

Summary
Large-scale genomic studies have identified multiple somatic aberrations in breast cancer, including copy number alterations, and point mutations. Still, identifying causal variants and emergent vulnerabilities that arise as a consequence of genetic alterations remain major challenges. We performed whole genome shRNA “dropout screens” on 77 breast cancer cell lines. Using a hierarchical linear regression algorithm to score our screen results and integrate them with accompanying detailed genetic and proteomic information, we identify vulnerabilities in breast cancer, including candidate “drivers,” and reveal general functional genomic properties of cancer cells. Comparisons of gene essentiality with drug sensitivity data suggest potential resistance mechanisms, effects of existing anti-cancer drugs, and opportunities for combination therapy. Finally, we demonstrate the utility of this large dataset by identifying BRD4 as a potential target in luminal breast cancer, and PIK3CA mutations as a resistance determinant for BET-inhibitors.
Introduction
Breast cancer is the second leading cause of cancer death in women. Better detection and therapy have led to >85% 5-year survival, yet half of affected women die from their disease. This outcome reflects incomplete understanding of the molecular alterations, heterogeneity, and determinants of drug response in breast tumors. Genetic and epigenetic abnormalities in breast cancer have been defined, but identifying causal defects and exploiting them for target discovery remain challenging.
“Breast cancer” actually comprises molecular subtypes that predict prognosis and drug response. Early profiling studies identified “intrinsic subtypes”: luminal A and B, basal-like (basal), HER2+ and normal-like (Perou et al., 2000; Sorlie et al., 2001). These were joined by a “claudin-low” subtype which, like basal breast cancer, is typically Estrogen Receptor-negative (ER−), Progesterone Receptor-negative (PR−), and HER2-negative (HER2−) (Hennessy et al., 2009; Prat et al., 2010). Basal and luminal B tumors have the worst prognosis; claudin-low tumors have intermediate outcome (Prat et al., 2010). Clinically, intrinsic subtypes can be defined by the “PAM50” classifier (Parker et al., 2009).
These molecular subtypes complement, but do not fully overlap, pathologic classification by ER, PR and HER2 status (Parker et al., 2009). Luminal tumors are typically ER+/PR+, and basal tumors are usually “triple negative” (ER−, PR−, HER2−). Breast cancer cell lines generally fall into four subtypes: basal A or B, HER2+, and luminal (Neve et al., 2006; Prat et al., 2010). Basal A lines resemble “basal” tumors; basal B lines are enriched for claudin-low genes.
Recent large scale RNA and proteomic profiling studies have further divided luminal and “triple negative” breast cancer (TNBC) into at least ten subtypes (Curtis et al., 2012; Lehmann et al., 2011; TCGA, 2012), and next-generation sequencing (NGS) has identified multiple aberrations in breast tumors (Banerji et al., 2012; Ellis et al., 2012; Shah et al., 2012; Stephens et al., 2012; TCGA, 2012). Whether breast cancer lines represent these new categories and have mutational profiles like tumors remains unresolved.
Moreover, genomics often cannot distinguish “passenger” mutations from “drivers” that promote tumorigenesis and might be therapeutic targets. Highly recurrent defects (e.g., HER2 amplification) point to drivers, and some have led to “targeted therapies” (e.g., Trastuzumab). Many other abnormalities, some clearly oncogenic, occur at low frequency, and some drivers are difficult to target (e.g., MYC, RAS). However, the collateral genotoxic, proteotoxic, and metabolic stresses caused by the abnormal tumor genome can cause “emergent dependencies,” potentially providing alternate therapeutic options.
Functional genomics, partnered with genomic data, can identify targets coupled to biomarkers (Zender et al., 2008). Pooled shRNA libraries enable genome-wide “drop-out” screens, which can identify cancer drivers and context-dependent events. Several groups have performed shRNA screens (Cheung et al., 2011; Marcotte et al., 2012), but most surveyed relatively few cell lines of the same cancer type, and none represented the diversity of neoplasms such as breast cancer. Here we report the results of genome-wide shRNA screens of >75 breast cancer lines with genomic, transcriptomic and proteomic annotation. Employing an improved statistical framework (siMEM), we provide an integrated map of subtype- and context-dependent essentiality in breast cancer cells.
Results
Breast cancer lines are reasonable models
We performed genomic and proteomic analysis on 78 breast cancer and 4 immortalized mammary cell lines (Table S1A). Copy number abnormalities (CNAs) were similar (r=0.7) in lines and breast tumors, with all major CNAs represented (Figures 1A, S1A). RNAseq and non-negative matrix factorization (NMF) yielded seven clusters (Figure 1B, S1B). Compared with the Neve classification (Neve et al., 2006), we found four basal, two luminal/HER2-, and one mixed cluster(s). The extra basal clusters mainly sub-divided the basal A and B subtypes (Figures 1B, S1C), and resembled the additional subgroups seen in an extensive survey of TNBC (Lehmann et al., 2011). Most luminal/HER2 cell lines fell into Clusters 6 and 7, which were distinguished by ERBB2 and ESR1 expression, respectively. The NMF clusters also related to specific METABRIC “iClusters” (Curtis et al., 2012). Every iCluster was present in the panel, although iClusters 2 and 7 each were represented by less than five lines (Figure S1C). Lines defined as “basal” by PAM50 generally fell into our basal clusters and those of Lehmann (Lehmann et al., 2011), but PAM50-derived signatures did not place luminal/HER2 lines into subgroups similar to those seen by NMF or the Curtis classification.
Figure 1.
Genomic/proteomic characterization. A) CNA profiles of breast tumors (top) from TCGA and cell lines (bottom). B) NMF clustering of RNAseq data for breast cancer lines. ESR1 (ER), ERBB2 (HER2), PGR (PR), and AR (AR) expression are represented by black squares. Lines were assigned to published subtypes (colored boxes). C) NMF clustering of RPPA data. D) Frequency of indicated mutations in cell lines and tumors, grouped into basal and luminal/HER2 subtypes. Tumor data are from COSMIC. Also see Figure S1 and Table S1.
The top 50% variable proteins by reverse-phase protein array (RPPA) formed 9 clusters by NMF (Figures 1C, S1D). With few exceptions, RPPA-(R) and RNA clusters differed markedly. Most (13/18) HER2+ lines fell into R-Cluster 9. R-Cluster 8 consisted mainly of expression-derived Cluster 7 lines and was driven by ERα, GATA3, and BCL2. Two small R-clusters were enriched for luminal/HER2 lines: R-Cluster 3 was mainly ER−/AR+ and featured high p-AKT (pT308 and pS473) and p–AMPKα (pT172). R-Cluster 7 (3 lines) was distinguished by high G6PD, p-4EBP, and reactivity to a VHL antibody that cross-reacts with Epiplakin. The other R-clusters were enriched for basal lines. R-Cluster-1 contained three of the four “normal breast” lines, and was driven by NDRG1, MYC, TAZ, and p-YAP. R-Cluster 2 also had high NDRG1, MYC, TAZ and p-YAP, as well as high PAI-1 and phospho- and total EGFR (Table S1B). R-Cluster-4, the largest, was a default basal cluster.
Exome sequencing of genes mutated in ≥3% of breast tumors in COSMIC and TCGA (Table S1C) showed that all frequent somatic mutations in breast cancer were found in our cell line panel. TP53 and PIK3CA mutations (23% and 26%, respectively, in tumors) were seen in 63% and 33% of lines, respectively. TP53 is mutated more often in TNBC/basal tumors (80% vs. 26%, TCGA 2012), but its mutation frequency was similar in basal and luminal/HER2 lines. For most genes, mutation rate and distribution were comparable in tumors and lines (Figure 1D).
We also profiled miRNAs by NanoString. ERα is the major determinant of miRNA levels in breast tumors (Dvinge et al., 2013; Riaz et al., 2013). Similarly, unsupervised clustering revealed three miRNA groups in cell lines, two basal and one luminal (Figure S1E). Overall, we conclude that a sufficiently large cell line panel represents the genomic and proteomic landscape of breast tumors and provides a reasonable template for identifying context-dependent essential genes.
Improved prediction of gene essentiality
To identify genes required for proliferation/survival (“essentials”), we used pooled lentiviral shRNA dropout screens (Marcotte et al., 2012). Nearly all (77/82) lines gave satisfactory data (Table S1A). Using our earlier metric, zGARP, we scored 402 genes as essential in at least 50% of lines (Table S2A). These included most (261/297 and 218/291, respectively) genes defined earlier as “general essential” or “core essential” in ovarian, pancreas, and selected breast cancer lines (Hart et al., 2014; Marcotte et al., 2012). Not surprisingly, genes annotated as having “housekeeping” roles (e.g., translation, splicing, proteasome, cell cycle) were prominent general essentials (Table S2B).
By contrast, neither zGARP, nor other algorithms [ATARIS (Shao et al., 2013), RIGER (Barbie et al., 2009), RSA (Konig et al., 2007)], identified known subtype-specific essential genes from our large dataset. Such methods summarize replicate shRNA measurements into single “hairpin” or “gene” scores, which are compared between subtypes by t tests or similar statistics. This approach leads to loss of information about measurement variance, limiting statistical power to detect biological differences.
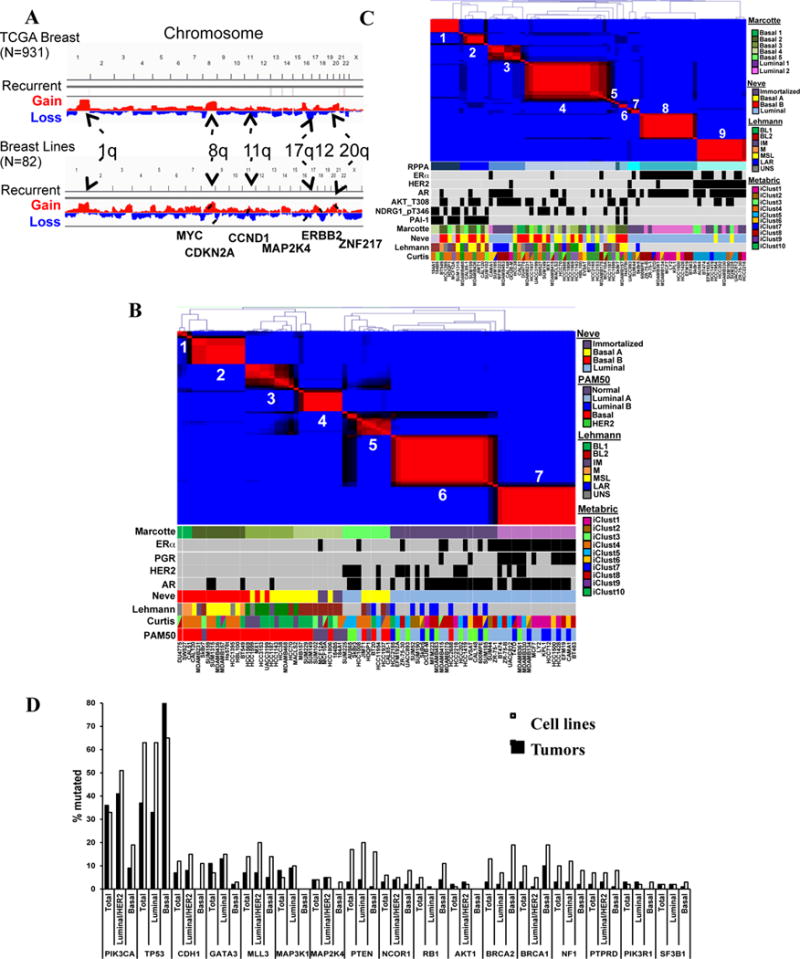
Hierarchical (“mixed-effect”) linear models allow systematic measurement effects, such as hairpin differences or heterogeneous genetic contexts, to be specified and used in significance calculations. Such a model could increase sensitivity for detecting biological differences in screens by avoiding information loss, while limiting false positives. We therefore developed the si/shRNA Mixed-Effect Model (siMEM), which considers the level of each shRNA to be a regression function of its initial abundance, baseline trend in abundance over time, and difference in abundance trend between samples sharing a common feature (Figure 2A, S2A–B and Extended Experimental Procedures).
Figure 2.
siMEM overview. A) Experimental scheme. Samples were hybridized to microarrays and dropout was quantified. Hierarchical linear regression summarizes data as a combination of initial measurement intensity, baseline trend, and difference in essentiality associated with changes in a genomic covariate (light blue vs. dark blue). B) Volcano plot of zGARP (left) and siMEM (right) essentiality differences associated with HER2+ lines. Dotted lines show FDR cutoff. C) siMEM produces the best p-values for known positives. D) BRAF, PIK3CA or KRAS mutant vs. normal, and expression vs. essentiality analyses of the Achilles dataset (N=102). Also see Figure S2, Table S2 and Extended Experimental Procedures.
Using siMEM and previous metrics, we sought genes selectively required in HER2+ lines (N=17). Reassuringly, siMEM detected known HER2+-associated essentials (“known positives”), such as ERBB2, its dimerization partner ERBB3, PI3K/mTOR pathway members (PIK3CA, AKT1/2, RHEB, MTOR), CDC37 (encodes an ERBB2 co-chaperone), and two transcription factors (TFAP2C, YBX1) in the HER2 (ERBB2) pathway. Almost none of these survived false discovery rate (FDR) correction using GARP or ATARIS (Figure 2B; Table S2C). Only siMEM predicted “known positives” from the data in our earlier screen (Marcotte et al., 2012), and it greatly improved their prediction rankings and p-values (Figure 2C, S2C). When classes (normal/HER2+) were shuffled randomly for each gene, siMEM p-values were close to the expected uniform distribution (Figure S2D). Regression structures that ignored systematic measurement effects produced many (incorrectly) significant p-values (Figures S2E–F). By contrast, siMEM produced the best fit and ranking of known positives (Figure S2B, G–H). Finally, we applied siMEM and ATARIS to the “Achilles” dataset (Cheung et al., 2011): siMEM was better at predicting BRAF, KRAS, or PIK3CA essentiality in cognate mutant cells and in finding genes more essential with increased expression, which are enriched for drivers (Figure 2D; also see below).
Breast- and subtype-specific essential genes
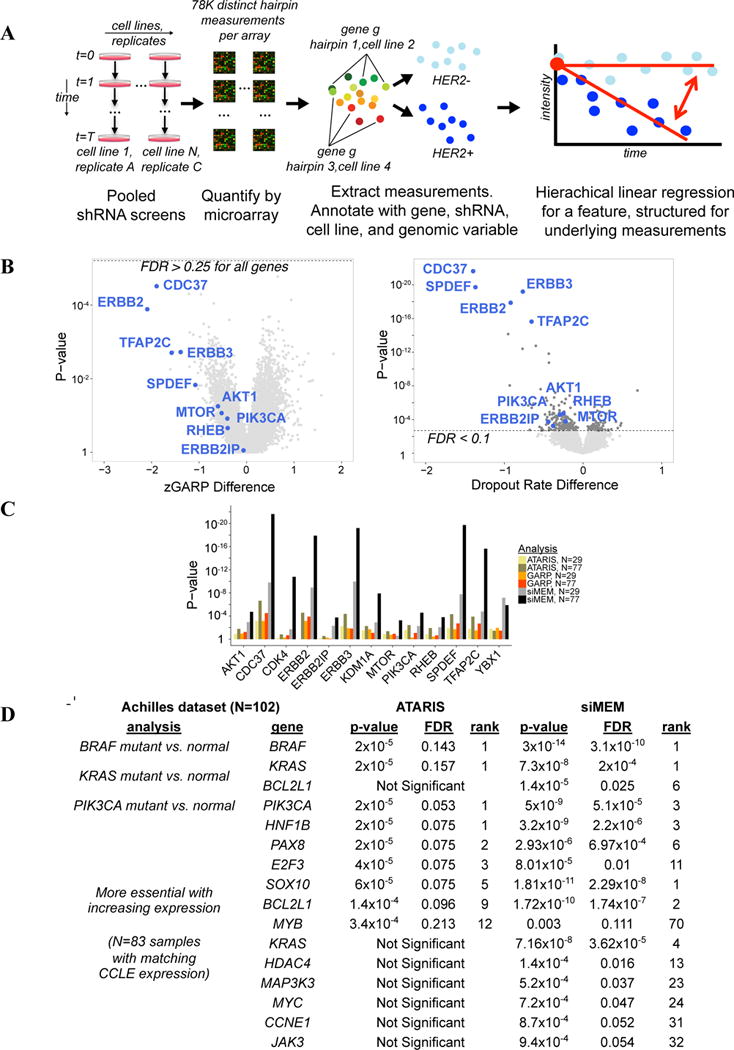
We focus here on gene essentiality relative to the Neve classification, which most closely resembles clinical subtypes, but Tables S3A–G provide essentiality data for each subtype in Figures 1B–C. Comparing basal with luminal/HER2 cell lines, we found 975 and 985 subtype-specific essentials (FDR < 0.1), respectively (Figure 3A, Table S3F–G). The top luminal/HER2-essentials were FOXA1, a pioneer factor for ERα (Lupien et al., 2008), SPDEF, which promotes luminal differentiation and survival of ERα + cells (Buchwalter et al., 2013), CDK4 and CCND1, which form a complex targeted by Palbociclib in ER+ breast cancer (Dhillon, 2015), and TFAP2C, which directs ERBB2 expression (Bosher et al., 1995). Other “expected” luminal/HER2-essential genes included PI3K/mTOR pathway components (PIK3CA, PDPK1, AKT1/2, RHEB, MTOR) and ER-interacting proteins/co-activators (KMT2D, EP300, GATA3, KDM1A, DNM1L, NCOA2).
Figure 3.
Subtype-specific essential genes. A) Volcano plot of basal- and luminal/HER2-specific essentials. B) Heat map shows % proliferation-inhibition, compared to general essential RPL9 (100% inhibition), after pooled siRNA treatment (p-values: 1-sided t test). C) Knockdown efficiency (by q-RT-PCR) of siRNAs for genes in (B). D) Subtype-specific pathways. Each node represents a process; functionally similar nodes are grouped and labeled by enriched function. Nodes are colored according to the subtype in which the process is enriched; processes enriched in more than one subtype have multiple colors. Red: basal B; Orange: basal A; Green: HER2+; Blue: luminal. E) PPI networks for subtype-specific genes. Nodes represent genes, and are multi-colored if present in multiple subtypes; edges represent interactions. Also see Figure S3 and Table S3.
The top basal-selective essentials, PSMB3 and PSMA6, encode proteasome subunits (Table S3F), a dependency seen earlier (Petrocca et al., 2013). The next most essential basal-specific gene was ATP6V1B2, which encodes a component of the vacuolar ATPase required for lysosomal acidification that is the target of Bafilomycin A1 (BafA1). Notably, basal lines were 5-fold, and basal A lines 7-fold, more sensitive to BafA1 than luminal/HER2 lines (Figure S3AB). Other genes reputedly more important in basal breast cancer scored as “basal-essential,” including PLK1, EGFR, FZD7, SLC7A11, CTNNB1, LRP5, FZD8, and TWIST2 (Jamdade et al., 2015; Maire et al., 2013; Timmerman et al., 2013), but we also saw other potential vulnerabilities (Table S3F).
We selected several subtype-specific genes for orthogonal testing with siRNAs. Multiple basal-, luminal-, and HER2-specific genes validated and demonstrated the predicted subtype preference (Figure 3B, Table S3L). Overall, the validation rate was ~70%, with most siRNAs showing >80% knockdown (Figure 3C and data not shown).
The genomics of basal breast cancer and high-grade serous ovarian cancer (HGSC) are very similar (TCGA, 2011, 2012). Remarkably, in a pairwise comparison with luminal-specific (this screen) or HGSC- or pancreatic cancer-specific essentials (Marcotte et al., 2012), only 20 essential genes differed between basal breast cancer and HGSC. By contrast, thousands of differences were seen in all other comparisons (Figure S3C).
We analyzed subtype-specific essential gene sets for preferred pathways and protein-protein interactions (PPIs) (Figure 3D–E; Table S3H–K). As expected, HER2-specific essential pathways included EGF, PI3K, and mTOR signalling. Other functions important in this subtype included regulation of eIF2, aerobic ATP synthesis/TCA cycle, chromatin-modifying enzymes, “response to gamma radiation” (including YAP1, ATR, and ATM), as well as an EP300/BRCA1 PPI sub-network (Table S3J). EP300 is a BRCA1 co-activator (Pao et al., 2000), and BRCA1 is phosphorylated via the PI3K/AKT pathway, which also is required in HER2+ lines (Figure 2C, Tables S2C, S3B). Notably, ATM is essential for HER2+ tumors (Stagni et al., 2015), and it also phosphorylates BRCA1 (Cortez et al., 1999; Gatei et al., 2000). Preferential sensitivity to loss of DNA damage sensors might explain the observed synergy of chemotherapy and Trastuzumab.
Top enriched pathways and PPIs for basal A lines were dominated by genes for splicing, the proteasome and mitosis (Figures 3D–E; Table S3H). Other required functions included the COP9 signalosome (CSN) and a PPI sub-network defined by CAND1/NEDD8 (Figure 3E). CSN and CAND1/NEDD8 regulate SKP1/CUL1/F-box (SCF) complexes (Flick and Kaiser, 2013). While the core SKP1/CUL1 complex showed no subtype specificity, several F-box genes were selectively essential in basal A lines, including FBXW11/β-TrCP2 (Table S3B). FBXL6 and FBXO15 were more essential in basal B or luminal/HER2 lines, respectively (Table S3B and data not shown). Hence, F-box proteins might impart subtype-specific functions to SKP1/CUL1.
Lack of functional annotation (<50% of genes annotated) resulted in a relative paucity of basal B and luminal nodes when compared to basal A- and HER2- nodes (>65% of genes annotated, Figure S3D–F). Nevertheless, essential pathways and PPI networks for luminal lines included epithelial development, MDM2, PI3K and hormone receptor (ESR1) signalling (Figures 3D–E). The latter two are targets of known drugs for luminal breast cancer. Less expected “luminal-enriched” pathways/PPIs included redox-related (SOD1, SOD2, ENOX1) and mitochondrial (e.g., electron transport chain, mitochondrial ribosome) proteins. By contrast, basal B-essentials were enriched for genes related to polarity (PARD3, PAR3D), cell-cell junctions and adhesion (CDH2, CLDN1, CLDN4, ITGA4, ITGAV, ITGB5), embryonic development, organ morphogenesis, fatty acid metabolism, and T-cell immunity (Figures 3D–E). Some of these genes, such as SOX9 (Guo et al., 2012a), KLF4 (Yu et al., 2011), and ALOX5AP (Kim et al., 2005), have reported roles in breast cancer, although not specifically in basal B tumors.
Cis- and trans-essential interactions with common CNAs
There are hundreds of CNAs in breast cancer (Curtis et al., 2012; TCGA, 2012), yet for most, the key driver gene(s) is unclear. METABRIC defines 30 regions of copy number gain and 15 deletions (Curtis et al., 2012). ISAR, being more sensitive for small amplicons, identifies 83 recurrent CNAs (Sanchez-Garcia et al., 2014). We predicted significant (FDR<0.2) cis-essential genes (more essential in amplicon+ lines) for 9/83 ISAR regions. Four corresponded to genes in a METABRIC amplicon (Figure S4A and Table S4A): EGFR (ISAR(I)-34/METABRIC(M)-10), CCND1 (I-52/M-21), ERBB2 (I-70/M-35), and TFAP2C (I-81/M-42). The others were unique to ISAR-defined regions (Table S4B): CTSS (I-6), ESR1 (I-30), RALGAPA1 (I-62), FOXA1 (I-63), and BCL2 (I-76).
Even for known drivers (or for deletions), targeting the key gene can be difficult. “Trans-essential” genes can suggest alternative strategies. Combining all METABRIC regions, we identified 2,560 trans-essentials, an average of 58 per CNA (range 0–285; Figures 4A and S4A; Table S4A). Only 61 (~3%) trans-essentials showed significantly increased or decreased expression in sensitive lines (Figure S4B and Extended Experimental Procedures); hence, most would not be found by gene expression surveys. Expected trans-essentials were seen for the CCND1 (CDK4, USP18) (Guo et al., 2012b) and ERBB2 (ERBB3, CDC37, PIK3CA) amplicons and for CDKN2A deletions (CCND1, CDK6) (Figure 2B, S4A and Table S4A). It can be difficult to know if a trans-essential is “expected” for deletions, especially if the cognate tumor suppressor is undefined. Even so, we saw intriguing associations with “druggable” targets for region 27, containing RB1 (more sensitive to MAP2K2 depletion), region 11 (more sensitive to TLK2, BRD4, or ACVR1B depletion), and region 40 (more sensitive to PTK6 or MAP2K4 depletion) (Table S4A).
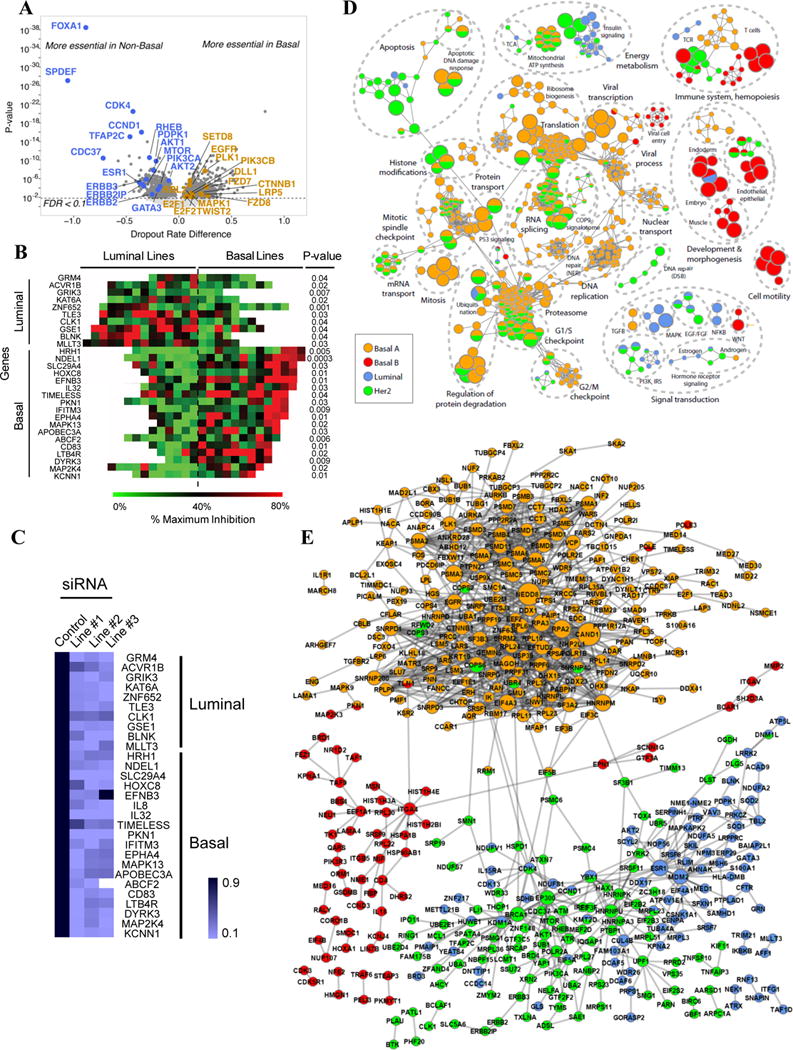
Figure 4.
cis- and trans-essential genes for CNAs. A) Heat map showing 8q24 amplification (METABRIC-14, containing MYC) in cell lines. Red=amplification, blue=deletion. Bar graph shows average zGARP score for genes in the amplified region in amplicon+ lines. CIRCOS plot depicts top 20 significant genes (by siMEM) in amplicon+ vs –amplicon− cells. B) GSEA of trans-essential genes for MYC targets (FDR < 0.0001). C) Validation of 8q24 trans-essential genes with siRNAs. Y-axis: % maximum inhibition Bar graphs: Knockdown efficiency (by qRT-PCR) of siRNAs. D) Correlation between published HELIOS scores (Y-axis) (Sanchez-Garcia et al. 2014) and new scores (X-axis) obtained using our screen data. Circled genes deviate from earlier score and represent potential new amplified drivers. E) Validation of HELIOS genes with siRNAs. Y-axis: % maximum inhibition Bar graphs: Knockdown efficiency of siRNAs. p-values were calculated by 1-sided t test. Also see Figure S4 and Table S4.
METABRIC region 14 includes MYC, which is generally deemed “undruggable”. Notably, MYC was the most essential gene in region 14-amplified lines (Figure 4A), but was not differentially essential by FDR, probably because of its requirement in most tumor cells (Dang, 2012). Pathway analysis of the 91 region 14 trans-essentials (FDR < 0.2; Table S4A) revealed genes for mitosis, DNA replication and RNA metabolism (Table S4C), all known MYC functions (Dang 2012). MYC transcriptional targets (Figure 4B) and genes encoding MYC-interacting proteins (Table S4D) also were strongly enriched: 46% of MYC trans-essential genes were MYC transcriptional targets/interactors. We tested two MYC trans-essentials potentially amenable to drug discovery; indeed, amplified lines were preferentially sensitive to MINK1 or USP5 depletion (Figure 4C). We also validated YAP1 and BRCA1 as trans-essential for METABRIC regions 35 (contains ERBB2), and 36 (putative driver: ZNF652), respectively (Figures S4C–D).
HELIOS integrates CNA, expression, mutation, and essentiality into a single score that predicts cis-essential genes (Sanchez-Garcia et al., 2014). The initial HELIOS report, using data from our earlier screen, identified and validated 10 potential drivers. Using our expanded dataset, the HELIOS score increased for most known drivers and previously validated genes (Figure 4D and Table S4E). We also tested two new predictions and found that amplicon+ lines were more sensitive to siRNA-mediated depletion (Figure 4E).
Functional genomic clustering reveals groups not captured by expression profiling
Using NMF clustering, we grouped lines based on shared dependencies (“functional genomic clustering”; (Marcotte et al., 2012)). Six “functional clusters” (fClusters) were observed, two containing lines designated as basal by expression profiling (fCluster-4, -5), two luminal/HER2 clusters (fCluster-2, -6), and two (fCluster-1, -3) comprising a mix of basal and luminal/HER2 lines (Figure 5A). Thus, as we saw earlier (Marcotte et al., 2012), “basal” and “luminal/HER2” lines have distinct patterns of gene dependency. Yet while there was little additional separation in our earlier study, with our expanded panel, HER2 (mainly fCluster-2) and ER+ (fCluster-6) lines largely segregated into distinct fClusters. Genes determining the ER+ (fCluster-6), HER2+ (fCluster-2), and basal (fCluster-4) clusters (Table S5A) overlapped substantially with luminal-, HER2- or basal- essential genes, respectively (Figure 3). fCluster-1 was enriched for genes curated as H3K27-trimethylated, neuroactive peptides or as involved in cytokine-cytokine interactions. fCluster-3 was enriched for annotations for cell cycle (G1/S and mitosis), DNA replication, and immune system genes, whereas fCluster-5 was enriched for genes involved in the immune system, lipid metabolism and NGF signaling (Table S5A).
Figure 5.
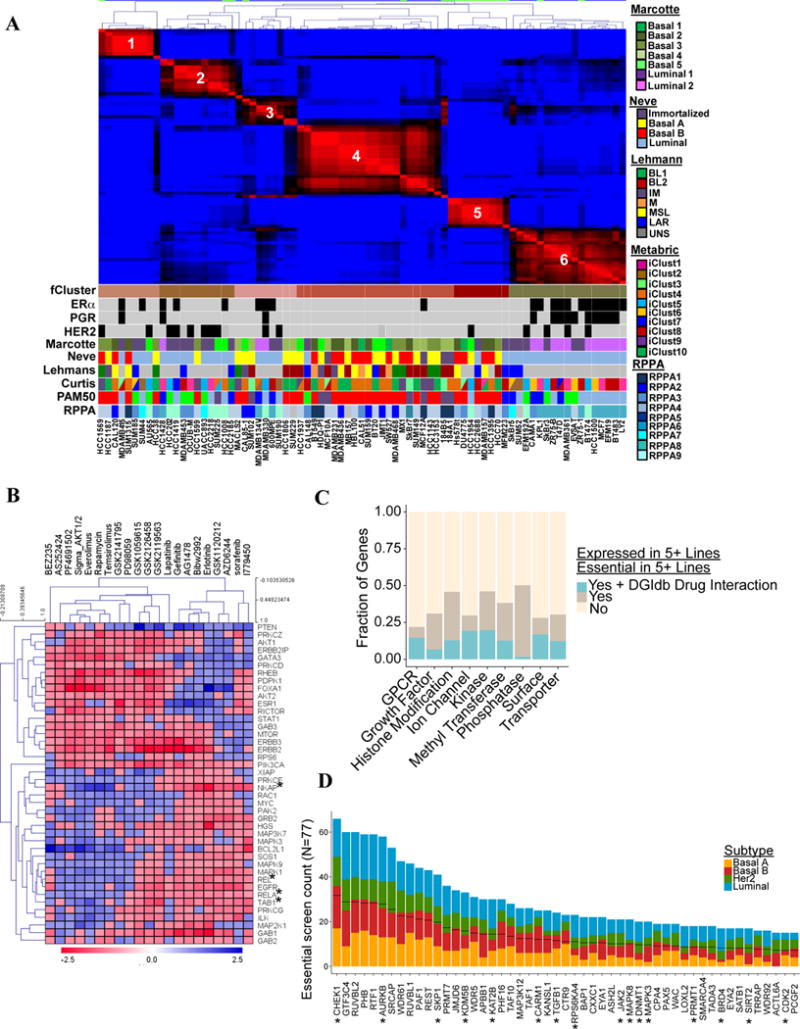
Screen refines classification and pathway identification. A) NMF clustering of screen results (zGARP). ESR1, ERBB2, and PGR expression are shown by black squares. Colored boxes indicate major published sub-categories. B) Unsupervised analysis of essential genes implicated in PI3K/mTOR or EGFR/MEK/ERK pathways. Heat map shows association of essentiality for each gene (this study) with sensitivity to drugs targeting these pathways (Daemen et al. 2013). C) Fraction of essential genes overlapping with reported “druggable” gene categories or gene-drug interactions (DGIdb). D) Top-ranked histone-modifying enzymes deemed essential in our screen, by breast cancer subtype. *Reported gene-drug interaction in DGIdb. Black lines represent 50% of lines in which the gene is essential. Also see Figure S5 and Table S5.
Drug sensitivity and gene essentiality
We also compared gene essentiality and sensitivity data for 90 drugs tested against 84 breast cancer lines (Daemen et al., 2013), most of which (69) were included in our panel. Using siMEM, we identified genes whose essentiality correlated with sensitivity to mTOR/PI3K/ERBB2/AKT or EGFR/MEK/ERK inhibitors. Hierarchical clustering revealed distinct positive (red) and negative (blue) correlation clusters associated with drug sensitivity (Figure 5B and Extended Experimental Procedures). Reassuringly, genes for PI3K/AKT pathway components were required in lines sensitive to the cognate inhibitors. Sensitivity also correlated with essentiality of the luminal markers ESR1, FOXA1, and GATA3, consistent with the known sensitivity of luminal tumors to these agents. Likewise, EGFR/MEK/ERK inhibitor response correlated with sensitivity to EGFR, GRB2, SOS1, MAPK1, MAPK3, or MAP2K1 depletion. Interestingly, response to EGFR/MEK/ERK inhibitors correlated with dependence on the NF-kB pathway: RELA, REL, and NKAP were more essential in such cells. These results comport with reports of NF-kB activation in response to EGFR, RAS, RAF, or MEK activation (Pan and Lin, 2013), and suggest that NF-kB inhibitors might be effective in basal breast cancer.
Drug sensitivity/essentiality comparisons also identified negative regulatory/tumor suppressor pathways. For example, PTEN was more essential in lines that were insensitive to mTOR/PI3K/ERBB2/AKT or EGFR/MEK/ERK inhibitors, consistent with the effects of PTEN deletion/inactivation (Worby and Dixon, 2014). Likewise, MDM2 and TP53 essentiality were associated with sensitivity or resistance to MDM2 and Nutlin-3A treatment, respectively.
Unsupervised analysis of the whole gene essentiality/drug sensitivity dataset revealed 5 clusters. Most drugs with a similar mechanism of action fell into the same cluster, and pathway analysis confirmed that essentiality clusters were enriched for genes implicated in the pathways targeted by their respective agents (Figure S5A, Table S5B–C). Unanticipated clusters also emerged. For example, sensitivity to 11 drugs, which included alkylating agents, topoisomerase inhibitors, and cell cycle/cell cycle checkpoint inhibitors, correlated with essentiality of genes “associated with the H3K27me3 mark” (e.g., PRDM13, NKX2-5, HOXC8, PAX7, HES2) and for “neuropeptides and neurotransmitter signalling” (Figure S5B, box #2, S5C–D). Notably, we had validated one of these genes, HOXC8, in our siRNA assays (Figures 3B–C).
Screen/drug sensitivity data might suggest drug combinations to kill resistant cells and/or negative regulators associated with drug resistance. For example, drugs targeting the PI3K/mTOR pathway (Cluster-1) strongly anti-correlated with BCL2L1 essentiality (i.e., cell lines resistant to PI3K/mTOR inhibitors required BCL2L1). Interestingly, drug combinations targeting the PI3K/mTOR pathway and BCL-XL are reported for several malignancies (Muranen et al., 2012; Rahmani et al., 2013). Another known combination predicted by our data is EGFR plus HDAC inhibitors (Zhang et al., 2015). Suggested combinations awaiting validation include RAF/MEK and CDK4 inhibitors, EGFR inhibitors with Cluster-5 drugs, BET-Is with Cluster-4 drugs, especially epirubicin and vinorelbine, PLK1 inhibitors with Nutlin-3A or PI3K/AKT inhibitors or Nutlin-3A with Cluster-5 drugs (Table S5B).
We also used DGIdb to identify essential genes that are potentially “druggable” (Griffith et al., 2013). Genes for kinases, phosphatases, and histone modifying enzymes were the most frequently essential, although other categories were represented (Figure 5C, Table S5D). Inhibitors exist for only a small fraction of most potential targets, especially the histone modifiers; a larger percentage of essential kinases had a known inhibitor (Figures 5C–D, S5E).
Additional functional genomic properties of cancer cells
For most genes, essentiality decreased as expression increased (Figure 6A, right); such genes are enriched for housekeeping functions (Table S6A). A smaller set of genes became more essential with increased expression (Figure 6A, left): 16 of the 20 top-ranked genes in this group are known drivers in breast or other cancers (Table S6B). We suspected that other genes whose essentiality increased with increased expression might be drivers, and tested several using siRNAs (Figures 6B, S6A). Indeed, 11/20 (55%) were more essential in lines with increased expression (R>0.3). Genes more essential with increased expression showed lower expression overall than genes whose essentiality lessened with increased expression (Figure S6B). The former were more variably expressed, though, consistent with the behavior of known oncogenes (e.g., ESR1, ERBB2).
Figure 6.
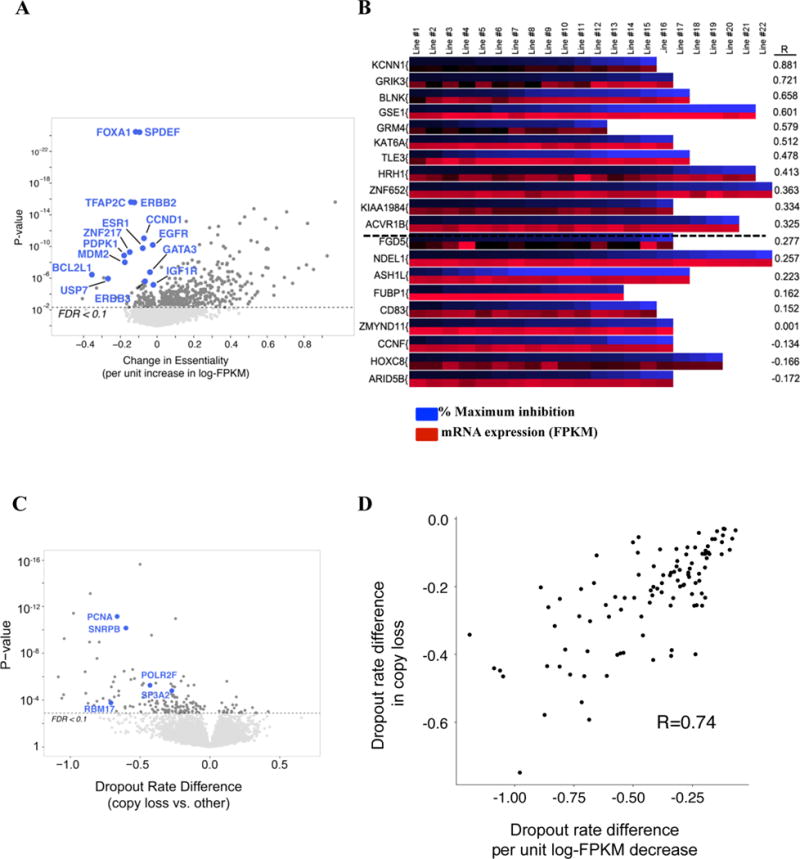
Additional features of shRNA screens. A) Volcano plot of relationship between essentiality and gene expression. X-axis: change in dropout rate per unit increase in expression log-FPKM; Y-axis: p-value. B) Heat map showing % inhibition of proliferation following knockdown by siRNA in cell lines. For each gene, the upper row (blue) represents maximum growth inhibition, while the lower row (red) represents mRNA levels of the same gene in each line. R=Pearson correlation. C) Vulnerabilities associated with genomic loss (CYCLOPS genes). D) Strong agreement (Spearman rho=0.74, p-value < 2.2 × 10−16) between genes more essential with heterozygous loss (FDR < 0.25) and genes whose essentiality changes significantly with expression (FDR < 0.25). Also see Figure S6 and Table S6.
“CYCLOPS” (Nijhawan et al., 2012) and “GO” (Solimini et al., 2012) genes show increased essentiality upon heterozygous deletion of their cognate genomic regions. We identified 224 genes (FDR < 0.2) that were more essential with copy number loss (Figure 6C, Table S6C); their essentiality also correlated strongly with decrease in their expression (Figure 6D; Spearman rho=0.74). These genes overlapped significantly with CYCLOPS and GO genes, only five showed homozygous deletion in any line, and their protein products were enriched for housekeeping functions (Figure S6C, Table S6C and Supplemental Experimental Procedures). Thus, our data validate the CYCLOPS/GO concept and provide many other candidate members of this class of genes.
PIK3CA mutations drive resistance to BET-I
BRD4, encoding a BET bromodomain-containing co-activator (Shi and Vakoc, 2014), was preferentially essential in luminal/HER2 lines (Figure 7A, Table S3G). Moreover, luminal/HER2 lines were more sensitive to BRD4 depletion by siRNAs (Figure 7B, S7B), and expression of shRNA-resistant BRD4 cDNA abrogated inhibition by BRD4 shRNA (Figure S7C).
Figure 7.
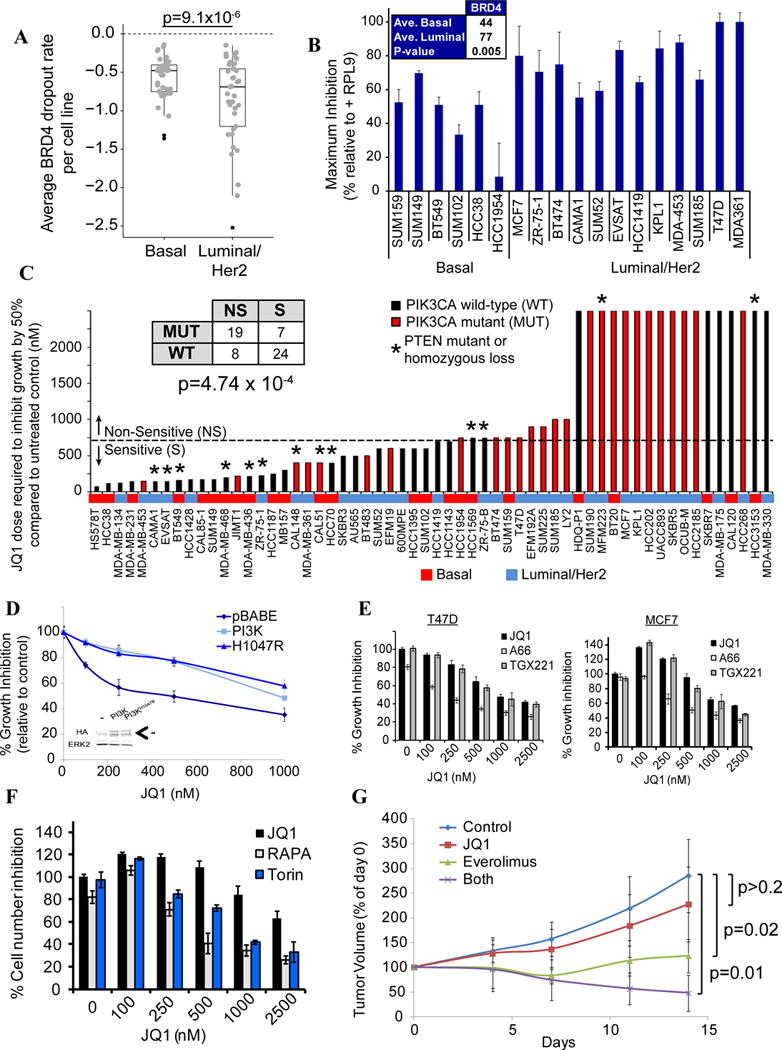
BRD4 is luminal-essential, and PIK3CA mutations cause BET-I resistance. A) Box plot showing BRD4 dropout in each line, by subtype. B) BRD4 siRNAs confirm pooled screen results. Averages are maximum percent inhibition (p=0.005, Student’s t-test). C) Effect of JQ1 on breast cancer lines. Table (inset) shows number of lines, grouped by JQ1 sensitivity (NS=non-sensitive, S=sensitive) and PIK3CA status (mut=mutated, wt=wild-type). Red shading shows lines with PIK3CA mutations. Mutant lines were more likely to be JQ1-resistant (p < 4.7×10−4, chi-square test). Sensitive lines have GI50 < 750nM. *Lines with PTEN mutation/homozygous deletion. D) WT or mutant PIK3CA (H1047R) render JQ1-sensitive SkBr3 line resistant to JQ1. Inset: Immunoblot showing expression of PIK3CA-p110α. Arrow indicates the specific band. E) JQ1 cooperates with PIK3CA (A66; 1 μM), but not with PIK3CB (TGX; 1μM) inhibitors to decrease MCF7 and T47D proliferation. “0” JQ1 represents A66 or TGX alone. F) JQ1 cooperates with mTOR inhibitors (Rapamycin; 0.5nM, Torin; 50nM) to decrease MCF7 proliferation. “0” represents Rapamycin or Torin alone. G) JQ1 and Everolimus cooperatively inhibit xenograft growth. MCF7 cells (2 × 106) were injected into mammary fat pads of athymic nude mice bearing a slow release estrogen pellet. When tumors were 5×5mm (~21 days), mice were grouped into: 1) control, 2) JQ1 (50mg/kg/day IP), 3) Everolimus (5mg/kg/day by gavage), and 4) JQ1+Everolimus daily. Tumors were measured with calipers every 3–4 days. p value: 1-sided Student’s t-test. Also see Figure S7.
We tested the BET domain inhibitor (BET-I) JQ1 on a subset of our lines, expecting greater sensitivity in luminal/HER2 cells. Cell line GI50s ranged from low nM (<100) to μM (>2.5), with lines that showed high JQ1 sensitivity undergoing apoptosis, while resistant lines had slower cell cycle progression (Figures S7D–F). However, many luminal/HER2 lines sensitive to BRD4 knockdown were JQ1-resistant. By contrast, most basal lines that were sensitive to BRD4 knockdown were JQ1-sensitive (Figure 7C and data not shown). In contrast to previous studies (Shi and Vakoc, 2014), JQ1 sensitivity did not reflect impaired MYC expression: sensitive and resistant cell lines displayed similar decreases in MYC mRNA (Figure S7G), and exogenous MYC did not convert JQ1-sensitive lines to JQ1-resistance (Figure S7H–I).
Instead, integrative analysis revealed a strong correlation between JQ1 resistance and PIK3CA mutation (Figure 7C). Over-expression of wild type or mutant PIK3CA conferred JQ1 resistance on JQ1-sensitive SkBR3 cells (Figure 7D), establishing a causal relationship between PI3K and resistance. Moreover, A66, a PIK3CA-specific inhibitor, but not TGX-221 (PIK3CB-specific), increased the JQ1 sensitivity of resistant cells, as did the mTOR inhibitors rapamycin or Torin (Figures 7E–F). The one basal line (SUM159) sensitive to BRD4 depletion but JQ1-resistant also has a PIK3CA mutation, and PIK3CA inhibitor treatment sensitized these cells to JQ1 (Figure S7J). Finally, combining JQ1 and Everolimus enhanced their respective anti-tumor effects (Figure 7G). In concert, these data indicate that BRD4 has bromodomain (BrD)-dependent and –independent effects in breast cancer cells, and establish PIK3CA mutations as a BET-I resistance mechanism.
Discussion
Most dropout screens analyze relatively few lines of any single cancer histotype. By constrast, we provide gene essentiality data for a large set of breast cancer lines with genomic, proteomic, and drug response annotation, and an analytic tool, siMEM, that more precisely measures differential essentiality. Our results identify and provide initial validation of synthetic lethal relationships with expression subtypes and CNAs, yield insight into essential pathways that correlate with anti-cancer drug response, and reveal general features of functional genomic screens. Illustrating the utility of combining genomic/functional genomic data, we identify and validate BRD4 as a luminal/HER2-selective essential gene, uncover BET-independent requirements for BRD4 in luminal/HER2 cells, and reveal PIK3CA mutations as a potential resistance mechanism to BET-Is in vitro and in vivo.
The breadth of our screen has several advantages. Many have argued that breast cancer lines only partly reflect tumor heterogeneity (Hollestelle et al., 2010; Kao et al., 2009; Neve et al., 2006). But there are at least ten breast cancer subtypes (Curtis et al., 2012; Lehmann et al., 2011; TCGA, 2012); only a large panel could possibly represent such heterogeneity (Figures 1, S1). Our screen identified nearly all known breast cancer drivers, linked to the appropriate biomarker (Figures 3A, 4A, S4A). The increased power of our dataset also revises the identification of putative targets of some breast cancer amplicons and strengthens the identification of others by HELIOS (Figure 4D; Table S4E). Thus, if enough cell lines are tested, they provide valid surrogates for probing core cancer cell properties, such as proliferation/survival.
Conventional algorithms for sh/siRNA screens generate hairpin- and/or gene-level scores that summarize multiple measurements, and fail to identify known differential essential genes. By contrast, siMEM greatly improves detection of essentials associated with CNAs, gene expression, somatic mutations, or cancer subtype without increasing the false positive rate. “Hits” suggested by siMEM have a high validation rate (~60–70%) (Figures 3B, 6B), and an analogous approach can be applied to any pooled screen (e.g., CRISPR/Cas9 screens).
Our screen identified “general” and “context-specific” essentials. As expected, general essentials are enriched for housekeeping functions, yet some show a gradient of essentiality tied to specific genetic parameters. For example, specific splicing factors (data not shown, but see (Hsu et al., 2015)) and proteasome genes are preferentially required in basal lines (Table S3F). A splicing inhibitor is in clinical trials (E7107; NCT00459823), and several proteasome inhibitors are approved drugs (Dou and Zonder, 2014) and could be repurposed for breast cancer therapy.
Our data provide strong confirmation of earlier work suggesting distinct subtype-specific vulnerabilities. The pivotal roles of hormone receptors in luminal breast cancer, of ERBB2 signaling in HER2+ disease, and of EGFR and WNT signalling in basal breast cancer are confirmed by our screen hits (Figures 3A, 3D–E, Table S3F–G). We also identify several “druggable” targets, including EFNB3/EPHA4, MAP2K4, MAPK13 and IL32, for basal breast cancer, the most lethal form of the disease. How these genes promote basal breast cancer is unclear. EFNB3/EPHA4 are a ligand/receptor pair that promotes neuronal proliferation and survival (Furne et al., 2009; Takemoto et al., 2002). MAP2K4 phosphorylates and activates MAPK13 (O’Callaghan et al., 2014); MAPK13 and IL32 are linked to IL-1 signaling (Netea et al., 2005; Yousif et al., 2013), which also is basal-specific in our screen. Basal A cells are preferentially susceptible to CAND1-NEDD8 depletion. A NEDD8 inhibitor, MLN4924, is in phase 1 trials (NCT00677170, NCT01862328); TNBC patients might benefit from this agent.
Basal B cell lines are claudin-low-like, represent a unique TNBC subset, and have EMT-, cancer stem cell-, and mammary stem cell-like gene signatures (Lim et al., 2010), like those seen in chemotherapy-resistant cells (Creighton et al., 2009). Basal B lines also showed unique essentialities: basal B-essentials are enriched for motility-, immune-related, developmental and neuronal-, and cell junction and adhesion genes, several of which validate in siRNA experiments (Figure 3B). We also find marked functional similarity between basal breast cancer and HGSC (Figure S3C). Our results and the shared genomics of these tumors (TCGA, 2011, 2012) argue for similar treatment strategies and drug discovery efforts.
Consistent with earlier work (Davoli et al., 2103; Solimini et al., 2012), our results suggest that for many amplicons, multiple genes contribute to increased fitness. For some amplicons, no clear cis-essential gene was identified. Failure to identify such genes might be technical (e.g., insufficient amplicon+ lines). More likely, these amplicons select for multiple weak drivers, miRNAs/lncRNAs, or genes dispensable for proliferation/survival, but mediating other cancer hallmarks. For other amplicons, the key gene(s) cannot be targeted directly, nor can deleted tumor suppressor genes be restored. “Trans-essentials” provide insight into pathways perturbed by CNAs and can suggest more tractable drug targets. For example, METABRIC region 14, containing MYC, confers dependency on a MYC-regulated functional network. Two genes in this network, MINK1 and USP5, are potential drug targets and validate by siRNA. Potentially druggable trans-associations also exist for common deletions: e.g., RB1-deleted lines are more sensitive to MAP2K2 depletion, whereas CDKN2A-deleted lines rely more on KAT6B, ADRBK1, SYK, and DNMT3A.
As expected, genes encoding targets of known anti-cancer drugs are more essential in lines sensitive to those agents. But other genes, without known or obvious connections to the target pathway, also show essentiality strongly correlated with specific drug sensitivity. Also, gene essentiality can anti-correlate with drug sensitivity. Such genes might mediate therapy resistance, and suggest potential combination strategies.
BRD4 was implicated in cancer by studies of NUT midline carcinoma, which often harbors a BRD4-NUT translocation (French et al., 2003). Subsequently, BRD4 emerged as a potential target for many other neoplasms (Shi and Vakoc, 2014). We identified BRD4 as more essential in luminal/HER2 lines (Figures 7A–B, Table S3G). In hematologic malignancies, BET-I sensitivity correlates with MYC down-regulation and is antagonized by exogenous MYC expression (Shi and Vakoc, 2014). Very recently, mouse basal-like breast tumors caused by MYC overexpression and mutant PIK3CA were found to be sensitive to combined BET/PI3K inhibition, as was a human basal line, SUM159 (Stratikopoulos et al., 2015). However, we saw no correlation between JQ1 sensitivity and basal MYC levels or the ability of JQ1 to inhibit MYC expression. Nor does forced MYC expression alter JQ1 sensitivity (Figure S7G–I).
Instead, using our genomic data, we found that PIK3CA mutations are biomarkers of BET-I resistance. Moreover, they are functional biomarkers, as treating cell lines or xenografts with a BET-I/mTOR inhibitor combination improves efficacy (Figure 7F–G). Our results have clear clinical implications, as Everolimus is approved for ER+ breast cancer, and BET-Is are in clinical trials. PIK3CA mutations are most frequent in luminal tumors, so such patients would likely benefit most from BET-I/mTOR-I combinations. But our results and those of Stratikopoulos et al. also suggest a role for BET-Is as single agents in basal tumors. Surprisingly, and for unclear reasons, in basal lines, PTEN mutation/homozygous deletion predicts BET-I sensitivity (Figure 7C and data not shown).
Finally, as breast cancer lines can be JQ1-insensitive, but BRD4-dependent, BRD4 must have (a) BrD-independent function(s). Although the detailed mechanism is unclear, mutant PIK3CA confers JQ1-resistance, so PI3K pathway activation can selectively abrogate BrD-dependent, but not BrD-independent functions of BRD4. Thus, our integrated functional genomic approach not only can suggest new treatment strategies for breast tumor subtypes, but also reveals new features of breast cancer biology.
Experimental Procedures
For additional details and computational methods, see Extended Experimental Procedures.
Cell Lines
Cell lines were from ATCC, Asterands, DZMS or were available in-house (Table S1).
Genomics/Proteomics
SNP-Arrays
Genomic DNA was amplified with the Illumina Infinium Genotyping kit, hybridized to Human Omni-Quad Beadchips, and analyzed on an iScan (Illumina). Data were quantified in GenomeStudio Version 2010.2 (Illumina) using Omni-Quad Multiuse_H manifest (April 2011 release), containing data from GenomeBuild 37, Hg19.
RNA-seq
RNA was reverse transcribed using the Illumina TruSeq Stranded mRNA kit. Libraries were sized (Agilent Bioanalyzer), normalized and pooled (6 each), and loaded onto an Illumina cBot. Paired-end sequencing (50-cycles) was performed on an Illumina HiSeq 2000.
Targeted Sequencing
DNA for 126 genes (1.264Mbp) mutated at ≥3% frequency in breast or ovarian carcinoma was captured using Agilent SureSelect XT, loaded onto the cBot and subjected to paired-end sequencing (100 cycles).
miRNA
miRNA expression was assessed by using the nCounter® Human V2 miRNA Assay Kit (Cat# GXA-MIR2-48) and a NanoString® counter.
RPPA
RPPAs were generated and analyzed as described (Tibes et al., 2006). For all lines, fresh media was added at 80–90% confluency, and cells were harvested 16 hours later.
shRNA/siRNA Experiments
Pooled screens with the TRC-II library were performed as described (Marcotte et al., 2012). HCC712, ZR-75-30, MDA-MB-175VII, UACC812, and UACC3199 failed quality control. For validation, cells (1,000–3,000) seeded in 96-well plates for 24 hr were transfected with Dharmacon SMARTPOOL™ siRNAs (10nM) using Lipofectamine RNAimax (Life Technologies). After 7 days, cells were stained with Alamar blue (Life Technologies), which measures redox activity and is as a surrogate for cell number. Percent maximum inhibition, corrected for transfection efficiency, was determined using siRNAs for the general essential RPL9.
Xenografts
MCF7 cells (5×106) were mixed 1:1 with growth factor-reduced Matrigel (BD Biosciences, NJ, USA), and injected into mammary fat pads of athymic nude mice (Charles River, MA, USA). When tumors were 5×5mm, mice were separated into control and drug-treated groups. JQ1 was synthesized (Filippakopoulos et al., 2010). Everolimus was purchased from Selleckchem.
RNAseq and screen data are deposited in GEO (Accessions GSE73526 and GSE74702). Genomics and proteomics data are available at http://azinsayad.github.io/brca_shrna_data/. All code is available by request from A.S. and siMEM code will be posted at https://github.com/azinsayad/simem.
Supplementary Material
Acknowledgments
This work was supported by the Canadian Foundation for Innovation (J.M.) and the Ontario Research Fund (B.G.N., J.M). B.G.N. was a Canada Research Chair, Tier 1, and work in his laboratory was supported in part by the Princess Margaret Cancer Foundation. R.M. was partly funded by a postdoctoral fellowship from the Canadian Breast Cancer Foundation. This work was also supported by U.S. National Institutes of Health grants R37 CA49132 (B.G.N.), P41 GM103504, R01 GM070743, U41 HG006623 (G.D.B.), and PO1 CA099031 (G.B.M), a Komen SAC and Promise grant (G.B.M), and the M.D. Anderson CCSG functional proteomics core (CA16672).
Footnotes
Author Contributions
R.M., A.S., J.M. and B.G.N. designed the study. R.M. and M.H. performed experiments. G.B.M. performed RPPAs. A.S. designed/implemented siMEM, with input from J.M. and K.R.B. K.R.B., C.V., R.M., and A.S. performed genomic and statistical analyses. J.R. and G.D.B. performed pathway enrichment and PPI analyses. F.S.G. and D.P. implemented HELIOS. J.B. provided JQ1. R.M., A.S., and B.G.N. wrote the paper, with help from all authors.
References
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosher JM, Williams T, Hurst HC. The developmentally regulated transcription factor AP-2 is involved in c-erbB-2 overexpression in human mammary carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:744–747. doi: 10.1073/pnas.92.3.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwalter G, Hickey MM, Cromer A, Selfors LM, Gunawardane RN, Frishman J, Jeselsohn R, Lim E, Chi D, Fu X, et al. PDEF promotes luminal differentiation and acts as a survival factor for ER-positive breast cancer cells. Cancer cell. 2013;23:753–767. doi: 10.1016/j.ccr.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science (New York, NY) 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daemen A, Griffith OL, Heiser LM, Wang NJ, Enache OM, Sanborn Z, Pepin F, Durinck S, Korkola JE, Griffith M, et al. Modeling precision treatment of breast cancer. Genome Biol. 2013;14:R110. doi: 10.1186/gb-2013-14-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S. Palbociclib: first global approval. Drugs. 2015;75:543–551. doi: 10.1007/s40265-015-0379-9. [DOI] [PubMed] [Google Scholar]
- Dou QP, Zonder JA. Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Current cancer drug targets. 2014;14:517–536. doi: 10.2174/1568009614666140804154511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Git A, Graf S, Salmon-Divon M, Curtis C, Sottoriva A, Zhao Y, Hirst M, Armisen J, Miska EA, et al. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature. 2013;497:378–382. doi: 10.1038/nature12108. [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick K, Kaiser P. Set them free: F-box protein exchange by Cand1. Cell research. 2013;23:870–871. doi: 10.1038/cr.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer research. 2003;63:304–307. [PubMed] [Google Scholar]
- Furne C, Ricard J, Cabrera JR, Pays L, Bethea JR, Mehlen P, Liebl DJ. EphrinB3 is an anti-apoptotic ligand that inhibits the dependence receptor functions of EphA4 receptors during adult neurogenesis. Biochimica et biophysica acta. 2009;1793:231–238. doi: 10.1016/j.bbamcr.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatei M, Scott SP, Filippovitch I, Soronika N, Lavin MF, Weber B, Khanna KK. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer research. 2000;60:3299–3304. [PubMed] [Google Scholar]
- Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, Koval J, Das I, Callaway MB, Eldred JM, et al. DGIdb: mining the druggable genome. Nature methods. 2013;10:1209–1210. doi: 10.1038/nmeth.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012a;148:1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Chinyengetere F, Dolinko AV, Lopez-Aguiar A, Lu Y, Galimberti F, Ma T, Feng Q, Sekula D, Freemantle SJ, et al. Evidence for the ubiquitin protease UBP43 as an antineoplastic target. Molecular cancer therapeutics. 2012b;11:1968–1977. doi: 10.1158/1535-7163.MCT-12-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Brown KR, Sircoulomb F, Rottapel R, Moffat J. Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Molecular systems biology. 2014;10:733. doi: 10.15252/msb.20145216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer research. 2009;69:4116–4124. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollestelle A, Nagel JH, Smid M, Lam S, Elstrodt F, Wasielewski M, Ng SS, French PJ, Peeters JK, Rozendaal MJ, et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast cancer research and treatment. 2010;121:53–64. doi: 10.1007/s10549-009-0460-8. [DOI] [PubMed] [Google Scholar]
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525:384–388. doi: 10.1038/nature14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamdade VS, Sethi N, Mundhe NA, Kumar P, Lahkar M, Sinha N. Therapeutic targets of triple-negative breast cancer: a review. British journal of pharmacology. 2015;172:4228–4237. doi: 10.1111/bph.13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, Kwei KA, Hernandez-Boussard T, Wang P, Gazdar AF, et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PloS one. 2009;4:e6146. doi: 10.1371/journal.pone.0006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Hubbard NE, Ziboh V, Erickson KL. Attenuation of breast tumor cell growth by conjugated linoleic acid via inhibition of 5-lipoxygenase activating protein. Biochimica et biophysica acta. 2005;1736:244–250. doi: 10.1016/j.bbalip.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Konig R, Chiang CY, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y, et al. A probability-based approach for the analysis of large-scale RNAi screens. Nature methods. 2007;4:847–849. doi: 10.1038/nmeth1089. [DOI] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E, Wu D, Pal B, Bouras T, Asselin-Labat ML, Vaillant F, Yagita H, Lindeman GJ, Smyth GK, Visvader JE. Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res. 2010;12:R21. doi: 10.1186/bcr2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maire V, Nemati F, Richardson M, Vincent-Salomon A, Tesson B, Rigaill G, Gravier E, Marty-Prouvost B, De Koning L, Lang G, et al. Polo-like kinase 1: a potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer research. 2013;73:813–823. doi: 10.1158/0008-5472.CAN-12-2633. [DOI] [PubMed] [Google Scholar]
- Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JL, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2:172–189. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, Brugge JS. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer cell. 2012;21:227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Azam T, Ferwerda G, Girardin SE, Walsh M, Park JS, Abraham E, Kim JM, Yoon DY, Dinarello CA, et al. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1beta and IL-6 production through a caspase 1-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16309–16314. doi: 10.1073/pnas.0508237102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D, Zack TI, Ren Y, Strickland MR, Lamothe R, Schumacher SE, Tsherniak A, Besche HC, Rosenbluh J, Shehata S, et al. Cancer vulnerabilities unveiled by genomic loss. Cell. 2012;150:842–854. doi: 10.1016/j.cell.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan C, Fanning LJ, Barry OP. p38delta MAPK: Emerging Roles of a Neglected Isoform. International journal of cell biology. 2014;2014:272689. doi: 10.1155/2014/272689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Lin X. Epithelial growth factor receptor-activated nuclear factor kappaB signaling and its role in epithelial growth factor receptor-associated tumors. Cancer journal (Sudbury, Mass) 2013;19:461–467. doi: 10.1097/PPO.0000000000000001. [DOI] [PubMed] [Google Scholar]
- Pao GM, Janknecht R, Ruffner H, Hunter T, Verma IM. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1020–1025. doi: 10.1073/pnas.97.3.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Petrocca F, Altschuler G, Tan SM, Mendillo ML, Yan H, Jerry DJ, Kung AL, Hide W, Ince TA, Lieberman J. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer cell. 2013;24:182–196. doi: 10.1016/j.ccr.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani M, Aust MM, Attkisson E, Williams DC, Jr, Ferreira-Gonzalez A, Grant S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer research. 2013;73:1340–1351. doi: 10.1158/0008-5472.CAN-12-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz M, van Jaarsveld MT, Hollestelle A, Prager-van der Smissen WJ, Heine AA, Boersma AW, Liu J, Helmijr J, Ozturk B, Smid M, et al. miRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation-specific miRNAs. Breast Cancer Res. 2013;15:R33. doi: 10.1186/bcr3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Garcia F, Villagrasa P, Matsui J, Kotliar D, Castro V, Akavia UD, Chen BJ, Saucedo-Cuevas L, Rodriguez Barrueco R, Llobet-Navas D, et al. Integration of genomic data enables selective discovery of breast cancer drivers. Cell. 2014;159:1461–1475. doi: 10.1016/j.cell.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao DD, Tsherniak A, Gopal S, Weir BA, Tamayo P, Stransky N, Schumacher SE, Zack TI, Beroukhim R, Garraway LA, et al. ATARiS: computational quantification of gene suppression phenotypes from multisample RNAi screens. Genome Res. 2013;23:665–678. doi: 10.1101/gr.143586.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Molecular cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solimini NL, Xu Q, Mermel CH, Liang AC, Schlabach MR, Luo J, Burrows AE, Anselmo AN, Bredemeyer AL, Li MZ, et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012;337:104–109. doi: 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagni V, Manni I, Oropallo V, Mottolese M, Di Benedetto A, Piaggio G, Falcioni R, Giaccari D, Di Carlo S, Sperati F, et al. ATM kinase sustains HER2 tumorigenicity in breast cancer. Nature communications. 2015;6:6886. doi: 10.1038/ncomms7886. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, Parsons R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer cell. 2015;27:837–851. doi: 10.1016/j.ccell.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto M, Fukuda T, Sonoda R, Murakami F, Tanaka H, Yamamoto N. Ephrin-B3-EphA4 interactions regulate the growth of specific thalamocortical axon populations in vitro. The European journal of neuroscience. 2002;16:1168–1172. doi: 10.1046/j.1460-9568.2002.02166.x. [DOI] [PubMed] [Google Scholar]
- TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Molecular cancer therapeutics. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, Hu M, Chan DA, Ethier SP, van ’t Veer LJ, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer cell. 2013;24:450–465. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby CA, Dixon JE. Pten. Annual review of biochemistry. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- Yousif NG, Al-Amran FG, Hadi N, Lee J, Adrienne J. Expression of IL-32 modulates NF-kappaB and p38 MAP kinase pathways in human esophageal cancer. Cytokine. 2013;61:223–227. doi: 10.1016/j.cyto.2012.09.022. [DOI] [PubMed] [Google Scholar]
- Yu F, Li J, Chen H, Fu J, Ray S, Huang S, Zheng H, Ai W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–2172. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, Zender P, Kubicka S, Luk JM, Schirmacher P, et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell. 2008;135:852–864. doi: 10.1016/j.cell.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang Y, Mehta A, Boufraqech M, Davis S, Wang J, Tian Z, Yu Z, Boxer MB, Kiefer JA, et al. Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget. 2015;6:9073–9085. doi: 10.18632/oncotarget.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.