

Abstract
Islet amyloid polypeptide (IAPP) is a peptide hormone whose pathological self-assembly is a hallmark of the progression of type II diabetes. IAPP–membrane interactions catalyze its higher-order self-assembly and also underlie its toxic effects toward cells. While there is great interest in developing small molecule reagents capable of altering the structure and behavior of oligomeric, membrane-bound IAPP, the dynamic and heterogeneous nature of this ensemble makes it recalcitrant to traditional approaches. Here, we build on recent insights into the nature of membrane-bound states and develop a combined computational and experimental strategy to address this problem. The generalized structural approach efficiently identified diverse compounds from large commercial libraries with previously unrecognized activities toward the gain-of-function behaviors of IAPP. The use of appropriate computational prescreening reduced the experimental burden by orders of magnitude relative to unbiased high-throughput screening. We found that rationally targeting experimentally derived models of membrane-bound dimers identified several compounds that demonstrate the remarkable ability to enhance IAPP–membrane binding and one compound that enhances IAPP-mediated cytotoxicity. Taken together, these findings imply that membrane binding per se is insufficient to generate cytotoxicity; instead, enhanced sampling of rare states within the membrane-bound ensemble may potentiate IAPP’s toxic effects.
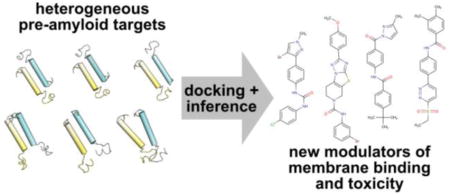
Islet amyloid polypeptide (IAPP, or amylin) is a small (37 residue) peptide hormone that forms fibrillar amyloid aggregates relevant to the pathology of type II, and treatment of type I, diabetes.1 IAPP is predominantly unstructured in solution, but weakly samples α-helical states which are then strongly stabilized upon binding phospholipid bilayers.2,3 The structured domain spans residues 1–22, with α-helical sampling dominating between residues 5 and 19.3–7 It has been suggested that oligomeric self-assembly mediated by this structural transition is associated with cytotoxicity and dysfunction of the insulin-secreting β-cells of the pancreas.1,8–11 In vitro, membrane-bound oligomeric states can accelerate amyloid formation12 and induce membrane leakage.13,14 Here, we describe a computational and experimental strategy to modulate these membrane-bound states of IAPP, and their downstream biochemical effects, by identifying previously unrecognized small molecule ligands from large commercial libraries.
While the mechanisms of IAPP-mediated toxicity are complex and may progress through several interrelated pathways,15 membrane disruption induced by prefibrillar oligomeric forms of IAPP is plainly a crucial component. Recent comparative studies of the amyloid-forming human isoform of IAPP (hIAPP) and the nonamyloidogenic rat isoform (rIAPP) highlight the contribution of helical states (as opposed to β-sheet-rich conformations exclusively) to this phenomenon: both hIAPP and rIAPP can cause membrane leakage13,14 and dose-dependent cytotoxicity,16–18 with the latter attributed to mitochondrial localization and dysfunction.19–21 Importantly, mutagenic diminution of α-helical propensity disrupts the cytotoxic potential of the human isoform.21 In addition, small molecules rationally designed to target helical states effectively inhibit IAPP-mediated cytotoxicity.22 These data all indicate that preamyloid α-helical membrane-bound oligomers are either themselves cytotoxic or enhance the sampling of cytotoxic states. hIAPP and rIAPP differ at 6 out of 37 residues, of which one substitution (H18R) occurs in the helical subdomain and preserves its amphipathic topology. Both isoforms appear to sample broadly similar membrane-bound monomeric structures, although the variety of different detergent micelle and bicelle systems used as model membranes in these NMR studies3,5–7 make direct comparisons difficult. The effects of synthetic inhibitors on rIAPP membrane binding, hIAPP-mediated cytotoxicity, and lipid-catalyzed hIAPP amyloidogenesis are strongly correlated,23,24 further suggesting that both isoforms sample similar membrane-bound states. The observed differences in cytotoxic potency between hIAPP and rIAPP thus appear to result from differences in the energetics of membrane binding and cooperative self-assembly25 rather than fundamentally distinct cytotoxic mechanisms.21 Given the striking commonalities in conformation and pathological activity of the two isoforms, rIAPP serves as a powerful and widely used model system to characterize prefibrillar states of hIAPP without experimental complications due to protein aggregation.
IAPP is one of a number of intrinsically disordered proteins for which membrane binding can result in the induction of a subset of pathological structures. Others include amyloid-β from Alzheimer’s disease and α-synuclein from Parkinson’s disease.8 A central challenge in developing insights in these systems is the heterogeneity and dynamic nature of the membrane-bound ensemble. In previous work, we addressed this by using sub-binding affinity concentrations of rIAPP in the presence of Nanodisc model membranes.26 This created an environment where only a small fraction (~0.5%) of protein samples a membrane-bound oligomeric state. Using single-pair Förster resonance energy transfer (spFRET), we were able to detect the formation of dimers with no progression to higher-order species. SpFRET-derived restraints were used in a computational refinement protocol to develop a set of α-helical models representative of the membrane-bound ensemble of rIAPP dimers.
These models present us with the opportunity to modulate cooperative membrane-mediated self-assembly: we expect that selective ligands targeting these experimentally derived conformations should alter the sampling of the initial membrane-bound dimeric ensemble so as to increase or decrease the overall fraction of membrane-bound IAPP. Rational design targeting monomeric α-helical IAPP has already been used to potently inhibit membrane binding,22,27,28 so any inhibitors we develop could act solely by interacting with monomeric states. In contrast, any bona fide enhancers of membrane binding must act by binding to membrane-bound dimers (or dimer-like states within membrane-bound oligomers) and enhancing their sampling. Such ligands would thus directly validate our spFRET-derived models and could also serve as pharmacological reagents to better understand the pathological self-assembly process of IAPP. The challenge in developing such compounds lies in effectively and rationally targeting a heterogeneous collection of states rather than a single well-defined structure. In this article, we develop and demonstrate a novel and efficient approach to this problem that encompasses computational docking, statistical inference of ligand activity, and robust experimental assays of compound effects on IAPP–membrane binding and IAPP-mediated cytotoxicity.
METHODS
Human and rat isoforms of IAPP were synthesized using standard Fmoc methods at the Keck Biotechnology Resource Laboratory at Yale University (New Haven, CT) or purchased from Elim Biopharmaceuticals (Hayward, CA). Fluorescent dyes were obtained from Life Technologies (Carlsbad, CA). 1,2-Dioleoylphosphatidylglycerol (DOPG) was purchased as powder from Avanti Polar Lipids (Alabaster, AL). Screening compounds were obtained from ChemDiv, Inc. (San Diego, CA), Maybridge (Waltham, MA), ChemBridge Corp. (San Diego, CA), or via the Yale Center for Molecular Discovery (YCMD, New Haven, CT). Other reagents were obtained from Sigma-Aldrich, unless otherwise stated.
Computational Prediction of Binding Selectivity
All small molecule structures were energy-minimized using the UFF force field29 implemented in Open Babel 2.230 prior to docking. The targets for computational docking were one monomeric, three antiparallel dimeric, and three parallel dimeric α-helical models of IAPP, generated by incorporating spFRET measurements into Rosetta31,32 computational refinement as previously described.26 These models are available in PDB format upon request. Models were prepared for docking using MGLTools 1.5.4,33 and docking was performed using Autodock Vina 1.1.2,34 running on local workstations and Yale High Performance Computing clusters.
Autodock results were used to train a fingerprint-based partial least-squares regression (PLSR) statistical inference algorithm. We implemented both feature-based fingerprints, which encode the presence or absence of specific chemical features as bit values of 1 or 0, respectively, in a binary vector, and topological fingerprints, which encode the various patterns of chemical connectivity within a compound by setting a particular combination of bits to take value 1. Fingerprints generated by either method are degenerate in that distinct (but similar) compounds may share the same fingerprint. Feature-based Molecular ACCess System (MACCS) fingerprints35 were generated using Open Babel 2.2, while topological 2048-bit Extended-Connectivity FingerPrints (ECFP)36 and feature-based 881-bit CACTVS37 keys were generated using ChemFP 1.0a1.38 PLSR inference was performed using custom Python scripts employing the SciPy module39 following the Nonlinear Iterative PArtial Least-Squares (NIPALS) algorithm40 as previously implemented41 and was used to predict the Autodock scores of a given small molecule binding to each of the seven IAPP models. Selectivity was defined as the difference between the lowest docking score for any of the three antiparallel dimers and the lowest docking score for any of the three parallel dimers such that negative values indicate preferential binding to antiparallel states. An iterative scheme was used to extend the training set into relevant regions of chemical space sampled by the NIH Molecular Libraries Small Molecule Repository (MLSMR) collection, as described in the Results section. The optimized training set was then applied to the YCMD compound collections. The 4000 compounds predicted to have the most positive and most negative selectivity scores (2000 each) were docked to the seven IAPP models, and the 80 most selective compounds from this step were then purchased and used for experimental screening, along with 20 compounds selected at random from the YCMD set.
Fluorescence Correlation Spectroscopy (FCS) Measurements of IAPP Binding and Modulation of Membrane Binding
IAPP was fluorescently labeled by standard amine coupling: carboxytetramethylrhodamine-succinimidylester (TAMRA-SE) was added in 5-fold molar excess to 250 μM rIAPP in 10 mM potassium phosphate, pH 7.2, and incubated for 2 h at room temperature. Peptide was separated from free dye using HiTrap G-25 desalting columns (GE Healthcare, Piscataway, NJ), and the extent of labeling was verified by mass spectrometry (Waters LCT, Waters Corp., Milford, MA). Labeled protein was flash-frozen and stored at −20 or −80 °C until use. Large unilamellar vesicles (LUVs) were prepared by drying a film of DOPG under a stream of argon, followed by lyophilization for 2 h, rehydration for 1 h in buffer (20 mM Tris, pH 7.4, 100 mM NaCl), and extrusion through a 100 nm diameter filter (Whatman, GE Healthcare, Piscataway, NJ). FCS samples contained 10 nM TAMRA-labeled rIAPP in 20 mM Tris, pH 7.4, 100 mM NaCl, 1% DMSO, in the presence or absence of 1.6 μM DOPG LUVs, and 10 μM of a given compound. Tests on representative compounds showed that the addition of 10 μM unlabeled rIAPP (for a final compound:protein ratio of 1:1) had minimal effects on the results (Figure S2b).
FCS measurements were performed on a home-built instrument as previously described.42 The output from a 561 nm, 50 mW continuous-wave laser (Newport Corp., Stratford, CT) was adjusted to a power of 5 μW using neutral-density filters, and directed into an Olympus IX-71 inverted microscope (Olympus America, Center Valley, PA) with a 60×/1.2 NA water-immersion objective. Emitted fluorescence was isolated using a 585 nm long-pass dichroic mirror and a 600 nm long-pass emission filter (Chroma Technology Corp., Bellows Falls, VT) and transmitted via a 50 μm multimode fiber (Oz Optics, Ottawa, Canada) to an avalanche photodiode detector (PerkinElmer, Waltham, MA) coupled to a Flex03-LQ-12 hardware correlator (Correlator.com, Bridgewater, NJ). Analysis of autocorrelation traces was performed using custom scripts in Matlab (Mathworks, Natick, MA). FCS data were fit to a model of a single molecular species diffusing in three dimensions, where the normalized autocorrelation G(τ) is given by
| (1) |
where N is the mean number of labeled particles in the detection volume, s is a structure factor that describes the dimensions of the observation volume, τ is the delay time, and τD is a diffusion time that is directly proportional to the hydrodynamic radius (i.e., it increases along with particle size). At 1.6 μM DOPG, IAPP exists as a mixture of lipid-bound and free states and therefore displays an apparent diffusion time intermediate between the solution (~0.25 ms) and completely LUV-bound (~7 ms) value. Here, we simply report an apparent diffusion time that is a nonlinear combination of these two values. Compounds that enhance lipid binding induce an increase in the apparent τD, while inhibitors decrease this parameter.
Compound Effects on IAPP Cytotoxicity
Rat insulinoma INS-1 cells (832/13, Dr. Gary W. Cline, Department of Internal Medicine, Yale University) were cultured at 37 °C and 5% CO2 in phenol red free RPMI 1640 media supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin (all from Life Technologies, Carlsbad, CA), and 2% INS-1 stock solution (0.5 M HEPES, 100 mM L-glutamine, 100 mM sodium pyruvate, and 2.5 mM β-mercaptoethanol). Cells were passaged upon reaching ~95% confluence (0.25% Trypsin-EDTA, Life Technologies), propagated, and/or used in experiments. Cells used in experiments were pelleted and resuspended in fresh media with no Trypsin-EDTA.
Cell viability was measured using the CellTiter Blue (CTB) fluorescence-based assay. CTB reagent (Promega, Madison, WI) comprises nonfluorescent resazurin, which is metabolically reduced to fluorescent resorufin in living cells. We have previously shown that for INS-1 cells the CTB readout directly corresponds to cell loss measured by direct counting.9 Cells were plated at a density of 20 000 cells/well (500 μL/well) in 24-well plates (BD Biosciences, San Diego, CA). After culturing for 48 h, media was replaced with fresh media containing hIAPP and small molecules premixed at the desired concentration. Cells were incubated at 37 °C and 5% CO2 with peptide and small molecules for an additional 48 h for experiments measuring the effect of computationally selected small molecules. After the incubation period, CTB reagent (100 μL) was added to each well and incubated at 37 °C and 5% CO2 for 2.5–4 h. Fluorescence of the resorufin product was measured on a FluoDia T70 fluorescence plate reader (Photon Technology International, Birmingham, NJ). All solutions included 0.1% DMSO and 1% H2O to account for the addition of peptide and small molecules vehicle to sample wells. Wells that included vehicle but not peptide or small molecule served as the negative control (100% viable), and wells containing 10% DMSO were the positive control (0% viable). Percent viability, V, was calculated using the following equation
| (2) |
Each independent variable is the average fluorescence of four technical replicates from the negative control (〈N〉), positive control (〈P〉), and samples (〈S〉).
RESULTS
The premise of this work is that ensembles of preferentially sampled states from a heterogeneous system are viable targets for rational screening and the development of novel biophysically and biologically active ligands. Our overall approach (shown schematically in Figure 1) begins as a superficially simple effort to dock a compound library to a structural model. However, this is anything but routine for a heterogeneously sized, partially structured, dynamic membrane-bound system such as IAPP. To our knowledge, the only other successful comparable effort is from an academic–industrial collaboration targeting monomeric states of α-synuclein that identified two active compounds from a 33 000 member library.43 To target membrane-bound states of IAPP, we perform computational docking to a small set of dimeric models and, through an iterative inference protocol, select putative hits from large compound libraries. Hits identified in this way are then validated using synthetic membrane binding assays and cytotoxicity measures.
Figure 1.

(a) Sequences of human (hIAPP) and rat (rIAPP) isoforms of IAPP. Differences between the two isoforms are in bold. The gray region indicates the approximate extent of the structured domain in the membrane-bound state. Stars indicate residue positions labeled in our previous spFRET study of membrane-bound IAPP dimers.26 (b) One representative spFRET-derived model of an IAPP dimer. The gray region indicates the hypothesized orientation of the membrane relative to the dimer. (c) Schematic of a combined computational and experimental approach to pharmacologically target conformationally plastic proteins based on an ensemble of targets (see text for details).
Our previously published spFRET-constrained ensemble predominantly sampled states with the helical domains of two IAPP monomers arranged in an antiparallel fashion, although a small population of parallel helix–helix dimers was also evident. In contrast, simulations from that work in which experimental constraints were omitted generated parallel and antiparallel dimers in approximately equal numbers. In order to define a sufficiently diverse panel of targets for this effort, we selected the three most stable of the antiparallel and parallel dimer models from these ensembles (shown in the first panel of Figure 1). The three antiparallel models represent stable conformations of rIAPP dimers that are fully consistent with all experimental observations of these states,26 whereas the three parallel models are not evident in the experimental data but are locally stable and may well be transiently sampled within the membrane-bound ensemble.
Computational Prediction of Binding Selectivity
It is computationally demanding to use a docking algorithm, such as Autodock,34 on multiple targets. Here, we are docking libraries on the order of 105 to 106 compounds to seven targets (i.e., one monomer, three antiparallel dimers, and three parallel dimer models). Moreover, as a strategy for manipulating intrinsically heterogeneous structures, small molecule docking to a modeled structural ensemble will often benefit from much larger sets of targets and decoys. We therefore investigated the capacity of fingerprint-based inference methods to efficiently predict Autodock scores of compounds binding the above arrangements of IAPP.
A fingerprint is a compact one-dimensional representation of the topological structure and/or chemical features of a compound. The conversion of diverse chemical structures to a common one-dimensional format facilitates the use of statistical inference methods to predict properties of interest. Here, we use a PLSR algorithm40,44 that is closely related to principal components analysis and is capable of efficiently identifying linear relationships between pairs of matrices. In our case, one matrix consists of fingerprints for each small molecule in a database, whereas the other contains the corresponding Autodock scores. Once optimized using a suitable training set, PLSR can then predict new rows of one matrix (in this context, Autodock scores) given the corresponding rows of the other matrix (i.e., fingerprints of new compounds not in the training set).
To generate an initial training data set, we performed docking calculations with the panel of IAPP targets described above and 2210 compounds from the NIH Clinical Collection and Microsource Gen-Plus and Natural Products libraries. We then randomly selected 20 compounds from this panel as a test set and used the remaining 2190 compounds as a training set for PLSR. We compared various fingerprinting algorithms and evaluated them based on their accuracy in predicting the test set Autodock scores for monomeric IAPP and the best (lowest-scoring) antiparallel and parallel dimer models. We found that 881-bit CACTVS fingerprints37 outperformed the widely used 166-bit MACCS keys35 and topological ECFP fingerprints,36 achieving high accuracy for the training set (R2 = 0.79) and reasonable accuracy for the test set (R2 = 0.61), as shown in Figures 2a and S1. CACTVS fingerprints are therefore sufficiently faithful representations of compound structures to enable rapid prediction of binding selectivity for membrane-bound IAPP states.
Figure 2.
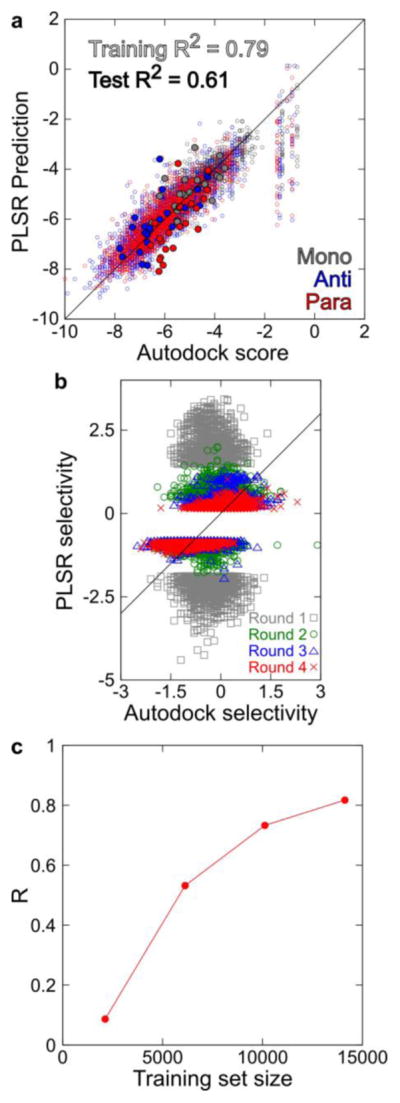
Statistical inference of IAPP–compound binding selectivity. (a) PLSR predictions compared with Autodock-derived affinity scores for 2190 compounds in the training set (small open circles) and 20 compounds in the test set (large closed circles) binding monomeric IAPP (red), the lowest-scoring antiparallel dimer (blue), and the lowest-scoring parallel dimer (cyan). (b) Comparison of PLSR predictions and Autodock scores obtained through the iterative extension of the training set. In each iteration, the 4000 most selective compounds from PLSR predictions were docked to a panel of IAPP models and added to the training set. (c) The correlation between PLSR predictions and Autodock scores increased progressively and asymptotically as the training set size was increased.
The predictive capacity of this approach is dependent on the extent to which the training set of small molecules covers similar regions of chemical space as the test compounds under investigation. We therefore developed an approach to extend the training set into regions of chemical space more likely to be populated by compounds capable of selectively interacting with our models. This was achieved by iteratively performing PLSR predictions on MLSMR, a highly diverse collection of ~350 000 structures available through PubChem. Using the prediction matrix obtained from the initial 2210 compounds, we predicted Autodock scores for the full MLSMR compound set. A set of 4000 compounds was identified and predicted to be selective for either antiparallel or parallel IAPP constructs (2000 compounds each). Explicit Autodock calculations for these 4000 test compounds were determined, and a new training set composed of the initial 2210 compounds and the 4000 new compounds was created. This whole process was repeated, with each iteration producing a new training set more tightly focused on the regions of chemical space enriched in antiparallel- and parallel-selective compounds. Test set prediction accuracy increased progressively (Figure 2b,c) until it matched the predictive power observed for the initial 20 test compounds (Figure 2a).
This final training set was used to predict Autodock scores for 139 584 compounds from commercial libraries available through the Yale Small Molecule Discovery Center (collectively denoted the SMDC set). Explicit Autodock simulations were then performed on 8000 SMDC compounds predicted to have the highest degree of selectivity (4000 each of antiparallel- and parallel-selective compounds). The final Autodock selectivity scores were then used to choose 81 compounds (41 antiparallel- and 40 parallel-specific, respectively) for subsequent experimental screening.
Compound Effects on Membrane Binding
Selected compounds show a range of activities in an IAPP–membrane binding assay. We used FCS to characterize the diffusive properties of fluorescently labeled rIAPP. FCS measures the diffusion time (τD), which is directly proportional to the size of the diffusing particles. We leveraged the large increase in the diffusion time of free IAPP upon binding to a liposome42 to characterize IAPP–membrane interactions. By measuring the apparent diffusion time of a mixture of fluorescently labeled IAPP and unlabeled lipid vesicles in the presence and absence of each compound, we were able to identify modulators that enhanced (increased τD) or inhibited (decreased τD) membrane binding (Figure S2a).
Computational prescreening dramatically enhances the sampling of modulators of membrane binding. As a negative control, 20 compounds were selected at random from the SMDC set and compared to the set of 81 computationally selected compounds, with respect to their capacity to modulate IAPP diffusion in the presence of liposomes (Figure 3a). In general, randomly selected compounds had little effect on IAPP diffusion or membrane binding, generating mean fold changes in τD of 0.8 ± 0.2. Of the 81 compounds selected above, 8 compounds generated an increase in τD more than 3 standard deviations above this range (a hit rate of ~10%), as compared to 0 compounds from the random set. Our computational preselection approach has thus unambiguously and substantially improved the odds of finding a compound with biophysical activity over random chance (i.e., unbiased screening). Closer examination reveals that 7 of the 8 hits targeted the experimentally inferred antiparallel states, while the eighth targeted an alternative parallel state (Figure 3b,c).
Figure 3.

FCS-based characterization of diverse new modulators of IAPP membrane binding. (a) Histogram of effects on IAPP–membrane binding for compounds selected by our screening protocol (gray bars) or at random (white bars) from the SMDC set. Enhanced membrane binding results in an increase in measured diffusion time. Compounds that slow the diffusion of IAPP in solution by more than 2-fold, and hence may exert their effects by mechanisms other than targeting membrane-bound states, are indicated by lighter shading in this and subsequent panels. (b) Histogram as in (a), showing only compounds with enhanced selectivity for antiparallel dimer targets. This set is highly enriched in enhancers of binding, including compounds 1–3. (c) Histogram as in (a), showing only compounds with enhanced selectivity for parallel dimer targets. (d) Structures of membrane-binding enhancers 1–3 and toxicity enhancer 4. Representative poses of these compounds docked to IAPP dimer models are shown in Figure S3.
In separate measurements, we assayed the effect of compounds on the diffusion of IAPP in the absence of lipid. While small increases or decreases in τD would indicate expansion or compaction, respectively, of the disordered solution state of IAPP induced by ligand binding, several compounds (indicated by lightly shaded bars in the histograms of Figure 3) induced larger (>2-fold) increases in τD. Such slow diffusion of IAPP in the absence of lipid could result from IAPP oligomerization or IAPP binding to small molecule micelles, possibly as a result of poor solubility. These two possibilities are distinguishable by additionally assessing the per-particle brightness parameter from the FCS measurement.45 None of the 13 compounds that markedly increased τD induced an increase in per-particle brightness that would be expected from the presence of multiple IAPPs. The observation of slowly diffusing IAPP is, therefore, either due to a ligand simultaneously inducing protein aggregation and quenching label fluorescence or (more probably) the result of solubility limitations under these conditions (10 μM compound), a costly and widespread source of false positives in screening.46 This observation highlights the ease with which FCS can be used to identify these potential artifacts at an early stage in the screening process. While it is possible that some of these 13 putative aggregate formers are instead exerting biologically informative effects on the solution-phase IAPP oligomeric ensemble, for the purposes of this work, we exclude them from further analysis.
Compound Effects on Toxicity
A cell-based assay was used to determine how identified modulators of membrane binding affect IAPP-induced toxicity. INS-1 cells were exposed to a concentration of human IAPP that reduced colorimetrically assessed viability by ~50% (8–12 μM, depending on the batch of synthetic peptide), in the presence and absence of 10 and 30 μM of the selected compounds. We assayed 13 screen-derived compounds, including enhancers and inhibitors of membrane binding from both parallel and antiparallel searches, as well as seven randomly selected compounds (Figure 4). Several compounds induced >25% toxicity at 30 μM in the absence of hIAPP, with two showing similar compound-only toxicity even at 10 μM. These data points were excluded from further analysis.
Figure 4.
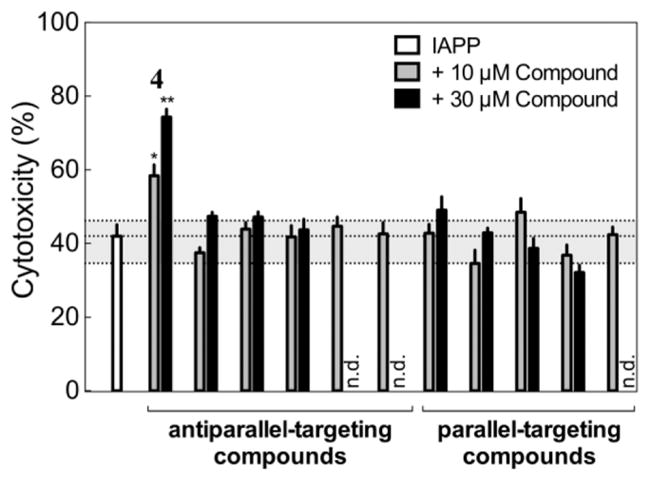
Effects of selected compounds at 10 μM (gray) and 30 μM (black) concentrations on IAPP-mediated toxicity in cell culture experiments. Antiparallel-selective compound 4 robustly enhances toxicity (*, P < 0.01; **, P < 0.0001). The dashed gray region spans the complete range of effects observed for seven compounds randomly selected from the SMDC set at a concentration of 10 μM, and n.d. indicates compounds that induced >25% toxicity even in the absence of IAPP.
Compound 4 clearly and significantly enhanced IAPP-mediated toxicity. In the presence of 10 μM 4, the cytotoxicity induced by IAPP increased from 42 ± 3.1% to 58 ± 3.0%, a toxicity enhancement of ~16%. The effect was more pronounced at 30 μM 4, with IAPP-mediated cytotoxicity increasing to 74 ± 2.2%, an enhancement of ~32%. Three additional compounds modulated IAPP activity to significant but smaller extents at 30 μM but not at 10 μM; tested compounds ranged from ~10% rescue to >30% toxicity enhancement. In comparison, toxicity measurements of the seven randomly selected compounds at 10 μM showed minor effects on IAPP toxicity (3.1 ± 4.3% rescue). These control measurements enable us to estimate that the probability of finding a hit at least as active as 4 from 13 trial compounds by random chance is ~5 × 10−5 (see Supporting Information for details). Clearly, our computational strategy is capable of identifying small molecule reagents that are biologically active in cell culture.
DISCUSSION
Tools such as computational docking have transformed the capacity of medicinal chemists to identify lead compounds that bind established protein targets.47 An exciting possibility, however, is that these tools have reached a level of refinement such that they can instead directly participate in the process of elucidating molecular mechanisms relevant to disease states. The membrane-bound oligomeric ensemble of IAPP represents one such system in which a diversity of conformations and oligomeric species governs the development of pathology. Subsets of these states and/or their interconversion are responsible for one or more gains-of-function. These include amyloid nucleation, membrane translocation, membrane leakage, cytotoxicity, cooperative membrane binding, and mitochondrial localization.15,48 Interactions with cations,49,50 binding partners such as insulin,51,52 or specific receptors15 may all play important roles in modulating these phenomena. Conventional computational screening efforts that might target such a diverse collection of gains-of-function are made particularly challenging by the ever-increasing sizes of small molecule libraries (both real and computed) and the intrinsically heterogeneous nature of the putative targets.
The target preferences of compounds that modulate IAPP membrane binding simultaneously inform our understanding of the membrane-bound IAPP ensemble and validate our earlier structural effort.26 Small molecules predicted to be selective for antiparallel forms of IAPP have a significantly enhanced propensity to increase membrane binding of IAPP. In fact, three antiparallel-targeting ligands (compounds 1–3 in Figure 3d) increase the IAPP diffusion time by at least a factor of 2 in the presence of lipid while minimally affecting τD in the absence of lipid, compared to none of the parallel-targeting compounds. This effect directly supports the spFRET-based observation of membrane-bound antiparallel states: selective drug binding to these states could enhance their population so as to cooperatively increase IAPP’s overall affinity for the membrane. In contrast, inhibition of membrane binding appears to be less dependent on the choice of target, with 3/41 antiparallel-targeting compounds and 4/40 parallel-targeting compounds decreasing the equivalent τD by at least 2-fold. The lack of selectivity with regard to inhibition indicates that these compounds may competitively bind to the membrane-binding interface of IAPP in either a dimeric or monomeric state.
The screening approach employed here possesses certain important limitations. First, a set of coarse-grained models of IAPP dimers cannot capture the full diversity of structures sampled by IAPP. A trade off must be made between the number of docking targets included, the resolution of structural information about the system, and the computational resources available. In this case, a set of three antiparallel targets and three parallel decoys was sufficient to generate multiple active hits. Second, neither the published spFRET-constrained model refinement26 nor our current docking calculations included a simulated membrane, due to the technical difficulty in accurately simulating lipid–protein and lipid–small molecule interactions at reasonable computational cost. Hydrophilic ligands could, in theory, be hindered from binding to the membrane-facing surface of IAPP dimers, thereby biasing our results. However, the set of 81 experimentally characterized compounds is significantly but not overwhelmingly lipophilic (calculated octanol/water log P values30 of 4.1 ± 1.5), suggesting that they, in general, ought to be able to access membrane-facing surfaces via the lipid phase.
Despite these caveats, our approach has enabled the identification of several novel active compounds that include three potent agonists of membrane binding and one agonist of cytotoxicity. The observed activity of compound 4 is unexpected and particularly intriguing because, despite its enhancement of IAPP-mediated toxicity, it is a fairly potent inhibitor of membrane binding, reducing τD in the presence of lipid by >2-fold. By comparison, the membrane-binding enhancers 1–3 either minimally affected cytotoxicity or caused levels of compound-only toxicity that precluded insight into their true effects on IAPP-mediated cytotoxicity. These findings clearly demonstrate that membrane binding alone is insufficient for IAPP to exert its cytotoxic effects. This reinforces the idea that numerous gains-of-function, beyond membrane leakage alone, may be relevant to toxicity.8,15,48 Recently described assays of compound effects on IAPP-induced membrane leakage,13 lipid-catalyzed IAPP amyloidogenesis,53 and intra-cellular localization21 could help to determine the particular mechanisms at play. Another important consideration is that membrane charge, curvature, and composition can all modulate the lipid-binding affinity of IAPP12,54 and could potentially perturb the membrane-bound structural ensemble. Proteinaceous or other nonlipid components of biologically derived membranes can also dramatically affect IAPP–membrane interactions.55 Also of note is the observation that over-stabilization of helical states, by an excess of either fluorinated solvents56 or membranes,12 can instead slow the time scale of fibril formation. Here, we used DOPG vesicles for our FCS experiments for consistency with earlier spFRET experiments.26 Subsequent NMR and spFRET experiments with appropriate, physiologically relevant membrane models could be used to refine structural models of prefibrillar membrane-bound IAPP for future screening and design efforts.
Moreover, the observation that 4 can bias the membrane-bound ensemble so as to simultaneously disfavor binding and enhance the sampling of toxic conformations suggests that particular, rare states within the ensemble are likely to be responsible for the generation of the relevant toxic IAPP species. It should be emphasized that our results do not imply that IAPP dimers are themselves the toxic species; instead, the toxic species may be the downstream products of an as-yet unknown assembly mechanism. Our results also do not indicate that hIAPP and rIAPP sample identical conformations in identical proportions but, rather, that the two populate sufficiently similar structures that compounds targeting the helical region of one peptide can exert effects on the other despite differences in activity16,57 and assembly pathways.58,59 In general, toxic species in amyloid diseases are thought to comprise a small fraction of the total precursor, and sensitive techniques such as autoradiography have been necessary to directly identify this subpopulation in systems such as amyloid-β.60 Agonists such as 4 might serve as pharmacological chaperones to aid in the identification and characterization of toxic states by enriching their population, mirroring the many contexts in which deleterious familial mutations have advanced our understanding of amyloid pathogenesis and toxicity.61–64 Future studies will focus on understanding how the interactions between IAPP and compounds 1–4 mediate their varied effects on membrane binding and toxicity. We anticipate that this approach will allow dissection of the mechanisms of membrane-mediated IAPP self-assembly and the consequent development of pathology in type II diabetes.
We have demonstrated that bioactive compounds can be identified from large libraries for targets as challenging as the transient, membrane-bound and heterogeneous oligomeric ensemble of an intrinsically disordered protein. To date, successful pharmacological modulation of amyloid assembly pathways has involved either structure-aided drug design targeting a well-defined state,65,66 conventional high-throughput screening unaided by structural insights,67 or computational docking to monomeric states derived from unconstrained68 or constrained43 molecular dynamics simulations. Our approach bridges these strategies, by using structural insights about relevant oligomeric states to strongly bias screening efforts, while also taking into account the potential for intrinsic structural heterogeneity in the target pool. Our combined computational and experimental strategy for the identification of novel, active small molecule modulators of the IAPP system entails relatively minor computational costs and tractable experimental effort. In particular, the use of fingerprint-based inference reduces the computational cost by >85% relative to docking alone; furthermore, the computational components enable experimental screening to focus on a relatively small region of chemical space enriched in active compounds. The protocol generated a diverse set of previously unrecognized modulators, which enhanced membrane binding or toxicity and could aid in the study of pathologically relevant rare states in the IAPP oligomeric ensemble. Analogous strategies may prove to be useful for similar targets including α-synuclein (implicated in Parkinson’s disease), amyloid-β, and tau (both implicated in Alzheimer’s disease). They may also enable the modulation of the numerous cell signaling pathways that involve the association of intrinsically disordered proteins with diverse binding partners.69,70
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Institutes of Health under grants GM102815 (to E.R. and A.D.M.) and GM094693 to A.D.M. and an American Diabetes Association Postdoctoral Fellowship to D.E.S.
We thank Drs. J. Hebda and M. Magzoub for helpful discussions. We also thank Dr. Gary W. Cline (Department of Internal Medicine, Yale University) for the gift of INS-1 cells and technical assistance with cell culture. This work also benefited from the facilities and staff of the Yale University Faculty of Arts and Sciences High Performance Computing Center, the Keck Biotechnology Research Laboratory at Yale, and the Yale Center for Molecular Discovery.
Footnotes
Notes
The authors declare no competing financial interest.
Methods for the estimation of enhancement in overall screening efficiency due to our computational protocol, results of PLSR predictions using the MACCS and ECFP fingerprints, control FCS measurements, and representative docked poses of compounds 1–4 with IAPP dimers. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00052.
References
- 1.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 2.Jayasinghe SA, Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 3.Williamson JA, Loria JP, Miranker AD. Helix stabilization precedes aqueous and bilayer-catalyzed fiber formation in islet amyloid polypeptide. J Mol Biol. 2009;393:383–396. doi: 10.1016/j.jmb.2009.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williamson JA, Miranker AD. Direct detection of transient alpha-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nanga RPR, Brender JR, Xu J, Hartman K, Subramanian V, Ramamoorthy A. Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. J Am Chem Soc. 2009;131:8252–8261. doi: 10.1021/ja9010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nanga RP, Brender JR, Vivekanandan S, Ramamoorthy A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim Biophys Acta. 2011;1808:2337–2342. doi: 10.1016/j.bbamem.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patil SM, Xu S, Sheftic SR, Alexandrescu AT. Dynamic alpha-helix structure of micelle-bound human amylin. J Biol Chem. 2009;284:11982–11991. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hebda JA, Miranker AD. The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: insights from type II diabetes. Annu Rev Biophys. 2009;38:125–152. doi: 10.1146/annurev.biophys.050708.133622. [DOI] [PubMed] [Google Scholar]
- 9.Cao P, Marek P, Noor H, Patsalo V, Tu LH, Wang H, Abedini A, Raleigh DP. Islet amyloid: from fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 2013;587:1106–1118. doi: 10.1016/j.febslet.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayasinghe SA, Langen R. Membrane interaction of islet amyloid polypeptide. Biochim Biophys Acta. 2007;1768:2002–2009. doi: 10.1016/j.bbamem.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 11.Engel MFM. Membrane permeabilization by islet amyloid polypeptide. Chem Phys Lipids. 2009;160:1–10. doi: 10.1016/j.chemphyslip.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J Mol Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 13.Last NB, Rhoades E, Miranker AD. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc Natl Acad Sci USA. 2011;108:9460–9465. doi: 10.1073/pnas.1102356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao P, Abedini A, Wang H, Tu LH, Zhang X, Schmidt AM, Raleigh DP. Islet amyloid polypeptide toxicity and membrane interactions. Proc Natl Acad Sci USA. 2013;110:19279–19284. doi: 10.1073/pnas.1305517110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abedini A, Schmidt AM. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013;587:1119–1127. doi: 10.1016/j.febslet.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry. 2008;47:12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Last NB, Miranker AD. Common mechanism unites membrane poration by amyloid and antimicrobial peptides. Proc Natl Acad Sci USA. 2013;110:6382–6387. doi: 10.1073/pnas.1219059110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomasello MF, Sinopoli A, Attanasio F, Giuffrida ML, Campagna T, Milardi D, Pappalardo G. Molecular and cytotoxic properties of hIAPP17-29 and rIAPP17-29 fragments: a comparative study with the respective full-length parent polypeptides. Eur J Med Chem. 2014;81:442–455. doi: 10.1016/j.ejmech.2014.05.038. [DOI] [PubMed] [Google Scholar]
- 19.Gurlo T, Ryazantsev S, Huang C, Yeh MW, Reber HA, Hines OJ, O’Brien TD, Glabe CG, Butler PC. Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol. 2010;176:861–869. doi: 10.2353/ajpath.2010.090532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li XL, Chen T, Wong YS, Xu G, Fan RR, Zhao HL, Chan JCN. Involvement of mitochondrial dysfunction in human islet amyloid polypeptide-induced apoptosis in INS-1E pancreatic beta cells: an effect attenuated by phycocyanin. Int J Biochem Cell Biol. 2011;43:525–534. doi: 10.1016/j.biocel.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 21.Magzoub M, Miranker AD. Concentration-dependent transitions govern the subcellular localization of islet amyloid polypeptide. FASEB J. 2011;26:1228–1238. doi: 10.1096/fj.11-194613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hebda JA, Saraogi I, Magzoub M, Hamilton AD, Miranker AD. A peptidomimetic approach to targeting pre-amyloidogenic states in type II diabetes. Chem Biol. 2009;16:943–950. doi: 10.1016/j.chembiol.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Miranker AD. A foldamer approach to targeting membrane bound helical states of islet amyloid polypeptide. Chem Commun. 2013;49:4749–4751. doi: 10.1039/c3cc41452c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar S, Schlamadinger DE, Brown MA, Dunn JM, Mercado B, Hebda JA, Saraogi I, Rhoades E, Hamilton AD, Miranker AD. Islet amyloid-induced cell death and bilayer integrity loss share a molecular origin targetable with oligopyridylamide-based α-helical mimetics. Chem Biol. 2015;22:369–378. doi: 10.1016/j.chembiol.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound alpha-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 26.Nath A, Miranker AD, Rhoades E. A membrane-bound antiparallel dimer of rat islet amyloid polypeptide. Angew Chem, Int Ed. 2011;50:10859–10862. doi: 10.1002/anie.201102887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar S, Brown MA, Nath A, Miranker AD. Folded small molecule manipulation of islet amyloid polypeptide. Chem Biol. 2014;21:775–781. doi: 10.1016/j.chembiol.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saraogi I, Hebda JA, Becerril J, Estroff LA, Miranker AD, Hamilton AD. Synthetic alpha-helix mimetics as agonists and antagonists of islet amyloid polypeptide aggregation. Angew Chem, Int Ed. 2010;49:736–739. doi: 10.1002/anie.200901694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rappe AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J Am Chem Soc. 1992;114:10024–10035. [Google Scholar]
- 30.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J. Practically useful: what the Rosetta protein modeling suite can do for you. Biochemistry. 2010;49:2987–2998. doi: 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaudhury S, Lyskov S, Gray JJ. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26:689–691. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durant JL, Leland BA, Henry DR, Nourse JG. Reoptimization of MDL keys for use in drug discovery. J Chem Inf Model. 2002;42:1273–1280. doi: 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- 36.Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 37.Ihlenfeldt WD, Gasteiger J. Hash codes for the identification and classification of molecular structure elements. J Comput Chem. 1994;15:793–813. [Google Scholar]
- 38.Dalke A. The FPS fingerprint format and chemfp toolkit. J Cheminf. 2013;5:P36. [Google Scholar]
- 39.Oliphant TE. Python for scientific computing. Comput Sci. 2007;9:10–20. [Google Scholar]
- 40.Abdi H. Partial least squares (PLS) regression. In: Lewis-Beck M, Bryman A, Futing T, editors. Encyclopedia of Social Science Research Methods. Sage; Thousand Oaks, CA: 2003. pp. 792–795. [Google Scholar]
- 41.Nath A, Zientek MA, Burke BJ, Jiang Y, Atkins WM. Quantifying and predicting the promiscuity and isoform specificity of small-molecule cytochrome P450 inhibitors. Drug Metab Dispos. 2010;38:2195–2203. doi: 10.1124/dmd.110.034645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nath A, Trexler AJ, Koo P, Miranker AD, Atkins WM, Rhoades E. Single-molecule fluorescence spectroscopy using phospholipid bilayer nanodiscs. Methods Enzymol. 2010;472:89–117. doi: 10.1016/S0076-6879(10)72014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tóth G, Gardai SJ, Zago W, Bertoncini CW, Cremades N, Roy SL, Tambe MA, Rochet JC, Galvagnion C, Skibinski G, Finkbeiner S, Bova M, Regnstrom K, Chiou SS, Johnston J, Callaway K, Anderson JP, Jobling MF, Buell AK, Yednock TA, Knowles TP, Vendruscolo M, Christodoulou J, Dobson CM, Schenk D, McConlogue L. Targeting the intrinsically disordered structural ensemble of α-synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLoS One. 2014;9:e87133. doi: 10.1371/journal.pone.0087133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wold S, Sjöström M, Eriksson L. PLS-regression: a basic tool of chemometrics. Chemom Intell Lab Syst. 2001;58:109–130. [Google Scholar]
- 45.Haustein E, Schwille P. Fluorescence correlation spectroscopy: novel variations of an established technique. Annu Rev Biophys Biomol Struct. 2007;36:151–169. doi: 10.1146/annurev.biophys.36.040306.132612. [DOI] [PubMed] [Google Scholar]
- 46.Shoichet BK. Screening in a spirit haunted world. Drug Discovery Today. 2006;11:607–615. doi: 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villoutreix BO, Eudes R, Miteva MA. Structure-based virtual ligand screening: recent success stories. Comb Chem High Throughput Screening. 2009;12:1000–1016. doi: 10.2174/138620709789824682. [DOI] [PubMed] [Google Scholar]
- 48.Brender JR, Salamekh S, Ramamoorthy A. Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc Chem Res. 2012;45:454–462. doi: 10.1021/ar200189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brender JR, Krishnamoorthy J, Messina GML, Deb A, Vivekanandan S, La Rosa C, Penner-Hahn JE, Ramamoorthy A. Zinc stabilization of prefibrillar oligomers of human islet amyloid polypeptide. Chem Commun. 2013;49:3339–3341. doi: 10.1039/c3cc40383a. [DOI] [PubMed] [Google Scholar]
- 50.Sciacca MFM, Milardi D, Messina GML, Marletta G, Brender JR, Ramamoorthy A, La Rosa C. Cations as switches of amyloid-mediated membrane disruption mechanisms: calcium and IAPP. Biophys J. 2013;104:173–184. doi: 10.1016/j.bpj.2012.11.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knight JD, Williamson JA, Miranker AD. Interaction of membrane-bound islet amyloid polypeptide with soluble and crystalline insulin. Protein Sci. 2008;17:1850–1856. doi: 10.1110/ps.036350.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brender JR, Lee EL, Hartman K, Wong PT, Ramamoorthy A, Steel DG, Gafni A. Biphasic effects of insulin on islet amyloid polypeptide membrane disruption. Biophys J. 2011;100:685–692. doi: 10.1016/j.bpj.2010.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hebda JA, Magzoub M, Miranker AD. Small molecule screening in context: lipid-catalyzed amyloid formation. Protein Sci. 2014;23:1341–1348. doi: 10.1002/pro.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caillon L, Lequin O, Khemtémourian L. Evaluation of membrane models and their composition for islet amyloid polypeptide-membrane aggregation. Biochim Biophys Acta. 2013;1828:2091–2098. doi: 10.1016/j.bbamem.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 55.Schlamadinger DE, Miranker AD. Fiber-dependent and -independent toxicity of islet amyloid polypeptide. Biophys J. 2014;107:2559–2566. doi: 10.1016/j.bpj.2014.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Higham CE, Jaikaran ETAS, Fraser PE, Gross M, Clark A. Preparation of synthetic human islet amyloid polypeptide (IAPP) in a stable conformation to enable study of conversion to amyloid-like fibrils. FEBS Lett. 2000;470:55–60. doi: 10.1016/s0014-5793(00)01287-4. [DOI] [PubMed] [Google Scholar]
- 57.Green J, Goldsbury C, Mini T, Sunderji S, Frey P, Kistler J, Cooper G, Aebi U. Full-length rat amylin forms fibrils following substitution of single residues from human amylin. J Mol Biol. 2003;326:1147–1156. doi: 10.1016/s0022-2836(02)01377-3. [DOI] [PubMed] [Google Scholar]
- 58.Dupuis NF, Wu C, Shea JE, Bowers MT. The amyloid formation mechanism in human IAPP: dimers have β-strand monomer–monomer interfaces. J Am Chem Soc. 2011;133:7240–7243. doi: 10.1021/ja1081537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Young LM, Cao P, Raleigh DP, Ashcroft AE, Radford SE. Ion mobility spectrometry-mass spectrometry defines the oligomeric intermediates in amylin amyloid formation and the mode of action of inhibitors. J Am Chem Soc. 2014;136:660–670. doi: 10.1021/ja406831n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA. 2013;110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia a, Dutra a, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou a, Papapetropoulos T, Johnson WG, Lazzarini aM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 62.Gessel MM, Bernstein S, Kemper M, Teplow DB, Bowers MT. Familial Alzheimer’s disease mutations differentially alter amyloid β-protein oligomerization. ACS Chem Neurosci. 2012;3:909–918. doi: 10.1021/cn300050d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wolfe MS. Tau mutations in neurodegenerative diseases. J Biol Chem. 2009;284:6021–6025. doi: 10.1074/jbc.R800013200. [DOI] [PubMed] [Google Scholar]
- 64.Rowczenio DM, Noor I, Gillmore JD, Lachmann HJ, Whelan C, Hawkins PN, Obici L, Westermark P, Grateau G, Wechalekar AD. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35:E2403–2412. doi: 10.1002/humu.22619. [DOI] [PubMed] [Google Scholar]
- 65.Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Münch J, Baker D, Eisenberg D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475:96–100. doi: 10.1038/nature10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421:185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McKoy AF, Chen J, Schupbach T, Hecht MH. A novel inhibitor of amyloid β (Aβ) peptide aggregation: from high throughput screening to efficacy in an animal model of Alzheimer disease. J Biol Chem. 2012;287:38992–39000. doi: 10.1074/jbc.M112.348037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu M, De Simone A, Schenk D, Toth G, Dobson CM, Vendruscolo M. Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J Chem Phys. 2013;139:035101. doi: 10.1063/1.4811831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Metallo SJ. Intrinsically disordered proteins are potential drug targets. Curr Opin Chem Biol. 2010;14:481–488. doi: 10.1016/j.cbpa.2010.06.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uversky VN. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin Drug Discovery. 2012;7:475–488. doi: 10.1517/17460441.2012.686489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.