

Abstract
Protein kinases are an important class of enzymes involved in the phosphorylation of their targets, which regulate key cellular processes and are typically mediated by a specificity for certain residues around the target phospho-acceptor residue. While efforts have been made to identify such specificities, only ∼30% of human kinases have a significant number of known binding sites. We describe a computational method that utilizes functional interaction data and phosphorylation data to predict specificities of kinases. We applied this method to human kinases to predict substrate preferences for 57% of all known kinases and show that we are able to reconstruct well-known specificities. We used an in vitro mass spectrometry approach to validate four understudied kinases and show that predicted models closely resemble true specificities. We show that this method can be applied to different organisms and can be extended to other phospho-recognition domains. Applying this approach to different types of posttranslational modifications (PTMs) and binding domains could uncover specificities of understudied PTM recognition domains and provide significant insight into the mechanisms of signaling networks.
Phosphorylation is a prominent protein posttranslational modification (PTM)1, which involves the transfer of a phosphate group (PO43-) to different amino-acids, including serine (Ser), threonine (Thr), or tyrosine (Tyr) residues of proteins. In human, this process is catalyzed by over 500 protein kinases (1), which can regulate protein function by inducing conformational changes in the protein structure, promote or disrupt protein interactions, or alter protein localization or expression. This process is crucial for the regulation of many biological pathways, including cell division, apoptosis, differentiation, and the response to stress. By phosphorylating other kinases and proteins, kinases form complex signaling networks. However, our understanding of the architecture of these networks remains limited.
The mode by which protein kinases recognize specific target residues depends on the accessibility of these residues, and more importantly, kinases have been shown to have preferences for certain amino-acids flanking the central phospho-acceptor Ser/Thr/Tyr site. These preferences define the kinase substrate specificity, often referred to as the kinase substrate “motif.” These motifs were initially defined by searching for consensus sequences among a set of known target sites. For example, the cyclin dependent kinase (CDK2) is known to preferentially target the motif [SerThr]ProX[ArgLys] (a proline at position +1, any amino acid at position +2, and Arg/Lys at position +3) (2). Computational approaches were then developed to combine known kinase-target sites and their flanking regions to model kinase specificity and successfully predict novel target phosphosites (3, 4). Understanding preferences of kinases toward their substrates therefore offers significant insight into the mechanisms of signaling networks.
Over the past decade, there has been an ever-increasing quantity of phosphorylation site data, typically identified using mass spectrometry (MS), with over 100,000 sites identified in human (5–7). Yet, these phosphoproteomic experiments have provided us with a large number of phosphosites for which we do not known the upstream regulatory kinase. Compendiums of kinase-site relationships curated from the literature (5–7) currently associate roughly 6% (6,320/107,444) of known phosphosites to one or more kinases. The experimental characterization of kinase target sites allows for the discovery of specificities for many kinases, but they are typically expensive, time consuming, and are not possible to perform on kinases that are difficult to work with.
Several methods aim at modeling kinase specificity using the collections of known targets sites. These include scan-x (8), Scansite (4), NetPhorest (3), GPS (9), KinasePhos (10), and many more. However, these methods depend on the availability of many known target sites for each modeled kinase. Here, we aim to tackle a more difficult challenge of predicting the specificity of kinases, without any direct knowledge of its experimentally determined target sites. The prediction tool Predikin (11) takes such an approach by trying to predict kinase specificity by examining 3D models of kinases bound to their substrate peptides. This analysis has identified residues in the kinases catalytic domain referred to as substrate determining residues (SDRs), which confer a preference for residues in the phosphosites flanking regions. Predikin uses these SDRs sites to match a new kinase sequence to a kinase with known specificity. In this way, Predikin also makes use of known kinase target site information. In addition, Predikin depends on the availability of protein structures and therefore cannot be easily scaled to kinase families without 3D structures nor to other PTM recognition domains.
We decided to take an alternative approach at predicting kinase specificity. Previous studies have shown that it is possible to use information regarding the interaction partners of a peptide-binding protein to identify potential motifs mediating these interactions (12). We reasoned that putative interaction partners of a kinase are more likely than random proteins to be phosphorylated by that kinase. Thus, phosphosites occurring on interaction partners of kinases should confer a bias in amino acid composition toward the kinase's specificity, which can be revealed by motif enrichment (Fig. 1). We tested this on human kinases to identify already known specificities, as well as other understudied kinases. We experimentally determined peptide targets of four understudied kinases and showed that predicted models closely resemble the experimentally identified sites. We extended our analysis to show that specificities can be predicted, not only for kinases, but also for other phospho-residue binding domains, such as 14-3-3 proteins and to an acetyl-lysine binding bromodomain. We also applied our method to mouse and showed that the predicted specificities of some kinases are conserved. We show here that it is possible to combine large-scale PTM data with protein network information to derive the specificity of PTM regulators and believe that this approach can be widely applicable to different PTM types.
Fig. 1.
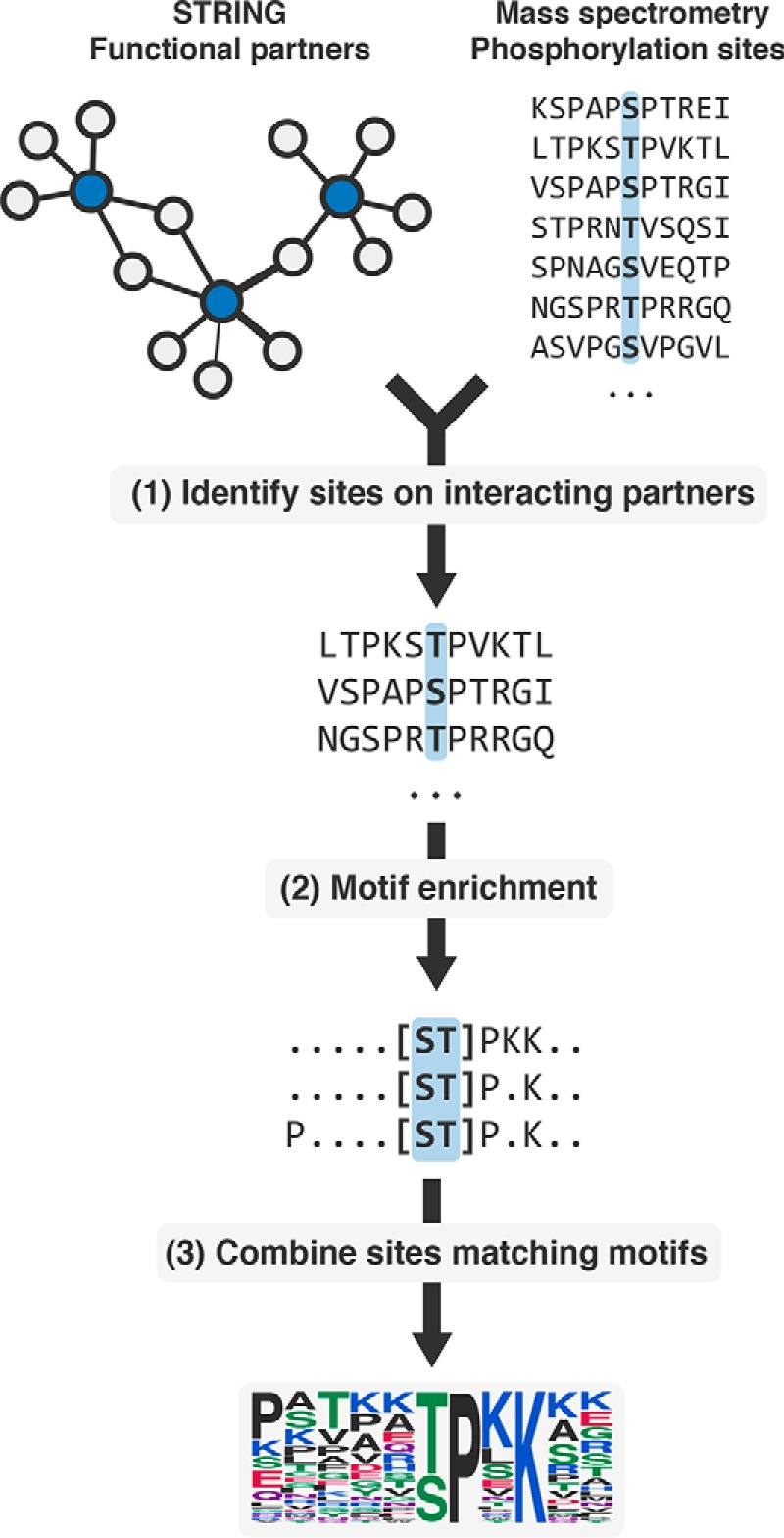
Overview of the method. (1) Experimentally identified phosphosites on functional interaction partners of a kinase are collected. (2) The sites are then subject to motif enrichment to identify overrepresented motifs, likely representing the kinase specificity. (3) Phosphosites matching the top k significant motifs are then retained and used to construct a specificity model.
EXPERIMENTAL PROCEDURES
Data Sources for Phosphosites and Functional Interactions
Functional interaction data were collected from STRING (v9.1). Phosphorylation sites were collected from public databases, including PhosphoSitePlus (5), PhosphoELM(6), and HPRD (7) and from a study of mouse tissues (13). Phosphosites were then mapped to protein sequences provided by STRING. Kinase orthologs for 471/493 (95%) kinases were obtained from InParanoid v8.0 (14).
Kinase Domain Prediction
Given a protein sequence, we used Kinomer (15), which uses multilevel hidden Markov models and HMMER (16) to identify protein kinases, and classify them into their appropriate kinase family. E-value cutoffs for each family were used as defined in (15). If a kinase was assigned more than one predicted family, the one with the highest E-value was used. These families were also used to determine if the kinase is Ser/Thr-specific or Tyr-specific. We assume that a kinase is either Ser/Thr-specific or Tyr-specific and do not account for dual specificity kinases.
Motif Enrichment
To identify motifs enriched within a set of phosphosites, compared with a background, we used the motif-x algorithm (17). Here, we used two background sets, defined as 10,000 15-mers centered on nonphosphorylated Ser/Thr or Tyr residues, depending on if the kinase is Ser/Thr-specific or Tyr-specific. All enrichments were carried out for ±7 residues surrounding the central residue with occurrences ≥ 10 and p < 10−6. Since the motif-x tool was only available via an online webserver, we reimplemented the tool for the R programming language, which can be found here: https://github.com/omarwagih/rmotifx.
Kinase Specificity Models
Specificity models were constructed as PWMs, which are commonly used to model specificities of linear motifs (18). PWMs can then be used to score peptides. We use an adapted version of the matrix similarity score (MSS), originally developed in the MATCH algorithm (19), as described in (20). The MSS ranges from 0–1, where 0 represents no predicted binding, and 1 represents perfect predicted binding.
The performance of a given PWM was evaluated as the area under the ROC curve (AUC), which is the curve representing the relationship between the false positive rate and true positive rate as the MSS score cutoff is varied:
 |
Here, FP, TP, TN, FN represent the number of false positives, true positives, true negatives, and false negatives, respectively. The PWMs were used to score positive and negative sequences in order to generate these values. For a kinase of interest, we define the positive sequences as the set of phosphosites annotated to the kinase and the negative sequences as phosphosites annotated to any kinase not belonging to the same kinase family, as defined by Manning et al. (1).
In the case where the performance of experimental models were evaluated (i.e. using the gold-standard sequences), we performed 10-fold cross validation in which the kinase sequences are randomly split into 10 bins. Each bin is iteratively used as the test set, while the remaining nine are used to construct the PWM. This results in 10 AUCs, which are averaged to provide an unbiased proxy of the PWM's prediction power.
We supply a resource that contains all of the information used for the specificity predictions of each kinase. This can be accessed from http://evocellnet.github.io/kpred/. For each kinase, the user can find the specificity logo as well as the list of interacting partners and phosphosites used to generate the prediction. We also provide an R package to allow others to more easily use this method for their own PTM and species of interest. The package and a tutorial on how to use it are available via the help section of the prediction website.
Profiling In Vitro Kinase Substrates
Identification of in vitro kinase substrates was conducted as previously described (21). Briefly, lysate proteins were extracted from HeLa S3 cells at about 80% confluence in 15 cm dishes, and the total protein amount was measured by a BCA protein assay kit. Dephosphorylation was then carried out with TSAP (Promega, Madison, MI, USA) at 37 °C for 1 h, and TSAP was inactivated by heating to 75 °C for 30 min. For in vitro kinase reaction, each 100 μg of dephosphorylated proteins (1 μg/μl) was reacted with 1 μl of each recombinant kinase (0.5 μg/μl) or distilled water as a control at 37 °C in kinase reaction buffer (40 mm Tris-HCl (pH 7.5), 20 mm MgCl2, 1 mm ATP) for 3 h. AKT2, catalytic domain [120–481(end), accession NP_001617.1], full-length EIF2AK4 [1–1649(end) accession Q9P2K8.2], full-length HIPK2 [1–1198(end) accession Q9H2X6] and full-length SRPK2 [1–688(end) accession NP_872633.1] were obtained from Carna Biosciences Inc. (Kobe, Japan). The kinases were expressed as N-terminal GST-fusion protein using baculovirus expression system with SF9 cells and were purified using glutathione Sepharose chromatography. The reaction was stopped by heating to 95 °C for 5 min. After protein reduction/alkylation, Lys-C/trypsin digestion (1/100 w/w) was performed and phosphopeptides were enriched by TiO2-based hydroxyl-acid-modified metal oxide chromatography (22).
Phosphopeptides were desalted by StageTips and analyzed by nanoLC-MS/MS using a self-pulled analytical column (150 mm length × 100 μm inner diameter) packed with ReproSil-Pur C18-AQ materials (3 μm, Dr. Maisch, Ammerbuch, Germany). An Ultimate 3000 pump (Thermo Fisher Scientific, Germering, Germany) and an HTC-PAL autosampler (CTC Analytics, Zwingen, Switzerland) were used coupled to an LTQ-Orbitrap XL (Thermo Fisher Scientific). A spray voltage of 2,400 V was applied. The MS scan range was m/z 300–1,500. The top 10 precursor ions were selected in MS scan by the Orbitrap with r = 60,000 for MS/MS scans and the ion trap in the automated gain control (AGC) mode, where automated gain control values of 5.00 × 105 and 1.00 × 104 were set for full MS and MS/MS, respectively. To minimize repetitive MS/MS scanning, a dynamic exclusion time was set at 20 s with a repeat count of 1 and an exclusion list size of 500. The normalized CID was set to be 35.0. Mass Navigator v1.2 (Mitsui Knowledge Industry, Tokyo, Japan) with the default parameters for the LTQ-Orbitrap XL was used to create peak lists on the basis of the recorded fragmentation spectra. Peptides and proteins were identified by automated database searching using Mascot v2.3 (Matrix Science, London, UK) against SwissProt release 2010_11 (02/11/2010, 522,019 entries) with a precursor mass tolerance of 3 ppm, a fragment ion mass tolerance of 0.8 Da, and strict trypsin specificity allowing for up to two missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionines; phosphorylation of serine, threonine, and tyrosine were allowed as variable modifications. Peptides were considered identified if the Mascot score was over the 95% confidence limit based on the “identity” score of each peptide and if at least three successive y- or b-ions with a further two or more y-, b-, and/or precursor-origin neutral loss ions were observed, based on the error-tolerant peptide sequence tag concept. After identification, phosphopeptides identified from the control samples were rejected. A randomized decoy database created by a Mascot Perl program gave a 1% false-discovery rate for identified peptides with these criteria. Phosphosite localization was evaluated using a site-determining ion combination method based on the presence of site-determining y- or b-ions in the peak lists of the fragment ions, which supported the phosphosites unambiguously.
RESULTS
Network-Based Prediction of Kinase-Substrate Specificity
We hypothesized that the interaction network of a protein kinase should be enriched in its target proteins. This hypothesis was confirmed by the observation of a very significant enrichment of known kinase targets in the functional interaction or physical interaction partners of kinases (Supplemental Fig. 1). In order to predict kinase specificities, we then combined information on human protein interaction data and phosphorylation data derived from large-scale MS studies. Given that kinases bind their target proteins transiently, we used functional interactions derived from STRING (23) as a source for potential kinase interactors. We collected a total of 2,425,314 interactions in 22,523 proteins and compiled experimentally determined phosphorylation sites from three public databases (PhosphoSitePlus (5), PhosphoELM (6) and HPRD (7)). Phosphosites were mapped back to proteins having information in STRING, resulting in 107,444 sites in 12,207 proteins. We identified 493 kinases in this reference proteome (81% serine/threonine and 19% tyrosine kinases) using the Kinomer prediction tool (15) (Methods). For a given kinase, all phosphosites occurring on the STRING partners of a kinase were collected and enriched for motifs using the motif-x algorithm (17) (Fig. 1, Methods). A random sample of 10,000 unphosphorylated Ser/Thr/Tyr sites were used as the background for enrichment. Phosphosites matching the most significant extracted motifs were then used to build a position weight matrix (PWM), which highlights the predicted specificity of the kinase and can be used to score novel phosphosites (Fig. 1).
A survey of all known phosphorylation sites revealed a strong enrichment for prolines at position +1 (Pro+1) (Supplemental Fig. 2). This results in consistent enrichment of Pro+1 motifs (Supplemental Fig. 3, Supplementary Results). To circumvent this, we require to know if a kinase is proline-directed (i.e. prefers Pro+1). We found that kinases of the CMGC family, including CDKs, MAPKs, GSKs, and CDK-like kinases have Pro+1 motifs as shown in their experimental binding sites (Supplemental Fig. 4). Also, of all the non-CMGC kinases (with ≥20 known targets) only 1.57% (1/68) were found to be proline directed. Thus, if a kinase is not predicted as CMGC, phosphosites containing Pro+1 are removed from foreground and background sets prior to motif enrichment.
There are two variable parameters in our method: the cutoff for the functional-interaction prediction score from STRING and the top k number of significant motifs extracted during the enrichment. To determine the best thresholds to use, we tested the predicted kinase specificity models against a set of 9,595 gold-standard kinase–substrate relationships. We carried out the benchmarking using a set of nine well-studied kinases from a diverse set of kinase families (ABL1, AKT1, ATR, AURKB, CDK2, CSNK2A1, GSK3B, MAPK1, and PRKACA) with well-recognized specificities in the literature. We varied the STRING cut-off, and the top k motifs extracted. The performance of the resulting PWM in each case was evaluated using the area under receiver operating characteristic (AUC) (Methods). We found that increasing the STRING score threshold, overall, resulted in higher AUCs (Supplemental Fig. 5). However, in most cases, we did not see a significant increase after a score cutoff of 400, and therefore, we used that cut-off throughout our analysis. We chose to select the top five motifs for two reasons. First, the AUCs among varied k values did not vary considerably. Second, overselecting motifs could mask the predicted specificity of the kinase (Supplemental Fig. 6). We also tested if difference sources of evidence (e.g. text mining, coexpression, interaction data) within the STRING database resulted in a different performance. Overall, using different evidence provided by the STRING database did not provide a significant increase in the AUCs, and a larger number of kinases can predicted using the combination of all evidence types (Supplemental Fig. 7).
Next, we checked to see how likely random models constructed without the network information performed in comparison to our predicted models. If a given kinase has n STRING interactions, and among those interactors there are m phosphosites (s1, s2, …, sm), then m random phosphosites are selected from all known phosphosites in public databases (r1, r2, …, rm). Specificity is then predicted, as previously described, using these sites. Random models, constructed using the top five motifs, were compared against the predicted models in their discriminative power against the gold-standard sequences, as measured by the AUC. We found that our predicted models for most of the nine kinases, with the exception of AKT1 and MAPK1, performed significantly better than random (Fig. 2A and Supplemental Fig. 8). This does not mean that the AKT1 model is incorrect since it performs very well at predicting known AKT1 target sites (AUC = 0.90, Fig. 2A). However, some kinases like AKT1 have specificities that are well modeled by the most represented motifs across all sites. Thus, in these cases, the network information appears to provide almost no gain compared with random sampling. In opposition to these kinases, ATR has a specificity that is very uncommon with a preference for glutamine at position +1 that is very well recovered by this approach (Figs. 2A and 2F) but very unlikely to be observed in a random pool of phosphosites.
Fig. 2.

Benchmarking of the method. (A) The performance of each predicted model compared with models predicted using random phosphosites. Seven of nine cases perform better than random (.p < .01, *p < .05, **p < .01, *** p < .001, one sided z-test). Error bars represent the median absolute deviation for 1,000 random models. 85 kinases with 20 or more known substrates were used as the gold standard. (B) Performance of predicted models across different families. The gray line denotes near-random performance. (C) Performance of gold-standard models is compared with that of the predicted models. A strong correlation suggests a relationship between the specificity of the kinase and predictability of a specificity model. (D-G) Examples of predicted specificity models. The top and middle panel of each example shows the specificity of the kinase as constructed from known substrates and as predicted by our method, respectively. The bottom panel shows the top five extracted motifs and the number of phosphosites matching them.
These results demonstrate the ability to integrate protein interaction information with large-scale data on protein phosphorylation to derive kinase specificity models.
Prediction of Kinase Specificity across All Human Kinases
Our method was applied to all kinases, resulting in predictions for 282/493 (57%) of kinases (Supplementary data). Kinases that did not result in a prediction either had a low number of partners or a scarcity of phosphosites on partners. We selected 85 kinases with ≥20 known target phosphosites as well as a prediction and compared how well the predicted models performed with respect to the kinase family (Fig. 2B). The average AUC across all kinases was 0.64 with 32% (27/85) of kinases having an AUC greater than 0.7 (Fig. 2B). We found that CMGC, PIKK, and AGC families performed best, whereas TKL, STE and TK kinases had a larger fraction of poorly performing models. Excluding the TKL, STE, and TK kinases, the average AUC increases to 0.68 with 44% of kinases (27/61) scoring higher than 0.7 (Fig. 2B). We speculated that the differences in performance across the different kinase families reflect different degrees of specificity in the kinase–substrate recognition. For example, many tyrosine kinases have additional targeting domains (i.e. PTB and SH2 domains). Also, several STE kinases are known to have an additional interaction surface for a “docking motif” (24, 25). For these kinases, targeting is achieved by multiple interfaces or often aided by other mechanisms (see Discussion), and therefore, they might be less specific in the recognition of sequences around the phosphosite. In line with this reasoning, we found that kinases that had additional protein domains also had worse-performing models (p < 1.88 × 10−3, Wilcoxon signed-rank test; Supplemental Fig. 9). We tested this notion more explicitly by comparing our predicted models with a proxy for kinase promiscuity. For the same set of kinases, we built PWMs using the gold-standard target sites and computed an experimental AUC using 10-fold cross-validation (Methods), which reflects how well the gold-standard models perform at distinguishing their own sites. Interestingly, we found that AUCs of predicted models showed a strong correlation to that of the experimental (r = 0.757, p < 2 × 10−16; Fig. 2C), suggesting that kinases with high true specificity are more likely to have high predictability.
We selected a few example kinases to demonstrate the specificity determinants captured by our approach (Figs. 3D–3G). The predicted specificities strongly resemble that of the experimental and, in many cases, are much more apparent. For example, the pronounced preference for glutamine at +1 for ATR is recovered (Fig. 2F). The known Akt preference for an arginine at -3 and -5 is fully recovered in the predicted model. In addition, there is a more apparent preference for [ArgSerThr] at -2, and arginine at -1 that is not as apparent in the experimental specificity (Fig. 2G).
Fig. 3.

Experimental validation. (A) Workflow for identifying phosphorylation sites. (B–E) Predicted specificity models of four experimentally validated kinases. The top and middle panel of each example shows the specificity of the kinase as constructed from the target sites of these kinases identified via MS and as predicted by our method, respectively. The bottom panel shows the top five extracted motifs and the number of phosphosites matching them. (F) The performance of each predicted model compared with models predicted using random phosphosites.
In attempt to identify features comprised by better performing models, we searched for relationships between the AUC of the predicted model and (1) the number of functional interacting partners, (2) the number of phosphosites on interacting partners, (3) the distribution of information content, and (4) the number of extracted motifs. We observed weak correlations (r < 0.361, Supplemental Fig. 10) for each of the individual features. However, we were able to achieve a higher correlation by combining a number of features using a linear regression model (r = 0.542, p = 8.37 × 10−8). This model can thus be used to assign a quantitative measure of confidence related to the truth of predicted specificity, which we use to rank our predictions (Supplementary Data).
We tested several alterations of the method, such as using different background sets for motif enrichment as well as using only high confidence phosphosites, and overall did not observe a strong improvement in the AUC (Supplementary Results, Supplemental Figs. 11–12). For example, to obtain a list of higher confidence sites, we tried excluding phosphosite positions that are supported only by one study or excluding from the analysis highly abundant proteins (Supplemental Fig. 11). To test the impact of removing the Pro+1 sites, we used an alternative strategy whereby we did not filter Pro+1 peptides but used as background all phosphosites instead of phospho-acceptor residues. Although we were still able to retrieve many correct predictions, the performance was lower in this alternative implementation.
Mass-Spectrometry-Based Validation of Kinase Specificity
We selected four kinases with few known target sites in the literature, across different kinases subfamilies for which we had a predicted model (CMGC kinases SRPK2 and HIPK2, AGC kinase AKT2, and PEK kinase EIF2AK4). For each of these kinases, we identified in vivo target sites using the phosphoproteomic approach described by Imamura et al. (21) (Fig. 3A). Briefly, HeLa cell extracts were treated with phosphatase to remove any existing phosphosites, and kinases were added in separate experiments. Phosphorylated extract were then subjected to trypsin digestion, phosphopeptide enrichment, and nanoLC-MS/MS (Fig. 3A). We identified a total of 483 novel phosphosites for these kinases (AKT2, n = 248; EIF2AK4, n = 91; HIPK2, n = 106; SRPK2, n = 38, available in Supplementary Data). The identified target sites were then compared with our predicted models for these kinases (Figs. 3B–3E). We found that all predicted models performed significantly better than random, and three of the four had an AUC ≥ 0.7 at classifying the experimentally identified sites (Fig. 3F). These results are in line with the benchmarks performed and further support the validity of the approach described here. We note that the SRPK2 kinase was predicted to have a strong preference for serines and arginines at several positions. This motif was unusual given previously described models, though several elements of this motif are confirmed by the experimental sites (Fig. 3D). The kinase specificity of SRPK2 was very recently determined using a chemical genetic approach (26) that provides further validation for the predicted specificity model for this kinase.
Prediction of PTM Binding Specificities
To demonstrate the extendibility of our method to other types of linear motif specificities, we applied the same method to 14-3-3 proteins. 14-3-3 proteins are conserved single domain proteins capable of binding a phospho-serine or threonine and are responsible for tight regulation of several important pathways such as cell death, cell cycle control, and signal transduction (27). Previous studies have shown that binding sites these proteins have specificities toward their target phosphosites (28) (Fig. 4A). We applied the method to all seven human 14-3-3 proteins (Figs. 4A–4C, Supplemental Fig. 13) and similarly show that the recovered models are very good predictors of known binding sites (AUC>0.80) and perform significantly better than random (Fig. 4D). We recovered the well-known determinants such as arginine at position -3 and some preference for proline at position +2. We found that there is little to no overlap between sites used to construct the models for each 14-3-3 protein, despite showing similar predicted specificity (Supplemental Fig. 14). This suggests that the same motif is recovered in each case from a different source of partner sites, adding to the confidence of the recovered models.
Fig. 4.
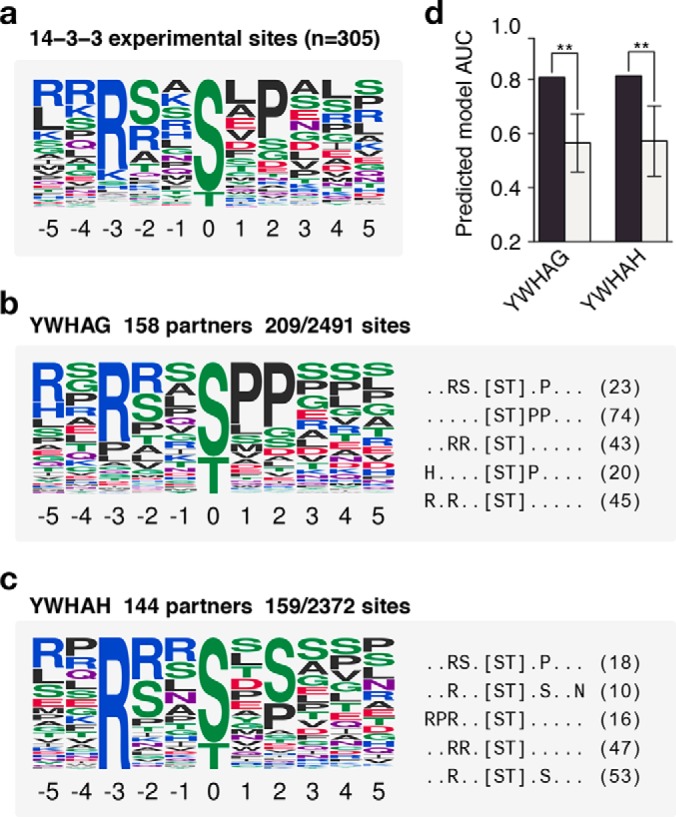
Prediction of 14-3-3 domain specificities. (A) The specificity of 14-3-3 domains as constructed by experimentally verified substrates. (B and C) Prediction of specificities for two 14-3-3 proteins. Each example shows a logo representing the predicted specificity (left) and the top five extracted motifs and the number of phosphosites matching them (right). (C) The performance of each predicted model compared with models predicted using random phosphosites. All cases perform better than random sampling of phosphosites (.p < .01, *p < .05, **p < .01, *** p < .001, one-sided z-test). Error bars represent the median absolute deviation of 1,000 random models.
To highlight the broader usefulness of the method, we selected p300 that contains a bromodomain that binds acetylated lysines and its binding specificity has been well characterized (29). We then obtained a collection of human lysine acetylation sites and used the same network-based motif enrichment to predict the specificity of p300's bromodomain. We found that the predicted specificity (Supplemental Fig. 13) is very similar to the known preference for KXXK or KXXXK (where X is any amino acid and both lysines are acetylated).
These results again show that PTM recognition specificity can be predicted by combining network information with PTM data.
Conservation of Kinase–Substrate Specificity
We next sought to predict kinase specificities in a different organism. We applied the same approach to mouse (Mus musculus), which contained 29,732 phosphosites and 2,425,424 STRING interactions. Using human kinases that had an AUC > 0.6, we identified one-to-one ortholog kinases in mouse, using the InParanoid resource (30). By applying our method to these kinases, we found a close resemblance of the specificity determinants of human and many of their corresponding mouse orthologs (Fig. 5). For 56 mouse kinases analyzed in this way, 19 (34%) showed a similar or better performance at predicting the known human kinase sites than the orthologous human model. This suggests that at least these 19 kinase pairs have very conserved kinase preferences. For the remaining cases, we cannot confidently say that there is a divergence in specificity since we cannot rule an incorrect prediction.
Fig. 5.

Conservation of kinase specificity. (A–F) Six examples showing the comparison of predicted human versus mouse models. Each example shows logos for human gold-standard specificity (top) and the predicted specificity model in human (middle) and mouse (bottom).
DISCUSSION
The advances in MS have expanded tremendously our knowledge of exact protein modifications sites for a number of different PTM types. However, there is almost no information regarding the regulatory interactions connecting regulators to target proteins. Determining the recognition preferences for PTM enzymes and binding domains in large scale is still an open problem and remains a limiting factor in achieving this goal. We used phospho-regulation as a model system and showed that it is possible to combine PTM information with interaction network data to derive accurate models of enzymes and binding domains. Predicted kinase motifs for 59% of human kinases are provided in supplementary material. In addition, a resource that contains all of the information used for the specificity predictions of each kinase can be accessed from http://evocellnet.github.io/kpred/. The code required to apply this approach can be found in the help page along with a tutorial.
We note that even though some models do not perform better than models created by random sampling of sites, this does not necessarily reflect the reliability of the predicted model. Some kinase specificities are well modeled by the most common motifs that are recovered from a random sample. For these cases, the added information from the network data does not result in a model that is more accurate than random. The power of our approach is therefore more obvious for regulators that have specificities that are less common such as the DNA damage kinase ATR. For this kinase, the recovered model is both accurate (AUC = 0.94) and performs much better than models produced by random sampling of sites.
In the current implementation of this approach, we assume that kinases that are not CMGC tend not to be proline directed and remove Pro+1 phosphosites. This may result in mispredicting cases where a non-CMGC kinases is proline directed and also cases where CMGC kinases are proline directed. We tested an alternative approach that does not require the removal of Pro+1 phosphosites, but this resulted in a lower overall performance. However, we note that, even when using Pro+1 peptides, we can still obtain predictions that do not have Pro+1. For example, the CK2a1 kinase is a casein kinase and therefore part of the CMGC group. For this group, we allow Pro+1 peptides to be included in the predictions and we still obtain a strong bias for an acidic residue at the +1 position. Additionally, we require to know the class of the kinase: serine/threonine or tyrosine to filter only phosphosites matching the class of the kinase. This is because phosphotyrosine is in many regards a different PTM from phosphoserine and phosphothreonine. In particular, it occurs at much lower frequency, so if we would not discriminate between these two types, the predicted specificities would be dominated by phospho-Ser/Thr.
It is important to take into account that most phosphosite information was retrieved from phosphoproteomics experiments that have used trypsin for protein digestion. Given that trypsin cleaves C-terminal to arginine and lysine residues, it is very possible to expect a bias for Arg/Lys residues in the phosphopeptides. However, we do not think this bias is a strong influence on the recovered motifs. If it was a strong influence, we would expect any bias to be equally possible at positions before or after the target site and also not specifically biased for Arg or Lys. Instead, arginine determinants are more frequent than Lys determinants and Arg determinants are not symmetrically distributed. Of the 202 Arg determined positions (defined as having >0.25 relative frequency at the position), 96% (194/202) are found before the phosphosite and 0.039% (8/202) are found after the phosphosite. There are only 19 positions where Lys is the major determinant, and these tend to be more distributed with 42.1% (8/19) occurring before and 57.89% (11/19) occurring after the phosphosite.
Different kinase families show different average performance in their predictions and that the degree of kinase specificity for the known target sites is correlated with the accuracy of the predicted models. These observations highlight the inherent limitation of the approach proposed here. PTM-interacting proteins that recognize their target sites mostly by residues flanking the target site will be more amenable to this approach than those that use multiple recognition mechanisms. These include docking motifs, colocalization, coexpression, and scaffolding interactions [see (2) for a full review]. In addition this approach assumes that the recognition occurs in a linear epitope at the PTM position. However, it has recently been shown that kinase targeting can occur also in a 3D epitope instead of a linear motif (31). If a PTM enzyme or binding domain often recognizes the target site by a 3D epitope, then this linear motif enrichment strategy will not be appropriate. These observations should be taken into account for future use of this method for other PTM recognition domains. We show that this method can be applied to different modes of site-directed motif-binding domains, such as 14-3-3 domains and bromodomains, suggesting that the method could thus be extended further to analyze specificities of other PTM recognition domains. Finally, we applied this approach to study the conservation of kinase specificity between human and mouse kinases. For the cases that we analyzed, at least 34% appear to have conserved specificity. We suggest that this approach, in combination with an analysis of potential mutations in specificity-determining residues, could be used to identify PTM recognition domains with diverged specificities across species. Given that these regulators interact with many different target PTMs, it is expected that their specificity diverges slowly. This is in contrast to the fast changes in the PTMs targeted by these proteins that can diverge quickly (32, 33). Conserved regulator specificity with diverged target sites is a scenario that is analogous to what is observed in transcriptional regulation (34). However, there have been cases described for divergence of transcription-factor specificity (35), so analogous cases of divergence of PTM recognition are likely to exist. In addition to studying the evolution of specificity, applying this method to different organisms could lend further confidence to the true specificity of a PTM recognition domain since models trained in different species could contribute complementary specificity determinants and ultimately be combined to provide better models.
In summary, we describe here a novel approach to predict PTM recognition motifs, and we believe it can be applicable to a wide range of recognition domains and contribute significantly to our understanding of these signaling systems.
Supplementary Material
Footnotes
Author contributions: O.W., N.S., Y.I., and P.B. designed the research; O.W. and N.S. performed the research; O.W., N.S., Y.I., and P.B. analyzed data; and O.W., Y.I., and P.B. wrote the paper.
* This work was funded by various grants. PB is funded by European Research Council Starting Grant ERC-STG-2014 638884 and Human Frontier Science Program career development award CDA00069/2013.
 This article contains supplemental material Supplemental Figs. 1–14.
This article contains supplemental material Supplemental Figs. 1–14.
1 The abbreviations used are:
- PTM
- posttranslational modification
- MS
- mass spectrometry
- AUC
- area under the ROC curve
- GPS
- group-based prediction system
- STRING
- search tool for the retrieval of interacting genes/proteins
- HPRD
- human protein reference database.
REFERENCES
- 1.Manning G., Whyte D. B., Martinez R., Hunter T., and Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 2.Ubersax J. A., and Ferrell J. E. Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 3.Miller M. L., Jensen L. J., Diella F., Jørgensen C., Tinti M., Li L., Hsiung M., Parker S. A., Bordeaux J., Sicheritz-Ponten T., Olhovsky M., Pasculescu A., Alexander J., Knapp S., Blom N., Bork P., Li S., Cesareni G., Pawson T., Turk B. E., Yaffe M. B., Brunak S., and Linding R. (2008) Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal. 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obenauer J. C., Cantley L. C., and Yaffe M. B. (2003) Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., and Sullivan M. (2012) PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinkel H., Chica C., Via A., Gould C. M., Jensen L. J., Gibson T. J., and Diella F. (2011) Phospho.ELM: A database of phosphorylation sites–Update 2011. Nucleic Acids Res. 39, D261–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keshava Prasad T. S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., Balakrishnan L., Marimuthu A., Banerjee S., Somanathan D. S., Sebastian A., Rani S., Ray S., Harrys Kishore C. J., Kanth S., Ahmed M., Kashyap M. K., Mohmood R., Ramachandra Y. L., Krishna V., Rahiman B. A., Mohan S., Ranganathan P., Ramabadran S., Chaerkady R., and Pandey A. (2009) Human Protein Reference Database–2009 update. Nucleic Acids Res. 37, D767–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz D., Chou M. F., and Church G. M. (2009) Predicting protein post-translational modifications using meta-analysis of proteome scale data sets. Mol. Cell. Proteomics 8, 365–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xue Y., Ren J., Gao X., Jin C., Wen L., and Yao X. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H.-D., Lee T.-Y., Tzeng S.-W., and Horng J.-T. (2005) KinasePhos: A web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 33, W226–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saunders N. F., and Kobe B. (2008) The Predikin webserver: Improved prediction of protein kinase peptide specificity using structural information. Nucleic Acids Res. 36, W286–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neduva V., Linding R., Su-Angrand I., Stark A., de Masi F., Gibson T. J., Lewis J., Serrano L., and Russell R. B. (2005) Systematic discovery of new recognition peptides mediating protein interaction networks. PLos Biol. 3, e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., and Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Brien K. P., Remm M., and Sonnhammer E. L. L. (2005) InParanoid: A comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 33, D476–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin D. M., Miranda-Saavedra D., and Barton G. J. (2009) Kinomer v. 1.0: A database of systematically classified eukaryotic protein kinases. Nucleic Acids Res. 37, D244–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finn R. D., Clements J., and Eddy S. R. (2011) HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, W29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou M. F., and Schwartz D. (2011) Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinformatics 13, Unit 13.15–24 [DOI] [PubMed] [Google Scholar]
- 18.Stormo G. D., Schneider T. D., Gold L., and Ehrenfeucht A. (1982) Use of the “Perceptron” algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 10, 2997–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kel A. E., Gössling E., Reuter I., Cheremushkin E., Kel-Margoulis O. V., and Wingender E. (2003) MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 31, 3576–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reimand J., Wagih O., and Bader G. D. (2013) The mutational landscape of phosphorylation signaling in cancer. Sci. Rep. 3, 2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imamura H., Sugiyama N., Wakabayashi M., and Ishihama Y. (2014) Large-scale identification of phosphorylation sites for profiling protein kinase selectivity. J. Proteome Res. 13, 3410–3419 [DOI] [PubMed] [Google Scholar]
- 22.Sugiyama N., Masuda T., Shinoda K., Nakamura A., Tomita M., and Ishihama Y. (2007) Phosphopeptide enrichment by aliphatic hydroxy acid-modified metal oxide chromatography for nano-LC-MS/MS in proteomics applications. Mol. Cell. Proteomics 6, 1103–1109 [DOI] [PubMed] [Google Scholar]
- 23.Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., and Jensen L. J. (2013) STRING v9.1: Protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanoue T., Adachi M., Moriguchi T., and Nishida E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2, 110–116 [DOI] [PubMed] [Google Scholar]
- 25.Sharrocks A. D., Yang S. H., and Galanis A. (2000) Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem. Sci. 25, 448–453 [DOI] [PubMed] [Google Scholar]
- 26.Lipp J. J., Marvin M. C., Shokat K. M., and Guthrie C. (2015) SR protein kinases promote splicing of nonconsensus introns. Nat. Struct. Mol. Biol. 22, 611–617 [DOI] [PubMed] [Google Scholar]
- 27.Fu H., Subramanian R. R., and Masters S. C. (2000) 14-3-3 proteins: Structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647 [DOI] [PubMed] [Google Scholar]
- 28.Johnson C., Crowther S., Stafford M. J., Campbell D. G., Toth R., and MacKintosh C. (2010) Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem. J 427, 69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delvecchio M., Gaucher J., Aguilar-Gurrieri C., Ortega E., and Panne D. (2013) Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 20, 1040–1046 [DOI] [PubMed] [Google Scholar]
- 30.Ostlund G., Schmitt T., Forslund K., Köstler T., Messina D. N., Roopra S., Frings O., and Sonnhammer E. L. L. (2010) InParanoid 7: New algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 38, D196–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duarte M. L., Pena D. A., Nunes Ferraz F. A., Berti D. A., Paschoal Sobreira T. J., Costa-Junior H. M., Abdel Baqui M. M., Disatnik M.-H., Xavier-Neto J., Lopes de Oliveira P. S., and Schechtman D. (2014) Protein folding creates structure-based, noncontiguous consensus phosphorylation motifs recognized by kinases. Sci. Signal. 7, ra105. [DOI] [PubMed] [Google Scholar]
- 32.Beltrao P., Albanèse V., Kenner L. R., Swaney D. L., Burlingame A., Villén J., Lim W. A., Fraser J. S., Frydman J., and Krogan N. J. (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landry C. R., Levy E. D., and Michnick S. W. (2009) Weak functional constraints on phosphoproteomes. Trends Genet. 25, 193–197 [DOI] [PubMed] [Google Scholar]
- 34.Moses A. M., and Landry C. R. (2010) Moving from transcriptional to phospho-evolution: Generalizing regulatory evolution? Trends Genet. 26, 462–467 [DOI] [PubMed] [Google Scholar]
- 35.Baker C. R., Tuch B. B., and Johnson A. D. (2011) Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proc. Natl. Acad. Sci. U.S.A. 108, 7493–7498 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.