

Abstract
Diabetes mellitus in early pregnancy causes birth defects by disturbing metabolic homeostasis and increasing programmed cell death in the embryo. Over-activation of phospholipase Cβ3 and γ1 suggests disturbed phospholipid metabolism, which is an important in regulation of cell signaling and activity. Metabolomic examinations reveal significant changes in the profile of phospholipid metabolism. Among the metabolites, levels of phosphatidylinositol bisphosphate (PIP2) are increased. PIP2 effector PTEN (phosphatase and tensin homolog deleted on chromosome 10) is activated. Activation of protein kinase Bα (PKBα, or AKT1) and mTOR (mechanistic target of rapamycin) is decreased. Inhibition of PLCs and PTEN suppresses over-generation of reactive oxygen species and inhibition of PLCs prevents fragmentation of mitochondria in neural stem cells cultured in high glucose. These observations suggest that maternal hyperglycemia disrupts phospholipid metabolism, leading to perturbation of mitochondrial dynamics and redox homeostasis and suppression of the PKB-mTOR cell survival signaling in the embryos.
Keywords: Diabetic embryopathy, Phospholipid, PTEN, PKB, AKT, mTOR, Reactive oxygen species, Mitochondrion
1. Introduction
Diabetes mellitus in early pregnancy causes congenital birth defects in newborn infants, a complication known as diabetic embryopathy [1]. The abnormalities in the central nervous system, including exencephaly and spina bifida, are a result of incomplete closure of the neural tube during embryogenesis, and are, therefore, known as neural tube defects (NTDs) [2].
Maternal hyperglycemia perturbs intracellular metabolic homeostasis and organelle function, generating intracellular stress conditions in the embryo [3]. Under the stress conditions, molecular cascades are activated to cause excessive programmed cell death (apoptosis) in the neural epithelium, resulting in failure in neural tube closure [4].
Phospholipid metabolism is an important process in regulation of cell signaling and activities [5]. It is regulated by members of the phospholipase (PL) A, C, and D families [5, 6]. Activation of cytosolic PLA2 (cPLA2) and peroxidation of arachidonic acid (AA) to generate isoprostanes (e.g., 8-iso-PGF2α) have been shown to be involved in diabetic embryopathy [7, 8]. Phospholipid metabolism is a complex process, involving multiple factors and metabolic pathways. Therefore, systematic characterization of global metabolic profile is essential for identification of novel intracellular signaling in regulation of cell apoptosis in diabetic embryopathy.
PLCs are important enzymes to generate second messengers to regulation intracellular signaling [6]. They act on phosphatidylinositol (PI) monophosphate and PI biphosphate (PIP2) [6]. Cleavage of PIP2 generates second messengers, diacylglycerol (DAG) and inositol triphosphate (IP3) [9]. PIP2 can be further phosphorylated to become PI triphosphate (PIP3) [9, 10]. PIP3 activates protein kinase Bs (PKBs), also known as AKTs. PKBs activate mTOR (mechanistic target of rapamycin) to regulate cell survival and proliferation [11]. PIP2 itself can activate PTEN (phosphatase and tensin homolog deleted on chromosome 10). PTEN dephosphorylates (deactivates) PIP3 and PKBs, and thus, suppresses the PKB-mTOR cell survival signaling [12].
The PKB-mTOR system controls cell survival and apoptosis by regulating pro- and anti-apoptotic factors in mitochondria [13]. Perturbation of the apoptotic regulators disturbs mitochondrial morphological dynamics and membrane activities, leading to generation of high levels of reactive oxygen species (ROS) and, resultant oxidative stress [14, 15].
In this study, we characterized the global profiles of phospholipid metabolism in diabetic embryopathy, demonstrated effects of metabolites on mitochondrial dynamics and intracellular redox homeostasis, and identified a novel signaling cascade, involving PIP2, PTEN, PKBs, and mTOR, which regulate cell survival in diabetic embryopathy.
2. Materials and Methods
2.1. Diabetic animal model
The use of animals was approved by the Institutional Animal Care and Use Committee of University of Maryland, Baltimore. Generation of diabetic mice was described previously [16]. Briefly, female mice (C57BL/6J) were induced diabetic (DM; blood glucose ≥ 250 mg/dl or 14 mM) via intravenous injection of streptozotocin (in citrate buffer; 65 mg/kg body weight). Non-diabetes (ND) control mice were injected with citrate buffer. The female mice were paired with normal male mice after euglycemia (~8 mM) was restored by subcutaneous implantation of insulin pellets. Insulin implants were removed at embryonic (E) day E5.5 to become hyperglycemic again from E6.5 before neurulation begins [17]. At E10.5, embryos were collected for examination of NTDs (open neural tube) and metabolic and molecular changes.
2.2. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)
Details are given in Supplementary Materials and Methods.
2.3. Ultra performance liquid chromatography (UPLC) and electrospray quadruple time-of-flight mass spectrometry (QTOF-MS)
The neural tubes of the embryos were isolated in cold phosphate-buffered saline (pH 7.4) and individually collected. Protein-free samples were prepared from tissue homogenates in 50% methanol containing internal standards (10 μl of 1 mg/ml debrisoquine and 50 μl of 1 mg/ml 4-Nitrobenzoic acid) and assayed using UPLC-QTOF-MS, using Acquity UPLC (Xevo; Waters Corporation) and G2-QTOF-MS systems (Waters Corporation). Detailed procedures are given in Supplementary Materials and Methods
2.4. Western blot assay
Detailed procedures are given in Supplementary Materials and Methods. Primary antibodies to the following proteins were used in this study: PLCβ3, phospho-PLCβ3 (Ser1105), PLCγ1, phospho-PLCγ1 (Tyr783), PTEN, phospho-PTEN (Ser380/Thr382/383), non-phospho-PTEN, PKBα, phospho-PKBα (Ser473), mTOR, phospho-mTOR (Ser2448) (Cell Signaling Technology, Beverly, MA) and β-actin (Abcam, Cambridge, MA).
2.5. Cell-based ROS assay
Neural stem cells (NE-4C; American Type Culture Collection), derived from E9 mouse embryos, were plated on 96-well plates (2 × 104 cells/well) in Dulbecco’s Modified Eagle Medium (DMEM, Life Technologies) containing 10% fetal bovine serum and a normal concentration of D-glucose (NG; 6 mM) at 37°C for 16 hours. The cells were treated with high glucose (HG; 33 mM) containing PLC inhibitor U73122 (HG+U73122) or PTEN inhibitor SF1670 (HG+SF1670), along with control groups [NG+vehicle (VEH, 0.1% dimethyl sulfoxide), HG+VEH, and L-Glucose (6 mM D-glucose, 27 mM L-glucose)+VEH]. After 24 hours of treatment, the cells were loaded with fluorescent dyes, 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; 5 μM), Hoechst 33342 (2 μM), and propidium iodide (1 μg/ml; Life Technologies) for 10 minutes at 37°C. After washing twice with FluoroBite DMEM (Life Technologies), the levels of fluorescence were measured using a Biotek Synergy microplate reader at 480 nm, 360 nm, and 540 nm. The ratio of the fluorescent values between H2DCFDA (480 nm) and Hoechst (360 nm) indicates the level of ROS per a number of cells. The ratio between the values of propidium iodide (540 nm) and Hoechst 33342 (360 nm) indicates cell viability. The experiments were repeated three times with six duplicates in each experiment.
2.6. Mitochondrial morphology imaging
The neural stem cells (NE-4C) were plated on chambered glass slides (4 × 104 cells/chamber) and cultured in NG for 16 hours. The cells were treated with NG-VEH, HG-VEH, and HG-U73122 at 37°C for 24 hours. The cells were loaded with MitoTracker Red (1 μM; Life Technology) at 37°C for 10 minutes to label mitochondria, washed with FluoroBite DMEM, fixed with 4% paraformaldehyde in PBS, and examined under a fluorescent microscope.
2.7. Statistical analyses
Ratios of fluorescence intensity at two different wavelengths and ratios of band density of interest to that of β-actin on Western blots, presented as Mean ± standard deviation (SD), were analyzed using the Student t-test. A p-value of < 0.05 was considered statistically significant.
3. Results
3.1. NTDs and apoptosis in embryos of diabetic mice
Maternal hyperglycemia disrupts neural tube fusion during the period of neurulation (E8.5 to E11.5) [17]. We examined NTDs at E10.5, which is the late stage of neurulation. The neural tube of the embryos of the ND group was closed (Fig. 1A). However, in the DM group, the neural tube was still open in the brain and/or spinal cord region (Fig. 1B).
Fig. 1. NTDs and apoptosis in E10.5 embryos.
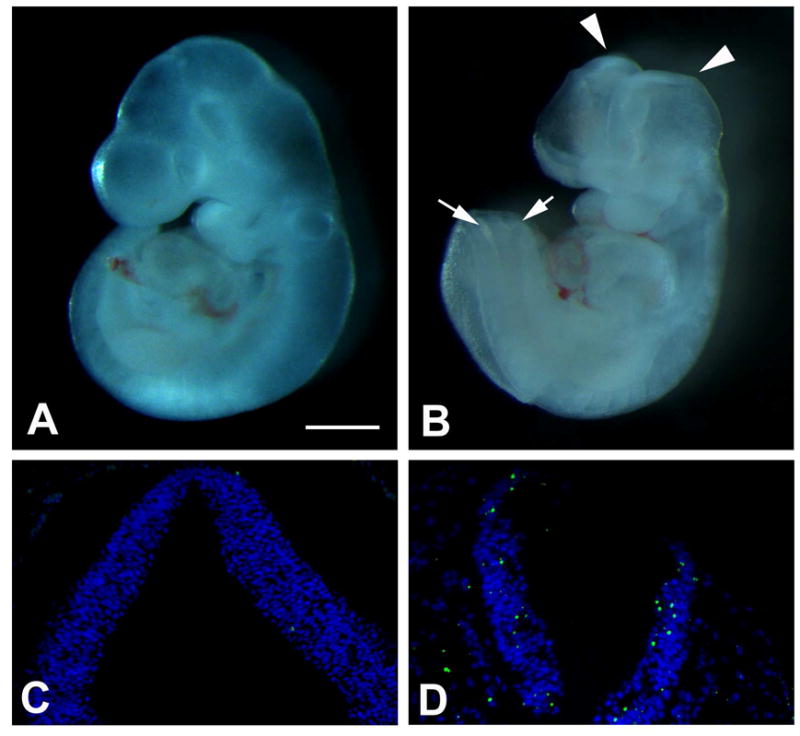
(A,C) ND. (B,D) DM. (A,B) Embryos. (C,D) TUNEL assay of tissue sections of the neural tube. TUNEL-positive signals are green. DAPI conterstaining is blue. Arrowheads and arrows in B indicate open brain and spinal cord, respectively. Scale bar = 5 mm in A, B; 100 μm in C, D.
We also examined apoptosis in the neural tissues of the embryos. In the ND group, very few TUNEL-positive signals were seen in the neural tube (Fig. 1C). In the DM group, much higher levels of TUNEL-positive apoptotic bodies were present in the neural epithelium of the open neural tube (Fig. 1D).
3.2. Phospholipid metabolism in embryos of diabetic mice
To gain systematic insights into the impact of phospholipid metabolism, we examined profiles of lipid metabolome of the embryos of ND and DM groups, using a high resolution UPLC-MS. Comparisons of the profiles showed evident differences between the ND and DM groups (Supplementary Figs. S1,2). Statistical and bioinformatic analyses revealed metabolites that showed significant increases or decreases in the DM group, compared with those in the ND group (Table 1). Significant perturbations were seen in lipids, phospholipids, amino acids, and co-enzyme As (Table 1; Supplementary Table S1).
Table 1.
Metabolomic profiles in diabetic embryopathy
| Name | M/Z | RT | FC (DM/ND) | p-value | Mode |
|---|---|---|---|---|---|
| Glycoursodeoxycholic acid | 450.3566 | 380.1778 | 4.9155 | 0.0303 | Positive |
| CE(20:3(8Z,11Z,14Z)) | 675.6755 | 556.3150 | 4.3483 | 0.0487 | Positive |
| Cer(d18:1/26:1(17Z)) | 676.6788 | 556.3023 | 4.2716 | 0.0491 | Positive |
| Geranylgeranylcysteine | 408.3078 | 504.8262 | 4.1764 | 0.0129 | Positive |
| Cholic acid | 409.3107 | 504.9104 | 4.0164 | 0.0118 | Positive |
| TG(24:1(15Z)/24:0/o-18:0) | 1044.6090 | 504.1836 | 3.7078 | 0.0261 | Negative |
| Cepanone | 211.1700 | 504.8546 | 3.6538 | 0.0161 | Positive |
| Geranylgeranylcysteine | 408.3084 | 138.7383 | 3.5957 | 0.0157 | Positive |
| PE(18:3/P-16:0) | 698.6586 | 241.0325 | 3.5759 | 0.0488 | Positive |
| Orotic acid | 156.9953 | 724.6285 | 3.4963 | 0.0317 | Positive |
| Lysyl-Proline | 244.1866 | 504.2698 | 3.3884 | 0.0110 | Positive |
| Metanephrine | 198.1859 | 503.9074 | 3.3789 | 0.0296 | Positive |
| N-Arachidonoyl glycine | 362.3021 | 137.1551 | 3.0794 | 0.0258 | Positive |
| Acetomenaphthone | 259.0975 | 46.1024 | 2.9574 | 0.0264 | Positive |
| Leucyl-Proline | 229.1929 | 504.1836 | 2.9560 | 0.0493 | Positive |
| PIP2(20:4/18:1(11Z)) | 1045.6037 | 502.9756 | 2.9042 | 0.0461 | Positive |
| Cysteinyl-Asparagine | 236.1623 | 506.7862 | 2.8581 | 0.0404 | Positive |
| Thiamine | 266.1833 | 136.5271 | 2.6693 | 0.0400 | Positive |
| SM(d18:0/12:0) | 651.4778 | 248.2837 | 0.3489 | 0.0359 | Positive |
| Melitin | 903.1448 | 517.2036 | 0.3039 | 0.0418 | Positive |
| PGP(18:0/20:3) | 881.1298 | 526.6806 | 0.3018 | 0.0377 | Positive |
| (2E)-Dodecenoyl-CoA | 944.6834 | 806.9919 | 0.2975 | 0.0484 | Positive |
| PI(22:4/18:0) | 915.6454 | 489.3901 | 0.2765 | 0.0437 | Negative |
| PE(15:0/14:0) | 650.4675 | 248.2485 | 0.2719 | 0.0208 | Negative |
| PC(24:0/P-18:0) | 859.1142 | 537.1303 | 0.2698 | 0.0340 | Positive |
| TG(14:1(9Z)/22:5/20:4) | 899.6532 | 817.4480 | 0.2564 | 0.0360 | Positive |
| PE(24:0/24:0) | 916.6511 | 490.5900 | 0.2475 | 0.0466 | Positive |
| PC(22:0/24:1(15Z)) | 928.1835 | 865.1219 | 0.2460 | 0.0410 | Positive |
| PC(16:1(9Z)/24:0) | 844.5977 | 417.0910 | 0.2433 | 0.0288 | Positive |
| TG(18:1(9Z)/15:0/o-18:0) | 834.1120 | 829.4199 | 0.2429 | 0.0374 | Positive |
| TG(24:1(15Z)/18:1(9Z)/22:2) | 1024.2350 | 829.3107 | 0.2251 | 0.0249 | Positive |
| TG(20:4/20:2n6/22:6) | 965.6926 | 798.0809 | 0.2161 | 0.0314 | Positive |
| TG(18:2/20:3n6/20:4) | 943.6815 | 807.3675 | 0.2102 | 0.0225 | Positive |
| TG(22:4/20:5/20:4) | 977.1785 | 373.3367 | 0.2062 | 0.0447 | Positive |
| PC(24:1(15Z)/20:3) | 894.6373 | 506.1557 | 0.2046 | 0.0439 | Positive |
| TG(18:4/20:5/20:4) | 921.6669 | 812.4027 | 0.2037 | 0.0254 | Positive |
| PGP(16:0/16:1(9Z)) | 801.0729 | 433.9451 | 0.1938 | 0.0288 | Positive |
| (3′-sulfo)Galbeta-Cer(d18:0/22:0(2OH)) | 880.6211 | 396.0516 | 0.1806 | 0.0319 | Positive |
| TG(22:4/24:0/o-18:0) | 1010.2210 | 749.1838 | 0.1791 | 0.0179 | Positive |
| TG(14:1(9Z)/15:0/20:5) | 810.0757 | 334.0019 | 0.1787 | 0.0287 | Positive |
| PGP(18:1(9Z)/20:3) | 879.6190 | 396.0207 | 0.1769 | 0.0394 | Positive |
| TG(22:0/18:1(11Z)/o-18:0) | 932.6513 | 386.7160 | 0.1643 | 0.0318 | Positive |
| PC(20:0/22:4) | 866.6123 | 408.3092 | 0.1604 | 0.0152 | Positive |
| Triiodothyronine glucuronide | 828.0993 | 547.1272 | 0.1549 | 0.0345 | Positive |
| TG(18:4/20:5/18:4) | 893.6341 | 504.7999 | 0.1480 | 0.0229 | Positive |
| PC(24:1(15Z)/24:1(15Z)) | 955.1662 | 379.9027 | 0.1478 | 0.0209 | Positive |
| Salicyl CoA | 886.1400 | 671.3439 | 0.1466 | 0.0194 | Positive |
| TG(24:0/22:0/o-18:0) | 1018.2164 | 567.8181 | 0.1199 | 0.0156 | Positive |
| PIP(16:1(9Z)/18:0) | 889.1267 | 399.8242 | 0.1180 | 0.0213 | Positive |
| TG(22:0/22:1/18:3) | 996.2041 | 584.8505 | 0.1119 | 0.0211 | Positive |
| TG(18:4/14:0/22:6) | 871.6229 | 519.4851 | 0.1041 | 0.0114 | Positive |
| 3-Oxooctanoyl-CoA | 908.1547 | 650.9502 | 0.1022 | 0.0135 | Positive |
| TG(20:0/22:5/18:3) | 959.6771 | 466.3990 | 0.0993 | 0.0227 | Positive |
| PG(16:1/18:3) | 743.0342 | 352.9815 | 0.0953 | 0.0197 | Negative |
| TG(18:1/22:1/o-18:0) | 930.1656 | 632.2212 | 0.0913 | 0.0075 | Negative |
| OPC4-CoA | 988.2058 | 768.9839 | 0.0802 | 0.0104 | Positive |
| (S)-3-Hydroxydodecanoyl-CoA | 966.1959 | 797.8451 | 0.0768 | 0.0116 | Positive |
| TG(24:0/18:0/18:1(11Z)) | 974.1922 | 599.4445 | 0.0672 | 0.0118 | Positive |
| Octanoyl-CoA | 894.1380 | 505.3860 | 0.0641 | 0.0151 | Positive |
| (S)-Hydroxydecanoyl-CoA | 938.1642 | 478.7701 | 0.0580 | 0.0242 | Positive |
| TG(22:0/22:4/o-18:0) | 982.1895 | 454.2675 | 0.0579 | 0.0349 | Positive |
| PG(20:3/22:2) | 853.5988 | 321.2533 | 0.0576 | 0.0205 | Positive |
| TG(18:1(11Z)/22:1(13Z)/20:4) | 963.1634 | 298.0084 | 0.0568 | 0.0454 | Positive |
| PC(P-18:1(9Z)/18:4) | 765.0458 | 345.7993 | 0.0567 | 0.0298 | Positive |
| TG(15:0/o-18:0/18:0) | 836.0951 | 417.6578 | 0.0556 | 0.0171 | Positive |
| TG(14:0/18:0/o-18:0) | 822.0772 | 333.9968 | 0.0527 | 0.0495 | Positive |
| TG(14:0/22:0/22:1(13Z)) | 946.1623 | 368.8135 | 0.0509 | 0.0422 | Positive |
| TG(20:4/22:1/o-18:0) | 952.1783 | 617.0061 | 0.0496 | 0.0102 | Positive |
| PE(P-16:0/22:5) | 750.5430 | 274.8144 | 0.0492 | 0.0499 | Positive |
| PIP(20:3/16:0) | 941.1504 | 302.3774 | 0.0469 | 0.0495 | Positive |
| TG(20:5/16:1(9Z)/20:5) | 897.6255 | 311.8055 | 0.0445 | 0.0383 | Positive |
| TG(22:1(13Z)/18:4/o-18:0) | 924.1475 | 377.3902 | 0.0431 | 0.0378 | Positive |
| SM(d16:1/24:1(15Z)) | 787.0600 | 340.0357 | 0.0368 | 0.0256 | Positive |
| PC(24:1(15Z)/22:5) | 919.1381 | 307.0278 | 0.0336 | 0.0360 | Negative |
| PE(22:6/24:1(15Z)) | 874.6095 | 316.4193 | 0.0327 | 0.0329 | Negative |
| PC(20:2/24:1(15Z)) | 896.6226 | 311.8084 | 0.0299 | 0.0344 | Negative |
| PI(16:0/16:1(9Z)) | 809.0733 | 333.4410 | 0.0189 | 0.0212 | Positive |
| PS(14:1(9Z)/14:1(9Z)) | 676.3967 | 178.1948 | 0.0097 | 0.0463 | Positive |
Cer, ceramide. DM, diabetic. FC, fold change. M/Z, mass-to-charge ratio. ND, non-diabetic. PA, phosphatidic acid. PC, phosphatidylcholines. PE, phosphatidylethanolamine. PG, phosphatidylglycerol. PGP, phosphatidylglycerolphosphate. PI, phosphatidylinositol. PIP, phosphatidylinositol phosphate. PS, phosphatidylserine. RT, retention time. SM, sphingomyelin. TG, triglyceride.
3.3. Activation of phospholipase Cs in the embryos of diabetic mice
Phospholipid metabolism regulated by PLCs plays an important in regulation of intracellular signaling [6]. We examined the activation (phosphorylation) PLCs in the neural tube of the embryos from ND and DM dams. Significant increases in phosphorylation of PLCβ3 and PLCγ1 were detected in the embryos of the DM group, compared with those in the ND group (Figs. 2A, B).
Fig. 2. Activation of PLCs, PTEN, PKBα, and mTOR in the neural tube of embryos.

Western blot assay of total and phosphorylated (p) proteins from E10.5 neural tissues in non-diabetic (ND) and diabetic (DM) groups. (A) PLCβ3; (B) PLCγ1; (C) PTEN; (D) PKBα; (E) mTOR; β-actin, loading control. Data: Mean ± SD, * p<0.05, n = 4 (p-PLCβ3, p-PLCγ1, p-PKBα, p-mTOR); n = 6 (p-PTEN, Non-p-PTEN).
3.4. Activation of PTEN, PKB, and mTOR in diabetic embryopathy
The metabolic assay revealed significant increases in PIP2, which can activate PTEN [12]. We examined the activation of PTEN in the neural tissues of embryos from ND and DM mice. When the C-terminus of PTEN is phosphorylated at the S380, T382, and T383 positions, it blocks the activity of the enzyme. Upon stimulation by PIP2, PTEN loses these phosphate groups and translocates to the membrane to exert phosphatase activity [18, 19]. We detected decreases in phosphorylated and increases in non-phosphorylated PTEN in embryos of the DM group, compared with those in the ND group (Fig. 2C).
PTEN regulates PKB-mTOR cell survival signaling [19]. To investigate whether PKB and mTOR are involved in diabetic embryopathy, we assessed the changes in activation of these factors. We observed that levels of phosphorylation of PKBα and mTOR in the DM group were significantly lower than those in the ND group (Figs. 2D, E)
3.5. Effect of PLC and PTEN inhibition on ROS generation in neural stem cells
To determine whether hyperglycemia-activated PLCs and PTEN affect cellular homeostasis, we used the PLC inhibitor U73122 (0.1 μM and 0.2 μM) and the PTEN inhibitor SF1670 (0.1 μM and 0.5 μM) to block their activity and assessed the effect on ROS generation in neural stem cells cultured in high glucose (33 mM).
We first tested if high glucose exerts osmotic effect on ROS generation, by comparing ROS levels between L-glucose+VEH group (6 mM D-glucose, 27 mM L-glucose) and NG+VEH group (6 mM D-glucose), and found no significant difference (data not shown). When the cells were treated with high D-glucose (33 mM; HG+VEH), levels of ROS were significantly elevated, compared with NG+VEH group (Fig. 3). The PLC and PTEN inhibitors in HG+U73122 and HG+SF1670 groups significantly decreased the ROS levels in the cells, to the level in the NG+VEH group (Fig. 3).
Fig. 3. Effects of PLC and PTEN inhibition on ROS generation in embryonic neural stem cells.

(A) PLC inhibition (U73122). (B) PTEN inhibition (SF1670). NG, normal glucose; HG, high glucose. VEH, vehicle. Data: Mean ± SD, *p<0.05 between HG+VEH and each of the other groups, n = 6, three repeats.
3.6. Effect of PLC inhibition on mitochondrial morphology
Generation of excess ROS is associated with mitochondrial dysfunction [20]. To address the questions whether high glucose alters mitochondrial morphology and PLC-mediated phospholipid metabolism affects mitochondrial dynamics under high glucose conditions, we examined changes in mitochondrial morphology in neural stem cells treated with HG+VEH and HG+U73122. In the cells of NG+VEH group, mitochondria showed long thread morphology and formed clusters in the cytoplasm (Fig. 4A). In contrast, in the cells of HG+VEH group, mitochondria were round and smaller (fragmented) (Fig. 4B). Treatment with the PLC inhibitor U73122 (HG+U73122) prevented high glucose-caused mitochondrial fragmentation (Fig. 4C).
Fig. 4. Effect of PLC inhibition on mitochondrial morphology in high glucose.

(A,B,C) MitoTracker Red. (D,E,F) Merged images of MitoTracker Red and DAPI (blue). (A,D) NG+VEH. (B,E) HG+VEH. (C,F) HG+U73122 (0.5 μM). Arrows in A and arrowheads in B indicate long and fragmented mitochondria, respectively. Scale bar, 10 μm for all images.
4. Discussion
Hyperglycemia in diabetic pregnancy perturbs intracellular metabolic homeostasis, leading to increased cell apoptosis and decreases cell proliferation, resulting embryonic malformations [3]. However, the triggers of these events, i.e., the early intracellular responses to hyperglycemia, remain to be identified. In the present study, we show that maternal hyperglycemia disturbs phospholipid metabolism and generates second messengers to suppress cell survival and promote apoptosis in the embryos of diabetic mice. High levels of PIP2 activate PTEN. PTEN suppresses the PKB-mTOR signaling in embryonic cells. Disruption of this cell survival signaling system leads to alteration of mitochondrial morphology and ROS production.
Dramatic metabolomic changes suggest that various intracellular activities are disturbed in the embryos of diabetic mice. For example, (S)-3-hydroxydodecanoyl-CoA, 3-oxooctanoyl-CoA, octanoyl-CoA, and (2E)-sodecenoyl-CoA, are essential for lipid synthesis and oxidation in mitochondria [21]. Depletion of these lipid derivatives suggests a compromise in mitochondrial membrane permeability and function in the embryos of diabetic mice.
The endogenous levels of phosphatidylglycerolphosphate (PGP) are also significantly decreased in the embryos diabetic mice. PGPs, having a net charge of −1 at physiological pH, are localized in the inner membrane of mitochondrion and influence mitochondrial respiration [22]. Two PGPs form a diphosphatidylglycerol, also known as cardiolipin, which is responsible for Barth syndrome with cardiac anomalies and growth retardation [23].
Phospholipid metabolism plays an important role in regulation of intracellular signaling and cellular activities [6]. Using a metabolomic approach, we observed significant changes in phospholipid metabolites in embryos of diabetic mice, including decreases in triacylglycerols (TGs), phosphocholine (PC), phosphoethanolamine (PE), phosphoinositol (PI), phosphatidylserine (PS), and sphingomyelins (SMs).
The mechanisms underlying the reduction of these phospholipid derivatives, either via increased lipid peroxidation or impaired phospholipid biosynthesis, remain to be delineated. However, the implication of the decreases is significant. For example, PC is a major source of choline synthesis in cells [24]. Choline is involved in protection of cells against extracellular and intracellular insults and important for fetal development [25]. The depletion of intracellular PC in embryos of diabetic mice suggests an attenuation of cellular defense capability.
SMs have been shown to have anti-apoptotic activity in cells under stress conditions [26] ; however, such activity appears to be diminished in diabetic embryopathy. SMs can be converted into ceramide (CER) [27]. CER, on the other hand, induces apoptosis [28]. In the embryos of diabetic mice, the decreases in SMs and increases in CER suggest a shifted balance from SM-mediated cell survival to CER-induced apoptosis in diabetic embryopathy.
Phosphatitylinositols (PIs), substrates of PLCs, are important membrane components and cell signaling molecules [29, 30]. The activation of PLCs suggests that the metabolism of PIs is altered by maternal hyperglycemia. The consequence may be multi-fold. The metabolites can affect various intracellular signaling systems. For example, DAG can activate protein kinase Cs [31]. It has been shown that some PKC isoforms are involved in diabetic embryopathy [32, 33]. In addition, disruption of PI metabolism can lead to membrane dysfunction, intracellular protein trafficking, and endo/exocytosis [29, 34]. Furthermore, in addition to the cell membrane, PLCs also act in organelles and the nucleus [35]. All these suggest that PLCs may play multiple and important roles in diabetic embryopathy.
As a signaling factor, PIP2 activates PTEN, which, in turn, represses the PKB-mTOR cell survival signaling [12]. Conditional deletion of the pten gene in neural progenitor cells demonstrates the role in PTEN in neural development by suppressing cell proliferation and survival [36]. Thus, in diabetic embryopathy, activated PTEN may act via the similar mechanisms to cause embryonic malformation.
One of the potential PTEN effector signaling pathways is the PKB/mTOR system [37], which enhances neural cell survival during embryogenesis [38, 39]. In diabetic embryopathy, the significant decreases in PKBα and mTOR activation implicate an attenuation of cell survival in the embryos. The PTEN-PKB-mTOR cell survival pathway in diabetic embryopathy deserves further investigation.
Cell survival capacity is diminished, in diabetic embryopathy, under intracellular stress conditions [3]. The imbalance between high levels of ROS and low levels of antioxidants manifests oxidative stress. Inhibition of PLCs or PTEN, which significant blunts high glucose-stimulated ROS increase in neural stem cells, suggests that PLC-mediated phospholipid metabolism and PTEN-induced signaling disturb intracellular redox homeostasis in embryonic cells of diabetic pregnancies.
One of the major sources of ROS in the embryos of diabetic pregnancy is mitochondrion [3]. Dysfunction of mitochondria is associated with their dynamic changes in morphology. In general, mitochondrial fusion/elongation (potentially associated with high energy production) and fission/fragmentation (defects in energy production and membrane integrity) are associated with cell survival and apoptosis, respectively [40]. In neural stem cells treated with high glucose, we observed mitochondrial fragmentation. Such effect can be prevented by inhibition of PLCs. The observations suggest that hyperglycemia alters mitochondrial dynamics, leading to dysfunction and ROS overproduction. Such adverse effects involve PLC-induced phospholipid metabolism.
Maternal hyperglycemia induces embryonic malformations through multiple molecular cascades [3]. To reduce embryonic abnormalities requires targeting multiple molecular events simultaneously [41, 42]. Inhibition of PLCs and PTEN to restore cell survival signaling is potentially a feasible approach. Selective inhibitors for these factors have been developed and employed in clinical applications for other diseases [43, 44]. Future work is aimed to explore the feasibility of this approach to prevention of birth defects in diabetic pregnancies.
Supplementary Material
Highlights.
Maternal hyperglycemia activates phospholipase Cs and alters phospholipid metabolism in embryo.
High levels of PIP2 activate PTEN.
The PKBα-mTOR cell survival signaling is suppressed.
PLCs and PTEN regulate high glucose-induced over-production of ROS in neural stem cells.
Inhibition of PLCs prevents mitochondrial fragmentation.
Acknowledgments
The authors thank Hua Li for technical assistance, and Dr. Julie Wu for critical reading and editing of the manuscript. This work was supported by grants R01HD076245 (to Z.Z.) and R03HD075995 (to Z.Z.) from National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Correa A, Gilboa SM, Besser LM, Botto LD, Moore CA, Hobbs CA, Cleves MA, Riehle-Colarusso TJ, Waller DK, Reece EA. Diabetes mellitus and birth defects. Am J Obstet Gynecol. 2008;199:237, e231–239. doi: 10.1016/j.ajog.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greene ND, Copp AJ. Neural tube defects. Annu Rev Neurosci. 2014;37:221–242. doi: 10.1146/annurev-neuro-062012-170354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao Z, Reece EA. New concepts in diabetic embryopathy. Clin Lab Med. 2013;33:207–233. doi: 10.1016/j.cll.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang P, Reece EA, Wang F, Gabbay-Benziv R. Decoding the oxidative stress hypothesis in diabetic embryopathy through proapoptotic kinase signaling. Am JObstet Gynecol. 2015;212:569–579. doi: 10.1016/j.ajog.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kent C, Carman GM, Spence MW, Dowhan W. Regulation of eukaryotic phospholipid metabolism. FASEB J. 1991;5:2258–2266. doi: 10.1096/fasebj.5.9.1860617. [DOI] [PubMed] [Google Scholar]
- 6.Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 7.Wentzel P, Welsh N, Eriksson UJ. Developmental damage, increased lipid peroxidation, diminished cyclooxygenase-2 gene expression, and lowered prostaglandin E2 levels in rat embryos exposed to a diabetic environment. Diabetes. 1999;48:813–820. doi: 10.2337/diabetes.48.4.813. [DOI] [PubMed] [Google Scholar]
- 8.Cederberg J, Basu S, Eriksson UJ. Increased rate of lipid peroxidation and protein carbonylation in experimental diabetic pregnancy. Diabetologia. 2001;44:766–774. doi: 10.1007/s001250051686. [DOI] [PubMed] [Google Scholar]
- 9.Kadamur G, Ross EM. Mammalian Phospholipase C. Annu Rev Physiol. 2012 doi: 10.1146/annurev-physiol-030212-183750. [DOI] [PubMed] [Google Scholar]
- 10.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 11.Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4:a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Worby CA, Dixon JE. Pten. Annu Rev Biochem. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 13.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Z, Eckert RL, Reece EA. Reduction in embryonic malformations and alleviation of endoplasmic reticulum stress by nitric oxide synthase inhibition in diabetic embryopathy. Reprod Sci. 2012;19:823–831. doi: 10.1177/1933719111434543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Copp AJ, Greene ND, Murdoch JN. The genetic basis of mammalian neurulation. Nat Rev Genet. 2003;4:784–793. doi: 10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- 18.Ross AH, Gericke A. Phosphorylation keeps PTEN phosphatase closed for business. Proc Natl Acad Sci. 2009;106:1297–1298. doi: 10.1073/pnas.0812473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 20.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 21.Tahiliani AG. Dependence of mitochondrial coenzyme A uptake on the membrane electrical gradient. J Biol Chem. 1989;264:18426–18432. [PubMed] [Google Scholar]
- 22.Stuhne-Sekalec L, Stanacev NZ. Modification of the biosynthesis and composition of polyglycerophosphatides in outer and inner mitochondrial membranes by cytidine liponucleotides. Membr Biochem. 1989;8:165–175. doi: 10.3109/09687688909025829. [DOI] [PubMed] [Google Scholar]
- 23.Schlame M, Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006;580:5450–5455. doi: 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 24.Fagone P, Jackowski S. Phosphatidylcholine and the CDP-choline cycle. Biochim Biophys Acta. 2013;1831:523–532. doi: 10.1016/j.bbalip.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeisel SH. Choline: critical role during fetal development and dietary requirements in adults. Annu Rev Nutr. 2006;26:229–250. doi: 10.1146/annurev.nutr.26.061505.111156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakraborty M, Jiang XC. Sphingomyelin and its role in cellular signaling. Adv Exp Med Biol. 2013;991:1–14. doi: 10.1007/978-94-007-6331-9_1. [DOI] [PubMed] [Google Scholar]
- 27.Mullen TD, Hannun YA, Obeid LM. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem J. 2012;441:789–802. doi: 10.1042/BJ20111626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolesnick RN, Kronke M. Regulation of ceramide production and apoptosis. Annu Rev Physiol. 1998;60:643–665. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- 29.Schink KO, Raiborg C, Stenmark H. Phosphatidylinositol 3-phosphate, a lipid that regulates membrane dynamics, protein sorting and cell signalling. Bioessays. 2013;35:900–912. doi: 10.1002/bies.201300064. [DOI] [PubMed] [Google Scholar]
- 30.Riehle RD, Cornea S, Degterev A. Role of phosphatidylinositol 3,4,5-trisphosphate in cell signaling. Adv Exp Med Biol. 2013;991:105–139. doi: 10.1007/978-94-007-6331-9_7. [DOI] [PubMed] [Google Scholar]
- 31.Zeng L, Webster SV, Newton PM. The biology of protein kinase C. Adv Exp Med Biol. 2012;740:639–661. doi: 10.1007/978-94-007-2888-2_28. [DOI] [PubMed] [Google Scholar]
- 32.Ma Y, Chen X, Sun M, Wan R, Zhu C, Li Y, Zhao Y. DNA cleavage function of seryl-histidine dipeptide and its application. Amino Acids. 2008;35:251–256. doi: 10.1007/s00726-007-0597-x. [DOI] [PubMed] [Google Scholar]
- 33.Cao Y, Zhao Z, Eckert RL, Reece EA. The essential role of protein kinase Cδ in diabetes-induced neural tube defects. J Matern Fetal Neonatal Med. 2012;25:2020–2024. doi: 10.3109/14767058.2012.677963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillooly DJ, Simonsen A, Stenmark H. Cellular functions of phosphatidylinositol 3-phosphate and FYVE domain proteins. Biochem J. 2001;355:249–258. doi: 10.1042/0264-6021:3550249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manzoli L, Martelli AM, Billi AM, Faenza I, Fiume R, Cocco L. Nuclear phospholipase C: involvement in signal transduction. Prog Lipid Res. 2005;44:185–206. doi: 10.1016/j.plipres.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 36.Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- 37.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 38.Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, Hemmings BA. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem. 2003;278:32124–32131. doi: 10.1074/jbc.M302847200. [DOI] [PubMed] [Google Scholar]
- 39.Hentges KE, Sirry B, Gingeras AC, Sarbassov D, Sonenberg N, Sabatini D, Peterson AS. FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc Natl Acad Sci. 2001;98:13796–13801. doi: 10.1073/pnas.241184198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reece EA, Wu YK, Zhao Z, Dhanasekaran D. Dietary vitamin and lipid therapy rescues aberrant signaling and apoptosis and prevents hyperglycemia-induced diabetic embryopathy in rats. Am J Obstet Gynecol. 2006;194:580–585. doi: 10.1016/j.ajog.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 42.Reece EA, Wu YK. Prevention of diabetic embryopathy in offspring of diabetic rats with use of a cocktail of deficient substrates and an antioxidant. Am J Obstet Gynecol. 1997;176:790–797. doi: 10.1016/s0002-9378(97)70602-1. discussion 797–798. [DOI] [PubMed] [Google Scholar]
- 43.Hou C, Kirchner T, Singer M, Matheis M, Argentieri D, Cavender D. In vivo activity of a phospholipase C inhibitor, 1-(6-((17beta-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-di one (U73122), in acute and chronic inflammatory reactions. J Pharmacol Exp Ther. 2004;309:697–704. doi: 10.1124/jpet.103.060574. [DOI] [PubMed] [Google Scholar]
- 44.Spinelli L, Lindsay YE, Leslie NR. PTEN inhibitors: an evaluation of current compounds. Adv Biol Regul. 2015;57:102–111. doi: 10.1016/j.jbior.2014.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.