

Abstract
Background
The cultivated peanut (Arachis hypogaea L.) is an important oil and food crop in the world. Pod- and kernel-related traits are direct factors involved in determining the yield of the peanut. However, the genetic basis underlying pod- and kernel-related traits in the peanut remained largely unknown, which hampered the improvement of peanut through marker-assisted selection. To understand the genetic basis underlying pod- and kernel-related traits in the peanut and provide more useful information for marker-assisted breeding, we conducted quantitative trait locus (QTL) analysis for pod length and width and seed length and width by use of two F2:3 populations derived from cultivar Fuchuan Dahuasheng × ICG 6375 (FI population) and cultivar Xuhua 13 × cultivar Zhonghua 6 (XZ population) in this study.
Results
Two genetic maps containing 347 and 228 polymorphic markers were constructed for FI and XZ populations respectively. In total, 39 QTLs explaining 1.25–26.11 % of the phenotypic variations were detected in two populations. For the FI population, 26 QTLs were detected between the two environments, among which twelve were not mapped before. For the XZ population, thirteen QTLs were detected, among which eight were not reported before. One QTL for pod width was repeatedly mapped between the two populations.
Conclusion
The QTL analyses for pod length and width and seed length and width were conducted in this study using two mapping populations. Novel QTLs were identified, which included two for pod length, four for pod width, five for seed length and one for seed width in the FI population, and three for pod length, three for pod width and two for seed width in the XZ population. Our results will be helpful for improving pod- and seed-related traits in peanuts through marker-assisted selection.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-016-0337-x) contains supplementary material, which is available to authorized users.
Keywords: Peanut (Arachis hypogaea L.), QTL analysis, Pod length, Pod width, Seed length, Seed width
Background
The cultivated peanut (Arachis hypogaea L.), also known as groundnut, is an allotetraploid (2n = 4x =40) legume that is widely grown in semi-arid regions in the world as an important oil or food crop. In 2013, the global yield of the peanut was estimated to be 45.65 million tonnes [1]. The actual yield of peanut cultivars in the farmers’ fields is far below their yield potential. Breeding peanut cultivars with a high yield is one of major objectives in peanut-breeding programs. Pod- and kernel-related traits are direct factors involved in yield determination [2]. The improvement of pod- and kernel-related traits is important for the development of peanut cultivars with a high yield performance.
Quantitative trait locus (QTL) mapping has been widely conducted for various crops to detect the genomic regions controlling important agronomic traits [3–7]. By use of this method, molecular markers tightly linked to the QTL can be developed and further deployed in marker-assisted breeding to improve the efficiency of conventional breeding. Due to the low level of genetic diversity of the peanut germplasm [8], QTL mapping in the peanut has been slow in the past. In recent years, progress has been achieved in the molecular mapping of the peanut. Gomez Selvaraj et al. [9] reported five SSR markers that were associated with seed length, pod length, number of pods per plant, 100-seed weight, maturity and oil content by use of a bulked segregant analysis. Despite non-classical QTL mapping with genetic linkage map, this paper is the first report to attempt to identify QTLs controlling pod- and kernel-related traits in the cultivated peanut. Khedikar et al. [10] mapped 11 QTLs for late leaf spot (LLs) and 12 for rust by use of a recombinant inbred line (RIL) population. Wang et al. [11] reported 23 QTLs, which included one for thrips, nine for tomato spotted wilt virus (TSWV), and thirteen for LLS through a RIL population derived from the cultivar Tifrunner × GT-C20. In a study focusing on drought tolerance related traits, Ravi et al. [12] reported 53 main-effect and 8 epistatic QTLs, among which four main-effect QTLs for pod weight and two main-effect QTLs for seed weight were identified. Shirasawa et al. [13] identified a total of 23 QTLs for different agronomic traits, including twelve QTLs for pod- and seed-related traits. In these two papers, the QTLs for pod weight, pod length and seed weight were commonly mapped on linkage group (LG) A5.
Although some QTLs associated with pod and seed related traits were reported [2, 12], more loci need to be identified to provide helpful information for marker-assisted selection in peanut breeding. Compared to ICG 6375 and cultivar (cv) Zhonghua 6, cv Fuchuan Dahuasheng and cv Xuhua 13 had larger pods and kernels. The objectives of this study were to identify QTLs controlling pod length (PL) and width (PW) and seed length (SL) and width (SW) in cv Fuchuan Dahuasheng and cv Xuhua 13 through two F2:3 populations derived from cv Fuchuan Dahuasheng × ICG 6375 and cv Xuhua 13 × cv Zhonghua 6, respectively.
Results
Phenotypic variation
The pod length and width and the seed length and width were evaluated in the FI and XZ populations. Both of the populations showed a large genetic variation in these four traits among the F2:3 progenies (Tables 1, 2, Fig. 1). The normality test by the Shapiro–Wilk (w) indicated that the phenotypic data of seed length for the FI population in Wuhan and the phenotypic data of pod length, pod width and seed length for the XZ population were normally distributed, while others were not. The broad-sense heritability of the phenotypic data for each trait was calculated based upon the analysis of variance of family means. The values ranged from 0.63 to 0.93. The Pearson correlation coefficients of the phenotypic data among four traits for the two populations ranged from 0.32 (P < 0.0001) between pod length and seed width for the FI population in Wuhan to 0.79 (P < 0.0001) between pod length and seed length for the FI population in Wuhan (Table 3).
Table 1.
Descriptive statistical analysis of the four traits
| Pop | Env | Trait | P1 | P2 | Max (cm) | Min (cm) | Mean (cm) | SD | H 2 | Shapiro-Wilk(w) | Kurt | Skew |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FI | Wuhan | PL | 3.05 | 1.82 | 3.66 | 1.39 | 2.52 | 0.33 | 0.89 | 0.98(0.03) | 1.12 | 0.27 |
| PW | 1.43 | 1.01 | 1.63 | 0.80 | 1.15 | 0.13 | 0.84 | 0.96(<0.0001) | 1.18 | 0.75 | ||
| SL | 1.52 | 0.96 | 1.71 | 0.98 | 1.31 | 0.15 | 0.63 | 0.99(0.2) | −0.14 | 0.26 | ||
| SW | 0.71 | 0.69 | 0.94 | 0.6 | 0.73 | 0.07 | 0.86 | 0.96(<0.0001) | 1.38 | 0.66 | ||
| FI | Yangluo | PL | 2.93 | 1.77 | 3.45 | 1.65 | 2.43 | 0.28 | 0.98 | 0.98(0.005) | 0.64 | 0.55 |
| PW | 1.34 | 0.95 | 1.59 | 0.92 | 1.16 | 0.12 | 0.85 | 0.98(0.002) | 0.46 | 0.56 | ||
| SL | 1.41 | 0.98 | 1.75 | 1.00 | 1.29 | 0.15 | 0.93 | 0.98(0.03) | 0.01 | 0.39 | ||
| SW | 0.75 | 0.72 | 1.13 | 0.59 | 0.75 | 0.07 | 0.77 | 0.94(<0.0001) | 3.72 | 0.96 | ||
| XZ | Wuhan | PL | 3.60 | 3.28 | 3.99 | 2.19 | 3.13 | 0.32 | 0.82 | 0.99(0.57) | −0.04 | −0.08 |
| PW | 1.50 | 1.34 | 1.81 | 1.01 | 1.34 | 0.14 | 0.79 | 0.99(0.56) | 0.10 | 0.36 | ||
| SL | 1.76 | 1.72 | 2.27 | 1.27 | 1.72 | 0.17 | 0.84 | 0.99(0.36) | −0.04 | 0.02 | ||
| SW | 1.08 | 0.88 | 1.31 | 0.74 | 0.95 | 0.08 | 0.81 | 0.99(0.02) | 1.35 | 0.50 |
Pop Population, Env Environments, P1 female parent, Fuchuan Dahuasheng in FI population and Xuhua 13 in XZ population, P2 male parent, ICG 6375 in FI population and Zhonghua 6 in XZ population, PL pod length, PW pod width, SL seed length, SW seed width, SD standard deviation, H 2 broad-sense heritability on entry-mean basis, Kurt kurtosis, Skew, skewness
Table 2.
Variance analysis of the four traits in FI population between Wuhan and Yangluo environment
| Sum of square | df | Mean square | F value | P-value | |
|---|---|---|---|---|---|
| Pod Length | 0.906 | 1 | 0.906 | 10.41 | 0.001 |
| Pod Width | 0.002 | 1 | 0.002 | 0.12 | 0.730 |
| Seed Length | 0.086 | 1 | 0.086 | 4.10 | 0.044 |
| Seed Width | 0.063 | 1 | 0.063 | 12.51 | 0.000 |
Fig. 1.
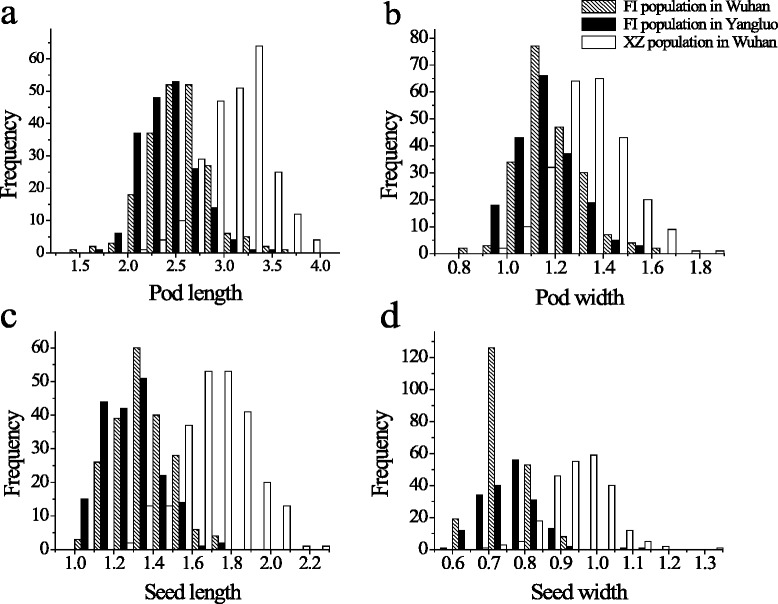
Distribution of pod length, pod width, seed length and seed width. a distribution of pod length, b distribution of pod width, c distribution of seed length, d distribution of seed width
Table 3.
Correlation analyses for the four traits
| Population | Environment | Phenotypea | PL | PW | SL | SW |
|---|---|---|---|---|---|---|
| FI | Wuhan | PL | ||||
| PW | 0.62** | |||||
| SL | 0.79** | 0.58** | ||||
| SW | 0.32** | 0.53** | 0.41** | |||
| FI | Yangluo | PL | ||||
| PW | 0.72** | |||||
| SL | 0.66** | 0.52** | ||||
| SW | 0.33** | 0.52** | 0.55** | |||
| XZ | Wuhan | PL | ||||
| PW | 0.71** | |||||
| SL | 0.72** | 0.66** | ||||
| SW | 0.48** | 0.66** | 0.72** |
** Indicated P < 0.01
a PL pod length, PW pod width, SL seed length, SW seed width
Molecular markers and genetic maps
For the FI population, 420 out of 3227 of the SSR markers (Additional file 1) were polymorphic between cv. Fuchuan Dahuasheng and ICG 6375. In total, 347 markers were successfully mapped on 22 LGs, which spanned 1675.6 cM with an average distance of 5.2 cM. The shortest linkage fragment, LG FB4, covering 36.1 cM, had only six markers, and the longest linkage group, LG FA6, had 26 markers, covering 131.5 cM (Table 4, Additional file 2). For the XZ population, 253 out of 2434 of the SSR markers (Additional file 1) were polymorphic between Xuhua 13 and Zhonghua 6. In addition, 228 polymorphic markers were utilized to construct 22 LGs that totally covered a 1337.7 cM genetic distance. The average distance between the adjacent loci was 6.5 cM, and the length of the linkage groups ranged from 21.6 to 111.4 cM (Table 4, Additional file 2).
Table 4.
Descriptions of the genetic linkage maps of FI and XZ population
| FI genetic map | XZ genetic map | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LGsa | Locus No.b | cM | cM/locus | LGs by Shirasawa | Common marker No. | LGs | Locus No. | cM | cM/locus | LGs by Shirasawa | Common marker No. |
| FA1 (LGF1) | 22 | 84.1 | 3.8 | A01 | 15 | XA1 (LGX5) | 11 | 78.7 | 7.2 | A01 | 4 |
| FA2 (LGF2) | 10 | 74.9 | 7.5 | A02 | 5 | XA2 (LGX6) | 13 | 57.9 | 4.5 | A02 | 2 |
| FA3 (LGF3) | 16 | 72.2 | 4.5 | A03 | 9 | XA3 (LGX7) | 9 | 80.7 | 9.0 | A03 | 5 |
| FA4 (LGF4) | 9 | 74.4 | 8.3 | A04 | 6 | XA4 (LGX8) | 20 | 93.7 | 4.7 | A04 | 9 |
| FA5 (LGF5) | 22 | 61.2 | 2.8 | A05 | 15 | XA5 (LGX9) | 9 | 60.6 | 6.7 | A05 | 2 |
| FA6 (LGF6) | 13 | 131.5 | 10.1 | A06 | 10 | XA6 (LGX10) | 12 | 43.6 | 3.6 | A06 | 5 |
| FA7 (LGF7) | 25 | 72.1 | 2.9 | A07 | 21 | XA7 (LGX11) | 17 | 68.8 | 4.0 | A07 | 9 |
| FA8 (LGF8) | 6 | 71.8 | 12.0 | A08 | 4 | XA8 (LGX12) | 10 | 94.9 | 9.5 | A08 | 3 |
| FA9 (LGF9) | 17 | 98.1 | 5.8 | A09 | 13 | XA9 (LGX13) | 14 | 56.5 | 4.0 | A09 | 5 |
| FA10 (LGF10) | 20 | 71.1 | 3.6 | A10 | 20 | XA10 (LGX14) | 10 | 46.2 | 4.6 | A10 | 6 |
| FB1 (LGF11) | 19 | 77.4 | 4.1 | B01 | 17 | - | - | - | |||
| FB2 (LGF12) | 24 | 98.3 | 4.1 | B02 | 21 | XB2 (LGX15) | 7 | 36.6 | 5.2 | B02 | 2 |
| FB3 (LGF13) | 20 | 95.8 | 4.8 | B03 | 15 | XB3 (LGX16) | 24 | 104.0 | 4.3 | B03 | 7 |
| FB4 (LGF14) | 7 | 36.1 | 5.2 | B04 | 7 | XB4 (LGX17) | 5 | 30.0 | 6.0 | B04 | 2 |
| FB5 (LGF15) | 18 | 74.5 | 4.1 | B05 | 16 | XB5 (LGX18) | 20 | 111.4 | 5.6 | B05 | 5 |
| FB6 (LGF16) | 19 | 66.2 | 3.5 | B06 | 17 | XB6 (LGX19) | 6 | 70.7 | 11.8 | B06 | 2 |
| FB7 (LGF17) | 9 | 75.0 | 8.3 | B07 | 8 | - | - | - | |||
| FB8 (LGF18) | 17 | 93.8 | 5.5 | B08 | 16 | - | - | - | |||
| FB9 (LGF19) | 26 | 72.0 | 2.8 | B09 | 22 | XB9 (LGX20) | 3 | 29.5 | 9.8 | B09 | 2 |
| FB10 (LGF20) | 16 | 75.3 | 4.7 | B10 | 15 | XB10 (LGX21) | 17 | 29.9 | 1.8 | B10 | 8 |
| FA7a (LGF21) | 6 | 47.8 | 8.0 | A07 | 4 | XB3a (LGX22) | 7 | 93.0 | 13.3 | B03 | 2 |
| FB7a (LGF22) | 6 | 52.0 | 8.7 | B07 | 5 | LGX1 | 3 | 49.7 | 16.6 | 0 | |
| - | - | - | LGX2 | 4 | 21.6 | 5.4 | 0 | ||||
| - | - | - | LGX3 | 4 | 56.9 | 14.2 | 0 | ||||
| - | - | - | LGX4 | 3 | 22.8 | 7.6 | 0 | ||||
| Total | 347 | 1675.6 | 5.7 | Total | 228 | 1337.7 | 7.2 | ||||
aThe initial “F” and “X” represented the FI population and XZ population, respectively
bThe number of loci on each linkage group
QTLs identified in the FI and XZ population
In total, 39 QTLs, explaining 1.25–26.11 % of the phenotypic variations, were detected in the two populations (Table 5). For the FI population, a total of 18 QTLs were detected in the Wuhan environment. Among them, 4 QTLs were detected for pod length with a 5.7–26.11 % phenotypic variation explained (PVE), 6 QTLs for pod width with a 7.42–16.14 % PVE, 5 QTLs for seed length with a 5.66–20.8 % PVE and 3 QTLs for seed width with a 7.42–12.6 % PVE, respectively. The QTLs on LG A5, qPLA5.1a, qPLA5.1b and qPLA5.1c, together explained more than 24 % of the phenotypic variation for pod length. Other QTLs were detected in the same region, which included qPLA5.1a for pod length and qPWA5.1a for pod width, qPLA5.1a for pod length and qSLA5.1a for seed length, qPLA5.1b for pod length and qSLA5.1b for seed length, qPLA7.1 for pod length and qSLA7.1a for seed length, qSLA5.1b for seed length and qSWA5.1a for seed width, qSLA5.1c for seed length and qSWA5.1b for seed width (Table 5, Additional file 3). Eleven QTLs were detected on LG A5, which included 3 for pod length, 3 for pod width, 3 for seed length and 2 for seed width (Table 5, Additional file 3). All of these 11 QTLs were mapped in a similar region on LG FA5 (Table 5).
Table 5.
Positions, effects, and phenotypic variation explained by QTLs for 4 agronomic traits detected in the 2 populations of 2 environments
| Population | Environment | Traits | QTL | LG | Position | Marker interval | LOD | Additive effect | Dominant effect | R 2 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| FI | Wuhan | PL | qPLA5.1a | FA5 | 24.51 | AHGS1341—pPGPseq9A7 | 8.47 | 0.19 | −0.09 | 24.24 |
| qPLA5.1b | FA5 | 35.41 | TC2B9—Ah4-26 | 9.22 | 0.18 | −0.11 | 24.29 | |||
| qPLA5.1c | FA5 | 47.61 | PM45—GNB533-2 | 6.63 | 0.17 | −0.16 | 26.11 | |||
| qPLA7.1 | FA7 | 34.51 | AHGS1980—pPGPseq3A1 | 3.35 | 0.10 | 0.00 | 5.70 | |||
| PW | qPWA3.1 | FA3 | 38.31 | AY232—AHGS0132 | 3.11 | 0.03 | −0.05 | 8.49 | ||
| qPWA5.1a | FA5 | 22.31 | AHGS1904-2—AHGS1341 | 5.76 | 0.06 | −0.03 | 16.14 | |||
| qPWA5.1b | FA5 | 30.21 | ARS715—TC2B9 | 3.26 | 0.05 | −0.02 | 9.42 | |||
| qPWA5.1c | FA5 | 41.71 | Ah4-26—POCR413 | 2.73 | 0.03 | −0.04 | 7.50 | |||
| qPWA8.1 | FA8 | 55.31 | ARS120—AHGS2319 | 3.93 | −0.04 | 0.04 | 9.85 | |||
| qPWA10.1 | FA10 | 42.61 | AHGS1606—AHGS1566 | 3.62 | 0.05 | −0.01 | 7.42 | |||
| SL | qSLA5.1a | FA5 | 24.51 | AHGS1341—pPGPseq9A7 | 7.38 | 0.08 | −0.04 | 20.80 | ||
| qSLA5.1b | FA5 | 35.41 | TC2B9—Ah4-26 | 6.70 | 0.07 | −0.04 | 16.97 | |||
| qSLA5.1c | FA5 | 42.71 | PM45—GNB533-2 | 5.98 | 0.07 | −0.05 | 19.32 | |||
| qSLA7.1a | FA7 | 29.41 | AHGS1475—pPGPseq3A1 | 4.36 | 0.05 | −0.04 | 11.15 | |||
| qSLA7.1b | FA7 | 41.31 | AHGS2022—AHGS2413 | 2.53 | 0.04 | −0.02 | 5.66 | |||
| SW | qSWA5.1a | FA5 | 35.41 | TC2B9—Ah4-26 | 2.94 | 0.01 | −0.03 | 7.42 | ||
| qSWA5.1b | FA5 | 42.71 | PM45—GNB533-2 | 3.04 | 0.02 | −0.04 | 9.43 | |||
| qSWA10.1 | FA10 | 22.81 | AHGS1939-1—TC1G4 | 3.93 | 0.02 | −0.03 | 12.60 | |||
| FI | Yangluo | PL | qPLA5.2 | FA5 | 9.71 | ARS760—TC6E1-1 | 2.85 | 0.08 | −0.06 | 7.92 |
| qPLA7.2 | FA7 | 47.81 | AHGS0346—AHGS1692 | 3.43 | 0.10 | −0.03 | 8.61 | |||
| PW | qPWA5.2 | FA5 | 37.41 | TC2B9 —Ah4-26 | 3.66 | 0.05 | 0.02 | 5.16 | ||
| qPWA10.2 | FA10 | 51.01 | GM2084—ARS710 | 2.95 | 0.04 | −0.03 | 8.36 | |||
| SL | qSLA10.2a | FA10 | 13.01 | pPGPseq3E10-1—AHGS1314-2 | 4.23 | 0.04 | −0.10 | 12.81 | ||
| qSLA10.2b | FA10 | 19.31 | AHGS1939-1—AHGS1314-1 | 4.30 | 0.06 | −0.06 | 15.75 | |||
| qSLA10.2c | FA10 | 26.41 | AHGS1314-1—pPGPseq4H11 | 3.71 | 0.05 | −0.04 | 12.37 | |||
| SW | qSWA5.2 | FA5 | 55.11 | ARS702—pPGPseq11C8 | 2.65 | −0.02 | 0.04 | 14.43 | ||
| XZ | Wuhan | PL | qPLA5.3 | XA5 | 0.01 | 0—GM1577 | 3.32 | 0.09 | 0.07 | 1.25 |
| qPLA9.3a | XA9 | 16.41 | EM87—ARS768 | 3.41 | 0.11 | 0.02 | 4.09 | |||
| qPLA9.3b | XA9 | 22.41 | AGGS1925—AGGS2572 | 3.78 | 0.11 | 0.01 | 5.10 | |||
| qPLA9.3c | XA9 | 39.41 | ARS205—TC1D2 | 3.83 | 0.11 | −0.04 | 7.79 | |||
| PW | qPWA5.3 | XA5 | 0.01 | 0—GM1577 | 3.90 | 0.05 | 0.01 | 4.48 | ||
| qPWA8.3 | XA8 | 93.81 | AGGS2186—TC9F10 | 4.21 | 0.05 | −0.02 | 8.78 | |||
| qPWA9.3a | XA9 | 16.41 | EM87—ARS768 | 3.44 | 0.05 | 0.00 | 5.35 | |||
| qPWA9.3b | XA9 | 22.51 | AGGS1925—AGGS2572 | 3.75 | 0.05 | −0.01 | 6.79 | |||
| SL | qSLA5.3 | XA5 | 0.01 | 0—GM1577 | 2.60 | 0.05 | 0.01 | 3.03 | ||
| qSLA6.3 | XA6 | 13.41 | GC47—ARS816 | 3.19 | −0.06 | 0.00 | 4.87 | |||
| SW | qSWA6.3 | XA6 | 13.41 | GC47—ARS816 | 3.71 | −0.03 | −0.01 | 3.77 | ||
| qSWA8.3a | XA8 | 78.11 | ARS120—AGGS2186 | 3.67 | 0.03 | 0.00 | 6.44 | |||
| qSWA8.3b | XA8 | 91.81 | AGGS2186—TC9F10 | 3.97 | 0.03 | −0.01 | 9.76 |
LG the linkage group the QTL located in, Marker interval the flanking marker nearest the 95 % confidence interval, R 2 percentage of the phenotypic variation explained by the QTLs
For the FI population in Yangluo, two QTLs were detected for pod length with a 7.92–8.61 % PVE, 2 for pod width with a 5.16–8.36 % PVE, 3 for seed length with a 12.37–15.75 % PVE and 1 for seed width with a 14.43 % PVE. For seed length, qSLA10.2a, qSLA10.2b and qSLA10.2c were mapped in similar regions (Table 5, Additional file 3). For the genetic loci between Wuhan and Yangluo, QTLs qPWA5.1c and qPWA5.2 for pod length, qPLA5.1a, qPLA5.1b, qPLA5.1c, qPLA7.1, qPLA5.2, qPLA7.2 for pod length, qPWA10.1, qPWA10.2 for pod width, and qSWA5.1a, qSWA5.1b, qSWA5.2 for seed width were repeatedly detected in overlapping or adjacent regions on the same linkage groups.
For the XZ population, 4 QTLs with a 1.25–7.79 % PVE for pod length, 4 QTLs with a 4.48 %–8.78 PVE for pod width, 2 QTLs with a 3.03–4.87 % PVE for seed length and 3 QTLs with a 3.77–9.76 % for seed width were detected. Among these loci, qPLA5.3 for pod length, qPWA5.3 for pod width and qSLA5.3 for seed length were mapped in the same region (Table 5, Additional file 4).
To compare the QTLs between the FI and XZ populations, we produced an integrated map because no common markers were linked to the detected QTLs between the two populations (Additional file 2). The results showed that qPWA5.1a in the FI population and qPWA5.3 in the XZ population overlapped on LG IA5 (Fig. 2, Table 5).
Fig. 2.
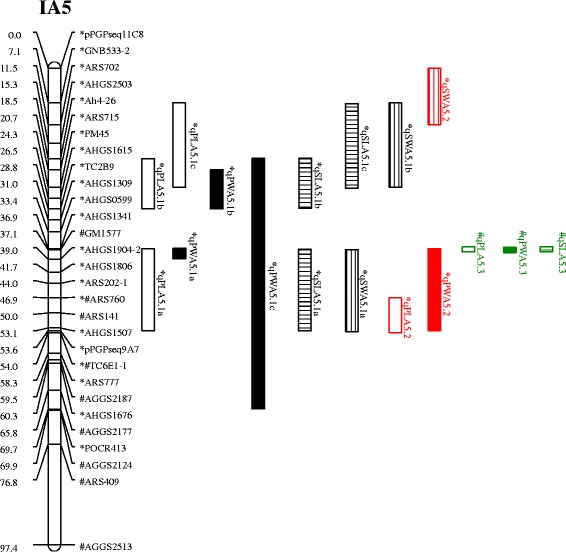
Positions of the QTLs on the integrated genetic linkage of FA5 and XA5. Scale bars on the left side describe the map distance in centimorgans. The markers on the linkage group FA5 of the FI population and XA5 of the XZ population were marked with “*” and “#”, respectively. The common markers between the two populations were marked with both “*” and “#”. The QTLs detected in the FI population were marked with “*”, and the QTLs in Wuhan and Yangluo were shown as black and red, respectively. The QTLs detected in the XZ population were marked with “#” and shown as green. The QTLs for pod length, pod width, seed length, and seed width were filled with none, pure colour, horizontal lines and vertical lines, respectively
Discussion
The broad-sense heritability estimated in this study was relatively high for pod and seed related traits in the two populations, which means a genetic component, rather than environmental conditions, plays a major role in the determination of these traits. The Pearson correlation coefficient analysis showed that pod length and pod width, as well as pod length and seed length, had a high positive correlation in the two populations, while pod length and seed width had a lower correlation, which was expected. Interestingly, the correlation between seed length and seed width for the FI population was obviously lower than that for the XZ population, which indicates that there may be a different mechanism underlying the determination of seed length and seed width between these two populations.
For both populations, the polymorphic markers were less than ten percent of all of the markers that were evaluated, which made trouble for the production of a high-density linkage map and accurate QTL results. A low level of genetic diversity in the peanut was also reported in other studies [14–16]. Recently, progress had been made on the production of high density linkage maps. Shirasawa et al. [17] constructed a dense integrated map with 3,693 markers on 20 linkage groups spanning 2,651 cM. Zhou et al. [18] construed the first SNP-based genetic linkage map in the cultivated peanut, which was comprised of 1,621 SNPs and 64 SSR markers on 20 linkage groups. Through the use of high-throughput sequencing technology, higher density linkage maps will undoubtedly be expected.
Two or more mapping populations have an advantage over a single population in order to identify more robust QTLs. In our study, two F2:3 mapping populations derived from cv Fuchuan Dahuasheng × ICG 6375 (FI) and cultivar Xuhua 13 × cultivar Zhonghua 6 (XZ) were used and the genomic region on the LG A5 were repeatedly mapped between these two populations. Combined with previous studies [12, 13], we believe that LG A5 may harbor important genes for pod and seed related traits, which can be deployed in marker-assisted selection in peanut breeding. Further research, such as fine mapping, should be conducted to investigate the genes responsible for the QTLs on LG A5. For the QTL results in the FI population between the two environments, Wuhan and Yangluo, although QTLs qPLA5.1a, qPLA5.1b, qPLA5.1c, qPLA7.1, qPLA5.2 and qPLA7.2 for pod length, qPWA10.1 and qPWA10.2 for pod width and qSWA5.1a, qSWA5.1b and qSWA5.2 for seed width did not overlap, they were mapped to adjacent regions on the same chromosomes. The inconsistency of the QTLs between the two environments might be attributed to the phenotypic deviations caused by environmental factors. QTLs qPLA7.1, qPLA7.2 for pod length, qPWA3.1, qPWA8.1, qPWA10.1, qPWA10.2 for pod width, qSLA7.1a, qSLA7.1b, qSLA10.2a, qSLA10.2b, and qSLA10.2c for seed length and qSWA10.1 for seed width had not been reported before, which provides more valuable sources of loci for the improvement of pod and seed related traits through marker-assisted selection in peanut breeding.
Thirteen QTLs were detected in the XZ population. Eight QTLs, such as qPLA9.3a, qPLA9.3b, qPLA9.3c, qPWA8.3, qPWA9.3a, qPWA9.3b, qSWA8.3a, and qSWA8.3b, were not reported before and might be novel. Because only phenotypic data from one environment was evaluated, we could not exclude the negative QTL caused by environmental factors. However, compared to the QTL results in the FI population through an integrated map, we found that the QTLs for pod width, qPWA5.3 in the XZ population and qPWA5.1a in FI the population, was repeatedly detected, which indicated that the QTL analyses in the XZ were reliable.
Despite the progress that has been made for genetic mapping in the peanut, QTL analyses are still few in comparison with the research that has been conducted on other crops. The peanut is an important oil or food crop worldwide. More genetic analyses of the genes controlling important agronomic traits, such as yield determining factors and resistance to diseases, will be helpful for improving the peanut through marker-assisted breeding in the future.
Conclusions
In this study, we conducted QTL analyses for pod and seed related traits in the peanut using two mapping populations, FI and XZ. For the FI population, in total, 26 QTLs were identified in the two environments, among which 12 QTLs were considered as novel loci. For the XZ population, 13 QTLs were detected. One QTL was commonly mapped between the FI and XZ populations. Our results will be helpful for improving pod and seed related traits in peanuts through marker-assisted selection.
Methods
Plant materials and phenotypic evaluation
Two F2 mapping populations, FI and XZ, were used to construct genetic linkage maps in this study. The FI population was comprised of 218 individuals derived from a cross of Fuchuan Dahuasheng × ICG 6375. Fuchuan Dahuasheng (A. hypogaea L. subsp. hypogaea L. var. hirsuta Kohle) was gathered from Fuchuan County, Guangxi province and is a cultivar with large pods and seeds. ICG 6375 (A. hypogaea L. subsp. fastigata Waldron var. vulgaris Harz) is a groundnut germplasm with small pods received from the International Crop Research Institute for the Semiarid Tropics (ICRISAT). The XZ population was comprised of 282 individuals derived from a cross of cv. Xuhua 13 × cv. Zhonghua 6. Xuhua 13 has large pods and seeds and Zhonghua 6 has small pods and seeds. Xuhua 13 (A. hypogaea L. subsp. hypogaea L. var. hypogaea) is a cultivar developed by the Xuzhou Agricultural Science Research Institute in 2002, and Zhonghua 6 (A. hypogaea L. subsp. fastigata Waldron var. vulgaris Harz.) is a cultivar developed by the Oil Crops Research Institute, Chinese Academy of Agricultural Sciences in 2000. All of the four parents were obtained from the National Mid-term Gene Bank for Oil Crops of China located in Wuhan. In this bank, the accession number of Fuchuan Dahuasheng, ICG 6375, Xuhua 13 and Zhonghua 6 were Zhh 2359, Zhh 7094, Zhh 7778 and Zhh 7629, respectively.
Genomic DNA was extracted from each F2 individuals for these two populations following a protocol described by Doyle [19]. The F2:3 progenies of each F2 individual were harvested and evaluated for pod length, pod width, seed length and seed width. The mean values of each trait of the F2:3 progenies were used to represent the phenotype of the F2 individuals. For the FI population, the F2:3 progenies were grown in two environments, Wuhan in 2012 and Yangluo 2013 in order to evaluate the environmental effects on the traits. Despite only 50 km from each other, the local climate and growing conditions of experimental fields between Wuhan and Yangluo were different. The fields with clay soils in Wuhan were located in the downtown with around 1–2 °C higher than the ones with sandy soil in Yangluo that were located in the countryside. These two experimental fields were owned and managed by Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences. For the XZ population, only Wuhan environment of 2012 was used because of the lack of seeds.
SSR marker analysis
For each SSR marker, PCR reactions were performed in a T100 Thermal Cycler in a volume of 10 μl, containing 20 ng of DNA template, 0.5 μM of each primer, 10 × PCR buffer, 1 mM MgCl2, 0.2 mM dNTPs and 0.5 U Taq polymerase. The PCR temperature profile was 95 °C for 4 min, 35 cycles of 55 s at 94 °C, 45 s at 55 °C and 1 min at 72 °C, and a final extension step of 7 min at 72 °C. The PCR products were visualized on 6 % polyacrylamide gels followed by silver staining. The fragment sizes of the PCR products were estimated by a comparison to a 50 bp DNA ladder.
Statistical analysis and linkage map
The phenotypic data for pod length and width and seed length and width were tested for normality using the PROC UNIVARIATE procedure of SAS 9.3 (SAS Institute, Cary, NY, USA). The Shapiro–Wilk (w) statistic was used to test the null hypothesis that the phenotypic data were normally distributed. Correlation coefficients among the four traits were estimated using the PROC CORR procedure of SAS. The broad-sense heritability for each trait was calculated by a method described by Wu et al. [20].
Genetic linkage maps were constructed using the Joinmap 3.0 software [21] with a maximum recombinant frequency of 0.4. The recombinant ratio was converted to genetic distance by Kossambi map function [22]. The linkage groups were aligned with the reference linkage maps based on the common markers. The Joinmap Combine Groups for Map Integration Module was used to integrate the linkage maps developed in this study.
QTL mapping
The software Windows QTL Cartographer 2.5 [23] was used to conduct the composite interval mapping (CIM) [24]. The LOD value chosen was 3.4 to declare a QTL significant based on a permutation test [25] with 1,000 runs to determine the P = 0.05 genome-wide significance level. To identify more potential QTLs, a QTL with a LOD value more than 2.5 was also presented. The nomenclature of the QTLs was similar to that described by Udall et al. [26] with codes 1 and 2 representing the QTLs detected in the Wuhan and Yangluo environments of the FI population, respectively, and code 3 representing the QTLs detected in the Wuhan environment of the XZ population. If two or more QTLs for the same trait were identified in the same linkage group in the same environment, an alphabetical letter was added at the end of the QTL name. For example, if two QTLs for seed length were detected on A3 in the F2 population, they were named as qSLA3.1a and qSLA3.1b.
Availability of supporting data
All the supporting data was included as Additional files.
Acknowledgements
This research was founded by the National Key Basic Research Special Foundation of China (2011CB109304), the National Natural Science Foundations of China (31271764, 31371662, 31471534 and 31461143022), and the Plant Germplasm Resources Sharing Platform [NB2011-2130135-29 (2)].
Additional files
Details of the primer sequences. (XLSX 33 kb)
Information for FI, XZ and the integrated map. (XLSX 27 kb)
Genetic map and positions of the QTLs of the FI population. Scale bars on the left side described the map distance in centimorgans, and QTLs detected in Wuhan and Yangluo were shown as black and red, respectively. The QTLs for pod length, pod width, seed length, and seed width were indicated by no shading, pure colour, horizontal lines and vertical lines, respectively. The vertical bars on the boxes show the regions over which significant LOD values were calculated by a permutations test (n = 1,000). (PPTX 141 kb)
Genetic map and positions of the QTLs of the XZ population. Scale bars on the left side described the map distance in centimorgans, and the QTLs detected in Wuhan and Yangluo were shown as green. The QTLs for pod length, pod width, seed length, and seed width were indicated by no shading, green, horizontal lines and vertical lines, respectively. The vertical bars on the boxes show the regions over which significant LOD values were calculated by a permutations test (n = 1,000). (PPTX 108 kb)
Footnotes
Weigang Chen, Yongqing Jiao, and Liangqiang Cheng are co-first authors.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WC, YJ and HJ analyzed the data, drafted and revised the manuscript. WC, LC and MT carried out the construction of the genetic maps and the investigation of the phenotypes. LH and BL helped to draft the manuscript. XR, XZ and YC provided technical support for the construction of the genetic maps and the detection of QTLs. HJ conceived of the study and participated in its design and coordination. All authors read and approved the final manuscript.
Authors’ information
Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences, Key Laboratory of Biology and Genetic Improvement of Oil Crops, Ministry of Agriculture, No.2 Xudong 2nd Rd, 430062 Wuhan, China.
Contributor Information
Weigang Chen, Email: wgchen2015@163.com.
Yongqing Jiao, Email: jiaoyongqing@caas.cn.
Liangqiang Cheng, Email: chengliangmei1984@126.com.
Li Huang, Email: huangli5100@126.com.
Boshou Liao, Email: lboshou@hotmail.com.
Mei Tang, Email: 15527050462@163.com.
Xiaoping Ren, Email: renxp1972@hotmail.com.
Xiaojing Zhou, Email: zhouxiaojing@caas.cn.
Yuning Chen, Email: ynchen126@126.com.
Huifang Jiang, Phone: 86-27-86712292, Email: peanutlab@oilcrops.cn.
References
- 1.Food and Agriculture Organization of the United Nations. http://faostat3.fao.org/browse/Q/QC/E. Accessed 8 Jun 2015.
- 2.Gomes RLF, Lopes AA. Correlations and path analysis in peanut. Crop Breeding and Applied Biotechnology. 2005;5(1):105–110. doi: 10.12702/1984-7033.v05n01a14. [DOI] [Google Scholar]
- 3.Buerstmayr H, Lemmens M, Hartl L, Doldi L, Steiner B, Stierschneider M, et al. Molecular mapping of QTLs for Fusarium head blight resistance in spring wheat. I. Resistance to fungal spread (Type II resistance) Theor Appl Genet. 2002;104(1):84–91. doi: 10.1007/s001220200009. [DOI] [PubMed] [Google Scholar]
- 4.Jiao Y, Vuong T, Liu Y, Li Z, Noe J, Robbins R, et al. Identification of quantitative trait loci underlying resistance to southern root-knot and reniform nematodes in soybean accession PI 567516C. Mol Breeding. 2015;35(6):1–10. doi: 10.1007/s11032-015-0330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foiada F, Westermeier P, Kessel B, Ouzunova M, Wimmer V, Mayerhofer W, et al. Improving resistance to the European corn borer: a comprehensive study in elite maize using QTL mapping and genome-wide prediction. Theor Appl Genet. 2015;128(5):875–891. doi: 10.1007/s00122-015-2477-1. [DOI] [PubMed] [Google Scholar]
- 6.Fan C, Xing Y, Mao H, Lu T, Han B, Xu C, et al. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet. 2006;112(6):1164–1171. doi: 10.1007/s00122-006-0218-1. [DOI] [PubMed] [Google Scholar]
- 7.Zhang T, Yuan Y, Yu J, Guo W, Kohel R. Molecular tagging of a major QTL for fiber strength in Upland cotton and its marker-assisted selection. Theor Appl Genet. 2003;106(2):262–268. doi: 10.1007/s00122-002-1101-3. [DOI] [PubMed] [Google Scholar]
- 8.Moretzsohn M, Hopkins M, Mitchell S, Kresovich S, Valls J, Ferreira M. Genetic diversity of peanut (Arachis hypogaea L.) and its wild relatives based on the analysis of hypervariable regions of the genome. BMC Plant Biol. 2004;4(1):11. doi: 10.1186/1471-2229-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomez Selvaraj M, Narayana M, Schubert AM, Ayers JL, Baring MR, Burow MD. Identification of QTLs for pod and kernel traits in cultivated peanut by bulked segregant analysis. Electronic Journal of Biotechnology. 2009;12(2):3–4. doi: 10.2225/vol12-issue2-fulltext-13. [DOI] [Google Scholar]
- 10.Khedikar Y, Gowda M, Sarvamangala C, Patgar K, Upadhyaya H, Varshney R. A QTL study on late leaf spot and rust revealed one major QTL for molecular breeding for rust resistance in groundnut (Arachis hypogaea L.) Theor Appl Genet. 2010;121(5):971–984. doi: 10.1007/s00122-010-1366-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Pandey MK, Qiao L, Qin H, Culbreath AK, He G et al. Genetic Mapping and Quantitative Trait Loci Analysis for Disease Resistance Using F2 and F5 Generation-based Genetic Maps Derived from ‘Tifrunner’ × ‘GT-C20’ in Peanut. The Plant Genome. 2013;6(3).
- 12.Ravi K, Vadez V, Isobe S, Mir R, Guo Y, Nigam S, et al. Identification of several small main-effect QTLs and a large number of epistatic QTLs for drought tolerance related traits in groundnut (Arachis hypogaea L.) Theor Appl Genet. 2011;122(6):1119–1132. doi: 10.1007/s00122-010-1517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shirasawa K, Koilkonda P, Aoki K, Hirakawa H, Tabata S, Watanabe M, et al. In silico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut. BMC Plant Biol. 2012;12(1):80. doi: 10.1186/1471-2229-12-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varshney R, Bertioli D, Moretzsohn M, Vadez V, Krishnamurthy L, Aruna R, et al. The first SSR-based genetic linkage map for cultivated groundnut (Arachis hypogaea L.) Theor Appl Genet. 2009;118(4):729–739. doi: 10.1007/s00122-008-0933-x. [DOI] [PubMed] [Google Scholar]
- 15.Hong Y, Chen X, Liang X, Liu H, Zhou G, Li S, et al. A SSR-based composite genetic linkage map for the cultivated peanut (Arachis hypogaea L.) genome. BMC Plant Biol. 2010;10(1):17. doi: 10.1186/1471-2229-10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin H, Feng S, Chen C, Guo Y, Knapp S, Culbreath A, et al. An integrated genetic linkage map of cultivated peanut (Arachis hypogaea L.) constructed from two RIL populations. Theor Appl Genet. 2012;124(4):653–664. doi: 10.1007/s00122-011-1737-y. [DOI] [PubMed] [Google Scholar]
- 17.Shirasawa K, Bertioli DJ, Varshney RK, Moretzsohn MC, Leal-Bertioli SC, Thudi M, et al. Integrated consensus map of cultivated peanut and wild relatives reveals structures of the A and B genomes of Arachis and divergence of the legume genomes. DNA Res. 2013;20(2):173–84. [DOI] [PMC free article] [PubMed]
- 18.Zhou X, Xia Y, Ren X, Chen Y, Huang L, Huang S, et al. Construction of a SNP-based genetic linkage map in cultivated peanut based on large scale marker development using next-generation double-digest restriction-site-associated DNA sequencing (ddRADseq) BMC Genomics. 2014;15(1):351. doi: 10.1186/1471-2164-15-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doyle JJ. Isolation of plant DNA from fresh tissue. Focus. 1990;12:13–15. [Google Scholar]
- 20.Wu X, Blake S, Sleper DA, Shannon JG, Cregan P, Nguyen HT. QTL, additive and epistatic effects for SCN resistance in PI 437654. Theor Appl Genet. 2009;118(6):1093–1105. doi: 10.1007/s00122-009-0965-x. [DOI] [PubMed] [Google Scholar]
- 21.Van Ooijen JW, Voorrips R. JoinMap® 3.0, Software for the calculation of genetic linkage maps. Plant research international, Wageningen 2001:1–51.
- 22.Kosambi D. The estimation of map distances from recombination values. Ann Eugen. 1943;12(1):172–175. doi: 10.1111/j.1469-1809.1943.tb02321.x. [DOI] [Google Scholar]
- 23.Wang S, Basten CJ, Zeng Z-B. Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. 2012.(http://statgen.ncsu.edu/qtlcart/WQTLCart.htm). Accessed 8 April 2012.
- 24.Zeng Z-B. Precision mapping of quantitative trait loci. Genetics. 1994;136(4):1457–1468. doi: 10.1093/genetics/136.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138(3):963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Udall JA, Quijada PA, Lambert B, Osborn TC. Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (Brassica napus L.): 2. Identification of alleles from unadapted germplasm. Theor Appl Genet. 2006;113(4):597–609. doi: 10.1007/s00122-006-0324-0. [DOI] [PubMed] [Google Scholar]