


Abstract
The sliding clamp enhances polymerase processivity and coordinates DNA replication with other critical DNA processing events including translesion synthesis, Okazaki fragment maturation and DNA repair. The relative binding affinity of the sliding clamp for its partners determines how these processes are orchestrated and is essential to ensure the correct processing of newly replicated DNA. However, while stable clamp interactions have been extensively studied; dynamic interactions mediated by the sliding clamp remain poorly understood. Here, we characterize the interaction between the bacterial sliding clamp (β-clamp) and one of its weak-binding partners, the DNA mismatch repair protein MutL. Disruption of this interaction causes a mild mutator phenotype in Escherichia coli, but completely abrogates mismatch repair activity in Bacillus subtilis. We stabilize the MutL-β interaction by engineering two cysteine residues at variable positions of the interface. Using disulfide bridge crosslinking, we have stabilized the E. coli and B. subtilis MutL-β complexes and have characterized their structures using small angle X-ray scattering. We find that the MutL-β interaction greatly stimulates the endonuclease activity of B. subtilis MutL and supports this activity even in the absence of the N-terminal region of the protein.
INTRODUCTION
Protein–protein and protein–nucleic acid interactions regulate virtually every cellular transaction. However, we only have a fragmented understanding of these regulatory processes due to the intrinsic challenges associated with studying short-lived interactions. Most well characterized protein–protein complexes form stable, high-affinity interactions (KD < 10−6 M). Weak-affinity (KD > 10−4 M) and transient interactions are equally important in the regulation of many cellular pathways (1), but they are poorly understood. Weak and transient interactions are especially difficult to study for proteins that coordinate multiple processes, because they interact with many binding partners often using a common interface. Allostery and conformational malleability are defining aspects of the hierarchy of these interactions (1,2), however most structural biology approaches fail to provide this type of information.
The sliding β-clamp (β) and its eukaryotic counterpart (PCNA) are a paradigm for proteins coordinating multiple interactions using a common binding site. They were first identified as processivity factors that tether the replicative polymerase to DNA during DNA replication. However, they also play critical roles coordinating DNA replication with other key cellular functions including Okazaki fragment maturation, polymerase switching during lesion bypass, DNA repair and DNA transposition (3–6). Sliding β-clamps form ring-shaped structures that are conserved at the structural, but not sequence, level. Their central cavity threads DNA and creates a topological link between their binding partners and DNA (7,8). They interact with their binding partners through a conserved hydrophobic groove located at the C-terminus of the protein (Figure 1). Reciprocally, clamp-binding partners contain a conserved linear motif known as PIP box (PCNA-interacting protein box) or β-binding motif that binds the hydrophobic groove of the clamp. β-binding motifs are not as well conserved as PIP boxes and, hence, β-clamp binding partners are difficult to identify (9). The sequence variability of this binding motif has been correlated to binding affinity, thereby allowing clamps to mediate both weak and strong interactions (10–14).
Figure 1.
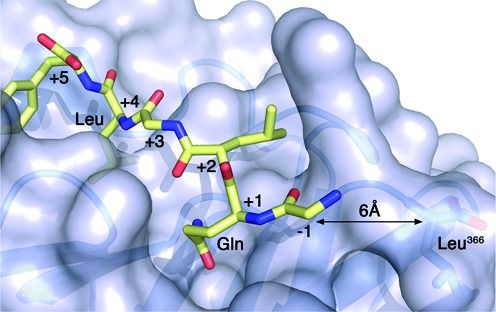
Crystal structure of the β-clamp bound to a polymerase II peptide. Surface representation of the E. coli β-clamp bound to the β-binding motif of polymerase II (color-coded sticks, PDB ID 3D1E). β has a well-defined groove near its C-terminus where partners interact. The conserved Gln and Leu residues at positions +1 and +4 of the β-binding motif are labeled. The C-terminal residue (Leu366) of β is also labeled and the distance to the N-terminal amino acid of the peptide is indicated.
One of the repair pathways orchestrated by the sliding β-clamp is DNA mismatch repair, a conserved post-replicative repair pathway that corrects replication errors introduced by DNA polymerase. The initiation of the mismatch repair response in Escherichia coli requires the coordinated action of three proteins: MutS, MutL and MutH. MutS recognizes base mismatches and small insertion or deletion loops and, once bound to the mismatch, it recruits MutL in an ATP-dependent manner (15,16). Together, MutS and MutL activate the MutH endonuclease that nicks the newly synthesized strand at a nearby hemi-methylated GATC site (16–18). While the DNA mismatch repair pathway is evolutionary conserved, MutH is only found in a subset of gamma-proteobacteria including E. coli. In organisms lacking MutH, MutL homologs harbour a weak endonuclease activity (19–24).
Bacterial MutL consists of two structurally conserved domains connected by a flexible linker (25). The N-terminal domain (NTD) supports DNA binding and ATPase activity (26,27). The C-terminal domain (CTD) mediates protein dimerization and harbours a metal binding motif associated to the endonuclease activity and a conserved β-binding motif (10,19–24,26–28).
Beyond, its role at the DNA synthesis step, the sliding β-clamp plays critical roles at earlier steps of the mismatch repair process. It interacts with MutS and recruits it to the mismatched sites (29,30). The bacterial β-clamp also interacts weakly, yet specifically, with the two structured domains of MutL (10,29), however the role of this interaction remains unclear. Mutation of the β-binding motif in the endonuclease domain of Bacillus subtilis MutL abrogates mismatch repair activity (24), whereas mutation of the same motif in E. coli MutL only causes a mild mutator phenotype (10,29), suggesting that this interaction is more important in organisms that lack the MutH endonuclease. PCNA stimulates the endonuclease activity of human MutLα and, due to its loading orientation, helps determine the strand that MutLα nicks (31). However, the direct interaction between PCNA and the endonuclease domain of MutLα has not been demonstrated biochemically.
In order to understand how the β-clamp regulates the activity of MutL in organisms containing or lacking MutH, we devised a strategy to stabilize the MutL-β clamp interaction using E. coli and B. subtilis proteins and analyzed the structure and function of these complexes. We find that binding to the β-clamp has a marginal effect on the activities of E. coli MutL, but it greatly stimulates the endonuclease activity of B. subtilis MutL. The interaction promotes endonuclease activity of B. subtilis MutL even in the absence of the ATPase domain of the protein, presumably by bypassing the DNA binding defect of the endonuclease domain of B. subtilis MutL. Based on these results, we propose a model to describe the role of the sliding β-clamp on the activation of MutL endonuclease activity of MutL.
MATERIALS AND METHODS
Cloning of the MutL and β-clamp cysteine variants
The expression plasmids encoding E. coli β-clamp and MutL were kind gifts from Dr Michael O'Donnell and Dr Wei Yang, respectively. Variants of both proteins, where surface exposed cysteines had been replaced by serines, were generated by overlap PCR. E. coli β (pAG 8769; residues 1–367) harboring mutations C260S, C333S, L366S and C367 was subcloned into a modified pET15b vector lacking the histidine tag. E. coli MutL (pAG 8814; residues 1–615), and its C-terminal domain MutLCTD (pAG 8768; residues 431–615), including mutations C61S, C446S, and C588S were subcloned in pET15b using NdeI and BamHI. Variants of E. coli MutL (eMutL*; pAG 8824) and MutLCTD (eMutLCTD*; pAG 8772) lacking the β-binding motif (482QPLLIP → 482ASAAA) were generated by overlap PCR.
Variants of B. subtilis MutL and β lacking surface exposed cysteine residues were generated similarly. B. subtilis β (pAG 8807; residues 1–380) including the Ser379–Cys380 dipeptide at the extreme C-terminus of the protein was subcloned into a modified pET15b vector including a TEV-removable histidine tag. B. subtilis MutL (pAG 8842; residues 1–627), and its C-terminal domain MutLCTD (pAG 8803; residues 433–627), including mutations C69S, C424S, E485C and C531S were subcloned in the pProExHTa vector. An inactive variant of MutLCTD including two additional point mutations (C573S and C604S), MutLCTDI (pAG 8927; residues 433–627), was generated by site-directed mutagenesis. Variants of B. subtilis MutL (bMutL*; pAG 8908), MutLCTD (bMutLCTD*; pAG 8909) and MutLCTDI (bMutLCTDI*; pAG 8929) lacking the β-binding motif (487QEMIVP → 487AEMAAP) were generated by overlap PCR. All mutants were verified by DNA sequencing (MOBIX, McMaster University).
Characterization of the MutL and β-clamp cysteine variants
E. coli and B. subtilis β and MutL cysteine-modified variants were over-produced and purified as described previously (10,24,27,28) and eluted from a size exclusion chromatography column at retention volumes similar to their native counterparts (Supplementary Figure S1). All purified cysteine-variants were monodisperse in solution as judged by dynamic light scattering using a Zetasizer Nano S (Malvern Instruments). Purified proteins (60 μM) were spun at 15 700 × g for 10 min and measurements were on 12 μl quartz cells at 4°C. Size distribution of the samples was calculated based on the correlation function provided by the Zetasizer software. Using Mie theory, the intensity-based distribution was transformed into a volume distribution to consider the relative proportion of potential multiple components in the sample (Supplementary Figure S1).
Thermal stability of the different samples was assessed by scanning differential fluorimetry using a CFX96 Real-Time System (BioRad) (32). Proteins were exchanged into 20 mM Tris pH 8.0, 150 mM NaCl, 5% glycerol (for E. coli proteins) or 20 mM Tris pH 7.6, 150 mM KCl, 10% glycerol (for B. subtilis proteins) using a Superdex-200 (GE Healthcare) gel filtration column. SYPRO Orange protein gel stain (4.8×) from Molecular Probes was incubated with 4.8 μM protein and the mixtures were subjected to a temperature gradient from 4 to 95°C over 90 min. Sample fluorescence was measured over time using the FRET setting. Each reaction was conducted in four replicates and representative curves are presented in Supplementary Figure S1.
Analysis of the function of the cysteine-variant of E. coli β in vivo
The ability of E. coli β to support viability was measured using a plasmid shuffle assay (33) (Table 1). Briefly, strain MS201 lacks a functional β-clamp gene (dnaN), due to a frameshift mutation (dnaN−1FS). Viability of strain MS201 relies on the ampicillin resistant plasmid pAMPdnaN+, which expresses physiological levels of the wild type β-clamp (34). MS201 bearing pAMPdnaN+ was transformed with the incompatible and chloramphenicol resistant pACM (negative control), pACMdnaN+ (eβWT) or pACMβCys (eβCys) plasmids, which contain the same origin of replication as pAMPdnaN+. Twenty randomly selected transformants were passaged ∼100 generations in Luria-Bertani (LB) media supplemented with 40 μg/ml chloramphenicol. The frequency of pAMPdnaN+ retention was measured by patching cells onto agar plates supplemented with either 150 μg/ml ampicillin (to score for pAMPdnaN+) or 40 μg/ml chloramphenicol (control). The presence of the dnaN−1FS alleles, and lack of the pAMPdnaN+ plasmid, in strains bearing pACMdnaN+ or pACMβCys was verified by diagnostic PCR and nucleotide sequence analysis of the chromosomal dnaN locus, as well as plasmid mini-prep and nucleotide sequence analysis, as described previously (33).
Table 1. Ability of βCys to support E. coli viability.
| Transforming plasmida | β-Clamp protein | Frequency of pAMPdnaN+ retentionb | E. coli viabilityc | β-Clamp expression levelsd | Doubling time (min)e |
|---|---|---|---|---|---|
| pACM | None (negative control) | 20/20 (100%) | – | NA | NA |
| pACMdnaN+ | eβWT (positive control) | 2/20 (10%) | + | 0.93 ± 0.08 (P = 0.26) | 37 ± 1.5 |
| pACMβCys | eβCys | 2/20 (10%) | + | 0.95 ± 0.13 (P = 0.56) | 42 ± 2.1 (P = 0.03) |
aStrain MS201 bearing plasmid pAMPdnaN+ was transformed with the incompatible plasmids pACM, pACMdnaN+ or pACMβCys, as indicated.
bThe frequencies of pAMPdnaN+ retention in 20 randomly selected pACM, pACMdnaN+ or pACMβCys transformants following ∼100 generations are indicated. Representative clones lacking pAMPdnaN+ were characterized further to verify they contained both the chromosomally-encoded dnaN−1FS as well as the indicated plasmid-encoded dnaN allele.
cViability refers to the ability of pACM, pACMdnaN+ or pACMβCys to support growth of E. coli in the absence of pAMPdnaN+.
dValues represent the average of triplicates ± one standard deviation, and are expressed relative to the level observed in the isogenic strain RW118. P values were calculated relative to the isogenic parent strain RW118 using the Student's t-test.
eResults shown are the average of three independent determinations ± one standard deviation. The P value for the strain bearing pACMβCys was calculated relative to the pACMdnaN+ strain using the Student's t-test.
Single isolates of the eβWT (VB100) and eβCys (VB101) strains were used for subsequent genetic analyses (Table 2). The frequency of spontaneous mutation of rpoB to rifampicin resistance (RifR) of strains VB100 and VB101 was measured (35), and 95% confidence limits were calculated as described previously (36). Mutation rates were calculated using FALCOR, a Ma-Sandri-Sarkar Maximum Likelihood Estimator (37). The Mann–Whitney U test (http://www.socscistatistics.com/tests/mannwhitney/Default.aspx) was used to determine whether mutation rate differences were statistically significant. Steady state levels of β-clamp proteins were measured using Western blot analysis (33). Doubling times for strains VB100 and VB101 grown at 37°C in liquid LB medium were calculated from linear portions of growth curves.
Table 2. Ability of E. coli βCys to support mismatch repair function in vivo.
| Straina | β-Clamp protein | Spontaneous mutation frequency (RifR)b | Spontaneous mutation rate (RifR)c |
|---|---|---|---|
| VB100 | eβWT | 1.79 × 10−9 (≤8.40 × 10−10 – 4.76 × 10−9) | 1.28 × 10−8 |
| VB101 | eβCys | 1.68 × 10−9 (8.40 × 10−10 – 3.36 × 10−9) | 1.57 × 10−8 (P = 0.79) |
aStrains VB100 and VB101 are representative plasmid shuffle isolates (derivatives of MS201 bearing the CamR plasmids) and express physiological levels of either wild type β-clamp (VB100) or βCys (VB101) as the sole clamp protein.
bMedian value from 14 (VB100) or 15 (VB101) independent cultures; 95% confidence intervals are in parentheses.
cSpontaneous RifR mutation rates were calculated from respective frequencies using the web tool FALCOR. The p value for strain VB101 was calculated relative to strain VB100 using the Mann-Whitney U test.
B. subtilis strains, bacteriology and western blotting for analysis of cysteine-variants of B. subtilis MutL and β in vivo
Each B. subtilis strain was created by first amplifying the mutant allele from plasmids (pAG 8807, pAG 8842). For JRR20, the mutant dnaN allele was placed into the pBGSC6 plasmid containing a chloramphenicol resistance marker and integrated into the native dnaN locus via single crossover. JRR21 was built by placing the mutant mutL allele into the pMiniMad plasmid with upstream and downstream sequence and integrating at the native mutL locus via double crossover. JRR28 was created by purifying genomic DNA from JRR20 and transforming the genomic DNA into competent JRR21 cells and selecting for chloramphenicol resistance. PB112 was made by placing upstream and downstream mutL sequence into pMiniMad and removing the gene via double crossover at the native locus. All strains were verified via DNA sequence analysis and are listed in Supplementary Table S3.
Growth curve analysis was done by growing each strain in culture in 3 ml of LB liquid and grown to an OD600 of between 0.4 and 0.6. Each culture was then back diluted to an OD600 of 0.05 and five 200 μl aliquots were distributed into wells of a sterile 96-well plate for each strain. This plate was incubated at 37°C, shaking overnight and the absorbance at 600 nm was taken every 0.5 h by an Omega plate reader. The standard deviation between the five aliquots was calculated and plotted.
Western blotting was performed by growing each strain to an OD600 of between 0.8 and 1. Cell number was then normalized to 1 ml of OD600 equal to 1 for each strain and cells were pelleted by centrifugation for 2 min at 14 000 rcf. The supernatant was aspirated and cells were resuspended in 50 μl of lysis buffer (50 mM Tris–HCl pH 8, 150 mM NaCl, 50 mM EDTA, 1× protease inhibitor cocktail (Thermoscientific), 10 mg/ml lysozyme). Cells were then incubated at 37°C for 30 min then left on ice for 10 min. The cell lysate was then centrifuged at 14 000 rcf for 5 min to remove cell debris. 50 μl of 10% SDS, and loading dye to 1× were then added and samples were heated to 100°C for 10 min before loading onto an 8% SDS-PAGE. The protein was transferred onto a nitrocellulose membrane and blocked for 30 min in TBST with 5% milk. The membrane was then incubated in the antiserum dilution (1/250 for α-MutL, 1/250 for α-DnaX, and 1/1000 α-DnaN) in TBST with 5% milk overnight at 4°C. The membrane was washed with TBST and incubated with a 1/15000 dilution of IR labeled secondary antibody (LiCOR) in TBST with 5% milk for 1 h at room temperature. The membrane was then washed again in TBST and visualized using an IR dye imager.
Determination of B. subtilis mutation rate
Mutation rate was performed and calculated essentially as described (38). Briefly, each strain was grown overnight for single colonies on LB agar with appropriate antibiotics at 30°C. Following overnight growth one colony was chosen for each strain per replicate and grown in 3 ml liquid LB culture to an OD600 between 0.8 and 1.2 (late exponential growth) at 37°C. 1.5 ml of each culture was removed and pelleted by centrifugation for 4 minutes at 8000 rcf. The supernatant was aspirated and cells were resuspended in 100 μl of a 0.85% saline solution. 1 μl of this solution was diluted 106 fold and 100 μl was plated on LB agar to obtain a count of total viable cells. 100 μl of the cell resuspension was then plated on 100 μg/ml rifampicin LB agar to determine the number of RifR colony forming units. Mutation rate analysis (39) was conducted using the Ma-Sandri-Sarkar maximum likelihood estimator (MSS-MLE) method described in (37). For the wild type control PY79, 32 replicates were analyzed, 14 replicates for ΔmutL, 31 for dnaNCys, 24 for mutLCys, and 9 for dnaNCys, mutLCys.
MutL-β complex formation
To form the MutL-β and MutLCTD-β complexes, β was incubated with either MutL or MutLCTD at a 1:1 ratio to a final concentration of 20 μM. The samples (1–2 ml) were dialyzed against 1 l of dialysis buffer A (20 mM Tris pH 8.0, 150 mM NaCl, 10 mM DTT, 5% glycerol for E. coli proteins; or 20 mM Tris pH 7.6, 150 mM KCl, 10 mM DTT, 10% glycerol for B. subtilis proteins) for 2 h at 4°C. The mixture was transferred into dialysis buffer B (same as A but with 5 mM DTT) for 1 h, followed by 1 h in dialysis buffer C (without DTT). The sample was then left overnight in dialysis buffer C. Complex formation was monitored over time by resolving the samples on 11% (E. coli MutL-β and B. subtilis MutLCTD-β) or 15% (E. coli MutLCTD-β) denaturing gels stained with Coomassie Brilliant Blue. Prior to forming the E. coli MutL−β complex, full-length MutL (43 μM) was pre-incubated in the absence and presence of 2 mM AMPPNP (Sigma) for 1 h at room temperature followed by an overnight incubation at 4°C. Association of the N-terminal domains of E. coli MutL was monitored as previously described (26,28).
SAXS data collection and analysis
E. coli MutLCTD-β samples were resolved over a Superdex-200 (GE Healthcare) size exclusion chromatography column. B. subtilis MutLCTDI-β samples were treated with 120 μM methyl methanethiosulfonate (MMTS) for 10 min at 22°C to sulfenylate unreacted thiol groups and resolved over a Mono Q 5/50 GL (GE Healthcare) ionic exchange column equilibrated with buffer D (20 mM Tris pH 7.6, 150 mM KCl, 0.5 mM EDTA, 1 mM MMTS, and 10% glycerol). B. subtilis MutLCTDI-β samples were exchanged into buffer D using a 100 kDa MWCO concentrator. Sample homogeneity was confirmed by dynamic light scattering (Malvern Instruments). Samples (35 μl) were spun at 15 700 x g for 10 min and scattering data was collected on a Rigaku BioSAXS-1000 instrument at 10°C. Consecutive scans of 10, 30, 60 and/or 180 min were collected over a range of protein concentrations (eβ: 47–186 μM; eMutLCTD: 91–364 μM; eMutLCTD-β (Day1): 1.8–2.0 μM; eMutLCTD-β (Day 2): 9.5–123 μM; bβ: 27–73 μM; bMutLCTDI: 88–218 μM; bMutLCTDI-β: 22–37 μM (Day 2)). SAXSLab 3.0.0r1 (Rigaku) was used to generate the scattering curves. Comparing 10-min exposures collected before and after data collection and resolving the samples on SDS-polyacrylamide gels before and after data collection confirmed sample integrity during data collection. The 1D scattering profile pairs were identical for all samples. Data quality was assessed by comparing scattering curves over a range of protein concentrations and exposure times using the ATSAS 2.6.0 program suite (Supplementary Figures S3–S5) (40). Radius of gyration and pair-distance distribution functions were determined using Primus and Gnom (Table 3 and Supplementary Table S2) (41). The highest quality estimate as determined by AutoRg and AutoGNOM functions was used to select the samples that were processed further. For the selected samples, twenty ab initio models were independently generated for each sample using DAMMIN, DAMMIF, or GASBOR (42–44). Ab initio models for bMutLCTDI, bβ and eMutLCTD-eβ were clustered based on the normalized spatial discrepancy using DAMCLUST (45) and the representative model of the dominant cluster is presented. Refined models for eβ, eMutLCTD and bMutLCTDI-bβ were generated with DAMMIN (43) using the damstart.pdb file as the starting search volume. Quaternary structure modeling of the bMutLCTD–β complex was generated with bMutLCTD (PDB 3KDK) and bβ (PDB 4TR6) using SASREF (46). The reported molecular weights were calculated based on the volume of correlation (47).
Table 3. SAXS data-collection and scattering-derived parameters for MutLCTDI-β.
| eMutLCTD-eβ | bMutLCTDI-bβ | ||
|---|---|---|---|
| Day 1 | Day 2 | Day 2 | |
| Data-collection parameters | |||
| Exposure time (min) | 180 | 180 | 180 |
| Concentration (μM) | 2 | 38.5 | 37 |
| Structural parameters | |||
| I0 (cm−1) [from Guinier] | 0.1 ± 0.0 | 1.3 ± 0.0 | 0.4 ± 0.0 |
| Rg (Å) [from Guinier] | 39.4 ± 2.1 | 35.1 ± 0.2 | 41.9 ± 1.3 |
| I0 (cm−1) [from P(r)] | 0.1 ± 0.0 | 1.3 ± 0.0 | 0.4 ± 0.0 |
| Rg (Å) [from P(r)] | 36.3 ± 0.4 | 35.3 ± 0.1 | 42.1 ± 0.3 |
| Dmax (Å) | 104 | 101 | 139 |
| Experimental MW [from QR]a | 152,490 Da | 143,778 Da | 137,052 Da |
| Calculated MW | 122,371 Da | 122,371 Da | 136,394 Da |
| Ab initio analysis | GASBOR | GASBOR | DAMMIF/DAMMIN |
| χ 2 of ab initio models | 1.0 | 1.2–1.3 | 1.0 |
aMW determined using ScÅtter (47).
MutL endonuclease assay
Mismatch-independent MutL endonuclease assays were performed as described previously (31) with minor modifications. The linear DNA substrate (200 base pairs) was amplified from the pUC19 vector (Invitrogen) using 32P end-labeled 5′-P-d(ATAGTTGCCTGACTCCCCGTCGTGTAGATAACTACG) and 5′-P-d(CGGCAACA ATTAATAGACTGGATGGAGGCG). MutL (152–606 nM), MutL* (152–303 nM), MutL-β (152–606 nM), and MutLCTD (1.15–2.3 μM) were incubated with DNA (10 nM) in the absence and presence of equimolar amounts of β in reaction buffer (20 mM Tris pH 7.6, 30 mM KCl, 0.5 mM ATP, 1 mM MnCl2, 1 mM MgCl2, 1 mM Zn(O2CCH3)2, 0.05 mg/ml BSA, 4% glycerol). Following a 1-h incubation at 37°C, the reaction was stopped with 25 mM EDTA and 1 mg/ml proteinase K (55°C for 20 min). Two times loading dye (90% formamide, 0.025% bromophenol blue, 0.025% xylene cyanol FF, 0.5 mM EDTA, 10% glycerol) was added and reaction mixtures were resolved on 8% polyacrylamide (8 M urea) gels in 0.5× tris–borate–EDTA buffer. Gels were visualized using the Typhoon Trio+ (GE Healthcare). All experiments were performed in triplicate.
ATP hydrolysis assay
ATP hydrolysis activity was performed as previously described (28) with minor modifications. E. coli MutL and MutL-β (1 μM) were incubated with MgCl2 (5 mM) and α-32P-labeled ATP (62.5–2000 μM) in reaction buffer (20 mM Tris pH 8.0, 90 mM KCl, 0.1 mg/ml BSA, 5% glycerol). Reactions (15 μl) were incubated at room temperature for 2 h and resolved by thin-layer chromatography in 0.75 M KH2PO4. ADP accumulation was visualized using the Typhoon Trio+ (GE Healthcare) and quantified using ImageJ (http://rsbweb.nih.gov/ij/). All experiments were performed in triplicate and error bars represent standard deviation of the mean.
DNA binding and helicase assays
The DNA substrate (250 base pairs) was amplified from the pUC19 vector (Invitrogen) using primers 5′-d(GCTTAATCAGTGAGGCACCTATCTCAGCG) and 32P 5′ end-labeled 5′-P-d(CGGCAACAATTAATAGACTGGATGGAGGCG). DNA (5 nM) was incubated with E. coli MutL and MutL-β complex from 40 to 320 nM in reaction buffer (20 mM Tris pH 8.0, 90 mM KCl, 15% glycerol). The E. coli MutL-β complex used for these assays was prepared by freezing a mixture of eMutL and eβ after incubation for 24 h in the absence of reducing agents (this is equivalent to the ‘Day 1’ sample used on the SAXS analysis). To confirm that the E. coli MutL–β complex binds DNA, the complex was incubated with DNA at 22°C for 10 min followed by 30 min on ice. Samples (15 μl) were resolved on 4.5% tris–borate–EDTA gels and visualized using the Typhoon Trio+ (GE Healthcare). All experiments were performed in triplicate.
Helicase assays were performed as described previously (28) with minor modifications. The UvrD expression vector (pWY 1365) was a kind gift from Dr Wei Yang. The 250 bp substrate described above was nicked near the center using Nb.BsrDI (New England Biolabs). The nicked DNA substrate (5 nM) was pre-incubated with increasing amounts of either E. coli MutL or MutL-β (5–80 nM) for 20 min at 22°C in reaction buffer (20 mM Tris pH 7.5, 50 mM NaCl, 3 mM MgCl2, 0.1 mg/ml BSA). Unwinding was initiated by addition of 5 nM UvrD. Reactions (10 μl) were incubated for 35 min at 37°C and resolved on 4.5% tris–borate–EDTA gels. Gels were visualized using the Typhoon Trio+ (GE Healthcare) and quantified using ImageJ (http://rsbweb.nih.gov/ij/). All experiments were performed in triplicate and error bars represent the standard deviation of the mean.
RESULTS AND DISCUSSION
Cysteine-modified variants of MutL and β are functional in vivo
We have previously shown that the conserved QXX[L/I]XP motif (482QPLLIP in E. coli and 487QEMIVP in B. subtilis) found in the dimerization domain of MutL is a genuine β-clamp binding motif (10). However, the complex between MutL and β is weak and, thus, difficult to study using structural biology techniques. To stabilize the complex, we exploited the presence of a naturally occurring cysteine in E. coli MutL (Cys480), located immediately upstream to the β-binding motif. This residue is not conserved in other MutL homologues (48), suggesting that it is likely dispensable for MutL function or its interaction with β. Furthermore, mutation of this residue does not affect DNA mismatch repair activity in vitro or in vivo (49). B. subtilis MutL has a glutamate residue at the equivalent position (Glu485). Based on the lack of conservation, and the fact that E. coli MutL tolerates a cysteine residue at this position, we generated a variant of B. subtilis MutL where a cysteine residue replaced Glu485.
In the crystal structure of the E. coli β-clamp bound to the β-binding peptide derived from DNA polymerase II, the C-terminal residue of the β-clamp (Leu366) is less than 10 Å away from the N-terminus of the polymerase II peptide (Figure 1). Therefore, we added a cysteine at the C-terminus of the E. coli β-clamp (Cys367) to promote the formation of a disulfide bridge with Cys480 found in MutL. We could not predict how far the C-terminus of B. subtilis β would be from Cys485 in B. subtilis MutL. Therefore, we engineered a cysteine at the C-terminus of B. subtilis β (Cys380) preceded by an additional residue (Ser379) to provide enough flexibility to this C-terminal extension and enhance the interaction with Cys485. Serine residues replaced other surface-exposed cysteine residues in MutL and the β-clamp to minimize the formation of unspecific complexes.
Kosinski et al. had previously shown that the single-cysteine variant of E. coli MutL (L480C) retains normal DNA mismatch repair activity in vivo (49). We also wanted to ensure that the cysteine-modified variants of B. subtilis MutL, as well as E. coli and B. subtilis β, retained normal function in vivo. The β-clamp plays a critical role in DNA replication, therefore we used a β-clamp plasmid shuffle assay to investigate the function of E. coli β in vivo. This assay measures the ability of a plasmid expressing physiological levels of wild type or cysteine-modified β to support viability of an E. coli strain harboring a frame shift in the endogenous β gene (dnaN) (33). The plasmid expressing cysteine-modified E. coli β was as efficient as wild-type β at replacing pACYCdnaN+, indicating the cysteine-modified variant of β was functional in replication (Table 1).
For subsequent analysis, we selected representative isolates of both the wild-type β (VB100) and cysteine-modified β (VB101) shuffle strains. Based on Western blotting with anti-β antibodies, cysteine-modified β was expressed at physiological levels (Table 1). The doubling time of the cysteine-modified β strain was slightly slower than that of the isogenic wild type strain (42 min versus 37 min; see Table 1), indicating the modification conferred a modest growth defect. To ensure that this variant of β supported DNA mismatch repair activity in vivo, we measured both the frequency and the rate of spontaneous mutation of rpoB to rifampicin resistance (RifR) for the wild type β or cysteine-modified β strains. Based on a previous study (10), disruption of the β clamp-MutL interaction resulted in a 12- to 43-fold increase in spontaneous mutation rate in RifR. Both the wild type β and βCys strains displayed similar mutation frequencies and rates (Table 2), indicating that cysteine-modified β supports wild type mismatch repair function in vivo.
Having shown that the cysteine-modified variants are active in E. coli, we performed similar experiments in B. subtilis to determine if the MutL and β-clamp variants were also functional in B. subtilis. The cysteine-modified variant of mutL and dnaN were integrated into their native chromosomal location to replace the wild type allele as described (see Materials and Methods). We performed a Western blot to probe for protein accumulation in vivo and found that cysteine-modified MutL and β-clamp accumulated to levels similar to the wild type protein using DnaX as a loading control (Figure 2A). Analysis of the growth curve showed that the single cysteine-modified MutL and β-clamp strains, and double mutant strain grew the same as the wild type control strain PY79 (Figure 2B). We then asked if the MutL and β-clamp variants were able to participate in mismatch repair in vivo. We assayed mutation rate by fluctuation analysis as an indicator for mismatch repair (38). We found that the cysteine variants of MutL and β-clamp, as well as the double mutant, conferred mutation rates within error of the wild type control (Figure 2C). In contrast, the ΔmutL control showed a 25-fold increase in mutation rate, which is similar to our previous measurements for defects in the mismatch repair pathway (38,50).
Figure 2.
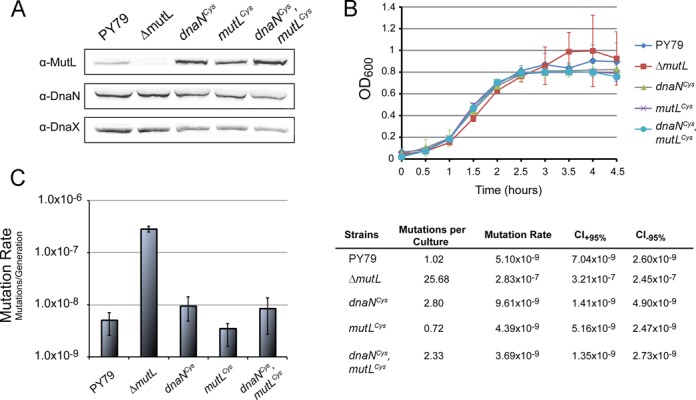
MutL and β-clamp cysteine variants are functional in B. subtilis. (A) Immunoblot analysis of the soluble fraction of B. subtilis cell lysate. The soluble fraction was probed for the presence of MutL and β-clamp (DnaN) cysteine variants. DnaX is a loading control. (B) Growth curves for wild type (PY79), null mutL(ΔmutL), MutL and β-clamp cysteine variants (mutLCys and dnaNCys), and double mutant (mutLCys, dnaNCys) B. subtilis strains. (C) Mutation rate analysis of wild type (PY79), null mutL(ΔmutL), MutL and β-clamp cysteine variants (mutLCys and dnaNCys), and double mutant (mutLCys, dnaNCys) B. subtilis strains.
Given that the cysteine-modified variants of MutL and the β-clamp were fully active in vivo, all subsequent experiments described in this manuscript were performed with these variants of MutL and β. Henceforth, for the remainder of the manuscript MutL, MutLCTD and β refer to the cysteine-modified variants of the proteins unless explicitly specified.
The C-terminal domain of MutL forms a specific complex with β
The cysteine-variants of E. coli and B. subtilis MutL and β were monodisperse in solution and eluted from a size exclusion chromatography column at retention volumes comparable to their native counterparts (Supplementary Figure S1). Furthermore, differential scanning fluorimetry revealed that all MutL and β variants, with the exception E. coli MutLCTD that had a melting temperature 14°C lower than native MutLCTD, had similar thermal stability to their wild-type counterparts (Supplementary Figure S1). Therefore, we presumed that the cysteine mutations not only preserved the function of MutL and β, but also their native conformations.
The C-terminal domain of E. coli MutL (eMutLCTD) was incubated with E. coli β-clamp (eβ) in the absence of reducing agents to promote the formation of a disulfide bridge when the two proteins interact. We resolved the reaction mixtures in SDS-polyacrylamide gels and found that a new species of ∼60 kDa immediately appeared (Figure 3A). This new species had a molecular weight consistent with the presence of a monomer of each protein and it accumulated at the same rate as free eMutLCTD and eβ disappeared. Furthermore, it dissociated in the presence of reducing agent, indicating that a disulfide bridge mediated its formation. To determine whether the eMutLCTD-eβ interaction was specific, we assembled the complex using a variant of eMutLCTD (eMutLCTD*) lacking the β-binding motif (10). Incubation of eMutLCTD* with eβ resulted in the accumulation of two new species of molecular weights ∼60 and ∼80 kDa in a denaturing polyacrylamide gel (Figure 3A). However, there was a significant delay in the accumulation of the 60 kDa species suggesting the β-binding motif enhances the formation of this species (Figure 3A). We reasoned that the 80 kDa molecular weight species could either be caused by the association of two eβ molecules or the interaction of a partially exposed cysteine (Cys180) present in eβ with an additional eMutLCTD* molecule. We favour the latter, because the relative amounts of free eMutLCTD and eMutLCTD* with respect to free eβ seem to suggest the formation of higher order complexes with eMutLCTD* (Figure 3A). Interestingly, the 80 kDa species does not form when the β-binding motif of MutL is present, reinforcing the idea that the formation of a specific complex prevails over non-specific crosslinked products.
Figure 3.
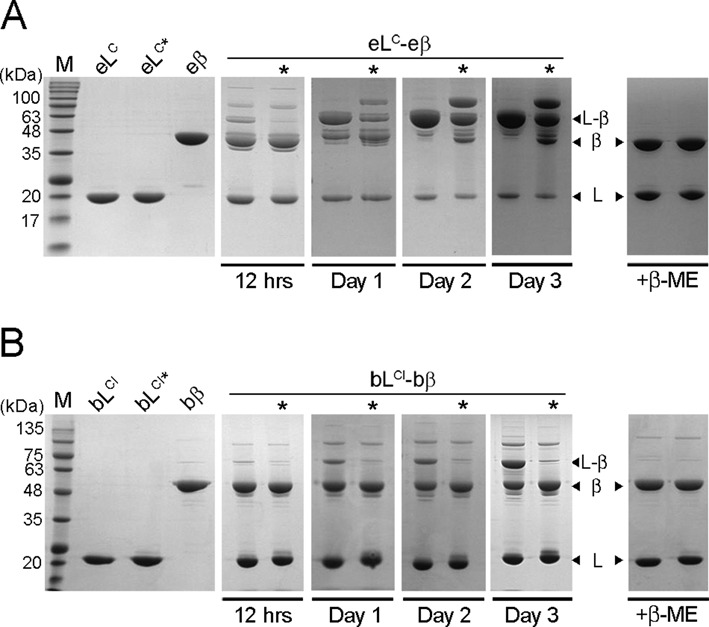
The C-terminal domain of MutL and β form a specific complex in solution. (A) E. coli MutLCTD (eLC), MutLCTD* (eLC*), and β (eβ) were purified and equimolar mixtures of either eLC-eβ or eLC*-eβ (lanes marked with an asterisk) were incubated in the absence of reducing agent. Samples withdrawn from the reaction at the indicated time points were resolved on denaturing gels in the absence of β-mercaptoethanol (β-ME). The right panel is a control gel run in the presence of β-ME. (B) The inactive variants of B. subtilis MutLCTD (bLCI), MutLCTD* (bLCI*), as well as β (bβ) were purified and equimolar mixtures of either bLCI-bβ or bLCI*-bβ (lanes marked with an asterisk) were incubated in the absence of reducing agent. Samples withdrawn from the reaction at the indicated time points were resolved on denaturing gels in the absence of β-mercaptoethanol (β-ME).
Incubation of an inactive variant of C-terminal domain of B. subtilis MutL (bMutLCTDI) with B. subtilis β-clamp (bβ) also yielded a major species at around 75 kDa, consistent with the interaction of one MutL and one β monomer (Figure 3B). However, the formation of this species was not as efficient as in the case of E. coli and longer incubations were required to accumulate significant amounts of this crosslinked product. In contrast to E. coli, formation of the 75 kDa species was almost completely abrogated when we assembled the B. subtilis MutLCTDI–β complex with a variant of MutL lacking the β-binding motif (bMutLCTDI*). The different behaviour of the E. coli and B. subtilis proteins seemed to suggest that the MutL-β complexes are different depending on whether MutL has endonuclease activity; therefore, we decided to characterize both complexes further.
Structural characterization of the MutLCTD–β complex
An intriguing difference between the B. subtilis and E. coli crosslinking experiments was the relatively slow formation of the B. subtilis MutLCTDI–β complex (Figure 3). E. coli MutLCTD–β complex starts accumulating as soon as reducing agents are removed from the buffer and two days later there are no free MutLCTD or β-clamp left in the solution (Figure 3A). Conversely, a significant amount of free B. subtilis MutLCTDI and β remain in solution even after three days without reducing agents (Figure 3B). We entertained the possibility that the E. coli proteins could form two distinct complexes, whereas the B. subtilis proteins could not. Therefore, we decided to characterize E. coli MutLCTD-β complex at two time points, herein referred to as ‘Day 1’ and ‘Day 2’ (see the third and fourth panels in Figure 3A). Conversely, we analyzed the B. subtilis MutLCTDI-β complex only at the later time point to maximize the amount of complex present in solution (Figure 3B, fourth panel).
We used small angle X-ray scattering (SAXS), a technique where scattering data is collected in solution, thereby facilitating the analysis of samples at different time points. Using dynamic light scattering, we confirmed that the E. coli and B. subtilis MutLCTD and β proteins were monodisperse (Supplementary Figure S1). Then, we collected scattering data of the individual proteins at a range of concentrations. None of the samples had concentration-dependent interparticle interactions (Supplementary Table S1 and Figure S3). Furthermore, the scattering curves for MutLCTD and β were also similar to the theoretical scattering profiles derived from the crystal structures of MutLCTD (PDB ID: 1X9Z and 3KDK) and β-clamp (PDB ID: 1MMI and 4RT6). The discrepancy between the theoretical and experimental scattering curves resulted in chi2 values of 1.1 (eMutLCTD), 1.0 (bMutLCTDI), 1.3 (eβ) and 1.5 (bβ) (Supplementary Figures S4 and S5, compare black lines to the experimental scattering curves). Accordingly, the pair-distance distribution functions were indicative of toroidal (eβ and bβ) and elongated (eMutLCTD and bMutLCTDI) particles (Figures 4 and 5 and Supplementary Figures S4 and S5).
Figure 4.
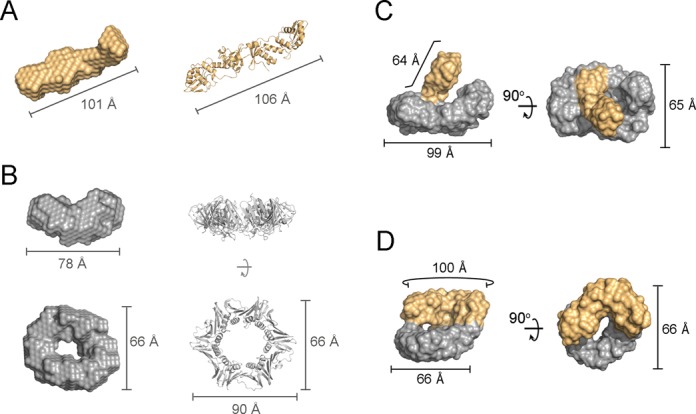
Structural models of the E. coli MutLCTD–β complexes. (A) Ab initio bead model of E. coli MutLCTD (eLC; orange) shown alongside its crystal structure (PDB ID: 1X9Z). (B) Orthogonal views of the refined ab initio model of the E. coli β-clamp (eβ; gray) compared to its crystal structure (PDB ID: 1MMI). (C and D) Orthogonal views of the representative models for each of the most populated clusters of eMutLCTD-eβ at ‘Day 1’ (C) and ‘Day 2’ (D). The moieties presumed to represent MutLCTD and β are highlighted in orange and grey. The overall dimensions of the bead models and crystal structures are indicated for reference.
Figure 5.
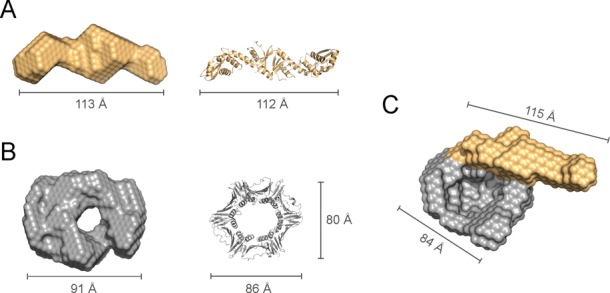
Structural model of the B. subtilis MutLCTD–β complex. (A) Ab initio bead model of B. subtilis MutLCTDI (bLCI; orange) shown alongside its crystal structure (PDB ID: 3KDK). (B) Orthogonal views of the most representative ab initio model of the B. subtilis β-clamp (bβ; grey) compared to its crystal structure (PDB ID: 4RT6). (C and D) Orthogonal views of the refined model for the bMutLCTDI-bβ at ‘Day 2’. The moieties presumed to represent MutLCTD and β are highlighted in orange and grey, respectively. The overall dimensions of the bead models and crystal structures are indicated for reference.
We then incubated eMutLCTD and eβ in the absence of reducing agents for either one (‘Day 1’) or two (‘Day 2’) days, resolved the mixtures by size exclusion chromatography and collected scattering data for both samples (Figure 4C and D and Table 3). The samples showed no signs of protein aggregation and the Kratky plots indicated the presence of globular structure (Supplementary Figure S4). They had similar pair-distance distribution functions and their estimated molecular weights were consistent with a dimer of MutLCTD interacting with one dimer of β (Figure 4 and Table 3). We generated twenty independent ab initio models for each sample and clustered them based on their normalized spatial discrepancy using DAMCLUST (45). We identified four different clusters for the sample at ‘Day 1’ and five different clusters for the sample at ‘Day 2’. However, in both cases one of the clusters was significantly more populated than the rest. Ten out of twenty models of ‘Day 1’ and eleven out of twenty models for ‘Day 2’ belonged to a single cluster. Therefore, we based our analysis on the representative model from the most populated cluster (Figure 4C-D).
The representative model for ‘Day 1’ forms a ring-shaped structure with a handle that resembles a hollow curling stone (Figure 4C). The toroidal moiety of the model is closely related to the SAXS model of eβ and the ‘handle’ has similar shape and dimensions to the SAXS model of eMutLCTD (Figure 4C). The β-binding motifs are found at both ends of the eMutLCTD dimer, therefore the ‘Day 1’ E. coli MutLCTD–β complex seems to adopt a structure where only one protomer of the MutL dimer is bound to the β ring (Figure 4C). Conversely, on the representative model for ‘Day 2’ both ends of eMutLCTD seem to be interacting with β (Figure 4D). The ‘handle’ of the model has collapsed on top the β moiety, suggesting that the two β-binding motifs of E. coli MutLCTD are bound to eβ (Figure 4D). The lack of free E. coli MutLCTD and β after two days of incubation also supports the idea that both subunits of eMutLCTD are bound to the eβ ring (Figure 3A, fourth panel). The differences between the two models, however, should be interpreted with caution. The samples for ‘Day 1’ were collected at much lower concentration than those for ‘Day 2’ and, therefore, the differences could also result from the different signal-to-noise ratio of the two scattering curves.
To understand the formation of the MutL-β complex further, we decided to perform the same experiment with B. subtilis MutLCTDI and β. We incubated the proteins in the absence of reducing agents for two days and collected scattering data (Figure 3B and Table 3). The samples showed no signs of protein aggregation and were folded (Supplementary Figure S5). The B. subtilis MutLCTDI–β complex had similar pair-distance distribution function to the E. coli MutLCTD-β samples (Supplementary Figures S4 and S5). The estimated molecular weight (137 kDa) is comparable to the calculated molecular weight (136 kDa) of the bMutLCTDI–β complex at a 1:1 ratio (Table 3). We generated 20 independent ab initio models and produced a refined ab initio model using DAMMIN (43). The general features of the B. subtilis MutLCTDI-β complex resembled more closely the ‘Day 1’ than the ‘Day 2’ E. coli MutLCTD-β complex, thereby suggesting that the B. subtilis MutL dimer cannot bind the two protomers of the β ring simultaneously (Figure 5). The presence of free B. subtilis MutLCTDI and β in the crosslinking experiments, even after three days of incubation, also supports this interpretation (Figure 3B, fifth panel).
Binding partners of the sliding β-clamp typically bind a single cleft on the ring (34,51–53). Therefore, the complex with a single protomer of the MutL dimer bound to the β ring may represent the functional form of the MutL-β complex (Figures 4 and 5), with the complex where both protomers of MutL are bound to β being an artifact caused by the presence of a reactive cysteine at the other end of the dimer. This explanation is appealing because B. subtilis MutLCTD only forms one complex with β (Figure 5). Furthermore, PCNA (the eukaryotic counterpart of β) stimulates the endonuclease activity of eukaryotic MutLα, which only have a single endonuclease site per dimer, implicitly suggesting that only one of the protomers needs to interact with the sliding clamp. However, we cannot rule out the possibility that both E. coli MutL-β complexes are indeed functional in vivo.
Full-length MutL binds a single cleft of the β-clamp
MutL undergoes a large conformational change upon ATP binding (20,26,28,54,55). Furthermore, ATP-binding greatly stimulates the endonuclease activity of bacterial and eukaryotic MutL (21,22,24). It is possible that the presence of the N-terminal domains of MutL or additional mismatch repair factors favour the formation of one of the complexes with β. Therefore, we repeated the crosslinking experiments with full-length E. coli MutL (eMutL). The cysteine-modified variant of E. coli MutL undergoes the characteristic nucleotide-dependent conformational change when purified over a size exclusion chromatography column (Supplementary Figure S6A).
To test whether eMutL favors the singly- or doubly-bound form of the MutL-β complex, we incubated it with equimolar amounts of eβ in the absence of reducing agents to promote cysteine-mediated crosslinking of the complex. In good agreement with previous experiments using the C-terminal domain of MutL, a new species consistent with the formation of a 1:1 complex readily appeared and accumulated over time (Figure 6). However, this new species stopped accumulating after ‘Day 1’ and a significant amount of free MutL and β remained in solution (Figure 6), thereby suggesting that MutL only binds to one cleft of the β-clamp ring.
Figure 6.
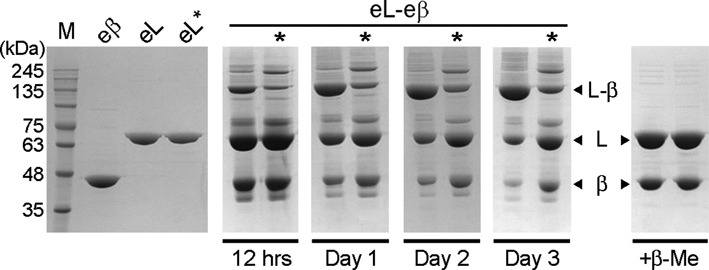
Full length MutL and β form a specific complex in solution. E. coli MutL (eL), MutL* (eL*), and β (eβ) were purified and equimolar mixtures of either eL-eβ or eL*-eβ were incubated in the absence of reducing agent. Samples withdrawn from the reaction at the indicated time points were resolved on denaturing gels in the absence of β-mercaptoethanol (β-ME).
Complex formation was significantly impaired when we used a variant of MutL lacking the β-binding motif (MutL*, 482QPLLI →482ASAAA). Mirroring the results obtained with MutLCTD*, incubation of MutL* with β resulted in the formation of two new species of molecular weights ∼130 and ∼180 kDa in a denaturing polyacrylamide gel (compare Figures 3A and 6). These two products were sensitive to the presence of reducing agent, indicating that disulfide bridges mediate these interactions. The lower molecular weight species is consistent with the presence of one monomer of eMutL and one monomer of eβ and it can presumably form because Cys480 (MutL) and Cys367 (β) can partially react even in the absence of the β-binding motif. The higher molecular weight species is consistent with the presence of two monomers of eMutL and one monomer of eβ (180.7 kDa). The eβ variant has a partially exposed cysteine (Cys180) that could mediate the interaction with the second eMutL monomer. It is worth noting that Cys180 (β) does not mediate the interaction with a second monomer of eMutL unless the β-binding motif has been disrupted, suggesting that the conformation of the 130 kDa species formed with eMutL* may not be identical to that formed with eMutL (Figure 6).
The presence of the N-terminal domain of MutL bias the interaction to a singly bound MutL–β complex and this effect is independent of the presence of nucleotide (Supplementary Figure S6B), indicating that the nucleotide-induced conformational change of MutL is not required to form a specific complex with β.
Functional implications of the MutL interaction with the β-clamp
We have previously shown that disruption of the β-binding motif found in MutL causes a severe mismatch repair defect in organisms lacking MutH, but only a moderate defect in those encoding a mutH gene (10,24). Mismatch recognition by MutS and MutS-dependent activation of MutL are common steps in MutH-dependent and MutH-independent mismatch repair pathways, suggesting that the E. coli β-clamp affects a function of MutL at or following nicking of the newly synthesized strand.
Since MutH, rather than MutL, is responsible for nicking the newly synthesized strand in E. coli mismatch repair, we explored whether β alters a function of E. coli MutL following DNA nicking. E. coli MutL has a role in repetitively loading UvrD onto DNA to facilitate unwinding of the nascent strand towards the mismatch (56) and this function has been linked to the C-terminal domain of MutL (28). We tested whether E. coli MutL stimulated the helicase activity of UvrD differently when bound to β. In good agreement with previously published data (28), we found that MutL stimulated the unwinding activity of UvrD when added in equimolar amounts to the reaction (Supplementary Figure S7A). However, we only observed a minimal increase in UvrD unwinding activity when the MutL-β complex replaced MutL and only when using long (>250 bp) DNA substrates (Supplementary Figure S7A). Since both MutL and MutL–β had similar ATPase and DNA binding activities (Supplementary Figure S7B-C), the interaction between MutL and β seems to be responsible for the enhanced stimulation of UvrD. The marginal effect would explain why disruption of the β-binding motif in E. coli MutL only causes a weak mutator phenotype (10).
The functional implications of this interaction in B. subtilis are starkly different. Human PCNA stimulates the endonuclease activity of human MutLα (21,22). Therefore, we tested whether B. subtilis β could stimulate the nicking activity of MutL on a linear substrate. To see the effect of bβ, we set the experiment at concentrations of bMutL that barely had nicking activity (Figure 7A). Addition of stoichiometric amounts of B. subtilis β greatly stimulated the nicking activity of MutL, an effect that was dependent on the availability of divalent metal ions (Figure 7A). The effect of the β-clamp was also dependent on the physical interaction between MutL and β because the endonuclease activity of bMutL* was not stimulated by bβ (Figure 7B). Interestingly, bβ stimulated the endonuclease activity of bMutL to a greater extend when it was mixed with MutL rather than crosslinked to MutL (Figure 7C). The difference may be attributed to the restricted flexibility of the crosslinked MutL-β complex, or the fact that the crosslinked complex may be a mixture of specific and non-specific complexes, thereby reducing the effective concentration of functional MutL-β complex. While the latter is likely a contributing factor based on the presence of unspecific products when the crosslinking experiments were done with the active cysteine variant of B. subtilis MutLCTD (compare Figure 3B and Supplementary Figure S8), the former is probably important because it has been shown that DNA must access the central cavity of β to exhibit binding (7), and the permanent presence of MutL bound to β may prevent DNA access to the central cavity. It is also tempting to speculate that the transient nature of the MutL-β interaction could provide an integral regulatory mechanism to prevent excessive nicking by MutL. However, this idea awaits validation.
Figure 7.
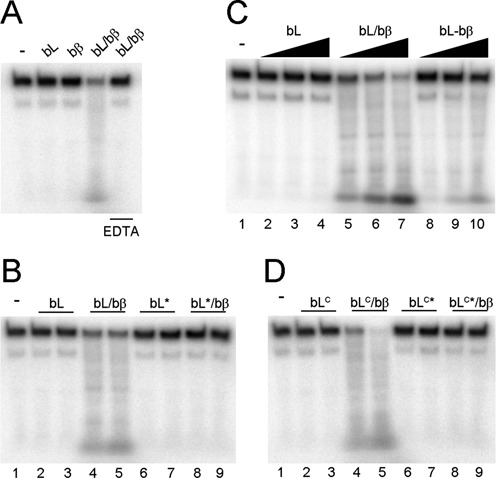
β stimulates the nicking activity of MutL and MutLCTD. (A) Full length B. subtilis MutL (bL; 303 nM) was incubated with a 200 base-pair radiolabelled DNA substrate (10 nM) in the absence or presence of equimolar β (bβ). The reaction was also performed in the presence of 2 mM EDTA where indicated. (B) bMutL and bMutL* (152–303 nM) were incubated with 200 base-pair DNA in the absence and presence of equimolar bβ. (C) bL, bL mixed with equimolar bβ, and crosslinked bL-bβ (152–606 nM) were incubated with 200 base-pair DNA. (D) bMutLCTD and bMutLCTD* (1.15–2.3 μM) were incubated with 200 base-pair DNA in the absence and presence of equimolar bβ.
We have previously shown that the endonuclease domain of B. subtilis MutL (bMutLCTD) does not have endonuclease activity because this region of the protein does not bind DNA (24). We presumed that β stimulates the nicking activity of MutL because it threads DNA onto the endonuclease site and, hence, predicted that the interaction with β may bypass the DNA-binding defect of the endonuclease domain. To probe this idea, we tested whether bβ stimulated the endonuclease activity of bMutLCTD and found that it, indeed, stimulated the nicking activity of bMutLCTD, but not that of the bMutLCTD* variant (Figure 7D). Interestingly, an 8-fold excess of bMutLCTD was required to observe comparable nicking activity to bMutL suggesting a role for the N-terminal domain in the endonuclease activity (Figure 7B and D, compare lane 4 in both gels). DNA bound at the N-terminus (26,57,58) likely enhances catalysis by increasing the frequency of MutL bound to DNA while the β-clamp may specifically feed DNA into its active site. Indeed, modelling the bMutLCTD–β complex using the solution scattering data and the X-ray data from bMutLCTD (PDB 3KDK) and bβ (PDB 4TR6) generated a model where the proximal endonuclease active site of MutLCTD is aligned with the central cavity of β (Supplementary Figure S9). Therefore, it is not surprising that mutations abrogating the MutL-β interaction result in strong mutator phenotypes in B. subtilis (24).
CONCLUSIONS
Characterization of the B. subtilis MutL endonuclease reveals that the β-binding motif in the endonuclease domain of MutL facilitates the β-dependent activation of MutL. The endonuclease domain of MutL is sufficient for catalysis when bound to the β-clamp, however, efficient nicking activity requires the full-length MutL protein (Figure 7B and D). Based on the characterization of the MutL endonuclease, we propose a model where efficient nicking of the nascent strand requires proper coordination between a series of signals. The mismatch recognition factor, MutS, interacts with the N-terminus of MutL (59–61) to communicate the presence of a replication error (Figure 8). In turn, this promotes a conformational change bringing DNA bound at the N-terminus of MutL towards the endonuclease domain. The β-binding motif encoded in the C-terminal domain of MutL mediates a weak interaction with the sliding clamp to activate and orient its nicking activity towards the nascent strand (10,21,31). Collectively, these signals promote transient activation of the MutL endonuclease in the presence of a mismatch (Figure 8).
Figure 8.
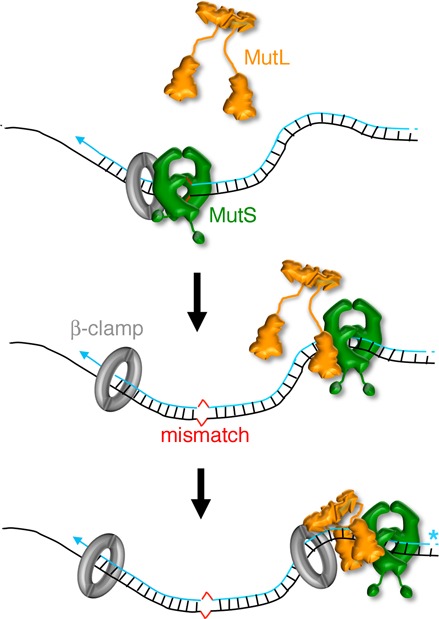
Model for the activation of the endonuclease activity of MutL. Upon encountering a mismatch (red), MutS (green) becomes a sliding clamp that can recruit MutL (orange). Coordinated interactions of MutS and the β-clamp (gray) with MutL enhance the endonuclease activity of MutL and direct it towards the strand containing a pre-existing nick (blue asterisk).
It is well documented how β binds and tethers its binding partners to DNA (5,62–66). However, the affinities for different β-containing complexes vary by several orders of magnitude. In this work, we engineered β-clamp variants in E. coli and B. subtilis that can be specifically crosslinked to one of its weak interacting partners, MutL. This variant allowed us to study the structural organization of the E. coli and B. subtilis MutL-β complexes. Characterization of these complexes explained the different relevance of this interaction in organisms that have or lack MutH. Given that most binding partners interact with the sliding β-clamp using the same molecular determinants, this approach can be easily translated to study the roles of the sliding β-clamp in other cellular processes.
Supplementary Material
Acknowledgments
We are grateful to Dr M. Junop, Dr D. Erie and members of the Guarné laboratory for stimulating discussions. We also thank Dr M. O'Donnell for providing the sliding β-clamp expression vector and Dr W. Yang for providing the MutL and UvrD expression vectors and helpful discussions.
Authors Contributions: M.C.P., A.G., V.M.P.B., M.D.S., J.R.R. and L.A.S. designed the experiments and analyzed data. M.C.P., J.C., V.M.P.B. and J.R.R. performed the experiments and prepared figures. M.C.P., A.G., M.D.S. and L.A.S. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Discovery Grant from the National Sciences and Engineering Research Council of Canada (NSERC) [288295 to A.G.]; Ontario graduate scholarship (to M.C.P.); Public Service Health Grant [GM066094 to M.D.S.]; National Science Foundation Grant [MCB1050948]; NIH Biotechnology Training Grant [T32 GM008353 to J.R.R.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Qin J., Gronenborn A.M. Weak protein complexes: challenges to study but essential for life. FEBS J. 2014;281:1948–1949. doi: 10.1111/febs.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nussinov R., Tsai C.J., Ma B. The underappreciated role of allostery in the cellular network. Ann. Rev. Biophys. 2013;42:169–189. doi: 10.1146/annurev-biophys-083012-130257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gomez M.J., Diaz-Maldonado H., Gonzalez-Tortuero E., Lopez de Saro F.J. Chromosomal replication dynamics and interaction with the beta sliding clamp determine orientation of bacterial transposable elements. Genome Biol. Evol. 2014;6:727–740. doi: 10.1093/gbe/evu052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parks A.R., Li Z., Shi Q., Owens R.M., Jin M.M., Peters J.E. Transposition into replicating DNA occurs through interaction with the processivity factor. Cell. 2009;138:685–695. doi: 10.1016/j.cell.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moldovan G.L., Pfander B., Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Lopez de Saro F.J., O'Donnell M. Interaction of the beta sliding clamp with MutS, ligase, and DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 2001;98:8376–8380. doi: 10.1073/pnas.121009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Georgescu R.E., Kim S.S., Yurieva O., Kuriyan J., Kong X.P., O'Donnell M. Structure of a sliding clamp on DNA. Cell. 2008;132:43–54. doi: 10.1016/j.cell.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stukenberg P.T., Studwell-Vaughan P.S., O'Donnell M. Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 1991;266:11328–11334. [PubMed] [Google Scholar]
- 9.Dalrymple B.P., Kongsuwan K., Wijffels G., Dixon N.E., Jennings P.A. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11627–11632. doi: 10.1073/pnas.191384398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pillon M.C., Miller J.H., Guarne A. The endonuclease domain of MutL interacts with the beta sliding clamp. DNA Repair. 2011;10:87–93. doi: 10.1016/j.dnarep.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maga G., Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 12.Bruning J.B., Shamoo Y. Structural and thermodynamic analysis of human PCNA with peptides derived from DNA polymerase-delta p66 subunit and flap endonuclease-1. Structure. 2004;12:2209–2219. doi: 10.1016/j.str.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 13.Yin Z., Kelso M.J., Beck J.L., Oakley A.J. Structural and thermodynamic dissection of linear motif recognition by the E. coli sliding clamp. J. Med. Chem. 2013;56:8665–8673. doi: 10.1021/jm401118f. [DOI] [PubMed] [Google Scholar]
- 14.Rolef Ben-Shahar T., Castillo A.G., Osborne M.J., Borden K.L., Kornblatt J., Verreault A. Two fundamentally distinct PCNA interaction peptides contribute to chromatin assembly factor 1 function. Mol. Cell. Biol. 2009;29:6353–6365. doi: 10.1128/MCB.01051-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Junop M.S., Obmolova G., Rausch K., Hsieh P., Yang W. Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol. Cell. 2001;7:1–12. doi: 10.1016/s1097-2765(01)00149-6. [DOI] [PubMed] [Google Scholar]
- 16.Acharya S., Foster P.L., Brooks P., Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol. Cell. 2003;12:233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 17.Giron-Monzon L., Manelyte L., Ahrends R., Kirsch D., Spengler B., Friedhoff P. Mapping protein-protein interactions between MutL and MutH by cross-linking. J. Biol. Chem. 2004;279:49338–49345. doi: 10.1074/jbc.M409307200. [DOI] [PubMed] [Google Scholar]
- 18.Joseph N., Sawarkar R., Rao D.N. DNA mismatch correction in Haemophilus influenzae: characterization of MutL, MutH and their interaction. DNA Repair. 2004;3:1561–1577. doi: 10.1016/j.dnarep.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 19.Duppatla V., Bodda C., Urbanke C., Friedhoff P., Rao D.N. The carboxy-terminal domain is sufficient for endonuclease activity of Neisseria gonorrhoeae MutL. Biochem. J. 2009;423:13. doi: 10.1042/BJ20090626. [DOI] [PubMed] [Google Scholar]
- 20.Fukui K., Nishida M., Nakagawa N., Masui R., Kuramitsu S. Bound nucleotide controls the endonuclease activity of mismatch repair enzyme MutL. J. Biol. Chem. 2008;283:12136–12145. doi: 10.1074/jbc.M800110200. [DOI] [PubMed] [Google Scholar]
- 21.Kadyrov F.A., Dzantiev L., Constantin N., Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 22.Kadyrov F.A., Holmes S.F., Arana M.E., Lukianova O.A., O'Donnell M., Kunkel T.A., Modrich P. Saccharomyces cerevisiae MutLa is a mismatch repair endonuclease. J. Biol. Chem. 2007;282:10. doi: 10.1074/jbc.M707617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mauris J., Evans T.C. Adenosine triphosphate stimulates Aquifex aeolicus MutL endonuclease activity. PLoS One. 2009;4:e7175. doi: 10.1371/journal.pone.0007175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pillon M.C., Lorenowicz J.J., Uckelmann M., Klocko A.D., Mitchell R.R., Chung Y.S., Modrich P., Walker G.C., Simmons L.A., Friedhoff P., et al. Structure of the endonuclease domain of MutL: unlicensed to cut. Mol. Cell. 2010;39:145–151. doi: 10.1016/j.molcel.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunkel T.A., Erie D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 26.Ban C., Junop M., Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell. 1999;97:85–97. doi: 10.1016/s0092-8674(00)80717-5. [DOI] [PubMed] [Google Scholar]
- 27.Ban C., Yang W. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell. 1998;95:541–552. doi: 10.1016/s0092-8674(00)81621-9. [DOI] [PubMed] [Google Scholar]
- 28.Guarné A., Ramon-Maiques S., Wolff E.M., Ghirlando R., Hu X., Miller J.H., Yang W. Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. EMBO J. 2004;23:4134–4145. doi: 10.1038/sj.emboj.7600412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez de Saro F.J., Marinus M.G., Modrich P., O'Donnell M. The beta sliding clamp binds to multiple sites within MutL and MutS. J. Biol. Chem. 2006;281:14340–14349. doi: 10.1074/jbc.M601264200. [DOI] [PubMed] [Google Scholar]
- 30.Simmons L.A., Davies B.W., Grossman A.D., Walker G.C. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol. Cell. 2008;29:291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pluciennik A., Dzantiev L., Iyer R.R., Constantin N., Kadyrov F.A., Modrich P. PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 2010;107:16066–16071. doi: 10.1073/pnas.1010662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boivin S., Kozak S., Meijers R. Optimization of protein purification and characterization using Thermofluor screens. Protein Expression Purif. 2013;91:192–206. doi: 10.1016/j.pep.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Babu V.M.P., Sutton M.D. A dnaN plasmid shuffle strain for rapid in vivo analysis of mutant Escherichia coli beta clamps provides insight into the role of clamp in umuDC-mediated cold sensitivity. PLoS One. 2014;9:e98791. doi: 10.1371/journal.pone.0098791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sutton M.D., Duzen J.M., Maul R.W. Mutant forms of the Escherichia colibeta sliding clamp that distinguish between its roles in replication and DNA polymerase V-dependent translesion DNA synthesis. Mol. Microbiol. 2005;55:1751–1766. doi: 10.1111/j.1365-2958.2005.04500.x. [DOI] [PubMed] [Google Scholar]
- 35.Sanders L.H., Devadoss B., Raja G.V., O'Connor J., Su S., Wozniak D.J., Hassett D.J., Berdis A.J., Sutton M.D. Epistatic roles for Pseudomonas aeruginosa MutS and DinB (DNA Pol IV) in coping with reactive oxygen species-induced DNA damage. PLoS One. 2011;6:e18824. doi: 10.1371/journal.pone.0018824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dixon W.J., Massey F.J. Introduction to Statistical Analysis. NY: McGraw-Hill; 1969. [Google Scholar]
- 37.Hall B.M., Ma C.X., Liang P., Singh K.K. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics. 2009;25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolz N.J., Lenhart J.S., Weindorf S.C., Simmons L.A. Residues in the N-terminal domain of MutL required for mismatch repair in Bacillus subtilis. J. Bacteriol. 2012;194:5361–5367. doi: 10.1128/JB.01142-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foster P.L. Methods for determining spontaneous mutation rates. Methods Enzymol. 2006;409:195–213. doi: 10.1016/S0076-6879(05)09012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konarev P.V., Petoukhov M.V., Volkov V.V., Svergun D.I. ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Cryst. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003;36:1277–1282. [Google Scholar]
- 42.Franke D., Svergun D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Svergun D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Svergun D.I., Petoukhov M.V., Koch M.H. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P.V., Svergun D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 2012;45:9. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petoukhov M.V., Svergun D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rambo R.P., Tainer J.A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kosinski J., Plotz G., Guarné A., Bujnicki J.M., Friedhoff P. The PMS2 subunit of human MutLalpha contains a metal ion binding domain of the iron-dependent repressor protein family. J. Mol. Biol. 2008;382:610–627. doi: 10.1016/j.jmb.2008.06.056. [DOI] [PubMed] [Google Scholar]
- 49.Kosinski J., Steindorf I., Bujnicki J.M., Giron-Monzon L., Friedhoff P. Analysis of the quaternary structure of the MutL C-terminal domain. J. Mol. Biol. 2005;351:895–909. doi: 10.1016/j.jmb.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 50.Walsh B.W., Bolz S.A., Wessel S.R., Schroeder J.W., Keck J.L., Simmons L.A. RecD2 helicase limits replication fork stress in Bacillus subtilis. J. Bacteriol. 2014;196:1359–1368. doi: 10.1128/JB.01475-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bubeck D., Reijns M.A., Graham S.C., Astell K.R., Jones E.Y., Jackson A.P. PCNA directs type 2 RNase H activity on DNA replication and repair substrates. Nucleic Acids Res. 2011;39:3652–3666. doi: 10.1093/nar/gkq980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heltzel J.M., Maul R.W., Scouten Ponticelli S.K., Sutton M.D. A model for DNA polymerase switching involving a single cleft and the rim of the sliding clamp. Proc. Natl. Acad. Sci. U.S.A. 2009;106:12664–12669. doi: 10.1073/pnas.0903460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakurai S., Kitano K., Yamaguchi H., Hamada K., Okada K., Fukuda K., Uchida M., Ohtsuka E., Morioka H., Hakoshima T. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005;24:683–693. doi: 10.1038/sj.emboj.7600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sacho E.J., Kadyrov F.A., Modrich P., Kunkel T.A., Erie D.A. Direct visualization of asymmetric adenine-nucleotide-induced conformational changes in MutL alpha. Mol. Cell. 2008;29:112–121. doi: 10.1016/j.molcel.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tran P.T., Liskay R.M. Functional studies on the candidate ATPase domains of Saccharomyces cerevisiae MutLalpha. Mol. Cell. Biol. 2000;20:6390–6398. doi: 10.1128/mcb.20.17.6390-6398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matson S.W., Robertson A.B. The UvrD helicase and its modulation by the mismatch repair protein MutL. Nucleic Acids Res. 2006;34:4089–4097. doi: 10.1093/nar/gkl450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hall M.C., Shcherbakova P.V., Fortune J.M., Borchers C.H., Dial J.M., Tomer K.B., Kunkel T.A. DNA binding by yeast Mlh1 and Pms1: implications for DNA mismatch repair. Nucleic Acids Res. 2003;31:2025–2034. doi: 10.1093/nar/gkg324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Junop M.S., Yang W., Funchain P., Clendenin W., Miller J.H. In vitro and in vivo studies of MutS, MutL and MutH mutants: correlation of mismatch repair and DNA recombination. DNA Repair. 2003;2:387–405. doi: 10.1016/s1568-7864(02)00245-8. [DOI] [PubMed] [Google Scholar]
- 59.Plotz G., Welsch C., Giron-Monzon L., Friedhoff P., Albrecht M., Piiper A., Biondi R.M., Lengauer T., Zeuzem S., Raedle J. Mutations in the MutSalpha interaction interface of MLH1 can abolish DNA mismatch repair. Nucleic Acids Res. 2006;34:6574–6586. doi: 10.1093/nar/gkl944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lenhart J.S., Pillon M.C., Guarne A., Simmons L.A. Trapping and visualizing intermediate steps in the mismatch repair pathway in vivo. Mol. Microbiol. 2013;90:680–698. doi: 10.1111/mmi.12389. [DOI] [PubMed] [Google Scholar]
- 61.Winkler I., Marx A.D., Lariviere D., Heinze R.J., Cristovao M., Reumer A., Curth U., Sixma T.K., Friedhoff P. Chemical Trapping of the Dynamic MutS-MutL Complex Formed in DNA Mismatch Repair in Escherichia coli. J. Biol. Chem. 2011;286:17326–17337. doi: 10.1074/jbc.M110.187641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jergic S., Horan N.P., Elshenawy M.M., Mason C.E., Urathamakul T., Ozawa K., Robinson A., Goudsmits J.M., Wang Y., Pan X., et al. A direct proofreader-clamp interaction stabilizes the Pol III replicase in the polymerization mode. EMBO J. 2013;32:1322–1333. doi: 10.1038/emboj.2012.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xing G., Kirouac K., Shin Y.J., Bell S.D., Ling H. Structural insight into recruitment of translesion DNA polymerase Dpo4 to sliding clamp PCNA. Mol. Microbiol. 2009;71:678–691. doi: 10.1111/j.1365-2958.2008.06553.x. [DOI] [PubMed] [Google Scholar]
- 64.Chapados B.R., Hosfield D.J., Han S., Qiu J., Yelent B., Shen B., Tainer J.A. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- 65.Burnouf D.Y., Olieric V., Wagner J., Fujii S., Reinbolt J., Fuchs R.P., Dumas P. Structural and biochemical analysis of sliding clamp/ligand interactions suggest a competition between replicative and translesion DNA polymerases. J. Mol. Biol. 2004;335:1187–1197. doi: 10.1016/j.jmb.2003.11.049. [DOI] [PubMed] [Google Scholar]
- 66.Sutton M.D. Coordinating DNA polymerase traffic during high and low fidelity synthesis. Biochim. Biophys. Acta. 2010;1804:1167–1179. doi: 10.1016/j.bbapap.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.