

Abstract
Strong or low-barrier hydrogen bonds have been often proposed in proteins to explain enzyme catalysis and proton transfer reactions. So far 1H chemical shifts and scalar couplings have been used as the main NMR spectroscopic signatures for strong H-bonds. In this work, we report simultaneous measurements of 15N and 1H chemical shifts and N-H bond lengths by solid-state NMR in 15N-labeled 1,8-bis(dimethylamino) naphthalene (DMAN), which contains a well known strong NHN H-bond. We complexed DMAN with three different counter anions to examine the effects of the chemical environment on the H-bond lengths and chemical shifts. All three DMAN compounds exhibit significantly elongated N-H distances compared to the covalent bond length, and the 1HN chemical shifts are larger than ~17 ppm, consistent with strong NHN H-bonds in the DMAN cation. However, the 15N and 1H chemical shifts and the precise N-H distances differ among the three compounds, and the 15N chemical shifts show opposite dependences on the proton localization from the general trend in organic compounds, indicating the significant effects of the counter anions on the electronic structure of the H-bond. These data provide useful NMR benchmarks for strong H-bonds, and caution against the sole reliance on chemical shifts for identifying strong H-bonds in proteins, since neighboring sidechains can exert similar influences on chemical shifts as the bulky organic anions in DMAN. Instead, N-H bond lengths should be measured, in conjunction with chemical shifts, as a more fundamental parameter of H-bond strength.
Keywords: Low-barrier hydrogen bonds, N-H bond lengths, influenza M2, DIPSHIFT, strong hydrogen bonds
Graphical abstract
Introduction
Hydrogen bonding is ubiquitous in biological systems, and strong hydrogen bonds (H-bonds) have been proposed to play important roles in enzyme catalysis 1–5 and proton transfer reactions in photoreceptors 6–9 and ion channels 10–11. However, the nature and function of strong H-bonds have been the subject of significant debate in the literature. Early NMR spectroscopic studies of the catalytic triad of α-chymotrypsin reported an unusually large 1H chemical shift of ~18 ppm for the proton bound to Nδ1 of His57 12. Combined with subsequent findings of low 1H/2H fractionation factors and large chemical shift perturbation, these data supported the existence of strong H-bonds in this and other enzymes 13–15.
The fundamental criterion for classifying H-bond strengths is the potential energy curve (Scheme 1). Strong H-bonds are characterized by the fact that the ground vibrational mode for proton motion lies either near or above the energy barrier for proton transfer 12, 16–17. Two types of strong H-bonds are especially noteworthy: single-well H-bonds, which have no barrier for proton transfer, and low-barrier H-bonds (LBHB), which has a double-well energy minimum (Scheme 1e, f). In both these strong H-bonds, the probability density function for proton position has a single maximum at the center of the two heteroatoms. A rigorous identification of the nature of H-bonds thus requires a study of the system’s electronic and energetic states, which have been conducted largely with computational methods 18–20.
Scheme 1.

Physical and energetic properties of NHN hydrogen bonds and proposed potentialenergy wells for the DMAN*HX salts studied here.
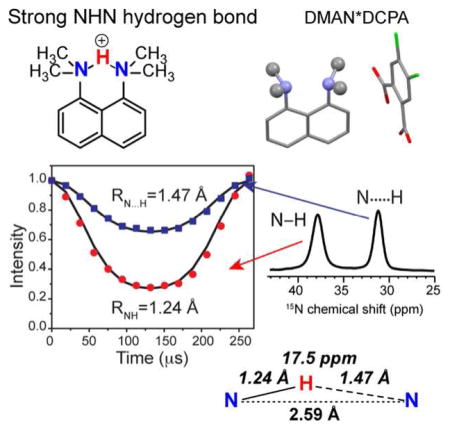
Since potential energies of H-bonds are difficult to measure experimentally, distances have been used as alternative criteria for classifying H-bonds. For NHN compounds, these are the distance between the donor and acceptor nitrogens, RNN, and the distances of the proton (HN) from the donor (RNH) and acceptor (RN---H). At one end of the hydrogen bonding spectrum are weak or regular H-bonds (Scheme 1a), in which the ground vibrational energy is much lower than the proton-transfer barrier, thus the probability density function for the proton has two maxima, centered near each heteroatom. Weak NHN H-bonds exhibit a much shorter RNH than RN---H, an RNN distance longer than 3.0 Å, and are primarily electrostatic in nature 21–22. As the H-bonding strength increases, the HN moves progressively towards the center of the two nitrogens, and RNN decreases, making the bond more covalent than electrostatic. Strong NHN H-bonds (Scheme 1c, e, f) have RNN ≤ 2.65 Å. The degree of covalency of the H-bonds is manifested by the chemical shift of the intervening proton and the two-bond J-couplings between the nitrogens, 2hJNN 23–27: both the 1H chemical shift and 2hJNN increase as the proton becomes more delocalized and the H-bond strength increases. For LBHBs (Scheme 1e), the shared proton between the donor and acceptor is expected to have a chemical shift above ~17 ppm 28–29.
We became interested in better understanding H-bonds in biological systems through our studies of the influenza A M2 protein 30. M2 forms a tetrameric proton channel in the virus envelope that is responsible for the acidification of the virion and the subsequent virus uncoating. The proton-selective residue in M2 is a single histidine, His37, in the transmembrane domain 31, which contains both a H-bond donor (N-H) and acceptor (N:) in the imidazole ring. Several mechanisms of proton conduction have been proposed. In the shuttle model, the His37 imidazole alternately protonates and deprotonates by proton transfer with water molecules 32. To generate the initial state for the next proton relay, the imidazole was thought to undergo ring flips and tautomerization, both of which have been observed experimentally 33–34. The H-bonds in this shuttle model are weak NHO H-bonds between histidine and water. A high-resolution crystal structure reported tightly bound water molecules in the vicinity of the H37 tetrad 35, and low-temperature imidazole HN chemical shifts measured by NMR under conditions where proton transfer is frozen are less than 15 ppm, supporting weak to moderate H-bonds. Moreover, at high temperature when proton exchange is active, the imidazole HN chemical shifts change to the water 1H chemical shift value of ~4.9 ppm, indicating that the H-bonding partner of His37 sidechain is water 36. These lines of evidence support the weak water-histidine H-bonded shuttle mechanism. In comparison, an alternative proton-conduction model posits that a neutral imidazole ring of one polypeptide chain forms a strong or low-barrier H-bond with a cationic imidazolium of a neighboring chain at mildly acidic pH, prior to channel activation at lower pH 10–11. This model was proposed to explain the observations that the first two pKa’s of the His37 tetrad are significantly elevated (8.2) compared to the typical pKa of histidines in solution and moreover are unresolved 10, suggesting that the M2 channel is able to store +2 charges before it becomes conductive. It is thought that a strong or low-barrier imidazole-imidazolium NHN H-bond can provide the mechanism for this charge stabilization. An implication of this LBHB model is that the tetramer consists of two structurally distinct units, or a dimer of dimers. Partial evidence for dimer formation was reported as doubled NMR chemical shifts for many residues in the protein 37–39; however, this chemical shift doubling was observed at high pH where all histidines are neutral, thus violating the requirement of an imidazole-imidazolium pair in the LBHB model.
These studies of H-bonding in M2 and other complex biological systems inspired us to obtain better and clearer solid-state NMR (SSNMR) signatures of strong H-bonds, to correlate multiple signatures, and to delineate the environmental factors that may affect the H-bond strengths or their manifestations. So far few studies have correlated the readily measureable 1H and 15N chemical shifts with the less easily measured but more definitive parameter of RNH distances. Given vibrational averaging effects and the low electron density of protons, N-H bond lengths obtained from X-ray crystallography are often imprecise and shorter than those measured by neutron diffraction and SSNMR. Thus, it is important to directly measure, in known strong H-bonds, RNH distances by NMR and correlate them with 15N and 1H chemical shifts. In this paper, we report a systematic study of these three NMR observables for the 1,8-bis(dimethylamino)-naphthalene (DMAN) family of strong H-bond compounds. X-ray and neutron diffraction of DMAN salts showed RNN distances of 2.55 Å – 2.63 Å 22, 40–43. For a small number of these compounds, RN-H and RN---H distances have been reported by X-ray crystallography and found to range from equivalent (1.31 Å) to off-center. 1H NMR chemical shifts of >18 ppm have been reported for DMAN salts containing small inorganic counterions 28, 43, but DMAN cations that are complexed with bulky organic anions, which better mimic protein sidechains, are less studied. We have thus chosen several DMAN compounds with organic counter anions, synthesized them with 15N and 13C labeling, and measured N-H bond lengths, 15N and 1H chemical shifts. The results should provide useful NMR fingerprints of strong H-bonds in biological macromolecules.
Experimental
Synthesis of 15N, 13C-labeled DMAN
DMAN was synthesized using modified literature procedures outlined in Scheme 2 44–47. Naphthalene (783 mg, 6.12 mmol), NH4 15NO3 (1 g, 12.3 mmol, 2.01 eq) and chloroform (6 mL) were added to a 50 mL round bottom flask and the mixture stirred. An addition funnel was added to the top of the flask and the whole system placed under N2 gas. Trifluoroacetic anhydride (5 mL, 35.4 mmol, 5.8 eq) was added to the addition funnel and introduced to the reaction flask via slow drip over 1.5 hours. The solution became homogeneous and changed from colorless to rose to yellow during the addition. The reaction was stirred overnight under N2. The resulting heterogeneous reaction mixture was then put on ice and 10 mL of H2O was slowly added to the flask by addition funnel to quench excess TFAA. The contents were poured into a separatory funnel containing 60 mL H2O and 20 mL CHCl3. The organic layer was extracted three more times with 20 mL CHCl3 and the combined organic fractions were dried over MgSO4, filtered and the solvent removed on a rotary evaporator to yield an orange-yellow crude mixture consisting of a 3 : 1 mixture of 15N2-1,8-dinitronaphthalene and 15N2-1,5-dinitronaphthalene (1.25 g, 93%). The 1,8 isomer was purified first with a basic Al2O3 column and eluted with benzene (1,8 isomer: Rf = 0.4; 1,5 isomer: Rf = 0.76). The 1,8-rich fractions were collected and recrystallized three times with EtOAc. The supernatant of each crystallization was collected and the solvent removed under reduced pressure. The resulting solid was again recrystallized with EtOAc. The recrystallization process was repeated one more time to give a total of 608 mg (65% recovery, 49% overall yield) 15N2-1,8-dinitronaphthalene as yellow plates. 1H NMR (400 MHz, CDCl3): δ 7.75 (t, 3JHH = 7.9 Hz, 2H), 8.24 (d, 3JHH = 8.3 Hz, 2H), 8.30 (dd, 3JHH = 7.6 Hz, 3JNH = 2 Hz, 2H).
Scheme 2.
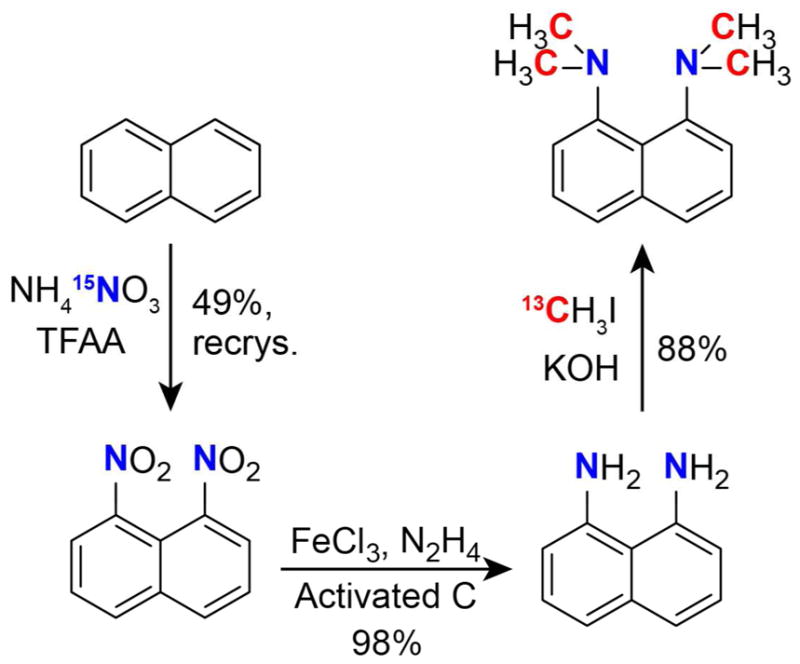
Synthetic route to DMAN.
15N2-labeled 1,8-dinitronaphthalene (604 mg, 2.74 mmol), FeCl3 hexahydrate (22 mg, 3 mol%) and 45 mg Nuchar SA activated carbon were combined in a 25 mL round bottom flask equipped with a stir bar and reflux condenser. MeOH (6 mL) and 50–60% N2H4 (464 μL, 5.45 eq) were added, the reaction stirred and heated to 65 °C. After 5.5 hrs an aliquot was removed and the reaction progress was checked by 1H NMR. There was 7% remaining starting material so two drops of hydrazine were added and stirred for another hour, after which no more starting material was observed. The reaction was cooled to room temperature, filtered over Celite and the filter cake-washed with MeOH. The filtrate was removed under reduced pressure to yield a crude oil (430 mg, 98%), which was the pure 15N2-1,8-diaminonaphthalene compound as verified by NMR. 15N2-1,8-diaminonaphthalene was used without purification for the next step. 1H NMR (400 MHz, CDCl3): δ 4.62 (bs, 4H, 15NH2), 6.60 (d, 3JHH = 7 Hz, 2H), 7.15–7.22 (m, 4H).
15N2-1,8-diaminonaphthalene (430 mg, 2.68 mmol) was added to a 10 mL round bottom flask equipped with a stir bar and placed under N2 gas. Anhydrous DMSO (4 mL) was quickly added, the contents stirred and then KOH (775 mg, 5.15 eq) was quickly added. The flask was placed in a room-temperature water bath, to which 13CHI (950 μL, 5.6 eq) was slowly added and stirred for 2.25 hrs. The temperature was then raised to 100 °C over 1 hr, at which point KOH (320 mg, 2.1 eq) was added. The reaction was stirred at 100 °C for another 40 minutes and then cooled to 50 °C. The reaction contents were quickly poured into a separatory funnel containing 150 mL 2 M NaOH and 50 mL Et2O. The organic layer was washed 3 × 50 mL 2 M NaOH and then collected. The combined aqueous layers were extracted with 2 × 50 mL Et2O. The combined organic layers were dried with MgSO4, filtered and the solvent removed under reduced pressure, resulting in a red oil (532 mg, 2.42 mmol, 90%) that crystallized into needles upon refrigerating and was pure by 1H NMR. δ 2.83 (dd, 1JCH = 138.7 Hz, 2JNH = 4.1 Hz, 12H), 6.96 (broad d, 3JHH = 7.4 Hz, 2H), 7.33 (t, 3JHH = 7.6 Hz, 2H), 7.38 (d, 3JHH = 7.7 Hz, 2H).
The 15N, 13C-labeled DMANH+ salts were recrystallized with three counterions: perchlorate (HClO4), 4,5-dichlorophthalic acid (DCPA), and furan-3,4-dicarboxylic acid (FDCA). DMAN*HClO4 was formed by dissolving DMAN in diethyl ether, stirring the solution and adding concentrated aq. HClO4 dropwise. DMAN*HClO4 precipitated as a brown material, and the addition of HClO4 was ceased when no more precipitate formed. The stirring was then stopped, the supernatant pipetted out, the precipitate washed three times with diethyl ether and then dried under vacuum. The resulting goo was recrystallized twice from a minimal amount of boiling acetonitrile. DMAN*DCPA and DMAN*FDCA were formed by adding DMAN and the respective acid as solids into a 1.5 mL HPLC vial, dissolving them in a minimal amount of hot acetonitrile and then letting the solution cool. Afterwards, the DMAN*DCPA and DMAN*FDCA solutions were allowed to evaporate slowly at room temperature. The resulting crystals were recrystallized once more, washed with a very small amount of cold acetonitrile, and dried. The structures of DMAN*HClO4 and DMAN*FDCA were solved using X-ray diffraction, since no structure existed for DMAN*HClO4 and, despite multiple attempts, the exact unit cell obtained by Wozniak for DMAN*FDCA could not be reproduced.42 Instead, a unique unit cell with the same molecular composition (DMAN + FDCA + water) was obtained. The unit cell of DMAN*DCPA matched the literature unit cell and no further X-ray analysis was performed.
Solid-state NMR spectroscopy
All experiments were conducted using Bruker 400 and 600 MHz solid-state NMR spectrometers equipped with 4 mm or 3.2 mm magic-angle-spinning (MAS) probes. Typical radiofrequency (rf) field strengths were 40–50 kHz for 15N and 80 – 100 kHz for 1H. The experiments were conducted at both ambient temperature and 95 K to investigate the degree of proton dynamics at ambient temperature. 15N cross-polarization (CP) spectra were measured under an MAS frequency of 7.58 kHz, while 1H spectra were measured under 15 kHz MAS. 15N chemical shifts were externally referenced to the N-acetyl-valine 15N chemical shift of 122.0 ppm on the liquid ammonium scale. 1H chemical shifts were referenced to the published values of the tripeptide formyl-MLF-OH 48. N-H bond lengths were measured using the dipolar-doubled 2D 15N-1H dipolar chemical-shift (DIPSHIFT) correlation experiment 49 under 3.79 kHz MAS. 1H homonuclear decoupling was conducted using the FSLG sequence 50, which has a theoretical scaling factor of 0.577. The experimental scaling factor was measured using formyl-MLF-OH 51–52 and found to be 0.577 at ambient temperature and 0.610 at 95 K. 2D 15N-1H HETCOR spectra were measured under 7.58 kHz MAS. Lee-Goldburg (LG) CP 53–54 with a 1 ms contact time and a 50 kHz effective spin-lock field was used to transfer the 1H polarization to 15N without 1H spin diffusion. 1H homonuclear decoupling during the t1 evolution period was carried out using the FSLG sequence with a transverse field strength of 80 kHz.
Results
X-ray diffraction data of selected DMAN compounds
We chose three compounds from the many known DMAN salts using two criteria 40, 42, 55–57. First, each recrystrallization should give a single crystalline form, since bulk separation of polymorphic crystals is often incomplete, which would complicate spectral interpretation. Second, DMAN salts with a highly delocalized proton as well as more localized protons are needed to compare the chemical shifts and bond lengths for different strengths of H-bonds. Based on the literature, we chose DMAN*DCPA and DMAN*FDCA as examples of salts with bulky organic counter anions (Fig. 1) 42 that contain partly localized HN protons and DMAN*HClO4 as a potential model system with a delocalized HN.
Figure 1.

Structures of the DMAN salts used in this study. (a) Chemical structures. (b–d) X-ray crystal structures. (b) DMAN*HClO4. (c) DMAN*DCPA. (d) DMAN*FDCA. Hydrogen atoms are omitted for clarity. The structure for DMAN*DCPA is reproduced from reference 42.
We first verified the structures of the recrystallized DMAN salts by X-ray diffraction (Fig. 1). DMAN*HClO4 has not been previously characterized by X-ray diffraction, thus a full atomic structure was determined. The unit cell for DMAN*DCPA matched the literature result while the unit cell of DMAN*FDCA differed significantly from the literature 42 (Table 1). Upon obtaining a full data set, we found that the FDCA in our crystal has a different spatial arrangement with respect to the DMAN cation. In addition, there is one equivalent of water in the DMAN*FDCA unit cell, which is absent in the other two salts, but the water molecule has a different position in our crystal from that of the literature.
Table 1.
Unit cell parameters of the crystal structures of the three DMAN salts used in this study.
| Crystal Group | DMAN*HClO4 | DMAN*DCPA | DMAN*FDCA |
|---|---|---|---|
| Fdd2 | P1 | P21/n | |
| α (°) | 90.00 | 75.13 | 90.00 |
| β (°) | 90.00 | 71.23 | 91.65 |
| γ (°) | 90.00 | 70.62 | 90.00 |
| a (Å) | 12.55 | 9.30 | 11.24 |
| b (Å) | 46.88 | 9.63 | 10.28 |
| c (Å) | 10.24 | 13.03 | 16.69 |
| Volume (Å3) | 6022.69 | 1027.63 | 1928.04 |
Comparison of the three DMAN crystal structures reveals small but important differences. First, despite the common cation, the RNN distance differs among the three compounds. The distance is the shortest for DMAN*HClO4 (2.55 Å) and increases to 2.59 Å for DMAN*DCPA and 2.62 Å for DMAN*FDCA. Second, the inorganic perchlorate anion rests proximally above the NHN H-bond, roughly equidistant to the two dimethylamino groups, whereas the organic DCPA and FDCA anions lie on one side of the NHN H-bond. The nearest heavy-atom distance between the nitrogen donor/acceptors and the counter anion is 3.30 Å for DMAN*HClO4, 3.85 Å for DMAN*DCPA, and 3.64 Å for DMAN*FDCA. The proximity and orientation of the anion to the NHN H-bond and the differences in RNN may affect the localization of the proton by changing the chemical environment at the nitrogen donors and acceptors.
15N chemical shifts and N-H distances of DMAN salts
Fig. 2 shows the 15N CP-MAS spectra of the three DMAN compounds at 283 K and 95 K. Consistent with the crystal structure, the compound with the shortest RNN distance and the highest unit cell symmetry, DMAN*HClO4, exhibits a single 15N peak at 35.4 ppm. In comparison, DMAN*DCPA and DMAN*FDCA display two 15N chemical shifts: 37.8 ppm and 31.2 ppm for the DCPA salt and 39.5 ppm and 29.3 ppm for the FDCA salt, suggesting that the proton is located asymmetrically between the two nitrogens. DMAN*DCPA has a smaller 15N chemical shift difference (6.6 ppm) than DMAN*FDCA (10.2 ppm), but the average of the two 15N chemical shifts, 34.4 ppm, is close to the single 15N chemical shift of DMAN*HClO4.
Figure 2.

15N CPMAS spectra of DMAN salts at 283 K and 95 K. (a) DMAN*HClO4. (b) DMAN*DCPA. (c) DMAN*FDCA. The N-H and N…H assignment is based on the DIPSHIFT data in Fig. 3.
In general, 15N chemical shifts of organic compounds follow the trend that larger (downfield) shifts correspond to more deprotonated nitrogens with longer RNH bond lengths, while smaller (upfield) chemical shifts indicate more protonated nitrogens with shorter RNH bond lengths. Interestingly, direct measurement of the N-H distance in the three DMAN compounds shows deviations from this rule. We used the dipolar-doubled DIPSHIFT experiment to measure the 15N-1H dipolar coupling and RNH distances (Fig. 3). Motion in these rigid organic crystals is negligible, thus the measured coupling constant directly indicates the internuclear distance. At ambient temperature, the single 15N peak of the perchlorate compound exhibits a 15N-1H dipolar coupling of 5.39 kHz, which translates to an RNH of 1.32 Å. This distance is in good agreement with the RNH of 1.31 Å in the crystal structure of DMAN salts that contain an equally shared proton, such as DMAN*HBr 56 and DMAN*tetrazole 40. In DMAN*DCPA, the downfield peak has a 6.50 kHz coupling, corresponding to an RNH of 1.24 Å, while the upfield 15N peak has a longer RNH of 1.47 Å. For DMAN*FDCA, the downfield 15N peak has an RNH distance of 1.23 Å while the upfield 15N peak has an RNH of 1.49 Å. Thus, for the DCPA and FDCA salts, the more deshielded (or downfield) 15N resonance has a stronger 15N-1H dipolar coupling or a shorter RNH bond length than the upfield 15N peak, opposite the general trend, suggesting that the two nitrogens’ spectroscopic signatures are significantly perturbed by the counter anions.
Figure 3.

15N-1H doubled-DIPSHIFT data of DMAN salts at 293 K (top) and 95 K (bottom). (a) DMAN*HClO4. (b) DMAN*DCPA. (c) DMAN*FDCA. The data were obtained under 3.79 kHz MAS. Solid lines are best-fit simulations. The listed N-H dipolar coupling strengths are true values after taking into account the experimental FSLG scaling factor and the doubling factor. The 15N chemical shift and the N-H distances are also indicated.
We repeated the 15N CP-MAS and N-H DIPSHIFT experiments at 95 K to determine if there is noticeable proton dynamics at ambient temperature and if the HN proton becomes more localized to one of the two nitrogens at low temperature. The 15N chemical shift showed negligible changes with temperature, but the linewidth broadened significantly for DMAN*HClO4, from 55 Hz at ambient temperature to 109 Hz at 95 K. In comparison, the DMAN*DCPA and DMAN*FDCA 15N peaks show only minor line broadening of ~10 Hz. The 15N-1H dipolar couplings remain unchanged within experimental uncertainty from 293 K to 95 K, indicating that the HN position is insensitive to temperature in this range.
1HN chemical shifts of DMAN salts
We next measured the acidic proton’s chemical shift to correlate with the 15N chemical shift and RNH bond lengths. The chemical shift differences between the HN and aliphatic protons are sufficiently large that 1D 1H MAS spectra are already effective in resolving them (Fig. 4). At 600 MHz under 15 kHz MAS, the 1D 1H spectra show sufficient resolution at both 283 K and 95 K. For DMAN*HClO4, the NHN+ proton resonates at 18.9 ppm while the methyl protons resonate at 3.50 ppm and 1.70 ppm, in good agreement with literature values 43. In addition, two resonances at 6.87 ppm and 5.48 ppm were observed and assigned to aromatic protons.
Figure 4.

1H MAS spectra of DMAN salts at 283 K (black) and 95 K (red) under 15 kHz MAS. Assignment is based on the general chemical shift trends of aromatic, HN, and aliphatic protons. Asterisks indicate spinning sidebands.
The 1H spectrum of DMAN*DCPA shows more resolved peaks. Two large 1H chemical shifts at 20.76 ppm and 17.52 ppm are detected and likely correspond to the OHO− proton of the DCPA anion and the NHN+ proton of the DMAN cation, respectively. The 8.59 ppm and 6.52 ppm peaks can be assigned to the naphthalene ring protons, while the 3.70 ppm and 1.41 ppm peaks are assigned to the N-methyl groups. DMAN*FDCA exhibited a single downfield 1H peak at 17.26 ppm, despite having a similar OHO− proton as in DMAN*DCPA. We attribute this to coincidental overlap of the NHN+ and OHO− chemical shifts, which is consistent with the high intensity of this peak compared to the aromatic and aliphatic signals. Similar to DMAN*DCPA, the aromatic 1H resonances are well resolved at 8.25 ppm and the methyl 1H signals span from 3.63 ppm to −0.47 ppm. The −0.47 ppm resonance likely results from the methyl group that is flanked by the π-cloud of the FDCA anion and the naphthalene ring of a neighboring DMANH+ cation in the crystal lattice, which causes additional shielding from the aromatic electrons.
For DMAN*DCPA, the 1D 1H MAS spectrum alone cannot unambiguously determine which of the two downfield signals belongs to the NHN+ H-bond. Thus, we measured 2D 15N-1H HETCOR spectra to make a definitive assignment. Fig. 5 shows two 1H-15N cross peaks at (17.4 ppm, 37.8 ppm) and (17.4 ppm, 31.2 ppm), thus confirming the assignment of the NHN+ proton to 17.4 ppm. The OHO− proton then resonates at 20.76 ppm, consistent with the fact that it lies in a very short and strong H-bond. The 2D HETCOR spectra of the other two DMAN compounds (not shown) also confirmed the 1H chemical shift assignment in the 1D 1H spectra.
Figure 5.

2D 15N-1H HETCOR spectrum of DMAN*DCPA at 293 K under 7.58 kHz MAS. The 1H chemical shift is the true value after taking into account the FLSG scaling factor of 0.577.
Discussion
While hydrogen bonding has undisputed importance in biological and synthetic compounds, rigorous studies of the NMR signatures of hydrogen bonding in the solid state are relatively scarce. McDermott and coworkers investigated NHN H-bonds in imidazole - imidazolium complexes with different counterions by measuring temperature-dependent 15N chemical shifts and correlating these with crystallographically determined RNN distances 58. The RNN were 2.65 – 2.68 Å, which are slightly longer than the cutoff for strong H-bonds. Their NMR data revealed the presence of fast proton exchange in some of the salts from 200 to 280 K, and the population of the two protonation states is strongly dependent on temperature and counter anions. At 210 K, these workers measured an RNH distance of 1.10 Å, which is close to the covalent bond length of RNH = 1.05 Å in imidazole 22. The temperature-dependent 15N chemical shifts, near-covalent RNH bond lengths, and the modest RNN distances, together indicate that the imidazole – imidazolium complexes contain weak to moderate H-bonds.
The intramolecular strong H-bonds formed by DMAN differ qualitatively from the intermolecular weak H-bonds in imidazoles. Fig. 6 summarizes the measured chemical shifts and RNH distances in our DMAN compounds. All three compounds show 1HN chemical shifts larger than ~17 ppm, which is generally accepted as the lower limit for strong H-bonds 27–29. Despite this large 1H chemical shift, the nitrogens in DMAN*DCPA and DMAN*FDCA are not chemically equivalent, as seen by two resolved 15N chemical shifts (Fig. 2). This is caused by the fact that the bulky organic DCPA and FDCA anions are positioned asymmetrically with respect to the DMANH+ cation, creating different chemical environments for the two nitrogens. In comparison, smaller counter anions, such as mineral acids, perchlorate, and tetrafluoroborate, do not exert as large an effect on the 15N chemical environment, as seen from X-ray and 15N NMR studies of Limbach and coworkers 41.
Figure 6.
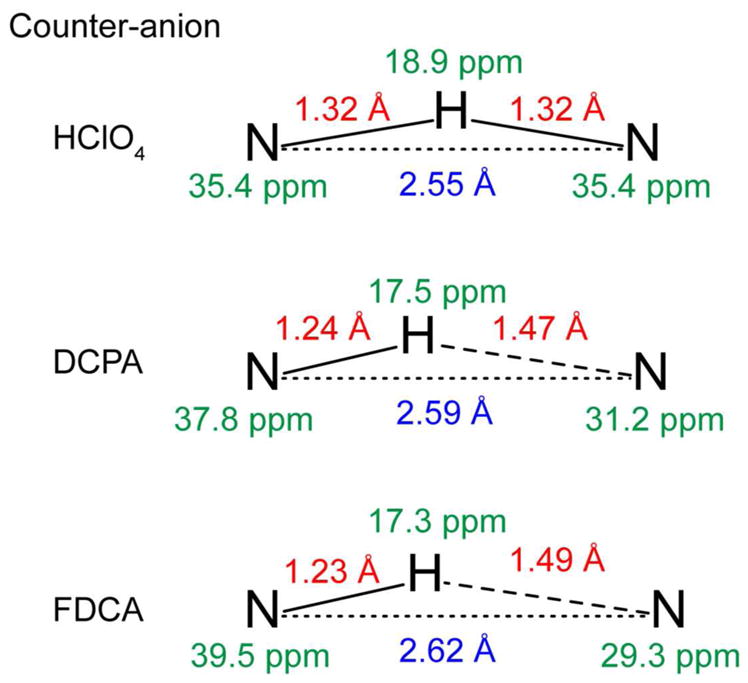
Summary of the 1H and 15N NMR chemical shifts (green), N-H bond lengths (red), and crystallographically determined N---N distances (blue) of the three DMAN salts. DMAN*HClO4 shows the most delocalized HN or the strongest H-bond, the largest 1H chemical shift and equivalent 15N chemical shifts. The two DMAN salts with bulky organic counter anions have different RNH’s, inequivalent 15N chemical shifts, and smaller 1H chemical shifts.
The distinct nitrogen environments affect the basicity of the nitrogen lone-pair electron and in turn the proton localization 27. Both DMAN*DCPA and DMAN*FDCA show a partially localized proton based on the measured RNH distances, and this proton localization correlates with a decrease in the 1H chemical shift (Fig. 6). This correlation is understandable, since a more covalently bonded proton is more shielded by the heteroatom’s electrons. Similar correlations between 1H chemical shifts and RNH have been observed for DMAN salts formed from mineral acids and organic acids 43. The mineral acids caused more equally shared HN protons, and the resulting HN chemical shifts ranged from 20.7 ppm to 18.8 ppm, whereas the organic acids caused more localized protons, and the 1H chemical shifts ranged from 18.6 to 16.9 ppm. The large effects of the counter anion on the charge density profiles of the DMAN cation have been previously observed in DMAN salts such as DMAN*picrate 41 and DMAN*PF6 42–43, 55, 59–60, and even minor polarization differences in the nitrogen lone pair have been shown to cause modest to large changes in RNH and RN---H.
While the two RNH distances in DMAN*DCPA and DMAN*FDCA are not equivalent, both distances are much longer than the covalent bond length of 1.1 Å, and both compounds also have relatively short RNN distances. These results indicate that DMAN complexed with DCPA and FDCA contain a strong NHN+ H-bond with an asymmetric double-well potential (Scheme 1d). In comparison, the NHN+ H-bond in DMAN*HClO4 has a symmetric double-well potential with a low barrier (Scheme 1e), because a single 15N peak and a single 1HN peak was observed at both high and low temperatures. Proton motion in this LBHB is instantaneous but over a very small distance, thus making the two RNH distances indistinguishable. That the DMAN*HClO4 potential energy curve still has a barrier, rather than being barrier-less (Scheme 1f), is manifested by the fact that the 15N and 1H peaks are both broadened at low temperature, while a barrier-less single-well potential should exhibit no temperature-dependent linewidths or chemical shifts.
In conclusion, the proton position along the H-bond and the strength of the H-bond in DMAN compounds are extremely sensitive to the distant effects of the anion. This sensitivity calls into question the likelihood of establishing and preserving strong H-bonds in complex macromolecules such as proteins. While strong and low-barrier H-bonds have been proposed in proteins such as chymotrypsin, ketosteroid isomerases and M2, the necessary distances are rarely measured. Instead, a large downfield 1H chemical shift is often used as proof of strong H-bonds. However, a large 1H chemical shift is not definitive proof of a strong H-bond, since sidechains and charged residues in close proximity to the H-bond of interest can exert significant influences on the 1H chemical shift. For example, the π-bond node of aromatic sidechains and electrostatic interactions between a downfield proton such as in carboxylic acids (around 12–14 ppm) and another group can cause large downfield 1H chemical shifts. On the other hand, small 1H chemical shifts can rule out the existence of strong H-bonds. In other words, large 1H chemical shifts are a necessary but not sufficient condition for strong or low-barrier H-bonds. For a strong H-bond to exist, the donor - acceptor distance must be significantly shorter than the sum of the van der Waals radii while the proton distance to the two heteroatoms must be significantly longer than the covalent bond length. While the heavy-atom distances can be readily measured by crystallography, the N-H or O-H distance cannot, thus NMR spectroscopy is an important tool for identifying strong H-bonds. In a true low-barrier H-bond, the proton in the H-bond should be equidistant between the donor and acceptor; in other words, the pKa of the donor and acceptor heteroatoms should be matched. Finally, as well documented in the literature, if the protein of interest functions in solution, then water further reduces the likelihood of LBHBs compared to the solid state because water oxygen can compete with the heteroatoms while the water hydrogen can compete for the lone pairs in the LBHB 61. For example, Yamaguchi recently reported crystallographic evidence for a LBHB between para-coumaric acid and Glu46 in the photoactive yellow protein; but in solution a small 1H chemical shift of 15.2 ppm was found for the OHO− proton, and computational modeling further supported a regular H-bond 5, 8–9. Thus, transient interactions between water and biomolecules substantially decrease the likelihood of LBHBs in solution. These considerations argue against the presence of LBHBs between the proton-selective histidines in the water-filled pore of the influenza M2 channel, in addition to the experimental evidence of imidazole HN chemical shifts near the water frequency and the X-ray crystal structure of a water cluster at the histidine tetrad.
Supplementary Material
Acknowledgments
This work is supported by NIH grant GM088204 to M.H. The authors thank Dr. Marc Caporini and Dr. Melanie Rosay at Bruker Biospin for making the 400 and 600 MHz NMR spectrometers available for experiments at 95 K. The authors would also like to thank Keith Fritzsching for insightful discussions on LBHBs and for initial exploratory experiments.
Footnotes
Supporting Information Available: Full author lists for references with more than ten authors. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cleland WW, Frey PA, Gerlt JA. The Low Barrier Hydrogen Bond in Enzymatic Catalysis. J Biol Chem. 1998;273:25529–25532. doi: 10.1074/jbc.273.40.25529. [DOI] [PubMed] [Google Scholar]
- 2.Langkilde A, Kristensen SM, Lo Leggio L, Molgaard A, Jensen JH, Houk AR, Poulsen JCN, Kauppinen S, Larsen S. Short Strong Hydrogen Bonds in Proteins: a Case Study of Rhamnogalacturonan Acetylesterase. Acta Crystallogr D Biol Crystallogr. 2008;64:851–863. doi: 10.1107/S0907444908017083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polgar L. The Catalytic Triad of Serine Peptidases. Cell Mol Life Sci. 2005;62:2161–2172. doi: 10.1007/s00018-005-5160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollack RM. Enzymatic Mechanisms for Catalysis of Enolization: Ketosteroid Isomerase. Bioorg Chem. 2004;32:341–353. doi: 10.1016/j.bioorg.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Sigala PA, Tsuchida MA, Herschlag D. Hydrogen Bond Dynamics in the Active Site of Photoactive Yellow Protein. Proc Natl Acad Sci USA. 2009;106:9232–9237. doi: 10.1073/pnas.0900168106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bondar AN, Fischer S, Smith JC. Water Pathways in the Bacteriorhodopsin Proton Pump. J Membrane Biol. 2011;239:73–84. doi: 10.1007/s00232-010-9329-3. [DOI] [PubMed] [Google Scholar]
- 7.Mak-Jurkauskas ML, Bajaj VS, Hornstein MK, Belenky M, Griffin RG, Herzfeld J. Energy Transformations Early in the Bacteriorhodopsin Photocycle Revealed by DNP-Enhanced Solid-State NMR. Proc Natl Acad Sci USA. 2008;105:883–888. doi: 10.1073/pnas.0706156105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nadal-Ferret M, Gelabert R, Moreno M, Lluch JM. Are There Really Low-Barrier Hydrogen Bonds in Proteins? The Case of Photoactive Yellow Protein. J Am Chem Soc. 2014;136:3542–3552. doi: 10.1021/ja4116617. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi S, Kamikubo H, Kurihara K, Kuroki R, Niimura N, Shimizu N, Yamazaki Y, Kataoka M. Low-Barrier Hydrogen Bond in Photoactive Yellow Protein. Proc Natl Acad Sci USA. 2009;106:440–444. doi: 10.1073/pnas.0811882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Histidines, Heart of the Hydrogen Ion Channel from Influenza A Virus: Toward an Understanding of Conductance and Proton Selectivity. Proc Natl Acad Sci USA. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma M, Yi MG, Dong H, Qin HJ, Peterson E, Busath DD, Zhou HX, Cross TA. Insight Into the Mechanism of the Influenza A Proton Channel from a Structure in a Lipid Bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frey PA, Whitt SA, Tobin JB. A Low-Barrier Hydrogen-Bond in the Catalytic Triad of Serine Proteases. Science. 1994;264:1927–1930. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 13.Lin J, Westler WM, Cleland WW, Markley JL, Frey PA. Fractionation Factors and Activation Energies for Exchange of the Low Barrier Hydrogen Bonding Proton in Peptidyl Trifluoromethyl Ketone Complexes of Chymotrypsin. Proc Natl Acad Sci USA. 1998;95:14664–14668. doi: 10.1073/pnas.95.25.14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loh SN, Markley JL. Hydrogen-Bonding in Proteins as Studied by Amide Hydrogen D/H-Fractionation Factors - Application to Staphylococcal Nuclease. Biochemistry. 1994;33:1029–1036. doi: 10.1021/bi00170a023. [DOI] [PubMed] [Google Scholar]
- 15.Markley JL, Westler WM. Protonation-State Dependence of Hydrogen Bond Strengths and Exchange Rates in a Serine Protease Catalytic Triad: Bovine Chymotrypsinogen A. Biochemistry. 1996;35:11092–11097. doi: 10.1021/bi961366k. [DOI] [PubMed] [Google Scholar]
- 16.GarciaViloca M, Gelabert R, GonzalezLafont A, Moreno M, Lluch JM. Is an Extremely Low-Field Proton Signal in the NMR Spectrum Conclusive Evidence for a Low-Barrier Hydrogen Bond? J Phys Chem A. 1997;101:8727–8733. [Google Scholar]
- 17.Garcia-Viloca M, Gelabert R, Gonzalez-Lafont A, Moreno M, Lluch JM. Temperature Dependence of Proton NMR Chemical Shift as a Criterion to Identify Low-Barrier Hydrogen Bonds. J Am Chem Soc. 1998;120:10203–10209. [Google Scholar]
- 18.Schutz CN, Warshel A. The Low Barrier Hydrogen Bond (LBHB) Proposal Revisited: The Case of the Asp…His Pair in Serine Proteases. Proteins-Structure Function and Bioinformatics. 2004;55:711–723. doi: 10.1002/prot.20096. [DOI] [PubMed] [Google Scholar]
- 19.Warshel A, Papazyan A. Energy Considerations Show That Low-Barrier Hydrogen Bonds Do Not Offer a Catalytic Advantage Over Ordinary Hydrogen Bonds. Proc Natl Acad Sci USA. 1996;93:13665–13670. doi: 10.1073/pnas.93.24.13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warshel A, Papazyan A, Kollman PA. On Low-Barrier Hydrogen-Bonds and Enzyme Catalysis. Science. 1995;269:102–104. doi: 10.1126/science.7661987. [DOI] [PubMed] [Google Scholar]
- 21.Emsley J. Very Strong Hydrogen-Bonding. Chemical Society Reviews. 1980;9:91–124. [Google Scholar]
- 22.Malarski Z, Sobczyk L, Grech E. Structure and IR Spectroscopic Behavior of NHN Hydrogen-Bonds. J Mol Struc. 1988;177:339–349. [Google Scholar]
- 23.Benedict H, Shenderovich IG, Malkina OL, Malkin VG, Denisov GS, Golubev NS, Limbach HH. Nuclear Scalar Spin-Spin Couplings and Geometries of Hydrogen Bonds. J Am Chem Soc. 2000;122:1979–1988. [Google Scholar]
- 24.Brown SP, Perez-Torralba M, Sanz D, Claramunt RM, Emsley L. The Direct Detection of a Hydrogen Bond in the Solid State by NMR Through the Observation of a Hydrogen-Bond Mediated N-15-N-15 J Coupling. J Am Chem Soc. 2002;124:1152–1153. doi: 10.1021/ja0172262. [DOI] [PubMed] [Google Scholar]
- 25.Dingley AJ, Masse JE, Peterson RD, Barfield M, Feigon J, Grzesiek S. Internucleotide Scalar Couplings Across Hydrogen Bonds in Watson-Crick and Hoogsteen Base Pairs of a DNA Triplex. J Am Chem Soc. 1999;121:6019–6027. [Google Scholar]
- 26.Pietrzak M, Limbach HH, Perez-Torralba M, Sanz D, Claramunt RM, Elguero J. Scalar Coupling Constants Across the Intramolecular NHN Hydrogen Bond of Symmetrically and Non-Symmetrically Substituted 6-Aminofulvene-1-aldimines. Magn Reson Chem. 2001;39:S100–S108. [Google Scholar]
- 27.Pietrzak M, et al. Symmetrization of Cationic Hydrogen Bridges of Protonated Sponges Induced by Solvent and Counteranion Interactions as Revealed by NMR Spectroscopy. Chemistry. 2010;16:1679–1690. doi: 10.1002/chem.200902259. [DOI] [PubMed] [Google Scholar]
- 28.Brycki B, Brzezinski B, Grech E, Malarski Z, Sobczyk L. H-1 and C-13 NMR-Studies of 1,8-Bis(Dimethylamino)Naphthalene Salts in Acetonitrile-D3. Magn Reson Chem. 1991;29:558–560. [Google Scholar]
- 29.Tobin JB, Whitt SA, Cassidy CS, Frey PA. Low-Barrier Hydrogen-Bonding in Molecular-Complexes Analogous to Histidine and Aspartate in the Catalytic Triad of Serine Proteases. Biochemistry. 1995;34:6919–6924. doi: 10.1021/bi00021a002. [DOI] [PubMed] [Google Scholar]
- 30.Hong M, DeGrado WF. Structural Basis for Proton Conduction and Inhibition by the Influenza M2 Protein. Protein Sci. 2012;21:1620–1633. doi: 10.1002/pro.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C, Lamb RA, Pinto LH. Activation of the M2 Ion Channel of Influenza Virus: A Role for the Transmembrane Domain Histidine Residue. Biophys J. 1995;69:1363–1371. doi: 10.1016/S0006-3495(95)80003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. A Functionally Defined Model for the M2 Proton Channel of Influenza A Virus Suggests a Mechanism for Its Ion Selectivity. Proc Natl Acad Sci USA. 1997;94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu F, Luo W, Hong M. Mechanisms of Proton Conduction and Gating by Influenza M2 Proton Channels from Solid-State NMR. Science. 2010;330:505–508. doi: 10.1126/science.1191714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu FH, Schmidt-Rohr K, Hong M. NMR Detection of pH-Dependent Histidine-Water Proton Exchange Reveals the Conduction Mechanism of a Transmembrane Proton Channel. J Am Chem Soc. 2012;134:3703–3713. doi: 10.1021/ja2081185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Acharya A, et al. Structural Mechanism of Proton Transport Through the Influenza A M2 Protein. Proc Natl Acad Sci U S A. 2010;107:15075–15080. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong M, Fritzsching KJ, Williams JK. Hydrogen-Bonding Partner of the Proton-Conducting Histidine in the Influenza M2 Proton Channel Revealed From H-1 Chemical Shifts. J Am Chem Soc. 2012;134:14753–14755. doi: 10.1021/ja307453v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andreas LB, Eddy MT, Pielak RM, Chou JJ, Griffin RG. Magic Angle Spinning NMR Investigation of Influenza A M2(18-60): Support for an Allosteric Mechanism of Inhibition. J Am Chem Soc. 2010;132:10958–10960. doi: 10.1021/ja101537p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andreas LB, Eddy MT, Chou JJ, Griffin RG. Magic-Angle-Spinning NMR of the Drug Resistant S31N M2 Proton Transporter From Influenza A. J Am Chem Soc. 2012;134:7215–7218. doi: 10.1021/ja3003606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colvin MT, Andreas LB, Chou JJ, Griffin RG. Proton Association Constants of His 37 in the Influenza-A M218-60 Dimer-of-Dimers. Biochemistry. 2014;53:5987–5994. doi: 10.1021/bi5005393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glowiak T, Malarski Z, Sobczyk L, Grech E. New Example of a Symmetrical Nhn Hydrogen-Bond in Protonated 1,8-Bis(Dimethylamino)Naphthalene (DMAN) J Mol Struc. 1992;270:441–447. [Google Scholar]
- 41.Lopez C, Claramunt RM, LlamasSaiz AL, FocesFoces C, Elguero J, Sobrados I, AguilarParrilla F, Limbach HH. X-Ray Diffraction and Solid State NMR Studies of 1,8-Bis(dimethylamino)naphthalene and Its Complexes with Picric and Hexafluorophosphoric Acids. New J Chemistry. 1996;20:523–536. [Google Scholar]
- 42.Parkin A, Wozniak K, Wilson CC. From Proton Disorder to Proton Migration: A Continuum in the Hydrogen Bond of a Proton Sponge in the Solid State. Crystal Growth & Design. 2007;7:1393–1398. [Google Scholar]
- 43.Wozniak K, He HY, Klinowski J, Barr TL, Milart P. ESCA and Solid-State NMR Studies of Ionic Complexes of 1,8-Bis(dimethylamino)naphthalene. J Phys Chem. 1996;100:11420–11426. [Google Scholar]
- 44.Crivello JV. Nitrations and Oxidations with Inorganic Nitrate Salts in Trifluoroacetic-Anhydride. J Org Chem. 1981;46:3056–3060. [Google Scholar]
- 45.Kurasov LA, Pozharskii AF, Kuzmenko VV. Peri-Naphthylenediamines .3. Convenient Method for the Alkylation of 1,8-Naphthylenediamines and Perimidines. Zhurnal Organicheskoi Khimii. 1981;17:1944–1947. [Google Scholar]
- 46.Lloyd-Jones GC, Harvey JN, Hodgson P, Murray M, Woodward RL. Scalar Coupling Between the N-15 Centres in Methylated 1,8-Diaminonaphthalenes and 1,6-Diazacyclodecane: To What Extent is (2H)J(NN) a Reliable Indicator of N-N Distance? Chemistry-a European Journal. 2003;9:4523–4535. doi: 10.1002/chem.200304890. [DOI] [PubMed] [Google Scholar]
- 47.Ni WH, Dongsheng, Shi Feng, Xue Dongjie, Yang Haizhou. Hydrazine-Hydrate Catalyzed Preparation of 1,8-Dimethylaminonapthalene from 1,8-Dinitronaphthalene. 2010 CN 101823968A. [Google Scholar]
- 48.Li S, Su Y, Luo W, Hong M. Water-protein Interactions of an Arginine-Rich Membrane Peptide in Lipid Bilayers Investigated by Solid-State Nuclear Magnetic Resonance Spectroscopy. J Phys Chem B. 2010;114:4063–4069. doi: 10.1021/jp912283r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hong M, Gross JD, Rienstra CM, Griffin RG, Kumashiro KK, Schmidt-Rohr K. Coupling Amplification in 2D MAS NMR and Its Application to Torsion Angle Determination in Peptides. J Magn Reson. 1997;129:85–92. doi: 10.1006/jmre.1997.1242. [DOI] [PubMed] [Google Scholar]
- 50.Bielecki A, Kolbert AC, Levitt MH. Frequency-Switched Pulse Sequences: Homonuclear Decoupling and Dilute Spin NMR in Solids. Chem Phys Lett. 1989;155:341–346. [Google Scholar]
- 51.Hong M, Griffin RG. Resonance Assignment for Solid Peptides by Dipolar-Mediated 13C/15N Correlation Solid-State NMR. J Am Chem Soc. 1998;120:7113–7114. [Google Scholar]
- 52.Rienstra CM, Hohwy M, Hong M, Griffin RG. 2D and 3D 15N-13C-13C NMR Chemical Shift Correlation Spectroscopy of Solids: Assignment of MAS Spectra of Peptides. J Am Chem Soc. 2000;122:10979–10990. [Google Scholar]
- 53.van Rossum BJ, de Groot CP, Ladizhansky V, Vega S, de Groot HJM. A Method for Measuring Heteronuclear (H-1-C-13) Distances in High Speed MAS NMR. J Am Chem Soc. 2000;122:3465–3472. [Google Scholar]
- 54.Hong M, Yao XL, Jakes K, Huster D. Investigation of Molecular Motions by Lee-Goldburg Cross-Polarization NMR Spectroscopy. J Phys Chem B. 2002;106:7355–7364. [Google Scholar]
- 55.Hoser AA, Dobrzycki L, Gutmann MJ, Wozniak K. Charge Densities of Two Polymorphs of Hydrated 1,8-Bis(dimethylamino)naphthalene Hydrochloride-Similarities and Differences. Crystal Growth & Design. 2010;10:5092–5104. [Google Scholar]
- 56.Pyzalska D, Pyzalski R, Borowiak T. Structure of 1,8-Bis(Dimethylamino)Naphthalene Hydrobromide Dihydrate. J Cryst Spectros Res. 1983;13:211–220. [Google Scholar]
- 57.Wozniak K, Krygowski TM, Kariuki B, Jones W, Grech E. Crystallographic Studies on Sterically Affected Chemical-Species .2. Molecular and Crystal-Structure of 1,8-Bis(Dimethylamino)-Naphthalene Tetrafluoroborate - Analysis of Distortion of Geometry in the Aromatic Part Due to Intramolecular Hydrogen-Bonding. J Mol Struc. 1990;240:111–118. [Google Scholar]
- 58.Song XJ, McDermott AE. Proton Transfer Dynamics and N-H Bond Lengthening in N-H Center dot Center dot Center dot N Model Systems: a Solid-State NMR Study. Magn Reson Chem. 2001;39:S37–S43. [Google Scholar]
- 59.Lopez C, Lorente P, Claramunt RM, Marin J, Foces-Foces C, Llamas-Saiz AL, Elguero J, Limbach HH. Localization of Hydrogen Bond Deuterons in Proton Sponges by Dipolar Solid State N-15 NMR Spectroscopy. Berichte Der Bunsen-Gesellschaft-Physical Chemistry Chemical Physics. 1998;102:414–418. [Google Scholar]
- 60.Mallinson PR, Smith GT, Wilson CC, Grech E, Wozniak K. From Weak Interactions to Covalent Bonds: A Continuum in the Complexes of 1,8-Bis(dimethylamino)naphthalene. J Am Chem Soc. 2003;125:4259–4270. doi: 10.1021/ja029389b. [DOI] [PubMed] [Google Scholar]
- 61.Frey PA, Cleland WW. Are There Strong Hydrogen Bonds in Aqueous Solutions? Bioorganic Chemistry. 1998;26:175–192. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.