

Abstract
Objective:
This review examines the evidence that: Diabetes is a state of DNA damage; pathophysiological factors in diabetes can cause DNA damage; DNA damage can cause mutations; and DNA mutation is linked to carcinogenesis.
Data Sources:
We retrieved information from the PubMed database up to January, 2014, using various search terms and their combinations including DNA damage, diabetes, cancer, high glucose, hyperglycemia, free fatty acids, palmitic acid, advanced glycation end products, mutation and carcinogenesis.
Study Selection:
We included data from peer-reviewed journals and a textbook printed in English on relationships between DNA damage and diabetes as well as pathophysiological factors in diabetes. Publications on relationships among DNA damage, mutagenesis, and carcinogenesis, were also reviewed. We organized this information into a conceptual framework to explain the possible causal relationship between DNA damage and carcinogenesis in diabetes.
Results:
There are a large amount of data supporting the view that DNA mutation is a typical feature in carcinogenesis. Patients with type 2 diabetes have increased production of reactive oxygen species, reduced levels of antioxidant capacity, and increased levels of DNA damage. The pathophysiological factors and metabolic milieu in diabetes can cause DNA damage such as DNA strand break and base modification (i.e., oxidation). Emerging experimental data suggest that signal pathways (i.e., Akt/tuberin) link diabetes to DNA damage. This collective evidence indicates that diabetes is a pathophysiological state of oxidative stress and DNA damage which can lead to various types of mutation to cause aberration in cells and thereby increased cancer risk.
Conclusions:
This review highlights the interrelationships amongst diabetes, DNA damage, DNA mutation and carcinogenesis, which suggests that DNA damage can be a biological link between diabetes and cancer.
Keywords: Cancer, Diabetes, DNA Damage, Mutation, Pathophysiological Factors
INTRODUCTION
Diabetes confers an increased risk for all-site cancer worldwide, except prostate cancer.[1] Among the Chinese population of Hong Kong, for example, the prevalence of diabetes was reported to be over 10% in mid 1990s with 30% higher risk for cancer in people with diabetes compared to the general population.[2,3] With aging and declining mortality from cardiovascular-renal disease, 25% of Chinese people with type 2 diabetes now died from cancer, mainly due to liver, pancreatic, colorectal, and breast cancer.[4] Despite the health care and socio-economic burden of diabetes and cancer, the mechanisms whereby diabetes favors cancer development remain unknown. Since insulin has pro-proliferative activity and activation of insulin signaling axis has been implicated in carcinogenesis,[5] some researchers proposed that the use of insulin by diabetic patients might contribute to cancer development.[1] However, this view is controversial, as some epidemiological studies do not show a correlation between use of insulin and all-site cancer[6] while some others have even reported that use of insulin might decrease the risk for cancer.[7]
One of the typical features in carcinogenesis is DNA mutation which can be caused by DNA damage often found in people with diabetes. In this review article, we searched the literature and summarized the evidence in support of a possible linking role of DNA damage between diabetes and cancer.
DIABETES IS A PATHOPHYSIOLOGICAL STATE OF DNA DAMAGE
DNA damage occurs in different forms through different mechanisms. Strand break and base modification are the most studied forms of DNA damage. DNA strand break can be detected using single cell gel electrophoresis or otherwise named comet assay. Base oxidation, i.e., the formation of 8-hydroxy-2’-deoxyguanosine (8-OHdG), the most studied form of base modification, can be quantified using specific antibody by enzyme-linked immunosorbent assay. These techniques are well-established, and assay kits are easily available and user-friendly. This review focuses on the two forms of DNA damage with relevance to diabetes and cancer.
Dandona et al. compared the levels of 8-OHdG in mononuclear cells amongst type 1 diabetic patients (n = 12), type 2 diabetic patients (n = 15) and healthy control subjects (n = 10). They found that both type 1 and type 2 diabetic patients had higher levels of 8-OHdG than the nondiabetic subjects. Production of reactive oxygen species by mononuclear cells was also significantly greater in diabetic patients than the control subjects.[8] Increased serum or urinary levels of 8-OHdG which correlated with poor glycemic control have been confirmed in both type 1 and type 2 diabetes.[9,10,11] In addition to DNA base oxidation, Collins, and co-workers used comet assays on white blood cells and reported higher levels of DNA strand break in people with type 1 diabetes (n = 10) compared to healthy controls (n = 10).[12] Subsequent studies have also confirmed elevated levels of DNA strand break in type 2 diabetes, which, similar to 8-OHdG levels, were correlated with poor glycemic control.[13,14,15]
Peripheral blood cells are often used for comet assay to detect DNA strand break while urine and serum samples are commonly used for 8-OHdG quantification. A key question is whether the results from the peripheral samples reflect the levels of DNA damage in other body tissues. To address this issue, Kushwaha et al. compared the levels of DNA strand break in lymphocytes, lung, liver, heart, aorta, kidney, and pancreas from diabetic and control rats. They found that DNA strand break was increased in all the tested tissues from diabetic rats, and the level of DNA strand break in lymphocytes was positively correlated with that in other tissues, suggesting that lymphocyte DNA strand break might be a suitable marker indicating DNA damage in internal organs.[16]
PATHOPHYSIOLOGICAL FACTORS IN DIABETES THAT CAN CAUSE DNA DAMAGE
Patients with type 1 or type 2 diabetes have increased plasma levels of glucose and advanced glycation endproducts (AGEs), which are diagnostic markers for diabetes and glycemic control. Type 2 diabetic patients often have increased plasma levels of free fatty acids and insulin, due to obesity-associated insulin resistance, especially during the early stage of the disease. These pathophysiological factors in diabetes have been found to cause DNA damage in vitro, which may explain why diabetic patients have increased level of DNA damage in vivo.
High glucose/hyperglycemia
Early in 1980s, researchers have reported that a high concentration of glucose (30 mmol/L) could cause DNA strand break in cultured human endothelial cells.[17] This observation has been confirmed in a subsequent study using the mouse and human renal cells as experimental models.[18] In addition to DNA strand break, high glucose could increase the level of 8-OHdG in endothelial and tubular cells.[19,20]
Since DNA damage is known to produce various mutations,[21] and that high glucose can promote DNA damage, the next question to ask is whether high glucose can cause or promote mutagenesis. Indeed, Zhang et al. examined the effect of high glucose on the genomic stability of phosphoribosyltransferase and thymidine kinase loci in human lymphoblastoid cell lines and reported a significant increase in mutations in both loci under high glucose.[22] Using mouse embryo as an experimental model, Lee et al. found that high glucose increased Lac I mutation in vitro and in vivo.[23] Furthermore, since high glucose can cause mutation and DNA mutation are believed to play an important role in carcinogenesis,[24] it is logical to speculate that high glucose can promote cancer development. To address this point, Berstein and Alexandrov injected carcinogen into pregnant rats via an intraperitoneal route and divided them into experimental and control groups. The rats in the experimental group drank 10% glucose in water till delivery, after which the rats and their progeny drank 5% glucose in water for 45 days. Rats in the control group and their progeny drank water alone. The authors found that the progeny of the rats in the experimental group had significantly higher incidence of tumors than those from the control group.[25] In a follow-up study, the authors investigated whether glucose could have an effect on carcinogen-induced mutation in fetal cells, as measured using in vivo/in vitro assay. They found that high glucose not only increased the frequency of mutation but also promoted the proliferation and survival of the fetal cells from the rats in the experimental group.[26] These data suggest that high glucose can promote carcinogenesis, which might be due to its DNA damaging and then mutagenic effect.
Advanced glycation endproducts
Hyperglycemia in diabetes promotes the formation of AGEs due to nonenzymatic reactions between reducing sugars and free amino groups of proteins. Subsequent reactions such as dehydration, oxidation, and condensation result in the irreversible formation of this heterogeneous group of products. Stopper et al. investigated whether AGEs were genotoxic using pig kidney cells as an experimental model, and found that AGEs could cause DNA strand break.[27] These findings were subsequently confirmed in human liver and colon cells[28] as well as mouse podocytes.[29] In addition to DNA strand break, AGEs can also trigger DNA base oxidation and promote the production of 8-OHdG in different types of cells.[30,31]
Free fatty acids and insulin
Palmitic acid is the most common saturated free fatty acid, which is often used to represent free fatty acids in experimental studies. Beeharry et al. found that palmitic acid caused DNA strand break and apoptosis in insulin-secreting cell line and primary human fibroblasts.[32] Under these experimental conditions, the DNA damage is the main significant event, and the clinical relevance of the small incremental trend in apoptosis remains uncertain. In a proof of concept study, we quantified 8-OHdG in the culture of HCT116 colon cancer cells treated with palmitic acid at concentrations lower than 75 μmol/L. These concentrations were not toxic to the cells but increased the production of 8-OHdG in a dose-dependent manner (Lee, unpublished date). In support of these data, obese subjects who had increased risk for cancer also had increased plasma levels of free fatty acids and 8-OHdG, supporting possible causal relationships among free fatty acid, DNA damage, and cancer.[33]
In subjects with obesity and prediabetes which are associated with insulin resistance, hyperinsulinemia is a frequent phenomenon.[34] Whether insulin can cause strand break remains to be examined. However, in colon cells and human lymphocytes, insulin was found to cause base oxidation by triggering the production of reactive oxygen species. In particular, for colon cancer cells, the lowest tested and lowest active concentration of insulin in vitro was 1 nmol/L for short time treatment (i.e., 2 h) and 0.5–1 nmol/L for longer exposure.[35] In healthy human subjects, plasma insulin concentrations are in the order of 0.04 nmol/L after fasting, which can increase to 0.2 nmol/L after a meal. Pathophysiological levels of insulin can reach 1 nmol/L. Thus, the experimental results showed that insulin in pathophysiological concentrations had DNA damaging effect.[35]
As summarized in Table 1, these common pathophysiological features in diabetes, that is, high glucose, high insulin, AGEs and free fatty acids, can individually cause DNA damage, strand break, and base oxidation, although the effect of insulin on base oxidation remains unknown. Since these pathophysiological factors frequently co-exist in type 2 diabetes during the clinical course, their potential synergistic effects on causing DNA damage is an interesting topic for exploration.
Table 1.
DNA damaging effects due to the pathophysiological factors in diabetes
| Items | High insulin | High glucose | AGEs | Free fatty acids (i.e., palmitic acid) |
|---|---|---|---|---|
| Strand break | ? | Yes | Yes | Yes |
| Base oxidation | Yes | Yes | Yes | Yes |
AGEs: Advanced glycation endproducts.
DIABETES CAN PROMOTE DNA DAMAGE BY DIFFERENT PATHWAY [FIGURE 1][36,37]
Figure 1.
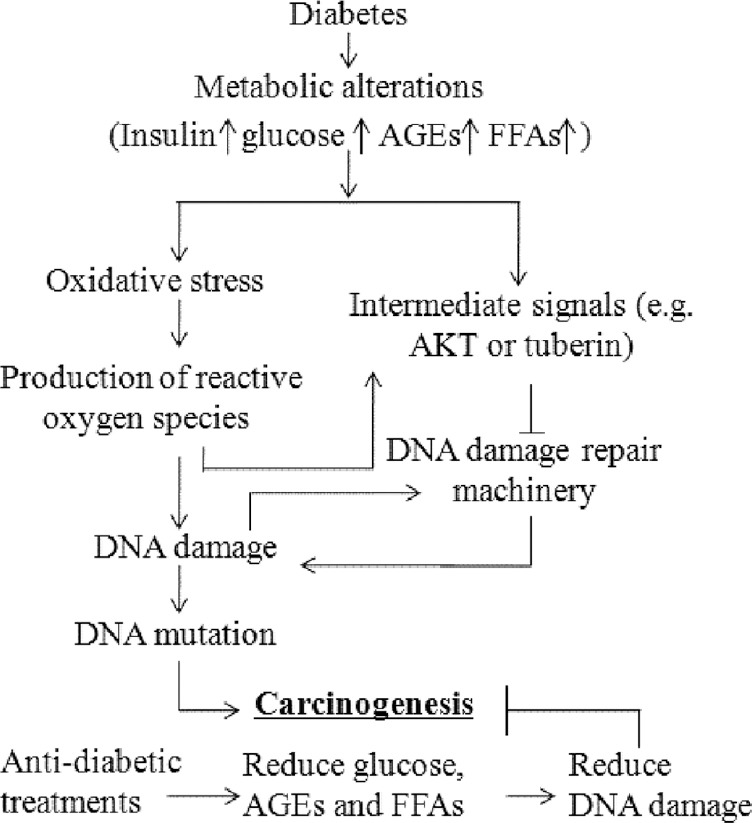
Pathways linking diabetes to DNA damage which is involved in carcinogenesis. AGEs: Advanced glycation end products; FFAs: Free fatty acids.
Direct (oxidative stress) pathway
Damage of DNA by reactive oxygen species can be considered as a direct pathway in diabetes-associated mutation. High serum levels of glucose, AGEs, free fatty acids, and insulin can all promote the production of reactive oxygen species found to be increased in type 2 diabetes compared to nondiabetic subjects.[38] Besides, people with diabetes had low anti-oxidative capacity such as reduced glutathione synthesis which might contribute to their proneness to oxidative damage.[39]
Despite the high heritability of diabetes, most of the genetic factors discovered by genome-wide association studies only explained small variance of the risk in the majority of people with type 2 diabetes.[40] Interestingly, Lai et al. found that genetic variants of peroxisome proliferator-activated receptor-γ coactivator-1α (PPARGC1A) were associated with increased risk of DNA damage (urine 8-OHdG) and diabetes.[41] In this light, PPARGC1A is known to regulate mitochondrial electron transport, which generates reactive oxygen species while at the same time, activates defending enzymes against reactive oxygen species. Imbalance between these two roles of PPARGC1A can lead to oxidative stress and possibly DNA damage. In Hong Kong Chinese, type 2 diabetes-related genetic variants of HHEX, TCF7L2, and CDKAL1 have also been reported to be associated with increased all-site cancer although the underlying mechanism remains to be explored.[42]
Signaling pathway
Indirectly, DNA damage in diabetes can occur through cell signal pathways. Simone et al.[36] reported the following observations in high glucose experimental models: (1) There were associations amongst Akt phosphorylation, tuberin phosphorylation, and 8-oxodG accumulation; (2) inhibition of Akt using PI3 kinase inhibitor reduced high glucose-induced tuberin phosphorylation, and (3) anti-oxidant inhibited reactive oxygen species generation, phosphorylation of Akt and tuberin, and 8-oxodG accumulation. These results indicated that the PI3 kinase-Akt-tuberin pathway might be important for DNA oxidation damage under hyperglycemic condition. In a recent study, Habib and Liang reported over-activation of Akt with decreased protein levels of tuberin and increased 8-OHdG concentration in kidney cancer tissues from diabetic patients, compared with cancer tissues from patients without diabetes.[43] Taken together, these data support a possible role of Akt/tuberin signaling in the occurrence of DNA damage in diabetes.
DNA repair pathway
Blasiak et al. compared the efficacy of removal of damaged DNA in peripheral blood lymphocytes between type 2 diabetic patients and healthy individuals, and reported reduced efficacy of repairing DNA damage in those with diabetes.[44] DNA is sensitive to damage caused by endogenous and exogenous factors, and as such, DNA damage occurs frequently in the absence of specific disease. Thus, an effective mechanism of DNA damage repair is crucial to maintain genomic integrity. Since defective DNA repair may determine the susceptibility to carcinogenesis,[44] decreased efficiency in DNA repair is another potential factor for cancer in diabetes.[45]
Activation of Akt/tuberin pathway might down-regulate DNA repair
The Akt pathway is an important pathway implicated in cell growth which can be activated by high levels of insulin, glucose,[36] AGEs[46] and free fatty acids (palmitic acid).[47] Here, insulin[48] and high glucose[36] activate Akt to phosphorylate tuberin which allows tuberin to interact with binding protein to perform its biological function.[48] Although there are reports on associations between DNA damage and changes in Akt activity, the mechanisms underlying Akt activation and DNA damage and repair is less clear. As reviewed by Xu and coworkers, Akt activation led to suppression of ATR/Chk1 signaling by direct phosphorylation of Chk1 or TopBP1. It could also inhibit recruitment of double-strand break resection factors (i.e., RPA, Brca1, and Rad51) to DNA damage sites, leading to compromised homologous recombination repair. Thus, Akt activation may be a potential cause of DNA repair inhibition and genomic instability.[49] In a study that used in vitro and in vivo experimental models, Akt activation was shown to phosphorylate Bim1, which led to increased genomic instability and increased oncogenic potential of Bim1.[50] In the case of tuberin, its phosphorylation or inactivation through activation of Akt could decrease the gene expression of OGG1, which was also downstream of Akt activation by reactive oxygen species, resulting in accumulation of DNA damage.[36,51,52] While these data support a regulatory role of Akt in DNA repair, the signal mediators downstream of Akt and their interactions remain to be defined.
DNA DAMAGE CAN CAUSE MUTATIONS
Genome stability is important for normal cell physiology. However, DNA bases are highly vulnerable to being damaged. Intrinsically, DNA is chemically unstable in an aqueous environment. Spontaneous reactions such as hydrolysis and de-amination can occur, resulting in DNA damage. Other endogenous factors such as metabolic products and exogenous factors such as environmental chemicals can also cause DNA damage. DNA strand breaks, in particular, double strand breaks, are potentially lethal. In surviving cells, double strand break triggers break-induced replication which is known to produce DNA mutations at high frequencies. The break-induced replication and related mechanisms can cause various DNA abnormalities which include loss of heterozygosity, telomere maintenance without telomerase, nonreciprocal translocation, copy number variation, and chromosomal rearrangements.[53]
Similarly, DNA base oxidation can be harmful.[54] Take 8-OHdG as an example, it can pair with A to form situ G to T substitution, and if incorporated into DNA, it can lead to mis-pairing with dA to form A to C substitution [Figure 2]. Moreover, 8-OHdG can change DNA conformation, leading to substitution mutations by mis-pairing with dA or dT. The presence of 8-OHdG in DNA can also have epigenetic consequences such as altering the gene transcription. Unfortunately, once mis-pairing occurs in DNA, it will not be repaired effectively in cells. Although 8-OHdG is the most frequently reported DNA damage in diabetes and other diseases, it should be kept in mind that all four DNA bases are susceptible to oxidative damage. Among these changes, modifications to GC base pairs tend to be more mutagenic while modifications on AT base pairs have weaker mutagenic potential.[55] In addition to base oxidation, lipid peroxidation is also harmful, which can cause the formation of reactive aldehydes such as malondialdehyde and 4-hydroxy-2-nonenal. These products are highly reactive to protein and DNA and have been shown to be mutagenic.[56]
Figure 2.
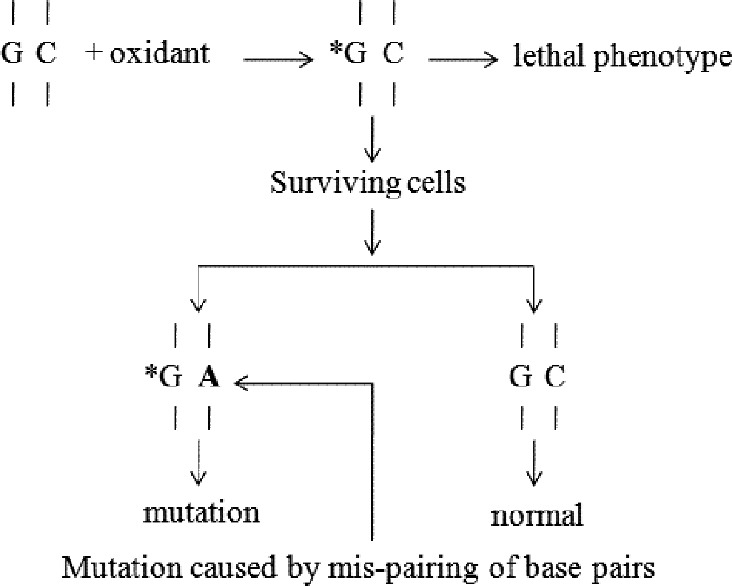
Mechanism of mutation caused by 8-hydroxy-2’-deoxyguanosine. *: Oxidized base; A: Mispaired base.
DNA MUTATION IS A TYPICAL FEATURE IN CARCINOGENESIS
About a century ago, Boveri speculated that cancer was a problem of cell proliferation due to chromosomal aberrations and/or mutations.[57] Indeed, with biotechnological advancement, large number of chromosomal abnormalities and DNA mutations had been demonstrated in various types of cancers. This leads to the proposal of “somatic mutation theory” to explain the mechanisms of cancer development, which considers the accumulation of somatic mutations as the origin of carcinogenesis and cancer progression. In animal models, carcinogenesis is found to involve multiple steps which include at least initiation, promotion, and progression stages.[58] DNA mutation is the main molecular feature of the initiation step which is irreversible characterized by micro-lesions such as base pair substitution and frame shift. Clinical cancer appears when cancerous cells enter the progression step where the karyotypic alteration of genomic instability and pathological features can be found. This step involves DNA macro-lesions such as DNA amplification and rearrangement.
CAN GLYCEMIC CONTROL AGENTS REDUCE CANCER RISK IN DIABETES?
If the pathophysiological factors of diabetes, notably, high blood glucose and AGEs, can contribute to the increased risk for all-site cancer in diabetes, then, we can expect that use of anti-diabetic drugs that reduce blood glucose and AGEs can also reduce cancer risk in diabetic patients. Indeed, this has been supported by epidemiological studies showing reduced cancer risk with metformin which is probably the most commonly used anti-diabetic drug.[1] In addition to its glucose-lowering effect, metformin itself can inhibit oxidative stress[59] and DNA damage.[60] Using the Hong Kong Diabetes Registry, we have also reported the association of reduced cancer risk with all anti-diabetic drugs as well as drugs such as renin-angiotensin system inhibitors and statins which have been shown to have anti-oxidant properties.[61,62] These epidemiological observations support a relationship among diabetes, DNA damage, and carcinogenesis.
CONCLUSION
The available data from the literature support the notion that pathophysiological metabolic factors in diabetes can cause DNA damage, making diabetes a state of DNA damage. Oxidative stress is likely the main mediator for DNA damage in diabetes. Inhibition of antioxidant capacity worsens the oxidative stress state while inhibition of DNA damage repair machinery contributes to DNA damage accumulation. DNA damage can cause mutation which plays key roles in carcinogenesis. Thus, DNA damage is possibly a biological link between diabetes and cancer. Apart from optimizing glycemic control, future cancer prevention in diabetic patients may target at inhibiting DNA damage using alternative drugs like antioxidants.
Footnotes
Edited by: Li-Shao Guo
Source of Support: This work was supported by direct grants to Dr. Shao Chin Lee, from Shanxi University and the Chinese University of Hong Kong, respectively.
Conflict of Interest: None declared.
REFERENCES
- 1.Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: A consensus report. Diabetes Care. 2010;33:1674–85. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan JC, Malik V, Jia W, Kadowaki T, Yajnik CS, Yoon KH, et al. Diabetes in Asia: Epidemiology, risk factors, and pathophysiology. JAMA. 2009;301:2129–40. doi: 10.1001/jama.2009.726. [DOI] [PubMed] [Google Scholar]
- 3.Yang X, So WY, Ma RC, Ko GT, Kong AP, Wang Q, et al. Predicting values of lipids and white blood cell count for all-site cancer in type 2 diabetes. Endocr Relat Cancer. 2008;15:597–607. doi: 10.1677/ERC-07-0266. [DOI] [PubMed] [Google Scholar]
- 4.Hong Kong Diabetes Registry. Yang X, So WY, Tong PC, Ma RC, Kong AP, et al. Development and validation of an all-cause mortality risk score in type 2 diabetes. Arch Intern Med. 2008;168:451–7. doi: 10.1001/archinte.168.5.451. [DOI] [PubMed] [Google Scholar]
- 5.Gupta K, Krishnaswamy G, Karnad A, Peiris AN. Insulin: A novel factor in carcinogenesis. Am J Med Sci. 2002;323:140–5. doi: 10.1097/00000441-200203000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Gu Y, Wang C, Zheng Y, Hou X, Mo Y, Yu W, et al. Cancer incidence and mortality in patients with type 2 diabetes treated with human insulin: A cohort study in Shanghai. PLoS One. 2013;8:e53411. doi: 10.1371/journal.pone.0053411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X, Ko GT, So WY, Ma RC, Yu LW, Kong AP, et al. Associations of hyperglycemia and insulin usage with the risk of cancer in type 2 diabetes: The Hong Kong diabetes registry. Diabetes. 2010;59:1254–60. doi: 10.2337/db09-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dandona P, Thusu K, Cook S, Snyder B, Makowski J, Armstrong D, et al. Oxidative damage to DNA in diabetes mellitus. Lancet. 1996;347:444–5. doi: 10.1016/s0140-6736(96)90013-6. [DOI] [PubMed] [Google Scholar]
- 9.Hinokio Y, Suzuki S, Hirai M, Chiba M, Hirai A, Toyota T. Oxidative DNA damage in diabetes mellitus: Its association with diabetic complications. Diabetologia. 1999;42:995–8. doi: 10.1007/s001250051258. [DOI] [PubMed] [Google Scholar]
- 10.Goodarzi MT, Navidi AA, Rezaei M, Babahmadi-Rezaei H. Oxidative damage to DNA and lipids: Correlation with protein glycation in patients with type 1 diabetes. J Clin Lab Anal. 2010;24:72–6. doi: 10.1002/jcla.20328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Aubaidy HA, Jelinek HF. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur J Endocrinol. 2011;164:899–904. doi: 10.1530/EJE-11-0053. [DOI] [PubMed] [Google Scholar]
- 12.Collins AR, Raslová K, Somorovská M, Petrovská H, Ondrusová A, Vohnout B, et al. DNA damage in diabetes: Correlation with a clinical marker. Free Radic Biol Med. 1998;25:373–7. doi: 10.1016/s0891-5849(98)00053-7. [DOI] [PubMed] [Google Scholar]
- 13.Tatsch E, Bochi GV, Piva SJ, De Carvalho JA, Kober H, Torbitz VD, et al. Association between DNA strand breakage and oxidative, inflammatory and endothelial biomarkers in type 2 diabetes. Mutat Res. 2012;732:16–20. doi: 10.1016/j.mrfmmm.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Dinçer Y, Akçay T, Ilkova H, Alademir Z, Ozbay G. DNA damage and antioxidant defense in peripheral leukocytes of patients with Type I diabetes mellitus. Mutat Res. 2003;527:49–55. doi: 10.1016/s0027-5107(03)00073-3. [DOI] [PubMed] [Google Scholar]
- 15.Sliwinska A, Blasiak J, Kasznicki J, Drzewoski J. In vitro effect of gliclazide on DNA damage and repair in patients with type 2 diabetes mellitus (T2DM) Chem Biol Interact. 2008;173:159–65. doi: 10.1016/j.cbi.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Kushwaha S, Vikram A, Trivedi PP, Jena GB. Alkaline, Endo III and FPG modified comet assay as biomarkers for the detection of oxidative DNA damage in rats with experimentally induced diabetes. Mutat Res. 2011;726:242–50. doi: 10.1016/j.mrgentox.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Lorenzi M, Montisano DF, Toledo S, Barrieux A. High glucose induces DNA damage in cultured human endothelial cells. J Clin Invest. 1986;77:322–5. doi: 10.1172/JCI112295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang S, Chintapalli J, Sodagum L, Baskin S, Malhotra A, Reiss K, et al. Activated IGF-1R inhibits hyperglycemia-induced DNA damage and promotes DNA repair by homologous recombination. Am J Physiol Renal Physiol. 2005;289:F1144–52. doi: 10.1152/ajprenal.00094.2005. [DOI] [PubMed] [Google Scholar]
- 19.Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: The role of protein kinase C and NAD (P) H-oxidase activation. Diabetes. 2003;52:2795–804. doi: 10.2337/diabetes.52.11.2795. [DOI] [PubMed] [Google Scholar]
- 20.Takao T, Horino T, Kagawa T, Matsumoto R, Shimamura Y, Ogata K, et al. Possible involvement of intracellular angiotensin II receptor in high-glucose-induced damage in renal proximal tubular cells. J Nephrol. 2011;24:218–24. doi: 10.5301/jn.2010.5785. [DOI] [PubMed] [Google Scholar]
- 21.Jena NR. DNA damage by reactive species: Mechanisms, mutation and repair. J Biosci. 2012;37:503–17. doi: 10.1007/s12038-012-9218-2. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Zhou J, Wang T, Cai L. High level glucose increases mutagenesis in human lymphoblastoid cells. Int J Biol Sci. 2007;3:375–9. doi: 10.7150/ijbs.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee AT, Reis D, Eriksson UJ. Hyperglycemia-induced embryonic dysmorphogenesis correlates with genomic DNA mutation frequency in vitro and in vivo. Diabetes. 1999;48:371–6. doi: 10.2337/diabetes.48.2.371. [DOI] [PubMed] [Google Scholar]
- 24.Frede J, Adams DJ, Jones PH. Mutation, clonal fitness and field change in epithelial carcinogenesis. J Pathol. 2014;234:296–301. doi: 10.1002/path.4409. [DOI] [PubMed] [Google Scholar]
- 25.Berstein LM, Alexandrov VA. Glucose consumption, newborn weight and nitrosomethylurea transplacental carcinogenesis in rats. Cancer Lett. 1984;25:171–6. doi: 10.1016/s0304-3835(84)80042-7. [DOI] [PubMed] [Google Scholar]
- 26.Donovan PJ, Smith GT, Riggs CW, Alexandrov VA. Effects of glucose on cloning efficiency and mutagenesis of fetal rat cells. Teratog Carcinog Mutagen. 2002;22:329–34. doi: 10.1002/tcm.10027. [DOI] [PubMed] [Google Scholar]
- 27.Stopper H, Schinzel R, Sebekova K, Heidland A. Genotoxicity of advanced glycation end products in mammalian cells. Cancer Lett. 2003;190:151–6. doi: 10.1016/s0304-3835(02)00626-2. [DOI] [PubMed] [Google Scholar]
- 28.Schupp N, Schinzel R, Heidland A, Stopper H. Genotoxicity of advanced glycation end products: Involvement of oxidative stress and of angiotensin II type 1 receptors. Ann N Y Acad Sci. 2005;1043:685–95. doi: 10.1196/annals.1333.079. [DOI] [PubMed] [Google Scholar]
- 29.Fukami K, Yamagishi S, Kaifu K, Matsui T, Kaida Y, Ueda S, et al. Telmisartan inhibits AGE-induced podocyte damage and detachment. Microvasc Res. 2013;88:79–83. doi: 10.1016/j.mvr.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 30.Mizutani K, Ikeda K, Nishikata T, Yamori Y. Phytoestrogens attenuate oxidative DNA damage in vascular smooth muscle cells from stroke-prone spontaneously hypertensive rats. J Hypertens. 2000;18:1833–40. doi: 10.1097/00004872-200018120-00018. [DOI] [PubMed] [Google Scholar]
- 31.Pazdro R, Burgess JR. Differential effects of a-tocopherol and N-acetyl-cysteine on advanced glycation end product-induced oxidative damage and neurite degeneration in SH-SY5Y cells. Biochim Biophys Acta. 2012;1822:550–6. doi: 10.1016/j.bbadis.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Beeharry N, Lowe JE, Hernandez AR, Chambers JA, Fucassi F, Cragg PJ, et al. Linoleic acid and antioxidants protect against DNA damage and apoptosis induced by palmitic acid. Mutat Res. 2003;530:27–33. doi: 10.1016/s0027-5107(03)00134-9. [DOI] [PubMed] [Google Scholar]
- 33.Donmez-Altuntas H, Sahin F, Bayram F, Bitgen N, Mert M, Guclu K, et al. Evaluation of chromosomal damage, cytostasis, cytotoxicity, oxidative DNA damage and their association with body-mass index in obese subjects. Mutat Res Genet Toxicol Environ Mutagen. 2014;771:30–6. doi: 10.1016/j.mrgentox.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Defronzo RA. Banting lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–95. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Othman EM, Leyh A, Stopper H. Insulin mediated DNA damage in mammalian colon cells and human lymphocytes in vitro. Mutat Res. 2013;745-746:34–9. doi: 10.1016/j.mrfmmm.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 36.Simone S, Gorin Y, Velagapudi C, Abboud HE, Habib SL. Mechanism of oxidative DNA damage in diabetes: Tuberin inactivation and downregulation of DNA repair enzyme 8-oxo-7,8-dihydro-2’-deoxyguanosine-DNA glycosylase. Diabetes. 2008;57:2626–36. doi: 10.2337/db07-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye C, Li X, Wang Y, Zhang Y, Cai M, Zhu B, et al. Diabetes causes multiple genetic alterations and downregulates expression of DNA repair genes in the prostate. Lab Invest. 2011;91:1363–74. doi: 10.1038/labinvest.2011.87. [DOI] [PubMed] [Google Scholar]
- 38.Orie NN, Zidek W, Tepel M. Reactive oxygen species in essential hypertension and non-insulin-dependent diabetes mellitus. Am J Hypertens. 1999;12:1169–74. doi: 10.1016/s0895-7061(99)00129-6. [DOI] [PubMed] [Google Scholar]
- 39.Seghrouchni I, Drai J, Bannier E, Rivière J, Calmard P, Garcia I, et al. Oxidative stress parameters in type I, type II and insulin-treated type 2 diabetes mellitus; insulin treatment efficiency. Clin Chim Acta. 2002;321:89–96. doi: 10.1016/s0009-8981(02)00099-2. [DOI] [PubMed] [Google Scholar]
- 40.McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010;363:2339–50. doi: 10.1056/NEJMra0906948. [DOI] [PubMed] [Google Scholar]
- 41.Lai CQ, Tucker KL, Parnell LD, Adiconis X, García-Bailo B, Griffith J, et al. PPARGC1A variation associated with DNA damage, diabetes, and cardiovascular diseases: The Boston Puerto Rican Health Study. Diabetes. 2008;57:809–16. doi: 10.2337/db07-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma RC, So WY, Tam CH, Luk AO, Ho JS, Wang Y, et al. Genetic variants for type 2 diabetes and new-onset cancer in Chinese with type 2 diabetes. Diabetes Res Clin Pract. 2014;103:328–37. doi: 10.1016/j.diabres.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 43.Habib SL, Liang S. Hyperactivation of Akt/mTOR and deficiency in tuberin increased the oxidative DNA damage in kidney cancer patients with diabetes. Oncotarget. 2014;5:2542–50. doi: 10.18632/oncotarget.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–32. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blasiak J, Arabski M, Krupa R, Wozniak K, Zadrozny M, Kasznicki J, et al. DNA damage and repair in type 2 diabetes mellitus. Mutat Res. 2004;554:297–304. doi: 10.1016/j.mrfmmm.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 46.Lan A, Du J. Potential role of Akt signaling in chronic kidney disease. Nephrol Dial Transplant. 2015;30:385–94. doi: 10.1093/ndt/gfu196. [DOI] [PubMed] [Google Scholar]
- 47.Pu J, Peng G, Li L, Na H, Liu Y, Liu P. Palmitic acid acutely stimulates glucose uptake via activation of Akt and ERK1/2 in skeletal muscle cells. J Lipid Res. 2011;52:1319–27. doi: 10.1194/jlr.M011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem. 2002;277:35364–70. doi: 10.1074/jbc.M205838200. [DOI] [PubMed] [Google Scholar]
- 49.Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: A double-edged sword in cell proliferation and genome stability. J Oncol 2012. 2012 doi: 10.1155/2012/951724. 951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nacerddine K, Beaudry JB, Ginjala V, Westerman B, Mattiroli F, Song JY, et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J Clin Invest. 2012;122:1920–32. doi: 10.1172/JCI57477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Habib SL, Phan MN, Patel SK, Li D, Monks TJ, Lau SS. Reduced constitutive 8-oxoguanine-DNA glycosylase expression and impaired induction following oxidative DNA damage in the tuberin deficient Eker rat. Carcinogenesis. 2003;24:573–82. doi: 10.1093/carcin/24.3.573. [DOI] [PubMed] [Google Scholar]
- 52.Habib SL. Molecular mechanism of regulation of OGG1: Tuberin deficiency results in cytoplasmic redistribution of transcriptional factor NF-YA. J Mol Signal. 2009;4:8. doi: 10.1186/1750-2187-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakofsky CJ, Ayyar S, Malkova A. Break-induced replication and genome stability. Biomolecules. 2012;2:483–504. doi: 10.3390/biom2040483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003;17:1195–214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 55.Retèl J, Hoebee B, Braun JE, Lutgerink JT, van den Akker E, Wanamarta AH, et al. Mutational specificity of oxidative DNA damage. Mutat Res. 1993;299:165–82. doi: 10.1016/0165-1218(93)90094-t. [DOI] [PubMed] [Google Scholar]
- 56.Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol. 2010;38:96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- 57.Soto AM, Sonnenschein C. One hundred years of somatic mutation theory of carcinogenesis: Is it time to switch? Bioessays. 2014;36:118–20. doi: 10.1002/bies.201300160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pitot HC. Fundamentals of Oncology. 4th ed. New York: Marcel Dekker; 2002. pp. 223–72. 335-71. [Google Scholar]
- 59.Kender Z, Fleming T, Kopf S, Torzsa P, Grolmusz V, Herzig S, et al. Effect of metformin on methylglyoxal metabolism in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2014;122:316–9. doi: 10.1055/s-0034-1371818. [DOI] [PubMed] [Google Scholar]
- 60.Na HJ, Park JS, Pyo JH, Lee SH, Jeon HJ, Kim YS, et al. Mechanism of metformin: Inhibition of DNA damage and proliferative activity in Drosophila midgut stem cell. Mech Ageing Dev. 2013;134:381–90. doi: 10.1016/j.mad.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 61.Kong AP, Yang X, So WY, Luk A, Ma RC, Ozaki R, et al. Additive effects of blood glucose lowering drugs, statins and renin-angiotensin system blockers on all-site cancer risk in patients with type 2 diabetes. BMC Med. 2014;12:76. doi: 10.1186/1741-7015-12-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang X, So WY, Ma RC, Kong AP, Xu G, Chan JC. Diabetes and cancer: The mechanistic implications of epidemiological analyses from the Hong Kong Diabetes Registry. Diabetes Metab Res Rev. 2012;28:379–87. doi: 10.1002/dmrr.2287. [DOI] [PubMed] [Google Scholar]