

Abstract
Diabetes and its complications are caused by chronic glucotoxicity driven by persistent hyperglycemia. In this article, we review the mechanisms of diabetic glucotoxicity by focusing mainly on hyperglycemic stress and carbon stress. Mechanisms of hyperglycemic stress include reductive stress or pseudohypoxic stress caused by redox imbalance between NADH and NAD+ driven by activation of both the polyol pathway and poly ADP ribose polymerase; the hexosamine pathway; the advanced glycation end products pathway; the protein kinase C activation pathway; and the enediol formation pathway. Mechanisms of carbon stress include excess production of acetyl-CoA that can over-acetylate a proteome and excess production of fumarate that can over-succinate a proteome; both of which can increase glucotoxicity in diabetes. For hyperglycemia stress, we also discuss the possible role of mitochondrial complex I in diabetes as this complex, in charge of NAD+ regeneration, can make more reactive oxygen species (ROS) in the presence of excess NADH. For carbon stress, we also discuss the role of sirtuins in diabetes as they are deacetylases that can reverse protein acetylation thereby attenuating diabetic glucotoxicity and improving glucose metabolism. It is our belief that targeting some of the stress pathways discussed in this article may provide new therapeutic strategies for treatment of diabetes and its complications.
Keywords: glucotoxicity, carbon stress, diabetes, hyperglycemic stress, reactive oxygen species, redox imbalance, pseudohypoxia
Introduction
Diabetes and its complications are diseases originated from impaired glucose metabolism [1-8]. As glucose metabolism is tightly regulated by insulin, aberrant glucose metabolism can also be regarded as the problems of insulin resistance or insulin deficiency [9-13]. In type 1 diabetes, there is an absolute deficiency or lack of insulin due to β cell destruction [14-16]. But in type 2 diabetes, it is more of an insulin resistance problem, at least at the early stage of the disease [11, 17-20]. As hyperglycemia persists, β cells attempt to secret more insulin to bring down the blood glucose levels [21]. This compensatory mechanism usually can't last long before eventual exhaustion of β cells as β cells cannot keep up with the ever increasing demand imposed by a persistent level of chronic hyperglycemia [22-24]. At this stage, insulin deficiency kicks in because of impaired β cell function, leading to frank type 2 diabetes [10, 21, 25, 26].
Regardless of which type of diabetes, the apparent manifestation of the disease is a high level of blood glucose [15, 27, 28]. While many pathways are activated or upregulated to dispose excess glucose, there is a price that the body has to pay, which is deterioration of cell or organ function caused by the toxicity of persistent hyperglycemia [2, 20, 29-32]. This glucose toxicity, often referred to as glucotoxicity, is mediated by many aberrant glucose metabolic pathways or signaling pathways that can eventually lead to cell death [33-36]. In this review article, we summarize these pathways that can be collectively placed under the umbrella of hyperglycemic stress or carbon stress. For hyperglycemic stress, after a brief overview of physiology and pathophysiology of insulin-mediated glucose metabolism, we discuss those stress pathways (Fig. 1) including the polyol pathway that contributes to reductive stress [36], the protein kinase C activation pathway, the hexosamine pathway, the advanced glycation end products (AGEs) pathway, and the enediol pathway that all culminate on oxidative stress [36, 37]. For carbon stress, we first discuss the sources and fates of acetyl-CoA and then expand on protein acetylation and succination [38, 39] that are mainly caused by the elevated levels of acetyl-CoA [40] fueled by increased utilization of fatty acids in diabetes [41]. It is our belief that understanding the mechanisms of hyperglycemic stress and carbon stress may help identify targets for controlling blood glucose levels and thus would benefit for combating diabetes.
Figure 1.

Major pathways upregulated by chronic hyperglycemia. These pathways include the polyol pathway, the hexosamine pathway, PKC activation, formation of advanced glycation end products (AGEs), and the enediol formation pathway. These pathways usually remain dormant under euglycemia conditions whereby majority of the body's glucose is combusted through glycolysis and TCA cycle.
Physiology and pathophysiology of insulin-mediated glucose metabolism
The process of glucose extraction from food is achieved in the gastrointestinal tract [42]. This is followed by release of glucose into the blood stream and glucose stimulation of β cell insulin secretion that promotes uptake of glucose by muscle and adipose tissues [43]. Any surplus glucose will be stored in the form of glycogen in the liver and skeletal muscle, and in the form of fat in adipose tissue (Fig. 2) [43, 44]. Insulin secretion and high level of blood glucose will also suppress gluconeogenesis in the liver, a process that is impaired in diabetes [44-46]. In type 2 diabetes, both muscle and adipose tissues can be insulin resistant, and will not take up more glucose like they do postprandially under euglycemic conditions [44, 47-49]. Interestingly, this phenomenon of resistance has been suggested to be a defense mechanism to prevent glucose toxicity to these tissues [50]. Indeed, diabetic complications in muscle and adipose tissues are very rare [50]. But the inability of these tissues, in particular, muscle, to take up more glucose, is the fundamental problem of glucotoxicity to other organs such as the brain, the kidney, and the lower limbs [51-54].
Figure 2.

Regulation of glucose homeostasis and pathophysiology of hyperglycemia. Glucose is extracted from food stuff in the gastrointestinal tract and is then released to the blood stream. High level of blood glucose stimulates insulin secretion from islet β cell in the pancreas, leading to uptake of glucose by muscle and adipose tissues. Insulin also suppresses the gluconeogenesis in the liver. Excess glucose is stored in the liver and the muscle as glycogen, and in the adipose tissue as fat. This glucose uptake and storage process and the overall control of glucose homeostasis are impaired in diabetes.
As mentioned above, insulin is tightly linked to glucose metabolism in the body [55-57]. Under normal physiological conditions, insulin stimulates numerous metabolic processes. As shown in Fig. 3, insulin triggers uptake of glucose by muscle and adipose tissue, stimulates fatty acid synthesis from acetyl-CoA, increases the activity of Na+/K+ pumps in muscle cells and adipocytes, and promotes glycogen synthesis in muscle and liver [58]. Insulin also promotes gene expression, protein synthesis, and amino acid uptake in all types of cells [58]. However, all these processes are perturbed in diabetes, leading to progressive glucotoxicity [2, 20, 37, 59] that includes hyperglycemic stress and carbon stress as is to be discussed in the following sections.
Figure 3.
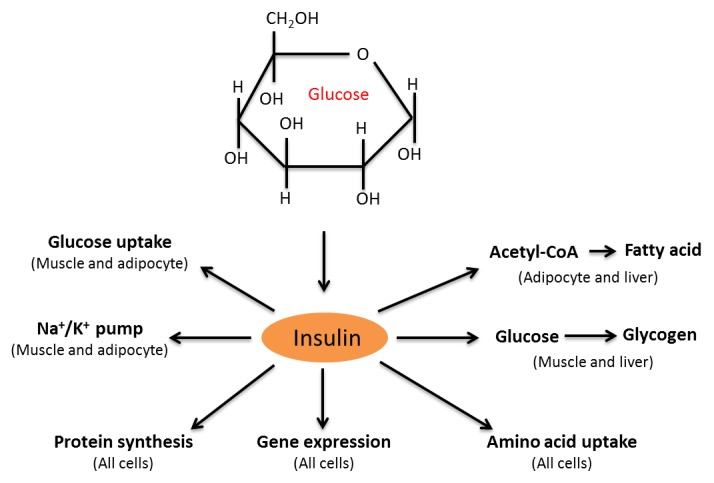
Summary of insulin-stimulated biological processes. Hyperglycemia-induced secretion of insulin can mediate numerous biological processes such as glucose uptake, activation of Na+/K+ pumps, synthesis of fatty acid from acetyl-CoA and glycogen from glucose, amino acid uptake, gene expression, and protein synthesis. Figure adapted from reference [58].
A. Hyperglycemic stress
A PubMed search indicates that the concept of “hyperglycemic stress” was first noted in diabetes in 1964 [60]. However, a further search for both “hyperglycemic stress” and “glucotoxicity” returned a zero result, demonstrating that a link between glucotoxicity and hyperglycemic stress has not been clearly and firmly established. Herein, we define hyperglycemic stress as one that mainly encompasses the pathways shown in Fig. 1 that all can be attributed to chronic hyperglycemia in diabetes. The mechanisms of these stresses are all related to elevated levels of ROS production and oxidative stress [37] and hence these stress pathways are highly interrelated.
1. Reductive stress
Reductive stress is defined as excess reducing equivalent in a cell and is usually expressed as an increased ratio between NADH and NAD+ (or NADPH and NADP+) [36, 61-65]. In chronic hyperglycemic situation, aldose reductase (AR), catalyzing the first reaction in the polyol pathway, is activated [66-69]. AR has a low affinity to glucose and hence is only active at high levels of glucose [54]. AR catalyzes reduction of glucose to sorbitol that is further oxidized to fructose by sorbitol dehydrogenase (the second reaction of the polyol pathway). As shown in Fig. 4, NADPH is consumed and NADH is produced, with accumulation of sorbitol and fructose that can affect cellular osmosis [70, 71]. While the activity of polyol pathway is usually negligible under euglycemic condition, it has been estimated that at least 30% of the body's glucose pool is disposed by this pathway under chronic hyperglycemic conditions [72]. Therefore, NADH level is highly elevated, leading to increased reducing equivalent reflected by an increased ratio between NADH and NAD+. As NAD+ level goes lower, cells undergo pseudohypoxia challenges [73-75].
Figure 4.

Glucose disposal via the polyol pathway under chronic hyperglycemic conditions in diabetes. This pathway includes two-step reactions. The first one is glucose reduction by aldose reductase to form sorbitol; while the second reaction is sorbitol oxidation by sorbitol dehydrogenase to form fructose. Reducing equivalent is transferred from NADPH to NADH, leading to elevated level of NADH and reductive stress. The glycolytic pathway is also shown.
Pseudohypoxia is different from hypoxia in that the former has low levels of NAD+ in the presence of normal level of tissue oxygen [76, 77] while the latter experiences a lower level of tissue oxygen [78-80]. Regardless, both hypoxia and pseudohypoxia will be manifested by impaired NADH oxidation or impaired NAD+ regeneration. In this sense, pseudohypoxic stress is equivalent to reductive stress as both are caused by a net decrease in the content of NAD+ [36, 74], a central molecule involved in metabolism, signal transduction, and stress response [81-84].
Does activation of the polyol pathway lead to depletion of NADPH (the first reaction) in diabetes? This has been repeatedly discussed in the literature and it has been assumed in certain studies that the level of NADPH goes lower in diabetes [85-87], which can diminish GSH content as GSH synthesis requires NADPH [88, 89]. This assumption probably needs to be examined in a tissue dependent manner as it has been reported that in the lens of diabetic rats, NADPH level was not decreased [90]. Moreover, it has been reported that the pentose phosphate pathway that makes NADPH is also upregulated by chronic hyperglycemia [91, 92], leading to a net transfer of excess reducing equivalent from NADPH to NADH [93]. Indeed, it has been demonstrated that NADPH depleted by the polyol pathway can be quickly replenished by the pentose phosphate pathway [64] and potentially by other pathways as well [94]. Therefore, the pentose phosphate pathway in diabetes could also contribute to reductive stress. Hence, reductive stress may be attributed to both the polyol pathway and the pentose phosphate pathway that are activated or upregulated by hyperglycemia in diabetes.
Additionally, as the second reaction of the polyol pathway consumes NAD+, the polyol pathway can compete for NAD+ with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [95], potentially down-regulating the glycolytic pathway. This competition, together with the fact that excess NADH will also inhibit GAPDH [67, 96, 97], can lead to more glucose being diverted to the non-conventional pathways as shown in Fig. 1, thereby aggravating glucotoxicity. Given the detrimental effects of the polyol pathway in diabetes, inhibition or disruption of this pathway has been demonstrated to ameliorate diabetes and its complications [98-100].
While there is an oversupply of NADH in diabetes due to persistent hyperglycemia and enhanced fatty acid β oxidation [101-104], there is also likelihood that NAD+ could be depleted. This is due to the activation of poly ADP ribose polymerase (PARP) by oxidative DNA damage during oxidative stress [105-108]. PARP uses NAD+ as its substrate and is a nuclear enzyme responsible for DNA repair after damage [109-111]. However, this enzyme, when over-activated, can deplete NAD+, which is often the case in diabetes [105, 112, 113]. Hence PARP activation has been demonstrated to be involved in cell death [114-117]. That over-activation of PARP contributes to the pathogenesis of diabetes has been further supported by evidence that PARP knockout or deficient animals are protected from chemical-induced diabetes [118-120] and that PARP inhibitors prevent development of diabetes and its complications [121-125].
Overall, the toxicity of reductive stress is generally reflected by an increased ratio between NADH and NAD+ or redox imbalance between NADH and NAD+, which can impair NAD-dependent enzyme function, deregulate energy metabolism and cell signaling pathways, increase cellular ROS production, and elevates oxidative damage to macromolecules.
2. The hexosamine pathway
As shown in Fig. 5, this pathway originates from fructose-6-P in the glycolytic pathway [126, 127]. It is another pathway that is significantly upregulated by chronic hyperglycemia [126-128]. Fructose-6-P is transformed to glucosamine-6-P by the enzyme glutamine fructose-6-P amidotransferase (GFAT), glucosamine then promotes the synthesis of uridine diphosphate-N-acetylhexosamine (UDP-GlcNAc) that then serves as a substrate for N- or O-glycation of numerous proteins [129, 130]. It should be noted that the mechanism by which hyperglycemia activates GFAT is poorly understood. This posttranslational modification can enhance glucotoxicity by impairing protein function [131, 132] and has been demonstrated to be involved in insulin resistance and pathogenesis of diabetes [133-136].
Figure 5.

Glucose disposal via the hexosamine pathway. This pathway involves activation of glutamine fructose-6-P amidotransferase that converts fructose 6-P to glucosamine 6-P. This is followed by the formation of UDP-GlcNAc that is the substrate for protein translational modifications. This pathway is known to be involved in insulin resistance and diabetes. The glycolytic pathway is also shown.
3. The protein kinase C (PKC) activation pathway
This pathway can originate from either fructose-6-P or glyceraldehyde-3-P in the glycolytic pathway (Fig. 1). The initial species formed is dihydroxyacetone that is further converted to glycerol-3-P. Glycerol-3-P then forms diacylglycerol (DAG) that can activate several isoforms of PKC [137, 138]. PKC then drives numerous signaling processes via protein phosphorylation that regulates signaling protein functions. One of the down-stream targets of PKC is known to be NADPH oxidase whose activation drives superoxide production and thus exacerbates oxidative damage to macromolecules thereby enhancing glucotoxicity [139, 140].
4. The advanced glycation end products (AGEs) pathway
There are two mechanisms by which advanced glycation end products (AGEs) can be formed. The first one is the one shown in Fig. 1 (pathway 4) via the formation of methylglyoxal from glyceraldehyde-3-P [37, 141]. Methylglyoxal then reacts with cysteine, lysine, and arginine residues of proteins, forming advanced glycation end products [142-145]. The second pathway is nonenzymatic, direct attachment of glucose to protein lysine residues via Schiff base formation [53, 146, 147]. The Schiff base then transforms slowly to form stable advanced glycation end products. Many proteins can form AGEs, such as HSP27 [148, 149] and hemoglobin (HbA1c) [27, 150], the quantitation of the latter is often used as an index to measure the progress of diabetes [16]. The glucotoxicity of this pathway has also been demonstrated by observations that the NF-KB signaling pathway can be activated to generate nitric oxide involved in inflammation that can drive the progression of diabetes [151, 152]. Moreover, formation of AGEs can also activate NADPH oxidase [153, 154], leading to superoxide production and oxidative stress. Hence, inhibition of NADPH oxidase could preserve islet β cell functions and lessens glucotoxicity [155-157].
5. The enediol pathway
Chronic hyperglycemia can also elevate the level of enediol, a by-product originated from autoxidation of glyceraldehyde-3-P in the glycolytic pathway [158]. This autoxidation process can generate alpha-ketoaldehyde and ROS, thereby elevating levels of oxidative stress [37]. Formation of enediol has been shown to be involved in pathogenesis of diabetes [159].
6. Oxidative stress
Oxidative stress has been thought to play a central role in the pathogenesis of diabetes and its complications [160-166]. It is induced by overwhelming production of ROS that can attack macromolecules including lipids, DNA, and proteins [28, 167-170]. ROS can be generated by a variety of systems such as mitochondrial electron transport chain [168, 171, 172], d-amino acid oxidase [173-175], dihydrolipoamide dehydrogenase [176-182], α-keto acid dehydrogenase complex [183-187], NADPH oxidase [188, 189], and xanthine oxidase [190, 191]. It should be noted that in the presence of nitric oxide, superoxide can react with nitric oxide to form peroxynitrite [192, 193], a highly reactive species that is known to exert cytotoxicity via modification of macromolecules [194-196] implicated in diabetes [197-201]. Given the role of oxidative stress in diabetes, a variety of antioxidants and phytochemicals have been evaluated for their protective or preventive effects on diabetes and its complications [202-211].
Each of the pathways shown in Fig. 1 can lead to oxidative stress [36, 37]. However, in this section, we would like to focus our discussions on oxidative stress that is preceded by reductive stress in diabetes [36]. As mentioned above, reductive stress is induced by redox imbalance between NADH and NAD+. Moreover, excess NADH can overwhelm mitochondrial complex I (NADH-ubiquinone oxidoreductase), a complex that has at least 45 subunits in mammalian cells and serves as the first electron entry point into the mitochondrial electron transport chain [212-216]. As polyol pathway is activated to increase NADH content and poly ADP ribose polymerase is activated to decrease NAD+ content [36], cells undergo persistent reductive stress as the overall ratio between NADH and NAD+ is increased [36]. One consequence of this redox imbalance is NADH overload of mitochondrial electron transport chain that is known to be capable of generating most of the ROS under pathological conditions [217, 218]. While the first three complexes (I to III) can all generate ROS via the formation of superoxide anion, complex I would be the major one that makes more ROS under NADH pressure as it is the only site in mitochondria that makes NAD+ from NADH [219, 220]. Furthermore, an inherent feature of complex I is that the more NADH it oxidizes, the more superoxide it would produce [219, 221]. Therefore, as shown in Fig. 6, complex I could be a pathogenic factor in diabetes [222-224] and could also be a promising target for lowering NADH level and ameliorating reductive stress or oxidative stress [225-229]. In fact, complex I has been the target for metformin in type 2 diabetes, whereby metformin inhibits complex I [230-232], reduces ATP production, and activates AMPK [223, 233-235], leading to improved metabolism in diabetes [236]. Importantly, the action of metformin seems to exert no cellular toxicity, presumably because fuel combustion via complex II can rescue cells from metformin toxicity [237] (Fig. 6).
Figure 6.

Summary of events leading to redox imbalance between NADH and NAD+ in diabetes. On one hand, the polyol pathway produces excess NADH; on the other hand, the activation of poly ADP ribose polymerase could potentially deplete NAD+, leading to great pressure on mitochondrial complex I that is in charge of NADH oxidation and NAD+ production. NADH overload on complex I can lead to more ROS production. Therefore, complex I could be a pathogenic factor in diabetes and could also be a target for diabetic therapies.
B. Carbon stress
A literature search in PubMed indicates that the concept of carbon stress was first mentioned in 1973 by Hopson and Sack in their studies of changes in cellular phosphorus associated with low carbon stress [238]. In the context of hyperglycemia and diabetes, however, we would like to focus on protein acetylation and succination induced, respectively, by excess acetyl-CoA and fumarate as both forms of posttranslational modifications have been implicated in diabetic glucotoxicity [38, 39, 239, 240]. It should be noted that over-consumption of alcohol can also lead to carbon stress via protein acetylation, which falls beyond the scope of this review [241, 242].
1. Protein acetylation
Excess acetyl-CoA can over-acetylate protein lysine or cysteine residues, leading to protein dysfunction or aberrant protein function [40, 239, 243]. Acetyl-CoA is a central intermediate in metabolism (Fig. 7). On one hand, there are many ways that acetyl-CoA can be produced in a cell. These include pyruvate decarboxylation by pyruvate dehydrogenase complex following glycolysis, β oxidation of fatty acids, deamination and oxidation of amino acids [244]. On the other hand, acetyl-CoA can be used as a source molecule for the synthesis of sterols and fatty acids, can enter into the TCA cycle for complete degradation to H2O and CO2, can form ketone bodies after long term fasting or starvation, and can be used as a substrate for modification of proteins, which is a chemical process that is largely independent of enzymes [241, 245-248]. In diabetes, persistent hyperglycemia itself can raise the level of acetyl-CoA, but persistent hyperglycemia can also enhance fatty acid oxidation that can generate a large amount of acetyl-CoA [44]. It is this chronic excess acetyl-CoA that starts attacking proteins via toxic acetylation mechanisms, thereby leading to impairment in protein function, a process contributing to carbon stress [249]. To cope with this carbon stress, cells have evolved mechanisms of detaching the acetyl groups from over-acetylated proteins, which is achieved by a class of enzymes called sirtuins [250-253] (Fig. 8).
Figure 7.

Sources and fates of acetyl-CoA. Acetyl-CoA is mainly generated by combustion of glucose, fatty acid, and proteins. When in excess, acetyl-CoA can be used to make sterols and fatty acids, and can also conjugate to proteins, forming acetylated protein products. In long term fasting or starvation, acetyl-CoA can be used to form ketone bodies that are needed for brain function [288, 289]. Under normal conditions, acetyl-CoA is metabolized to provide energy via TCA cycle and oxidative phosphorylation inside mitochondria.
Figure 8.

Excess acetyl-CoA produced by hyperglycemia and hyperlipidemia in diabetes can increase nonenzymatic acetylation of proteins via lysine residues. This modification can regulate protein function under stress conditions via sirtuins actions that remove the acetyl groups from the target proteins.
Sirtuins can be activated under certain stress conditions such as starvation and caloric restriction [254-256] to increase the efficiency of metabolism to cope with metabolic stress. However, sirtuins are usually less active under overnutrition conditions such as diabetes because of overproduction of NADH that can inhibit sirtuins activity [257, 258]. This can make acetylation widespread and toxic in the presence of elevated levels of acetyl-CoA [259, 260]. Therefore, sirtuins have been touted as promising targets for diabetic therapy if their activities can be enhanced [261, 262].
However, sirtuins are NAD+-dependent deacetylases and are unfortunately usually down-regulated in diabetes [251, 263-267]. If sirtuins have to be upregulated to cope with carbon stress, such approaches will certainly lead to more consumption of NAD+, the level of which is already low given the activation of poly ADP ribose polymerase and over production of NADH [36]. Therefore, it seems that upregulation of sirtuins in diabetes will compete for NAD+ with other NAD+-dependent enzymes such as PARP and CD38 [105, 268-270] and thus will aggravate the situation of pseudohypoxia. How this can be reconciled needs to be further investigated before sirtuins can be designed as therapeutic targets for diabetes [271, 272].
2. Protein succination
Protein succination [39, 273, 274], another form of carbon stress, originates from fumarate [240, 275-277] that is an intermediate in the TCA cycle. When the level of acetyl-CoA is elevated, so is the level of fumarate. Fumarate can then attack protein cysteine residues, resulting in protein succination [240, 278]. Succination can also occur to glutathione [279]. This modification can severely disrupt protein functions given that protein cysteine residues are intricately involved in protein function and redox signaling [280-287]. For example, GAPDH could be inactivated via succination in diabetes [39, 277]. Moreover, in mitochondria, increased succination has been linked to glucotoxicity under hyperglycemia or in diabetes [240]. It should be noted that given the observations that both acetylation of lysine residues and succination of cysteine residues are linked to glucotoxicity, carbon stress may also be placed conceptually under hyperglycemia stress.
Summary and perspectives
In this article, we have reviewed the concept of diabetic glucotoxicity. We classified diabetic glucotoxicity into two categories of stress: hyperglycemic stress and carbon stress. Under hyperglycemic stress, we discussed several mechanisms of glucotoxicity such as reductive or pseudohypoxic stress, the polyol pathway, the hexosamine pathway, the PKC pathway, the AGEs pathway, and the enediol pathway. We emphasize that all the pathways culminate on oxidative stress [37]. As redox imbalance between NADH and NAD+ is the precursor of oxidative stress [36], we further discussed the role of mitochondrial complex I in glucotoxicity as this complex can produce more ROS in the presence of hyperglycemia and excess NADH and is responsible for mitochondrial regeneration of NAD+. We think that complex I can be both a pathogenic factor and a potential therapeutic target in diabetes. Under carbon stress, we focused on protein acetylation and succination; both of which can manifest diabetic glucotoxicity. In the context of protein acetylation, we touched on sirtuins that are acetylases capable of improving metabolism by removing acetyl groups off from target proteins. Future in-depth studies targeting these hyperglycemic- and carbon stress pathways may help design novel strategies for treatment of diabetes and its complications.
Acknowledgements
L.J.Y. was supported in part by National Institute of Neurological Disorders and Stroke (Grant: R01NS079792).
Conflict of interest
The authors declare that there is no conflict of interests
References
- [1].Bensellam M,Laybutt DR,Jonas JC (2012). The molecular mechanisms of pancreatic beta-cell glucotoxicity: recent findings and future research directions. Molecular and cellular endocrinology, 364: 1-27 [DOI] [PubMed] [Google Scholar]
- [2].Del Prato S (2009). Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabet Med, 26: 1185-1192 [DOI] [PubMed] [Google Scholar]
- [3].Dedoussis GV,Kaliora AC,Panagiotakos DB (2007). Genes, diet and type 2 diabetes mellitus: a review. The review of diabetic studies : RDS, 4: 13-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Somesh BP,Verma MK,Sadasivuni MK,Mammen-Oommen A,Biswas S,Shilpa PC, et al. (2013). Chronic glucolipotoxic conditions in pancreatic islets impair insulin secretion due to dysregulated calcium dynamics, glucose responsiveness and mitochondrial activity. BMC Cell Biol, 14: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Leibowitz G,Kaiser N,Cerasi E (2011). beta-Cell failure in type 2 diabetes. Journal of diabetes investigation, 2: 82-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leibowitz G,Bachar E,Shaked M,Sinai A,Ketzinel-Gilad M,Cerasi E, et al. (2010). Glucose regulation of beta-cell stress in type 2 diabetes. Diabetes, obesity & metabolism, 12 Suppl 2: 66-75 [DOI] [PubMed] [Google Scholar]
- [7].Barnett AH (2012) Type 2 diabetes, Oxford University Press [Google Scholar]
- [8].Ruderman NB,Carling D,Prentki M,Cacicedo JM (2013). AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest, 123: 2764-2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bacha F,Gungor N,Lee S,Arslanian SA (2013). Progressive deterioration of beta-cell function in obese youth with type 2 diabetes. Pediatr Diabetes, 14: 106-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gallwitz B,Kazda C,Kraus P,Nicolay C,Schernthaner G (2013). Contribution of insulin deficiency and insulin resistance to the development of type 2 diabetes: nature of early stage diabetes. Acta Diabetol, 50: 39-45 [DOI] [PubMed] [Google Scholar]
- [11].DeFronzo RA (1997). Insulin resistance: a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidaemia and atherosclerosis. The Netherlands journal of medicine, 50: 191-197 [DOI] [PubMed] [Google Scholar]
- [12].Abdul-Ghani MA,DeFronzo RA (2010). Pathogenesis of insulin resistance in skeletal muscle. Journal of biomedicine & biotechnology, 2010: 476279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gungor N,Arslanian S (2004). Progressive beta cell failure in type 2 diabetes mellitus of youth. J Pediatr, 144: 656-659 [DOI] [PubMed] [Google Scholar]
- [14].Funk SD,Yurdagul A Jr.,Orr AW (2012). Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes. International journal of vascular medicine, 2012: 569654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Eiselein L,Schwartz HJ,Rutledge JC (2004). The challenge of type 1 diabetes mellitus. ILAR journal / National Research Council, Institute of Laboratory Animal Resources, 45: 231-236 [DOI] [PubMed] [Google Scholar]
- [16].Tuch B,Dunlop M,Proietto J (2000) Diabetes Research: A guide for postgraduates, Harwood Academic Publishers [Google Scholar]
- [17].Larsen MO (2009). Beta-cell function and mass in type 2 diabetes. Danish medical bulletin, 56: 153-164 [PubMed] [Google Scholar]
- [18].Butler AE,Janson J,Bonner-Weir S,Ritzel R,Rizza RA,Butler PC (2003). Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes, 52: 102-110 [DOI] [PubMed] [Google Scholar]
- [19].Abdul-Ghani MA,DeFronzo RA (2008) Oxidative stress in type 2 diabetes. In Oxidative stress in aging (Miwa S,Beckman KB,Muller FL, eds) pp. 191-212, Humana Press [Google Scholar]
- [20].Kaiser N,Leibowitz G,Nesher R (2003). Glucotoxicity and beta-cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab, 16: 5-22 [DOI] [PubMed] [Google Scholar]
- [21].Szendroedi J,Phielix E,Roden M (2012). The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nature reviews. Endocrinology, 8: 92-103 [DOI] [PubMed] [Google Scholar]
- [22].Maedler K,Donath MY (2004). Beta-cells in type 2 diabetes: a loss of function and mass. Horm Res, 62 Suppl 3: 67-73 [DOI] [PubMed] [Google Scholar]
- [23].Chang-Chen KJ,Mullur R,Bernal-Mizrachi E (2008). Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord, 9: 329-343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Muoio DM,Newgard CB (2008). Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nature reviews. Molecular cell biology, 9: 193-205 [DOI] [PubMed] [Google Scholar]
- [25].Lee SA,Lee WJ,Kim EH,Yu JH,Jung CH,Koh EH, et al. (2011). Progression to insulin deficiency in Korean patients with Type 2 diabetes mellitus positive for anti-GAD antibody. Diabet Med, 28: 319-324 [DOI] [PubMed] [Google Scholar]
- [26].Cnop M,Welsh N,Jonas JC,Jorns A,Lenzen S,Eizirik DL (2005). Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes, 54 Suppl 2: S97-107 [DOI] [PubMed] [Google Scholar]
- [27].Gholap NN,Davies MJ,Mostafa SA,Khunti K (2013). Diagnosing type 2 diabetes and identifying high-risk individuals using the new glycated haemoglobin (HbA1c) criteria. The British journal of general practice : the journal of the Royal College of General Practitioners, 63: e165-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wu J,Luo X,Yan LJ (2015). Two dimensional blue native/SDS-PAGE to identify mitochondrial complex I subunits modified by 4-hydroxynonenal (HNE). Frontiers in Physiology, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Korsgren O,Jansson L,Sandler S,Andersson A (1990). Hyperglycemia-induced B cell toxicity. The fate of pancreatic islets transplanted into diabetic mice is dependent on their genetic background. J Clin Invest, 86: 2161-2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Poitout V,Robertson RP (2002). Minireview: Secondary beta-cell failure in type 2 diabetes--a convergence of glucotoxicity and lipotoxicity. Endocrinology, 143: 339-342 [DOI] [PubMed] [Google Scholar]
- [31].Roseman HM (2005). Progression from obesity to type 2 diabetes: lipotoxicity, glucotoxicity, and implications for management. Journal of managed care pharmacy : JMCP, 11: S3-S11 [PubMed] [Google Scholar]
- [32].Weir GC,Marselli L,Marchetti P,Katsuta H,Jung MH,Bonner-Weir S (2009). Towards better understanding of the contributions of overwork and glucotoxicity to the beta-cell inadequacy of type 2 diabetes. Diabetes, obesity & metabolism, 11 Suppl 4: 82-90 [DOI] [PubMed] [Google Scholar]
- [33].Poitout V,Robertson RP (2008). Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocrine reviews, 29: 351-366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kawahito S,Kitahata H,Oshita S (2009). Problems associated with glucose toxicity: role of hyperglycemia-induced oxidative stress. World J Gastroenterol, 15: 4137-4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Giaccari A,Sorice G,Muscogiuri G (2009). Glucose toxicity: the leading actor in the pathogenesis and clinical history of type 2 diabetes - mechanisms and potentials for treatment. Nutr Metab Cardiovasc Dis, 19: 365-377 [DOI] [PubMed] [Google Scholar]
- [36].Yan LJ (2014). Pathogenesis of Chronic Hyperglycemia: From Reductive Stress to Oxidative Stress. Journal of diabetes research, 2014: 137919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Robertson RP (2004). Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem, 279: 42351-42354 [DOI] [PubMed] [Google Scholar]
- [38].Iyer A,Fairlie DP,Brown L (2012). Lysine acetylation in obesity, diabetes and metabolic disease. Immunol Cell Biol, 90: 39-46 [DOI] [PubMed] [Google Scholar]
- [39].Frizzell N,Lima M,Baynes JW (2011). Succination of proteins in diabetes. Free Radic Res, 45: 101-109 [DOI] [PubMed] [Google Scholar]
- [40].Wagner GR,Hirschey MD (2014). Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell, 54: 5-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Abel ED (2010). Free fatty acid oxidation in insulin resistance and obesity. Heart Metab, 48: 5-10 [PMC free article] [PubMed] [Google Scholar]
- [42].Poretsky L (2010) Principles of diabetes mellitus, Springer, New York [Google Scholar]
- [43].de Koning L,Amlik VS,Hu FB (2014) Dietary carbohydrates and type 2 diabetes. In Nutrition and type 2 diabetes etiology and prevention(Pereira MA, ed) pp. 11-64, CRC Press, New York [Google Scholar]
- [44].Seely L,Olefsky JM (1993) Potential cellular and genetic mechanisms for insulin resistance in the common disorders of diabetes and obesity. In Insulin resistance (Moller DE, ed) pp. 187-252, Wiley, New York [Google Scholar]
- [45].Cook JR,Langlet F,Kido Y,Accili D (2015). On the Pathogenesis of Selective Insulin Resistance in Isolated Hepatocytes. J Biol Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sajan MP,Jurzak MJ,Samuels VT,Shulman GI,Braun U,Leitges M, et al. (2014). Impairment of insulin-stimulated glucose transport and ERK activation by adipocyte-specific knockout of PKC-lambda produces a phenotype characterized by diminished adiposity and enhanced insulin suppression of hepatic gluconeogenesis. Adipocyte, 3: 19-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Turner N,Cooney GJ,Kraegen EW,Bruce CR (2014). Fatty acid metabolism, energy expenditure and insulin resistance in muscle. J Endocrinol, 220: T61-79 [DOI] [PubMed] [Google Scholar]
- [48].Yang J (2014). Enhanced skeletal muscle for effective glucose homeostasis. Prog Mol Biol Transl Sci, 121: 133-163 [DOI] [PubMed] [Google Scholar]
- [49].Gustafson B,Hedjazifar S,Gogg S,Hammarstedt A,Smith U (2015). Insulin resistance and impaired adipogenesis. Trends Endocrinol Metab, 26: 193-200 [DOI] [PubMed] [Google Scholar]
- [50].Yki-Jarvinen H (2002). Insulin resistance in patients with IDDM. In: Insulin resistance in patients with IDDM. In: Hormone resistance and hypersensitivity states. Lippincott William & Wilkins, Baltimore, 175-185 [Google Scholar]
- [51].Groop L,Orho M (1998). Metabolic aspects of glycogen synthase activation. In: Metabolic aspects of glycogen synthase activation. In: Molecular and cell biology of type 2 diabetes and its complications. Karger, Basel, 47-55 [Google Scholar]
- [52].Stolar M (2010). Glycemic control and complications in type 2 diabetes mellitus. The American journal of medicine, 123: S3-11 [DOI] [PubMed] [Google Scholar]
- [53].Brownlee M (2001). Biochemistry and molecular cell biology of diabetic complications. Nature, 414: 813-820 [DOI] [PubMed] [Google Scholar]
- [54].Brownlee M (2005). The pathobiology of diabetic complications: a unifying mechanism. Diabetes, 54: 1615-1625 [DOI] [PubMed] [Google Scholar]
- [55].Finocchietto P,Barreyro F,Holod S,Peralta J,Franco MC,Mendez C, et al. (2008). Control of muscle mitochondria by insulin entails activation of Akt2-mtNOS pathway: implications for the metabolic syndrome. PLoS ONE, 3: e1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cheng Z,Tseng Y,White MF (2010). Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab, 21: 589-598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cline GW (2011). Fuel-Stimulated Insulin Secretion Depends upon Mitochondria Activation and the Integration of Mitochondrial and Cytosolic Substrate Cycles. Diabetes Metab J, 35: 458-465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].White MF,Khan CR (1993) Mechanisms of insulin action. In Insulin resistance (Moller DE, ed) pp. 9-47, Wiley, New York [Google Scholar]
- [59].Brunner Y,Schvartz D,Priego-Capote F,Coute Y,Sanchez JC (2009). Glucotoxicity and pancreatic proteomics. J Proteomics, 71: 576-591 [DOI] [PubMed] [Google Scholar]
- [60].Seltzer HS,Harris VL (1964). Exhaustion of Insulogenic Reserve in Maturity-Onset Diabetic Patients during Prolonged and Continuous Hyperglycemic Stress. Diabetes, 13: 6-13 [DOI] [PubMed] [Google Scholar]
- [61].Lipinski B (2002). Evidence in support of a concept of reductive stress. The British journal of nutrition, 87: 93-94; discussion 94 [DOI] [PubMed] [Google Scholar]
- [62].Teodoro JS,Rolo AP,Palmeira CM (2013). The NAD ratio redox paradox: why does too much reductive power cause oxidative stress? Toxicology Mechanisms and Methods, 23: 297-302 [DOI] [PubMed] [Google Scholar]
- [63].Pung YF,Chilian WM (2010). Corruption of coronary collateral growth in metabolic syndrome: Role of oxidative stress. World journal of cardiology, 2: 421-427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tilton RG (2002). Diabetic vascular dysfunction: links to glucose-induced reductive stress and VEGF. Microscopy research and technique, 57: 390-407 [DOI] [PubMed] [Google Scholar]
- [65].Valadi H,Valadi A,Ansell R,Gustafsson L,Adler L,Norbeck J, et al. (2004). NADH-reductive stress in Saccharomyces cerevisiae induces the expression of the minor isoform of glyceraldehyde-3-phosphate dehydrogenase (TDH1). Current genetics, 45: 90-95 [DOI] [PubMed] [Google Scholar]
- [66].Chung SS,Chung SK (2005). Aldose reductase in diabetic microvascular complications. Curr Drug Targets, 6: 475-486 [DOI] [PubMed] [Google Scholar]
- [67].Dunlop M (2000). Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int Suppl, 77: S3-12 [DOI] [PubMed] [Google Scholar]
- [68].Hodgkinson AD,Sondergaard KL,Yang B,Cross DF,Millward BA,Demaine AG (2001). Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney Int, 60: 211-218 [DOI] [PubMed] [Google Scholar]
- [69].Iwata K,Nishinaka T,Matsuno K,Kakehi T,Katsuyama M,Ibi M, et al. (2007). The activity of aldose reductase is elevated in diabetic mouse heart. J Pharmacol Sci, 103: 408-416 [DOI] [PubMed] [Google Scholar]
- [70].Yabe-Nishimura C (1998). Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev, 50: 21-33 [PubMed] [Google Scholar]
- [71].Tang WH,Martin KA,Hwa J (2012). Aldose reductase, oxidative stress, and diabetic mellitus. Frontiers in pharmacology, 3: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Fantus IG (2002). The pathogenesis of the chronic complications of the diabetes mellitus. Endocrinology Rounds, 2: 1-8 [Google Scholar]
- [73].Gomes AP,Price NL,Ling AJ,Moslehi JJ,Montgomery MK,Rajman L, et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell, 155: 1624-1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ido Y,Williamson JR (1997). Hyperglycemic cytosolic reductive stress 'pseudohypoxia': implications for diabetic retinopathy. Invest Ophthalmol Vis Sci, 38: 1467-1470 [PubMed] [Google Scholar]
- [75].Williamson JR,Chang K,Frangos M,Hasan KS,Ido Y,Kawamura T, et al. (1993). Hyperglycemic pseudohypoxia and diabetic complications. Diabetes, 42: 801-813 [DOI] [PubMed] [Google Scholar]
- [76].Hotta N (1997). New concepts and insights on pathogenesis and treatment of diabetic complications: polyol pathway and its inhibition. Nagoya J Med Sci, 60: 89-100 [PubMed] [Google Scholar]
- [77].Yasunari K,Kohno M,Kano H,Minami M,Yoshikawa J (2000). Aldose reductase inhibitor improves insulin-mediated glucose uptake and prevents migration of human coronary artery smooth muscle cells induced by high glucose. Hypertension, 35: 1092-1098 [DOI] [PubMed] [Google Scholar]
- [78].Chavez JC,Agani F,Pichiule P,LaManna JC (2000). Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J Appl Physiol (1985), 89: 1937-1942 [DOI] [PubMed] [Google Scholar]
- [79].Benderro GF,LaManna JC (2013). Kidney EPO expression during chronic hypoxia in aged mice. Adv Exp Med Biol, 765: 9-14 [DOI] [PubMed] [Google Scholar]
- [80].Li R,Luo X,Wu J,Thangthaeng N,Jung ME,Jing S, et al. (2015). Mitochondrial dihydrolipoamide dehydrogenase is upregulated in response to intermittent hypoxic preconditioning. Int. J. Med. Sci., 12: 432-440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Houtkooper RH,Canto C,Wanders RJ,Auwerx J (2010). The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocrine reviews, 31: 194-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Braidy N,Guillemin GJ,Mansour H,Chan-Ling T,Poljak A,Grant R (2011). Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One, 6: e19194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [83].Chiarugi A,Dolle C,Felici R,Ziegler M (2012). The NAD metabolome--a key determinant of cancer cell biology. Nat Rev Cancer, 12: 741-752 [DOI] [PubMed] [Google Scholar]
- [84].Houtkooper RH,Auwerx J (2012). Exploring the therapeutic space around NAD+. J Cell Biol, 199: 205-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chandra D,Jackson EB,Ramana KV,Kelley R,Srivastava SK,Bhatnagar A (2002). Nitric oxide prevents aldose reductase activation and sorbitol accumulation during diabetes. Diabetes, 51: 3095-3101 [DOI] [PubMed] [Google Scholar]
- [86].Lee AY,Chung SS (1999). Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J, 13: 23-30 [DOI] [PubMed] [Google Scholar]
- [87].Obrosova IG (2005). Increased sorbitol pathway activity generates oxidative stress in tissue sites for diabetic complications. Antioxid Redox Signal, 7: 1543-1552 [DOI] [PubMed] [Google Scholar]
- [88].Harding JJ,Blakytny R,Ganea E (1996). Glutathione in disease. Biochem Soc Trans, 24: 881-884 [DOI] [PubMed] [Google Scholar]
- [89].Yan LJ,Christians ES,Liu L,Xiao X,Sohal RS,Benjamin IJ (2002). Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J, 21: 5164-5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lou MF,Dickerson JE Jr.,Garadi R,York BM Jr., (1988). Glutathione depletion in the lens of galactosemic and diabetic rats. Experimental eye research, 46: 517-530 [DOI] [PubMed] [Google Scholar]
- [91].Sato T,Sasaki H,Watanabe R,Yoshinaga K (1988). Enhancement of pentose phosphate pathway in vascular intima from diabetic rabbit. Tohoku J Exp Med, 155: 97-100 [DOI] [PubMed] [Google Scholar]
- [92].Rosa AP,Jacques CE,de Souza LO,Bitencourt F,Mazzola PN,Coelho JG, et al. (2015). Neonatal hyperglycemia induces oxidative stress in the rat brain: the role of pentose phosphate pathway enzymes and NADPH oxidase. Mol Cell Biochem, 403: 159-167 [DOI] [PubMed] [Google Scholar]
- [93].Bast A,Haenen GR (2002) Lipoic acid: a multifunctional antioxidant. In Thiol metabolism and redox regulation of cellular function (Pompella A,Banhegyi G,Wellman-Rousseau M, eds) pp. 230-237, IOS Press, Amsterdam, Netherlands [Google Scholar]
- [94].Winkler BS,DeSantis N,Solomon F (1986). Multiple NADPH-producing pathways control glutathione (GSH) content in retina. Experimental eye research, 43: 829-847 [DOI] [PubMed] [Google Scholar]
- [95].Tang WH,Wu S,Wong TM,Chung SK,Chung SS (2008). Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radic Biol Med, 45: 602-610 [DOI] [PubMed] [Google Scholar]
- [96].Yang Y,Hayden MR,Sowers S,Bagree SV,Sowers JR (2010). Retinal redox stress and remodeling in cardiometabolic syndrome and diabetes. Oxid Med Cell Longev, 3: 392-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ussher JR,Jaswal JS,Lopaschuk GD (2012). Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ Res, 111: 628-641 [DOI] [PubMed] [Google Scholar]
- [98].Suzen S,Buyukbingol E (2003). Recent studies of aldose reductase enzyme inhibition for diabetic complications. Curr Med Chem, 10: 1329-1352 [DOI] [PubMed] [Google Scholar]
- [99].Reddy AB,Ramana KV (2010). Aldose reductase inhibition: emerging drug target for the treatment of cardiovascular complications. Recent Pat Cardiovasc Drug Discov, 5: 25-32 [DOI] [PubMed] [Google Scholar]
- [100].Tang J,Du Y,Petrash JM,Sheibani N,Kern TS (2013). Deletion of aldose reductase from mice inhibits diabetes-induced retinal capillary degeneration and superoxide generation. PLoS One, 8: e62081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Alzaid A,Rizza RA (1993). Insulin resistance and its role in the pathogenesis of impaired glucose tolerance and non-insulin-dependent diabetes mellitus: perspectives gained from in vivo studies. In: Insulin resistance. John Wiley & Sons Ltd, New York, 143-186 [Google Scholar]
- [102].Bevilacqua S,Buzzigoli G,Bonadonna R,Brandi LS,Oleggini M,Boni C, et al. (1990). Operation of Randle's cycle in patients with NIDDM. Diabetes, 39: 383-389 [DOI] [PubMed] [Google Scholar]
- [103].Nuutila P,Koivisto VA,Knuuti J,Ruotsalainen U,Teras M,Haaparanta M, et al. (1992). Glucose-free fatty acid cycle operates in human heart and skeletal muscle in vivo. J Clin Invest, 89: 1767-1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Ferrannini E,Barrett EJ,Bevilacqua S,DeFronzo RA (1983). Effect of fatty acids on glucose production and utilization in man. J Clin Invest, 72: 1737-1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Dolle C,Rack JG,Ziegler M (2013). NAD and ADP-ribose metabolism in mitochondria. FEBS J, 280: 3530-3541 [DOI] [PubMed] [Google Scholar]
- [106].Ying W (2008). NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal, 10: 179-206 [DOI] [PubMed] [Google Scholar]
- [107].Szabados E,Fischer GM,Gallyas F Jr.,Kispal G,Sumegi B (1999). Enhanced ADP-ribosylation and its diminution by lipoamide after ischemia-reperfusion in perfused rat heart. Free Radic Biol Med, 27: 1103-1113 [DOI] [PubMed] [Google Scholar]
- [108].Szabo C (2005). Roles of poly(ADP-ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol Res, 52: 60-71 [DOI] [PubMed] [Google Scholar]
- [109].Pittelli M,Felici R,Pitozzi V,Giovannelli L,Bigagli E,Cialdai F, et al. (2011). Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol Pharmacol, 80: 1136-1146 [DOI] [PubMed] [Google Scholar]
- [110].Szabo C,Zanchi A,Komjati K,Pacher P,Krolewski AS,Quist WC, et al. (2002). Poly(ADP-Ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation, 106: 2680-2686 [DOI] [PubMed] [Google Scholar]
- [111].Pacher P,Liaudet L,Soriano FG,Mabley JG,Szabo E,Szabo C (2002). The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes, 51: 514-521 [DOI] [PubMed] [Google Scholar]
- [112].Horvath EM,Magenheim R,Kugler E,Vacz G,Szigethy A,Levardi F, et al. (2009). Nitrative stress and poly(ADP-ribose) polymerase activation in healthy and gestational diabetic pregnancies. Diabetologia, 52: 1935-1943 [DOI] [PubMed] [Google Scholar]
- [113].Du X,Matsumura T,Edelstein D,Rossetti L,Zsengeller Z,Szabo C, et al. (2003). Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest, 112: 1049-1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Pacher P,Szabo C (2005). Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal, 7: 1568-1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Obrosova IG,Drel VR,Pacher P,Ilnytska O,Wang ZQ,Stevens MJ, et al. (2005). Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes, 54: 3435-3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Chiu J,Xu BY,Chen S,Feng B,Chakrabarti S (2008). Oxidative stress-induced, poly(ADP-ribose) polymerase-dependent upregulation of ET-1 expression in chronic diabetic complications. Can J Physiol Pharmacol, 86: 365-372 [DOI] [PubMed] [Google Scholar]
- [117].Puthanveetil P,Zhang D,Wang Y,Wang F,Wan A,Abrahani A, et al. (2012). Diabetes triggers a PARP1 mediated death pathway in the heart through participation of FoxO1. J Mol Cell Cardiol, 53: 677-686 [DOI] [PubMed] [Google Scholar]
- [118].Masutani M,Suzuki H,Kamada N,Watanabe M,Ueda O,Nozaki T, et al. (1999). Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc Natl Acad Sci U S A, 96: 2301-2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Pieper AA,Brat DJ,Krug DK,Watkins CC,Gupta A,Blackshaw S, et al. (1999). Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci U S A, 96: 3059-3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Virag L,Szabo C (2002). The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev, 54: 375-429 [DOI] [PubMed] [Google Scholar]
- [121].Long CA,Boulom V,Albadawi H,Tsai S,Yoo HJ,Oklu R, et al. (2013). Poly-ADP-ribose-polymerase inhibition ameliorates hind limb ischemia reperfusion injury in a murine model of type 2 diabetes. Ann Surg, 258: 1087-1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Sarras MP Jr.,Mason S,McAllister G,Intine RV (2014). Inhibition of poly-ADP ribose polymerase enzyme activity prevents hyperglycemia-induced impairment of angiogenesis during wound healing. Wound Repair Regen, 22: 666-670 [DOI] [PubMed] [Google Scholar]
- [123].Szkudelski T (2012). Streptozotocin-nicotinamide-induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med (Maywood), 237: 481-490 [DOI] [PubMed] [Google Scholar]
- [124].Fukaya M,Tamura Y,Chiba Y,Tanioka T,Mao J,Inoue Y, et al. (2013). Protective effects of a nicotinamide derivative, isonicotinamide, against streptozotocin-induced beta-cell damage and diabetes in mice. Biochem Biophys Res Commun, 442: 92-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Obrosova IG,Minchenko AG,Frank RN,Seigel GM,Zsengeller Z,Pacher P, et al. (2004). Poly(ADP-ribose) polymerase inhibitors counteract diabetes- and hypoxia-induced retinal vascular endothelial growth factor overexpression. Int J Mol Med, 14: 55-64 [PubMed] [Google Scholar]
- [126].Beyer AM,Weihrauch D (2012). Hexosamine pathway activation and O-linked-N-acetylglucosamine: novel mediators of endothelial dysfunction in hyperglycemia and diabetes. Vascul Pharmacol, 56: 113-114 [DOI] [PubMed] [Google Scholar]
- [127].Schleicher ED,Weigert C (2000). Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl, 77: S13-18 [DOI] [PubMed] [Google Scholar]
- [128].Yki-Jarvinen H,Daniels MC,Virkamaki A,Makimattila S,DeFronzo RA,McClain D (1996). Increased glutamine:fructose-6-phosphate amidotransferase activity in skeletal muscle of patients with NIDDM. Diabetes, 45: 302-307 [DOI] [PubMed] [Google Scholar]
- [129].Yki-Jarvinen H,Vogt C,Iozzo P,Pipek R,Daniels MC,Virkamaki A, et al. (1997). UDP-N-acetylglucosamine transferase and glutamine: fructose 6-phosphate amidotransferase activities in insulin-sensitive tissues. Diabetologia, 40: 76-81 [DOI] [PubMed] [Google Scholar]
- [130].Ma J,Hart GW (2013). Protein O-GlcNAcylation in diabetes and diabetic complications. Expert review of proteomics, 10: 365-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Issad T,Kuo M (2008). O-GlcNAc modification of transcription factors, glucose sensing and glucotoxicity. Trends Endocrinol Metab, 19: 380-389 [DOI] [PubMed] [Google Scholar]
- [132].Kuo M,Zilberfarb V,Gangneux N,Christeff N,Issad T (2008). O-GlcNAc modification of FoxO1 increases its transcriptional activity: a role in the glucotoxicity phenomenon? Biochimie, 90: 679-685 [DOI] [PubMed] [Google Scholar]
- [133].Hardiville S,Hart GW (2014). Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab, 20: 208-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Semba RD,Huang H,Lutty GA,Van Eyk JE,Hart GW (2014). The role of O-GlcNAc signaling in the pathogenesis of diabetic retinopathy. Proteomics. Clinical applications, 8: 218-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Lima VV,Spitler K,Choi H,Webb RC,Tostes RC (2012). O-GlcNAcylation and oxidation of proteins: is signalling in the cardiovascular system becoming sweeter? Clin Sci (Lond), 123: 473-486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Issad T,Masson E,Pagesy P (2010). O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes & metabolism, 36: 423-435 [DOI] [PubMed] [Google Scholar]
- [137].Xia L,Wang H,Munk S,Frecker H,Goldberg HJ,Fantus IG, et al. (2007). Reactive oxygen species, PKC-beta1, and PKC-zeta mediate high-glucose-induced vascular endothelial growth factor expression in mesangial cells. American journal of physiology. Endocrinology and metabolism, 293: E1280-1288 [DOI] [PubMed] [Google Scholar]
- [138].Xia L,Wang H,Munk S,Kwan J,Goldberg HJ,Fantus IG, et al. (2008). High glucose activates PKC-zeta and NADPH oxidase through autocrine TGF-beta1 signaling in mesangial cells. American journal of physiology. Renal physiology, 295: F1705-1714 [DOI] [PubMed] [Google Scholar]
- [139].Bey EA,Xu B,Bhattacharjee A,Oldfield CM,Zhao X,Li Q, et al. (2004). Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol, 173: 5730-5738 [DOI] [PubMed] [Google Scholar]
- [140].Fontayne A,Dang PM,Gougerot-Pocidalo MA,El-Benna J (2002). Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry, 41: 7743-7750 [DOI] [PubMed] [Google Scholar]
- [141].Thornalley PJ,Langborg A,Minhas HS (1999). Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J, 344 Pt 1: 109-116 [PMC free article] [PubMed] [Google Scholar]
- [142].Dmitriev LF,Dugin SF (2007). Aldehydes and disturbance of carbohydrate metabolism: some consequences and possible approaches to its normalization. Arch Physiol Biochem, 113: 87-95 [DOI] [PubMed] [Google Scholar]
- [143].Allaman I,Belanger M,Magistretti PJ (2015). Methylglyoxal, the dark side of glycolysis. Front Neurosci, 9: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Maessen DE,Stehouwer CD,Schalkwijk CG (2015). The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin Sci (Lond), 128: 839-861 [DOI] [PubMed] [Google Scholar]
- [145].Queisser MA,Yao D,Geisler S,Hammes HP,Lochnit G,Schleicher ED, et al. (2010). Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes, 59: 670-678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Wolff SP,Jiang ZY,Hunt JV (1991). Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Radic Biol Med, 10: 339-352 [DOI] [PubMed] [Google Scholar]
- [147].Tomlinson DR,Gardiner NJ (2008). Glucose neurotoxicity. Nat Rev Neurosci, 9: 36-45 [DOI] [PubMed] [Google Scholar]
- [148].Gawlowski T,Stratmann B,Stork I,Engelbrecht B,Brodehl A,Niehaus K, et al. (2009). Heat shock protein 27 modification is increased in the human diabetic failing heart. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme, 41: 594-599 [DOI] [PubMed] [Google Scholar]
- [149].Schalkwijk CG,van Bezu J,van der Schors RC,Uchida K,Stehouwer CD,van Hinsbergh VW (2006). Heat-shock protein 27 is a major methylglyoxal-modified protein in endothelial cells. FEBS Lett, 580: 1565-1570 [DOI] [PubMed] [Google Scholar]
- [150].Koga M,Murai J,Morita S,Saito H,Kasayama S (2013). Comparison of annual variability in HbA1c and glycated albumin in patients with type 1 vs. type 2 diabetes mellitus. Journal of diabetes and its complications, 27: 211-213 [DOI] [PubMed] [Google Scholar]
- [151].Hayden MR,Tyagi SC (2002). Islet redox stress: the manifold toxicities of insulin resistance, metabolic syndrome and amylin derived islet amyloid in type 2 diabetes mellitus. JOP : Journal of the pancreas, 3: 86-108 [PubMed] [Google Scholar]
- [152].Munoz A,Costa M (2013). Nutritionally mediated oxidative stress and inflammation. Oxid Med Cell Longev, 2013: 610950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Gao L,Mann GE (2009). Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res, 82: 9-20 [DOI] [PubMed] [Google Scholar]
- [154].Zhang M,Kho AL,Anilkumar N,Chibber R,Pagano PJ,Shah AM, et al. (2006). Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation, 113: 1235-1243 [DOI] [PubMed] [Google Scholar]
- [155].Newsholme P,Morgan D,Rebelato E,Oliveira-Emilio HC,Procopio J,Curi R, et al. (2009). Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia, 52: 2489-2498 [DOI] [PubMed] [Google Scholar]
- [156].Koulajian K,Desai T,Liu GC,Ivovic A,Patterson JN,Tang C, et al. (2013). NADPH oxidase inhibition prevents beta cell dysfunction induced by prolonged elevation of oleate in rodents. Diabetologia, 56: 1078-1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Weaver JR,Grzesik W,Taylor-Fishwick DA (2015). Inhibition of NADPH oxidase-1 preserves beta cell function. Diabetologia, 58: 113-121 [DOI] [PubMed] [Google Scholar]
- [158].Mira ML,Martinho F,Azevedo MS,Manso CF (1991). Oxidative inhibition of red blood cell ATPases by glyceraldehyde. Biochim Biophys Acta, 1060: 257-261 [DOI] [PubMed] [Google Scholar]
- [159].Wolff SP,Dean RT (1987). Glucose autoxidation and protein modification. The potential role of 'autoxidative glycosylation' in diabetes. Biochem J, 245: 243-250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Nishikawa T,Araki E (2013). Mechanism-based antioxidant therapies promise to prevent diabetic complications? Journal of diabetes investigation, 4: 105-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Wolff SP (1993). Diabetes mellitus and free radicals. Free radicals, transition metals and oxidative stress in the aetiology of diabetes mellitus and complications. Br Med Bull, 49: 642-652 [DOI] [PubMed] [Google Scholar]
- [162].Tiganis T (2011). Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends in pharmacological sciences, 32: 82-89 [DOI] [PubMed] [Google Scholar]
- [163].Bocci V,Zanardi I,Huijberts MS,Travagli V (2014). An integrated medical treatment for type-2 diabetes. Diabetes & metabolic syndrome, 8: 57-61 [DOI] [PubMed] [Google Scholar]
- [164].Giacco F,Brownlee M (2010). Oxidative stress and diabetic complications. Circ Res, 107: 1058-1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Shaw A,Doherty MK,Mutch NJ,MacRury SM,Megson IL (2014). Endothelial cell oxidative stress in diabetes: a key driver of cardiovascular complications? Biochem Soc Trans, 42: 928-933 [DOI] [PubMed] [Google Scholar]
- [166].Haldar SR,Chakrabarty A,Chowdhury S,Haldar A,Sengupta S,Bhattacharyya M (2015). Oxidative stress-related genes in type 2 diabetes: association analysis and their clinical impact. Biochem Genet, 53: 93-119 [DOI] [PubMed] [Google Scholar]
- [167].Yan LJ (2009). Analysis of oxidative modification of proteins. Curr Protoc Protein Sci, Chapter 14: Unit14 14. [DOI] [PubMed] [Google Scholar]
- [168].Ames BN,Shigenaga MK (1992). Oxidants are a major contributor to aging. Ann N Y Acad Sci, 663: 85-96 [DOI] [PubMed] [Google Scholar]
- [169].Yan LJ,Sohal RS (1998). Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci USA, 95: 12896-12901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Yan LJ,Levine RL,Sohal RS (1997). Oxidative damage during aging targets mitochondrial aconitase. Proc. Natl. Acad. Sci. USA, 94: 11168-11172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Starkov AA (2008). The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci, 1147: 37-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Lenaz G (2012). Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv Exp Med Biol, 942: 93-136 [DOI] [PubMed] [Google Scholar]
- [173].Pollegioni L,Molla G (2011). New biotech applications from evolved D-amino acid oxidases. Trends Biotechnol, 29: 276-283 [DOI] [PubMed] [Google Scholar]
- [174].Fang J,Sawa T,Akaike T,Maeda H (2002). Tumor-targeted delivery of polyethylene glycol-conjugated D-amino acid oxidase for antitumor therapy via enzymatic generation of hydrogen peroxide. Cancer Res, 62: 3138-3143 [PubMed] [Google Scholar]
- [175].Haskew-Layton RE,Payappilly JB,Smirnova NA,Ma TC,Chan KK,Murphy TH, et al. (2010). Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proc Natl Acad Sci U S A, 107: 17385-17390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Bando Y,Aki K (1991). Mechanisms of generation of oxygen radicals and reductive mobilization of ferritin iron by lipoamide dehydrogenase. J Biochem (Tokyo), 109: 450-454 [DOI] [PubMed] [Google Scholar]
- [177].Sreider CM,Grinblat L,Stoppani AO (1990). Catalysis of nitrofuran redox-cycling and superoxide anion production by heart lipoamide dehydrogenase. Biochem Pharmacol, 40: 1849-1857 [DOI] [PubMed] [Google Scholar]
- [178].Gazaryan IG,Krasnikov BF,Ashby GA,Thorneley RN,Kristal BS,Brown AM (2002). Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J Biol Chem, 277: 10064-10072 [DOI] [PubMed] [Google Scholar]
- [179].Tahara EB,Barros MH,Oliveira GA,Netto LE,Kowaltowski AJ (2007). Dihydrolipoyl dehydrogenase as a source of reactive oxygen species inhibited by caloric restriction and involved in Saccharomyces cerevisiae aging. Faseb J, 21: 274-283 [DOI] [PubMed] [Google Scholar]
- [180].Zhang Q,Zou P,Zhan H,Zhang M,Zhang L,Ge RS, et al. (2011). Dihydrolipoamide dehydrogenase and cAMP are associated with cadmium-mediated Leydig cell damage. Toxicol Lett, 205: 183-189 [DOI] [PubMed] [Google Scholar]
- [181].Kareyeva AV,Grivennikova VG,Cecchini G,Vinogradov AD (2011). Molecular identification of the enzyme responsible for the mitochondrial NADH-supported ammonium-dependent hydrogen peroxide production. FEBS Lett, 585: 385-389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Kareyeva AV,Grivennikova VG,Vinogradov AD (2012). Mitochondrial hydrogen peroxide production as determined by the pyridine nucleotide pool and its redox state. Biochim Biophys Acta [DOI] [PubMed] [Google Scholar]
- [183].Quinlan CL,Goncalves RL,Hey-Mogensen M,Yadava N,Bunik VI,Brand MD (2014). The 2-Oxoacid Dehydrogenase Complexes in Mitochondria Can Produce Superoxide/Hydrogen Peroxide at Much Higher Rates than Complex I. J Biol Chem, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Tretter L,Adam-Vizi V (2005). Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci, 360: 2335-2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Starkov AA,Fiskum G,Chinopoulos C,Lorenzo BJ,Browne SE,Patel MS, et al. (2004). Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci, 24: 7779-7788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Ambrus A,Tretter L,Adam-Vizi V (2009). Inhibition of the alpha-ketoglutarate dehydrogenase-mediated reactive oxygen species generation by lipoic acid. J Neurochem, 109 Suppl 1: 222-229 [DOI] [PubMed] [Google Scholar]
- [187].Ambrus A,Torocsik B,Tretter L,Ozohanics O,Adam-Vizi V (2011). Stimulation of reactive oxygen species generation by disease-causing mutations of lipoamide dehydrogenase. Hum Mol Genet, 20: 2984-2995 [DOI] [PubMed] [Google Scholar]
- [188].Manea A (2010). NADPH oxidase-derived reactive oxygen species: involvement in vascular physiology and pathology. Cell Tissue Res, 342: 325-339 [DOI] [PubMed] [Google Scholar]
- [189].Bylund J,Brown KL,Movitz C,Dahlgren C,Karlsson A (2010). Intracellular generation of superoxide by the phagocyte NADPH oxidase: how, where, and what for? Free Radic Biol Med, 49: 1834-1845 [DOI] [PubMed] [Google Scholar]
- [190].Harrison R (2004). Physiological roles of xanthine oxidoreductase. Drug Metab Rev, 36: 363-375 [DOI] [PubMed] [Google Scholar]
- [191].Agarwal A,Banerjee A,Banerjee UC (2011). Xanthine oxidoreductase: a journey from purine metabolism to cardiovascular excitation-contraction coupling. Crit Rev Biotechnol, 31: 264-280 [DOI] [PubMed] [Google Scholar]
- [192].Radi R (2013). Protein Tyrosine Nitration: Biochemical Mechanisms and Structural Basis of Functional Effects. Acc Chem Res, 46: 550-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Radi R,Cassina A,Hodara R,Quijano C,Castro L (2002). Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med, 33: 1451-1464 [DOI] [PubMed] [Google Scholar]
- [194].Landino LM (2008). Protein thiol modification by peroxynitrite anion and nitric oxide donors. Methods Enzymol, 440: 95-109 [DOI] [PubMed] [Google Scholar]
- [195].Singh IN,Sullivan PG,Hall ED (2007). Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J Neurosci Res, 85: 2216-2223 [DOI] [PubMed] [Google Scholar]
- [196].Alvarez B,Radi R (2003). Peroxynitrite reactivity with amino acids and proteins. Amino Acids, 25: 295-311 [DOI] [PubMed] [Google Scholar]
- [197].Son SM (2012). Reactive oxygen and nitrogen species in pathogenesis of vascular complications of diabetes. Diabetes Metab J, 36: 190-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Stavniichuk R,Shevalye H,Lupachyk S,Obrosov A,Groves JT,Obrosova IG, et al. (2014). Peroxynitrite and protein nitration in the pathogenesis of diabetic peripheral neuropathy. Diabetes/metabolism research and reviews, 30: 669-678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Hung LM,Huang JP,Liao JM,Yang MH,Li DE,Day YJ, et al. (2014). Insulin renders diabetic rats resistant to acute ischemic stroke by arresting nitric oxide reaction with superoxide to form peroxynitrite. J Biomed Sci, 21: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Li Y,Qi J,Liu K,Li B,Wang H,Jia J (2010). Peroxynitrite-induced nitration of cyclooxygenase-2 and inducible nitric oxide synthase promotes their binding in diabetic angiopathy. Mol Med, 16: 335-342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Liang JH,Li YN,Qi JS,Jia XX (2010). Peroxynitrite-induced protein nitration is responsible for renal mitochondrial damage in diabetic rat. J Endocrinol Invest, 33: 140-146 [DOI] [PubMed] [Google Scholar]
- [202].Wu CH,Hsieh HT,Lin JA,Yen GC (2013). Alternanthera paronychioides protects pancreatic beta-cells from glucotoxicity by its antioxidant, antiapoptotic and insulin secretagogue actions. Food Chem, 139: 362-370 [DOI] [PubMed] [Google Scholar]
- [203].Zhao WC,Zhang B,Liao MJ,Zhang WX,He WY,Wang HB, et al. (2014). Curcumin ameliorated diabetic neuropathy partially by inhibition of NADPH oxidase mediating oxidative stress in the spinal cord. Neurosci Lett, 560: 81-85 [DOI] [PubMed] [Google Scholar]
- [204].Alam MM,Meerza D,Naseem I (2014). Protective effect of quercetin on hyperglycemia, oxidative stress and DNA damage in alloxan induced type 2 diabetic mice. Life Sci, 109: 8-14 [DOI] [PubMed] [Google Scholar]
- [205].Niture NT,Ansari AA,Naik SR (2014). Anti-hyperglycemic activity of rutin in streptozotocin-induced diabetic rats: an effect mediated through cytokines, antioxidants and lipid biomarkers. Indian journal of experimental biology, 52: 720-727 [PubMed] [Google Scholar]
- [206].Erejuwa OO,Sulaiman SA,Wahab MS,Sirajudeen KN,Salleh MS,Gurtu S (2010). Antioxidant protection of Malaysian tualang honey in pancreas of normal and streptozotocin-induced diabetic rats. Annales d'endocrinologie, 71: 291-296 [DOI] [PubMed] [Google Scholar]
- [207].Parveen K,Khan MR,Mujeeb M,Siddiqui WA (2010). Protective effects of Pycnogenol on hyperglycemia-induced oxidative damage in the liver of type 2 diabetic rats. Chem Biol Interact, 186: 219-227 [DOI] [PubMed] [Google Scholar]
- [208].Ku CR,Lee HJ,Kim SK,Lee EY,Lee MK,Lee EJ (2012). Resveratrol prevents streptozotocin-induced diabetes by inhibiting the apoptosis of pancreatic beta-cell and the cleavage of poly (ADP-ribose) polymerase. Endocrine journal, 59: 103-109 [DOI] [PubMed] [Google Scholar]
- [209].Chanpoo M,Petchpiboonthai H,Panyarachun B,Anupunpisit V (2010). Effect of curcumin in the amelioration of pancreatic islets in streptozotocin-induced diabetic mice. Journal of the Medical Association of Thailand = Chotmaihet thangphaet, 93 Suppl 6: S152-159 [PubMed] [Google Scholar]
- [210].Ding Y,Zhang Z,Dai X,Jiang Y,Bao L,Li Y, et al. (2013). Grape seed proanthocyanidins ameliorate pancreatic beta-cell dysfunction and death in low-dose streptozotocin- and high-carbohydrate/high-fat diet-induced diabetic rats partially by regulating endoplasmic reticulum stress. Nutrition & metabolism, 10: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Ola MS,Aleisa AM,Al-Rejaie SS,Abuohashish HM,Parmar MY,Alhomida AS, et al. (2014). Flavonoid, morin inhibits oxidative stress, inflammation and enhances neurotrophic support in the brain of streptozotocin-induced diabetic rats. Neurol Sci, 35: 1003-1008 [DOI] [PubMed] [Google Scholar]
- [212].Hirst J,Carroll J,Fearnley IM,Shannon RJ,Walker JE (2003). The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim Biophys Acta, 1604: 135-150 [DOI] [PubMed] [Google Scholar]
- [213].Carroll J,Fearnley IM,Skehel JM,Shannon RJ,Hirst J,Walker JE (2006). Bovine complex I is a complex of 45 different subunits. J Biol Chem, 281: 32724-32727 [DOI] [PubMed] [Google Scholar]
- [214].Carroll J,Ding S,Fearnley IM,Walker JE (2013). Post-translational modifications near the quinone binding site of mammalian complex I. J Biol Chem, 288: 24799-24808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Andrews B,Carroll J,Ding S,Fearnley IM,Walker JE (2013). Assembly factors for the membrane arm of human complex I. Proc Natl Acad Sci U S A, 110: 18934-18939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Hirst J (2013). Mitochondrial complex I. Annu Rev Biochem, 82: 551-575 [DOI] [PubMed] [Google Scholar]
- [217].Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J, 417: 1-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].St-Pierre J,Buckingham JA,Roebuck SJ,Brand MD (2002). Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem, 277: 44784-44790 [DOI] [PubMed] [Google Scholar]
- [219].Coughlan MT,Thorburn DR,Penfold SA,Laskowski A,Harcourt BE,Sourris KC, et al. (2009). RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol, 20: 742-752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Papa S,Sardanelli AM,Scacco S,Petruzzella V,Technikova-Dobrova Z,Vergari R, et al. (2002). The NADH: ubiquinone oxidoreductase (complex I) of the mammalian respiratory chain and the cAMP cascade. J Bioenerg Biomembr, 34: 1-10 [DOI] [PubMed] [Google Scholar]
- [221].Hirst J,King MS,Pryde KR (2008). The production of reactive oxygen species by complex I. Biochem Soc Trans, 36: 976-980 [DOI] [PubMed] [Google Scholar]
- [222].Bridges HR,Jones AJ,Pollak MN,Hirst J (2014). Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J, 462: 475-487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Fontaine E (2014). Metformin and respiratory chain complex I: the last piece of the puzzle? Biochem J, 463: e3-5 [DOI] [PubMed] [Google Scholar]
- [224].Matsuzaki S,Humphries KM (2015). Selective inhibition of deactivated mitochondrial complex I by biguanides. Biochemistry, 54: 2011-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Hur JH,Bahadorani S,Graniel J,Koehler CL,Ulgherait M,Rera M, et al. (2013). Increased longevity mediated by yeast NDI1 expression in Drosophila intestinal stem and progenitor cells. Aging, 5: 662-681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Cho J,Hur JH,Graniel J,Benzer S,Walker DW (2012). Expression of yeast NDI1 rescues a Drosophila complex I assembly defect. PLoS One, 7: e50644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Marella M,Seo BB,Nakamaru-Ogiso E,Greenamyre JT,Matsuno-Yagi A,Yagi T (2008). Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson's disease. PLoS One, 3: e1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Santidrian AF,Matsuno-Yagi A,Ritland M,Seo BB,LeBoeuf SE,Gay LJ, et al. (2013). Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest, 123: 1068-1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Luo X,Li R,Yan LJ (2015). Roles of Pyruvate, NADH, and Mitochondrial Complex I in Redox Balance and Imbalance in β Cell Function and Dysfunction. Journal of diabetes research [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Brunmair B,Staniek K,Gras F,Scharf N,Althaym A,Clara R, et al. (2004). Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes, 53: 1052-1059 [DOI] [PubMed] [Google Scholar]
- [231].Sliwinska A,Drzewoski J (2015). Molecular Action of Metformin in Hepatocytes: an Updated Insight. Current diabetes reviews [DOI] [PubMed] [Google Scholar]
- [232].Owen MR,Doran E,Halestrap AP (2000). Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J, 348 Pt 3: 607-614 [PMC free article] [PubMed] [Google Scholar]
- [233].Masini M,Anello M,Bugliani M,Marselli L,Filipponi F,Boggi U, et al. (2014). Prevention by metformin of alterations induced by chronic exposure to high glucose in human islet beta cells is associated with preserved ATP/ADP ratio. Diabetes research and clinical practice, 104: 163-170 [DOI] [PubMed] [Google Scholar]
- [234].Viollet B,Guigas B,Sanz Garcia N,Leclerc J,Foretz M,Andreelli F (2012). Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond), 122: 253-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Pernicova I,Korbonits M (2014). Metformin--mode of action and clinical implications for diabetes and cancer. Nature reviews. Endocrinology, 10: 143-156 [DOI] [PubMed] [Google Scholar]
- [236].Jenkins Y,Sun TQ,Markovtsov V,Foretz M,Li W,Nguyen H, et al. (2013). AMPK activation through mitochondrial regulation results in increased substrate oxidation and improved metabolic parameters in models of diabetes. PLoS One, 8: e81870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Hinke SA,Martens GA,Cai Y,Finsi J,Heimberg H,Pipeleers D, et al. (2007). Methyl succinate antagonises biguanide-induced AMPK-activation and death of pancreatic beta-cells through restoration of mitochondrial electron transfer. Br J Pharmacol, 150: 1031-1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Hopson NE,Sack WA (1973). Cellular phosphorus changes under low-carbon stress. J Water Pollut Control Fed, 45: 85-96 [PubMed] [Google Scholar]
- [239].Kosanam H,Thai K,Zhang Y,Advani A,Connelly KA,Diamandis EP, et al. (2014). Diabetes induces lysine acetylation of intermediary metabolism enzymes in the kidney. Diabetes, 63: 2432-2439 [DOI] [PubMed] [Google Scholar]
- [240].Frizzell N,Thomas SA,Carson JA,Baynes JW (2012). Mitochondrial stress causes increased succination of proteins in adipocytes in response to glucotoxicity. Biochem J, 445: 247-254 [DOI] [PubMed] [Google Scholar]
- [241].Fritz KS,Galligan JJ,Hirschey MD,Verdin E,Petersen DR (2012). Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J Proteome Res, 11: 1633-1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Fritz KS,Green MF,Petersen DR,Hirschey MD (2013). Ethanol metabolism modifies hepatic protein acylation in mice. PLoS One, 8: e75868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Verdin E,Ott M (2015). 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nature reviews. Molecular cell biology, 16: 258-264 [DOI] [PubMed] [Google Scholar]
- [244].Cohn RM,Roth KS (1996) Biochemistry and diabetes, Williams & Wilkins, Baltimore [Google Scholar]
- [245].Paik WK,Pearson D,Lee HW,Kim S (1970). Nonenzymatic acetylation of histones with acetyl-CoA. Biochim Biophys Acta, 213: 513-522 [DOI] [PubMed] [Google Scholar]
- [246].Ramponi G,Manao G,Camici G (1975). Nonenzymatic acetylation of histones with acetyl phosphate and acetyl adenylate. Biochemistry, 14: 2681-2685 [DOI] [PubMed] [Google Scholar]
- [247].Baeza J,Smallegan MJ,Denu JM (2015). Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem Biol, 10: 122-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Wagner GR,Payne RM (2013). Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem, 288: 29036-29045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Muoio DM (2014). Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell, 159: 1253-1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Morris BJ (2013). Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med, 56: 133-171 [DOI] [PubMed] [Google Scholar]
- [251].Caton PW,Richardson SJ,Kieswich J,Bugliani M,Holland ML,Marchetti P, et al. (2013). Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia, 56: 1068-1077 [DOI] [PubMed] [Google Scholar]
- [252].Michan S (2014). Calorie restriction and NAD(+)/sirtuin counteract the hallmarks of aging. Front Biosci (Landmark Ed), 19: 1300-1319 [DOI] [PubMed] [Google Scholar]
- [253].Yang T,Sauve AA (2006). NAD metabolism and sirtuins: metabolic regulation of protein deacetylation in stress and toxicity. AAPS J, 8: E632-E643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Wang Y (2014). Molecular Links between Caloric Restriction and Sir2/SIRT1 Activation. Diabetes Metab J, 38: 321-329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Ramis MR,Esteban S,Miralles A,Tan DX,Reiter RJ (2015). Caloric restriction, resveratrol and melatonin: Role of SIRT1 and implications for aging and related-diseases. Mech Ageing Dev, 146-148C: 28-41 [DOI] [PubMed] [Google Scholar]
- [256].Kincaid B,Bossy-Wetzel E (2013). Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci, 5: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Canto C,Gerhart-Hines Z,Feige JN,Lagouge M,Noriega L,Milne JC, et al. (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature, 458: 1056-1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Kim HJ,Kim JH,Noh S,Hur HJ,Sung MJ,Hwang JT, et al. (2011). Metabolomic analysis of livers and serum from high-fat diet induced obese mice. J Proteome Res, 10: 722-731 [DOI] [PubMed] [Google Scholar]
- [259].Jing E,Emanuelli B,Hirschey MD,Boucher J,Lee KY,Lombard D, et al. (2011). Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A, 108: 14608-14613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Hirschey MD,Shimazu T,Jing E,Grueter CA,Collins AM,Aouizerat B, et al. (2011). SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell, 44: 177-190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Wang RH,Kim HS,Xiao C,Xu X,Gavrilova O,Deng CX (2011). Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest, 121: 4477-4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Cerutti R,Pirinen E,Lamperti C,Marchet S,Sauve AA,Li W, et al. (2014). NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab, 19: 1042-1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Balestrieri ML,Rizzo MR,Barbieri M,Paolisso P,D'Onofrio N,Giovane A, et al. (2015). Sirtuin 6 expression and inflammatory activity in diabetic atherosclerotic plaques: effects of incretin treatment. Diabetes, 64: 1395-1406 [DOI] [PubMed] [Google Scholar]
- [264].de Kreutzenberg SV,Ceolotto G,Papparella I,Bortoluzzi A,Semplicini A,Dalla Man C, et al. (2010). Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes, 59: 1006-1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Hou X,Zeng H,He X,Chen JX (2015). Sirt3 is essential for apelin-induced angiogenesis in post-myocardial infarction of diabetes. J Cell Mol Med, 19: 53-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Herskovits AZ,Guarente L (2013). Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res, 23: 746-758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Turkmen K,Karagoz A,Kucuk A (2014). Sirtuins as novel players in the pathogenesis of diabetes mellitus. World J Diabetes, 5: 894-900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Hassa PO,Haenni SS,Elser M,Hottiger MO (2006). Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev, 70: 789-829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Kaelin WG Jr.,McKnight SL (2013). Influence of metabolism on epigenetics and disease. Cell, 153: 56-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Mouchiroud L,Houtkooper RH,Auwerx J (2013). NAD(+) metabolism: a therapeutic target for age-related metabolic disease. Critical reviews in biochemistry and molecular biology, 48: 397-408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Huynh FK,Hershberger KA,Hirschey MD (2013). Targeting sirtuins for the treatment of diabetes. Diabetes Manag (Lond), 3: 245-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Kitada M,Kume S,Kanasaki K,Takeda-Watanabe A,Koya D (2013). Sirtuins as possible drug targets in type 2 diabetes. Curr Drug Targets, 14: 622-636 [DOI] [PubMed] [Google Scholar]
- [273].Lin H,Su X,He B (2012). Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem Biol, 7: 947-960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Piroli GG,Manuel AM,Walla MD,Jepson MJ,Brock JW,Rajesh MP, et al. (2014). Identification of protein succination as a novel modification of tubulin. Biochem J, 462: 231-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Merkley ED,Metz TO,Smith RD,Baynes JW,Frizzell N (2014). The succinated proteome. Mass Spectrom Rev, 33: 98-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Yang M,Ternette N,Su H,Dabiri R,Kessler BM,Adam J, et al. (2014). The Succinated Proteome of FH-Mutant Tumours. Metabolites, 4: 640-654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Blatnik M,Thorpe SR,Baynes JW (2008). Succination of proteins by fumarate: mechanism of inactivation of glyceraldehyde-3-phosphate dehydrogenase in diabetes. Ann N Y Acad Sci, 1126: 272-275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Nagai R,Brock JW,Blatnik M,Baatz JE,Bethard J,Walla MD, et al. (2007). Succination of protein thiols during adipocyte maturation: a biomarker of mitochondrial stress. J Biol Chem, 282: 34219-34228 [DOI] [PubMed] [Google Scholar]
- [279].Zheng L,Cardaci S,Jerby L,MacKenzie ED,Sciacovelli M,Johnson TI, et al. (2015). Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat Commun, 6: 6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Yan LJ (2014). Positive oxidative stress in aging and aging-related disease tolerance. Redox Biology, 2: 165-169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Yan LJ (2014). Protein Redox Modification as a Cellular Defense Mechanism against Tissue Ischemic Injury. Oxidative Medicine and Cellular Longevity, 2014: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Winterbourn CC,Hampton MB (2008). Thiol chemistry and specificity in redox signaling. Free Radic Biol Med, 45: 549-561 [DOI] [PubMed] [Google Scholar]
- [283].Ying J,Clavreul N,Sethuraman M,Adachi T,Cohen RA (2007). Thiol oxidation in signaling and response to stress: detection and quantification of physiological and pathophysiological thiol modifications. Free Radic Biol Med, 43: 1099-1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Forman HJ,Fukuto JM,Torres M (2004). Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol, 287: C246-256 [DOI] [PubMed] [Google Scholar]
- [285].Fomenko DE,Marino SM,Gladyshev VN (2008). Functional diversity of cysteine residues in proteins and unique features of catalytic redox-active cysteines in thiol oxidoreductases. Molecules and cells, 26: 228-235 [PMC free article] [PubMed] [Google Scholar]
- [286].Biswas S,Chida AS,Rahman I (2006). Redox modifications of protein-thiols: emerging roles in cell signaling. Biochem Pharmacol, 71: 551-564 [DOI] [PubMed] [Google Scholar]
- [287].Cai Z,Yan LJ (2013). Protein oxidative modifications: Beneficial roles in disease and health. Journal of Biochemical and Pharmacological Research, 1: 15-26 [PMC free article] [PubMed] [Google Scholar]
- [288].White H,Venkatesh B (2011). Clinical review: ketones and brain injury. Crit Care, 15: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Cotter DG,Schugar RC,Crawford PA (2013). Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol, 304: H1060-1076 [DOI] [PMC free article] [PubMed] [Google Scholar]