

Abstract
Protein function prediction is an active area of research in computational biology. Function prediction can help biologists make hypotheses for characterization of genes and help interpret biological assays, and thus is a productive area for collaboration between experimental and computational biologists. Among various function prediction methods, predicting binding ligand molecules for a target protein is an important class because ligand binding events for a protein are usually closely intertwined with the proteins’ biological function, and also because predicted binding ligands can often be directly tested by biochemical assays. Binding ligand prediction methods can be classified into two types: those which are based on protein-protein (or pocket-pocket) comparison, and those that compare a target pocket directly to ligands. Recently, our group proposed two computational binding ligand prediction methods, Patch-Surfer, which is a pocket-pocket comparison method, and PL-PatchSurfer, which compares a pocket to ligand molecules. The two programs apply surface patch-based descriptions to calculate similarity or complementarity between molecules. A surface patch is characterized by physicochemical properties such as shape, hydrophobicity, and electrostatic potentials. These properties on the surface are represented using three-dimensional Zernike descriptors (3DZD), which are based on a series expansion of a 3 dimensional function. Utilizing 3DZD for describing the physicochemical properties has two main advantages: 1) rotational invariance and 2) fast comparison. Here, we introduce Patch-Surfer and PL-PatchSurfer with an emphasis on PL-PatchSurfer, which is more recently developed. Illustrative examples of PL-PatchSurfer performance on binding ligand prediction as well as virtual drug screening are also provided.
Keywords: structure-function relationship, 3D Zernike descriptor, protein function prediction, Patch-Surfer, ligand-protein interaction, ligand binding pockets
1. Introduction
Understanding protein function is one of the central problems in modern biology, including molecular biology, genetics, biochemistry, and bioinformatics. Bioinformatics can make substantial contributions in elucidating function of proteins. Using various types of biological databases now available, computational methods can quickly make function prediction to a large number of query proteins. To date, a number of bioinformatics tools for function prediction have been proposed [1–3]. These existing approaches can be categorized based on types of information they use, which include sequence-based, genome-based, proteomics-based, pathway-based, and structure-based [2].
Sequence-based methods compare a query protein sequence to sequences of known function in a database. This is the most classical strategy of function prediction. Conventional methods, which are often called homology search methods [4–6], use the well-accepted concept of homology and transfer function from highly similar (and thus considered as homologous) sequences. The sensitivity of a search can be improved by employing a statistical algorithm, hidden Markov models [7,8]. Identifying short sequence patterns that are conserved at known functional sites supplements homology search and help in annotating protein sequences [9–11]. Recent sequence-based methods try to improve the prediction performance in terms of the accuracy and the coverage by using more elaborated algorithms [12–16].
Genome-based methods predict functional relationship of protein genes from conservation of gene orders in different genomes [17], domain fusion events [18], and the similarity of phylogenetic profile [19]. STRING [20] is a database that contains pre-computed predicted functional relationship of proteins from genome information.
Proteomics-based methods predict protein function in the context of protein-protein interaction (PPI) or gene expression. Proteins exhibit their functions by interacting with their partner molecules. Therefore, their functions can be inferred from an interaction graph drawn by PPI network data [21,22]. Gene expression data can also be a source for protein function annotation, because functionally related proteins are expected to have correlated expression patterns [23,24].
Pathway-based methods find missing genes in pathways of an organism, which make holes in pathway assignment of genes. Examples of holes in pathways can be observed, for example, at the KEGG pathway database [25], which maps genes or an organism to known pathways by homology search. Candidates of missing genes are unannotated genes in a genome. PathoLogic [26] employs Bayesian approach to match the gaps of pathways and uncharacterized proteins, while Chen and Vitkup [27] fill holes in the pathway by integrating phylogenetic profile and local structures of metabolic networks.
In this article, we introduce two of our methods, Patch-Surfer [28,29] and PL-PatchSurfer [30,31], which predict biological function, more precisely, the binding ligand for a query protein structure. These methods belong to structure-based function prediction methods. In general, structure-based methods are further classified into global structure-based methods and local structure-based methods. The former type compares the global fold of proteins as a strategy for finding distantly related proteins, using the observation that structures are more conserved than sequences [32,33]. Predicted structures of query proteins can be also used in global-structure based function assignment to achieve larger coverage in a genome-scale function assignment [34]. FINDSITE [35] and GalaxySite [36] predict active sites in a query protein structure by global structure matching. The latter, local-structure-based methods, search known functional sites in the global structure of a query protein [37–39] or compare a potential ligand binding site in a query structure to known ligand binding pockets [28–31,40–44]. Ligand binding sites in a protein structure can be predicted by considering geometric or energetic features of known binding pockets, which usually are cavities in protein surface [45–50]. Predicting binding ligands for a protein forms an important and interesting class of protein function predictions because it can often be directly tested by biochemical assays, and because it can provide useful information for drug design [51] and polypharmacology [52–54]. Therefore, conversely, ligand screening methods used for drug development, often called virtual screening methods, such as AutoDock [55], DOCK [56], and GLIDE [57], or pharmacophore search, e.g. LigandScout [58–60], can be applied for binding ligand prediction.
Recently, our group proposed two binding ligand prediction methods, one that performs pocket-pocket comparison, named Patch-Surfer [28,29] and the other one that compares a pocket against ligand molecules, named PL-PatchSurfer [30,31]. Patch-Surfer was designed specifically for binding ligand prediction as a way of predicting protein function while PL-PatchSurfer was developed for structure-based drug virtual screening. The two methods represent molecular surface and physicochemical properties of the surface using three-dimensional Zernike descriptors (3DZD) [61,62], a descriptor that is based on a mathematical series expansion of a three dimensional (3D) function. 3DZD compactly represents molecular surface as a vector of coefficients of a series expansion in a rotationally invariant fashion, which makes it a faster program than others in the field. Here, we introduce Patch-Surfer and PL-PatchSurfer with an emphasis on PL-PatchSurfer, which is more recently developed. Illustrative examples of PL-PatchSurfer performance on binding ligand prediction as well as virtual drug screening are provided.
2. Methods
2.1 Three-dimensional Zernike Descriptors (3DZD)
In this section, a brief introduction of 3DZD will be given. Details of 3DZD can be found on two papers [61,62]. 3DZD is a representation of three-dimensional function of Euclidean space using 3D Zernike polynomials [61,62]. 3D Zernike polynomials are shown in (1).
| (1) |
n, l, m are called as order, degree, and repetition, respectively. The three indices are integers that are subjected to –l < m < l, 0 ≤ l ≤ n, and (n – l) is even. Rnl(r) is a radial function while is a spherical harmonics.
To compute 3DZD of any properties, the values should be mapped on three-dimensional grid points. For example, shape of a molecule is represented as a binary function, which gives 1 for surface region and 0 otherwise. For representing the surface electrostatic potential, the electrostatic values are first mapped on the surface and 3DZD is computed separately for regions with positive values and those with negative values [63]. The grid points with values that are assigned are considered a three-dimensional function of Euclidean space, f(x), where x = {x, y, z}. The function is expanded as a series of terms of Zernike basis as (2).
| (2) |
Calculating Zernike moments, , taking the norm of the moments as (3), and collecting them yields (2l + 1) dimensional vector, a roationally invariant representation of three-dimensional functions.
| (3) |
The dimension of a 3DZD vector is determined by order n, which is related to the resolution of the representation. In Patch-Surfer and PL-PatchSurfer, introduced in following sections, n = 15 is used, yielding 72 invariants.
2.2 Patch-Surfer: A Binding Ligand Prediction by Patch-based Pocket-Pocket Comparison
Patch-Surfer predicts a binding ligand of a given query protein by comparing a query pocket of the protein with known pockets in a pre-constructed pocket database. It was developed for structure-based protein function prediction by pocket comparison to characterize protein structures with unknown function. Pockets are segmented to a set of surface patches. Each patch is represented with 3DZD. The advantages of 3DZD and local patch are: 1) 3DZD comparison is fast and rotationally invariant and 2) local patch matching captures the local similarity of pockets, which is good for predicting docking of flexible molecules [28,29]. Below we outline the algorithm as illustrated in the left half of Figure 1.
Figure 1.

Flowchart of Patch-Surfer and PL-PatchSurfer.
First, given a query protein with a potential ligand binding pocket, the surface of the pocket is generated using APBS [64], which constructs a molecular surface on a 3D grid and computes the electrostatic potential on the surface by solving the Poisson-Boltzmann equation. The pocket region of a query protein is defined by the atoms of the protein that are interacting with atoms of the ligand, if a ligand bound structure of the query protein is available. If the query structure is in apo form (without ligand) and binding sites are not known, a pocket can be predicted using various software [35–50]. After the surface is generated, hydrophobicity and concavity are also calculated and assigned on the surface. Then, the surface of the pocket is segmented into a number of overlapping patches. The segmentation is done as follows: 1) identifying the center of the ligand binding pocket; 2) casting rays from the center of the pocket to the protein surface to identify the boundary of the pocket; 3) distributing seed points on the pocket surface so that they are 3.0 Å away from each other; 4) generating a sphere with 5.0 Å radius centered at each seed point to define a patch; and 5) computing 3DZD for the four properties that characterize a patch: the shape, electrostatic potential, hydrophobicity, and the concavity.
To compute the similarity between a query pocket and a pocket in the database, a modified Auction algorithm [65] is implemented, which optimizes a target scoring function that consists of three parts: 1) Similarity of matched patches taken from the two pockets, which is quantified by the Euclidean distance of 3DZDs; 2) the geodesic distance difference of matched patch pairs in the two pockets, to make sure that corresponding patches in each pocket locate in similar relative locations within each pocket; and 3) difference of approximate position of corresponding patches in the two pockets, which is represented as a histogram of geodesic distance between the patch and other patches of the pocket.
Once the scores for a query pocket to all pockets in the database are computed and ranked by the score, prediction of binding ligands for the pocket is made using the following Pocket_Score (Equation 4). The score of a ligand type F is calculated as:
| (4) |
where n is the number of pockets in rank list, k is the number of top matches of similar ligands of F found by k-nearest neighbor classifier, i is the rank of the ligand, and wl(i),F is a two-dimensional similarity calculated by SIMCOMP [66].
The current pocket database used by Patch-Surfer contains 6547 pockets that bind to 2444 different ligand types. This database was constructed from protein-small-molecule database (PSMDB) [67,68]. Patch-Surfer is available as a webserver at http://kiharalab.org/patchsurfer2.0/.
2.2.1 Previous benchmark studies of Patch-Surfer
The performance of Patch-Surfer has been extensively tested since its initial development [28,29,69]. Firstly, it was compared with Pocket-Surfer [70], which uses 3DZD for describing the global shape of a pocket, and with four other similar mathematical series expansion, i.e. spherical harmonics, Legendre moments, 2DZD, and pseudo 2D Zernike descriptors [28]. Thus, the purpose of this benchmark was to evaluate the effect of using patch-representation over the global pocket representation by 3DZD (Pocket-Surfer) as well as evaluation of 3DZD relative to the similar choices of mathematical representations of a pocket. Patch-Surfer showed the highest performance among six programs when they were benchmarked on a dataset of 100 receptor pockets with nine ligand types [71]. Area Under Curve (AUC) values of Patch-Surfer, Pocket-Surfer, Legendre polynomial, pseudo Zernike, 2DZD, spherical harmonics are 0.81, 0.66, 0.53, 0.66, 0.66, 0.64, respectively when only pocket shape information was considered.
In addition, Patch-Surfer was further compared with four existing binding site comparison methods; eF-seek (which use graph matching) [72], SitesBase (geometric hashing) [73], PROSURFER (fingerprinting) [74], and XBSite2F (fingerprinting) [75]. These five methods were tested to 118 proteins bind with 18 different ligands [69]. The AUC value of Patch-Surfer is 0.86 while the other four methods show 0.49, 0.60, 0.57, and 0.55, respectively.
The latest systematic benchmark was done to compare the performance between Patch-Surfer2.0 [29] and APoc [40]. APoc searches similar binding pocket by structural alignment between query pocket and pockets in database. The benchmark set is composed of 348 binding sites with 15 different ligands. The average AUC values are 0.77 and 0.65 for Patch-Surfer and APoc, respectively.
To summarize, 3DZD, the mathematical base of the surface representation used in Patch-Surfer, performed better than the other similar moments, and moreover, the local patch representation was better than a global pocket shape representation employed in Pocket-Surfer. Moreover, Patch-Surfer showed best performance among other related binding site comparison methods.
2.3 PL-PatchSurfer: Virtual Screening and Binding Ligand Prediction by Patch-based Pocket-Ligand Complementarity Calculation
Recently, we have proposed a new protein-ligand screening method, called PL (Protein-Ligand)-PatchSurfer [30,31]. Instead of comparing a query pocket to known ligand binding pockets as Patch-Surfer performs, PL-PatchSurfer compares a pocket to ligand molecules and identifies the molecules that have high complementarity to the query pocket. Unlike Patch-Surfer, PL-PatchSurfer is originally designed for investigating ligand-protein interaction with an application of virtual screening of drug molecules. The flow of PL-PatchSurfer is illustrated on the right side in Figure 1.
The algorithm of PL-Patchsurfer is similar to that of Patch-Surfer, except that ‘ alternative conformations of a ligand molecule (usually up to 50 conformations) are explicitly generated with OMEGA [76] to fully incorporate the ligand flexibility. In the case of Patch-Surfer, we thought representing a pocket with a combination of local patches is sufficient to consider flexibility of proteins [77], but in the case of ligands, a small bond rotation in a molecule can cause a large conformational change relative to the overall conformation of the molecule. After generating multiple conformations for a ligand, surfaces of all the generated ligand conformations are computed by APBS and physicochemical properties are assigned on the surfaces. Physicochemical properties represented with 3DZDs in PL-PatchSurfer are shape, electrostatic potential, hydrophobicity, and hydrogen bonding acceptors and donors. To measure complementarity between protein and ligand, the properties on ligand surface assigned opposite sign of its original value so that the complementarity of the properties can be computed as the similarity of 3DZDs. For example, if an electrostatic potential on a ligand surface point is −0.1, then it is converted to +0.1 while the sign is kept the same as the original value on the protein surface.
The scoring function of PL-PatchSurfer is composed of four parts. Three of which are the same as Patch-Surfer. The fourth term compares the size of a ligand and pocket, which is quantified as the number of patches that cover the ligand and the pocket. Since multiple conformations are generated for a ligand, a query pocket is matched to each conformation and the best score among them is taken as the final score between the pocket and ligand. Finally, ligands in a library are ranked by their scores.
2.3.1 Previous benchmark studies of PL-PatchSurfer
Since PL-PatchSurfer was originally developed for virtual screening of ligand molecules for a query binding pocket, it was originally tested [30] for its ability of finding active drugs among decoy molecules using the directory of useful decoys (DUD) dataset [78], a commonly used dataset in this field. The DUD dataset is composed of 40 traditional drug target proteins (e.g. kinase, nuclear receptors, and metalloenzymes) and their active compounds that are known to bind to the target, and decoy compounds that have similar chemical properties with active compounds. We used 25 targets out of 40, excluding 15 targets that have cofactors or metal ions because these additional molecules cannot be easily handled by APBS to compute electrostatic potential. In this benchmark, the initial version of PL-PatchSurfer [30], which only considers the shape and electrostatic potential to represent local patches, was tested. The performance of screening was compared with PharmDock [79], a protein pharmacophore-based docking program that has been compared with six state-of-the-art programs, DOCK [56], FlexX [80], Glide [57], ICM [81], Surflex [82], and PhDock [83], and shown to have a comparable performance with ICM and FlexX, and better performance than DOCK and PhDock [79]. Using the average Enrichment Factor (EF) at 1%, 10%, and 20% as the evaluation metrics, PL-PatchSurfer performed better than PharmDock at EF of 1% (8.6 and 6.9, respectively) and at 10% (2.5 and 2.2, respectively), while at 20% the two programs showed the same value (1.7).
PL-PatchSurfer was also applied to compute the similarity between ligands (ligand-to-ligand comparison) [31]. From the Jain set [84], which is composed of 22 proteins and their active and decoy compounds, up to 22 active compounds and 845 decoy compounds for each target protein were selected as a benchmark set. The performance of the program was compared with Global 3DZD [85], USR [86], and ROCS [87]. Although ROCS performs best, PL-PatchSurfer showed a unique feature to find diverse active compounds from a ligand library, which was different from the other global similarity search methods [31].
3. Benchmark Setting
In this work we benchmarked PL-PatchSurfer in two settings. The first test was to evaluate its binding ligand prediction using datasets of ligand binding pockets. Next, we evaluated PL-PatchSurfer for virtual drug screening ability with an interest on its performance on apo form of target proteins. In what follows, first we explain the setting of the benchmark study and then report the results.
3.1 Datasets for binding ligand prediction
The binding ligand prediction was performed on two datasets. The first dataset is a compilation of 100 X-ray structures that bind either of nine ligand types. This dataset is called the Kahraman set [71]. The list of the PDB entries and the ligand structures are shown in Table 1A and Figure 2 (a). This set was manually curated using the following five criteria: 1) Structures should be determined with X-ray crystallography. 2) Binding sites of each ligand are not evolutionary related with each other and belong to different H-levels (homology-levels) in the CATH database [88]. 3) Modified, partial, or incorrectly labeled ligands are discarded. 4) Binding sites are only occupied by their cognate ligands. 5) Each ligand set has at least five members.
Table 1.
| A. PDB ID list of Kahraman data set. | |
|---|---|
| Ligand | PDB ID |
| AMP | 12AS, 1AMU, 1C0A, 1CT9, 1JP4, 1KHT, 1QB8, 1TB7, 8GBP |
| ATP | 1A0I, 1A49, 1AYL, 1B8A, 1DV2, 1DY3, 1E2Q, 1E8X, 1ESQ, 1GN8, 1KVK, 1O9T, 1RDQ, 1TID |
| FAD | 1CQX, 1E8G, 1EVI, 1H69, 1HSK, 1JQI, 1JR8, 1K87, 1POX, 3GRS |
| FMN | 1DNL, 1F6V, 1JA1, 1MVL, 1P4C, 1P4M |
| α-D-glucose | 1BDG, 1CQ1, 1K1W, 1NF5, 2GBP |
| HEME | 1D0C, 1D7C, 1DK0, 1EQG, 1EW0, 1GWE, 1IQC, 1NA 1NP4, 1PO5, 1PP9, 1QHU, 1QLA, 1QPA 1SOX, 2CPO |
| NAD | 1EJ2, 1HEX, 1B0, 1JQ5, 1MEW, 1MI3, 1O04, 1OG3, 1QAX, 1RLZ, 1S7B, 1T2D, 1TOX, 2AF5, 2NPX |
| PO4 | 1A6Q, 1B8O, 1BRW, 1CQJ, 1D1Q, 1DAK, 1E9G, 1EJD, 1EUC, 1EWC, 1FBT, 1GYP, 1H6L, 1HO5, 1L5W, 1L7M, 1LBY, 1LYV, 1QF5, 1TCO |
| Steroids | 1E3R, 1FDS, 1J99, 1LHU, 1QKT |
| B. PDB list of Chikhi set | |
|---|---|
| Ligand | PDB ID |
| AMP | 1AMU, 1QB8, 8GPB |
| ATP | 1A0I, 1KVK, 1O9T |
| FAD | 1EVI, 1POX, 3GRS |
| FMN | 1DNL, 1JA1, 1P4C |
| α-D-glucose | 1K1W, 1NF5, 2GBP |
| HEME | 1QPA, 1SOX, 2CPO |
| (NAD) | 1IB0, 1TOX, 2NPX |
| D-fructose-6-phosphate | 1UXR, 2BIF, 4PFK |
| β-D-galactose | 1XC6, 1Z45, 2GAL |
| Guanine | 1XE7, 2PUC, 2PUF |
| α-Methyl-D-mannose | 1LOB, 1MSA, 1MVQ |
| Palmitic acid | 1MZM, 1PZ4, 1SZ7 |
| C. PDB list of DUD set | |
|---|---|
| Protein | PDB ID |
| CDK2 | 1CKP, 1HCL |
| AChE | 1EVE, 1QIH |
| PPARγ | 1FM9, 1PRG |
| ERα (agonist) | 1L2I, 2B23 |
| NA | 1A4G, 1NSB |
| P38 MAP | 1KV2, 1P38 |
| RXRα | 1MVC, 1G1U |
| HIVPR | 1HPX,3PHV |
| SRC | 2SRC, 1FMK |
| Thrombin | 1BA8, 2AFQ |
Underlined PDB ID is an apo form.
Figure 2.

2D structures of ligands in A, the Kahraman benchmark set and B, the Chikhi set. The ligands in red box are common compounds of the both sets. Two ligands in blue circle are grouped as steroids.
The second dataset was 36 protein structures that bind with either of 12 ligands (the Chikhi dataset) [70]. Three proteins are selected for each ligand type. The PDB entries are listed in Table 1B and the 2D structure of ligands is shown in Figure 2 (b). These two benchmark sets were chosen because they have been used in our earlier studies so that we can compare PL-PatchSurfer’s performance with them [28,70].
The files of the datasets were prepared as follows: 1) Protein structure files were downloaded from the PDB database [89]. 2) Multiple conformations of each ligand were generated from its SMILES string using OMEGA [76]. The maximum number of conformations was set to 50. The ‘ewindow’ option, which sets the condition for maximum energy of conformation, was set to 15 kcal/mol. The RMSD cutoffs between conformations were set to 0.5 Å, 0.8 Å, and 1.0 Å for ligands with zero to five, six to ten, and more than ten free torsion angles, respectively. The other options of OMEGA were kept same as default values.
3.2 Dataset for virtual screening
The performance of PL-PatchSurfer on virtual screening was evaluated on ten targets in the DUD set [78]. One of the foci of this benchmark is to investigate how well PL-PatchSurfer performs on apo structures of the targets. The list of the apo structures were found in a paper by Fan et al. [90]. We selected ten targets that have crystallized holo and apo structures; three from nuclear receptors (PPARγ, ERα (agonist), RXRa), three from kinases (CDK2, SRC, P38 MAP), one from serine proteases (thrombin), and three from other enzymes (AChE, NA, HIVPR). The receptor PDB ID is listed in Table 1C.
The binding site of a target was defined as a set of residues that have any heavy atom that is closer or equal to 5.0 Å to ligand heavy atoms. Cα-RMSD between an apo and a holo form of a binding pocket of a target was computed after aligning them with TM-align [91]. Three targets, RXRα, HIVPR, and SRC, have Cα-RMSD higher than 2.0 Å.
Holo receptor structures and the ligand library were kept as the same as the original DUD dataset, except that ratio of the number of active compounds and decoys was changed to 1:29 from 1:36 in order to make the computation time shorter. To do so, if the target has more than 3000 decoy molecules, all the actives and decoys were selected randomly so that the ratio of actives and decoys to achieve the 1:29 ratio. If not, only decoys were randomly removed. The holo structures were included in the DUD dataset while the apo structures were obtained from PDB. The multiple conformations of ligands were generated by OMEGA with the same parameter as described in Section 3.1. These datasets are made available at http://www.kiharalab.org/ps_ligandset/.
3.3. Performance metric used in the virtual screening benchmark
The performances of the methods were evaluated with Enrichment Factor (EF) and Area-Under Receiver Operating Characteristics Curve (AUC). These two values can be computed after compounds in the dataset, both actives and decoys, are ranked according to the score of a method to be evaluated. The perfect performance of selecting actives from decoys is achieved when all the actives are ranked higher than any decoys. EF at top x% subset is calculated as follows:
| (5) |
where Activesx% is the number of actives found in the top x% subset of compounds ranked by the score of the method, and Compoundsx% represents the number of all the compounds within the top x% subset. In the similar way, ActivesLibrary and CompoundsLibrary are the number of actives and compounds that are contained in the whole library, respectively. EFx% = 1 means that the retrieval of actives by a program is in the same level as a random selection. In this benchmark we examined EFs at 1%, 2%, and 5%.
Receiver operating characteristic (ROC) curve shows the performance of a virtual screening program by plotting actives found rate (true positive rate) against decoys found rate (false positive rate). As a program performs better, the curve approaches the upper-left corner. AUC is the area under the ROC curve. If a program successfully finds active compounds without finding any decoy compounds, AUC value becomes one, while the program totally fails to find active compounds then the value becomes zero, and a value close to 0.5 indicates that the retrieval is random.
4. Result and Discussion
4.1 Binding ligand prediction
First, we tested PL-PatchSurfer for its performance on binding ligand prediction using the Kahraman set, which contains 100 binding pockets. Each pocket was considered as a query. The complementarities between a query pocket and ten ligands (nine ligand types, because prasterone and estradiol were considered as steroid) with multiple conformations are calculated. The ligand conformations were sorted according to PL-PatchSurfer score. The performance of a retrieval was evaluated by examining whether the top hit was the correct ligand type for the query pocket (top 1 accuracy) and also if the correct ligand was ranked within top three (top 3 accuracy). We used these metrics so that we can compare the performance of PL-PatchSurfer with our previous studies on Patch-Surfer [28] and Pocket-Surfer [70].
The summary of the results are given in Table 2. Overall, PL-PatchSurfer showed the best Top 1 prediction accuracy of 48.2% while Patch-Surfer performed better than PL-PatchSurfer when the best top 3 accuracy was considered. Examining results of each ligand type in Table 3, it is apparent that the larger overall average Top 1 accuracy by PL-PatchSurfer over Patch-Surfer comes mainly from better accuracy for ATP and NAD. For ATP, PL-PatchSurfer showed 78.6% and 100.0% for Top 1 and Top 3 accuracy, respectively, for which Patch-Surfer’s accuracy showed 28.6% and 85.7%, respectively. For NAD, PL-PatchSurfer’s Top 1 and Top 3 accuracy were 66.7% and 80.0%, while the corresponding values for Patch-Surfer were 6.7% and 80.0%, respectively. These differences might come from consideration of hydrogen bonding in PL-PatchSurfer. As mentioned in the previous section, PL-PatchSurfer additionally considers hydrogen-bonding acceptors and donors as one of the features of protein and ligand surfaces, which is not coded in Patch-Surfer. ATP has seven hydrogen bond donors and 18 hydrogen bond acceptors. Likewise, NAD has eight hydrogen bond donors and 19 hydrogen bond acceptors. Except for NAD and ATP, the eight ligands have 3.8 hydrogen bond donors and 8.3 hydrogen bond acceptors on average. To further examine the effect of considering hydrogen bonding, we ran PL-PatchSurfer without the hydrogen bonding scoring term. The result is shown in the second row of Table 2 and Table 3. Without considering hydrogen bonds, the Top 1 accuracy of PL-PatchSurfer dropped from 78.6% to 42.8% for ATP and 66.7% to 13.3% for NAD (Table 3), which reflected to the overall better accuracy of PL-PatchSurfer with the hydrogen term over the method without hydrogen bond term (weights for features of patches were trained without hydrogen bond term) (Table 2). However, Table 3 also shows that the accuracy of some other ligands decreased by adding the hydrogen bond feature, which implies that different ligands may have a different optimal setting of parameters.
Table 2.
Success rates of binding ligand prediction methods benchmarked on Kahraman set.
Table 3.
The success rate of binding ligand prediction on the Kahraman set.
| PL-PatchSurfer | PL-PatchSurfer (w/o H-bond) | PatchSurfera | ||||
|---|---|---|---|---|---|---|
| Top 1 (%) | Top 3 (%) | Top 1 (%) | Top 3 (%) | Top 1 (%) | Top 3 (%) | |
| AMP | 44.4 | 100.0 | 55.6 | 100.0 | 66.7 | 28.6 |
| ATP | 78.6 | 100.0 | 42.8 | 85.7 | 100.0 | 85.7 |
| FAD | 50.0 | 70.0 | 50.0 | 60.0 | 80.0 | 80.0 |
| FMN | 0.0 | 66.7 | 16.7 | 66.7 | 0.0 | 50.0 |
| α-D-Glucose | 40.0 | 60.0 | 40.0 | 60.0 | 40.0 | 100.0 |
| HEME | 68.8 | 87.5 | 68.8 | 93.8 | 87.5 | 100.0 |
| NAD | 66.7 | 80.0 | 13.3 | 80.0 | 6.7 | 80.0 |
| PO4 | 65.0 | 100.0 | 75.0 | 100.0 | 100.0 | 100.0 |
| Steroids | 20.0 | 80.0 | 40.0 | 80.0 | 0.0 | 80.0 |
| Average | 48.2 | 82.7 | 44.7 | 80.7 | 45.5 | 87.0 |
In the second half of Table 2, we compared PL-PatchSurfer with two other existing programs, eF-Seek [72] and SitesBase [73]. PL-PatchSurfer showed the highest success rates in both Top 1 and Top 3 predictions among them.
To summarize, PL-PatchSurfer performed comparably if not better than Patch-Surfer in binding ligand prediction, although its original purpose is different, virtual screening. It also performed better than two other approaches for structure-based binding ligand comparison. Considering the complementary nature of PL-PatchSurfer and Patch-Surfer, in that they compare a query pocket to different types of structural data, it would be beneficial to use either one of them or combine their results depending on the situation of scenarios users have. This is discussed further in Conclusions.
4.2 Results of the virtual screening experiments
Next, we tested the performance of PL-PatchSurfer on holo and apo structures of ten targets in the DUD dataset in comparison with two state-of-the-art protein-ligand docking programs, AutoDockVina [55] and DOCK6 [56]. These two programs were run with their default parameters. The center coordinates of the binding pocket box for both programs were set to the geometric center of the bound ligand in the crystal structure of target proteins. Protonation states and atomic charges of all targets and ligands were kept as same as given in the DUD dataset. The results are summarized in Table 3.
For the holo structure (ligand bound structure) set (the upper half of Table 4), PL-PatchSurfer showed overall highest EF1% and EF2% values among three programs (the left columns). For EF5%, DOCK6 showed a slightly higher value of 5.5 over 5.3 by PL-PatchSurfer. In terms of AUC, AutoDockVina showed the highest performance. These results imply that PL-PatchSurfer finds active compounds in earlier ranks than the other two programs.
Table 4.
Virtual screening results on the ten DUD targets by the three programs. The ten targets are further classified into two groups based on Cα-RMSD of binding sites.
| Holo Receptor Structures | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall | RMSD < 2.0 Å | RMSD>2.0 Å | ||||||||||
| Program | EF1% | EF2% | EF5% | AUC | EF1% | EF2% | EF5% | AUC | EF1% | EF2% | EF5% | AUC |
| PL-PatchSurfer | 14.3 | 9.5 | 5.3 | 0.578 | 12.0 | 7.8 | 4.4 | 0.550 | 19.7 | 13.5 | 7.5 | 0.643 |
| AutoDockVina | 7.1 | 6.7 | 5.4 | 0.689 | 5.5 | 5.5 | 4.3 | 0.650 | 10.8 | 9.4 | 8.2 | 0.780 |
| DOCK6 | 13.6 | 9.3 | 5.5 | 0.578 | 11.5 | 8.1 | 5.2 | 0.604 | 18.4 | 12.1 | 6.2 | 0.516 |
| Apo Receptor Structures | ||||||||||||
| Overall | RMSD < 2.0 Å | RMSD≥2.0 Å | ||||||||||
| Program | EF1% | EF2% | EF5% | AUC | EF1% | EF2% | EF5% | AUC | EF1% | EF2% | EF5% | AUC |
| PL-PatchSurfer | 12.8 | 8.7 | 4.6 | 0.562 | 12.3 | 7.6 | 4.3 | 0.547 | 14.1 | 11.1 | 5.3 | 0.596 |
| AutoDockVina | 2.8 | 2.8 | 2.3 | 0.546 | 3.4 | 3.2 | 1.8 | 0.521 | 1.3 | 1.7 | 3.4 | 0.604 |
| DOCK6 | 6.6 | 5.5 | 4.0 | 0.551 | 8.7 | 7.2 | 5.1 | 0.615 | 1.7 | 1.7 | 1.6 | 0.402 |
The values are averaged over the targets.
For the apo structure (ligand unbound structure) set (the bottom half in Table 4), overall PL-PatchSurfer gave the highest performance except for AUC (the left columns). All the values of all programs decreased from the holo set results as expected, because the apo structures are different from the ligand binding form. It is interesting that for the two conventional docking programs, EF values decreased about 50% or more for EF1% and EF2% for EF, whereas PL-PatchSurfer EF values did not deteriorate significantly. To investigate the effect of the receptor structure change between holo and apo forms, we classified the targets based on the pocket Cα-RMSD. For targets with Cα-RMSD > 2.0 Å (the right columns), the performances of AutoDockVina and DOCK6 decreased drastically. Their EF1% and EF2% values decreased by about 90% when compared with the holo form results. In contrast, PL-PatchSurfer results retained ~70% of those of the holo forms. The reason why PL-PatchSurfer was not severely affected by pocket structure change is probably because it uses the molecular surface description of local patches. This positive effect of the patch representation was also observed for Patch-Surfer in the previous works [28,29], where it was able to identify pockets of the same ligands that have largely different global shape.
The changes of enrichment factors of the individual targets are shown in Figure 3. Figure 3A shows the difference of enrichment factors (ΔEF = EFapo -EFholo). As binding site Cα-RMSD becomes larger, ΔEF tends to largely decrease for AutoDockVina and DOCK6. However, PL-PatchSurfer seems not to be greatly affected by the structural change of the receptors, as the decrement of PL-PatchSurfer is lower than 10 in all the cases. In Figure 3B, the ratio of the ΔEF relative to the enrichment factor of the holo forms (i.e. ΔEF/EFholo) were plotted. It is obvious also from this plot that PL-PatchSurfer is more tolerant to the structure change. Enrichment factors of PL-PatchSurfer did not decrease more than 70%, while those of the other two programs decreased by 100% in one or two targets. When the RMSD was smaller than 2.0 Å, PL-PatchSurfer somehow performed even better for the apo forms for a number of cases.
Figure 3.
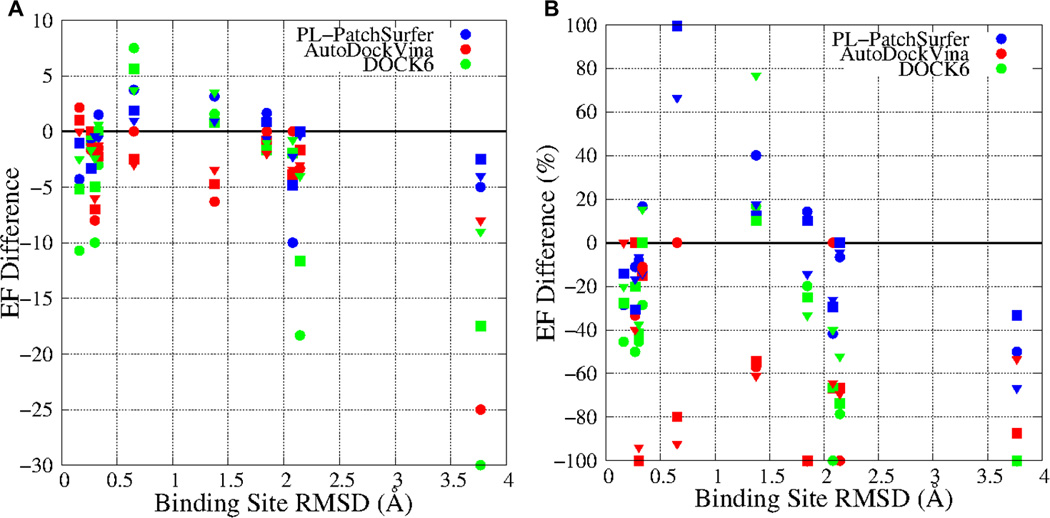
Differences of enrichment factor for holo and apo forms. A, Enrichment factor differences between the holo form and the apo form structure (y-axis, EFapo-EFholo) as a function of binding site Ca-RMSD (x-axis). B, the ratio of enrichment factor difference relatvie to the enrichment factor fo the holo form (y-axis, (EFapo-EFholo)/EFholo) as a function of Ca-RMSD (x-axis). PL-PatchSurfer, AutoDockVina, and DOCK6 are colored in blue, red, and green, respectively. The circle, square, and triangle represent EF1%, EF2%, and EF5%.
The largest structure variation occurs in RXRα, with a Cα-RMSD of 3.8 Å. Similar to other nuclear receptors, AF2-helix of RXRα changes its position by the state of the protein; apo form, agonist binding, and antagonist binding form [92]. The superimposition of the holo and the apo forms is shown in Figure 4A. When the holo form was used as a target structure, EF1% of PL-PatchSurfer, AutoDockVina, and DOCK6 were 10.0, 25.0, and 30.0, respectively. For the apo form, EF1% of PL-PatchSurfer decreased to 5.0 while that of the other two programs deteriorated to 0.0, which means that actives were not retrieved within 1.0% at all. To find the reason for this performance difference between PL-PatchSurfer and the other two programs, the highest ranked active compounds in the holo form were examined. When the holo structure was used as a target, both AutoDockVina (Figure 4C, left panel) and DOCK6 (Figure 4D, left panel) found the correct docked conformation of the ligand that were well-aligned with the cognate ligand. However, when the apo structure was used, the active ligands were located outside of the binding pocket due to the change of the pocket shape caused by the move of the AF-2 helix. These docked conformations cannot make hydrophobic contact with phenylalanines, which is important for ligand binding in the nucleus [93]. In comparison, in Figure 4B we show the docking poses of the top scoring active compound of PL-PatchSurfer (ZINC03834071). It is shown that the identified interacting local regions between the receptor and the ligand shown in the same colors are almost the same between the holo (upper panel) and the apo form (lower panel). These interacting residues to the ligand are composed of aromatic residues and hydrophobic residues. Thus, unlike the other two docking programs, PL-PatchSurfer could find the similar corresponding docking poses of the ligand in the binding pocket.
Figure 4.

Ligand docking of RXRα. A, Superimposed holo (gold) and apo form (green) of RXRa. The cognate ligand is colored in magenta. B, The top scored conformation of ZINC03834071, which is ranked 2nd(holo form) and 1st(apo form) by PL-PatchSurfer. Matched patch pairs of the pocket and the ligand are shown in the same colors. C, the docked structure of an active compound, ZINC01539579, which was ranked the 1st(holo form, left, pink) and the 8th(apo form, right, white) by AutoDockVina. The bound structrure of the compound is shown in magenta. D, the docked structure of ZINC03834076, which was ranked the 1st(holo form, left, cyan) and the 84th(apo form, right, orange) by DOCK6.
Similar to RXRα, the performance of the two conventional docking programs significantly dropped for CDK2 when the apo form was used as the query. The binding site Cα-RMSD between the holo and apo form is only 0.3 Å; however, two lysines change their rotameric state when the ligand binds (Figure 5A). When the holo form was used as a query, EF at 1% of PL-PatchSurfer, AutoDockVina, and DOCK6 were 24.0, 8.0, and 22.0, respectively. When the apo structure was used, the values were reduced to 22.0, 0.0, and 12.0 respectively. Although the conformational change between the holo and the apo form was small, EF1% values of AutoDockVina and Dock6 dropped substantially while PL-PatchSurfer almost maintained performance. To understand this different performance of the programs for the apo form, we examined docking poses of ZINC03814433, the ligand that ranked within top 30 (which corresponds to the top 2% rank) for the holo form by all the three programs. PL-PatchSurfer found corresponding patch pairs in the binding pocket for ligand patches in similar positions for the both holo and apo structures (Figure 5B). Purple and orange patches in the receptor structures are composed of hydrophobic residues, while blue patches correspond to polar residues (ASN for the holo form, ASP for the apo form). The other two programs (Figure 5C, D) found a correct position of the ligand, where half of the ligand is overlapped with the cognate ligand binding position, for the holo form. However, when the apo structure was used, the two programs could not find the correct docking position of the ligand, but rather located the ligand outside of the binding pocket. The ROC curves of CDK2 for the three programs are shown in Figure 6. In Figure 6, solid lines represent the holo structure virtual screening results, while dashed lines show apo structure results. It is shown that the performance of AutoDockVina and DOCK6 were substantially lowered for early recognition for the apo form.
Figure 5.

Ligand docking for CDK2. A, superimposition of the holo (gold) and the apo (green) form of the binding sites of CDK2. The cognate ligand is colored in magenta. The three key residues for the interaction, ASP86, ILE11, and LEU134 [94] (from left to right) are shown. B, the top scoring conformation of an active compound, ZINC03814433, which was ranked the 12th(holo form) and the 26th(apo form) by PL-PatchSurfer. Matched patch pairs of the pocket and the ligand are shown in the same colors. C, Docked structure of ZINC03814433, which was ranked at the 23rd(holo form, left, pink) and the 429th(apo form, right, white) by AutoDockVina. The docked confomration of the compound is shown in magenta. D, the docked structure of ZINC03814433, which was ranked the 10th(holo form, left, cyan) and the 60th(apo form, right, orange) by DOCK6.
Figure 6.

ROC of CDK2 virtual screening. Blue, red, and green lines are for PL-PatchSurfer, AutoDockVina, and DOCK6, respectively. Solid lines are results when the holo structure was used while dashed lines are results for the apo structure.
4.3. Computational Time of PL-PatchSurfer
The average computational times to screen the library of each target are 2.7, 51.7, and 54.4 hours for PL-PatchSurfer, DOCK6, and AutoDockVina, respectively. Thus, PL-PatchSurfer is ~20 times faster than the other two programs. The computational times were measured on a Linux machine with Intel i7–3820 3.60 GHz CPU and 64 GB RAM.
5. Conclusions
We introduced two binding ligand prediction programs, Patch-Surfer and PL-PatchSurfer. Both programs use 3DZD to describe the properties of surface patches of molecules. The advantage of 3DZD moments is that it allows fast comparison of surfaces, because it is a compact and rotationally invariant representation of surfaces. In addition, the programs also enjoy advantages from the local patch description, which is insensitive to subtle atomic position change of molecular surfaces, such as tautomeric shifts and conformational changes. PL-PatchSurfer performed in the similar level if not better to Patch-Surfer for the binding ligand predictions in the Kahraman dataset and performed better than the two state-of-the-art programs, AutoDockVina and Dock6, in the virtual screening benchmarks. The advantage of PL-PatchSurfer was evident over the two programs when apo forms of binding pockets were used as queries.
Although the algorithms of the two methods share a common architecture, they are fundamentally different in the reference data, against which a query pocket is compared. Patch-Surfer compares a query pocket against known ligand binding pockets, while the PL-PatchSurfer compares a pocket directly against 3D conformations of compounds in a library. These two strategies have both advantages and disadvantages: An advantage of the pocket-pocket comparison performed by Patch-Surfer is that generating conformations of compounds, which is known to be difficult and time consuming, is circumvented. Also, biological function (ligand binding) of a query protein can be easily inferred by similarity to known pockets. On the other hand, the types of ligands that can be predicted by Patch-Surfer are limited due to the limited availability of known crystal structures of binding pockets. On the other hand, PL-PatchSurfer has a larger coverage in the ligand space that can be predicted, because as long as the 2D structure of ligands are known, their 3D conformations can be generated and docked to a query pocket. However, a challenge in PL-PatchSurfer is the efficient and accurate sampling of conformational space of ligands. Knowing the differences and complementary nature of these two methods, they must be properly chosen or combined depending on the user’s prediction scenarios.
PatchSurfer predicts binding ligands for a query pocket by finding similar pockets.
PL-PatchSurfer finds binding ligands for a pocket by screening ligand library.
Due to local patch representation, good accuracy is retained for apo structures.
ACKNOWLEDGEMENTS
The authors are thankful to Josh McGraw and Lyman Monroe for proofreading the manuscript. This work is supported by a grant from the Lilly Research Award Program. DK also acknowledges funding from the National Institute of General Medical Sciences of the National Institutes of Health (R01GM097528) and the National Science Foundation (IIS1319551, DBI1262189, IOS1127027).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Watson JD, Laskowski RA, Thornton JM. Struct. Curr. Opin. Biol. 2005;15:275–284. doi: 10.1016/j.sbi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins T, Kihara D. J. Bioinform. Comput. Biol. 2007;5:1–30. doi: 10.1142/s0219720007002503. [DOI] [PubMed] [Google Scholar]
- 3.Konc J, Janezic D. Curr. Opin. Struct. Biol. 2014;25:34–39. doi: 10.1016/j.sbi.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 5.Pearson WR. Methods Enzymol. 1990;183:63–98. doi: 10.1016/0076-6879(90)83007-v. [DOI] [PubMed] [Google Scholar]
- 6.Pearson WR, Lipman DJ. Proc. Natl. Acad. Sci. USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR. Nucleic Acids Res. 2003;31:D138–D401. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Remmer M, Biegert A, Hauser A, Söding J. Nat. Methods. 2011;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 9.Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, de Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJA. Nucleic Acids Res. 2008;36:D245–D249. doi: 10.1093/nar/gkm977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, E de Castro, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, F Quinn A, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJA, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, -Y Yong S. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, Ponting CP, Bork P. Nucleic Acids Res. 2004;32:D142–D144. doi: 10.1093/nar/gkh088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hawkins T, Luban S, Kihara D. Protein Sci. 2006;15:1550–1556. doi: 10.1110/ps.062153506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan IK, Wei Q, Kihara D. Bioinformatics. 2015;31:271–272. doi: 10.1093/bioinformatics/btu646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Messih MA, Chitale M, Bajic VB, Kihara D, Gao X. Bioinformatics. 2012;28:i444–i450. doi: 10.1093/bioinformatics/bts398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wass MN, Sternberg MJ. Bioinformatics. 2008;24:798–806. doi: 10.1093/bioinformatics/btn037. [DOI] [PubMed] [Google Scholar]
- 16.Minneci F, Piovesan D, Cozzetto D, Jones DT. PLoS One. 2013;8:e63754. doi: 10.1371/journal.pone.0063754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kihara D, Kanehisa M. Genome Res. 2000;10:731–743. doi: 10.1101/gr.10.6.731. [DOI] [PubMed] [Google Scholar]
- 18.Yanai I, Derti A, DeLisi C. Proc. Natl. Acad. Sci. USA. 2001;98:7940–7945. doi: 10.1073/pnas.141236298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeats TO. Proc. Natl. Acad. Sci. USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. Nucleic Acids Res. 2003;31:258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwikowski B, Uetz P, Fields S. Nat. Biotechnol. 2000;18:1257–1261. doi: 10.1038/82360. [DOI] [PubMed] [Google Scholar]
- 22.Vasquez A, Flammini A, Maritan A, Vespignani A. Nat. Biotechnol. 2003;21:697–700. doi: 10.1038/nbt825. [DOI] [PubMed] [Google Scholar]
- 23.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, W Kane D, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barnett JC, Weinstein JN. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. Nat. Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- 25.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Nucleic. Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green ML, Karp PD. BMC Bioinform. 2004;5:76. doi: 10.1186/1471-2105-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Vitkup D. Genome Biol. 2006;7:R17. doi: 10.1186/gb-2006-7-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sael L, Kihara D. Proteins. 2012;80:1177–1195. doi: 10.1002/prot.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu X, Xiong Y, Kihara D. Bioinformatics. 2015;31:707–713. doi: 10.1093/bioinformatics/btu724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu B, Zhu X, Xiong Y, G Bures M, Kihara D. Int. J. Mol. Sci. 2014;15:15122–15145. doi: 10.3390/ijms150915122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin W-H, Zhu X, G Bures M, Kihara D. Molecules. 2015;20:12841–12862. doi: 10.3390/molecules200712841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chothia C, Lesk AM. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson CA, Kreychman J, Gerstein M. J. Mol. Biol. 2000;297:233–249. doi: 10.1006/jmbi.2000.3550. [DOI] [PubMed] [Google Scholar]
- 34.Kihara D, Skolnick J. Proteins. 2004;55:464–473. doi: 10.1002/prot.20044. [DOI] [PubMed] [Google Scholar]
- 35.Brylinski M, Skolnick J. PLoS Comput. Biol. 2009;5:e1000405. doi: 10.1371/journal.pcbi.1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heo L, Shin W-H, Lee MS, Seok C. Nucleic Acids Res. 2014;42:W210–W214. doi: 10.1093/nar/gku321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potter CT, Bartlett GJ, Thornton JM. Nucleic Acids Res. 2004;32:D129–D133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang MP, Banatao DR, Klein TE, L Brutlag D. Nucleic Acids Res. 2003;31:3324–3327. doi: 10.1093/nar/gkg553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinoshita K, Nakamura H. Bioinformatics. 2004;20:1329–1330. doi: 10.1093/bioinformatics/bth073. [DOI] [PubMed] [Google Scholar]
- 40.Gao M, Skolnick J. Bioinformatics. 2013;29:579–604. doi: 10.1093/bioinformatics/btt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brylinski M. PLoS Comput. Biol. 2014;10:e1003829. doi: 10.1371/journal.pcbi.1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HS, Im W. J. Chem. Inf. Model. 2012;52:2784–2795. doi: 10.1021/ci300178e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito J, Ikeda K, Yamada K, Mizuguchi K, Tomil K. Nucleic Acids Res. 2015;43:D392–D398. doi: 10.1093/nar/gku1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roy A, Yang J, Zhang Y. Nucleic Acids Res. 2012;40:W471–W477. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laurie AT, Jackson RM. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 46.Huang B, Schroeder M. BMC Struct. Biol. 2006;6:19. doi: 10.1186/1472-6807-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li B, Turuvekere S, Agrawal M, La D, Ramani K, Kihara D. Proteins. 2008;71:670–683. doi: 10.1002/prot.21732. [DOI] [PubMed] [Google Scholar]
- 48.Chen K, Mizianty MJ, Gao J, Kurgan L. Structure. 2011;19:613–621. doi: 10.1016/j.str.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 49.Le Guilloux V, Schmidtke P, Tuffery P. BMC Bioinformatics. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weisel M, Proschak E, Schneider G. Chem. Cent. J. 2007;1:7. doi: 10.1186/1752-153X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiong Y, Zhu X, Kihara D. In silico Drug Discovery and Design Techniques, Future Science, London, UK. 2013:204–220. [Google Scholar]
- 52.Keiser MJ, Roth BL, Ambruster BN, Ernsberger P, Irwin JJ. B. K. Shoichet. Nature Biotechnology. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 53.Milletti F, Vulpetti A. J. Chem. Inf. Model. 2010;50:1418–1431. doi: 10.1021/ci1001263. [DOI] [PubMed] [Google Scholar]
- 54.Anighoro A, Bojorath J, Rastelli G. J. Med. Chem. 2014;57:7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- 55.Trott O, Olson AJ. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, Rizzo RC. J. Comput. Chem. 2015;36:1132–1156. doi: 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 58.Wolber G, Langer T. J. Chem. Inf. Model. 2005;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 59.Cross S, Baroni M, Carosati E, Benedetti P, Clementi S. J. Chem. Inf. Model. 2010;50:1442–1450. doi: 10.1021/ci100221g. [DOI] [PubMed] [Google Scholar]
- 60.Abrahamian E, Fox PC, Naerum L, Christensen IR, Thøgersen H, Clark RD. J. Chem. Inf. Sci. 2003;43:458–468. doi: 10.1021/ci025595r. [DOI] [PubMed] [Google Scholar]
- 61.Canterakis N. Proceedings of 11th Scandinavian Conference on Image Analysis; 1999. pp. 85–93. [Google Scholar]
- 62.Novotni M, Klein R. Proceedings of eighth ACM symposium on Solid modeling and applications; 2003. pp. 216–225. [Google Scholar]
- 63.Sael L, La D, Li B, Rustamov R, Kihara D. Proteins. 2008;73:1–10. doi: 10.1002/prot.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bake NA, Sept D, Joseph S, Holst MJ, McCammon JA. Proc. Nat. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sael L, Kihara D. Int. J. Mol. Sci. 2010;11:5009–5026. doi: 10.3390/ijms11125009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hattori M, Okuno Y, Goto S, Kanehisa M. J. Am. Chem. Soc. 2003;125:11853–11865. doi: 10.1021/ja036030u. [DOI] [PubMed] [Google Scholar]
- 67.Wallach I, Lilien R. Bioinformatics. 2009;25:615–620. doi: 10.1093/bioinformatics/btp035. [DOI] [PubMed] [Google Scholar]
- 68.Sael L, Kihara D. BMC Bioinformatics. 2012;13:S7. doi: 10.1186/1471-2105-13-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kihara D, Sael L, Chikhi R, Esquivel-Rodriguez J. Curr. Protein Pept. Sci. 2011;12:520–530. doi: 10.2174/138920311796957612. [DOI] [PubMed] [Google Scholar]
- 70.Chikhi R, Sael L, Kihara D. Proteins. 2010;78:2007–2028. doi: 10.1002/prot.22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kahraman A, Morris RJ, Laskowski RA, Thornton JM. J. Mol. Biol. 2007;368:283–301. doi: 10.1016/j.jmb.2007.01.086. [DOI] [PubMed] [Google Scholar]
- 72.Kinoshita K, Murakami Y, Nakamura H. Nucleic Acids Res. 2007;35:W398–W402. doi: 10.1093/nar/gkm351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gold ND, Jackson RM. J. Mol. Biol. 2006;355:1112–1124. doi: 10.1016/j.jmb.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 74.Minai R, Matsuo Y, Onuki H, Hirota H. Proteins. 2008;72:367–381. doi: 10.1002/prot.21933. [DOI] [PubMed] [Google Scholar]
- 75.Xiong B, Wu J, Burk D, Xue M, Jiang H, Shen J. BMC Bioinformatics. 2010;11:47. doi: 10.1186/1471-2105-11-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kirchmair J, Wolber G, Laggner C, Langer T. J. Chem. Inf. Model. 2006;46:1848–1861. doi: 10.1021/ci060084g. [DOI] [PubMed] [Google Scholar]
- 77.Gaudreault F, Chartier M, Najmanovich R. Bioinformatics. 2012;28 doi: 10.1093/bioinformatics/bts395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang N, Shoichet BK, J Irwin J. J. Med. Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu B, Lill MA. J. Cheminform. 2014;4:14. doi: 10.1186/1758-2946-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kramer B, Rarey M, Lengauer T. Proteins. 1999;37:228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 81.Abagyan R, Totrov M, Kuznetsov D. J. Comput. Chem. 1994;15:488–506. [Google Scholar]
- 82.Jain A. J. Med. Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 83.Joseph-McCarthy D, Thomas BE, Belmarsh M, Moustakas D, Alvarez JC. Proteins. 2003;51:172–188. doi: 10.1002/prot.10266. [DOI] [PubMed] [Google Scholar]
- 84.Cleves AE, Jain AN. J. Med. Chem. 2006;49:2921–2938. doi: 10.1021/jm051139t. [DOI] [PubMed] [Google Scholar]
- 85.Venkatraman V, Chakravarthy P, Kihara D. J. Cheminf. 2009;1:19. doi: 10.1186/1758-2946-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ballester PJ, Richards WG. J. Comput. Chem. 2007;28:1711–1723. doi: 10.1002/jcc.20681. [DOI] [PubMed] [Google Scholar]
- 87.Hawkins PCD, Skillman AG, Nicholls A. J. Med. Chem. 2007;50:74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 88.Sillitoe I, Lewis TE, Cuff A, Das S, Ashford P, Dawson NL, Furnham N, Laskowski RA, Lee D, G Lees J, Lehtinen S, Studer RA, Thornton J, Orengo CA. Nucleic Acids Res. 2015;43:D376–D381. doi: 10.1093/nar/gku947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Berman HM, Westbrook J, Feng Z, G Gilliland T, Bhat N, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fan H, Irwin JJ, Webb BM, Klebe G, Shoichet BK, Sali A. J. Chem. Inf. Model. 2009;49:2512–2527. doi: 10.1021/ci9003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y, Skolnick J. Nucleic Acid Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bain DL, F Heneghan A, Connaghan-Jones KD, T Miura M. Annu. Rev. Physiol. 2007;69:201–220. doi: 10.1146/annurev.physiol.69.031905.160308. [DOI] [PubMed] [Google Scholar]
- 93.Beautrait A, Karaboga AS, Souchet M, Maigret B. Proteins. 2008;72:873–882. doi: 10.1002/prot.21977. [DOI] [PubMed] [Google Scholar]
- 94.Jiang Y, Zou J, Gui C. J. Mol. Model. 2005;11:509–515. doi: 10.1007/s00894-005-0263-8. [DOI] [PubMed] [Google Scholar]