

Abstract
Epigenetic information encoded in covalent modifications of DNA and histone proteins regulates fundamental biological processes through the action of chromatin regulators, transcription factors, and noncoding RNA species. Epigenetic plasticity enables an organism to respond to developmental and environmental signals without genetic changes. However, aberrant epigenetic control plays a key role in pathogenesis of disease. Normal epigenetic states could be disrupted by detrimental mutations and expression alteration of chromatin regulators or by environmental factors. In this primer, we briefly review the epigenetic basis of human disease and discuss how recent discoveries in this field could be translated into clinical diagnosis, prevention, and treatment. We introduce platforms for mapping genome-wide chromatin accessibility, nucleosome occupancy, DNA-binding proteins, and DNA methylation, primarily focusing on the integration of DNA methylation and chromatin immunoprecipitation–sequencing technologies into disease association studies. We highlight practical considerations in applying high-throughput epigenetic assays and formulating analytical strategies. Finally, we summarize current challenges in sample acquisition, experimental procedures, data analysis, and interpretation and make recommendations on further refinement in these areas. Incorporating epigenomic testing into the clinical research arsenal will greatly facilitate our understanding of the epigenetic basis of disease and help identify novel therapeutic targets.
Keywords: cancer, complex diseases, DNA methylation, epigenome, histone modification
Epigenetics is the study of mitotically or meiotically heritable changes in gene expression that occur without changes in DNA sequence and are mediated by postsynthetic modifications of DNA and histone proteins, histone variants, noncoding RNAs, and the proteins that regulate and interpret the modifications (1, 2). The term “epigenome” refers to the genome-wide distribution of these marks and regulators (3–5). Acting “above and beyond” the normally static genome, hundreds of epigenomes define the unique transcriptional programs of our cells throughout development and aging (3, 6) under the influence of stochastic and environmental factors (1, 7, 8). Epigenetic mechanisms have been directly implicated in disease susceptibility and progression (2, 3). The epigenome's ability to translate environmental exposures into gene regulation also provides a link to disease.
The development of assays based on microarrays and next-generation sequencing has allowed the identification of mutations in chromatin regulators and other epigenetic variation related to human disease. Since epigenetic information varies not only between individuals but also between cell types and their functional states, accurate detection of epigenetic changes in patients requires the generation of a large number of reference epigenomes. The International Human Epigenome Consortium (IHEC) aims to generate over 1,000 reference epigenomes within a decade (9). As part of the International Human Epigenome Consortium, the National Institutes of Health Roadmap Epigenomics Project and the National Human Genome Research Institute Encyclopedia of DNA Elements (ENCODE) Consortium have generated genome-wide maps for DNA methylation, histone modifications, transcription factor binding, and chromatin accessibility from over 200 cell or tissue types (10–12) (see Appendix). This joint effort has advanced our understanding of the interindividual variations and cell-type specificity of key epigenetic marks (13–15). Rapid development in epigenomic technology and the increasing body of genomic and epigenomic data offer an unprecedented opportunity to delineate how the interplay between genetic, environmental, and epigenetic components triggers complex diseases (16). In this review, we describe how high-throughput epigenomic technologies could be applied to population-based studies of diseases/traits. Focusing on DNA methylation and chromatin immunoprecipitation–sequencing (ChIP-seq) assays, we discuss challenges and possible solutions in experimental design, data analysis, and interpretation of genome-wide epigenomic studies.
EPIGENETIC MARKS AND REGULATORS
Chromatin structure and function
The nucleosome, the fundamental unit of chromatin, is composed of 147 base pairs of DNA wrapped around a histone octamer composed of 2 copies each of the 4 canonical histones or their variants (6, 17). DNA and histones are subject to covalent modifications, and there are at least 18 different functional groups (18). These modifications are dynamic and reversible, mediated by proteins that add (“writers”), interpret (“readers”), or remove (“erasers”) them (19, 20).
Several factors are responsible for shaping chromatin organization and DNA packaging (5, 21). ATP-dependent chromatin remodeling complexes regulate the spacing, positioning, deposition and ejection, and histone composition of nucleosomes (21–23). Regions occupied by densely packed nucleosomes block transcription factors from accessing the DNA, while nucleosome-free regions like these at the transcriptional start sites of actively transcribed genes are more “open” to transcription factor binding (24, 25). Several next-generation sequencing–based methods have been developed to map nucleosome positions and chromatin accessibility (5, 26) (Table 1). Furthermore, the chromatin fiber is folded into 3-dimensional structures, thus enabling physical interactions between gene regulatory elements that are often far away from each other on the linearized DNA but are regulated by the same mechanisms (27–29).
Table 1.
High-Throughput Epigenomic Assays
| Assay | Target(s) | Features | References |
|---|---|---|---|
| MNase-seq | Nucleosome occupancy and positioning | Maps both histone and nonhistone proteins | 180–182 |
| Localization of DNA-binding proteins | Needs high sequencing depth | ||
| Bias toward AT-rich regions | |||
| DNase-seq | Chromatin accessibility | Maps cis-regulatory regions | 73, 174, 183, 184 |
| Generates base-pair resolution footprints for some transcription factors | |||
| Requires high sequencing depth | |||
| DNase I cleavage bias | |||
| May miss some distal regulatory regions | |||
| FAIRE-seq | Chromatin accessibility | Maps cis-regulatory regions | 185–187 |
| Does not rely on any antibody or enzyme digestion | |||
| Relatively lower signal enrichment compared with DNase-seq | |||
| May miss some promoter regions | |||
| ATAC-seq | Chromatin accessibility | Maps nucleosome positioning, chromatin accessibility, and transcription factor binding sites simultaneously | 188, 189 |
| Localization of DNA-binding proteins | |||
| Nucleosome positioning | |||
| Requires fewer cells | |||
| Fast protocol | |||
| Primarily targets nucleosomes around regulatory regions | |||
| ChIP-seq | Localization of DNA-binding proteins | Maps DNA-binding proteins | 10, 12, 14, 77, 142, 144, 190, 191 |
| Often requires large quantities of starting material | |||
| Needs a high-quality antibody | |||
| WGBS | DNA methylation | Covers approximately 95% of CpGs | 13, 15, 32, 96 |
| Accurate quantification | |||
| Single-base resolution | |||
| High proportion of uninformative reads from non-CpG regions | |||
| Does not differentiate between 5meC and 5hmeC | |||
| High cost | |||
| RRBS | DNA methylation | Covers approximately 10%–20% of CpGs, largely in CpG islands | 32, 45, 192, 193 |
| Accurate quantification | |||
| Single-base resolution | |||
| Low cost | |||
| Limited coverage | |||
| Does not differentiate between 5meC and 5hmeC | |||
| MeDIP-seq | DNA methylation | Covers approximately 60%–90% of CpGs | 32, 123, 124, 139, 193, 194 |
| Can distinguish between 5meC and 5hmeC | |||
| Can target a large fraction of repeats regions | |||
| Low cost | |||
| Quantitative estimation | |||
| Low (approximately 150-base-pair) resolution | |||
| Difficult to identify small methylation changes | |||
| Reduced sensitivity to less methylated regions | |||
| MBD-seq | DNA methylation | Covers approximately 60% of CpGs | 32, 60, 126, 127 |
| Can target a large fraction of repeats regions | |||
| Low cost | |||
| Quantitative estimation | |||
| Low (approximately 150-base-pair) resolution | |||
| Difficult to identify small methylation changes | |||
| Reduced sensitivity to less methylated regions | |||
| 27K arraya | DNA methylation | Covers 27,578 CpGs | 61, 79, 84, 104, 120, 121, 193 |
| Single-base resolution | |||
| Low cost | |||
| Limited coverage | |||
| Does not differentiate between 5meC and 5hmeC | |||
| 450K arrayb | DNA methylation | Covers 482,421 CpGs | 48, 91, 105, 122 |
| Single-base resolution | |||
| Low cost | |||
| Ideal for screening large cohorts | |||
| Limited coverage | |||
| Does not estimate allele-specific methylation | |||
| Does not differentiate between 5meC and 5hmeC |
Abbreviations: A, adenine; ATAC-seq, assay for transposase-accessible chromatin using sequencing; C, cytosine; ChIP-seq, chromatin immunoprecipitation and sequencing; CpG, cytosine-guanine dinucleotide; DNase-seq, DNase I digestion and sequencing; FAIRE-seq, formaldehyde-assisted isolation of regulatory elements sequencing; G, guanine; 5hmeC, 5-hydroxymethylcytosine; MBD-seq, methyl-CpG-binding domain protein-based capture and sequencing; 5meC, 5-methylcytosine; MeDIP-seq, methylated DNA immunoprecipitation and sequencing; MNase-seq, micrococcal nuclease digestion and sequencing; RRBS, reduced representation bisulfite sequencing; T, thymine; WGBS, whole-genome bisulfite sequencing.
a Illumina Infinium HumanMethylation27 BeadChip array (Illumina, Inc., San Diego, California).
b Illumina Infinium HumanMethylation450 BeadChip array (Illumina, Inc.).
DNA methylation
DNA methylation refers to the addition of a methyl group (me) at position 5 of the cytosine base (5meC), occurring mostly in cytosine-guanine dinucleotides (CpG's) (30, 31). 5meC also occurs in non-CpG contexts in embryonic stem cells (32–34), adult neurons (35, 36), and some other types of cells (35, 36). DNA methylation is mediated by the DNA methyltransferase family. 5meC can be further modified by ten-eleven translocation (TET) family dioxygenases, which sequentially oxidize 5meC to 5-hydroxymethylcytosine (5hmeC), 5-formylcytosine (5fC), and 5-carboxycytosine (5caC) (11, 37, 38). Additional reactions can eventually restore unmodified cytosine (39). The exact function of these oxidation derivatives remains unknown. It has been suggested that 5hmeC may represent an intermediate product in an active demethylation pathway (37, 40, 41). 5hmeC is highly enriched in neurons and may serve as an epigenetic mark involved in neural development (41).
In normal human cells, 5meC occurs at approximately 70%–80% of CpGs, mostly in low-density CpG regions (42, 43). The so-called CpG islands, which have a high CpG and high (>50%) GC content, are infrequently methylated (33, 43, 44). CpG islands represent approximately 2% of the human genome (45) and occupy approximately 60% of promoter regions (33). While the majority of gene promoters are devoid of DNA methylation, marked methylation is observed within actively transcribed gene bodies (9, 46).
DNA methylation, which is highly divergent across different organisms and cell types (11, 34), plays essential roles in mammalian development, X chromosome inactivation, transposable element repression, and genomic imprinting (11, 32, 34, 37). DNA methylation is influenced by genetic, stochastic, and environmental factors and is involved in developmental disorders and cancer (11, 42, 46–49).
Histone modifications
The core histones (H2A, H2B, H3, and H4) each undergo a variety of modifications, primarily on their N-terminal “tails” (11, 50). The common modifications include lysine (K) mono-, di-, and trimethylation (Kme1/2/3), arginine (R) mono- and dimethylation (Rme1/2), and K acetylation (Kac) and ubiquitination, as well as serine/threonine/tyrosine phosphorylation (17, 18, 38, 51).
Histone modifications play fundamental roles in regulating gene transcription, DNA replication, and DNA repair by influencing chromatin states and serving as docking sites for other proteins like histone modification “readers” and “writers” (9, 38). Histone acetylation is generally associated with transcriptional activation, whereas methylation can activate or repress transcription, depending on the residue modified and the level of modification (9, 11, 52). Genome-wide profiling of histone modifications has revealed their association with distinct genomic regions such as enhancers, promoters, and gene bodies (11, 53) (Figure 1).
Figure 1.
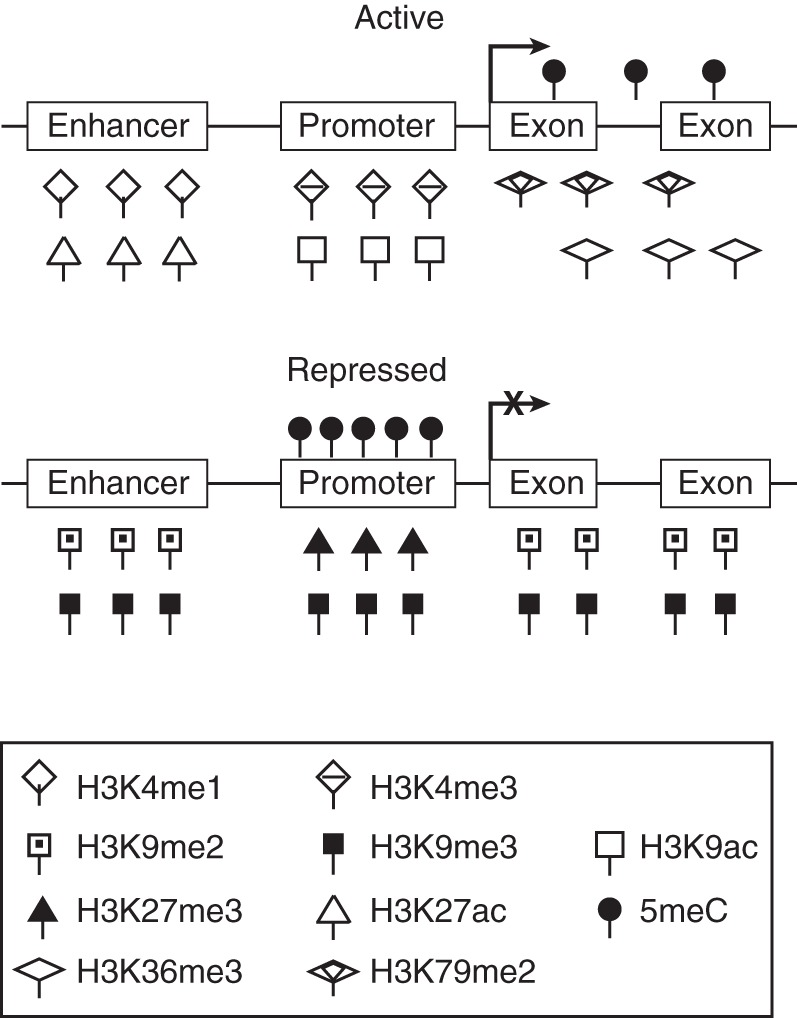
Epigenetic features that mark active and repressed genes. Active enhancers, promoters, and gene bodies are marked by H3K4me1+H3K27ac, H3K4me3+H3K9ac, and H3K79me2+ H3K36me3, respectively; 5-methylcytosine (5meC) is often detected in the gene bodies. Inactive enhancers, promoters, and gene bodies are marked by H3K9me2/3 and H3K27me3; 5meC also occurs in the promoters. H3K4me1, histone H3 lysine 4 monomethylation; H3K4me3, histone H3 lysine 4 trimethylation; H3K9ac, histone H3 lysine 9 acetylation; H3K9me2, histone H3 lysine 9 dimethylation; H3K9me3, histone H3 lysine 9 trimethylation; H3K27ac, histone H3 lysine 27 acetylation; H3K27me3, histone H3 lysine 27 trimethylation; H3K36me3, histone H3 lysine 36 trimethylation; H3K79me2, histone H3 lysine 79 dimethylation.
EPIGENETIC MECHANISMS OF DISEASE
Abnormal epigenetic profiles in disease
Epigenetic abnormalities have been detected in developmental syndromes, cancer (54), and nonmalignant diseases, including inflammatory (55, 56) and psychiatric (57) disorders. Epigenetic changes can occur locally, affecting individual genes, or globally, such as genome-wide DNA hypomethylation (33), loss of DNA methylation boundaries at CpG islands (58), and loss of balance between active and repressive histone modifications (59).
Dysregulation of DNA methylation is pervasive in disease (43, 60, 61). Cancer cells display both global DNA hypomethylation and focal hypermethylation (33, 37, 62). The former often happens in low-density CpG regions (43), repeats, satellite DNA (33, 62), and regions associated with nuclear envelope lamina (43, 44). DNA hypomethylation can spread over large blocks of DNA up to a few megabases (58, 59). In contrast, locus-specific DNA hypermethylation occurs in CpG islands and lower-CpG-density CpG island “shores” (within 2 kilobases of the CpG islands) (33, 43). Hypermethylation of CpG islands in promoters leads to silencing of genes, including tumor suppressors (37, 63, 64). Promoter hypomethylation can aberrantly activate oncogenes (33, 37, 64) and cause loss of imprinting (33, 47).
Global loss of active histone marks and global loss or gain of repressive marks have been identified across a variety of cancers. Histone acetylation and methylation are most commonly affected in cancer (51). Reduced binding has been reported for the active marks H3K9ac (acetylated lysine 9 of histone H3), H3K18ac, H3K4me1/2/3, H4K5ac, H4K8ac, and H4K16ac, as well as the repressive marks H4K20me3 and H4R3me2 (9, 33, 65). The repressive marks H3K27me3 and H3K9me2/3 show either increased or decreased binding (33, 65, 66).
Mechanisms of epigenetic alterations in disease
Multiple mechanisms are involved in the disruption of the epigenome in disease (54, 67). Altered cellular signaling can change the epigenome through the misregulation of epigenetic writers, readers, and erasers. Chromatin regulators, which often play roles in cancer as oncogenes or tumor suppressors (68), are frequently mutated (63, 69), translocated (65), or aberrantly expressed (33, 59) in various diseases. Mutations of transcription factors that are often cancer drivers (68) may also contribute to tumorigenesis (69). This topic has been extensively reviewed (19, 37, 59, 69–72).
Recent studies have suggested roles for epigenetic mechanisms in linking DNA variants to disease phenotypes (50). Genome-wide association studies (GWAS) have identified single-nucleotide polymorphisms (SNPs) associated with many complex human diseases or traits. Approximately 90% of the SNPs reside in noncoding regions, particularly in regions sensitive to DNase I cleavage, suggesting regulatory roles for SNPs in gene expression (50, 73). Specifically, variants lying within canonical binding motifs can directly affect transcription factor occupancy by disrupting these binding sites or creating new ones (52, 74–77). Alteration of transcription factor binding affinity may lead to changes in gene expression (76), possibly through the recruitment of histone-modifying enzymes (i.e., enzymes which catalyze the addition or removal of histone modifications) that determine local chromatin states (50, 52). Genetic variants can influence multiple layers of epigenomic information. For example, in lymphoblastoid cell lines, a single genetic variant has been shown to alter nucleosome position, chromatin accessibility, histone modifications, and RNA polymerase II occupancy (52). Thus, the interaction of genetic variation and epigenetic mechanisms increases susceptibility to disease (69, 72).
Aging is a key risk factor for many diseases (78). Some environmental and lifestyle factors, such as smoking (7, 55, 79), alcohol (80), diet (55), and life stress (37), have also been linked to aging-associated diseases. The epigenome is an emerging link between aging and disease and between environmental factors and disease (37, 47, 81). The plasticity of the epigenome enables an organism to respond to both external and internal stimuli (7, 9). The organism can accumulate these experiences over time, and such an “epigenetic memory” may be maintained for life (9, 82) or even transmitted to the next generation (37, 83).
Both aging (37, 84) and environmental factors (7, 55, 82) can alter DNA methylation and/or histone modifications. Aging is accompanied by DNA hypermethylation of gene promoters and global hypomethylation (16, 62), which may in part reflect the accumulation of environmental exposures over time (81). Ethanol affects DNA methylation (85) and/or histone modifications (80, 85), as do smoking (79, 86, 87) and nutritional factors (55, 88). The contribution of environmental exposures and lifestyles to epigenetic variation is being intensively investigated in large cohorts (2).
GENOME-WIDE DNA METHYLATION ASSAYS
Early studies inferred overall methylation status by digesting genomic DNA samples in parallel with both methylation-sensitive restriction enzymes (e.g., HpaII) and their methylation-insensitive isoschizomers (e.g., MspI). Later, sodium bisulfite, which converts unmethylated but not methylated or hydroxymethylated cytosines into uracil, was introduced (11). Here, the introduced sequence differences from bisulfite treatment allow the assessment of methylation status at individual cytosines (31, 89). Large-scale DNA methylation profiling is performed by microarray hybridization, affinity-based assays followed by sequencing, or sequencing of bisulfite-converted DNA (34, 81, 90–92) (Table 1). These approaches differ in cost, resolution, accuracy, genomic coverage, and specificity for 5meC versus 5hmeC (2). 5hmeC and 5-formylcytosine can be selectively assessed by means of affinity- and chemical modification-based approaches coupled with next-generation sequencing (93–95). Since these methods have not been utilized in epidemiologic studies, they are not discussed further here.
Bisulfite sequencing platforms
Whole-genome bisulfite sequencing (WGBS) aims to quantify the methylation status of each cytosine in both CpG and non-CpG contexts (13). It is considered the “gold standard” for methylation profiling. However, the required high coverage (approximately 30× coverage) and associated high cost have limited its application (2, 34). In addition, 70%–80% of WGBS reads are not informative because they provide little information about CpG methylation (96). These reads are mostly mapped to non-CpG regions, with a small proportion mapped to “static” regions showing no changes or small changes in CpG methylation between different tissues or cell types (96). Reduced representation bisulfite sequencing (RRBS) is much more cost-effective, because it selectively targets CpG-rich sites. In RRBS, genomic DNA is digested with a methylation-insensitive restriction enzyme (e.g., BglII, MspI) to enrich for genomic regions containing CpGs. The fragments are then subjected to bisulfite treatment. Thus, RRBS is biased toward CpG-rich sites such as CpG islands (9). In clinical settings, RRBS is very useful because it allows rapid screening of many patient samples in a cost-effective manner (9).
Bioinformatic analysis begins with checking sequence quality using software packages like FastQC (Babraham Institute, Cambridge, United Kingdom; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adaptor sequences attached during library preparation for sequencing can be removed using the cutadapt program (Department of Computer Science, TU Dortmund University, Dortmund, Germany; http://code.google.com/p/cutadapt/). Two alignment strategies, “wild-card” and “3-letter,” are available for mapping reads to the reference genome. The wild-card approach, such as BSMAP (97, 98), which uses the Short Oligonucleotide Alignment Program, replaces cytosines in the reference sequence with the wild-card letter Y, which is defined to match both cytosines (i.e., originally (hydroxy)methylated and, therefore, unconverted cytosines) and thymines (i.e., originally unmethylated cytosines, converted to uracils by bisulfite treatment and then to thymines during polymerase chain reaction (PCR) amplification in library preparation) in the reads (42). In the 3-letter approach, all cytosines are first converted into thymines and guanines to adenines in both the reads and the reference sequence; mapping is then performed using a standard aligner like Bowtie (99) (implemented in Bismark (100)). These alignment methods achieve a similar coverage of CpG sites (101).
To quantify the methylation level of individual CpGs and non-CpGs at a given base C on the plus strand, the total number of C-carrying and T-carrying reads is counted and the methylation ratio is estimated as C/(C+T). Similarly, for a given base G on the minus strand, the methylation ratio is estimated as G/(G+A) (102). The methylation level can also be calculated by summing up reads mapping to both strands (32). Several pipelines have been developed for streamlined analysis of WGBS and/or RRBS data (102, 103).
Hybridization-based platforms
The Illumina Infinium HumanMethylation27 (27K) (104) and HumanMethylation450 (450K) BeadChip arrays (Illumina, Inc., San Diego, California) (105) have been most widely used in epigenome-wide association studies, largely because of their relatively low cost and high reproducibility (11, 89, 91, 106). The methylation module of GenomeStudio software (Illumina, Inc.) provides options for signal normalization and background subtraction using control probes (42). DNA methylation is assessed by calculating the fraction of methylated cytosines (β values) at individual CpGs (89, 107). Several software packages have been developed for analysis of the array data (108), including lumi for both the 27K and 450K arrays (109) and minfi for the 450K array (110).
Affinity-based platforms
Methylated DNA immunoprecipitation and sequencing (MeDIP-seq) and methyl-CpG-binding domain protein-based capture and sequencing (MBD-seq) are cost-effective assays for genome-wide DNA methylation profiling, particularly for repetitive DNA regions (32, 111). In MeDIP-seq, genomic DNA is fragmented by sonication and an adaptor is ligated. The sample is denatured and immunoprecipitated with an anti-5meC antibody. The immunoprecipitated products are PCR-amplified, size-selected, and sequenced to a depth of about 30–50 million mapped reads (34, 45). DNA hydroxymethylation can be similarly assessed with an anti-5hmeC antibody (112).
MBD-seq is performed similarly but without denaturing (32). It uses recombinant methyl-CpG-binding domain from methyl-CpG-binding domain protein 2 (MBD2 or MeCP2) as the primary affinity reagent (60). In both methods, the local enrichment level is highly correlated with CpG density (113). MBD-seq is more effective in identifying methylated regions containing multiple methylated cytosines; in contrast, MeDIP-seq often recovers regions with sporadically methylated CpGs of presumably less biological relevance (60). Affinity enrichment methods only provide qualitative estimation of DNA methylation (9) and have low resolution (11, 34, 92). They are also less sensitive to methylated regions with low CpG density than bisulfite sequencing (11, 34) and are less powerful in detecting small changes (11).
Publicly available software packages for analyzing MeDIP-seq and MBD-seq data include Batman (114), MEDIPS (115), and BayMeth (part of the Repitools package) (113). For example, MEDIPS takes mapping output and assigns reads to predefined regions of interest or fixed-size windows. It checks overall enrichment and saturation, estimates relative methylation score after CpG density normalization using linear modeling, identifies differentially methylated regions (DMRs) using edgeR, and annotates methylated regions relative to genomic features (115).
Differential methylation analysis
Differential methylation analysis seeks to determine significant differences in methylation levels between groups of samples (107). It can be performed on individual CpGs or, for increased statistical power, on predefined regions (e.g., CpG islands, CpG island shores) or sliding windows (42). Software packages for identifying differentially methylated cytosines or DMRs from the above 3 platforms have been reviewed (42, 107, 108, 116).
For WGBS and RRBS data analysis, the methylKit package uses Fisher's exact test in the absence of biological replicates and logistic regression (β binomial model) when biological replicates are available (117). The BSmooth pipeline is tailored to the analysis of low-coverage WGBS data, including the function for identifying DMRs (118). Several packages provide functions to identify differentially methylated cytosines, differentially methylated positions, or DMRs from methylation arrays (107, 108, 116). The Wilcoxon rank-sum test (119), moderated t test (116), and F test (110, 116) are commonly applied to normalized log ratios of intensities. The IMA (119) and minfi (110) packages allow both site- and region-level analysis of differential methylation. For MeDIP-seq and MBD-seq data, DMRs can be identified using edgeR implemented in the MEDIPS pipeline (115).
Tissue-specific or disease-related DMRs are largely located at distal regulatory regions, CpG island shores, or lowCpG-density promoters rather than at promoter CpG islands (11, 59). It is important to verify and validate the identified DMRs (92). A simple way is to check some of the DMRs in a genome browser and look for signs of artifacts. A variety of quality control plots provide global views about the magnitude of the changes and genomic distribution of DMRs (42). In addition, it is indispensable to experimentally verify the reproducibility of the DMRs using a different assay and validate the findings in a different cohort (2, 42).
DNA methylation case studies
The Illumina Infinium BeadArrays have been the method of choice in epidemiologic studies (48, 84, 120–122). More recently, MeDIP-seq (123–125) and MBD-seq (126, 127) have begun to gain popularity, owing to their much wider genomic coverage and relatively lower cost.
Traditional epidemiologic study designs, like case-control and cohort studies, have been applied to epigenetic studies (2, 47). These designs need to include unrelated, age- and sex-matched normal controls (88) or case-control individuals (124). Monozygotic twins are genetically identical and thus provide a powerful approach for assessing the role of the epigenome in mediating environmental and lifestyle risk factors involved in complex diseases (7, 24, 37, 47, 92). Several ongoing large-scale cohort studies, such as Accessible Resource for Integrated Epigenomics Studies (ARIES), which uses a birth cohort (http://www.ariesepigenomics.org.uk/), and EpiTwin, which uses a twin cohort (http://www.epitwin.eu/index.html), aim to correlate methylation changes with environmental exposures and disease development (2, 125, 128).
These studies have identified DMRs or differentially methylated positions associated with complex diseases, including autoimmune diseases (88, 121), diabetes (123, 125, 129), heat pain (123), major depressive disorder (124), autism spectrum disorder (130), schizophrenia (126), and rheumatoid arthritis (122). Significant changes in DNA methylation also occur in response to environmental factors (2, 55, 131, 132) and during aging (48, 84, 120, 127, 133).
Challenges in DNA methylation studies
Despite their widespread use in epidemiology, DNA methylation studies face several challenges, from sample collection to data analysis and interpretation. Firstly, DNA methylation analysis often uses whole tissue containing several different cell types, each with its own unique DNA methylome (56, 134). Thus, some of the DMRs may simply reflect the differences in cell composition (42, 47). Indeed, much of the variability in DNA methylation previously thought to be associated with aging is actually caused by aging-dependent changes in cell content (49). In addition, true methylation differences may be averaged out if they exist only in a particular type of cell.
Algorithms have been developed for in silico deconvolution of array-based methylation signals in whole blood (49, 135), taking advantage of prior knowledge of the dominant cell types' methylation profiles (116). Briefly, cell-type-specific differentially methylated cytosines were first identified using 5–6 dominant cell populations generated by cell sorting (49, 135). The top differentially methylated cytosines were then used to build a regression model, which was finally used to estimate the relative proportions of these major cell types in new blood samples based on existing methylation profiles. This method, available in the minfi package (110), was used to adjust for cell mixture in epigenome-wide association studies (122). Recently, 2 packages were developed to correct for difference in cell composition without relying on DNA methylation profiles from the major cell types (136, 137). In addition, several experimental methods are available for reducing cellular heterogeneity in a tissue, such as microdissection, laser-capture microdissection, and fluorescence-activated cell sorting (2, 138). The applicability of these methods, however, is tissue-dependent.
Secondly, there are extensive variations in DNA methylation between different tissues—in fact, DNA methylation varies more between brain and blood from the same individual than between individuals in the same tissue (139). Therefore, in most cases it is critical to use the tissue most relevant to the disease under investigation (7, 30). Nevertheless, DNA methylation studies frequently use easily accessible surrogate tissues like whole blood (49, 56, 83, 91, 106). Although this practice remains controversial (55, 92, 140), it may well be justified if systemic involvement or immune/inflammatory etiology is suspected (141).
Thirdly, despite the development of platform-specific software packages, analyzing genome-wide DNA methylation data from large cohorts remains a challenge (2, 42, 116). In order to reduce artifacts and increase sensitivity, further efforts are needed to systematically benchmark existing tools and to develop more powerful analytical systems. Finally, since DNA methylation differences between cases and controls are usually quite small, especially in surrogate tissues (2), the findings need to be interpreted with caution.
CHROMATIN IMMUNOPRECIPITATION-SEQUENCING
ChIP-seq is a powerful method for identifying global protein-DNA interactions (142, 143), including transcription factor binding (5), histone modifications (144), chromatin remodeling complex subunits (145), and RNA polymerase occupancy (146). In this assay, chromatin is cross-linked by formaldehyde and sheared into approximately 150- to 300-base-pair fragments by sonication and/or micrococcal nuclease digestion. The size distribution of chromatin fragments should be verified by an appropriate method. Genomic regions of interest are enriched using a factor-specific antibody. Purified ChIP DNA is then subjected to adaptor ligation, PCR amplification, and sequencing (11, 143, 147). A control library—for example, input or ChIP obtained with a nonspecific antibody—needs to be generated and sequenced in parallel (148).
ChIP-seq experimental design
The outcome of a ChIP-seq experiment strongly depends on the optimization of a few key factors, such as antibody, amount of input material, tissue source and quality, and sequencing depth. Antibody quality is a key factor responsible for the reliability of the binding profile. Over 200 commercial antibodies against transcription factors, histone modifications, and chromatin regulators have been evaluated (148–151). While these public resources contain information about previously validated antibodies, assessing antibody quality before a ChIP experiment is highly recommended, since lot-to-lot variability is quite common.
If possible, a ChIP-seq experiment should include 2 biological replicates in order to assess reproducibility (26, 148). Most ChIP and control libraries are single-end sequenced to a length of approximately 50 base pairs. A minimum of 20 million uniquely mapped reads (10 million per replicate) is required for transcription factors and highly localized chromatin marks (148), whereas 40 million reads are needed for assessing marks with diffuse binding profiles (148, 152). Depending on mark abundance, conventional ChIP-seq experiments typically require 1–10 million cells (143, 153). A few new protocols have used approximately 10,000 cells in ChIP-seq targeting histone modifications (147, 154) and transcription factors (154). Notable examples are the indexing-first chromatin immunoprecipitation (iChIP) approach (154) and the high-throughput chromatin immunoprecipitation (HT-ChIP) method (147); the latter allows automatic and sensitive ChIP-seq in a 96-well format.
ChIP-seq data analysis
To identify binding sites, reads are first mapped back to the reference genome using a short-read aligner like BWA (155). Very often only uniquely mapped reads are kept and duplicates are filtered out. A peak caller is then used to identify binding sites and generate files for data visualization. Many packages have been developed for identifying peaks from discrete binding profiles, including the widely used model-based algorithm MACS (156). However, it remains challenging to define the boundary of broad peaks due to the discontinuity in signal distribution. SICER, which employs spatial clustering to address this issue, models read counts using the Poisson distribution (157). It scans for individual enriched windows, links nearby enriched windows (often allowing a gap of 3–5 windows) into a single domain, and then calculates statistics for domain enrichment over the control library. RSEG, which uses negative binomial distribution and corrects for mappability bias, shows performance comparable to that of SICER (158). Overall ChIP-seq quality can be assessed based on the quality metrics proposed by the Encyclopedia of DNA Elements Consortium (148, 159).
The resolution of ChIP-seq is limited by the size of sequenced fragments, often a few hundred base pairs. Generating high-resolution maps is important for defining DNA motifs preferred by sequence-specific transcription factors and other regulatory elements (5). Protein-DNA interactions can be mapped at base-pair resolution using ChIP-exo, which employs lambda exonuclease to digest immunoprecipitated chromatin not protected by protein–DNA crosslinks (160).
Differential binding analysis
A number of approaches have been developed to identify differential binding sites between groups of samples (161–164). The DiffBind (162) and DBChIP packages (161) start with peaks, while diffReps (163) uses 1-kilobase sliding windows. For the former, peaks from all libraries need to be consolidated into a single peak list (161, 162). The number of reads overlapping each window or merged peak is then tabulated for individual ChIP and input libraries and normalized. Several methods have been proposed for ChIP-seq data normalization, including global scaling based on total mapped reads or number of reads within peaks (26), linear modeling based on M (log ratio) and A (mean on a log scale) values of shared peaks (165), quantile normalization (166), and trimmed mean of M values (167). To choose an appropriate normalization method, it is better to first check whether global enrichment levels are comparable among different libraries. Recently, “spike-in” chromatin from a different organism was used as an internal control to facilitate data normalization across different samples (168, 169).
DiffBind can only be applied to data sets with biological replicates (162). It uses edgeR or DESeq to identify differential binding sites. DBChIP uses edgeR to estimate the dispersion parameter under the negative binomial distribution (161) and can also handle cases without biological replicates. In contrast, the sliding window-based diffReps selects significant windows that meet a predefined P-value cutoff, and then repeats the test of significance after merging neighboring significant windows into regions (163). An exact negative binomial test is used if there are biological replicates; otherwise, a G test or χ2 test is used.
ChIP-seq applications
ChIP-seq has been applied to a few case-control studies with a limited number of subjects (20 or less). These studies aimed to identify marks in regulatory regions that were associated with aging (134) or cancer development (162, 170). H3K4me1 and H3K27ac together mark active enhancers, and H3K4me1 alone marks poised enhancers. Compared with normal colon crypts, colorectal cancer is characterized by both gain of H3H4me1 sites in the open chromatin regions and loss of H3H4me1 sites from condensed chromatin regions, suggesting distinct chromatin changes in enhancer regions (170). Estrogen receptor α is a key transcription factor in the majority of breast cancers. ChIP-seq revealed distinct estrogen receptor α binding signatures that can separate patients with good clinical outcomes from those with poor outcomes, and the latter showed a global increase in estrogen receptor α binding (162).
ChIP-seq is heavily used in post-GWAS functional studies by mapping transcription factor binding and histone modifications to regulatory regions, thereby allowing the inference of causality for noncoding SNPs (171). This area of research is rapidly expanding, due in part to the massive amounts of epigenomic data generated by the International Human Epigenome Consortium (14, 29, 144) and the recognition that noncoding SNPs play regulatory roles (52, 75, 77). In fact, a significant portion of causal SNP candidates overlap enhancers marked by H3K4me1 and H3K27ac (73, 77).
There are frequent cross-talks between different marks (33, 44, 52, 63). For example, in one study, DNA methylation and H3K27me3 together contributed to the gene silencing in hindbrain ependymomas (172). Whole-genome or exome sequencing failed to identify significant recurrent mutations across a cohort of 47 hindbrain ependymoma patients; however, array hybridization- and WGBS-based DNA methylation assays identified DNA methylation target genes that showed significant overlap with H3K27me3 targets in the group A subtype (with poor prognosis), a pattern that was much less obvious in the group B subtype (with good prognosis) (172).
Therefore, the epigenetic basis of disease can be more readily revealed through integrative analysis of genomic and epigenomic data sets (29, 173). Public data generated from the same or relevant cell/tissue types represent good resources (12, 73, 144, 174). Correlating expression data with epigenomic data like transcription factor or histone modification ChIP-seq or DNA methylation will help identify the genes potentially targeted by an epigenetic mark (172). Furthermore, chromatin interaction maps generated by the chromosome conformation capture (3C)-based assays can be utilized to link gene promoters to the distal regulatory regions like enhancers (27–29). The combined analysis of variant calls with expression data, WGBS/RRBS data, DNase I hypersensitive sites, or ChIP-seq data can identify allele-specific events, thus pinpointing the disease-associated noncoding functional variants in linkage disequilibrium (14, 52, 77).
Challenges in ChIP-seq experiments
Unlike DNA methylation experiments, which use genomic DNA and generally work well in archived samples (2, 175, 176), ChIP-seq experiments require fresh or frozen samples containing high-quality chromatin. In addition, the conventional ChIP protocol often needs large amounts of starting material (106 cells or more) for a single mark, which limits its applicability to scarce clinical samples. In these cases, the indexing-first chromatin immunoprecipitation approach, which has high sensitivity and reproducibility, may become the method of choice (154).
Similarly to DNA methylation studies, the interpretation of ChIP-seq experiments performed on unfractionated tissues is complicated by the presence of multiple cell types. The ideal solution would be to use sorted cell populations (77, 154). Nevertheless, it is often difficult to obtain large numbers of homogeneous cells from cell sorting (140), and cell sorting itself might alter epigenetic states (3). Cell culturing also promotes epigenetic changes (3, 10), limiting its usefulness in enriching target cells (50, 81, 138). Lastly, given the variation in ChIP efficiency in different experiments (26, 159), it is difficult to directly compare data sets that were generated with antibodies from different sources, in different laboratories, or at different times. Further automation of the ChIP protocol is expected to reduce variability.
CONCLUSIONS AND OUTLOOK
Epigenetic changes very likely contribute to the pathogenesis of complex diseases by mediating gene-environment interactions. Epigenome-wide association studies and post-GWAS functional analyses support this notion by revealing disease-associated changes in DNA methylation, histone modifications, transcription factor binding, and noncoding RNA expression. However, development of epigenomics-based therapeutic strategies requires establishment of causality (55). Several hurdles need to be cleared to achieve this goal.
Firstly, given the strong tissue specificity and highly dynamic nature of the epigenome (2, 9, 37, 177), it is essential to generate reference epigenomes for additional tissue types and developmental stages (9, 12, 16). Cataloging epigenome variations across normal individuals will enhance the identification of site-specific variations associated with disease (2, 12, 57) (Appendix). Establishment of concordance between easily accessible tissues and inaccessible ones of greater disease relevance (2) would facilitate not only biomarker discovery (124) but also longitudinal evaluation of epigenetic changes in prospective cohort studies, which, in turn, should help differentiate cause from simple association (7, 56, 106, 175). These efforts should be accompanied by the development of more sensitive assay protocols (3), because current methods often require large quantities of tissues (38).
Analysis of large data sets represents another challenge. The Encyclopedia of DNA Elements Consortium has developed analytical guidelines for multiple types of epigenetic data (178). However, bioinformatic methods must also take into account the fact that technologies are frequently updated or replaced, and must offer solutions to improve compatibility across data generated over time or on different platforms (5, 16, 81). Additional tools for the integrative analysis of “multi-omic” data sets must be developed to enhance the dissection of disease mechanisms.
It is anticipated that progress in our understanding of disease epigenetics will lead to new disease diagnosis, prevention, and treatment (9, 61, 179). While the current exponential growth in this area of biomedical research supports this view, the path toward achieving this ambitious goal remains challenging.
ACKNOWLEDGMENTS
Author affiliations: Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, Minnesota (Huihuang Yan, Shulan Tian, Susan L. Slager, Zhifu Sun); Center for Individualized Medicine, Mayo Clinic, Rochester, Minnesota (Huihuang Yan, Zhifu Sun, Tamas Ordog); Department of Physiology and Biomedical Engineering, Mayo Clinic, Rochester, Minnesota (Tamas Ordog); and Division of Gastroenterology and Hepatology, Mayo Clinic, Rochester, Minnesota (Tamas Ordog).
This study was supported by the Center for Individualized Medicine at the Mayo Clinic and by the National Institutes of Health (grants R01 DK058185, P01 DK068055, and R21 CA191186 to T.O.).
Conflict of interest: none declared.
APPENDIX
Suggested Resources
National Institutes of Health Common Fund Epigenomics Program:
-
http://commonfund.nih.gov/epigenomics/grants
International Human Epigenome Consortium (the goal is to generate 1,000 reference epigenomes):
-
Encyclopedia of DNA Elements (ENCODE) Consortium (extensive epigenome data from cultured cell lines):
-
National Institutes of Health Roadmap Epigenomics Mapping Consortium (epigenomic maps for stem cells and primary ex vivo tissues):
-
http://egg2.wustl.edu/roadmap/web_portal/
European BLUEPRINT project (the goal is to generate approximately 100 reference epigenomes):
-
http://www.blueprint-epigenome.eu/
International Cancer Genome Consortium (include epigenomic maps in 50 different tumor types or subtypes):
-
The Cancer Genome Atlas:
-
https://tcga-data.nci.nih.gov/tcga/
Epigenomics mirror of the University of California, Santa Cruz, genome browser:
-
http://www.epigenomebrowser.org/
National Center for Biotechnology Information epigenomics portal:
-
http://www.ncbi.nlm.nih.gov/epigenomics
Galaxy (for data-intensive biomedical research):
REFERENCES
- 1.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;84:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mill J, Heijmans BT. From promises to practical strategies in epigenetic epidemiology. Nat Rev Genet. 2013;148:585–594. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein BE, Stamatoyannopoulos JA, Costello JF et al. . The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;2810:1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Satterlee JS, Schübeler D, Ng HH. Tackling the epigenome: challenges and opportunities for collaboration. Nat Biotechnol. 2010;2810:1039–1044. [DOI] [PubMed] [Google Scholar]
- 5.Zentner GE, Henikoff S. High-resolution digital profiling of the epigenome. Nat Rev Genet. 2014;1512:814–827. [DOI] [PubMed] [Google Scholar]
- 6.Sarda S, Hannenhalli S. Next-generation sequencing and epigenomics research: a hammer in search of nails. Genomics Inform. 2014;121:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;4657299:721–727. [DOI] [PubMed] [Google Scholar]
- 8.D'Urso A, Brickner JH. Mechanisms of epigenetic memory. Trends Genet. 2014;306:230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maunakea AK, Chepelev I, Zhao K. Epigenome mapping in normal and disease states. Circ Res. 2010;1073:327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu J, Adli M, Zou JY et al. . Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;1523:642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin J, Ming GL, Song H. Decoding neural transcriptomes and epigenomes via high-throughput sequencing. Nat Neurosci. 2014;1711:1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kundaje A, Meuleman W, Ernst J et al. . Integrative analysis of 111 reference human epigenomes. Nature. 2015;5187539:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lister R, Pelizzola M, Dowen RH et al. . Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;4627271:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ernst J, Kheradpour P, Mikkelsen TS et al. . Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;4737345:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lister R, Mukamel EA, Nery JR et al. . Global epigenomic reconfiguration during mammalian brain development. Science. 2013;3416146:1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Almouzni G, Altucci L, Amati B et al. . Relationship between genome and epigenome—challenges and requirements for future research. BMC Genomics. 2014;15:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends Genet. 2011;2710:389–396. [DOI] [PubMed] [Google Scholar]
- 18.Huang H, Sabari BR, Garcia BA et al. . SnapShot: histone modifications. Cell. 2014;1592:458–458.e451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chi P, Allis CD, Wang GG. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;107:457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marsh DJ, Shah JS, Cole AJ. Histones and their modifications in ovarian cancer—drivers of disease and therapeutic targets. Front Oncol. 2014;4:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;18398:627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell O, Tiwari VK, Thomä NH et al. . Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;128:554–564. [DOI] [PubMed] [Google Scholar]
- 23.Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;152:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milosavljevic A. Emerging patterns of epigenomic variation. Trends Genet. 2011;276:242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;203:267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer CA, Liu XS. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat Rev Genet. 2014;1511:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;261:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;146:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivera CM, Ren B. Mapping human epigenomes. Cell. 2013;1551:39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Birney E. Chromatin and heritability: how epigenetic studies can complement genetic approaches. Trends Genet. 2011;275:172–176. [DOI] [PubMed] [Google Scholar]
- 31.Szyf M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012;43:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris RA, Wang T, Coarfa C et al. . Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010;2810:1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;2810:1057–1068. [DOI] [PubMed] [Google Scholar]
- 34.Li D, Zhang B, Xing X et al. . Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods. 2015;72:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinney SE. Mammalian non-CpG methylation: stem cells and beyond. Biology (Basel). 2014;34:739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156(1-2):45–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brookes E, Shi Y. Diverse epigenetic mechanisms of human disease. Annu Rev Genet. 2014;48:237–268. [DOI] [PubMed] [Google Scholar]
- 38.Hyun BR, McElwee JL, Soloway PD. Single molecule and single cell epigenomics. Methods. 2015;72:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams K, Christensen J, Helin K. DNA methylation: TET proteins—guardians of CpG islands? EMBO Rep. 2012;131:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plongthongkum N, Diep DH, Zhang K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat Rev Genet. 2014;1510:647–661. [DOI] [PubMed] [Google Scholar]
- 41.Kinde B, Gabel HW, Gilbert CS et al. . Reading the unique DNA methylation landscape of the brain: non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci U S A. 2015;11222:6800–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012;1310:705–719. [DOI] [PubMed] [Google Scholar]
- 43.Weisenberger DJ. Characterizing DNA methylation alterations from the Cancer Genome Atlas. J Clin Invest. 2014;1241:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013;203:274–281. [DOI] [PubMed] [Google Scholar]
- 45.Gu H, Smith ZD, Bock C et al. . Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;64:468–481. [DOI] [PubMed] [Google Scholar]
- 46.Yang X, Han H, De Carvalho DD et al. . Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;264:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rakyan VK, Down TA, Balding DJ et al. . Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;128:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martino D, Loke YJ, Gordon L et al. . Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013;145:R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;152:R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Furey TS, Sethupathy P. Genetics driving epigenetics. Science. 2013;3426159:705–706. [DOI] [PubMed] [Google Scholar]
- 51.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;1501:12–27. [DOI] [PubMed] [Google Scholar]
- 52.McVicker G, van de Geijn B, Degner JF et al. . Identification of genetic variants that affect histone modifications in human cells. Science. 2013;3426159:747–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;188:1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;84:286–298. [DOI] [PubMed] [Google Scholar]
- 55.Relton CL, Davey Smith G. Epigenetic epidemiology of common complex disease: prospects for prediction, prevention, and treatment. PLoS Med. 2010;710:e1000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy TM, Mill J. Epigenetics in health and disease: heralding the EWAS era. Lancet. 2014;3839933:1952–1954. [DOI] [PubMed] [Google Scholar]
- 57.Lelièvre SA. Taking a chance on epigenetics. Front Genet. 2014;5:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansen KD, Timp W, Bravo HC et al. . Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;438:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;137:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Serre D, Lee BH, Ting AH. MBD-isolated genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;382:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu Z, Bolick SC, DeRoo LA et al. . Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J Natl Cancer Inst. 2013;10510:694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ulrich CM, Grady WM. Linking epidemiology to epigenomics—where are we today? Cancer Prev Res (Phila). 2010;312:1505–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;545:716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nabilsi NH, Deleyrolle LP, Darst RP et al. . Multiplex mapping of chromatin accessibility and DNA methylation within targeted single molecules identifies epigenetic heterogeneity in neural stem cells and glioblastoma. Genome Res. 2014;242:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;135:343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chervona Y, Costa M. Histone modifications and cancer: biomarkers of prognosis? Am J Cancer Res. 2012;25:589–597. [PMC free article] [PubMed] [Google Scholar]
- 67.Albert M, Helin K. Histone methyltransferases in cancer. Semin Cell Dev Biol. 2010;212:209–220. [DOI] [PubMed] [Google Scholar]
- 68.Suvà ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;3396127:1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;1526:1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet. 2013;1324:359–383. [DOI] [PubMed] [Google Scholar]
- 71.Plass C, Pfister SM, Lindroth AM et al. . Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. 2013;1411:765–780. [DOI] [PubMed] [Google Scholar]
- 72.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;1531:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maurano MT, Humbert R, Rynes E et al. . Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;3376099:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kilpinen H, Waszak SM, Gschwind AR et al. . Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science. 2013;3426159:744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee MN, Ye C, Villani AC et al. . Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science. 2014;3436175:1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ye CJ, Feng T, Kwon HK et al. . Intersection of population variation and autoimmunity genetics in human T cell activation. Science. 2014;3456202:1254665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Farh KK, Marson A, Zhu J et al. . Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;5187539:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Flanagan JM. Human methylome variation and the rise of epigenetic epidemiology. Curr Pharmacogenomics Person Med. 2010;82:89–91. [Google Scholar]
- 79.Wan ES, Qiu W, Baccarelli A et al. . Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012;2113:3073–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moghe A, Joshi-Barve S, Ghare S et al. . Histone modifications and alcohol-induced liver disease: are altered nutrients the missing link? World J Gastroenterol. 2011;1720:2465–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bakulski KM, Fallin MD. Epigenetic epidemiology: promises for public health research. Environ Mol Mutagen. 2014;553:171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heijmans BT, Tobi EW, Stein AD et al. . Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;10544:17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Relton CL, Davey Smith G. Is epidemiology ready for epigenetics? Int J Epidemiol. 2012;411:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rakyan VK, Down TA, Maslau S et al. . Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;204:434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zakhari S. Alcohol metabolism and epigenetics changes. Alcohol Res. 2013;351:6–16. [PMC free article] [PubMed] [Google Scholar]
- 86.Breton CV, Byun HM, Wenten M et al. . Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;1805:462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Breitling LP, Yang R, Korn B et al. . Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;884:450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Javierre BM, Fernandez AF, Richter J et al. . Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;202:170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mensaert K, Denil S, Trooskens G et al. . Next-generation technologies and data analytical approaches for epigenomics. Environ Mol Mutagen. 2014;553:155–170. [DOI] [PubMed] [Google Scholar]
- 90.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;113:191–203. [DOI] [PubMed] [Google Scholar]
- 91.Michels KB, Binder AM, Dedeurwaerder S et al. . Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013;1010:949–955. [DOI] [PubMed] [Google Scholar]
- 92.Tsai PC, Spector TD, Bell JT. Using epigenome-wide association scans of DNA methylation in age-related complex human traits. Epigenomics. 2012;45:511–526. [DOI] [PubMed] [Google Scholar]
- 93.Yu M, Hon GC, Szulwach KE et al. . Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;1496:1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Booth MJ, Branco MR, Ficz G et al. . Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;3366083:934–937. [DOI] [PubMed] [Google Scholar]
- 95.Booth MJ, Marsico G, Bachman M et al. . Quantitative sequencing of 5-formylcytosine in DNA at single-base resolution. Nat Chem. 2014;65:435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ziller MJ, Gu H, Müller F et al. . Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;5007463:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xi Y, Bock C, Müller F et al. . RRBSMAP: a fast, accurate and user-friendly alignment tool for reduced representation bisulfite sequencing. Bioinformatics. 2012;283:430–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Langmead B, Trapnell C, Pop M et al. . Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;103:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;2711:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kunde-Ramamoorthy G, Coarfa C, Laritsky E et al. . Comparison and quantitative verification of mapping algorithms for whole-genome bisulfite sequencing. Nucleic Acids Res. 2014;426:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sun Z, Baheti S, Middha S et al. . SAAP-RRBS: streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing. Bioinformatics. 2012;2816:2180–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun D, Xi Y, Rodriguez B et al. . MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 2014;152:R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bibikova M, Le J, Barnes B et al. . Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics. 2009;11:177–200. [DOI] [PubMed] [Google Scholar]
- 105.Bibikova M, Barnes B, Tsan C et al. . High density DNA methylation array with single CpG site resolution. Genomics. 2011;984:288–295. [DOI] [PubMed] [Google Scholar]
- 106.Paul DS, Beck S. Advances in epigenome-wide association studies for common diseases. Trends Mol Med. 2014;2010:541–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wilhelm-Benartzi CS, Koestler DC, Karagas MR et al. . Review of processing and analysis methods for DNA methylation array data. Br J Cancer. 2013;1096:1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morris TJ, Beck S. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods. 2015;72:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;2413:1547–1548. [DOI] [PubMed] [Google Scholar]
- 110.Aryee MJ, Jaffe AE, Corrada-Bravo H et al. . Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;3010:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maunakea AK, Nagarajan RP, Bilenky M et al. . Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;4667303:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ficz G, Branco MR, Seisenberger S et al. . Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;4737347:398–402. [DOI] [PubMed] [Google Scholar]
- 113.Riebler A, Menigatti M, Song JZ et al. . BayMeth: improved DNA methylation quantification for affinity capture sequencing data using a flexible Bayesian approach. Genome Biol. 2014;152:R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Down TA, Rakyan VK, Turner DJ et al. . A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;267:779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lienhard M, Grimm C, Morkel M et al. . MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 2014;302:284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Robinson MD, Kahraman A, Law CW et al. . Statistical methods for detecting differentially methylated loci and regions. Front Genet. 2014;5:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Akalin A, Kormaksson M, Li S et al. . methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;1310:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;1310:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang D, Yan L, Hu Q et al. . IMA: an R package for high-throughput analysis of Illumina's 450K Infinium methylation data. Bioinformatics. 2012;285:729–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bell JT, Tsai PC, Yang TP et al. . Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;84:e1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gervin K, Vigeland MD, Mattingsdal M et al. . DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: identification of epigenetically dysregulated genes. PLoS Genet. 2012;81:e1002454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu Y, Aryee MJ, Padyukov L et al. . Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;312:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bell JT, Loomis AK, Butcher LM et al. . Differential methylation of the TRPA1 promoter in pain sensitivity. Nat Commun. 2014;5:2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Davies MN, Krause L, Bell JT et al. . Hypermethylation in the ZBTB20 gene is associated with major depressive disorder. Genome Biol. 2014;154:R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yuan W, Xia Y, Bell CG et al. . An integrated epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic twins. Nat Commun. 2014;5:5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Aberg KA, McClay JL, Nerella S et al. . MBD-seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case-control samples. Epigenomics. 2012;46:605–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McClay JL, Aberg KA, Clark SL et al. . A methylome-wide study of aging using massively parallel sequencing of the methyl-CpG-enriched genomic fraction from blood in over 700 subjects. Hum Mol Genet. 2014;235:1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.van Dijk SJ, Molloy PL, Varinli H et al. . Epigenetics and human obesity. Int J Obes (Lond). 2015;391:85–97. [DOI] [PubMed] [Google Scholar]
- 129.Rakyan VK, Beyan H, Down TA et al. . Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011;79:e1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wong CC, Meaburn EL, Ronald A et al. . Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. 2014;194:495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tobi EW, Slagboom PE, van Dongen J et al. . Prenatal famine and genetic variation are independently and additively associated with DNA methylation at regulatory loci within IGF2/H19. PLoS One. 2012;75:e37933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lam LL, Emberly E, Fraser HB et al. . Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci U S A. 2012;109(suppl 2):17253–17260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Teschendorff AE, Menon U, Gentry-Maharaj A et al. . Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;204:440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cheung I, Shulha HP, Jiang Y et al. . Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci U S A. 2010;10719:8824–8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Houseman EA, Accomando WP, Koestler DC et al. . DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;3010:1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zou J, Lippert C, Heckerman D et al. . Epigenome-wide association studies without the need for cell-type composition. Nat Methods. 2014;113:309–311. [DOI] [PubMed] [Google Scholar]
- 138.Maze I, Shen L, Zhang B et al. . Analytical tools and current challenges in the modern era of neuroepigenomics. Nat Neurosci. 2014;1711:1476–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Davies MN, Volta M, Pidsley R et al. . Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;136:R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Revill K, Tycko B. Epigenomic insights into common disease. Genome Med. 2011;311:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gjoneska E, Pfenning AR, Mathys H et al. . Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 2015;5187539:365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Johnson DS, Mortazavi A, Myers RM et al. . Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;3165830:1497–1502. [DOI] [PubMed] [Google Scholar]
- 143.Kidder BL, Hu G, Zhao K. ChIP-Seq: technical considerations for obtaining high-quality data. Nat Immunol. 2011;1210:918–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;4897414:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shimbo T, Du Y, Grimm SA et al. . MBD3 localizes at promoters, gene bodies and enhancers of active genes. PLoS Genet. 2013;912:e1004028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods. 2009;6(11 suppl):S22–S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Blecher-Gonen R, Barnett-Itzhaki Z, Jaitin D et al. . High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat Protoc. 2013;83:539–554. [DOI] [PubMed] [Google Scholar]
- 148.Landt SG, Marinov GK, Kundaje A et al. . ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;229:1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Egelhofer TA, Minoda A, Klugman S et al. . An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2011;181:91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Harvard University. Antibody Validation Database. http://compbio.med.harvard.edu/antibodies/ Published 2010. Accessed July 7, 2015.
- 151.University of California, Santa Cruz. ENCODE Antibodies 2007–2012. https://genome.ucsc.edu/ENCODE/antibodies.html Accessed July 7, 2015. [Google Scholar]
- 152.Jung YL, Luquette LJ, Ho JW et al. . Impact of sequencing depth in ChIP-seq experiments. Nucleic Acids Res. 2014;429:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;1312:840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lara-Astiaso D, Weiner A, Lorenzo-Vivas E et al. . Chromatin state dynamics during blood formation. Science. 2014;3456199:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;2514:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang Y, Liu T, Meyer CA et al. . Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;99:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zang C, Schones DE, Zeng C et al. . A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;2515:1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Song Q, Smith AD. Identifying dispersed epigenomic domains from ChIP-Seq data. Bioinformatics. 2011;276:870–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Marinov GK, Kundaje A, Park PJ et al. . Large-scale quality analysis of published ChIP-seq data. G3 (Bethesda). 2014;42:209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;1476:1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liang K, Keles S. Detecting differential binding of transcription factors with ChIP-seq. Bioinformatics. 2012;281:121–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ross-Innes CS, Stark R, Teschendorff AE et al. . Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;4817381:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shen L, Shao NY, Liu X et al. . diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS One. 2013;86:e65598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lun AT, Smyth GK. De novo detection of differentially bound regions for ChIP-seq data using peaks and windows: controlling error rates correctly. Nucleic Acids Res. 2014;4211:e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shao Z, Zhang Y, Yuan GC et al. . MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 2012;133:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bardet AF, He Q, Zeitlinger J et al. . A computational pipeline for comparative ChIP-seq analyses. Nat Protoc. 2012;71:45–61. [DOI] [PubMed] [Google Scholar]
- 167.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;261:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bonhoure N, Bounova G, Bernasconi D et al. . Quantifying ChIP-seq data: a spiking method providing an internal reference for sample-to-sample normalization. Genome Res. 2014;247:1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Orlando DA, Chen MW, Brown VE et al. . Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 2014;93:1163–1170. [DOI] [PubMed] [Google Scholar]
- 170.Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O et al. . Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;3366082:736–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Paul DS, Soranzo N, Beck S. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays. 2014;362:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mack SC, Witt H, Piro RM et al. . Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;5067489:445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;117:476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Thurman RE, Rynes E, Humbert R et al. . The accessible chromatin landscape of the human genome. Nature. 2012;4897414:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Beyan H, Down TA, Ramagopalan SV et al. . Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res. 2012;2211:2138–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Aberg KA, Xie LY, Nerella S et al. . High quality methylome-wide investigations through next-generation sequencing of DNA from a single archived dry blood spot. Epigenetics. 2013;85:542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tobi EW, Lumey LH, Talens RP et al. . DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;1821:4046–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rosenbloom KR, Sloan CA, Malladi VS et al. . ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41(database issue):D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fang F, Turcan S, Rimner A et al. . Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;375:75ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schones DE, Cui K, Cuddapah S et al. . Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;1325:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Henikoff JG, Belsky JA, Krassovsky K et al. . Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci U S A. 2011;10845:18318–18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Valouev A, Johnson SM, Boyd SD et al. . Determinants of nucleosome organization in primary human cells. Nature. 2011;4747352:516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010;20102:pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.He HH, Meyer CA, Hu SS et al. . Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods. 2014;111:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Giresi PG, Kim J, McDaniell RM et al. . FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;176:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Song L, Zhang Z, Grasfeder LL et al. . Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;2110:1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Simon JM, Hacker KE, Singh D et al. . Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res. 2014;242:241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Buenrostro JD, Giresi PG, Zaba LC et al. . Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;1012:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Buenrostro JD, Wu B, Chang HY et al. . ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:21.29.21–21.29.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Barski A, Cuddapah S, Cui K et al. . High-resolution profiling of histone methylations in the human genome. Cell. 2007;1294:823–837. [DOI] [PubMed] [Google Scholar]
- 191.Leung D, Jung I, Rajagopal N et al. . Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature. 2015;5187539:350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Meissner A, Gnirke A, Bell GW et al. . Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;3318:5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bock C, Tomazou EM, Brinkman AB et al. . Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010;2810:1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jacinto FV, Ballestar E, Esteller M. Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylome. Biotechniques. 2008;441:35, 37, 39 passim. [DOI] [PubMed] [Google Scholar]