

Abstract
The Neuroplastin gene encodes two synapse-enriched protein isoforms, Np55 and Np65, which are transmembrane glycoproteins that regulate several cellular processes, including the genesis, maintenance, and plasticity of synapses. We found that an absence of Np65 causes early-onset sensorineural hearing loss and prevented the normal synaptogenesis in inner hair cells (IHCs) in the newly identified mouse mutant pitch. In wild-type mice, Np65 is strongly upregulated in the cochlea from around postnatal day 12 (P12), which corresponds to the onset of hearing. Np65 was specifically localized at the presynaptic region of IHCs. We found that the colocalization of presynaptic IHC ribbons and postsynaptic afferent terminals is greatly reduced in pitch mutants. Moreover, IHC exocytosis is also reduced with mutant mice showing lower rates of vesicle release. Np65 appears to have a nonessential role in vision. We propose that Np65, by regulating IHC synaptogenesis, is critical for auditory function in mammals.
SIGNIFICANCE STATEMENT In the mammalian cochlea, the sensory inner hair cells (IHCs) encode auditory information. They do this by converting sound wave-induced mechanical motion of their hair bundles into an electrical current. This current generates a receptor potential that controls release of glutamate neurotransmitter from their ribbon synapses onto the auditory afferent fiber. We show that the synapse-enriched protein Np65, encoded by the Neuroplastin gene, is localized at the IHC presynaptic region. In mutant mice, absence of Np65 causes early-onset sensorineural hearing loss and prevents normal neurotransmitter release in IHCs and colocalization of presynaptic ribbons with postsynaptic afferents. We identified Neuroplastin as a novel deafness gene required for ribbon synapse formation and function, which is critical for sound perception in mammals.
Keywords: exocytosis, hearing loss, inner hair cell, neuroplastin, ribbon synapse, synaptogenesis
Introduction
Sensory organs, such as the ear and eye, are supremely well adapted for detecting input from the external environment. The correct perception of sensory information requires a carefully coordinated formation and maturation of the sensory receptors and their neural networks during immature stages of development. The development of neuronal circuits depends upon a combination of genetic programs that are coordinated by specific microRNAs (Sun et al., 2013) and experience-independent or spontaneous electrical activity that occur during a critical period (Blankenship and Feller, 2010). In the mammalian auditory system, inner hair cells (IHCs) relay sound stimuli to the brain via the release of glutamate from ribbon synapses onto Type I spiral ganglion neurons (Glowatzki and Fuchs, 2002). Ribbon synapses, which are also found in the photoreceptors and vestibular hair cells, are specialized electron-dense organelles that tether a large pool of releasable synaptic vesicles and are thought to be important for coordinated vesicle release onto the auditory afferent terminals (Matthews and Fuchs, 2010). Currently, the molecular composition of the exocytotic machinery of auditory hair cells and the mechanisms involved in its maturation are largely unknown (Marcotti, 2012; Safieddine et al., 2012).
Neuroplastins are neuronal and synapse-enriched glycoproteins belonging to the immunoglobulin (Ig) superfamily of neural cell adhesion molecules (CAMs) (Beesley et al., 2014). They have been reported to mediate several cellular processes, including synaptic plasticity and neuronal differentiation (Smalla et al., 2000; Marzban et al., 2003; Owczarek et al., 2011; Beesley et al., 2014; Herrera-Molina et al., 2014). The Nptn gene transcript produces two Neuroplastin protein isoforms, Np55 and Np65, named according to their molecular weight (Langnaese et al., 1997). Np65 is expressed primarily in the brain, initially detected at low levels before P12 and then increasing rapidly and reaching a plateau of expression at 4–5 weeks postnatally (Beesley et al., 2014). Np65 has also been linked with ribbon synapse formation in the plexiform layers of the rat retina (Kreutz et al., 2001). So far, Neuroplastins have not been implicated in auditory function or directly linked to any disease in mammals. However, Np65 has been implicated as a risk factor in schizophrenia and has been proposed to play a role in recovery from ischemia (Ouchi et al., 2005; Saito et al., 2007; Beesley et al., 2014).
We have identified pitch, a mouse model of early-onset hearing loss, which results from an N-ethyl-N-nitrosourea (ENU)-induced lesion in the novel deafness gene Neuroplastin (Nptnpitch). Here we exploit Nptnpitch and two additional mouse models (NptnY219X and Nptn−) to identify the role of Neuroplastins in the visual and auditory systems. While Neuroplastins appear to have a nonessential role in the retina, the absence of Np65 leads to an early-onset profound hearing loss, which precedes sensory hair cell degeneration. In the cochlea, Np65 is selectively and highly expressed at the IHC synapse from around the onset of hearing. We demonstrate that Np65 is involved in regulating IHC synaptogenesis, with Np65-deficient mice showing reduced “matching” of presynaptic and postsynaptic ribbon synapse markers. In addition, IHCs also demonstrate reduced neurotransmitter release. This study establishes a critical role for the synaptic protein Np65 in the function of the mammalian auditory system.
Materials and Methods
Mice.
All animal studies were licensed by the Home Office under the Animals (Scientific Procedures) Act 1986, United Kingdom, and additionally approved by the relevant Institutional Ethical Review Committees.
The generation of ENU-induced mouse mutants has been described previously (Acevedo-Arozena et al., 2008). Briefly, for phenotype-driven screens, C57BL/6J mice were treated with ENU and bred with wild-type “sighted C3H” (C3H.Pde6b+) females (Hoelter et al., 2008). The resulting G1 males were bred with wild-type C3H.Pde6b+ females to produce G2 females, which were backcrossed to their G1 fathers to generate recessive G3 pedigrees that entered a phenotyping pipeline, including auditory screening. Once identified, the pitch mutant was backcrossed to C3H.Pde6b+ for several generations. All pitch characterization was performed on intercrossed mice from backcross 5 or greater.
To identify additional Nptn alleles, a gene-driven screen of the Harwell ENU DNA archive was undertaken. The archive consists of >10,000 DNA samples from individual G1 ENU mutagenized mice paralleled by frozen sperm samples (Coghill et al., 2002). Oligonucleotide primers were designed for each Nptn exon and amplicons assessed using a DNA melting analysis. Mutations are evident as a left-shifted melt curve resulting from the reduced melting temperature of the mis-matched nucleotide. Sanger sequencing was used to identify the nature of the ENU-induced heterozygous lesion. Sequencing data were assessed using Lasergene (DNASTAR). The identified NptnY219X allele was rederived using in vitro fertilization by the FESA core at the Mary Lyon Centre. NptnY219X mice were backcrossed to C3H.Pde6b+, ensuring loss of the C57BL/6J Cdh23ahl allele, and then intercrossed to maintain the line on a mixed C3H.Pde6b+ and C57BL/6J genetic background.
The two ENU-induced Nptn mutants (pitch and Y219X) are on a mixed genetic background involving the C3H.Pde6b+ strain. The presence of the Gpr179nob5 allele in this strain impacts upon retinal function and hence renders them unsuitable for electroretinography (ERG) studies (Hoelter et al., 2008; Balmer et al., 2013). As part of the International Mouse Phenotyping Consortium program, a Neuroplastin knock-out (Nptntm1b(EUCOMM)Hmgu, herein called Nptn−/−) model on a C57BL/6NTac genetic background has been generated, in which exons 5 and 6 of the Neuroplastin gene are deleted. These two exons code for the Ig3 and transmembrane (TM) domains common to both Np55 and Np65 (see Fig. 1C). Given the allele design, no full-length transcripts can be generated from the Nptn gene locus in the Nptn−/− mice; as such, no functional Np55 nor Np65 protein can be produced. This model has been subject to a broad-based phenotyping pipeline; and consistent with our findings for the pitch and Y219X models, homozygous (but not heterozygous) knock-out mice have profound hearing loss (http://www.mousephenotype.org/data/genes/MGI:108077). Although the C57BL/6NTac genetic background also contains an ocular-relevant mutation (retinal degeneration 8 (rd8) allele of the Crb1 gene) (Mehalow et al., 2003), the impact on ERG function is minimal. Retinal degeneration in rd8 mice is focal in nature, and global ERG function is essentially normal to >9 months of age (Aleman et al., 2011).
Figure 1.

Mapping, identification, and consequence of the pitch mutation. A, Ideogram showing the SNP mapped critical interval for the pitch mutation on chromosome 9. The interval contains 25 genes; 10 were prioritized for Sanger sequencing (marked with an asterisk). B, Detection of a coding mutation in the Nptn gene in pitch mice. DNA sequence analysis revealed an ENU-induced nucleotide transversion (c.943T>A, ENSMUST00000177292) at codon 315, thus altering the wild-type (WT) sequence TGT, encoding an cysteine (Cys, C), to the mutant (m) sequence AGT, encoding a serine (Ser, S). Electropherograms derived from a pitch mutant mouse (Nptnpitch/pitch) and a wild-type littermate (Nptn+/+) control showing the sequence surrounding Nptn nucleotide 943 (indicated by an arrow). C, Schematic representation of the genomic structure of the Nptn gene. The murine Nptn gene consists of 8 exons, spanning ∼69 kb of genomic DNA. Because of alternative splicing of the gene transcript, two protein isoforms are produced, Np55 and Np65. Np65 is a 397 amino acid cell adhesion molecule that contains a signal peptide sequence, three extracellular Ig-like domains (Ig1–3) (light gray bars), a transmembrane domain (TM) (dark gray bar), and a short intracellular domain. Whereas Np55 is a 281 amino acid cell adhesion molecule that contains only two Ig-like domains (Ig2 and Ig3), lacking Ig1 due to in-frame skipping of exon 2. The ATG (translation start) and the TAA (stop) sites are in exons 1 and 8, respectively, and the 5′ untranslated regions is shown in black. The location of the mutation identified in pitch by the present study is shown and numbered according to the Np65-coding transcript/protein. D, Conservation of the mutated cysteine residue across species. H. sapiens, ENSG00000156642; M. musculus, ENSMUSG00000032336; G. gallus, ENSGALG00000001741; T. rubripes, ENSTRUG00000010655; D. rerio, ENSDARG00000043864; X. tropicalis, ENSXETG00000005188. E, Crystal structure of the ectodomain of Np55 (PDB ID: 2WV3), showing Ig2 and Ig3, and the four cysteine residues required for disulphide bridge formation within these two domains as colored spheres. The cysteine residue mutated in pitch is colored red.
Genetic mapping and candidate gene analysis.
An initial linkage study was undertaken using DNA extracted from ear biopsies of affected G3 mice using single nucleotide polymorphism (SNP) markers, across all chromosomes, polymorphic between the parental strains C57BL/6 and C3H. After linkage to chromosome 9 was established, further SNP markers were selected and fine mapping undertaken to delineate an ∼1.6 Mb critical region between SNPs rs29835016 and rs3688614. The Ensembl database (http://www.ensembl.org/Mus_Musculus/Info/Index) was used to determine those genes located within the critical interval, and prioritized according to known/assumed protein function and/or expression profile. For each candidate gene, primers were designed to allow amplification of exonic regions and adjoining splice sites. DNA from an affected animal was used as template for PCR, and resulting amplicons were analyzed by Sanger sequencing. Sequencing data were assessed for variation using Lasergene (DNASTAR).
Auditory-evoked brainstem response (ABR).
Analysis of hearing function by ABR has been described previously (Hardisty-Hughes et al., 2010). Briefly, mice were anesthetized with ketamine (10%v/v) and xylasine (5% v/v) administered at the rate of 0.1 ml/10 g body mass. Animals were placed on a heated mat inside a sound-attenuated chamber, and electrodes were inserted subdermally: below the right pinnae, into the muscle mass below the left ear, and over the vertex. ABR responses were collected, amplified, and averaged using TDT System III (Tucker Davies Technology) in conjunction with SigCal and SigGen (Biosig) software. The click stimulus consisted of a 0.1 ms broadband click. Tone burst stimuli were of 7 ms duration and measured at the following frequencies: 8, 12, 20, and 26 kHz, in 5 dB decreasing steps starting at 95 dB. Animals were recovered using the anesthetic reversal agent antipamezole (Antisedan, 0.1 ml 1% v/v), unless under age of 4 weeks when procedure was performed terminally.
ERG.
Adult (9- to 10-week-old) mice were used to examine retinal function by ERG testing. All experiments were performed at the University of Oxford in compliance with the Association for Research in Vision and Ophthalmology statement for the Use of Animals in Ophthalmic and Vision Research. N = 7 homozygous Nptn knock-out mice were tested, with the same number of age- and sex-matched homozygous wild-type littermates used as controls. Before testing, mice were dark-adapted overnight (>16 h), and all experimental preparation was performed under dim red illumination. Animals were anesthetized via intraperitoneal injection of 80 mg/kg ketamine (Vetalar, Pfizer Animal Health) and 10 mg/kg xylazine (Rompun, Bayer HealthCare) in sterile water. Pupils were fully dilated with eye drops (Minims tropicamide 1% w/v and Minims phenylephrine hydrochloride 2.5% w/v, both Bausch & Lomb). Animals were placed on a heated platform, maintained at 38°C using a circulating pump-water bath. A DTL-type silver-coated nylon thread active electrode (DTL Plus Electrode; Diagnosys) modified to include a custom-made contact lens of clear Aclar film (Honeywell International, supplied by Agar Scientific) was positioned concentrically on the cornea with 1% hypromellose eye drops (Isopto alkaline, Alcon Laboratories). Platinum needle electrodes in the forehead and at the base of the tail served as reference and ground electrodes, respectively.
All recordings were made in a custom-made light-tight Faraday cage. ERG signals were differentially amplified and digitized at a rate of 5 kHz using an Espion E2 console (Diagnosys) that also generated and controlled the light stimuli, which were delivered in a Ganzfeld dome. For dark-adapted testing, responses were elicited by brief (4 ms), single flashes of white light on a dark background. Stimulus intensity was increased across a 7 log unit range (−6 to 1 log cd · s/m2) in log unit steps with the following response averaging and interstimulus interval (ISI) times used: −6 to −5 log cd.s/m2, 16 responses averaged with an ISI of 3 s; −4 to −3 log cd.s/m2, 9 responses, ISI = 4 s; −2 log cd.s/m2, 9 responses, ISI = 8 s; −1 log cd.s/m2, 4 responses, ISI = 16 s; 0 log cd.s/m2, 4 responses, ISI = 32 s; 1 log cd.s/m2, 4 responses, ISI = 64 s. Animals were then exposed to a full field 30 cd/m2 white background for at least 10 min (light-adapted), and responses to a bright white stimulus (1 log cd s/m2, 20 responses averaged, ISI = 500 ms) superimposed on the constant background were recorded. The amplitude and latency of the major ERG components (a- and b-waves for dark-adapted, b-wave only for light-adapted) were measured using automated and manual methods. By convention, b-wave amplitude is measured from the a-wave trough (where present), and a-wave amplitude is measured from baseline to a-wave trough. Implicit times of both waveforms as measured from stimulus onset to the peak amplitude.
Nptn expression in mouse cochleae.
Total RNA samples were extracted from wild-type C3H mouse whole cochleae at P4, P8, P12, P16, P21, and P27 using a Direct-zol RNA MiniPrep Kit (Zymo Research). Five separate samples were prepared for each age, comprising whole-cochlea mouse RNA extracted from both ears pooled at P4 and P8, and a single ear for the remaining later ages. In each case, quintuplicate samples from independent mice were analyzed for each stage. Complementary DNAs were generated from 1 μg total RNA using the High Capacity cDNA synthesis kit (Applied Biosystems) following the manufacturer's instructions. Amplifications were performed using TaqMan Gene Expression assays with Fast Universal PCR Master Mix. A β-actin primer/probe set (Mm00607939_s1) was used as an internal control, and two custom TaqMan assays were designed to allow the specific amplification of the two alternatively spliced Neuroplastin transcripts encoding Np55 or Np65 (using oligonucleotides spanning the exon junctions at the sites of alternative splicing), using the following forward and reverse primers, and FAM-MGB probes: Np55-F 5′-GCTCAGAACGAACCAAGAATTGT-3′, Np55-R 5′-CAGGAAGGAGGCTTTCTCGAA-3′, Np55-probe 5′-ACCAGTGAAGAGGTCA-3′, Np65-F 5′-GCTCAGAACGCTGGGTTTGT-3′, Np65-R 5′-CGTCCCCCGTGAGCTTAGT-3′, Np65-probe 5′-AAGTCGCCCATGTCAG-3′. PCRs were run in triplicate in the Applied Biosystems real-time PCR device (7500 Fast Real-Time PCR System) in 20 μl reactions containing 10 μl Fast TaqMan Master Mix, 1 μl TaqMan assay, 4 μl ddH2O, and 5 μl complementary DNA (5 ng/μl), using the following cycles: 95°C for 20 s and 40 cycles at 95°C for 3 s and 60°C for 30 s. Amplification data were recorded and relative quantification performed using the 7500 software version 2.0.6 software (Applied Biosystems) comparing threshold cycles (Ct). Nptn mRNA levels were first normalized to Actin (ΔCt = CtNptn − CtActin) for each Nptn isoform at each age, and changes in expression relative to P4 were calculated for each Nptn isoform as 2−(ΔCt−ΔCtP4).
In vitro assessment of the pitch mutation.
Full-length wild-type Np65 lacking the TAA stop codon was amplified using the primers Nptn-HindIII-F 5′-ACAAGCTTGATGTCGGGTTCGTCGCTGCCCAGC-3′ and Nptn-BamHI-R 5′-GTGGATCCGAATTTGTGTTTCTCTGGCGCAAG-3′, which contain restriction endonuclease recognition site overhangs for HindIII and BamHI, respectively, and cloned into pGEM-T vector (Promega). This was used as template for the generation of pitch mutant Np65 construct via site-directed mutagenesis (Quikchange Lightning, Agilent Technologies). The DNA sequences of all constructs were verified by Sanger sequencing. The wild-type and mutant Np65 constructs were excised and directionally subcloned in-frame into the mammalian expression vector pEGFP-N1 (BD Biosciences, Clontech) to yield Np65 tagged at the C terminus with EGFP. These plasmids were used to transiently transfect 1 × 105 HEK293T cells seeded in 6-well plates (1 μg plasmid/well). A mock transfection was used as a control. Forty-eight hours after transfection, cells were lysed using 125 μl RIPA buffer/well, and total protein (85 ng) assessed by Western blotting. Immunodetection was achieved using a goat anti-Np65 antibody (1:100, R&D Systems AF5360), followed by an HRP-conjugated rabbit anti-goat secondary antibody (1:10,000, Bio-Rad). To confirm equal loading the blot was stripped and reprobed with rabbit anti-β-actin antibody (1:1500, Sigma, A2066), followed by an HRP-conjugated goat anti-rabbit secondary antibody (1:3000, Bio-Rad). Finally, to confirm that the anti-Np65 antibody is specific, we again stripped the blot and reprobed with rabbit anti-GFP(FL) antibody (1:1500, Santa Cruz Biotechnology, sc-8334), followed by an HRP-conjugated goat anti-rabbit secondary antibody (1:3000, Bio-Rad).
Western blot analysis of cochlear and brain protein lysates.
Whole cochleae and brain were dissected from P19 Nptn+/+, Nptn+/pitch, and Nptnpitch/pitch mice. For each genotype, two pooled cochleae and a half brain were used and total protein lysates prepared using RIPA buffer. Total protein (110 ng/tissue/genotype) was assessed by Western blot and probed with a goat anti-Np65 antibody (1:100, R&D Systems, AF5360) and an HRP-conjugated rabbit anti-goat secondary antibody (1:10,000, Bio-Rad). To confirm equal loading, blots were stripped and reprobed with an anti-β-actin antibody (1:1000, Sigma, A2066), followed by an HRP-conjugated goat anti-rabbit secondary antibody (1:3000, Bio-Rad).
Cochlear whole-mount immunolabeling.
Animals were culled by cervical dislocation and inner ears removed into 4% PFA. Postfixation ears were fine dissected to expose the sensory epithelium. Cochleae were permeabilized using PBST (PBS+ 0.1% Tween 20), blocked in 10% donkey serum (Sigma-Aldrich), and incubated with primary antibodies overnight at 4°C. To allow detection of primary antibodies, cochleae were incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature. Cochleae were mounted on slides containing custom-made wells using Vectashield with, or without, DAPI. Primary antibodies: goat anti-Np65 (1:60, R&D Systems, AF5360), mouse anti-Otoferlin (1:100, Abcam, ab53233), rabbit anti-NF-200 (1:500, Sigma-Aldrich, N4142), mouse anti-NF-200 (1:150, Sigma-Aldrich, N0142), rabbit anti-Ribeye (1:500, Synaptic Systems, 192 103), mouse anti-GluR2 (1:100, Millipore MAB397), rabbit anti-parvalbumin (1:500, Swant, PV25), and sheep anti-NPTN (1:40, R&D Systems, AF5174). Secondary antibodies: AlexaFluor-647 donkey anti-rabbit, AlexaFluor-568 donkey anti-mouse, AlexaFluor-488 donkey anti-goat, AlexaFluor-488 donkey anti-rabbit (1:200, Invitrogen), and Phalloidin CF405 conjugate (1:200, Biotium). Samples were visualized using a Zeiss LSM 710 Multiphoton fluorescence confocal microscope. To assess ribbon synapses, an 80 μm section (x-axis) of sensory epithelium was imaged through the z-axis (0.46 μm steps) from the first apical quarter turn of the cochlear spiral. The image stack was then assembled in a z-projection (ImageJ), allowing the 3D image to be tilted and rotated. All Ribeye-positive and GluR2-positive puncta were counted to identify the total number of each. Additionally, Ribeye-positive puncta closely apposed to GluR2-positive puncta were also counted and classified as “matched” and assumed to represent a functional ribbon synapse.
Scanning electron microscopy.
Animals were killed by cervical dislocation and the inner ears removed into 2.5% gluteraldehyde in 0.1 m sodium phosphate buffer for 4 h at 4°C. Postfixation ears were placed in 4.3% EDTA for 48 h at 4°C. Once decalcified, ears were dissected to expose the organ of Corti. To preserve stereocilia structure, samples were processed with alternating 1% osmium tetroxide (O) and 1% thiocarbohydrazide (T) treatments (OTO processing) and then dehydrated in increasing concentrations of ethanol at 4°C. Samples were critical point dried with liquid CO2 and mounted on stubs using silver paint. The mounted samples were sputter coated using a platinum target and visualized using a JEOL JSM 6010LV Scanning Electron Microscope.
Single-hair cell electrophysiology.
Apical-coil IHCs and outer hair cells (OHCs) from the pitch mutant mouse and their littermate controls were studied in acutely dissected cochlea from postnatal day 6 (P6) to P22, where the day of birth is P0. Cochleae were dissected in normal extracellular solution (in mm) as follows: 135 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 5.6 d-glucose, 10 HEPES-NaOH. Sodium pyruvate (2 mm), MEM amino acids solution (50×, without l-glutamine), and MEM vitamins solution (100×) were added from concentrates (Fisher Scientific). The pH was adjusted to 7.5 (osmolality ∼308 mmol/kg). The dissected cochleae were transferred to a microscope chamber and viewed using an upright microscope (Leica DMLFS). Patch-clamp recordings were performed using an Optopatch (Cairn Research) amplifier. Patch pipettes were made from soda glass capillaries (Harvard Apparatus) and had a typical resistance in extracellular solution of 2–3 mΩ. To reduce the electrode capacitance, patch electrodes were coated with surf wax (Mr Zoggs SexWax). Data acquisition was controlled by pClamp software using a Digidata 1440A interface (Molecular Devices). Recordings were low-pass filtered at 2.5 kHz (8-pole, Bessel), sampled at 5 kHz, and stored on computer for off-line analysis (Origin: OriginLab).
Mechanoelectrical transducer (MET) currents were elicited by stimulating the hair bundles of OHCs using a fluid jet from a pipette (tip diameter 8–10 μm) driven by a piezoelectric disc (Johnson et al., 2012). Mechanical stimuli were applied as force-steps or 50 Hz sinusoids (filtered at 0.25 kHz, 8-pole Bessel) with driving voltages of ±40 V. MET currents were recorded with a patch pipette solution containing the following (in mm): 106 Cs-glutamate, 20 CsCl, 3 MgCl2, 1 EGTA-CsOH, 5 Na2ATP, 0.3 Na2GTP, 5 HEPES-CsOH, 10 sodium phosphocreatine, pH 7.3. Membrane potentials were corrected for the liquid junction potential (−11 mV).
Real-time changes in membrane capacitance (ΔCm) were measured using the Optopatch as previously described (Johnson et al., 2010, 2013). All experiments were performed at near body temperature (34°C–37°C) and using 1.3 mm extracellular Ca2+. Briefly, a 4 kHz sine wave of 13 mV RMS was applied to IHCs from −81 mV and was interrupted for the duration of the voltage step. The capacitance signal from the Optopatch was amplified (×50), filtered at 250 Hz, and sampled at 5 kHz. ΔCm was measured by averaging the Cm trace over a 200 ms period following the voltage step and subtracting from prepulse baseline. For these experiments, the Cs-glutamate-based intracellular solution was used (see above). ΔCm and Ca2+ current recordings were performed in the presence of 30 mm TEA and 15 mm 4-AP (Fluka) and Linopirdine (80 μm: Tocris Bioscience) to block the K+ currents (Johnson et al., 2007).
Statistical comparisons of means were made by the two-tailed t test or, for multiple comparisons, ANOVA, one-way ANOVA followed by the Tukey test. Two-way ANOVA, followed by the Bonferroni test, was used to compare datasets from apical and basal IHCs. Data are mean ± SEM. p < 0.05 indicates statistical significance.
Results
Identification of pitch, a mouse model of recessive hearing loss
The pitch mutant was identified from a recessive ENU screen undertaken at the Medical Research Council Harwell Mammalian Genetics Unit. Within the original G3 cohort, 18 of 91 mice (19.8%) showed a reduced or absent click box response at 3 weeks of age, indicating impaired hearing function. No hearing-impaired mice were observed in the outcrossed G4 litters, but in the intercrossed G5 litters 41 of 174 mice (23.4%) showed reduced hearing, indicating that the line displays recessive inheritance. In addition, hearing-impaired pitch mice also displayed a late-onset head tilt, trunk curling, impaired righting reflex, and abnormal locomotion in swim testing, suggesting a vestibular deficit.
Neuroplastin (Nptn) is a novel deafness gene
A genome scan using G3 mice showed linkage to chromosome 9. Subsequent SNP mapping of affected G5 mice narrowed the critical interval to an ∼1.6 Mb region containing 25 genes (Fig. 1A). Based on reported function and expression data, 10 genes were prioritized for Sanger sequencing using primers designed to amplify exons and adjacent splice sites. DNA prepared from an affected animal was used as template for PCR amplification and analyzed for sequence variations. This identified a T-to-A transversion at nucleotide 943 of Neuroplastin (Nptn) (ENSMUST00000177292), causing a nonsynonymous cysteine-to-serine substitution at residue 315 (Fig. 1B). The cysteine residue is highly conserved across evolution and is common to both Neuroplastin protein isoforms: Np65 is encoded by a longer 8 exon transcript (Cys315) and Np55 is encoded by a shorter 7 exon transcript (Cys199), skipping exon 2 that encodes Ig-like module 1 (Ig1) (Fig. 1C,D). The mutated cysteine residue is located within the common extracellular Ig-like module 3 (Ig3) and has been reported (Owczarek et al., 2010) to form part of a disulphide bridge between the two β-sheets that comprise the domain (Fig. 1E). Indeed, in vitro studies comprising transient transfections of plasmids containing full-length wild-type and pitch mutant Neuroplastin-tagged constructs demonstrated that, whereas the wild-type Np55 and Np65 proteins localized to the plasma membrane, the mutant Np55 and Np65 were retained in the endoplasmic reticulum (data not shown), indicating that pitch is a loss-of-function allele affecting both Nptn splice isoforms. To confirm our identification of Nptn as the pitch causative gene, we sought additional mutant alleles from the Harwell ENU DNA Archive (Coghill et al., 2002), identifying a nonsense allele in exon 4 (c.657T>A, p.Y219X). This mutation also affects both Nptn transcripts and, if translated, will lead to production of truncated Np55 and Np65 proteins lacking part of Ig2, all of Ig3, and the transmembrane domain (Fig. 1C). These domains are essential for Np55 and Np65 to act as cell adhesion molecules; therefore, the Y219X allele will lead to loss of function.
The contribution of Nptn to hearing was investigated by measuring in vivo auditory function of pitch (missense mutant), Y219X (nonsense mutant), and compound genotypes. ABR was performed using 8-week-old mice by applying click and tone-burst stimuli. Nptnpitch/pitch, NptnY219X/Y219X, and Nptnpitch/Y219X mice display very elevated thresholds (≥90 dB SPL) at all frequencies tested: 8, 12, 20, and 26 kHz (Fig. 2A). However, Nptnpitch/+ and NptnY219X/+ mice exhibit thresholds comparable with those of wild-type (Nptn+/+) littermates (∼20–35 dB SPL) (Fig. 2A), demonstrating the absence of a heterozygous auditory phenotype. To establish the onset of auditory impairment in Nptnpitch/pitch mice, we performed ABR measurements at P16, which is just after the onset of hearing, and at P27. At P16, Nptnpitch/pitch mice display elevated hearing thresholds (≥70 dB SPL) compared with their littermate controls (Fig. 2B). At P27, Nptnpitch/pitch mice display slightly increased auditory thresholds compared with those at P16 (≥85 dB SPL) (Fig. 2B), suggesting a progressive element to the hearing loss phenotype (Fig. 2B).
Figure 2.

Loss-of-function Neuroplastin mutations cause early-onset progressive hearing loss in the mouse. A, ABR measurements from 8-week-old Nptnpitch/pitch (n = 7), NptnY219X/Y219X (n = 5), and Nptnpitch/Y219X (n = 5) mice showing very elevated thresholds (≥85 dB SPL) at all frequencies tested. Control littermates (Nptn+/+, n = 7; Nptnpitch/+, n = 11; NptnY219X/+, n = 10) display thresholds within the normal range (∼20–40 dB SPL). These results indicate that Nptn is the single gene causing deafness in the pitch mutant. Data analyzed using one-way ANOVA with Tukey post hoc test to correct for multiple comparisons, with p < 0.05 set as the detection limit for significance. B, ABR recordings from P16 (n = 5) and P27 (n = 5) Nptnpitch/pitch mice showing elevated thresholds at all frequencies tested (≥70 dB SPL), whereas littermate controls (P16: Nptnpitch/+ and Nptn+/+, n = 5; P27: Nptnpitch/+, n = 7; Nptn+/+, n = 5) display thresholds within the normal range. These data indicate that Nptnpitch/pitch mice exhibit profound auditory impairment at very early stages (P16), which is just after the onset of hearing at ∼P12. Analysis of hearing thresholds of Nptnpitch/pitch mice at P16 and P27 using a single tailed Student's t test (p < 0.05 detection limit for significance) suggests progressive hearing deterioration at the higher frequencies (12, 20, and 26 kHz). *p < 0.05. **p < 0.01. ***p < 0.0001. Data are mean ± SD.
Neuroplastins are nonessential for retinal function
Previously, the Neuroplastins, Np55 and Np65, have been reported to be expressed in the two synaptic layers of the rat retina (the inner and outer plexiform layers) at a time that coincides with synaptogenesis (Kreutz et al., 2001). We found a similar expression profile for Np65 in the wild-type mouse retina (Fig. 3A). Although Np65 was absent from Nptnpitch/pitch mouse retina, we did not observe any gross retinal dysmorphology (Fig. 3B). To ascertain whether Neuroplastins are required for retinal function, we undertook ERG recordings from Neuroplastin knock-out (Nptn−/−) mice, which lack functional Np55 and Np65 protein (see Materials and Methods). Compared with wild-type littermate mice (Nptn+/+), no significant abnormalities in retinal function were detected in Neuroplastin-deficient mice (Nptn−/−) (Fig. 3C–G).
Figure 3.
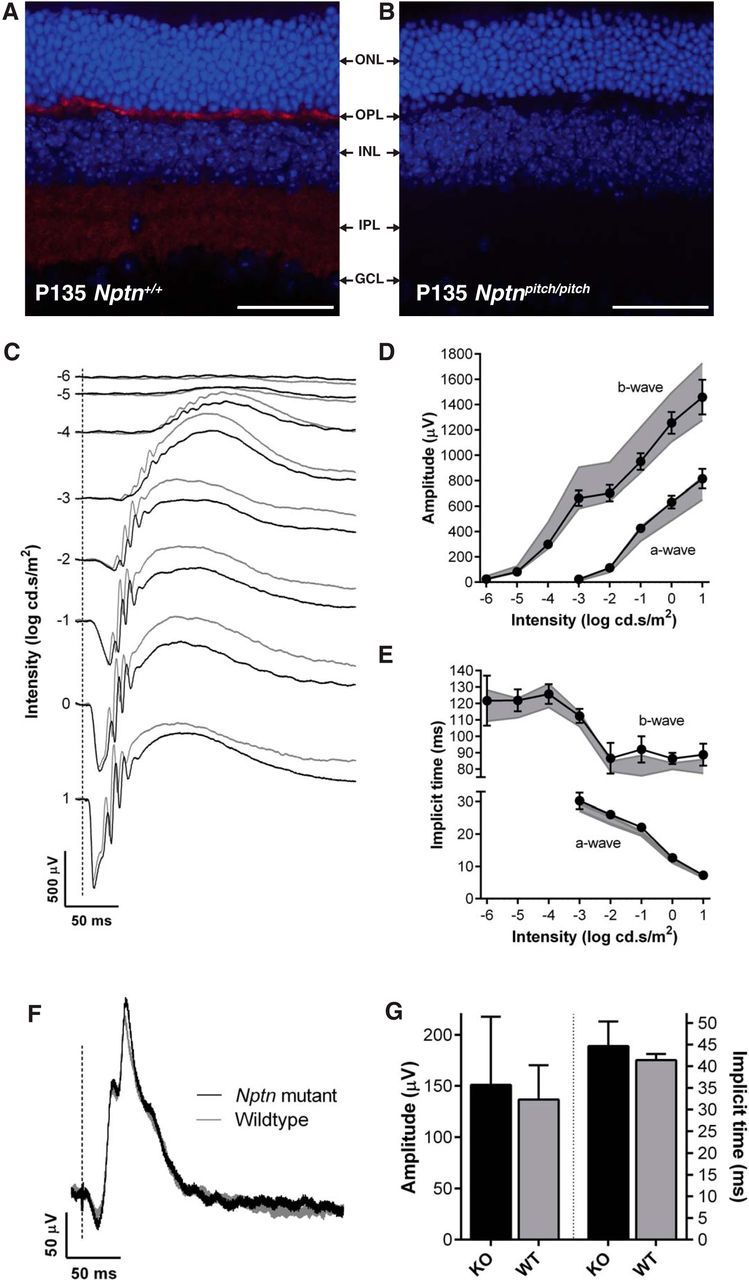
Neuroplastins are not required for retinal function. Retinal immunolabeling was undertaken using pitch mice, however, due to a strain-specific genetic anomaly of the eye in the C3H.Pde6b+ line, ERGs were recorded from Nptn−/− (n = 7) and Nptn+/+ (n = 7) mice, on a C57BL/6NTac genetic background, between 9 and 10 weeks of age. A, B, Np65 immunolabeling (red) of retinal cryosections from P135 Nptn+/+ (A) and Nptnpitch/pitch (B) mice, using DAPI as a nuclei stain. Scale bar, 50 μm. Although no gross retinal dysmorphology was observed in Nptnpitch/pitch mice, Np65 labeling was only observed in the plexiform layers of Nptn+/+ mice (OPL and IPL). C, Representative dark-adapted, flash ERG traces for Nptn−/− (black) and Nptn+/+ (gray) mice. Flash intensity increases from top to bottom and is indicated as log cd.s/m2 along left edge of responses. D, Quantification of the b-wave (left) and a-wave (bottom right) amplitudes across the stimulus intensity range. In this and the following panels, values from knock-out mice are shown as black circles/solid line (means ± 95% confidence limits); Nptn+/+ values are indicated as a gray area representing means ± 95% confidence limits. E, Quantification of the b-wave (left) and a-wave (bottom right) implicit times across the stimulus intensity range. Responses in both genotypes were intensity-dependent and not significantly different. F, Representative light-adapted, flash ERG traces from Nptn−/− and Nptn+/+ mice in response to 1 log cd.s/m2 stimulus. G, Quantification of the b-wave amplitudes (left) and implicit times (right) of light-adapted, flash ERG responses. Values from Nptn−/− (black) and Nptn+/+ (gray) are plotted as means (± 95% confidence limits as error bar) and were found not to be significantly different. ONL, Outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Np65 expression increases in the postnatal mouse inner ear
The elevated hearing thresholds measured in the Nptnpitch/pitch, NptnY219X/Y219X, and Nptnpitch/Y219X mice are indicative of a sensorineural hearing impairment. Consistent with this hypothesis, no gross morphological abnormalities were observed in the outer or middle ears of these mice (data not shown). To establish whether Nptn is expressed in the inner ear and whether this is temporally regulated, we undertook a qPCR analysis of total RNA prepared from wild-type whole cochleae at several postnatal ages. Using primers specific for each Nptn isoform, we measured mRNA levels for Np55 and Np65 at postnatal day 8 (P8), P12, P16, P21, and P27 relative to their expression in the whole cochlea at P4 (Fig. 4A). We found that the relative mRNA level for Np55 remains stable between P4 and P27 (Fig. 4A). However, the relative mRNA level for Np65 increased ∼7-fold at P12 compared with the level at P4 (Fig. 4A). Between P12 and P27 (the latest age tested), the relative Np65 mRNA level remains higher compared with the early postnatal levels. These data indicate that only the expression of the Nptn transcript encoding the Np65 protein isoform is regulated in the cochlea. Interestingly, the upregulation of Np65 occurs at around the onset of hearing (P12), a time when IHCs undergo their functional maturation (Marcotti, 2012).
Figure 4.

Time-dependent expression of Neuroplastins in the postnatal cochlea. A, Level of mRNA for Np55 and Np65 (measured using qPCR) in whole cochleae at P8, P12, P16, P21, and P27. Values were calculated relative to mRNA expression at P4. While the relative mRNA level for Np55 remained relatively stable between P4 and P27, that of Np65 increases significantly between P8 and P12 and remains higher at least up to P27, which was the oldest age tested. Statistical significance is given above the bar for comparison of the five biological replicate datasets obtained for each age using an unpaired Student's t test. B, Western blot analyses of total protein lysates prepared from HEK293T cells transiently transfected with EGFP-tagged wild-type Np65 or pitch mutant Np65, or an untransfected control. Immunodetection with an anti-Np65 antibody shows that the antibody is able to bind both wild-type and pitch mutant Np65 protein. To confirm equal loading of protein lysates, the blot was stripped and reprobed with an anti-β actin antibody. To confirm the identity of the bands detected by the anti-Np65 antibody, the blot was again stripped and probed with an anti-GFP antibody. C, Western blot analyses of cochlea and brain total protein lysates from P19 pitch mice. Immunodetection with an anti-Np65 antibody identified that Np65 protein was only present in cochlea and brain lysates from Nptn+/+ and Nptn+/pitch mice. No Np65 was detected in lysates prepared from Nptnpitch/pitch mice. As a control, blots were stripped and reprobed with an anti-β actin antibody to confirm equal loading of total protein lysate between genotypes. D, Mid-modiolar cochlear section from a P16 Nptnpitch/+ mouse labeled with DAPI (blue), anti-Np65 (red), and anti-NF-200 (green). Np65 was only found localized to the cuticular plate of the IHCs and OHCs and the basolateral region of the IHCs (arrow and inset). E, F, Cochlear whole mount from P16 Nptn+/+ (E) and Nptnpitch/pitch (F) mice immunolabeled with anti-Np65 (red), anti-otoferlin (blue), and phalloidin (green). Np65 labeling was only observed at the basolateral membrane of IHCs from Nptn+/+ mice (arrow). *p < 0.05. **p < 0.01. ****p < 0.0001. PCs, Pillar cells.
To facilitate investigation of Np65 protein expression and localization within the cochlea, we first wanted to establish the specificity of a commercially available anti-Np65 antibody. We generated C-terminally EGFP-tagged constructs for wild-type and pitch mutant Np65, transiently transfected these into cells, and assessed total protein lysates by Western blotting. In this overexpression system, wild-type and pitch mutant protein could be detected using the anti-Np65 antibody (Fig. 4B). Interestingly, immunodetection of the wild-type transfected lysate gave two bands, whereas the pitch mutant-transfected lysate gave only one band. The lower molecular weight band common to both the wild-type and pitch mutant-transfected lysates likely corresponds to the core Np65 protein (predicted ∼47 kDa) with the EGFP tag (predicted ∼27 kDa). The higher molecular weight band observed in the wild-type transfected lysate likely corresponds to glycosylated Np65 plus the EGFP tag. These data are consistent with our in vitro protein localization studies showing that while wild-type Np65 is correctly trafficked to the plasma membrane, pitch mutant Np65 protein is retained within the endoplasmic reticulum and so is unable to be fully modified post-translation.
To assess the level of Np65 protein in pitch mice, Western blot analysis of inner ear and brain total protein lysates was undertaken. At P19, Np65 protein was detected in cochlea and brain lysates from Nptn+/+ and Nptn+/pitch mice, but not Nptnpitch/pitch mice (Fig. 4C). This suggests that the mutant pitch protein, which cannot form a structural disulphide bridge in Ig3, is rapidly targeted for degradation in vivo.
The localization of Np65 protein within the inner ear was investigated by performing immunolabeling studies of cochlear sections and whole-mount tissues. At P16, Np65 labeling was localized to the cuticular plate of the IHCs and OHCs and the basolateral region of the IHCs of Nptnpitch/+ mice (Fig. 4D). We did not find Np65 labeling in other cochlear structures (e.g., spiral ganglion, stria vascularis, or spiral ligament). Whole-mount immunolabeling of cochlea from P16 Nptn+/+ mice confirms Np65 localization to the basolateral portion of IHCs (Fig. 4E). Np65 immunolabeling was not present in the P16 Nptnpitch/pitch cochlea, which is consistent with the Western blot analysis of cochlea total protein (Fig. 4C), indicating an absence of Np65 protein in the IHCs of pitch mutant mice (Fig. 4F). Np55 was not detected in P16 wild-type mice (data not shown).
Neuroplastin is not required for stereocilia bundle formation or mechanoelectrical transduction
The expression of Np65 at the cuticular plate of hair cells from wild-type mice prompted us to determine whether it was required for the formation of the stereociliary bundle and for mechanoelectrical transduction. Scanning electron microscopy analysis of the apical surface of IHCs and OHCs from P16 Nptnpitch/pitch mice revealed a similar ultrastructure to that in Nptn+/+ littermates, with normal looking stereocilia bundles in all regions along the length of the cochlea (Fig. 5A). IHC counts at P16 and P27 were similar between Nptnpitch/pitch and Nptn+/+ mice (Fig. 5B). The number of OHCs was significantly reduced in the basal coil of the cochlea of mutant mice at both ages tested (Fig. 5C). However, this loss of OHCs would not account for the profound hearing loss measured at all frequency regions of mutant mice (Fig. 2A,B).
Figure 5.

Np65 is not required for the normal development of the MET apparatus. A, Scanning electron micrographs of the apical and basal coil of the cochlea from P16 Nptn+/+ (left) and Nptnpitch/pitch (right) showing that the stereocilia bundle structure of IHCs and OHCs appears similar between the two genotypes. B, Number of apical and basal IHCs per 10 pillar cells from Nptn+/+ (white bar) and Nptnpitch/pitch (black bar) mice at P16 and P27. We found no significant IHC loss in the mutant mice compared with wild-type controls at both ages tested. C, Apical and basal OHC counts per 10 pillar cells from Nptn+/+ and Nptnpitch/pitch mice at P16 and P27. A significant loss of OHCs was only observed in the basal turn of the cochlea from Nptnpitch/pitch mice at both ages: *p < 0.05; **p < 0.01. D, E, Saturating MET currents recorded from a P6 control (D) and pitch mutant (E) apical-coil OHC by applying voltage steps from −121 mV to 99 mV in 20 mV increments (holding potential −81 mV). For clarity, only two voltage steps are shown. During the voltage steps, hair bundles were displaced by applying a 50 Hz sinusoidal force stimuli (the driver voltage [DV] to the fluid jet is shown above the traces). Negative deflections of the DV are inhibitory. Arrows indicate the closure of the transducer channels (i.e., disappearance of the resting current) during inhibitory bundle displacements. Dashed lines indicate the holding current, which is the current at the holding potential of −81 mV. F, Peak-to-peak current–voltage curves obtained from 10 controls and eight pitch mutant OHCs (P6). The fits through the data are according to a single-energy-barrier model (Géléoc et al., 1997): I(V) = k [exp ((1 − γ) (V − Vr)/Vs) −exp (−γ(V − Vr)/Vs)], where k is a proportionality constant, Vr is the reversal potential, Vs is a measure for the steepness of the rectification, and γ is the fractional distance within the membrane's electrical field of an energy barrier, as measured from the outside. The values are as follows: control k = 445 ± 37, Vr = 0.4 ± 0.1 mV, Vs = 39 ± 3 mV, γ = 0.42 ± 0.01; pitch mutant k = 428 ± 31, Vr = 0.1 ± 0.2 mV, Vs = 38 ± 2 mV, γ = 0.42 ± 0.01.
MET currents were recorded from postnatal day 6 OHCs by displacing their hair bundles in the excitatory and inhibitory direction using a piezo-driven fluid-jet (Johnson et al., 2011, 2012). At hyperpolarized membrane potentials, the displacement of the hair bundle in the excitatory direction (i.e., toward the taller stereocilia) elicited a large inward MET current in OHCs from both control (Fig. 5D, +/+) and mutant (Fig. 5E, pitch/pitch) mice. The maximal MET current in control OHCs (−1614 ± 69 pA at −121 mV, n = 10) was not significantly different to that recorded in mutant (−1616 ± 52 pA n = 8) OHCs. The resting current flowing through open MET channels in the absence of mechanical stimulation was reduced when bundles were moved in the inhibitory direction (i.e., away from the taller stereocilia) in all OHCs tested (Fig. 5D,E, arrows). Because the MET current reverses near 0 mV (Ohmori, 1985), it became outward when excitatory bundle stimulation was applied during voltage steps positive to its reversal potential (Fig. 5F). At positive membrane potentials, excitatory bundle stimulation now elicited an outward MET current with larger resting amplitudes (arrows), which are due to an increased open probability of the transducer channel resulting from a reduced driving force for Ca2+ influx (Crawford et al., 1989).
Neuroplastin is required for IHC synaptogenesis
Given the localization of Np65 at the basolateral aspect of IHCs and the reported function of Neuroplastins in synaptogenesis (Marzban et al., 2003; Beesley et al., 2014; Herrera-Molina et al., 2014), we investigated for the presence of IHC auditory ribbon synapses. We performed cochlear whole-mount immunolabeling studies using an anti-Ribeye antibody (a component protein of the presynaptic IHC ribbon) and an anti-GluR2 antibody (AMPA receptor localized to the afferent postsynaptic density). In apical coil IHCs, we found a similar number of presynaptic ribbons (Ribeye-positive puncta) and postsynaptic densities (GluR2-positive puncta) between P16 pitch mutant (n = 5) and wild-type mice (n = 5) (Fig. 6A–C). We then investigated whether the presynaptic and postsynaptic sites were juxtaposed as previously described in IHCs from rodents (Brandt et al., 2005; Zampini et al., 2013). Dual-labeling of Nptn+/+ and Nptnpitch/pitch cochleae demonstrated a significantly reduced number (∼30%: p < 0.05) of matched presynaptic and postsynaptic puncta in the Nptnpitch/pitch mutant mice compared with Nptn+/+ control mice (Fig. 6C). To assess this further, the number of mismatched Ribeye and GluR2 puncta were calculated as a percentage of the total number of Ribeye and GluR2 puncta, respectively (Fig. 6D). We found a significantly increased number of mismatched puncta present in Nptnpitch/pitch mutants compared with Nptn+/+ control mice (Ribeye: from ∼9% to ∼30%, p < 0.01; GluR2 from ∼13% to ∼40%, p < 0.001).
Figure 6.

Reduced presynaptic and postsynaptic colocalization in IHCs from mutant pitch mice. Cochlear whole mounts prepared from P16 Nptn+/+ and Nptnpitch/pitch mice were labeled for the IHC presynaptic ribbon marker Ribeye (green) and the afferent neuron postsynaptic density marker GluR2 (red) and imaged using confocal microscopy. A, B, Three-dimensional perspectives of z-stack image showing closely apposed Ribeye/GluR2-labeled punctum (matched) in IHCs from both Nptn+/+ (A) and Nptnpitch/pitch (B) mice. Note the larger number of single-label punctum (mismatched) in the Nptnpitch/pitch cochlea (insets). C, Quantification of the total number of Ribeye-positive and GluR2-positive puncta (matched + mismatched), and the total number of matched puncta in Nptn+/+ and Nptnpitch/pitch cochleae. Although there is no difference in the total number of Ribeye- or GluR2-labeled puncta between Nptn+/+ and Nptnpitch/pitch cochleae, there is a reduction in the number of matched Ribeye/GluR2-labeled puncta in Nptnpitch/pitch cochleae compared with Nptn+/+. D, The percentage of mismatched Ribeye and GluR2 puncta were determined in relation to the total number of Ribeye and GluR2 puncta, respectively. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar: 20 μm.
Neuroplastin is required for exocytosis in adult IHCs
Having identified synaptic mismatching in Nptn-deficient mice, we next investigated presynaptic function (i.e., exocytosis) of IHCs from adult pitch mice. This was assessed by measuring the change in cell membrane capacitance (ΔCm) following cell depolarization, which provides an indication of vesicle fusion into the plasma membrane at presynaptic active zones (Moser and Beutner, 2000; Johnson et al., 2008). We have previously shown that, at the onset of hearing, the synaptic machinery of IHCs becomes more sensitive to Ca2+ influx, causing docked vesicles to be released linearly with increases in Ca2+ current (ICa) (Johnson et al., 2007, 2010, 2013) and requires synaptotagmin IV (Johnson et al., 2010). The process of linearization is under direct control of Ca2+ action potentials in immature cells (Johnson et al., 2013). We found that, in the absence of functional Neuroplastin, the progression in the functional maturation of the synaptic machinery in IHCs was prevented. ICa and corresponding ΔCm were obtained by applying 50 ms voltage steps, which is known to recruit the readily releasable pool (RRP) (Johnson et al., 2008). Adult pitch mutant IHCs (n = 9) had a slightly larger maximal ICa (p < 0.02) but similar corresponding ΔCm compared with control littermates (n = 7; Fig. 7A–C). The exocytotic Ca2+ dependence, defined as the change in ΔCm as a function of ICa and measured using the synaptic transfer function (Johnson et al., 2010), was significantly less linear (power of 3.2 ± 0.4, n = 11) than that in control IHCs (power of 1.1 ± 0.1, n = 10, p < 0.0001; Fig. 7D).
Figure 7.

Exocytosis does not fully mature in pitch IHCs. A, B, ICa and changes in ΔCm in adult control (P19) and pitch mutant (P21) IHCs. Recordings were obtained in response to 50 ms voltage steps from −81 mV in 10 mV increments. A, Only maximal responses are shown. C, Maximal peak ICa (top) and ΔCm (bottom) values for adult control and mutant IHCs. D, Synaptic transfer curves obtained by plotting ΔCm against the corresponding ICa between −71 mV and −11 mV (Johnson et al., 2010, 2013). Fits are according to a power function: ΔCm = cICaN where c is a scaling coefficient and the exponent N is the power. Mutant IHCs show a steeper intrinsic Ca2+ dependence than control cells. E, Average ΔCm from seven control and nine mutant IHCs (P16-P22) in response to voltage steps from 2 ms to 2 s (to ∼−11 mV) showing the RRP and SRP. F, ΔCm elicited using repetitive voltage steps to −11 mV of 1 s in duration to elicit the SRP. The interstep interval was 200 ms. For clarity, only the first few steps are shown. The voltage protocol used is shown above the traces. G, Average cumulative ΔCm values obtained in response to the 1 s (35 step) protocol, from four control and six mutant IHCs. H, Individual ΔCm values from control and mutant IHCs measured following each voltage step from G.
The rate of neurotransmitter release in adult IHCs was studied by measuring ΔCm in response to depolarizing voltage steps to −11 mV of varying duration (2 ms to 2 s: interstep interval was at least 11 s), which allowed us to investigate the emptying of different synaptic vesicle pool populations (Fig. 7E). While relatively short stimuli (≤100 ms), when using 1.3 mm extracellular Ca2+ and body temperature (Johnson et al., 2008), reveal the number of vesicles docked at the active zones (RRP), longer steps induce the release of vesicles from a secondarily releasable pool (SRP) that is located further away from the Ca2+ channels (von Gersdorff et al., 1996; Moser and Beutner, 2000; Johnson et al., 2008). In agreement with the results shown above (Fig. 7C), the size of the isolated RRP was similar between the two genotypes and consisted of ∼600 vesicles when using a conversion factor of 37 aF/vesicle (Lenzi et al., 1999). However, the size of the SRP was largely reduced in pitch mutant IHCs (Fig. 7E). We then investigated whether the reduced vesicle release from the SRP in pitch mutant IHCs was limited by the relative pool filling rates using repetitive stimulation. Figure 7F shows ΔCm when a train of 1 s steps to −11 mV were applied to investigate the depletion of the SRP. The cumulative ΔCm showed that SRP release saturation was much more pronounced in pitch mutant than control IHCs (Fig. 7G). The earlier depletion of the SRP in pitch mutant IHCs, compared with control cells, was also evident from the individual ΔCm (Fig. 7H).
Discussion
We have shown that the absence of Neuroplastin 65 leads to an early-onset hearing loss in pitch mice. The loss of Np65 causes synaptic mis-matching between IHCs and afferent neurons of the auditory nerve. Our results identify Np65 as a novel deafness gene required for ribbon synapse formation and vesicle exocytosis in cochlear IHCs.
Neural CAMs are required for the genesis, maintenance, and plasticity of synapses. Neuroplastin, together with Basigin and Embigin, forms a distinct family of neural CAMs belonging to the Ig superfamily. Embigin has been shown to be involved in neuromuscular synapse formation and plasticity (Lain et al., 2009), and Basigin is reported to have important roles in fetal, neuronal, lymphocyte, and extracellular matrix development (Iacono et al., 2007). Both Neuroplastin 55 and 65 have previously been identified in the plexiform layers of the rat retina (Kreutz et al., 2001), where an increase in Np65 expression has been observed to correlate with the formation of ribbon synapses between photoreceptors and the postsynaptic complex formed by horizontal and bipolar cells (Kreutz et al., 2001). In addition, Np65, which has been shown to be mainly expressed in the brain, have been reported to mediate several cellular processes including cell–cell adhesion, neurite outgrowth, synaptic plasticity, and long-term potentiation in the hippocampus (Smalla et al., 2000; Kreutz et al., 2001; Empson et al., 2006; Owczarek et al., 2010, 2011; Owczarek and Berezin, 2012).
Np65 is required for hearing
Despite the involvement of Neuroplastins in several crucial physiological mechanisms (Beesley et al., 2014), they have yet to be directly linked to diseases in mammals, including humans. The pitch mouse was identified in a recent ENU-mutagenesis, phenotype-driven screen as a recessive model of profound sensorineural deafness. We found that pitch has a loss-of-function coding mutation in the novel deafness-related gene Neuroplastin (Nptn). ABR analyses identified that pitch mutant mice have severely impaired hearing from P16, and most likely never achieve normal hearing at the onset of hearing (∼P12). However, compared with hearing thresholds of P27 and 8-week-old mutant animals (Fig. 2), it appears that there may be some hearing function at P16, albeit very slight, which deteriorates by 8 weeks of age.
We found that Np65 transcript is upregulated in the mouse inner ear from at or just before P12. In contrast, Np55 was present at a basal level throughout this postnatal period. Immunolabeling studies demonstrated that, at the protein level, Np65 is primarily localized to the basolateral portion of IHCs (Fig. 4), which is consistent with previous reports of Np65 expression at synapses located in the brain and the retina (Smalla et al., 2000; Kreutz et al., 2001; Owczarek et al., 2011). Np65 protein was absent in pitch mutant mice, which reflects the in silico and in vitro data showing that the pitch mutation is a loss-of-function allele. In addition, pitch mutants display a very similar hearing loss phenotype to that displayed by mice homozygous for either the Nptn nonsense allele (NptnY219X/Y219X; see Results) or to that reported for the Nptn knock-out allele (Nptn−/−; see Materials and Methods and www.mousephenotype.org), both of which are loss-of-function alleles. Histological and ultrastructural studies show no gross morphological changes within the cochlea of P16 pitch mutant mice (Fig. 5). Therefore, the overlap between the onset of Np65 expression and the onset of hearing at ∼P12, support the hypothesis that Np65 is critical for IHC function.
Despite the fact that Np65 has been shown to be present within the synaptic layers of the rat (Kreutz et al., 2001) and mouse (present study) retina, retinal function, as assessed using ERG, was found to be unaffected by the absence of Np55 and Np65 (Fig. 3), indicating a nonessential role for Neuroplastins in the mouse retina. Indeed, the related-neural CAM Basigin has been shown to be crucial for retinal function, with Basigin knock-out mice exhibiting impaired visual function from the time of eye-opening (Ochrietor et al., 2002).
Np65 is required for the formation of synapses in mature IHC
Our morphological and electrophysiology findings show that the MET complex that projects from the IHC's apical surface develops and function normally in the absence of Np65. However, we found that IHC ribbon synapses fail to form correctly, with an increased number of mismatched presynaptic ribbons (Ribeye) and postsynaptic densities (GluR2) identified in pitch mutant compared with wild-type mice (Fig. 6). This observation, together with the Neuroplastin expression data and Np65 immunolabeling studies, indicates that Neuroplastin is required for the tight coupling of IHCs and afferent neurons, which normally occurs with maturation. Recently, it has been shown that, in hippocampal cultures from Nptn-deficient mice, the number of newly formed glutamatergic (excitatory) synapses was unchanged but showed increased mismatch between presynaptic and postsynaptic terminals compared with wild-type-derived cultures (Herrera-Molina et al., 2014). However, with maturation, the number of excitatory synapses was reduced in Nptn-deficient cultures. These morphological defects were reported to be linked to the absence of the neural CAM Np65, but not Np55, and likely a result of synaptic instability caused by loss of trans-homodimerization via the Np65-specific Ig1 domain (Herrera-Molina et al., 2014). The strikingly similar morphological defects in the synapses of both Nptn-deficient hippocampal neurons and pitch IHCs suggest a common acute synaptic function for this neural CAM. Many CAMs are able to signal physiological cues via interaction with fibroblast growth factor receptors (FGFRs) (Hansen et al., 2008). Np65 has been shown to regulate neurite outgrowth (Owczarek et al., 2011) and synaptic plasticity (Empson et al., 2006) through interaction with FGFR1, which triggers intracellular cascades mediated by p38 MAPKs and Ca2+/calmodulin-dependent protein kinase. Ca2+/calmodulin-dependent protein kinase has been linked to the mobilization reserve vesicle pool in hippocampal neurons (Chi et al., 2003). Although FGF signaling in the cochlea has so far been linked to early hair cell differentiation (Jacques et al., 2012), its possible role in the adult cochlea is still unknown. Our observation that the release from the reserve vesicle pool was reduced in pitch mutant IHCs (Fig. 7) suggests that a similar mechanism to that of the hippocampus could be present in IHCs.
In conclusion, we demonstrate that the neural CAM Neuroplastin is required for ribbon synaptogenesis and vesicle exocytosis, which are prerequisite for IHC function and auditory transduction.
Footnotes
This work was supported by Medical Research Council MC_U142684175 to S.D.M.B. and Wellcome Trust 102892 to W.M. L. Carrott and L. Chessum are Medical Research Council PhD students. S.L.J. is a Royal Society University Research Fellow. We thank L. Vizor, T. Osborne, and L. Ireson for assistance with the breeding of pitch animals; D. Williams for help with the qRT-PCR experiments; J. Sanderson for help with the immunolabeling; and T. Moser and J. Neef for technical advice regarding imaging and quantification of ribbon synapses.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Acevedo-Arozena A, Wells S, Potter P, Kelly M, Cox RD, Brown SD. ENU mutagenesis, a way forward to understand gene function. Annu Rev Genomics Hum Genet. 2008;9:49–69. doi: 10.1146/annurev.genom.9.081307.164224. [DOI] [PubMed] [Google Scholar]
- Aleman TS, Cideciyan AV, Aguirre GK, Huang WC, Mullins CL, Roman AJ, Sumaroka A, Olivares MB, Tsai FF, Schwartz SB, Vandenberghe LH, Limberis MP, Stone EM, Bell P, Wilson JM, Jacobson SG. Human CRB1-associated retinal degeneration: comparison with the rd8 Crb1-mutant mouse model. Invest Ophthalmol Vis Sci. 2011;52:6898–6910. doi: 10.1167/iovs.11-7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer J, Ji R, Ray TA, Selber F, Gassmann M, Peachey NS, Gregg RG, Enzmann V. Presence of the Gpr179(nob5) allele in a C3H-derived transgenic mouse. Mol Vis. 2013;19:2615–2625. [PMC free article] [PubMed] [Google Scholar]
- Beesley PW, Herrera-Molina R, Smalla KH, Seidenbecher C. The Neuroplastin adhesion molecules: key regulators of neuronal plasticity and synaptic function. J Neurochem. 2014;131:268–283. doi: 10.1111/jnc.12816. [DOI] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB. Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci. 2010;11:18–29. doi: 10.1038/nrn2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt A, Khimich D, Moser T. Few CaV1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J Neurosci. 2005;25:11577–11585. doi: 10.1523/JNEUROSCI.3411-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/S0896-6273(03)00151-X. [DOI] [PubMed] [Google Scholar]
- Coghill EL, Hugill A, Parkinson N, Davison C, Glenister P, Clements S, Hunter J, Cox RD, Brown SD. A gene-driven approach to the identification of ENU mutants in the mouse. Nat Genet. 2002;30:255–256. doi: 10.1038/ng847. [DOI] [PubMed] [Google Scholar]
- Crawford AC, Evans MG, Fettiplace R. Activation and adaptation of transducer currents in turtle hair cells. J Physiol. 1989;419:405–434. doi: 10.1113/jphysiol.1989.sp017878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empson RM, Buckby LE, Kraus M, Bates KJ, Crompton MR, Gundelfinger ED, Beesley PW. The cell adhesion molecule neuroplastin-65 inhibits hippocampal long-term potentiation via a mitogen-activated protein kinase p38-dependent reduction in surface expression of GluR1-containing glutamate receptors. J Neurochem. 2006;99:850–860. doi: 10.1111/j.1471-4159.2006.04123.x. [DOI] [PubMed] [Google Scholar]
- Géléoc GS, Lennan GW, Richardson GP, Kros CJ. A quantitative comparison of mechanoelectrical transduction in vestibular and auditory hair cells of neonatal mice. Proc Biol Sci. 1997;264:611–621. doi: 10.1098/rspb.1997.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA. Transmitter release at the hair cell ribbon synapse. Nat Neurosci. 2002;5:147–154. doi: 10.1038/nn796. [DOI] [PubMed] [Google Scholar]
- Hansen SM, Berezin V, Bock E. Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-cadherin. Cell Mol Life Sci. 2008;65:3809–3821. doi: 10.1007/s00018-008-8290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardisty-Hughes RE, Parker A, Brown SD. A hearing and vestibular phenotyping pipeline to identify mouse mutants with hearing impairment. Nat Protoc. 2010;5:177–190. doi: 10.1038/nprot.2009.204. [DOI] [PubMed] [Google Scholar]
- Herrera-Molina R, Sarto-Jackson I, Montenegro-Venegas C, Heine M, Smalla KH, Seidenbecher CI, Beesley PW, Gundelfinger ED, Montag D. Structure of excitatory synapses and GABAA receptor localization at inhibitory synapses are regulated by neuroplastin-65. J Biol Chem. 2014;289:8973–8988. doi: 10.1074/jbc.M113.514992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelter SM, Dalke C, Kallnik M, Becker L, Horsch M, Schrewe A, Favor J, Klopstock T, Beckers J, Ivandic B, Gailus-Durner V, Fuchs H, Hrabé de Angelis M, Graw J, Wurst W. “Sighted C3H” mice: a tool for analysing the influence of vision on mouse behaviour? Front Biosci. 2008;13:5810–5823. doi: 10.2741/3118. [DOI] [PubMed] [Google Scholar]
- Iacono KT, Brown AL, Greene MI, Saouaf SJ. CD147 immunoglobulin superfamily receptor function and role in pathology. Exp Mol Pathol. 2007;83:283–295. doi: 10.1016/j.yexmp.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques BE, Dabdoub A, Kelley MW. Fgf signaling regulates development and transdifferentiation of hair cells and supporting cells in the basilar papilla. Hear Res. 2012;289:27–39. doi: 10.1016/j.heares.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Adelman JP, Marcotti W. Genetic deletion of SK2 channels in mouse inner hair cells prevents the developmental linearization in the Ca2+ dependence of exocytosis. J Physiol. 2007;583:631–646. doi: 10.1113/jphysiol.2007.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Forge A, Knipper M, Münkner S, Marcotti W. Tonotopic variation in the calcium dependence of neurotransmitter release and vesicle pool replenishment at mammalian auditory ribbon synapses. J Neurosci. 2008;28:7670–7678. doi: 10.1523/JNEUROSCI.0785-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Franz C, Kuhn S, Furness DN, Rüttiger L, Münkner S, Rivolta MN, Seward EP, Herschman HR, Engel J, Knipper M, Marcotti W. Synaptotagmin IV determines the linear Ca2+ dependence of vesicle fusion at auditory ribbon synapses. Nat Neurosci. 2010;13:45–52. doi: 10.1038/nn.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Eckrich T, Kuhn S, Zampini V, Franz C, Ranatunga KM, Roberts TP, Masetto S, Knipper M, Kros CJ, Marcotti W. Position-dependent patterning of spontaneous action potentials in immature cochlear inner hair cells. Nat Neurosci. 2011;14:711–717. doi: 10.1038/nn.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Kennedy HJ, Holley MC, Fettiplace R, Marcotti W. The resting transducer current drives spontaneous activity in prehearing mammalian cochlear inner hair cells. J Neurosci. 2012;32:10479–10483. doi: 10.1523/JNEUROSCI.0803-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Kuhn S, Franz C, Ingham N, Furness DN, Knipper M, Steel KP, Adelman JP, Holley MC, Marcotti W. Presynaptic maturation in auditory hair cells requires a critical period of sensory-independent spiking activity. Proc Natl Acad Sci U S A. 2013;110:8720–8725. doi: 10.1073/pnas.1219578110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutz MR, Langnaese K, Dieterich DC, Seidenbecher CI, Zuschratter W, Beesley PW, Gundelfinger ED. Distribution of transcript and protein isoforms of the synaptic glycoprotein neuroplastin in rat retina. Invest Ophthalmol Vis Sci. 2001;42:1907–1914. [PubMed] [Google Scholar]
- Lain E, Carnejac S, Escher P, Wilson MC, Lømo T, Gajendran N, Brenner HR. A novel role for embigin to promote sprouting of motor nerve terminals at the neuromuscular junction. J Biol Chem. 2009;284:8930–8939. doi: 10.1074/jbc.M809491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langnaese K, Beesley PW, Gundelfinger ED. Synaptic membrane glycoproteins gp65 and gp55 are new members of the immunoglobulin superfamily. J Biol Chem. 1997;272:821–827. doi: 10.1074/jbc.272.2.821. [DOI] [PubMed] [Google Scholar]
- Lenzi D, Runyeon JW, Crum J, Ellisman MH, Roberts WM. Synaptic vesicle populations in saccular hair cells reconstructed by electron tomography. J Neurosci. 1999;19:119–132. doi: 10.1523/JNEUROSCI.19-01-00119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W. Functional assembly of mammalian cochlear hair cells. Exp Physiol. 2012;97:438–451. doi: 10.1113/expphysiol.2011.059303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzban H, Khanzada U, Shabir S, Hawkes R, Langnaese K, Smalla KH, Bockers TM, Gundelfinger ED, Gordon-Weeks PR, Beesley PW. Expression of the immunoglobulin superfamily neuroplastin adhesion molecules in adult and developing mouse cerebellum and their localisation to parasagittal stripes. J Comp Neurol. 2003;462:286–301. doi: 10.1002/cne.10719. [DOI] [PubMed] [Google Scholar]
- Matthews G, Fuchs P. The diverse roles of ribbon synapses in sensory neurotransmission. Nat Rev Neurosci. 2010;11:812–822. doi: 10.1038/nrn2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehalow AK, Kameya S, Smith RS, Hawes NL, Denegre JM, Young JA, Bechtold L, Haider NB, Tepass U, Heckenlively JR, Chang B, Naggert JK, Nishina PM. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum Mol Genet. 2003;12:2179–2189. doi: 10.1093/hmg/ddg232. [DOI] [PubMed] [Google Scholar]
- Moser T, Beutner D. Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci U S A. 2000;97:883–888. doi: 10.1073/pnas.97.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochrietor JD, Moroz TP, Clamp MF, Timmers AM, Muramatsu T, Linser PJ. Inactivation of the Basigin gene impairs normal retinal development and maturation. Vision Res. 2002;42:447–453. doi: 10.1016/S0042-6989(01)00236-X. [DOI] [PubMed] [Google Scholar]
- Ohmori H. Mechano-electrical transduction currents in isolated vestibular hair cells of the chick. J Physiol. 1985;359:189–217. doi: 10.1113/jphysiol.1985.sp015581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi Y, Kubota Y, Kuramasu A, Watanabe T, Ito C. Gene expression profiling in whole cerebral cortices of phencyclidine- or methamphetamine-treated rats. Brain Res Mol Brain Res. 2005;140:142–149. doi: 10.1016/j.molbrainres.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Owczarek S, Berezin V. Neuroplastin: cell adhesion molecule and signaling receptor. Int J Biochem Cell Biol. 2012;44:1–5. doi: 10.1016/j.biocel.2011.10.006. [DOI] [PubMed] [Google Scholar]
- Owczarek S, Kiryushko D, Larsen MH, Kastrup JS, Gajhede M, Sandi C, Berezin V, Bock E, Soroka V. Neuroplastin-55 binds to and signals through the fibroblast growth factor receptor. FASEB J. 2010;24:1139–1150. doi: 10.1096/fj.09-140509. [DOI] [PubMed] [Google Scholar]
- Owczarek S, Soroka V, Kiryushko D, Larsen MH, Yuan Q, Sandi C, Berezin V, Bock E. Neuroplastin-65 and a mimetic peptide derived from its homophilic binding site modulate neuritogenesis and neuronal plasticity. J Neurochem. 2011;117:984–994. doi: 10.1111/j.1471-4159.2011.07269.x. [DOI] [PubMed] [Google Scholar]
- Safieddine S, El-Amraoui A, Petit C. The auditory hair cell ribbon synapse: from assembly to function. Annu Rev Neurosci. 2012;35:509–528. doi: 10.1146/annurev-neuro-061010-113705. [DOI] [PubMed] [Google Scholar]
- Saito A, Fujikura-Ouchi Y, Kuramasu A, Shimoda K, Akiyama K, Matsuoka H, Ito C. Association study of putative promoter polymorphisms in the neuroplastin gene and schizophrenia. Neurosci Lett. 2007;411:168–173. doi: 10.1016/j.neulet.2006.08.042. [DOI] [PubMed] [Google Scholar]
- Smalla KH, Matthies H, Langnäse K, Shabir S, Böckers TM, Wyneken U, Staak S, Krug M, Beesley PW, Gundelfinger ED. The synaptic glycoprotein neuroplastin is involved in long-term potentiation at hippocampal CA1 synapses. Proc Natl Acad Sci U S A. 2000;97:4327–4332. doi: 10.1073/pnas.080389297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun AX, Crabtree GR, Yoo AS. MicroRNAs: regulators of neuronal fate. Curr Opin Cell Biol. 2013;25:215–221. doi: 10.1016/j.ceb.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Vardi E, Matthews G, Sterling P. Evidence that vesicles on the synaptic ribbon of retinal bipolar neurons can be rapidly released. Neuron. 1996;16:1221–1227. doi: 10.1016/S0896-6273(00)80148-8. [DOI] [PubMed] [Google Scholar]
- Zampini V, Johnson SL, Franz C, Knipper M, Holley MC, Magistretti J, Masetto S, Marcotti W. Burst activity and ultrafast activation kinetics of CaV1.3 Ca(2) (+) channels support presynaptic activity in adult gerbil hair cell ribbon synapses. J Physiol. 2013;591:3811–3820. doi: 10.1113/jphysiol.2013.251272. [DOI] [PMC free article] [PubMed] [Google Scholar]