

Abstract
Ageing and lifespan are strongly affected by metabolism. The maximal possible uptake of oxygen is not only a good predictor of performance in endurance sports, but also of life expectancy. Figuratively speaking, healthy ageing is a competitive sport. Although the root cause of ageing is damage to macromolecules, it is the balance with repair processes that is decisive. Reduced or intermittent nutrition, hormones and intracellular signalling pathways that regulate metabolism have strong effects on ageing. Homeostatic regulatory processes tend to keep the environment of the cells within relatively narrow bounds. On the other hand, the body is constantly adapting to physical activity and food consumption. Spontaneous fluctuations in heart rate and other processes indicate youth and health. A (homeo)dynamic aspect of homeostasis deteriorates with age. We are now in a position to develop computational models of human metabolism and the dynamics of heart rhythm and oxygen transport that will advance our understanding of ageing. Computational modelling of the connections between dietary restriction, metabolism and protein turnover may increase insight into homeostasis of the proteins in our body. In this way, the computational reconstruction of human physiological processes, the Physiome, can help prevent frailty and age-related disease.
Keywords: endurance, oxygen transport, heart rate variability, metabolism, computational modelling, homeodynamics
1. Introduction
Old age comes with frailty and disease, or does it? While most older people suffer from severely diminished endurance, there are some who complete marathons and even Ironman events (3.9 km swimming, 180 km cycling and 42 km running). They are slower of course, but winning times for 70 year olds are less than 60% over those of the best athletes aged 30. Ageing is thought to be a very complex phenomenon in which interactions of many processes at several levels are involved. In this perspective paper, we examine how computational models of physiological processes may help to understand ageing. Understanding all aspects of ageing would require considering many processes, and we will therefore mainly focus on a few physiological processes which play important roles in ageing: changes in metabolism, the decline in physical endurance with age and heart rate variability (HRV). This choice does not mean that we consider other processes unimportant for ageing. However, we propose that metabolism and endurance are two key physiological processes that can be fruitfully investigated with computational modelling approaches in the coming years, given the current state of knowledge and availability of measurement technologies.
Human endurance can be quantified well and involves the coordinated effort of many physiological systems such as breathing, the pump function of the heart, transport of oxygen and nutrients in the circulating blood, mitochondrial energy generation, muscle contraction, nutrition, metabolism and integrated control of the human system by nerves and hormones. Many subsystems in the body interact in complex ways to make an athlete perform at maximal speed. Maximal oxygen consumption is a good predictor of life expectancy. Studying the physiology of endurance therefore forms an attractive test bed to find out how to study the physiology of ageing by integrative computer modelling of the human body.
Metabolism affects ageing and longevity to a remarkable degree, and forms the second aspect of ageing which we focus on in this perspective paper. Increased metabolism supplies the energy for physical exercise, but we will see below that sufficient metabolic capacity is also a key requirement for healthy cell function in general. In a certain sense, keeping the body healthy is a highly competitive sport, even if we do not run the marathon. Metabolic processes support brain and heart function, immune cells and tissue repair, among others. Changes in metabolism brought about by interventions in nutrition or in hormonal regulation turn out to have big effects on lifespan, ageing and health.
Metabolism provides not only energy by burning the carbohydrates and fats in our food, but also small molecular building blocks, such as amino acids and fatty acids, which are used for synthesis of the macromolecules such as proteins and lipids. Metabolism is therefore of enormous importance for muscle building and energy turnover. The dark side of metabolism is that it can be the source of oxygen radicals and other by-products which damage macromolecules. To counteract damage, metabolism supports detoxifying processes. It is the balance between damage and repair rather than their individual rates that is important for ageing. It is striking that hormones which regulate the link between food intake and metabolism, such as insulin, appear to have a strong effect on longevity [1,2]. Signals from hormones and nerves impinging on the cells stimulate intracellular signalling networks, including the allocation of metabolic resources between activities such as maintenance and growth. Some proteins in these intracellular networks, such as insulin receptors and mTOR, also affect ageing strongly. This suggests that signalling networks which coordinate metabolism and nutrient uptake throughout the whole body modulate ageing. Top-down signals which coordinate whole-body physiology therefore play roles in ageing and the decline of endurance.
The interplay between many regulatory networks is involved in dynamic adaptation of the body to varying circumstances. Especially in young persons this creates a flexible and usually fluctuating physiological system, as seen in the variability of heart rate which declines with age. Rather than static regulation of body processes (homeostasis), very dynamic, sometimes chaotic patterns of regulation are seen (‘homeodynamics’) which form the third aspect of ageing treated in this perspective paper.
Considering that metabolic pathways consist of thousands of distinct enzymatic reactions and that multiple hormones and neural signals affect ageing via complex networks of intracellular signalling, it is no wonder that multiple factors have been proposed to play a role in ageing and that interactions of many processes (metabolism, molecular damage and repair, hormonal regulation, intracellular signalling, tissue homeostasis, etc.) must be considered to understand ageing. Making use of computer modelling to sort, arrange and explore such intricate and extensive systems is critical to understanding interactions between molecules, cells, organs, etc. Mathematical reconstructions of the relationships among system elements can be used to describe interactions among modules, i.e. independently operable units, even across several levels. Our physiome, the functional or operational behaviour of our molecules, cells, organs and bodies as a whole, is multiscalar in nature, long recognized in the teaching of physiology and medicine and nowadays including molecular biology and the regulation of transcription from the genome.
The goal of the ‘Human Physiome project’ is to define precisely the interactions occurring within the human body and to integrate the knowledge of the essential interactions in quantitative computer models. Making human physiology ‘computable’ provides insight into how the body functions as a whole. The physiome approach contrasts with genomic projects to determine the sequence of bases in the DNA and to catalogue all the molecules found in the human body in databases, e.g. proteins in the proteome and metabolites in the metabolome. At the subcellular, molecular level, the science that addresses the study of molecular interactions in large networks is often termed ‘systems biology’, but the human body includes elements at several scales (molecules, cells, tissues, organs, whole body). The Human Physiome not only addresses processes that traditionally were studied in physiology, but necessarily incorporates the molecular and cell levels, the intracellular organelles and the signalling pathways that regulate the manufacturing of new proteins from the recipes in the DNA and the clearing out of damaged proteins from the cell. At larger scales, the Physiome further defines the functioning of cells, tissues, organs and the body as a whole. Essential aspects of functioning at these larger scales can usually be understood through knowing only selective molecular details, not everything at all levels. For instance to understand why human postural control declines with ageing [3], we do not need to know all molecular details. Computational model analysis shows that even if the parameters that govern molecular processes are imprecisely known, it is often still possible to calculate relatively precise predictions at the level of cell or body functions [4,5].
In this paper, we discuss how the decrease of endurance and the changes in metabolism with ageing may be understood better in the near future by using computer simulations and quantitative analyses. The human body is a large system consisting of so many parts and interactions that its complexity overwhelms the capacity of an ordinary human for understanding. This is illustrated by the chain of physiological processes from breathing to mitochondria that play a role in oxygen transport and consumption and therefore in human endurance. The factors which limit endurance gradually shift during ageing. The complexity of the system is reflected in the complex temporal structure of physiological signals such as heart rate or breathing pattern. Quantitative computer models will be helpful to understand and prevent ageing processes that affect endurance. Understanding the bases for endurance and the robustness of metabolic systems in some aged individuals should be an achievable goal towards the description of the Ageing Human Physiome. This should be useful for understanding and prevention of frailty and age-related disease.
2. Ageing: the balance of damage and repair
Ageing can be described as a process of intrinsic deterioration with time that causes decreases in strength, endurance and fecundity, and increases in disease susceptibility and likelihood of death. Walter Cannon opened his classic book on human physiology with the sentence: ‘Our bodies are made of extraordinarily unstable material’ [6, p. 19]. It is therefore to be expected on the one hand that the body is affected by its hostile environment and slowly degrades, much like man-made objects, while on the other hand it is the efficacy of maintenance and repair processes which determines how long it takes for this eventual degradation to occur. It is generally thought that the ultimate cause of ageing is damage to the macromolecules that constitute the human body [7–12]. If not repaired, this damage will gradually accumulate, and for instance lead to increasing oxidation of DNA. The proteins that form roughly three quarters of the dry weight of the human body are subjected to oxidative or other damage as well. The damage may seem unavoidable when we consider that the energy which makes the human body work is derived from the ‘burning’ of nutrients, mainly carbohydrates and fat which react with oxygen. The violence of the combustion process has been harnessed in the cells by multistep enzymatic processes which produce small, convenient parcels of energy in the form of ATP molecules. Unfortunately, this does not completely prevent damage to the macromolecules by dangerous metabolic by-products, for instance generated during the splitting of glucose or during the transfer of electrons from nutrients to oxygen. Note that the macromolecular building blocks of the body are under threat from both the reducing power of sugars like glucose and also the oxidative power of oxygen. The danger of glucose is demonstrated by the damage to blood vessels occurring during uncontrolled diabetes which results in high blood sugar levels. The danger of oxygen is demonstrated by toxic effects in the lungs, eyes and central nervous system when pure oxygen is inhaled. On the one hand, the transfer of electrons from sugars to oxygen is a major source of energy supply for cellular function and required for normal brain function, on the other hand sugar and oxygen are potentially damaging.
Damage to macromolecules is repaired continuously throughout the body. It is the balance of damage and repair that is important. Several biochemical systems repair damaged DNA [11]. Proteins are constantly broken down and replaced by newly synthesized proteins. Proteins which have lost their correct folding structure are also refolded. Refolding and breakdown followed by resynthesis together form the process of proteostasis which preserves the content of undamaged proteins in the cell. However, the balance of molecular injury and repair is apparently such that at certain moments there is still net accumulation of damaged molecules, and the question therefore is whether damage can be minimized or whether the repair processes can be made more efficient. Apparently evolution has chosen not to invest in perfecting cell and tissue repair processes. By analogy, very old cars can be repaired and kept running although at substantial costs, but why bother when you can buy a new car with novel and improved technology for the same amount of money or less? Perhaps evolution has made a similar choice for animals and humans. This does not mean that repair and maintenance processes in our body cannot be made optimal by human ingenuity and intervention in order to prevent frailty and age-related disease.
Damage is often due to the generation of oxygen radicals [8,12] which react with DNA, proteins and other macromolecules. However, proteins are also changed in other ways, by removal of nitrogen-containing side-groups from amino acids (deamidation), rearrangement of chemical bonds (racemization) and breaks in the chains of amino acids that form the proteins [13]. These forms of damage may in particular be found in proteins that persist in tissue for tens of years without turnover, such as the proteins forming the lens in the eye or elastic fibres in lungs and blood vessels. This non-oxidative damage may be increased by the heat generated as a side effect of metabolism in the human body. Proteins are continuously broken down and re-synthesized through transcription, replacing damaged ones [14]. The proteins of the heart turn over at an average rate that would replace all proteins in the course of a month, although each protein has its own half-life [15]. On the other hand, some proteins in the human body, in particular those with structural functions, are not replaced for tens of years. Examples are the proteins crystallin in the lens of the eye, elastin in the lungs and dentin in the teeth. People born at the time of nuclear bomb testing in 1945–1962 still had high amounts of radioactive carbon in these proteins in later years, showing that these molecules have not been replaced since that time [13]. Measurements of radioactive isotopes in the cortex of the brain also show lack of turnover of DNA. Some macromolecules therefore accumulate damage: the loss of the ability to adjust the shape of the lens to deliver sharp vision correlates with the accumulation of modified amino acids in the proteins in the lens [13]. To what extent similar changes in structural proteins in the lungs and the walls of blood vessels determine decline of function remains to be determined.
Not only oxygen radicals cause damage, but sugars and methylglyoxal, a by-product formed during glycolytic breakdown of glucose, react with macromolecules and damage them by forming glycation end-products [16] which accumulate during ageing and at a particularly high rate during diabetes. Last but not least, an often neglected by-product of metabolism, heat, may cause problems due to failure of efficient temperature regulation in older people [6] increasing damage caused by high body temperature. Environmental toxic chemicals may also affect ageing [17]. More so than visible damage (e.g. fractures, cuts, bacterial damage to teeth), microscopic damage to macromolecules may drive the ageing process.
The damage to macromolecules gradually affects cellular subsystems, e.g. the mitochondria. The mitochondrial free radical theory of ageing proposes that reactive oxygen species (ROS), leaking from the mitochondria during normal oxidative metabolism, cause damage to the cellular macromolecules which accumulates in the course of life [18]. The accumulation of damage to mitochondrial DNA is thought to form an important cause of ageing. As a consequence of oxidative damage to the mitochondria, increased generation of oxygen radicals may result, resulting in a vicious cycle. Increased levels of 4-hydroxynonenal, resulting from oxidative damage to lipids, were found in liver and muscle of aged mice [19]. Clearance of damaged mitochondria by autophagy, a process which removes parts of the cell by ‘self-eating’, accompanied by re-synthesis of new mitochondria does not provide wholly adequate repair in the long run. However, not all measurements support extensive and increasing oxidative damage with age: only modest increases in carbonyl content, indicating oxidation of proteins, were found in the brain and liver from old rats, but no difference was observed in lipid peroxidation products in the brain beyond six months of age and in the liver beyond one month [20], which is short relative to the 2–3.5 year lifespan of a rat. Many studies have been inspired by the free radical theory of ageing, but a recent review reports results that have often failed to support this theory [21]. However, if oxygen radicals reach their targets via short pathways that are not accessible to antioxidants, contradictions between the theory and the experiments may be resolved [9]. It is clear that oxygen radical generation and its damaging effects on the mitochondria are closely linked to metabolism. Rather than just causing random damage, ROS are thought to play a role in signalling as specific regulators of cellular defence mechanisms which increase stress resistance and promote longevity, a response termed ‘mitochondrial hormesis' or ‘mitohormesis' [22]. In any case, if ROS are involved, they will probably not be the only drivers of the ageing process [9].
Damage to DNA, proteins and mitochondria are not the only processes to explain ageing. Erosion of telomeres, the end caps of chromosomes, is considered an important cause of the arrest of cell division in senescent cells [23]. Errors arising in nuclear DNA during replication in haematopoietic stem cells may play a role [24] and clonal somatic mutations in haematopoietic stem cells predispose to disease and increase mortality with ageing [25,26]. Increases in inflammation, innate immune cell activity and senescence may lead to failure of proteostasis, at least in model organisms [11]. Systems for molecular integrity control appear to have limits of perfection: they may manage to control damage for a certain time but fail eventually. Interconnections between telomere dynamics, metabolism and stem cell function [27,28] entail that DNA damage may for instance lead to mitochondrial dysfunction and vice versa. The processes mentioned above provide many hypotheses on the causes of ageing [7]. However, these may not be mutually exclusive and may be different aspects of the complete story. Although computational models of damage processes have been published, we barely touch on this topic, in order to focus on the modelling of changes in metabolism and endurance. However, metabolism is on the one hand linked to damage, for instance via oxygen radical generation, glycation and heat generation, and on the other hand linked to repair, for instance via protein breakdown and synthesis and detoxification of oxygen radicals. Damage and repair are important for ageing. Metabolism is in turn important for damage and repair [6,9,16–22].
Given the damage to molecules and organelles inside the cell, at some time the damage is recognized at the whole cell level. In skeletal and cardiac muscle, this can lead to decreasing mass and force, resulting in diminished endurance. Some dysfunctional cells die or commit a kind of cellular suicide termed ‘apoptosis', while other become ‘senescent’, failing to divide or function normally and affecting surrounding cells negatively, leading to diminished function of the tissues. In the young, defective cells can often be replaced by new ones to maintain the tissue in good condition. Muscle is regenerated after injury from satellite cells, which are specialized muscle stem cells. This regeneration starts to fail in older mice. Mutated stem cells may emerge and proliferate with DNA damage accompanying replication [29,30]. However, the failing stem cell system can be rescued by bringing the tissue in contact with blood from younger mice or by providing the protein growth differentiation factor GDF11 in the blood [31]. This suggests that hormones determine stem cell function and failure of cell replacement is an important cause of ageing. However, also in stem cells metabolism may play a very important role. Haematopoietic stem cells with faulty mitochondria have been shown to function badly [29] and metabolism may be an important determinant of stem cell function. The immune system also plays an important role: declining function has consequences for immune surveillance of organs, increased risk of infections, changes in the gut microbiome with consequences for metabolism [32] and autoimmune diseases. Regulatory T cells accumulate in fat during ageing and the activity of these cells worsens defects in metabolism [33].
In this perspective, we focus on metabolism. The effects of metabolism on ageing and health may for an important part be due to the effects of metabolic processes on the balance of damage and repair. Disruption of this balance may affect endurance, for instance by causing defects in tissue repair in muscle or in the maintenance of the mitochondria that provide the energy for muscle contraction.
3. Athletic performance, activity and age
It is important to relate athletic performance to age for both recreational and competitive athletes. One of the many physiological factors known to change with age is maximal oxygen consumption (VO2max). VO2max is a generally accepted overall best measure of whole body work. It represents the maximum amount of oxygen that the body can take up per minute per kilogram body mass. Sid Robinson [34] was the first to record VO2max as a function of age, showing that it increased only slightly in boys, per unit mass, as they grew, peaking at almost 50 ml kg−1 min−1 in the early 20s and then declining to half that at age 75. Many other researchers followed, reporting on various aspects of the relationship. The relationship between age, gender and VO2max is important to athletic enthusiasts at all levels of participation to understand their maximal metabolic function and in the designing of exercise prescriptions with the goal of improving their endurance capacity. Today, the information is critically important to competitive athletes as endurance events are often won by time differences measured in seconds.
Recently, Malkinson [35,36] has reported the results of his investigation into the relationships between lifelong male and female VO2max and age-division marathon and Ironman triathlon performance. Figure 1a shows the linear decline in VO2max that occurs with age in male and female sedentary individuals. Men consumed significantly more oxygen than women in all 5-year age-divisions, indicating that they are doing more work to reach their maxima. For endurance-trained people, figure 1b, the decline in VO2max also occurs with age, but only after a peak in the late 20s. Endurance-trained athletes have greater capacity to process more oxygen at all ages: training enhances VO2max. Many factors contribute to the decline and to the difference between males and females. The question is whether the limitation occurs exclusively at the cellular level by changes in cellular energetics and mitochondrial efficiency. There may be other impediments to oxygen uptake and transport from the alveolus to the body tissues.
Figure 1.

Maximal oxygen consumption (VO2max) in sedentary (a) and endurance-trained (b) males (diamonds) and females (squares). Data collected by Malkinson [35,36]. Sedentary: n = 22 063 men and 17 445 women; Endurance-trained: n = 18 763 men and 8868 women. (Online version in colour.)
Endurance athletics statistics tells us that over time there are accumulations of skill, knowledge or physical capabilities that lead to peak performance being reached in the early thirties, as shown for both marathon (figure 2a) and Ironman triathlon performance (figure 2b). Event performance data can be compared with the lifelong changes in VO2max in endurance-trained athletes presented in figure 1. Maximal aerobic capacity occurred in the age-division 25–29 and maximum event performance occurred in the age-division 30–34.
Figure 2.

Marathon run (a) and Ironman Triathlon (b) performance in males (diamonds) and females (squares). Times are the averages of the first three finishing times. Data from Malkinson: Marathon: male (n = 3988) and female (n = 3552); Ironman Triathlon: male (n = 875) and female (n = 699). (Online version in colour.)
These data raise the question of the physiological basis for the changes over time. They also raise the issue of what this means in terms of health and daily functioning. Tanaka & Seals [37] expressed it graphically, exposing a view that aerobic capacity is a measure of the safety margin keeping our system out of ‘frailty’. Frailty is defined as the manifestation of decline in physiological functions and the inability to maintain homeostasis [38]. In figure 3, aerobic capacity is plotted versus age of women aerobically trained for at least 2 years with road-racing competition (closed circles) versus age of sedentary women (open squares), who performed no regular exercise and had body mass indexes (BMIs) more than 35 kg m−2. The provocative analysis shows that the average VO2max at age 80 for the athletic women and for the sedentary women at age 50 is about equal. With regard to aerobic exercise capacity, athletic women are 30 years younger. At age 50–55 exercisers had more capacity than sedentary 20 year olds. Is there a relationship between level of physical activity and health? Not only were effects found of extensive endurance training, but also a relation has even been found between sitting time and premature death. In short, TV viewing is rather deadly. Katzmarzyk et al. [39], Dunstan et al. [40] and Stamatakis et al. [41] showed associations between sitting time and mortality. In 20 000 Australian men and women, 1 h d–1 increase in viewing time was associated with an 18% increase in cardiovascular disease mortality, and an 80% increase for viewing time more than 4 h d−1 when compared with less than 2 h d−1. These differences were independent of other known influences on risk: smoking, high blood pressure, elevated cholesterol, fatty diet, large waist circumference and too little leisure-time exercise. Even low-intensity muscle contraction helps, showing favourable improvements on plasma glucose in glucose tolerance tests. Stamatakis et al. [41] postulated that sitting lowers skeletal muscle blood flow, lowering shear stress on vascular endothelial cells and decreasing endothelial nitric oxide synthase (NOS) expression. They also noted that the low-grade inflammatory marker, C-reactive protein, was twice as high in those with more than 4 h d−1 viewing compared with those less than 2 h d−1. Watson et al. [42] noted that hind limb unloading of rats decreased protein synthesis rates within the first 6 h. Even passive bending of bone changes gene expression [43]. This is to be expected in a system in which the regulation of transcription is changing, often dramatically, with each change in physiological activity.
Figure 3.
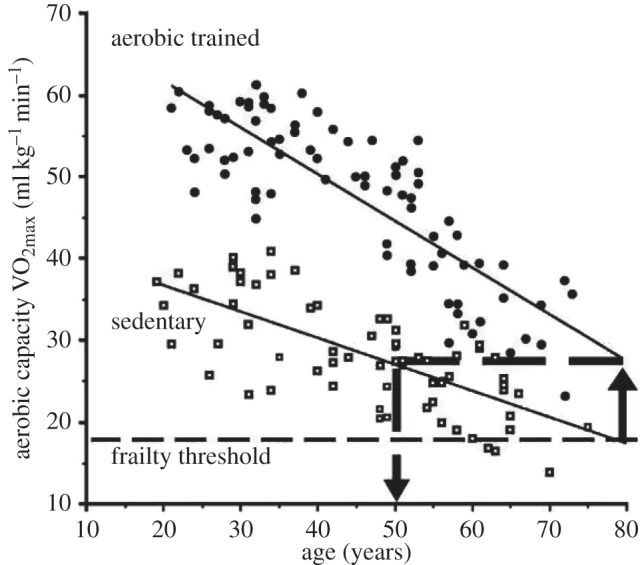
Aerobic capacity versus age in sedentary (squares) and trained women (circles). Best-fit linear regressions are shown for aerobic capacities of the two groups as a function of their increasing chronological age. At the chronological age of 80 years, a horizontal line is extended from the endurance-trained line to the left where it intersects the sedentary line at age 50 years. Adapted with permission from [37].
The inference from figure 3 is that low maximal aerobic capacity (VO2max) is a biomarker, not for imminent death, but for shorter life expectancies, perhaps by many years. To be sure, the decline with age seems inevitable. Even so, it is clear that how we treat our bodies can have an influence, as demonstrated by the outstanding performances of elderly people in figure 2. A truly exceptional case, Olga Kotelko, aged 93, is the subject of Grierson's book ‘What makes Olga run?’ [44]. Olga is competing in multiple events, from javelin and the sprints to the shot put and the high jump; she holds 23 world records, albeit for her age groups. There are genes associated with athletic abilities, e.g. ACTN3, which comes in two forms, fast for muscular power and speed, and slow for endurance, and Olga is +/+ for the fast form, but otherwise there seems to be nothing special about her genes. What is special, however, is her desire to compete and her ability to get joy out of hard training. Competitive people like Olga seem to thrive mentally as well as physically.
Is the message that exercise and even mild physical activity inhibit cellular and mitochondrial degeneration? That is undoubtedly too simplistic a view. Endurance depends on lung and heart function and blood vessels to transport oxygen and nutrients to the muscle and to remove waste products such as carbon dioxide, lactic acid and heat. Though it is the cell that drives transcription, and the cell that makes the shopping list of which items to pull from the shelves of the DNA store, the cell has to sort messages coming from the humours of the body, emanating from other tissues and organs, and from the brain, by neural transmission, often reflecting the state of the milieu exterieure. Below we will examine how computational modelling may help to determine which components in the human physiological system play a role in the decline and how they contribute quantitatively to the limitation of endurance. Whether enhanced cardiorespiratory fitness also affects metabolism and resilience positively in parts of the body beyond the muscular system is another intriguing question to be answered.
4. Homeostasis
4.1. The classic story
A central idea in human physiology is that regulatory processes keep the state of the internal environment within a relatively narrow working range despite large changes in the external environment. This is termed ‘homeostasis' [6]. An example of such natural regulation is that the temperature in the core of the resting body is kept relatively close to 37°C, even at high or low air temperatures. Homeostatic processes are also very active when disturbances come from within the body, for instance heat generation in the muscles during exercise. About 1600 watt of heat can be generated in a pro cyclist climbing a mountain road [45]. Humans have the capability to hunt animals in hot conditions by having greater endurance and better temperature regulation than their prey, something probably important to human evolution [46]. This would not be possible without temperature homeostasis which prevents the proteins in our body from coagulating during exercise like in a boiled egg [6]. Another well-known example of homeostasis is the regulation of blood sugar level in healthy people by insulin. Large amounts of sugar are used as fuel for the muscles and metabolic homeostasis is therefore important during exercise. Owing to metabolic homeostasis the cells are supplied with sufficient nutrition, even when the last meal was hours ago. On the other hand, the body is not overloaded with too many harmful sugar molecules after a copious dessert. Although homeostatic processes appear to be very efficient, it is striking that the regulatory capacity of our body ultimately always fails against one condition: the progress of time which makes us age and die.
Claude Bernard, a key figure in the history of physiology, wrote already in the middle of the nineteenth century: ‘The human machine shall be more perfect the better that it defends itself against the penetration of influences from the external environment; when the organism ages and grows weaker, it becomes more sensitive to the external influences of cold, heat, humidity, as well as to all climatic influences in general’ [47, p. 137]. In his experimental studies, published in 1865, Bernard found that the understanding of physiology lay in examining the intact functioning organism and in figuring out the mechanisms by which the behaviours of the parts were integrated [47]. His modesty, integrity and openness in performing and interpreting experiments helped him to find explanations. Through his experiments, he unravelled one mystery after another in his early years, and with the depth and elegance in his writings on the scientific method [47] he transformed biological and medical science: ‘Je crois avoir le premier insisté sur cette idée qu'il y a pour l'animal réellement deux milieux: un milieu extérieur dans lequel est placé l'organisme, et un milieu intérieur dans lequel vivent les éléments des tissus.’. He emphasized ‘La fixité du milieu intérieur est la condition de la vie libre, indépendante’ for the milieu intérieur maintained all the conditions necessary for the life of the body's anatomic elements, from cells to organs. He demonstrated that levels of glucose, oxygen, carbon dioxide and other solutes varied over time, though constrained. Bernard emphasized the constancy of the internal environment in the body, ‘le milieu intérieur’. It is clear that this constant environment requires regulatory processes to achieve constancy of the fluids bathing the body cells. In Walter Cannon's words [6, p. 24] homeostasis means ‘ … the coordinated physiological processes which maintain most of the steady states in the organism … The word does not imply something set and immobile … (but rather) a condition which may vary, but which is relatively constant’. Important disturbances of the balance may come from inside the body when during exercise energy turnover goes up drastically, leading to intense heat generation and manifold increases in energy and oxygen demand.
Cannon examined ageing of homeostatic mechanisms and noted that ‘in temperature regulation, in the storage and use of sugar in the body, and in the maintenance of the acid–base balance of the blood the homeostatic mechanisms, when subjected to stress, are revealed to be more and more limited … as life advances into the last decade.’ [6, p. 214]. He also noted: ‘There are few stars in sport after age 40’ [6, p. 214], which is still true more than 80 years after Cannon wrote this. This not only applies to endurance but also to brain functions, although they may decline more slowly: the present chess world champion is not yet 26; the oldest chess world champion ever was William Steinitz, who was 58 years when he lost his title.
Regulation of body processes is based on input from sensors throughout the body. There are for instance temperature sensors in the brain and skin. This provides input for regulation via the nervous system that sends signals to the skin to stimulate sweating or increase blood flow to the skin. If the body temperature rises too high, these cooling processes are stimulated and provide negative feedback. Another example of homeostasis is the regulation of blood glucose levels. The beta cells in the pancreas sense the glucose level, and excrete insulin in case blood glucose levels get too high which stimulates glucose uptake in the cells. This is an example of feedback employing hormones. Feedback that is not strong enough may cause problems, but feedback that is too strong or too delayed is a cause of oscillation and instability, well known in engineering. A lot of spontaneous fluctuation is seen in normal physiological processes; extremes, as in the exaggerated oscillation of Cheyne–Stokes breathing or the lack of fluctuation in heart rate with age, are signs of loss of appropriate control.
4.2. The dynamic side of homeostasis
Homeostatic processes do not keep key variables in the human body absolutely constant. The core temperature shows diurnal variation and goes up substantially during intense exercise. The glucose concentration in the blood increases temporarily after a meal before being regulated back to resting levels. Variability and fluctuation are parts of the normal state of the human body. While Claude Bernard's statement ‘La fixité du milieu intérieur … ’ stimulated Cannon in 1932 [6] to think of ‘homeostasis' as characterizing the milieu intérieur, it is safer philosophically, and truer scientifically to think of the system as being ‘homeodynamic’. We can define a homeodynamic system as one in which there are many controllers of elements of the system whereby the system is loosely regulated, but sturdy, with robust responses that maintain the milieu within broad limits. The beauty of the homeodynamics is that the regulation is softer, more flexible and less subject to injury. Marey [48] in 1863 reported the wanderings of the normal blood pressure, with slow undulations over uneven length periods lasting tens of seconds to minutes, seemingly a dynamical system. ‘Tight control’, forcing a system to operate within very narrow limits, is energetically expensive and inevitably fragile [49]. Dynamic control in response to a dynamic, fast-changing environment is a hallmark of healthy human physiology.
It may even be that certain forms of variability are hallmarks of youth and health. This may indicate that not only cellular function, but also control systems age. One piece of evidence is a decrease in HRV and its dynamic complexity with age. Approximate Entropy, ApEn, provides a measure of this complexity derived by plotting successive RR intervals in two- and three-dimensional space and calculating the distances. The analysis by Pincus (1991, unpublished data, given in [50]) showed that with age the complexity decreases in both men and women (figure 4a). The interpretation is that the variation diminishes because there are fewer controllers. For example, young people have striking changes in heart rate with each breath: as described by Shepherd [52] ‘lung inflation causes a reflex decrease in blood pressure as a result of dilatation of systemic vessels, bradycardia and a negative inotropic effect on the ventricles. This lung inflation-vasodepressor reflex is due to activation of low-threshold pulmonary stretch receptors, subserved by vagal afferents’. This is the Hering–Breuer reflex, described in 1868, one of a host of regulatory responses to perturbations. Closely related is the ‘Bainbridge reflex’, an increase in heart rate with filling of the right atrium as enhanced during inspiration. Shepherd's review [52] portrays the complexity of the interaction of these reflexes with other elements of cardiovascular control. With age, the sensitivity of these reflexes diminishes and heart rate varies less.
Figure 4.

(a) Approximate entropy for heart rate versus age in healthy subjects at rest. The line is the mean linear regression. Triangles indicate males; circles females. (From SM Pincus, IM Gladstone and RA Ehrenkranz 1991, unpublished data, with permission. Figure from ‘Fractal Physiology’ [50]). (b) Cumulative survival of patients having had myocardial infarction (MI) over a total follow-up period as a function of HRV expressed as standard deviation (s.d.) of the beat-to-beat interval. Survival curves were calculated by the method of Kaplan and Meier. Those with s.d. < 50 ms differed from both other groups (p < 0.0001) (adapted from [51]).
A typical measure of HRV from 24 h Holter recordings, as used by Kleiger et al. [51], was to take the variance of the RR interval over a 60 s period. They observed in patients after a myocardial infarction that death was much more likely if the HRV was small (figure 4b). These observations gave credence to Goldberger's idea that decreased HRV gave a premonition of sudden cardiac death [53]. The explanation proposed is that high HRV is inherently healthy, for it is due to a multiplicity of controllers, respiratory arrhythmia, body and mental activity, variations in sympathetic and parasympathetic activity. With illness and ageing, controllers are lost or become less efficient, so the variability diminishes. And in the case of Kleiger's patients who had myocardial infarctions and inevitably had abnormalities in the spread of excitation across the ventricles, the small variance presaged sudden death. HRV is easily measured and provides a convenient window to look at homeodynamics in the human body. HRV analysis has also been applied to assess athletic training [54]. Decline of HRV during ageing may therefore be considered for computational modelling.
4.3. Metabolism and homeostasis
Nutrients, absorbed in the gut and delivered to the cells via the blood, are extensively processed by a large number of biochemical reactions in the body which together form human metabolism. Metabolism enables cell growth, division, maintenance and supplies the energy for muscle work. Cell growth and intracellular turnover of energy and molecules are coordinated with the direct tissue environment to fulfil the body's requirements, such as repair of injured tissue or build-up of muscle mass during exercise training.
It is remarkable how often metabolism is mentioned in the scientific literature as a factor that modulates ageing. Caloric restriction, which means restriction of the amount of food that is consumed, is one of the few interventions proven to increase lifespan in many organisms [55]. However, studies in rhesus monkeys gave contradictory results with regard to survival outcomes, although health effects were generally positive [56,57]. Studying the effect of caloric restriction on the survival of human subjects is of course difficult, and requires very long-term studies; nevertheless, a 2-year study suggests a positive influence of caloric restriction on correlates of survival and disease-risk factors [58]. Lifespan is also increased by disruption of signalling via insulin and insulin-like growth factor (IGF) or using the drug rapamycin to inhibit mTOR, a protein that signals the status of nutrient supply [55]. Indeed, ‘a hallmark of ageing is dysfunction in nutrient signalling pathways that regulate glucose homeostasis' [1,11].
Amino acids absorbed from food or synthesized in the body are the building blocks to synthesize proteins which form about three-quarters of the dry mass of the body. Protein synthesis also requires substantial amounts of ATP, which forms another link to small-molecule metabolism. Despite constant renewal and proteolysis there is an age-dependent deterioration of the balance: the proteome, the complement of all the different proteins in the cells [59], does age. However, although changes in proteins are large in roundworms, a favourite model in ageing research, in mice such changes during ageing are modest [60]. Given the substantial effect of caloric restriction and of genetic and pharmacologic manipulation of signalling molecules which regulate metabolism, understanding the role of metabolism during ageing should be a prime target of quantitative computational modelling [61]. Physical exertion leads to a large increase in oxygen consumption and ATP synthesis, and protein synthesis in muscle is stimulated by insulin and IGF [62], signals linked to metabolism that affect ageing strongly. In order to investigate the effect of ageing on endurance, modelling of metabolism will be indispensable.
Mitochondria play an important role in metabolism and are powerful producers of ATP which provides the energy for contraction in heart and skeletal muscle and for information processing in the nervous system. However, ATP is also needed for breakdown and synthesis of proteins and other macromolecules, cell growth and maintenance. The contribution of mitochondria to ageing is often discussed in terms of their contribution to oxygen radical formation. However, in mice whose mitochondrial DNA replication is faulty because of genetic interventions, ATP synthesis may be compromised. Not only does skeletal muscle show low ATP levels in these mice, without increase in oxidative damage [63], but even haematopoietic stem cells function badly, although they contain only few mitochondria [29]. Mitochondria may be causal players in cellular senescence [64,65]. A mitochondrial cascade hypothesis has been proposed to explain late-onset Alzheimer's disease, stating that a decline in mitochondrial function precipitates the disease and enhances amyloid-beta accumulation and regional brain atrophy. Given the decline in glucose uptake and oxygen consumption which precedes clinical symptoms, the role of energy metabolism in Alzheimer's merits serious consideration. Given the widespread effects of energy metabolism and its link to endurance, the investigation of its role in ageing is desirable.
4.4. Reconstructing and modelling human metabolism
Human metabolism is being reconstructed computationally. The present compilation of known biochemical reactions in the human body comprises about 7440 reactions [66] and has been termed Recon 2 (‘recon-struction’ of metabolism). Although the number of reactions is large, Recon 2 is probably not complete or error-free and will require continuous improvement. Recon 2 sums up the type and number of molecules consumed and produced by each biochemical reaction in computer-readable form. This biochemical information is linked to the gene(s) for the protein(s) which form the enzyme that catalyses the reaction. The products of one reaction form the input for other reactions, which implies that a large metabolic network can be assembled from the reconstruction. Figure 5 shows an example of a metabolic network representing brain metabolism. The network's mathematical representation enables calculation of the metabolic fluxes (rates of the biochemical reactions). For the calculation, the assumption is often made that production and consumption of intracellular metabolites is balanced over time intervals which are not too short [68]. This approach, termed flux balance analysis (FBA), does not preclude slow changes in metabolite levels during ageing or disease, as long as the rate of change of the metabolite pools is negligible compared with the fluxes into and out of a pool. The balance assumption can be combined with knowledge about those reactions which are irreversible, meaning that they run only in one direction. When the exchange fluxes of metabolites between blood and an organ have been measured they can be imposed as additional constraints, and possible flux distributions in the metabolic network can be predicted. When metabolite exchange measurements are not available, one can optimize a cost or profit function which is assumed to represent the functional goal for the metabolic system, e.g. making the cell grow as quickly as possible, or making as much energy available to the cell as possible given a limited nutrient supply. For such FBA approaches, linear programming or quadratic programming methods are applied [68].
Figure 5.

Example of part of a human metabolic network selected from a large reconstruction of human metabolism. Here the core of central energy metabolism in brain cells is shown, including multiple steps involved in the transfer of electrons from glucose to oxygen leading to the production of ATP. Arrows represent biochemical reactions which connect metabolite pools, represented by abbreviated metabolite names. Next to the arrows abbreviated names are given for the reactions. Many reactions run in both directions, but many others are unidirectional. Transport across the cell membrane (outer rounded rectangle) and the mitochondrial inner membrane (inner rounded rectangle) is also represented by arrows. Reproduced with permission from [67], where details can be found. (Online version in colour.)
An example of the prediction of fluxes starts from the measured exchange of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids in the brain of young and elderly people [69]. These measured exchange rates form the input for a prediction of the flux distribution in a model of brain metabolism [67,70]. Because of the many degrees of freedom involved in large biochemical systems, the distribution of metabolic fluxes inside the network can usually not be uniquely determined and a range of reaction velocities are still possible. Monte Carlo methods can then be employed to explore the full solution space compatible with the measured input and output to the network [67,68], generating a random selection of possible flux patterns. Uncertainty quantification, the UQ paradigm for the accuracy of prediction, is of particular importance with relatively unconstrained analysis like FBA [71]. It is also possible to determine for each reaction in the network what the maximal and minimal fluxes are under the boundary conditions imposed on the model, an approach which is termed Flux Variability Analysis. Computer simulations can help to determine which additional measurements are needed to uniquely determine the full flux pattern. Using such methods, the changes in metabolism during ageing may be quantified. Developing and validating specific models for the many cell types and organs in the human body will be a time-consuming task, but one that may be worth the effort given the importance of metabolism for ageing and health.
The downside of FBA is that metabolite concentrations and consequences of changes in enzyme properties cannot be predicted without further assumptions. The FBA may for instance show how much of a harmful metabolite is formed per hour, but does not by itself provide a reliable prediction of the blood or tissue concentration of the metabolite. The FBA approach has, however, one important advantage: the precise kinetic equations, which contain the many parameters that govern the rate of enzymatic reactions, do not have to be known to calculate the distribution of fluxes in the metabolic network. This is an advantage because the properties of many enzymes are insufficiently known. Enzyme parameters are usually measured in an artificial environment in the test tube, and it is doubtful that this reflects the situation in the cells where a dense, structured environment exists. Enzymes and other proteins are often chemically modified inside the cells, transported to other intracellular compartments and form dynamic complexes. Different versions of an enzyme, isoforms, are often found in different tissues and at different stages of development or disease. Complex experiments in the intact human body and in human cell or organ cultures will be needed to fine-tune the equations for metabolism and define the dynamic contribution of various forms of the enzymes.
The distribution of fluxes has been predicted for large metabolic networks, for instance for brain and cancer cells [70,72,73] or for cells in the gut [74]. Changes in brain metabolism during Alzheimer's disease have initially been predicted based on the reduction of the maximal flux measured for one enzyme (2-oxoglutarate dehydrogenase) that catalyses a reaction midway in the Krebs or tricarboxylic acid cycle) [70]. Changes in expression of genes coding for the enzymes in a metabolic network model are now also used for the analysis of network fluxes. The levels of gene transcripts for many enzymes, measured in post-mortem samples from patients, were analysed with the network model of metabolism (figure 5), which led to the prediction of large changes in metabolic fluxes in the brains of Alzheimer's disease patients [67].
The human metabolic system is extensively and dynamically regulated by modifying the properties and levels of enzymes. In response to hormones arriving at the cell surface, phosphate groups are for instance attached to specific sites on the enzymes which alter the enzyme's catalytic properties. Such regulation of multiple sites in the metabolic pathways is very powerful. The kinetic equations for such regulatory processes are even less well known than for the biochemical reactions. In addition, the amount of an enzyme present in the cell can change strongly under control of regulatory processes which alter the rate of synthesis or breakdown of the enzyme. It is still very difficult to accurately and comprehensively model regulation of metabolism. The same is true for processes related to metabolism which lead to cellular damage. An attempt was made to model energy metabolism, feedback loops and stress responses during ageing using a fuzzy logic approach [75]. Note that this approach does not require very precise quantitative relations for molecular interaction processes, with obvious consequences for the results. While quantitative modelling of complex regulatory processes is difficult, comprehensive modelling of flux distributions in the (near) steady state using FBA can yield useful results and is potentially applicable to large networks. Quantitative analysis of flux distributions may therefore be a useful approach to determine changes in metabolism during ageing.
4.5. Mitochondrial degradation and homeodynamics
Mitochondria play a key role in metabolism, and the question is how the changes in capacity and properties of mitochondria can be incorporated in metabolic models of ageing. Diminished physical activity will lead to diminished muscle cell size, chronic under-stimulation of mitochondrial ATP production, net protein and enzyme degradation, and reduced capacity to respond to high demand. In vivo measurements of mitochondrial metabolism in ageing individuals are necessary to provide data for the models. A particular example is provided by Conley and co-workers [76–78]. They examined the status of high-energy phosphates in exercising people using magnetic resonance spectroscopy. Figure 6 shows that the time course or recovery of phosphocreatine, PCr, an energy store and cytosolic buffer for ATP, after a 2 min contraction of the muscle is slower in the elderly than in younger men. The rate constant for PCr recovery (KPCr), reflecting oxidative capacity in the muscle, diminished with age.
Figure 6.

(a) Phosphocreatine concentration [PCr] recovery in quadriceps muscle following muscle stimulation via the femoral nerve for 2 min. Open symbols denote younger adult; closed symbols, an elderly person. Mono-exponential fits provide rate constants of recovery, KPCr. (b) Recovery rate constant, KPCr, in younger adults (25–48 years: open squares) versus elderly (65–80 years: closed squares). Higher KPCr values reflect higher mitochondrial capacity for mitochondrial resynthesis of ATP (adapted with permission from [79]).
Conley [79] found that mitochondrial volume density was significantly lower (2.9 ± 0.15% of volume) in elderly compared with younger adult muscle (3.6 ± 0.11%). The oxidative capacity per mitochondrial volume is also reduced, indicating diminished mitochondrial efficiency. In more recent studies [77], they also used optical spectroscopy to non-invasively measure oxygen uptake and ATP turnover in the tibialis anterior muscle in the leg, and from those calculated the ratio of number of oxygen molecules used to produce one ATP. The muscle was made ischaemic by an occlusion cuff, and the haemoglobin (oxygen carrying molecule in the blood) and myoglobin (oxygen buffer inside muscle cells) spectra were analysed to provide the rate of utilization of O2. When the oxygen was totally depleted so that ATP could not be generated, the rate of ATP depletion was estimated by measuring the decay slope of the PCr signal measured by NMR spectroscopy. The results were clear: ATP flux was definitively less in the elderly and the ratio of ATP formation to oxygen uptake was low in the elderly [77]. This indicates reduced efficiency in generating ATP, possibly due to proton leak across the mitochondrial inner membrane. The elderly are also less efficient at another level: the demand for oxygen is higher per watt of energy output by the muscles. In older people not only loss of mitochondrial volume, but also loss of mitochondrial performance per unit volume is seen, probably caused by degradative accumulations of molecular and membrane damage in aged mitochondria and cells. How can this be prevented?
Conley suggests that one can improve mitochondrial function and elevate gene expression levels by exercise training [78]. Signalling pathways that influence mitochondrial synthesis and function have been explored in increasing detail over the past two decades. Peroxisome proliferator-activated receptor-γ coactivator (PGC-1α), a member of a family of transcription coactivators, plays a central role in mitochondrial remodelling, the regulation of fatty acid oxidation and the transcription of antagonists of ROS, all influencing the progress of ageing [80]. PGC-1α stimulates mitochondrial formation and recycling, and the remodelling of muscle tissue to a fibre-type composition that is metabolically more oxidative and less glycolytic in nature. By upregulating lipid metabolism it participates in the downregulation of carbohydrate usage, meaning it is playing a role in substrate switching. It probably also plays a role in fibre-type switching in skeletal muscle, between fast twitch glycolytic fibres and slow twitch oxidative fibres. PGC-1α is an important node in extensive nutrient sensing signalling networks and improves mitochondrial function.
The accumulation of defective mitochondria, degradation of damaged organelles and building of new ones during ageing is being modelled computationally [81,82], but many facets are still undetermined. The fission–fusion cycling of mitochondria, not yet well understood, appears to be important in maintaining mitochondrial quality [83]. It is hypothesized that reactive oxygen and nitrogen species are involved in the regulation of mitochondrial shape and function with hydroxyl and superoxide anions as local messengers and hydrogen peroxide and nitric oxide as messengers which diffuse farther into the cytosol and out of the cell. Two additional intracellular homeostatic regulatory circuits were proposed for redox homeostasis within mitochondria and cytosol [83]. While the severity of oxidative stress is variable, it is countered by the inhibition of ROS generation by substances produced in response to PGC-1α, resulting in antioxidant upregulation. Mitochondrial deterioration with ageing implies diminished success in replacing damaged proteins, transcription regulators or genes. Some of the damage may be simply the accumulation of infrequent errors in proofreading during transcription, but ROS and even radiation from outside the body are more likely causes.
4.6. Metabolism and proteostasis
The turnover of protein, monthly or so [15], is throughout the body, even in cells which are not dividing any more. Homeostasis of the protein content of the cells has been termed proteostasis. The balance between protein degradation and synthesis depends on several mechanisms for protein degradation; these are usually well regulated to remove damaged proteins [11,15]. Nevertheless, during ageing there are progressive changes in the proteome, and damaged proteins as well as protein aggregates accumulate. Clumps of damaged proteins will hinder cell function and aggregates of misfolded proteins play a role in neurodegeneration during Alzheimer's and Parkinson's disease. Renewal of the protein content is directly linked to metabolism [15] and is regulated by hormone systems such as insulin, IGF and mTOR which are also linked to metabolism [62]. Signals generated during the metabolism of glucose appear to regulate protein turnover in the heart [15,84].
Although physico-chemical characteristics of oxidation of proteins have been studied [85], computational models of protein damage and repair are scarce, if existing at all. It will be a challenge to couple models of metabolic pathways to levels of damaged proteins and levels of regulatory molecules. It should be possible to build separate protein breakdown and protein synthesis reactions in metabolic models to analyse protein turnover. These can be linked to experimental measurements of the protein turnover and to simultaneous measurements of metabolism. In particular, the synthesis of non-essential amino acids and the uptake of essential amino acids, which the body cannot synthesize, may be analysed to unravel the links between protein turnover and metabolism during ageing.
4.7. Chain of physiological processes for oxygen and nutrient supply
A chain of physiological processes determines endurance. Oxygen transport and consumption in the tissue is a very important factor. If lung capacity becomes low, oxygen transport between air and blood may become limiting. If the heart pumps weakly, then cardiac output may become a limiting factor. In the case of anaemia, the oxygen carrying capacity of the blood may become a problem. The illegal erythropoietin usage of pro cyclists and runners illustrates that the haematocrit or haemoglobin content of the blood form determinants of endurance. Capillary blood flow and diffusion of oxygen between blood and muscle cells are further steps in the chain. The mitochondrial capacity in the muscle cells to turn nutrients and oxygen into ATP necessary for contraction is potentially an important factor which determines endurance (see above). None of these links in the chain may form the absolute unique limiting step, but the contribution to limitation of endurance may be distributed over several of these links.
Understanding the effects of changes in the various links in the chain during ageing requires computational modelling. An example of a model of oxygen uptake including circulatory interactions was used by Benson et al. [86] to fit experimental data on the dynamic adaptation to exercise. The distribution of energy conversion and temperature regulation during cycling has also been modelled [87]. Such models may be fine-tuned by doing experiments in various age groups, measuring oxygen uptake in the exercise laboratory, muscle oxygenation with near-infrared spectroscopy, muscle perfusion with NMR methods, changes in metabolite levels and fluxes as well as mitochondrial function with NMR spectroscopy.
Similar considerations apply to uptake and transport of nutrients via digestion, uptake across the gut wall, molecular processing in the liver, transport in the blood, uptake and storage in tissue, etc. The microbes in the large intestine play an important role in the processing of food. There are so many species of microorganisms in the gut that detailed in silico modelling of total gut metabolism will be difficult and use of in vitro models of the human gut microbiota provides an alternative [88]. On the other hand, modelling of metabolism in the human gut microbiota has recently been attempted by restricting the model description to five example microorganisms that are representative of the hundreds of species actually found in the gut [89]. Digestive and transport processes which precede intracellular metabolism can be captured in computational models which can be tested and fine-tuned based on experimental data. For brevity, we have restricted ourselves mainly to the discussion of oxygen transport as an example.
4.8. Multiscale homeostasis
Regulatory interactions involving transfer of information and physical transport of molecules and heat take place in the human body at many levels, both inside cells and across large distances between organs. Many complex signalling systems operate between and inside the cells to adapt functioning at the intracellular molecular level to the needs of the body. The function of organelles such as the mitochondria must be coordinated with overall cellular function. The process of ageing is not restricted to just one or a few of these systems. Many intersecting molecular processes play a role (figure 7).
Figure 7.

Intracellular molecular processes involved in ageing. Both short-term adaptation of energy metabolism to dynamic cell work (ATP breakdown for muscle contraction, nerve firing, etc.) and long-term adaptation (protein turnover and build-up of protein mass) are interlinked. ATP production by the mitochondria is coupled to oxygen consumption. This cellular level is connected to processes at higher levels, such as the supply chains for oxygen and nutrients and hormonal signalling. ROS may damage DNA in the cell nucleus and the mitochondria. Damaged DNA leads to altered and dysfunctional protein molecules.
Nine tentative hallmarks for ageing have been nominated [11], among them loss of proteostasis, deregulated nutrient sensing and mitochondrial dysfunction. Given that no single process seems to dominate the ageing process, the interaction of many slow deterioration processes may determine ageing. Does the body behave, for instance, like a system that becomes 50% weaker if three of its parts each individually become 10% weaker? That would mean as far as damage is concerned, ‘the whole is more than the sum of its parts'. Or do homeostatic mechanisms enhance robustness and prevent that whole body performance becomes equally weak as individual parts until such compensation eventually breaks down? Computational modelling of the interaction among the processes involved in ageing, taking processes at all levels into account, may help to find the answer to such questions. In any case, oxygen transport, nutrient supply and hormonal and neural signalling link the body systems involved in metabolism, endurance and ageing across many scales to determine the ‘milieu intérieur’ in which the intracellular processes of ageing take place. Homeostatic processes above the cell level therefore need to be taken into account in the modelling process. A limitation is that despite the large amount of experimental information, the properties of molecular interactions in the signalling pathways are often not yet completely quantifiable and therefore of limited use for mechanistic and predictive modelling.
5. Possible contributions to the quantitative human physiome: endurance and metabolism during ageing
The human body consists of many components. There are for instance about 22 000 genes that code for proteins. This vast amount of information at the molecular level is overwhelming, making it difficult to see the forest for the trees. Many interactions at many levels are candidates for investigation and inevitably choices have to be made, depending on available information and access to measurements to build useful models. There are, however, some areas where valuable insight may be gained if computational modelling and targeted experimental data collection are combined.
Metabolism plays a key role in ageing and exercise. There is a lot of information on the components of metabolism, from the genes involved in metabolism found in the genome and from accumulated knowledge of about a century of biochemical laboratory work. This knowledge is presently already incorporated in digital reconstructions, such as Recon 2 [66]. There are efficient ways to use this information to predict the distribution of metabolic fluxes in cells by using the flux balance principle. On the other hand, models for the metabolism of specific cell types in the body are still scarce and incomplete. There are about 200 cell types in the body, organized in tissues and organs that communicate by exchanging metabolites and hormones. Given the importance of metabolism for human physiology and the good knowledge base, it should be a high priority for Physiome research to model and quantify the flow of metabolites in the human system. This applies not only to intracellular biochemical reactions but also to uptake in the gut and transport via the blood. This requires not only a computational effort, but definitely also extensive measurements.
Metabolite levels in blood and tissue can be measured, even quantifying many metabolites simultaneously with mass spectrometry or NMR spectroscopy. Exchange of metabolites between blood and tissue can be determined by measuring the concentrations in arterial and venous blood and multiplying their difference with the local blood flow [69]. NMR spectroscopy makes it possible to measure metabolites and metabolic fluxes in tissue non-invasively, although with limited sensitivity [90,91]. Providing the body with nutrients that have been labelled with stable isotopes makes it possible to follow metabolism by taking samples from blood, gut content [88], tissue samples [92–95] or measuring metabolic fluxes non-invasively by positron emission tomography or NMR spectroscopy [96]. Almost completely carbon-13 enriched plants are for instance available to serve as a source of nutrition labelled with a stable (non-radioactive) isotope, meaning it can be determined how the carbon atoms travel in the metabolic networks and even making quantitation of fluxes possible. These studies of human metabolism can be used to determine shifts in metabolism during ageing. The shifts in metabolic fluxes should then be determined during caloric restriction and dietary interventions which are of interest because of their apparently beneficial effects on health and ageing. The shifts in metabolism in people who exercise regularly should also be investigated, not only during exercise but also in the resting state. The dynamics of human physiology is of key importance and the dynamics of metabolic adaptation should be integral to the study, for instance by studying adaptation during cycles of fasting and eating. This may give clues as to why regular exercise not only improves the circulatory and muscle system, but also systems that seem not to be directly affected by exercise, such as the brain and immune cells. Given the effects of insulin and related hormones on ageing in lower organisms and the development of diabetes, studying the effects of these hormones in humans by comprehensively mapping shifts in the metabolic system would be highly desirable, although the risks of studies on healthy subjects during interventions in these hormonal systems should be carefully considered. The life expectancy of women is consistently greater than of men by several years which is not explained by social and lifestyle factors. It is therefore desirable to study the changes in metabolism with age both in women and in men.
Flux balance models are very suited to study large human metabolic networks because they deal efficiently with available information. Full kinetic models will be possible for only part of all biochemical pathways in the short run. Approaches are therefore necessary to link fluxes to metabolite levels.
To begin computational modelling to understand the physiology of ageing, three starting points are in principle possible: a bottom-up, top-down or middle-out view of the system. The investigation of metabolism alluded to in the previous paragraph may be viewed as a bottom-up approach from the molecular metabolic level to integrated body function. However, we should realize that this is not quite the bottom because details at the level of enzymes are ignored. Details of the regulation of metabolism via networks of interacting signalling proteins [55] are of great interest, although probably harder to quantify at this point in time than the regulated metabolic fluxes per se. The regulatory details at the bottom level are presently difficult to integrate in a robust way. On the other, attempts have been made to translate changes at the gene expression level to changes in metabolic flux distribution [67].
Large robust fluxes can be assessed, enabling the study of changes in the use of major nutrients, such as carbohydrates, lipids, amino acids and proteins. The subtle fluxes connected with oxidative damage form a greater challenge. A ‘middle-out’ approach [97,98], where computational models and experiments are developed starting at the mitochondrial or cell level, then stretch through the tissue and organ level towards the whole body, may be a viable competitor to the bottom-up and top-down approaches. Although hypotheses on oxidative damage as the cause of ageing and disease have generated an enormous amount of publications [18], the importance of oxidative damage remains controversial [21]. The difficulty of measuring short-lived oxygen radicals may have caused this [9]. We propose here that detailed molecular modelling of mitochondrial metabolism, in particular of electron flow in the respiratory chain, may be helpful to understand oxygen radical generation. The conditions that increase oxygen radical generation in isolated mitochondria or sub-mitochondrial particles could be systematically investigated by a close interplay between experiments and modelling. We propose the hypothesis that oscillations in ATP synthesis and low ADP levels stimulate oxygen radical generation. Cyclic ATP synthesis, O2 consumption, electron flow, and reduction and oxidation of electron carriers, for instance during cyclic contraction in heart and skeletal muscle might stimulate oxygen radical formation [5]. When activities of the buffering enzyme creatine kinase are lower during heart failure, there is an increased amplitude of oscillation and a lower minimal concentration of ADP during heart muscle contraction [5,91] which may induce higher oxygen radical formation that in turn may cause heart failure. We propose that investigation of oxygen radical formation in mitochondrial systems in vitro in combination with modelling at the molecular level may make it possible to understand the regulation of oxygen radical formation better. Modelling at the molecular level of how ROS diffuse and react in the immediate environment where they are formed [9] would complement this. The combination will enhance our understanding of the role of oxygen radicals in ageing.
To complement the proposed bottom-up and middle-out approaches, a top-down approach is an attractive alternative possibility. Responses to exercise in ageing people can be characterized with physiological measurements and interpreted with quantitative models. Examples of measurements are oxygen uptake, heart rate, metabolite levels in blood and tissue, temperature, blood flow, etc. The time course of the response will be especially informative. Changes in the potential bottlenecks for endurance performance (lung, heart, blood vessels, muscle mass, mitochondrial metabolism, other metabolic pathways) can be followed during ageing. We may then be able to predict how exercise training and nutritional interventions can make the bottlenecks wider during the ageing process. An important question to address is how it is possible that a high VO2max correlates with survival. The oxygen that is measured under maximal exercise conditions is mainly taken up by skeletal muscle, while other organs and cell types seem more important for prevention of disease and death than the maximal performance of skeletal muscle during exercise.
The response of the circulatory system to changing cardiac workload may help reveal the intersecting systems that regulate heart rate and its natural variability. Advanced models of the electrophysiology of the heart including pacemaker activity already exist [98] and may be extended with the effects of regulatory loops that affect the heart rhythm. This may allow us to understand HRV and improve its predictive possibilities.
The gap between the bottom-up and top-down approaches can be gradually closed to obtain comprehensive computational models of changes in endurance and metabolism during ageing. One may for instance incorporate models at the level of the mitochondria in the muscle cells (figure 7) into the model of oxygen transport to investigate endurance. Relevant components of the system can be added gradually to such a model. One can think of oxygen transport to the mitochondria, glycolysis to supply the mitochondria with products from carbohydrates, lipid metabolism, the cycle of removing damaged mitochondria and rebuilding them, fusion and fission of the mitochondria, etc.
Will studying how and why endurance and metabolism change during ageing throw light on the general causes of ageing? The least we can say is that endurance and metabolism provide well-quantifiable variables which can be related to ageing. Endurance and metabolism reflect dynamic aspects of human physiology and provide a window to look at ageing processes and therefore a great opportunity to apply Quantitative Physiome approaches to study human health. The metabolic networks in the human body, mitochondrial oxygen radical generation and the dynamic response to exercise appear amenable to in depth modelling and analysis with a physiome approach. Of course there are many systems that may be important for ageing. Modelling of DNA replication stress, telomere shortening, non-oxidative damage to proteins, replacement of damaged cells by stem cells, etc., are highly relevant. Quantitative investigation of stem cell viability and function and its link to mitochondria and metabolism would be highly desirable, combining suitable experimental systems with model analysis. In the description of questions addressable with physiome approaches above, we have given priority to a few systems that not only are important for ageing, but for which also sufficient prior knowledge and methodology is presently available to make them suitable targets for in depth analysis in the very near future.
5.1. Hypothetical scenario
Having a hypothesis on what may be an important process in ageing may help to focus thought and effort during the development of a physiome model of metabolism. A partial explanation of the effect of caloric restriction on ageing was formulated by Spindler [99], who stated that caloric restriction enhances and maintains protein turnover and renewal which would normally decline with ageing. He found in mice that messenger RNAs for enzymes that are needed for protein turnover in the body are upregulated during caloric restriction. After meals, glucose levels in the blood go up and are subsequently downregulated via glucose uptake in tissues, which is stimulated by insulin excretion from the pancreas. This is accompanied by a wave of protein synthesis. Between meals glucose levels in the blood are maintained, among others via glucose synthesis in the liver: the material needed for such synthesis is derived from protein breakdown in peripheral tissues. This cycle forms an example of metabolic homeostasis with very dynamic aspects. During caloric restriction there could be enhanced turnover of protein which improves clearance of damaged protein and in that way promotes health and ageing.
However, this hypothesis does not by itself explain how caloric restriction may bring about changes in the ‘waves' of protein synthesis and breakdown between meals. Is the amplitude of the waves of protein synthesis and breakdown really higher during caloric restriction? If so, what is the mechanism underlying the increased amplitude? Is this wave stimulated by exercise? Does this mechanism also explain why deletion of the insulin and IGF pathways increases longevity in animal experiments? Can this hypothesis explain the finding that intermittent fasting for 1 or 2 days promotes health and longevity [2]. Beneficial effects of such intermittent fasting are even found when total food intake is not diminished. At first glance, larger amplitudes of the waves of protein synthesis and breakdown during intermittent fasting would be in line with the hypothesis. Protein synthesis and breakdown can be included in the metabolic measurements and model building which can therefore be applied to investigate the ‘wave’ hypothesis.
Exercise has been found to benefit health in many ways which appears to involve regulatory pathways of metabolism [100]. Exercise is performed in bouts and stimulates metabolism strongly. Do effects of exercise on metabolism and health have mechanisms in common with caloric restriction? Are there other cycles of breakdown and synthesis, e.g. of the mitochondria or lipids, which show a similar pattern and a similar effect on ageing? In this way, the explanation of increased waves of protein breakdown and synthesis as a mechanism that ameliorates ageing raises many questions that may be answered with the aid of ‘physiomic’ modelling approaches.
6. Conclusion
Investigating the problem of ageing will benefit from the synergy of computational modelling of the dynamics of human metabolism with extensive measurements. Endurance provides a nicely quantifiable measure of the performance of the human system that may help reveal mechanisms of ageing. Dynamic variations in homeostasis are important for the whole body and for the mitochondria, and probably for maintaining physiological fitness in general. Because of the complex nature of ageing, the Quantitative Human Physiome approach provides the key to integrating the physiology of ageing, including the regulation of metabolism and intracellular maintenance.
Acknowledgement
We thank Dr Riekelt Houtkooper (Academic Medical Centre, Amsterdam, the Netherlands) for critically reading the manuscript and for helpful suggestions.
Authors' contributions
J.H.v.B .drafted the general outline and the manuscript, while J.B.B. drafted the initial versions of several sections. T.B.L.K. contributed discussion of molecular damage and repair and revised the manuscript critically for intellectual content. All authors contributed to revision of the manuscript and gave final approval for publication.
Competing interests
We have no competing interests.
Funding
We received no funding for this study.
References
- 1.Riera CE, Dillin A. 2015. Tipping the metabolic scales towards increased longevity in mammals. Nat. Cell Biol. 17, 196–203. ( 10.1038/ncb3107) [DOI] [PubMed] [Google Scholar]
- 2.Fontana L, Partridge L. 2015. Promoting health and longevity through diet: from model organisms to humans. Cell 161, 106–118. ( 10.1016/j.cell.2015.02.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matheson A. 1999. Further evidence for age-related deficits in human postural function. J. Vestib. Res. 9, 261–264. [PubMed] [Google Scholar]
- 4.Gutenkunst RN, Waterfall JJ, Casey FP, Brown KS, Myers CR, Sethna JP. 2007. Universally sloppy parameter sensitivities in systems biology models. PLoS Comput. Biol. 3, 1871–1878. ( 10.1371/journal.pcbi.0030189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hettling H, van Beek JH. 2011. Analyzing the functional properties of the creatine kinase system with multiscale 'sloppy' modeling. PLoS Comput. Biol. 7, e1002130 ( 10.1371/journal.pcbi.1002130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cannon WB. 1947. (first edition 1932) The wisdom of the body. London, UK: Paul, Trechn, Trubner. [Google Scholar]
- 7.Kirkwood TBL. 2011. Systems biology of ageing and longevity. Phil. Trans. R. Soc. B 366, 64–70. ( 10.1098/rstb.2010.0275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirkwood TB. 2005. Understanding the odd science of aging. Cell 120, 437–447. ( 10.1016/j.cell.2005.01.027) [DOI] [PubMed] [Google Scholar]
- 9.Kirkwood TB, Kowald A. 2012. The free-radical theory of ageing—older, wiser and still alive: modelling positional effects of the primary targets of ROS reveals new support. Bioessays 34, 692–700. ( 10.1002/bies.201200014) [DOI] [PubMed] [Google Scholar]
- 10.Fontana L, Partridge L, Longo VD. 2010. Extending healthy life span—from yeast to humans. Science 328, 321–326. ( 10.1126/science.1172539) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153, 1194–1217. ( 10.1016/j.cell.2013.05.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Partridge L, Gems D. 2002. Mechanisms of ageing: public or private? Nat. Rev. Genet. 3, 165–175. ( 10.1038/nrg753) [DOI] [PubMed] [Google Scholar]
- 13.Truscott RJ. 2011. Macromolecular deterioration as the ultimate constraint on human lifespan. Ageing Res. Rev. 10, 397–403. ( 10.1016/j.arr.2010.12.001) [DOI] [PubMed] [Google Scholar]
- 14.Guggenheim KY. 1991. Rudolf Schoenheimer and the concept of the dynamic state of body constituents. J. Nutr. 121, 1701–1704. [DOI] [PubMed] [Google Scholar]
- 15.Taegtmeyer H, Lubrano G. 2014. Rethinking cardiac metabolism: metabolic cycles to refuel and rebuild the failing heart. F1000Prime Rep. 6, 90 ( 10.12703/P6-90) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thornalley PJ. 2008. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems—role in ageing and disease. Drug Metabol. Drug Interact. 23, 125–150. ( 10.1515/DMDI.2008.23.1-2.125) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sorrentino JA, Sanoff HK, Sharpless NE. 2014. Defining the toxicology of aging. Trends Mol. Med. 20, 375–384. ( 10.1016/j.molmed.2014.04.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beckman K, Ames B. 1998. The free radical theory of aging matures. Physiol. Rev. 78, 548–581. [DOI] [PubMed] [Google Scholar]
- 19.Houtkooper RH et al. 2011. The metabolic footprint of aging in mice. Sci. Rep. 1, 134 ( 10.1038/srep00134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian L, Cai Q, Wei H. 1998. Alterations of antioxidant enzymes and oxidative damage to macromolecules in different organs of rats during aging. Free Radic. Biol. Med. 24, 1477–1484. ( 10.1016/S0891-5849(98)00025-2) [DOI] [PubMed] [Google Scholar]
- 21.Stuart J, Maddalena L, Merilovich M, Robb E. 2014. A midlife crisis for the mitochondrial free radical theory of aging. Longevity Healthspan 3, 1–15. ( 10.1186/2046-2395-3-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ristow M, Schmeisser S. 2011. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 51, 327–336. ( 10.1016/j.freeradbiomed.2011.05.010) [DOI] [PubMed] [Google Scholar]
- 23.Blackburn EH. 2000. Telomere states and cell fates. Nature 408, 53–56. ( 10.1038/35040500) [DOI] [PubMed] [Google Scholar]
- 24.Flach J, et al. 2014. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202. ( 10.1038/nature13619) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaiswal S, et al. 2014. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498. ( 10.1056/NEJMoa1408617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genovese G, et al. 2014. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487. ( 10.1056/NEJMoa1409405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahin E, Depinho RA. 2010. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464, 520–528. ( 10.1038/nature08982) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahin E, et al. 2011. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470, 359–365. ( 10.1038/nature09787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahlqvist KJ, Suomalainen A, Hamalainen RH. 2015. Stem cells, mitochondria and aging. Biochim. Biophys. Acta 1847, 1380–1386. ( 10.1016/j.bbabio.2015.05.014) [DOI] [PubMed] [Google Scholar]
- 30.Adams PD, Jasper H, Rudolph KL. 2015. Aging-induced stem cell mutations as drivers for disease and cancer. Cell Stem Cell 16, 601–612. ( 10.1016/j.stem.2015.05.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha M, Jang YC, Oh J, Khong D, Wu EY, Goodyear LJ, Rosner B, Lee RT, Wagers AJ. 2014. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 344, 649–652. ( 10.1126/science.1251152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. 2011. Human nutrition, the gut microbiome and the immune system. Nature 474, 327–336. ( 10.1038/nature10213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maillard I, Saltiel AR. 2015. Metabolism: inflammation keeps old mice healthy. Nature 528, 44–46. ( 10.1038/nature15648) [DOI] [PubMed] [Google Scholar]
- 34.Robinson SM. 1938. Experimental studies of physical fitness in relation to age. Arbeitsphysiologie 10, 251–323. ( 10.1007/bf02011412) [DOI] [Google Scholar]
- 35.Malkinson TJ. 2009. Maximal aerobic power in endurance trained and sedentary men and women, 10-74 years of age. In Science and Technology for Humanity (TIC-STH): 2009 IEEE Toronto Int. Conf., pp. 18–21. Toronto, Canada.
- 36.Malkinson TJ. 2014. Male and female age division VO2max–comparison with age division marathon and ironman triathlon performance. Med. Sci. Sports Med. 46, 892–893. [Google Scholar]
- 37.Tanaka H, Seals DR. 2003. Invited review: dynamic exercise performance in masters athletes: insight into the effects of primary human aging on physiological functional capacity. J. Appl. Physiol. 95, 2152–2162. ( 10.1152/japplphysiol.00320.2003) [DOI] [PubMed] [Google Scholar]
- 38.Lang PO, Michel JP, Zekry D. 2009. Frailty syndrome: a transitional state in a dynamic process. Gerontology 55, 539–549. ( 10.1159/000211949) [DOI] [PubMed] [Google Scholar]
- 39.Katzmarzyk PT, Church TS, Craig CL, Bouchard C. 2009. Sitting time and mortality from all causes, cardiovascular disease, and cancer. Med. Sci. Sports Exerc. 41, 998–1005. ( 10.1249/MSS.0b013e3181930355) [DOI] [PubMed] [Google Scholar]
- 40.Dunstan DW et al. 2010. Television viewing time and mortality: the Australian Diabetes, Obesity and Lifestyle Study (AusDiab). Circulation 121, 384–391. ( 10.1161/circulationaha.109.894824) [DOI] [PubMed] [Google Scholar]
- 41.Stamatakis E, Hamer M, Dunstan DW. 2011. Screen-based entertainment time, all-cause mortality, and cardiovascular events: population-based study with ongoing mortality and hospital events follow-up. J. Amer. College Cardiol. 57, 292–299. ( 10.1016/j.jacc.2010.05.065) [DOI] [PubMed] [Google Scholar]
- 42.Watson PA, Stein JP, Booth FW. 1984. Changes in actin synthesis and alpha-actin-mRNA content in rat muscle during immobilization. Amer. J. Physiol. 247, C39–C44. [DOI] [PubMed] [Google Scholar]
- 43.Reijnders CMA, van Essen HW, van Rens BTTM, van Beek JHGM, Ylstra B, Blankenstein MA, Lips P, Bravenboer N. 2013. Increased expression of matrix extracellular phosphoglycoprotein (MEPE) in cortical bone of the rat tibia after mechanical loading: identification by oligonucleotide microarray. PLoS ONE 8, e79672 ( 10.1371/journal.pone.0079672) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grierson B. 2014. What makes Olga run? New York, NY: Henry Holt and Co. [Google Scholar]
- 45.van Beek JH, Supandi F, Gavai AK, de Graaf AA, Binsl TW, Hettling H. 2011. Simulating the physiology of athletes during endurance sports events: modelling human energy conversion and metabolism. Phil. Trans. R. Soc. A 369, 4295–4315. ( 10.1098/rsta.2011.0166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carrier D. 1984. The energetic paradox of human running and hominid evolution. Curr. Anthropol. 25, 483–495. ( 10.1086/203165) [DOI] [Google Scholar]
- 47.Bernard C. 1865. Introduction à l'étude de la médecine expérimentale. Paris: J. B. Baillière. [Google Scholar]
- 48.Marey E-J. 1863. Physiologie médicale de la circulation du sang, basée sur l’étude de graphique des mouvements du coeur and du pouls arteriel avec application aux maladies de l'appareil circulatoire. Paris: A. Delahaye viii. [Google Scholar]
- 49.Chandra FA, Buzi G, Doyle JC. 2011. Glycolytic oscillations and limits on robust efficiency. Science 333, 187–192. ( 10.1126/science.1200705) [DOI] [PubMed] [Google Scholar]
- 50.Bassingthwaighte JB, Liebovitch LS, West BJ. 1994. Fractal physiology. Oxford, UK: Oxford University Press. [Google Scholar]
- 51.Kleiger RE, Miller JP, Bigger JT Jr, Moss AJ. 1987. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Amer. J. Cardiol. 59, 256–262. ( 10.1016/0002-9149(87)90795-8) [DOI] [PubMed] [Google Scholar]
- 52.Shepherd JT. 1981. The lungs as receptor sites for cardiovascular regulation. Circulation 63, 1–10. ( 10.1161/01.CIR.63.1.1) [DOI] [PubMed] [Google Scholar]
- 53.Goldberger AL, Rigney DR, Mietus J, Antman EM, Greenwald S. 1988. Nonlinear dynamics in sudden cardiac death syndrome: heartrate oscillations and bifurcations. Experientia 44, 983–987. ( 10.1007/BF01939894) [DOI] [PubMed] [Google Scholar]
- 54.Buchheit M. 2014. Monitoring training status with HR measures: do all roads lead to Rome? Front. Physiol. 5, 73 ( 10.3389/fphys.2014.00073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Houtkooper RH, Williams RW, Auwerx J. 2010. Metabolic networks of longevity. Cell 142, 9–14. ( 10.1016/j.cell.2010.06.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mattison JA, et al. 2012. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 489, 318–321. ( 10.1038/nature11432) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM. 2014. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 5, 3557 ( 10.1038/ncomms4557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ravussin E, et al. 2015. A 2-year randomized controlled trial of human caloric restriction: feasibility and effects on predictors of health span and longevity. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1097–1104. ( 10.1093/gerona/glv057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walther DM, et al. 2015. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161, 919–932. ( 10.1016/j.cell.2015.03.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walther DM, Mann M. 2011. Accurate quantification of more than 4000 mouse tissue proteins reveals minimal proteome changes during aging. Mol. Cell Proteomics 10, M110004523. ( 10.1074/mcp.M110.004523) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. 2013. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. ( 10.1038/nature12188) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goldspink DF. 1991. Exercise-related changes in protein turnover in mammalian striated muscle. J. Exp. Biol. 160, 127–148. [DOI] [PubMed] [Google Scholar]
- 63.Hiona A, et al. 2010. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 5, e11468 ( 10.1371/journal.pone.0011468) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Correia-Melo C, Passos JF. 2015. Mitochondria: are they causal players in cellular senescence? Biochim Biophys Acta 1847, 1373–1379. ( 10.1016/j.bbabio.2015.05.017) [DOI] [PubMed] [Google Scholar]
- 65.Passos JF, Von Zglinicki T. 2012. Mitochondrial dysfunction and cell senescence—skin deep into mammalian aging. Aging 4, 74–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thiele I, et al. 2013. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 31, 419–425. ( 10.1038/nbt.2488) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gavai AK, Supandi F, Hettling H, Murrell P, Leunissen JA, van Beek JH. 2015. Using bioconductor package BiGGR for metabolic flux estimation based on gene expression changes in brain. PLoS ONE 10, e0119016 ( 10.1371/journal.pone.0119016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Palsson BO. 2005. Systems biology. Properties of reconstructed networks. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 69.Lying-Tunell U, Lindblad BS, Malmlund HO, Persson B. 1981. Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol. Scand. 63, 337–350. ( 10.1111/j.1600-0404.1981.tb00788.x) [DOI] [PubMed] [Google Scholar]
- 70.Lewis NE, et al. 2010. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 28, 1279–1285. ( 10.1038/nbt.1711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bassingthwaighte JB. 2014. New and notable: uncertainty quantification. Biophys. J. 107, 2481–2483. ( 10.1016/j.bpj.2014.10.040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cakir T, Alsan S, Saybasili H, Akin A, Ulgen KO. 2007. Reconstruction and flux analysis of coupling between metabolic pathways of astrocytes and neurons: application to cerebral hypoxia. Theor. Biol. Med. Model 4, 48 ( 10.1186/1742-4682-4-48) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lewis NE, Abdel-Haleem AM. 2013. The evolution of genome-scale models of cancer metabolism. Front. Physiol. 4, 237 ( 10.3389/fphys.2013.00237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sahoo S, Thiele I. 2013. Predicting the impact of diet and enzymopathies on human small intestinal epithelial cells. Hum. Mol. Genet. 22, 2705–2722. ( 10.1093/hmg/ddt119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kriete A, Bosl WJ, Booker G. 2010. Rule-based cell systems model of aging using feedback loop motifs mediated by stress responses. PLoS Comput. Biol. 6, e1000820 ( 10.1371/journal.pcbi.1000820) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conley KE, Jubrias SA, Cress ME, Esselman P. 2013. Exercise efficiency is reduced by mitochondrial uncoupling in the elderly. Exp. Physiol. 98, 768–777. ( 10.1113/expphysiol.2012.067314) [DOI] [PubMed] [Google Scholar]
- 77.Amara CE, Shankland EG, Jubrias SA, Marcinek DJ, Kushmerick MJ, Conley KE. 2007. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc. Natl Acad. Sci. USA 104, 1057–1062. ( 10.1073/pnas.0610131104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Conley KE, Jubrias SA, Cress ME, Esselman PC. 2013. Elevated energy coupling and aerobic capacity improves exercise performance in endurance-trained elderly subjects. Exp. Physiol. 98, 899–907. ( 10.1113/expphysiol.2012.069633) [DOI] [PubMed] [Google Scholar]
- 79.Conley KE, Jubrias SA, Esselman PC. 2000. Oxidative capacity and ageing in human muscle. J. Physiol. 526, 203–210. ( 10.1111/j.1469-7793.2000.t01-1-00203.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Austin S, St-Pierre J. 2012. PGC1α and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 125, 4963–4971. ( 10.1242/jcs.113662) [DOI] [PubMed] [Google Scholar]
- 81.Kowald A, Kirkwood TB. 2000. Accumulation of defective mitochondria through delayed degradation of damaged organelles and its possible role in the ageing of post-mitotic and dividing cells. J. Theor. Biol. 202, 145–160. ( 10.1006/jtbi.1999.1046) [DOI] [PubMed] [Google Scholar]
- 82.Kowald A, Kirkwood TBL. 2014. Transcription could be the key to the selection advantage of mitochondrial deletion mutants in aging. Proc. Natl Acad. Sci. USA 111, 2972–2977. ( 10.1073/pnas.1314970111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. 2015. Redox homeostasis and mitochondrial dynamics. Cell Metab. 22, 207–218. ( 10.1016/j.cmet.2015.06.006) [DOI] [PubMed] [Google Scholar]
- 84.Sen S, et al. 2013. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J. Am. Heart Assoc. 2, e004796 ( 10.1161/JAHA.113.004796) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davies MJ. 2005. The oxidative environment and protein damage. Biochim. Biophys. Acta 1703, 93–109. ( 10.1016/j.bbapap.2004.08.007) [DOI] [PubMed] [Google Scholar]
- 86.Benson AP, Grassi B, Rossiter HB. 2013. A validated model of oxygen uptake and circulatory dynamic interactions at exercise onset in humans. J. Appl. Physiol. (1985) 115, 743–755. ( 10.1152/japplphysiol.00184.2013) [DOI] [PubMed] [Google Scholar]
- 87.van Beek JH. 2015. Computer modelling of energy turnover and body temperature in elite cyclists during climbing. Cold, colder, Gavia. Steep, steeper, Angliru. J. Sci. Cycling. 4, 35. [Google Scholar]
- 88.Binsl TW, De Graaf AA, Venema K, Heringa J, Maathuis A, De Waard P, Van Beek JH. 2010. Measuring non-steady-state metabolic fluxes in starch-converting faecal microbiota in vitro. Benef. Microb. 1, 391–405. ( 10.3920/BM2010.0038) [DOI] [PubMed] [Google Scholar]
- 89.Shoaie S, et al. 2015. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 22, 320–331. ( 10.1016/j.cmet.2015.07.001) [DOI] [PubMed] [Google Scholar]
- 90.Albers MJ, et al. 2008. Hyperpolarized 13C lactate, pyruvate, and alanine: noninvasive biomarkers for prostate cancer detection and grading. Cancer Res. 68, 8607–8615. ( 10.1158/0008-5472.CAN-08-0749) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Weiss K, Bottomley PA, Weiss RG. 2015. On the theoretical limits of detecting cyclic changes in cardiac high-energy phosphates and creatine kinase reaction kinetics using in vivo (3)(1)P MRS. NMR Biomed. 28, 694–705. ( 10.1002/nbm.3302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Binsl TW, Alders DJ, Heringa J, Groeneveld AB, van Beek JH. 2010. Computational quantification of metabolic fluxes from a single isotope snapshot: application to an animal biopsy. Bioinformatics 26, 653–660. ( 10.1093/bioinformatics/btq018) [DOI] [PubMed] [Google Scholar]
- 93.van Beek JH, van Mil HGJ, King RB, de Kanter FJ, Alders DJ, Bussemaker J. 1999. A 13C NMR double-labeling methods to quantitate local myocardial O2 consumption using frozen tissue samples. Amer. J. Physiol. Heart Circul. Physiol. 277, 1630–1640. [DOI] [PubMed] [Google Scholar]
- 94.Alders DJ, Groeneveld AB, Binsl TW, van Beek JH. 2015. Progressively heterogeneous mismatch of regional oxygen delivery to consumption during graded coronary stenosis in pig left ventricle. Am. J. Physiol. Heart Circ. Physiol. 309, H1708–H1719. ( 10.1152/ajpheart.00657.2014) [DOI] [PubMed] [Google Scholar]
- 95.Hettling H, Alders DJ, Heringa J, Binsl TW, Groeneveld AB, Van Beek JH. 2013. Computational estimation of tricarboxylic acid cycle fluxes using noisy NMR data from cardiac biopsies. BMC Syst. Biol. 7, 1–12. ( 10.1186/1752-0509-7-82) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hyder F, Fulbright RK, Shulman RG, Rothman DL. 2013. Glutamatergic function in the resting awake human brain is supported by uniformly high oxidative energy. J. Cereb. Blood Flow Metab. 33, 339–347. ( 10.1038/jcbfm.2012.207) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Noble D. 2012. A theory of biological relativity: no privileged level of causation. Interface Focus 2, 55–64. ( 10.1098/rsfs.2011.0067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bassingthwaighte J, Hunter P, Noble D. 2009. The Cardiac Physiome: perspectives for the future. Exp. Physiol. 94, 597–605. ( 10.1113/expphysiol.2008.044099) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Spindler SR. 2001. Calorie restriction enhances the expression of key metabolic enzymes associated with protein renewal during aging. Ann. NY Acad. Sci. 928, 296–304. ( 10.1111/j.1749-6632.2001.tb05659.x) [DOI] [PubMed] [Google Scholar]
- 100.Watson K, Baar K. 2014. mTOR and the health benefits of exercise. Semin. Cell Dev. Biol. 36, 130–139. ( 10.1016/j.semcdb.2014.08.013) [DOI] [PubMed] [Google Scholar]