytic NADPH oxidase Duox1 mediates a positive feedback loop during T cell receptor signaling. Sci Signaling. 2010;3:ra59. doi: 10.1126/scisignal.2000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Seo JH, Ahn Y, Lee S-R, Yeol Yeo C, Chung Hur K. The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol Biol Cell. 2005;16:348–357. doi: 10.1091/mbc.E04-05-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dagnell M, Frijhoff J, Pader I, Augsten M, Boivin B, Xu J, et al. Selective activation of oxidized PTP1B by the thioredoxin system modulates PDGF-β receptor tyrosine kinase signaling. Proc Natl Acad Sci USA. 2013;110:13398–13403. doi: 10.1073/pnas.1302891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Colicelli J. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signaling. 2010;3:re6–re6. doi: 10.1126/scisignal.3139re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wang JYJ. The capable ABL: what is its biological function? Mol Cell Biol. 2014;34:1188–1197. doi: 10.1128/MCB.01454-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Alvarez AR, Sandoval PC, Leal NR, Castro PU, Kosik KS. Activation of the neuronal c-Abl tyrosine kinase by amyloid-β-peptide and reactive oxygen species. Neurobiol Dis. 2004;17:326–336. doi: 10.1016/j.nbd.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 203.Klein A, Maldonado C, Vargas LM, Gonzalez M, Robledo F, Perez de Arce K, et al. Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann-Pick type C neurons. Neurobiol Dis. 2011;41:209–218. doi: 10.1016/j.nbd.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 204.Wen ST, Etten RAV. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997;11:2456–2467. doi: 10.1101/gad.11.19.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Takeuchi K, Morizane Y, Kamami-Levy C, Suzuki J, Kayama M, Cai W, et al. AMP-dependent kinase inhibits oxidative stress-induced caveolin-1 phosphorylation and endocytosis by suppressing the dissociation between c-Abl and Prdx1 proteins in endothelial cells. J Biol Chem. 2013;288:20581–20591. doi: 10.1074/jbc.M113.460832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Cao C, Leng Y, Huang W, Liu X, Kufe D. Glutathione peroxidase 1 is regulated by the c-Abl and Arg tyrosine kinases. J Biol Chem. 2003;278:39609–39614. doi: 10.1074/jbc.M305770200. [DOI] [PubMed] [Google Scholar]
- 207.Cao C, Leng Y, Liu X, Yi Y, Li P, Kufe D. Catalase is regulated by ubiquitination and proteosomal degradation: role of the c-Abl and Arg tyrosine kinases. Biochemistry. 2003;42:10348–10353. doi: 10.1021/bi035023f. [DOI] [PubMed] [Google Scholar]
- 208.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tobiume K, Saitoh M, Ichijo H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J Cell Physiol. 2002;191:95–104. doi: 10.1002/jcp.10080. [DOI] [PubMed] [Google Scholar]
- 210.Fujino G, Noguchi T, Matsuzawa A, Yamauchi S, Saitoh M, Takeda K, et al. Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol Cell Biol. 2007;27:8152–8163. doi: 10.1128/MCB.00227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Takeda K, Noguchi T, Naguro I, Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu Rev Pharmacol Toxicol. 2008;48:199–225. doi: 10.1146/annurev.pharmtox.48.113006.094606. [DOI] [PubMed] [Google Scholar]
- 212.Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, et al. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci USA. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Cosentino-Gomes D, Rocco-Machado N, Meyer-Fernandes JR. Cell signaling through protein kinase C oxidation and activation. Int J Mol Sci. 2012;13:10697–10721. doi: 10.3390/ijms130910697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, et al. Phosphorylation sites of protein kinase C δ in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc Natl Acad Sci USA. 2001;98:6587–6592. doi: 10.1073/pnas.111158798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 216.Rosse C, Linch M, Kermorgant S, Cameron AJM, Boeckeler K, Parker PJ. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol. 2010;11:103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- 217.Tan SL, Parker PJ. Emerging and diverse roles of protein kinase C in immune cell signalling. Biochem J. 2003;376:545. doi: 10.1042/BJ20031406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.El-Benna J, Dang PMC, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med. 2009;41:217–225. doi: 10.3858/emm.2009.41.4.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 220.Lv P, Miao SB, Shu YN, Dong LH, Liu G, Xie XL, et al. Phosphorylation of smooth muscle 22α facilitates angiotensin II-induced ROS production via activation of the PKCδ-P47phox axis through release of PKCδ and actin dynamics and is associated with hypertrophy and hyperplasia of vascular smooth muscle cells in vitro and in vivo. Circ Res. 2012;111:697–707. doi: 10.1161/CIRCRESAHA.112.272013. [DOI] [PubMed] [Google Scholar]
- 221.Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–44682. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 222.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 223.Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am J Physiol Cell Physiol. 2003;285:C334–C342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- 224.Rains JL, Jain SK. Oxidative stress, insulin signalling and diabetes. Free Radic Biol Med. 2011;50:567–575. doi: 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bashan N, Kovsan J, Kachko I, Ovadia H, Rudich A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol Rev. 2009;89:27–71. doi: 10.1152/physrev.00014.2008. [DOI] [PubMed] [Google Scholar]
- 226.Meher AK, Sharma PR, Lira VA, Yamamoto M, Kensler TW, Yan Z, et al. Nrf2 deficiency in myeloid cells is not sufficient to protect mice from high-fat diet-induced adipose tissue inflammation and insulin resistance. Free Radic Biol Med. 2012;52:1708–1715. doi: 10.1016/j.freeradbiomed.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhang YKJ, Wu KC, Liu J, Klaassen CD. Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet. Toxicol Appl Pharmacol. 2012;264:305–314. doi: 10.1016/j.taap.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Aleksunes LM, Reisman SA, Yeager RL, Goedken MJ, Klaassen CD. Nuclear factor erythroid 2-related factor 2 deletion impairs glucose tolerance and exacerbates hyperglycemia in type 1 diabetic mice. J Pharmacol Exp Ther. 2010;333:140–151. doi: 10.1124/jpet.109.162271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Denison MS, Fisher JM, Whitlock JP. Protein-DNA interactions at recognition sites for the dioxin-Ah receptor complex. J Biol Chem. 1989;264:16478–16482. [PubMed] [Google Scholar]
- 231.Denison MS, Fisher JM, Whitlock JP. The DNA recognition site for the dioxin-Ah receptor complex: nucleotide sequence and functional analysis. J Biol Chem. 1988;263:17221–17224. [PubMed] [Google Scholar]
- 232.Ma Q, Kinneer K, Bi Y, Chan JY, Kan YW. Induction of murine NAD(P)H: quinone oxidoreductase by 2,3,7,8-tetrachlorodibenzo-p-dioxin requires the CNC (cap “n” collar) basic leucine zipper transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2): cross-interaction between AhR (aryl hydrocarbon receptor) and Nrf2 signal transduction. Biochem J. 2004;377:205–213. doi: 10.1042/BJ20031123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem. 2005;280:20340–20348. doi: 10.1074/jbc.M412081200. [DOI] [PubMed] [Google Scholar]
- 234.Burczynski ME, Lin HK, Penning TM. Isoform-specific induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: implications for the alternative pathway of PAH activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res. 1999;59:607–614. [PubMed] [Google Scholar]
- 235.Burczynski ME, Penning TM. Genotoxic polycyclic aromatic hydrocarbon ortho-quinones generated by aldo-keto reductases induce CYP1A1 via nuclear translocation of the aryl hydrocarbon receptor. Cancer Res. 2000;60:908–915. [PubMed] [Google Scholar]
- 236.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Rushworth SA, Zaitseva L, Murray MY, Shah NM, Bowles KM, MacEwan DJ. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood. 2012;120:5188–5198. doi: 10.1182/blood-2012-04-422121. [DOI] [PubMed] [Google Scholar]
- 238.Sanderson LM, Boekschoten MV, Desvergne B, Müller M, Kersten S. Transcriptional profiling reveals divergent roles of PPARα and PPARβ/δ in regulation of gene expression in mouse liver. Physiol Genomics. 2010;41:42–52. doi: 10.1152/physiolgenomics.00127.2009. [DOI] [PubMed] [Google Scholar]
- 239.Watai Y, Kobayashi A, Nagase H, Mizukami M, McEvoy J, Singer JD, et al. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells. 2007;12:1163–1178. doi: 10.1111/j.1365-2443.2007.01118.x. [DOI] [PubMed] [Google Scholar]
- 240.Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci USA. 2004;101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 242.Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 246.Devling TWP, Lindsay CD, McLellan LI, McMahon M, Hayes JD. Utility of siRNA against Keap1 as a strategy to stimulate a cancer chemopreventive phenotype. Proc Natl Acad Sci USA. 2005;102:7280–7285. doi: 10.1073/pnas.0501475102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Canning P, Cooper CDO, Krojer T, Murray JW, Pike ACW, Chaikuad A, et al. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J Biol Chem. 2013;288:7803–7814. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Ogura T, Tong KI, Mio K, Maruyama Y, Kurokawa H, Sato C, et al. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc Natl Acad Sci USA. 2010;107:2842–2847. doi: 10.1073/pnas.0914036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 250.Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, et al. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves Keap1 disulfide formation. J Biol Chem. 2010;285:8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, et al. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol. 2008;29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.McMahon M, Lamont DJ, Beattie KA, Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci USA. 2010;107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Fujii S, Sawa T, Ihara H, Tong KI, Ida T, Okamoto T, et al. The critical role of nitric oxide signaling, via protein S-guanylation and nitrated cyclic GMP, in the antioxidant adaptive response. J Biol Chem. 2010;285:23970–23984. doi: 10.1074/jbc.M110.145441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Nioi P, Nguyen T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem Biophys Res Commun. 2007;362:816–821. doi: 10.1016/j.bbrc.2007.08.051. [DOI] [PubMed] [Google Scholar]
- 256.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 257.Hansen RE, Roth D, Winther JR. Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci USA. 2009;106:422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Salsbury FR, Knutson ST, Poole LB, Fetrow JS. Functional site profiling and electrostatic analysis of cysteines modifiable to cysteine sulfenic acid. Protein Sci. 2008;17:299–312. doi: 10.1110/ps.073096508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Fukutomi T, Takagi K, Mizushima T, Ohuchi N, Yamamoto M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol Cell Biol. 2014;34:832–846. doi: 10.1128/MCB.01191-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol. 2006;26:2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2004;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Baird L, Llères D, Swift S, Dinkova-Kostova AT. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc Natl Acad Sci USA. 2013;110:15259–15264. doi: 10.1073/pnas.1305687110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci USA. 2005;102:10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Li Y, Paonessa JD, Zhang Y. Mechanism of chemical activation of Nrf2. PLoS One. 2012;7:e35122. doi: 10.1371/journal.pone.0035122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Takaya K, Suzuki T, Motohashi H, Onodera K, Satomi S, Kensler TW, et al. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic Biol Med. 2012;53:817–827. doi: 10.1016/j.freeradbiomed.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Burk RF, Hill KE, Nakayama A, Mostert V, Levander XA, Motley AK, et al. Selenium deficiency activates mouse liver Nrf2-ARE but vitamin E deficiency does not. Free Radic Biol Med. 2008;44:1617–1623. doi: 10.1016/j.freeradbiomed.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Carlson BA, Novoselov SV, Kumaraswamy E, Lee BJ, Anver MR, Gladyshev VN, et al. Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J Biol Chem. 2004;279:8011–8017. doi: 10.1074/jbc.M310470200. [DOI] [PubMed] [Google Scholar]
- 271.McLeod R, Ellis EM, Arthur JR, Neal GE, Judah DJ, Manson MM, et al. Protection conferred by selenium deficiency against aflatoxin B1 in the rat is associated with the hepatic expression of an aldo-keto reductase and a glutathione S-transferase subunit that metabolize the mycotoxin. Cancer Res. 1997;57:4257–4266. [PubMed] [Google Scholar]
- 272.Mostert V, Hill KE, Ferris CD, Burk RF. Selective induction of liver parenchymal cell heme oxygenase-1 in selenium-deficient rats. Biol Chem. 2003;384:681–687. doi: 10.1515/BC.2003.076. [DOI] [PubMed] [Google Scholar]
- 273.Suzuki T, Kelly VP, Motohashi H, Nakajima O, Takahashi S, Nishimura S, et al. Deletion of the selenocysteine tRNA gene in macrophages and liver results in compensatory gene induction of cytoprotective enzymes by Nrf2. J Biol Chem. 2008;283:2021–2030. doi: 10.1074/jbc.M708352200. [DOI] [PubMed] [Google Scholar]
- 274.Patterson AD, Carlson BA, Li F, Bonzo JA, Yoo MH, Krausz KW, et al. Disruption of thioredoxin reductase 1 protects mice from acute acetaminophen-induced hepatotoxicity through enhanced NRF2 activity. Chem Res Toxicol. 2013;26:1088–1096. doi: 10.1021/tx4001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 276.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A non-canonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Ichimura Y, Waguri S, Sou Y, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 278.Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013;17:73–84. doi: 10.1016/j.cmet.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 279.Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, et al. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol. 2013;33:2436–2446. doi: 10.1128/MCB.01748-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T, et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci USA. 2012;109:13561–13566. doi: 10.1073/pnas.1121572109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lee OH, Jain AK, Papusha V, Jaiswal AK. An auto-regulatory loop between stress sensors INrf2 and Nrf2 controls their cellular abundance. J Biol Chem. 2007;282:36412–36420. doi: 10.1074/jbc.M706517200. [DOI] [PubMed] [Google Scholar]
- 282.Lo SC, Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem. 2006;281:37893–37903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- 283.Jiang Z-Y, Chu H-X, Xi M-Y, Yang T-T, Jia J-M, Huang J-J, et al. Insight into the intermolecular recognition mechanism between Keap1 and IKKβ combining homology modelling, protein-protein docking, molecular dynamics simulations and virtual alanine mutation. PLoS One. 2013;8:e75076. doi: 10.1371/journal.pone.0075076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Lee D-F, Kuo H-P, Liu M, Chou C-K, Xia W, Du Y, et al. Keap1 E3 ligase-mediated downregulation of NF-κB signaling by targeting IKKβ. Mol Cell. 2009;36:131–140. doi: 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Takeda K, Komuro Y, Hayakawa T, Oguchi H, Ishida Y, Murakami S, et al. Mitochondrial phosphoglycerate mutase 5 uses alternate catalytic activity as a protein serine/threonine phosphatase to activate ASK1. Proc Natl Acad Sci USA. 2009;106:12301–12305. doi: 10.1073/pnas.0901823106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Lo SC, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res. 2008;314:1789–1803. doi: 10.1016/j.yexcr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Gamble C, McIntosh K, Scott R, Ho KH, Plevin R, Paul A. Inhibitory κB Kinases as targets for pharmacological regulation. Br J Pharmacol. 2012;165:802–819. doi: 10.1111/j.1476-5381.2011.01608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 289.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 290.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 291.Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-β-catenin complex: destruction motif binding and lysine specificity of the SCF(β-TrCP1) ubiquitin ligase. Mol Cell. 2003;11:1445–1456. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- 292.Putters J, Slotman JA, Gerlach JP, Strous GJ. Specificity, location and function of βTrCP isoforms and their splice variants. Cell Signalling. 2011;23:641–647. doi: 10.1016/j.cellsig.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 293.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 294.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, et al. Crystal structure of glycogen synthase kinase 3β: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 296.Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- 297.ter Haar E, Coll JT, Austen DA, Hsiao H-M, Swenson L, Jain J. Structure of GSK3β reveals a primed phosphorylation mechanism. Nat Struct Mol Biol. 2001;8:593–596. doi: 10.1038/89624. [DOI] [PubMed] [Google Scholar]
- 298.Wu G, He X. Threonine 41 in β-catenin serves as a key phosphorylation relay residue in β-catenin degradation. Biochemistry. 2006;45:5319–5323. doi: 10.1021/bi0601149. [DOI] [PubMed] [Google Scholar]
- 299.Rojo AI, Rada P, Mendiola M, Ortega-Molina A, Wojdyla K, Rogowska-Wresinska A, et al. The PTEN/NRF2 axis promotes human carcinogenesis. Antioxid Redox Signaling. 2014;21:2498–2514. doi: 10.1089/ars.2014.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851. doi: 10.1074/jbc.M513737200. [DOI] [PubMed] [Google Scholar]
- 301.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JPY. DJ-1, a cancer-and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci USA. 2006;103:15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Bitar MS, Liu C, Ziaei A, Chen Y, Schmedt T, Jurkunas UV. Decline in DJ-1 and decreased nuclear translocation of Nrf2 in Fuchs endothelial corneal dystrophy. Invest Ophthalmol Visual Sci. 2012;53:5806–5813. doi: 10.1167/iovs.12-10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Aleyasin H, Rousseaux MWC, Marcogliese PC, Hewitt SJ, Irrcher I, Joselin AP, et al. DJ-1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc Natl Acad Sci USA. 2010;107:3186–3191. doi: 10.1073/pnas.0914876107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell. 2005;7:263–273. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 305.Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Hayes JD, Chowdhry S, Dinkova-Kostova AT, Sutherland C. Dual regulation of transcription factor Nrf2 by Keap1 and the combined actions of β-TrCP and GSK-3. Biochem Soc Trans. 2015;43:611–620. doi: 10.1042/BST20150011. [DOI] [PubMed] [Google Scholar]
- 307.Rada P, Rojo AI, Offergeld A, Feng GJ, Velasco-Martín JP, González-Sancho JM, et al. WNT-3A regulates an Axin1/NRF2 complex that regulates antioxidant metabolism in hepatocytes. Antioxid Redox Signaling. 2015;22:555–571. doi: 10.1089/ars.2014.6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Jain AK, Jaiswal AK. GSK-3β acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282:16502–16510. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- 309.Kaspar JW, Jaiswal AK. Tyrosine phosphorylation controls nuclear export of Fyn, allowing Nrf2 activation of cytoprotective gene expression. FASEB J. 2011;25:1076–1087. doi: 10.1096/fj.10-171553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Han D, Li SJ, Zhu YT, Liu L, Li MX. LKB1/AMPK/mTOR signaling pathway in non-small-cell lung cancer. Asian Pac J Cancer Prev. 2013;14:4033–4039. doi: 10.7314/apjcp.2013.14.7.4033. [DOI] [PubMed] [Google Scholar]
- 311.Hardie DG. AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annu Rev Nutr. 2014;34:31–55. doi: 10.1146/annurev-nutr-071812-161148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Hardie DG. AMPK—sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939–952. doi: 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008;57:945–957. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- 314.Eyre TA, Collins GP, Goldstone AH, Cwynarski K. Time now to TORC the TORC? New developments in mTOR pathway inhibition in lymphoid malignancies. Br J Haematol. 2014;166:336–351. doi: 10.1111/bjh.12945. [DOI] [PubMed] [Google Scholar]
- 315.Masui K, Cavenee WK, Mischel PS. mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol Metab. 2014;25:364–373. doi: 10.1016/j.tem.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Zarogoulidis P, Lampaki S, Turner J, Huang H, Kakolyris S, Syrigos K, et al. mTOR pathway: a current, up-to-date mini-review. Oncol Lett. 2014;8:2367–2370. doi: 10.3892/ol.2014.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 318.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 320.Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22:169–176. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–921. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kang HJ, Hong YB, Kim HJ, Bae I. CR6-interacting factor 1 (CRIF1) regulates NF-E2-related factor 2 (NRF2) protein stability by proteasome-mediated degradation. J Biol Chem. 2010;285:21258–21268. doi: 10.1074/jbc.M109.084590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Fishel ML, Wu X, Devlin CM, Logsdon DP, Jiang Y, Luo M, et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J Biol Chem. 2014;290:3057–3068. doi: 10.1074/jbc.M114.621995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Baba K, Morimoto H, Imaoka S. Seven in absentia homolog 2 (Siah2) is a regulator of NF-E2-related factor 2 (Nrf2) J Biol Chem. 2013;288:18393–18405. doi: 10.1074/jbc.M112.438762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Malloy MT, McIntosh DJ, Walters TS, Flores A, Goodwin JS, Arinze IJ. Trafficking of the transcription factor Nrf2 to promyelocytic leukemia-nuclear bodies: implications for degradation of NRF2 in the nucleus. J Biol Chem. 2013;288:14569–14583. doi: 10.1074/jbc.M112.437392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–99. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Maher J, Yamamoto M. The rise of antioxidant signaling—the evolution and hormetic actions of Nrf2. Toxicol Appl Pharmacol. 2010;244:4–15. doi: 10.1016/j.taap.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 328.Zucker SN, Fink EE, Bagati A, Mannava S, Bianchi-Smiraglia A, Bogner PN, et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol Cell. 2014;53:916–928. doi: 10.1016/j.molcel.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Brewer AC, Mustafi SB, Murray TVA, Rajasekaran NS, Benjamin IJ. Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid Redox Signaling. 2013;18:1114–1127. doi: 10.1089/ars.2012.4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kannan S, Muthusamy VR, Whitehead KJ, Wang L, Gomes AV, Litwin SE, et al. Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc Res. 2013;100:63–73. doi: 10.1093/cvr/cvt150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Rajasekaran NS, Varadharaj S, Khanderao GD, Davidson CJ, Kannan S, Firpo MA, et al. Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxid Redox Signaling. 2011;14:957–971. doi: 10.1089/ars.2010.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, et al. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS One. 3(2008) doi: 10.1371/journal.pone.0003791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Dinkova-Kostova AT, Talalay P, Sharkey J, Zhang Y, Holtzclaw WD, Wang XJ, et al. An exceptionally potent inducer of cytoprotective enzymes: elucidation of the structural features that determine inducer potency and reactivity with Keap1. J Biol Chem. 2010;285:33747–33755. doi: 10.1074/jbc.M110.163485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol. 2003;23:8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Osburn WO, Yates MS, Dolan PD, Liby KT, Sporn MB, Taguchi K, et al. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol Sci. 2008;104:218–227. doi: 10.1093/toxsci/kfn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Okawa H, Motohashi H, Kobayashi A, Aburatani H, Kensler TW, Yamamoto M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys, Res Commun. 2006;339:79–88. doi: 10.1016/j.bbrc.2005.10.185. [DOI] [PubMed] [Google Scholar]
- 338.Jiang T, Chen N, Zhao F, Wang XJ, Kong B, Zheng W, et al. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 2010;70:5486–5496. doi: 10.1158/0008-5472.CAN-10-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J, et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008;68:7975–7984. doi: 10.1158/0008-5472.CAN-08-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Kim YC, Yamaguchi Y, Kondo N, Masutani H, Yodoi J. Thioredoxin-dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene. 2003;22:1860–1865. doi: 10.1038/sj.onc.1206369. [DOI] [PubMed] [Google Scholar]
- 341.Tanito M, Masutani H, Kim YC, Nishikawa M, Ohira A, Yodoi J. Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in mice. Invest, Ophthalmol Visual Sci. 2005;46:979–987. doi: 10.1167/iovs.04-1120. [DOI] [PubMed] [Google Scholar]
- 342.Harvey CJ, Thimmulappa RK, Singh A, Blake DJ, Ling G, Wakabayashi N, et al. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic Biol Med. 2009;46:443–453. doi: 10.1016/j.freeradbiomed.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Varma S, Chandrasekaran K, Kovtun S. Sulforaphane-induced transcription of thioredoxin reductase in lens: possible significance against cataract formation. Clin Ophthalmol. 2013;2013:2091–2098. doi: 10.2147/OPTH.S52678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Wattenberg LW. Chemoprevention of cancer. Cancer Res. 1985;45:1–8. [PubMed] [Google Scholar]
- 345.Stiborová M, Frei E, Schmeiser HH, Arlt VM, Martínek V. Mechanisms of enzyme-catalyzed reduction of two carcinogenic nitro-aromatics, 3-nitrobenzanthrone and aristolochic acid. I. Experimental and theoretical approaches. Int J Mol Sci. 2014;15:10271–10295. doi: 10.3390/ijms150610271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Hayes JD, Nguyen T, Judah DJ, Petersson DG, Neal GE. Cloning of cDNAs from fetal rat liver encoding glutathione S-transferase Yc polypeptides: the Yc2 subunit is expressed in adult rat liver resistant to the hepatocarcinogen aflatoxin B1. J Biol Chem. 1994;269:20707–20717. [PubMed] [Google Scholar]
- 347.Hayes JD, Judah DJ, McLellan LI, Kerr LA, Peacock SD, Neal GE. Ethoxyquin-induced resistance to aflatoxin B1 in the rat is associated with the expression of a novel Alpha-class glutathione S-transferase subunit, Yc2, which possesses high catalytic activity for aflatoxin B1-8,9-epoxide. Biochem J. 1991;279:385–398. doi: 10.1042/bj2790385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Kelly VP, Ellis EM, Manson MM, Chanas SA, Moffat GJ, McLeod R, et al. Chemoprevention of aflatoxin B1 hepatocarcinogenesis by coumarin, a natural benzopyrone that is a potent inducer of aflatoxin B1-aldehyde reductase, the glutathione S-transferase A5 and P1 subunits, and NAD(P)H:quinone oxidoreductase in rat liver. Cancer Res. 2000;60:957–969. [PubMed] [Google Scholar]
- 349.Liby K, Yore MM, Roebuck BD, Baumgartner KJ, Honda T, Sundararajan C, et al. A novel acetylenic tricyclic bis-(cyano enone) potently induces phase 2 cytoprotective pathways and blocks liver carcinogenesis induced by aflatoxin. Cancer Res. 2008;68:6727–6733. doi: 10.1158/0008-5472.CAN-08-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Primiano T, Egner PA, Sutter TR, Kelloff GJ, Roebuck BD, Kensler TW. Intermittent dosing with oltipraz: relationship between chemoprevention of aflatoxin-induced tumorigenesis and induction of glutathione S-transferases. Cancer Res. 1995;55:4319–4324. [PubMed] [Google Scholar]
- 351.Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, Sutter TR, et al. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res. 2006;66:2488–2494. doi: 10.1158/0008-5472.CAN-05-3823. [DOI] [PubMed] [Google Scholar]
- 352.Ellis EM, Judah DJ, Neal GE, O’Connor T, Hayes JD. Regulation of carbonyl-reducing enzymes in rat liver by chemoprotectors. Cancer Res. 1996;56:2758–2766. [PubMed] [Google Scholar]
- 353.Ellis EM, Judah DJ, Neal GE, Hayes JD. An ethoxyquin-inducible aldehyde reductase from rat liver that metabolizes aflatoxin B1 defines a subfamily of aldo-keto reductases. Proc Natl Acad Sci USA. 1993;90:10350–10354. doi: 10.1073/pnas.90.21.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Ellis EM, Hayes JD. Substrate specificity of an aflatoxin-metabolizing aldehyde reductase. Biochem J. 1995;312:535–541. doi: 10.1042/bj3120535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Kozma E, Brown E, Ellis EM, Lapthorn AJ. The high resolution crystal structure of rat liver AKR7A1: understanding the substrate specificities of the AKR7 family. Chem Biol Interact. 2003;143–144:289–297. doi: 10.1016/s0009-2797(02)00186-2. [DOI] [PubMed] [Google Scholar]
- 356.Sherratt PJ, Manson MM, Thomson AM, Hissink EA, Neal GE, van Bladeren PJ, et al. Increased bioactivation of dihaloalkanes in rat liver due to induction of class theta glutathione S-transferase T1-1. Biochem J. 1998;335:619–630. doi: 10.1042/bj3350619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Hayes JD, Judah DJ, Neal GE, Nguyen T. Molecular cloning and heterologous expression of a cDNA encoding a mouse glutathione S-transferase Yc subunit possessing high catalytic activity for aflatoxin B1-8,9-epoxide. Biochem J. 1992;285:173–180. doi: 10.1042/bj2850173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Ilic Z, Crawford D, Vakharia D, Egner PA, Sell S. Glutathione S-transferase A3 knockout mice are sensitive to acute cytotoxic and genotoxic effects of aflatoxin B1. Toxicol Appl Pharmacol. 2010;242:241–246. doi: 10.1016/j.taap.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Jowsey IR, Jiang Q, Itoh K, Yamamoto M, Hayes JD. Expression of the aflatoxin B1-8,9-epoxide-metabolizing murine glutathione S-transferase A3 subunit is regulated by the Nrf2 transcription factor through an antioxidant response element. Mol Pharmacol. 2003;64:1018–1028. doi: 10.1124/mol.64.5.1018. [DOI] [PubMed] [Google Scholar]
- 360.Kensler KH, Slocum SL, Chartoumpekis DV, Dolan PM, Johnson NM, Ilic Z, et al. Genetic or pharmacologic activation of Nrf2 signaling fails to protect against aflatoxin genotoxicity in hypersensitive GSTA3 knockout mice. Toxicol Sci. 2014;139:293–300. doi: 10.1093/toxsci/kfu056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.McLellan LI, Kerr LA, Cronshaw AD, Hayes JD. Regulation of mouse glutathione S-transferases by chemoprotectors: molecular evidence for the existence of three distinct Alpha-class glutathione S-transferase subunits, Ya1, Ya2, and Ya3, in mouse liver. Biochem J. 1991;276:461–469. doi: 10.1042/bj2760461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.McLellan LI, Hayes JD. Differential induction of class Alpha glutathione S-transferases in mouse liver by the anticarcinogenic antioxidant butylated hydroxyanisole: purification and characterization of glutathione S-transferase Ya1Ya1. Biochem J. 1989;263:393–402. doi: 10.1042/bj2630393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004;64:6424–6431. doi: 10.1158/0008-5472.CAN-04-1906. [DOI] [PubMed] [Google Scholar]
- 364.Ramos-Gomez M, Dolan PM, Itoh K, Yamamoto M, Kensler TW. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene–DNA adducts and tumor yield in mice. Carcinogenesis. 2003;24:461–467. doi: 10.1093/carcin/24.3.461. [DOI] [PubMed] [Google Scholar]
- 365.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Bains OS, Takahashi RH, Pfeifer TA, Grigliatti TA, Reid RE, Riggs KW. Two allelic variants of aldo-keto reductase 1A1 exhibit reduced in vitro metabolism of daunorubicin. Drug Metab Dispos. 2008;36:904–910. doi: 10.1124/dmd.107.018895. [DOI] [PubMed] [Google Scholar]
- 367.Steckelbroeck S, Oyesanmi B, Jin Y, Lee S-H, Kloosterboer HJ, Penning TM. Tibolone metabolism in human liver is catalyzed by 3α/3β-hydroxysteroid dehydrogenase activities of the four isoforms of the aldo-keto reductase (AKR)1C subfamily. J Pharmacol Exp Ther. 2006;316:1300–1309. doi: 10.1124/jpet.105.091587. [DOI] [PubMed] [Google Scholar]
- 368.Steckelbroeck S, Jin Y, Oyesanmi B, Kloosterboer HJ, Penning TM. Tibolone is metabolized by the 3α/3β-hydroxysteroid dehydrogenase activities of the four human isozymes of the aldo-keto reductase 1C subfamily: inversion of stereospecificity with a Δ5(10)-3-ketosteroid. Mol Pharmacol. 2004;66:1702–1711. doi: 10.1124/mol.104.004515. [DOI] [PubMed] [Google Scholar]
- 369.Jin Y, Duan L, Chen M, Penning TM, Kloosterboer HJ. Metabolism of the synthetic progestogen norethynodrel by human ketosteroid reductases of the aldo-keto reductase superfamily. J Steroid Biochem Mol Biol. 2012;129:139–144. doi: 10.1016/j.jsbmb.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Martin HJ, Maser E. Role of human aldo-keto-reductase AKR1B10 in the protection against toxic aldehydes. Chem Biol Interact. 2009;178:145–150. doi: 10.1016/j.cbi.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 371.Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: generation of reactive and redox active o-quinones. Chem Res Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- 372.Birtwistle J, Hayden RE, Khanim FL, Green RM, Pearce C, Davies NJ, et al. The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: implications for leukemogenesis. Mutat Res. 2009;662:67–74. doi: 10.1016/j.mrfmmm.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 373.Aleksunes LM, Yeager RL, Klaassen CD. Application of multivariate statistical procedures to identify transcription factors that correlate with MRP2, 3, and 4 mRNA in adult human livers. Xenobiotica. 2009;39:514–522. doi: 10.1080/00498250902952514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Berg JM, Tymoczko JL, Stryer L. Biochemistry. 7th. Freeman; New York: 2011. [Google Scholar]
- 375.Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest. 2013;123:2921–2934. doi: 10.1172/JCI66353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 377.Sugimoto H, Okada K, Shoda J, Warabi E, Ishige K, Ueda T, et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2010;298:G283–294. doi: 10.1152/ajpgi.00296.2009. [DOI] [PubMed] [Google Scholar]
- 378.Zhang YKJ, Yeager RL, Tanaka Y, Klaassen CD. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol Appl Pharmacol. 2010;245:326–334. doi: 10.1016/j.taap.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Rakhshandehroo M, Knoch B, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010;2010:1–20. doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Xu J, Donepudi AC, Moscovitz JE, Slitt AL. Keap1-knockdown decreases fasting-induced fatty liver via altered lipid metabolism and decreased fatty acid mobilization from adipose tissue. PLoS One. 2013;8:e79841. doi: 10.1371/journal.pone.0079841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Bauer AK, Cho HY, Miller-Degraff L, Walker C, Helms K, Fostel J, et al. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS One. 2011;6:e26590. doi: 10.1371/journal.pone.0026590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Hayes JD, McMahon M. NRF2 and Keap1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 384.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Lewis KN, Wason E, Edrey YH, Kristan DM, Nevo E, Buffenstein R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc Natl Acad Sci USA. 2015;112:3722–3727. doi: 10.1073/pnas.1417566112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Lewis KN, Mele J, Hayes JD, Buffenstein R. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr Comp Biol. 2010;50:829–843. doi: 10.1093/icb/icq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Kwak MK, Kensler TW. Targeting NRF2 signaling for cancer chemoprevention. Toxicol Appl Pharmacol. 2010;244:66–76. doi: 10.1016/j.taap.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Slocum SL, Kensler TW. Nrf2: control of sensitivity to carcinogens. Arch Toxicol. 2011;85:273–284. doi: 10.1007/s00204-011-0675-4. [DOI] [PubMed] [Google Scholar]
- 389.Yang L, Zahid M, Liao Y, Rogan EG, Cavalieri EL, Davidson NE, et al. Reduced formation of depurinating estrogen–DNA adducts by sulforaphane or Keap1 disruption in human mammary epithelial MCF-10A cells. Carcinogenesis. 2013;34:2587–2592. doi: 10.1093/carcin/bgt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Wattenberg LW. Inhibition of chemical carcinogen-induced pulmonary neoplasia by butylated hydroxyanisole. J Natl Cancer Inst. 1973;50:1541–1544. doi: 10.1093/jnci/50.6.1541. [DOI] [PubMed] [Google Scholar]
- 391.Benson AM, Batzinger RP, Ou SY, Bueding E, Cha YN, Talalay P. Elevation of hepatic glutathione S-transferase activities and protection against mutagenic metabolites of benzo(a)pyrene by dietary antioxidants. Cancer Res. 1978;38:4486–4495. [PubMed] [Google Scholar]
- 392.Lam LK, Sparnins VL, Hochalter JB, Wattenberg LW. Effects of 2- and 3-tert-butyl-4-hydroxyanisole on glutathione S-transferase and epoxide hydrolase activities and sulfhydryl levels in liver and forestomach of mice. Cancer Res. 1981;41:3940–3943. [PubMed] [Google Scholar]
- 393.Whysner J, Williams GM. Butylated hydroxyanisole mechanistic data and risk assessment: conditional species-specific cytotoxicity, enhanced cell proliferation, and tumor promotion. Pharmacol Ther. 1996;71:137–151. doi: 10.1016/0163-7258(96)00066-6. [DOI] [PubMed] [Google Scholar]
- 394.Williams GM, Iatropoulos MJ, Whysner J. Safety assessment of butylated hydroxyanisole and butylated hydroxytoluene as antioxidant food additives. Food Chem Toxicol. 1999;37:1027–1038. doi: 10.1016/s0278-6915(99)00085-x. [DOI] [PubMed] [Google Scholar]
- 395.Ito N, Hirose M, Fukushima S, Tsuda H, Shirai T, Tatematsu M. Studies on antioxidants: their carcinogenic and modifying effects on chemical carcinogenesis. Food Chem Toxicol. 1986;24:1071–1082. doi: 10.1016/0278-6915(86)90291-7. [DOI] [PubMed] [Google Scholar]
- 396.Dinkova-Kostova AT, Jenkins SN, Fahey JW, Ye L, Wehage SL, Liby KT, et al. Protection against UV-light-induced skin carcinogenesis in SKH-1 high-risk mice by sulforaphane-containing broccoli sprout extracts. Cancer Lett. 2006;240:243–252. doi: 10.1016/j.canlet.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 397.Liby K, Risingsong R, Royce DB, Williams CR, Ma T, Yore MM, et al. Triterpenoids CDDO-methyl ester or CDDO-ethyl amide and rexinoids LG100268 or NRX194204 for prevention and treatment of lung cancer in mice. Cancer Prev Res. 2009;2:1050–1058. doi: 10.1158/1940-6207.CAPR-09-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.To C, Ringelberg CS, Royce DB, Williams CR, Risingsong R, Sporn MB, et al. Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis. 2015;36:769–781. doi: 10.1093/carcin/bgv061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Kim EH, Deng C, Sporn MB, Royce DB, Risingsong R, Williams CR, et al. CDDO-methyl ester delays breast cancer development in BRCA1-mutated mice. Cancer Prev Res. 2012;5:89–97. doi: 10.1158/1940-6207.CAPR-11-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Liby KT, Royce DB, Risingsong R, Williams CR, Maitra A, Hruban RH, et al. Synthetic triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer. Cancer Prev Res. 2010;3:1427–1434. doi: 10.1158/1940-6207.CAPR-10-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional Keap1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Li QK, Singh A, Biswal S, Askin F, Gabrielson E. Keap1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. J Hum Genet. 2011;56:230–234. doi: 10.1038/jhg.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 404.Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, Hirohashi S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135:1358–1368. 1368.e1–4. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 405.Yoo NJ, Kim HR, Kim YR, An CH, Lee SH. Somatic mutations of the Keap1 gene in common solid cancers. Histopathology. 2012;60:943–952. doi: 10.1111/j.1365-2559.2012.04178.x. [DOI] [PubMed] [Google Scholar]
- 406.Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol. 2010;220:446–451. doi: 10.1002/path.2653. [DOI] [PubMed] [Google Scholar]
- 407.Kovac M, Navas C, Horswell S, Salm M, Bardella C, Rowan A, et al. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nat Commun. 2015;6:6336. doi: 10.1038/ncomms7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Ooi A, Dykema K, Ansari A, Petillo D, Snider J, Kahnoski R, et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013;73:2044–2051. doi: 10.1158/0008-5472.CAN-12-3227. [DOI] [PubMed] [Google Scholar]
- 409.Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia. 2011;13:864–873. doi: 10.1593/neo.11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci USA. 2008;105:13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1–Nrf2 system in cancers: stress response and anabolic metabolism. Mol, Cell Oncol. 2012;2:200. doi: 10.3389/fonc.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20:511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 418.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in Keap1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 420.Homma S, Ishii Y, Morishima Y, Yamadori T, Matsuno Y, Haraguchi N, et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin Cancer Res. 2009;15:3423–3432. doi: 10.1158/1078-0432.CCR-08-2822. [DOI] [PubMed] [Google Scholar]
- 421.Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional over-expression in human lung carcinoma (A549) cells. J Biol Chem. 2002;277:24799–24808. doi: 10.1074/jbc.M112424200. [DOI] [PubMed] [Google Scholar]
- 422.Palackal NT, Burczynski ME, Harvey RG, Penning TM. The ubiquitous aldehyde reductase (AKR1A1) oxidizes proximate carcinogen trans-dihydrodiols to o-quinones: potential role in polycyclic aromatic hydrocarbon activation. Biochemistry. 2001;40:10901–10910. doi: 10.1021/bi010872t. [DOI] [PubMed] [Google Scholar]
- 423.Park JH, Mangal D, Tacka KA, Quinn AM, Harvey RG, Blair IA, et al. Evidence for the aldo-keto reductase pathway of polycyclic aromatic trans-dihydrodiol activation in human lung A549 cells. Proc Natl Acad Sci USA. 2008;105:6846–6851. doi: 10.1073/pnas.0802776105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Atalla A, Breyer-Pfaff U, Maser E. Purification and characterization of oxidoreductases-catalyzing carbonyl reduction of the tobacco-specific nitrosamine 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (NNK) in human liver cytosol. Xenobiotica. 2000;30:755–769. doi: 10.1080/00498250050119826. [DOI] [PubMed] [Google Scholar]
- 425.Breyer-Pfaff U, Martin HJ, Ernst M, Maser E. Enantioselectivity of carbonyl reduction of 4-methylnitrosamino-1-(3-pyridyl)-1-butanone by tissue fractions from human and rat and by enzymes isolated from human liver. Drug Metab Dispos. 2004;32:915–922. [PubMed] [Google Scholar]
- 426.Upadhyaya P, Carmella SG, Guengerich FP, Hecht SS. Formation and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers in vitro in mouse, rat and human tissues. Carcinogenesis. 2000;21:1233–1238. [PubMed] [Google Scholar]
- 427.Stiborová M, Dracínská H, Mizerovská J, Frei E, Schmeiser HH, Hudecek J, et al. The environmental pollutant and carcinogen 3-nitrobenzanthrone induces cytochrome P450 1A1 and NAD(P)H:quinone oxidoreductase in rat lung and kidney, thereby enhancing its own genotoxicity. Toxicology. 2008;247:11–22. doi: 10.1016/j.tox.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 428.Stiborová M, Dracínská H, Hájková J, Kaderábková P, Frei E, Schmeiser HH, et al. The environmental pollutant and carcinogen 3-nitrobenzanthrone and its human metabolite 3-aminobenzanthrone are potent inducers of rat hepatic cytochromes P450 1A1 and -1A2 and NAD(P)H:qui-none oxidoreductase. Drug Metab Dispos. 2006;34:1398–1405. doi: 10.1124/dmd.106.009373. [DOI] [PubMed] [Google Scholar]
- 429.Penning TM, Lerman C. Genomics of smoking exposure and cessation: lessons for cancer prevention and treatment. Cancer Prev Res. 2008;1:80–83. doi: 10.1158/1940-6207.CAPR-08-0047. [DOI] [PubMed] [Google Scholar]
- 430.Quinn AM, Harvey RG, Penning TM. Oxidation of PAH trans-dihydrodiols by human aldo-keto reductase AKR1B10. Chem Res Toxicol. 2008;21:2207–2215. doi: 10.1021/tx8002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Martin HJ, Breyer-Pfaff U, Wsol V, Venz S, Block S, Maser E. Purification and characterization of akr1b10 from human liver: role in carbonyl reduction of xenobiotics. Drug Metab Dispos. 2006;34:464–470. doi: 10.1124/dmd.105.007971. [DOI] [PubMed] [Google Scholar]
- 432.Hayes JD, Wolf CR. Molecular mechanisms of drug resistance. Biochem J. 1990;272:281–295. doi: 10.1042/bj2720281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Leslie EM, Deeley RG, Cole SPC. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204:216–237. doi: 10.1016/j.taap.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 434.McLellan LI, Wolf CR. Glutathione and glutathione-dependent enzymes in cancer drug resistance. Drug Resist, Updates. 1999;2:153–164. doi: 10.1054/drup.1999.0083. [DOI] [PubMed] [Google Scholar]
- 435.Powis G, Kirkpatrick DL. Thioredoxin signaling as a target for cancer therapy. Curr Opin Pharmacol. 2007;7:392–397. doi: 10.1016/j.coph.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 436.Selenius M, Rundlöf AK, Olm E, Fernandes AP, Björnstedt M. Selenium and the selenoprotein thioredoxin reductase in the prevention, treatment and diagnostics of cancer. Antioxid Redox Signaling. 2010;12:867–880. doi: 10.1089/ars.2009.2884. [DOI] [PubMed] [Google Scholar]
- 437.Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994;54:4313–4320. [PubMed] [Google Scholar]
- 438.Bhat PJ, Darunte L, Kareenhalli V, Dandekar J, Kumar A. Can metabolic plasticity be a cause for cancer? Warburg-Waddington legacy revisited. Clin Epigenet. 2011;2:113–122. doi: 10.1007/s13148-011-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Ladewig J, Koch P, Brüstle O. Leveling Waddington: the emergence of direct programming and the loss of cell fate hierarchies. Nat Rev Mol Cell Biol. 2013;14:225–236. doi: 10.1038/nrm3543. [DOI] [PubMed] [Google Scholar]
- 440.Menendez JA, Alarcón T. Metabostemness: a new cancer hallmark. Front, Oncol. 2014;4:262. doi: 10.3389/fonc.2014.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Jang J, Wang Y, Kim HS, Lalli MA, Kosik KS. Nrf2, a regulator of the proteasome, controls self-renewal and pluripotency in human embryonic stem cells. Stem Cells. 2014;32:2616–2625. doi: 10.1002/stem.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 443.Velmurugan GV, Sundaresan NR, Gupta MP, White C. Defective Nrf2-dependent redox signalling contributes to microvascular dysfunction in type 2 diabetes. Cardiovasc Res. 2013;100:143–150. doi: 10.1093/cvr/cvt125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Shin S, Wakabayashi J, Yates MS, Wakabayashi N, Dolan PM, Aja S, et al. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-Imidazolide. Eur J Pharmacol. 2009;620:138–144. doi: 10.1016/j.ejphar.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Xu J, Kulkarni SR, Donepudi AC, More VR, Slitt AL. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes. 2012;61:3208–3218. doi: 10.2337/db11-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Cheng X, Siow RC, Mann GE. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid Redox Signaling. 2011;14:469–487. doi: 10.1089/ars.2010.3283. [DOI] [PubMed] [Google Scholar]
- 447.Yoh K, Hirayama A, Ishizaki K, Yamada A, Takeuchi M, Yamagishi S, et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells. 2008;13:1159–1170. doi: 10.1111/j.1365-2443.2008.01234.x. [DOI] [PubMed] [Google Scholar]
- 448.Pi J, Zhang Q, Fu J, Woods CG, Hou Y, Corkey BE, et al. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol Appl Pharmacol. 2010;244:77–83. doi: 10.1016/j.taap.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.More VR, Xu J, Shimpi PC, Belgrave C, Luyendyk JP, Yamamoto M, et al. Keap1 knockdown increases markers of metabolic syndrome after long-term high fat diet feeding. Free Radic Biol Med. 2013;61:85–94. doi: 10.1016/j.freeradbiomed.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Jiang T, Huang Z, Lin Y, Zhang Z, Fang D, Zhang DD. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:850–860. doi: 10.2337/db09-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.He X, Ma Q. Disruption of Nrf2 synergizes with high glucose to cause heightened myocardial oxidative stress and severe cardiomyopathy in diabetic mice. J Diabetes Metab. 2012;S7 doi: 10.4172/2155-6156.S7-002. http://www.omicsonline.org/disruption-of-nrf2-synergizes-with-high-glucose-to-cause-heightened-myocardial-oxidative-stress-and-severe-cardiomyopathy-in-diabetic-mice-2155-6156.S7-002.php?aid=7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Xu Z, Wei Y, Gong J, Cho H, Park JK, Sung ER, et al. NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia. 2014;57:204–213. doi: 10.1007/s00125-013-3093-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Bai Y, Cui W, Xin Y, Miao X, Barati MT, Zhang C, et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J Mol Cell Cardiol. 2013;57:82–95. doi: 10.1016/j.yjmcc.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 454.Cui W, Bai Y, Miao X, Luo P, Chen Q, Tan Y, et al. Prevention of diabetic nephropathy by sulforaphane: possible role of Nrf2 upregulation and activation. Oxid Med Cell Longevity. 2012;2012:821936. doi: 10.1155/2012/821936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Miao X, Bai Y, Sun W, Cui W, Xin Y, Wang Y, et al. Sulforaphane prevention of diabetes-induced aortic damage was associated with the up-regulation of Nrf2 and its down-stream antioxidants. Nutr Metab (London) 2012;9:84. doi: 10.1186/1743-7075-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Zheng H, Whitman SA, Wu W, Wondrak GT, Wong PK, Fang D, et al. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes. 2011;60:3055–3066. doi: 10.2337/db11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Liu ZZ, Zhao XZ, Zhang XS, Zhang M. Promoter DNA demethylation of Keap1 gene in diabetic cardiomyopathy. Int J Clin Exp Pathol. 2014;7:8756–8762. [PMC free article] [PubMed] [Google Scholar]
- 458.Chartoumpekis DV, Kensler TW. New player on an old field; the keap1/Nrf2 pathway as a target for treatment of type 2 diabetes and metabolic syndrome. Curr Diabetes Rev. 2013;9:137–145. doi: 10.2174/1573399811309020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, et al. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15:713–724. doi: 10.1016/j.cmet.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Maiese K, Chong ZZ, Shang YC, Wang S. Novel directions for diabetes mellitus drug discovery. Expert Opin Drug Discovery. 2013;8:35–48. doi: 10.1517/17460441.2013.736485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Joshi G, Johnson JA. The Nrf2-ARE pathway: a valuable therapeutic target for the treatment of neurodegenerative diseases. Recent Patents CNS Drug Discovery. 2012;7:218–229. doi: 10.2174/157488912803252023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol. 2013;100:30–47. doi: 10.1016/j.pneurobio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem. 2008;104:1116–1131. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol. 2013;106–107:17–32. doi: 10.1016/j.pneurobio.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Lesage S, Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–R59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- 467.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 468.Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Barone MC, Sykiotis GP, Bohmann D. Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis Model Mech. 2011;4:701–707. doi: 10.1242/dmm.007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, et al. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proc Natl Acad Sci USA. 2009;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Innamorato NG, Jazwa A, Rojo AI, García C, Fernández-Ruiz J, Grochot–Przeczek A, et al. Different susceptibility to the Parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS One. 2010;5:e11838. doi: 10.1371/journal.pone.0011838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Jakel RJ, Townsend JA, Kraft AD, Johnson JA. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007;1144:192–201. doi: 10.1016/j.brainres.2007.01.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Rojo AI, Innamorato NG, Martín-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010;58:588–598. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- 474.Burton NC, Kensler TW, Guilarte TR. In vivo modulation of the Parkinsonian phenotype by Nrf2. NeuroToxicology. 2006;27:1094–1100. doi: 10.1016/j.neuro.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 475.Galuppo M, Iori R, De Nicola GR, Bramanti P, Mazzon E. Anti-inflammatory and anti-apoptotic effects of (RS)-glucoraphanin bioactivated with myrosinase in murine sub-acute and acute MPTP-induced Parkinson’s disease. Bioorg Med Chem. 2013;21:5532–5547. doi: 10.1016/j.bmc.2013.05.065. [DOI] [PubMed] [Google Scholar]
- 476.García E, Santana-Martínez R, Silva-Islas CA, Colín-González AL, Galván-Arzate S, Heras Y, et al. S-allyl cysteine protects against MPTP-induced striatal and nigral oxidative neurotoxicity in mice: participation of Nrf2. Free Radic Res. 2014;48:159–167. doi: 10.3109/10715762.2013.857019. [DOI] [PubMed] [Google Scholar]
- 477.Jazwa A, Rojo AI, Innamorato NG, Hesse M, Fernández-Ruiz J, Cuadrado A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid Redox Signaling. 2011;14:2347–2360. doi: 10.1089/ars.2010.3731. [DOI] [PubMed] [Google Scholar]
- 478.Kaidery NA, Banerjee R, Yang L, Smirnova NA, Hushpulian DM, Liby KT, et al. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid Redox Signaling. 2013;18:139–157. doi: 10.1089/ars.2011.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Kim SS, Lim J, Bang Y, Gal J, Lee SU, Cho YC, et al. Licochalcone E activates Nrf2/antioxidant response element signaling pathway in both neuronal and microglial cells: therapeutic relevance to neurodegenerative disease. J Nutr Biochem. 2012;23:1314–1323. doi: 10.1016/j.jnutbio.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 480.Wang XL, Xing GH, Hong B, Li XM, Zou Y, Zhang XJ, et al. Gastrodin prevents motor deficits and oxidative stress in the MPTP mouse model of Parkinson’s disease: involvement of ERK1/2–Nrf2 signaling pathway. Life Sci. 2014;114:77–85. doi: 10.1016/j.lfs.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 481.Williamson TP, Johnson DA, Johnson JA. Activation of the Nrf2-ARE pathway by siRNA knockdown of Keap1 reduces oxidative stress and provides partial protection from MPTP-mediated neurotoxicity. Neurotoxicology. 2012;33:272–279. doi: 10.1016/j.neuro.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Woo SY, Kim JH, Moon MK, Han SH, Yeon SK, Choi JW, et al. Discovery of vinyl sulfones as a novel class of neuroprotective agents toward Parkinson’s disease therapy. J Med Chem. 2014;57:1473–1487. doi: 10.1021/jm401788m. [DOI] [PubMed] [Google Scholar]
- 483.Yang L, Calingasan NY, Thomas B, Chaturvedi RK, Kiaei M, Wille EJ, et al. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS One. 2009;4:e5757. doi: 10.1371/journal.pone.0005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Dinkova-Kostova AT, Baird L, Holmström KM, Meyer C, Abramov AY. The spatiotemporal regulation of the Keap1-Nrf2 pathway and its importance in cellular bioenergetics. Biochem Soc Trans. 2015;43:602–610. doi: 10.1042/BST20150003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.von Otter M, Bergström P, Quattrone A, Marco EVD, Annesi G, Söderkvist P, et al. Genetic associations of Nrf2-encoding NFE2L2 variants with Parkinson’s disease—a multicenter study. BMC Med Genet. 2014;15 doi: 10.1186/s12881-014-0131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.von Otter M, Landgren S, Nilsson S, Celojevic D, Bergström P, Håkansson A, et al. Association of Nrf2-encoding NFE2L2 haplotypes with Parkinson’s disease. BMC Med Genet. 2010;11:36. doi: 10.1186/1471-2350-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci. 2010;114:237–246. doi: 10.1093/toxsci/kfp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Lee DH, Gold R, Linker RA. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: therapeutic modulation via fumaric acid esters. Int J Mol Sci. 2012;13:11783–11803. doi: 10.3390/ijms130911783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 490.Blake DJ, Singh A, Kombairaju P, Malhotra D, Mariani TJ, Tuder RM, et al. Deletion of Keap1 in the lung attenuates acute cigarette smoke-induced oxidative stress and inflammation. Am J Respir Cell Mol Biol. 2010;42:524–536. doi: 10.1165/rcmb.2009-0054OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Marzec JM, Christie JD, Reddy SP, Jedlicka AE, Vuong H, Lanken PN, et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007;21:2237–2246. doi: 10.1096/fj.06-7759com. [DOI] [PubMed] [Google Scholar]
- 492.Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol Sci. 2013;34:340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]