

SUMMARY
It is known that internal physiological state, or interoception, influences central nervous system function and behavior. However, the neurons and mechanisms that integrate sensory information with internal physiological state remain largely unknown. Here, we identify C. elegans body cavity neurons called URX(L/R) as central homeostatic sensors that integrate fluctuations in oxygen availability, with internal metabolic state. We show that depletion of internal body fat reserves increases the tonic activity of URX neurons, which influences the magnitude of the evoked sensory response to oxygen. These responses are integrated via intracellular cGMP and Ca2+. The extent of neuronal activity thus reflects the balance between the perception of oxygen, and available fat reserves. The URX homeostatic sensor ensures that neural signals that stimulate fat loss are only deployed when there are sufficient fat reserves to do so. Our results uncover an interoceptive neuroendocrine axis that relays internal state information to the nervous system.
Graphical Abstract

INTRODUCTION
The central nervous system is a major regulator of body fat and energy balance, independent of its effects on food intake. With respect to the sensory nervous system, examples of broad sensory dysfunctions that are accompanied by profound obesity are prevalent in many species. For example, Bardet Biedl Syndrome is characterized by defects in sensory processing and extreme obesity stemming from nervous system dysfunction in humans and in model systems (Mykytyn et al., 2002; Davis et al., 2007; Lee et al., 2011). Enhanced sensory environments have also been shown to improve metabolic homeostasis (Cao et al., 2011). However, the mechanisms by which a discrete sensory modality is connected to peripheral lipid metabolism have been difficult to elucidate, in part due to the heterogeneity of sensory dysfunction in mammalian systems. Thus, the role of sensory systems in regulating organismal metabolic control has remained under-appreciated.
A body of evidence suggests that in addition to external sensory cues, interoception or the sensitivity to stimuli originating inside the body, is also perceived by the central nervous system (Cannon, 1932; Craig, 2002). Internal state information is used to modulate behavior in many species. For example, internal sensing of blood glucose regulates feeding behavior (Wang et al., 2008; Mighiu et al., 2013). Intestinal fatty acids are also sensed by the nervous system in mice, D. melanogaster and C. elegans, and modulate behavior and physiology (Wingrove and O'Farrell, 1999; Kniazeva et al., 2004; Lam et al., 2005; Srinivasan et al., 2008). It follows then that sensory and interoceptive information is integrated by the central nervous system for an organism to function as a cohesive entity. The complexity and redundancy of sensory and homeostatic functions in mammalian nervous systems makes it challenging to decipher the underlying neuronal sites, cellular mechanisms and the fundamental principles by which this integration occurs. The genetically tractable nematode C. elegans is an excellent model system for the study of neural circuits and their role in governing physiology. Many behaviors have been attributed to individual neurons, and their mechanisms of action revealed (Bargmann, 2006). Despite these tremendous advances, neural sites of integration between sensory and metabolic information have remained unknown.
Food availability is perhaps one of the most salient external sensory cues in an animal's environment (Libert and Pletcher, 2007; Berthoud and Morrison, 2008). In C. elegans, food sensory cues influence nearly all aspects of behavior and physiology including sensory functions, locomotion, reproduction, metabolism and lifespan (Lemieux and Ashrafi, 2015; Srinivasan, 2015). Food presence is encoded by two major neuroendocrine systems: serotonin (5-hydroxytryptamine, 5-HT) and transforming growth factor beta (TGF-β) (Entchev et al., 2015). 5-HT synthesis and signaling from a single pair of chemosensory neurons called ADF(L/R) regulates a complex cascade of whole-body metabolic responses that drive peripheral lipid metabolism and fat loss (Srinivasan et al., 2008; Noble et al., 2013). In contrast, food absence is encoded in part by oxygen sensation. In a lab setting on agar plates, worms consume live bacteria whose respiration drops the local oxygen concentration from 21% (atmospheric) to 10-13% (Sylvia, 1998; Gray et al., 2004). Thus, worms avoid 21% oxygen and prefer an intermediate oxygen concentration in the range of 10-13% to remain in the presence of food (Scott, 2011). The avoidance of 21% oxygen is regulated by a quartet of neurons called the body cavity neurons: AQR, PQR and the bilaterally symmetric URX pair (Gray et al., 2004; Cheung et al., 2005; Chang et al., 2006).
Body cavity neurons have a unique anatomical feature: their cell bodies and ciliated dendrites are positioned within the coelomic fluid, which functions as the circulatory system for C. elegans (White et al., 1986). Thus, the body cavity neurons have the capacity to send and receive endocrine signals from other organs. Interestingly, food presence encoded by 5-HT signaling from the ADF neurons impinges on the body cavity neurons and URX neurons receive direct synaptic input from the serotonergic ADF neurons. These neurons also regulate body size and lifespan via distinct signaling pathways (Mok et al., 2011; Liu and Cai, 2013). Despite the importance of the body cavity neurons in the regulation of C. elegans behavior and physiology, many questions remain. First, a role for the body cavity neurons in regulating lipid metabolism, a hallmark of organismal state, and the underlying cellular mechanism of action, has not been defined. Second, with respect to the body cavity neurons, the extent to which neural mechanisms of oxygen sensing impinge upon metabolic outcomes, is not known. Finally, despite the many suggestions that body cavity neurons function as homeostatic sensors, there is no direct evidence showing that these neurons respond to changes in internal state. Addressing these questions will define the precise role of the body cavity neurons in detecting and regulating fat stores, and allow the investigation of mechanisms of integration of external sensory cues, with internal metabolic state.
In the present study, we report that the URX body cavity neurons function as homeostatic sensors that integrate internal metabolic state with external oxygen availability. The integration of internal and external signals occurs in the URX neurons via the second messenger cGMP. The net activation status of the URX neurons in turn dictates the magnitude of fat loss in the periphery. Our results reveal a homeostatic loop in which neural signals to stimulate fat loss are only deployed when two conditions are met: oxygen availability and the presence of sufficient body fat reserves. Our results suggest one mechanism underlying the self-limiting nature of homeostatic systems.
RESULTS
G protein signaling from the body cavity neurons stimulates body fat loss
To investigate the role of the sensory nervous system in regulating body fat, we conducted a screen of the 19 viable Gα protein null mutants. We focused on the heterotrimeric G proteins because they are a well-conserved family of signaling proteins that control second messengers and cellular activity (Bastiani and Mendel, 2006). The Gα subunits of heterotrimeric G proteins are regulatory in nature, and relative to mammals, this family is elaborated in C. elegans, perhaps reflecting the functional diversification of GPCRs. Added advantages of studying this family of signaling proteins are that the majority of null mutants are viable, and the anatomical locations of each of the Gα proteins have been precisely defined (Jansen et al., 1999). We measured the extent to which each of the viable Gα mutants led to a change in body fat (Figure S1A). We found that gpa-8 null mutants had a robust decrease in body fat as judged by oil red O staining (Figure 1A) and by quantitation of biochemically-extracted triglycerides (Figure 1B). The decrease in body fat in gpa-8 mutants was not accompanied by a change in food intake (Figure S1B) or increased movement (Yemini et al., 2013), suggesting that a selective shift in fat metabolism rather than feeding or locomotion behavior, was responsible for the decreased body fat phenotype of gpa-8 mutants.
Figure 1. G protein signaling from the body cavity neurons stimulates body fat loss.
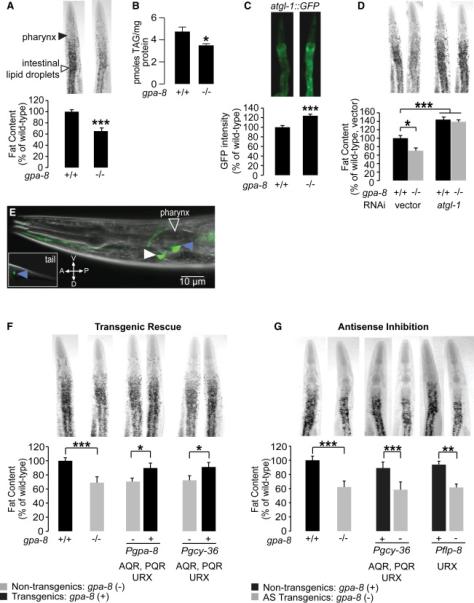
(A) Representative images are shown of wild-type and gpa-8 animals fixed and stained with oil Red O (upper panels). Fat deposition in the intestinal cells is visible as stained lipid droplets (white arrowhead). Animals are oriented facing upwards with the pharynx (black arrowhead) at the anterior end. Fat content was quantified for each genotype and is expressed as a percentage of wild-type animals ± SEM (lower panels; n=20). ***, p<0.001 by Student's t-test.
(B) Biochemical extraction and quantitation of triglycerides was conducted for wild-type and gpa-8 animals. gpa-8 animals have a significant reduction in triglycerides compared to wild-type animals. *, p<0.05 by Student's t-test.
(C) Representative images are shown of wild-type animals and gpa-8 mutants bearing an integrated atgl-1::GFP transgene (upper panels). The fluorescence intensity of atgl-1 expression was quantified and is expressed as a percentage of wild-type animals ± SEM (lower panels; n=10). ***, p<0.001 by Student's t-test.
(D) Representative images are shown of RNAi-treated wild-type animals and gpa-8 mutants fixed and stained with oil Red O (upper panels). Fat content was quantified for each genotype and condition and is expressed as a percentage of wild-type animals grown on vector RNAi ± SEM (lower panels; n=12). *, p<0.05 and ***, p<0.001 by two-way ANOVA.
(E) Fluorescent image of a transgenic animal bearing a gpa-8::GFP transgene. Blue arrowheads indicate expression in AQR and PQR neurons and the white arrowhead indicates expression in URX. A, anterior; P, posterior; V, ventral; D, dorsal.
(F) Representative images are shown of wild-type animals and gpa-8 mutants fixed and stained with oil Red O (upper panels). For each transgenic line bearing gpa-8 expression using the indicated promoter, non-transgenic animals (−) and transgenic animals (+) are shown. Relative to non-transgenic controls (gray bars), transgenic animals (black bars) bearing the gpa-8 transgene under the control of the endogenous gpa-8 and the heterologous gcy-36 promoters restore body fat content to that seen in wild-type animals. Data are expressed as a percentage of body fat in wild-type animals ± SEM (lower panels; n=20-24). *, p<0.05 and ***, p<0.001 by Student's t-test.
(G) Representative images are shown of animals bearing antisense (AS)-mediated inactivation of gpa-8. For each transgenic line bearing gpa-8 antisense using the indicated promoter, nontransgenic animals (+) and transgenic animals (−) are shown. Relative to non-transgenic controls (black bars), transgenic animals (gray bars) bearing gpa-8 antisense under the heterologous gcy-36 and flp-8 promoters recapitulate the decreased body fat seen in gpa-8 mutants. Data are expressed as a percentage of body fat in wild-type animals ± SEM (n=10-12). **, p<0.01 and ***, p<0.001 by Student's t-test. See also Figure S1.
To explore the relationship between loss of gpa-8 and decreased body fat, we crossed the gpa-8 null mutants into a transgenic line bearing the adipocyte triglyceride lipase (atgl-1) promoter fused to GFP. ATGL-1 is a rate-limiting enzyme that generates free fatty acids from stored triglycerides in eukaryotes, which are then oxidized in the mitochondria for the production of energy (Salway, 1999; Zimmermann et al., 2004). We previously showed that C. elegans atgl-1 is expressed in the intestine, and is transcriptionally induced in response to neural signals that stimulate fat loss (Noble et al., 2013). Although atgl-1 null mutants are not viable, RNAi-mediated inactivation of atgl-1 leads to increased fat retention in adults (Noble et al., 2013). Relative to wild-type animals, gpa-8 mutants have an approximately 25% induction of atgl-1 expression in the intestine (Figure 1C). In addition, RNAi-mediated inactivation of atgl-1 abrogated the reduced body fat of gpa-8 mutants (Figure 1D). Our results indicate that increased fat utilization via induction of triglyceride hydrolysis underlies the reduced body fat of gpa-8 mutants.
GPA-8 is expressed in four neurons: AQR, PQR and the bilaterally symmetric URX pair (Figure 1E) (Jansen et al., 1999). In gpa-8 null mutants, we restored gpa-8 cDNA to the AQR, PQR and URX neurons using the endogenous gpa-8 promoter, and the heterologous gcy-36 promoter that confers expression in the same neurons. Relative to gpa-8 mutants and nontransgenic animals, both promoters conferred near-complete restoration of body fat (Figure 1F). Previous reports have shown that the URX pair alone is sufficient for the behaviors this quartet of neurons regulates (Coates and de Bono, 2002; Zimmer et al., 2009). To test for necessity of GPA-8 function in the URX neurons, we generated transgenic lines bearing antisense-mediated inhibition of gpa-8, under the control of the gcy-36 (AQR, PQR, URX) and flp-8 (URX, and occasional expression in AUA and PVM) promoters. Inactivation of gpa-8 in the URX neurons recapitulated the gpa-8 mutant phenotype to the same extent as its inactivation in AQR, PQR and URX neurons (Figure 1G). Thus, GPA-8 function in the URX neurons is necessary and sufficient to maintain normal body fat reserves.
cGMP signaling in the body cavity neurons regulates the extent of body fat loss
The body cavity neurons are oxygen sensors in which two genes play dominant roles (Gray et al., 2004; Zimmer et al., 2009). First, the soluble guanylate cyclase GCY-36 expressed in the AQR, PQR and URX neurons, binds molecular oxygen to generate cGMP. Second, the cyclic nucleotide-gated (CNG) channel TAX-4 activates URX via Ca2+ influx. Imaging and behavioral studies have shown that GCY-36 and TAX-4 are key regulators of cGMP-mediated URX function (Cheung et al., 2004; Cheung et al., 2005; Zimmer et al., 2009). An additional guanylate cyclase called GCY-35 is thought to function as a heterodimer with GCY-36 in the body cavity neurons, however it is also expressed in several other neuron pairs. gcy-35 and gcy-36 null mutants have similar defects in oxygen sensation (Gray et al., 2004), and we found that gcy-35 mutants did not have altered body fat (not shown).
To test whether cGMP-signaling genes that control oxygen sensing in URX neurons also regulate body fat, we generated gpa-8;gcy-36 and gpa-8;tax-4 mutants. Although the gcy-36 single mutants did not show an appreciable difference in body fat, they fully suppressed the body fat loss of the gpa-8 mutants, and gpa-8;gcy-36 double mutants retained body fat to the same extent as gcy-36 single mutants (Figure 2A). tax-4 mutants retained significantly greater body fat than wild-type animals (~125% of wild-type; Figure 2A) and gpa-8;tax-4 double mutants retained body fat to the same extent as tax-4 single mutants, fully suppressing the decreased body fat of gpa-8 mutants (Figure 2A). The genetic epistasis experiments indicate that GPA-8 and the URX-cGMP signaling genes have opposing effects on body fat, and that GCY-36 and TAX-4 are required to manifest the GPA-8 phenotype. To examine the effect of cGMP on body fat directly, we treated worms with a non-hydrolyzable analog of cGMP, 8-(para-Chlorophenylthio)-guanosine 3’5’-cyclic monophosphate (8-pCPT-cGMP). In wild-type animals, exogenous treatment of 8-pCPT-cGMP led to decreased body fat, resembling gpa-8 mutants (Figure 2B). Exogenous treatment of 8-pCPT-cGMP also led to decreased body fat in gcy-36 and gpa-8;gcy-36 mutants because the treatment bypasses GCY-36, but not gpa-8 mutants (Figure 2B). Finally, tax-4 and gpa-8;tax-4 mutants fully suppressed the fat loss induced by 8-pCPT-cGMP, suggesting that the observed cGMP effects on body fat require TAX-4 function (Figure 2B). tax-4 is also expressed in other sensory neurons in addition to the body cavity neurons (Coburn and Bargmann, 1996), therefore we wanted to measure the extent to which rescuing tax-4 in the oxygen sensing neurons restored body fat levels. In tax-4 mutants, we re-expressed tax-4 cDNA under the control of the gcy-36 promoter. We found that relative to tax-4 mutants and non-transgenic controls, transgenic animals lost the additional body fat and resembled wild-type animals (Figure 2C).
Figure 2. cGMP signaling in the body cavity neurons regulates the extent of body fat loss.
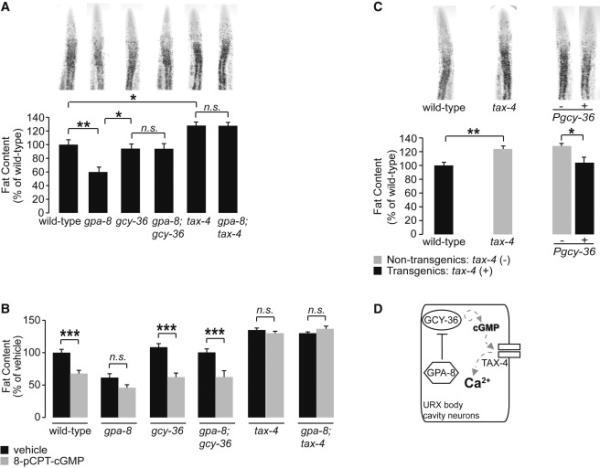
(A) Representative images are shown of animals fixed and stained with oil Red O (upper panels). Fat content was quantified for each genotype and is expressed as a percentage of wild-type animals ± SEM (lower panels; n=20-22). *, p<0.05; **, p<0.01; n.s., not significant by oneway ANOVA.
(B) For each genotype, animals were grown on plates containing either vehicle (10% DMSO) or 200 μM 8-(para-Chlorophenylthio)-guanosine 3’5’-cyclic monophosphate (8-pCPT-cGMP). Fat content was quantified for each genotype and is expressed as a percentage of vehicle-treated wild-type animals ± SEM (n=16-21). *** p<0.001 and n.s., not significant by two-way ANOVA.
(C) Representative images are shown of wild-type animals and tax-4 mutants fixed and stained with oil Red O (upper panels). For each transgenic line bearing tax-4 expression using the indicated promoter, non-transgenic animals (−) and transgenic animals (+) are shown. Relative to non-transgenic controls (gray bars), transgenic animals (black bars) bearing the tax-4 transgene under the control of the heterologous gcy-36 promoter restore body fat content to wild-type. Data are expressed as a percentage of body fat in wild-type animals ± SEM (lower panels; n=20-22). *, p<0.05 and **, p<0.01 by Student's t-test.
(D) Schematic depiction of a signaling pathway in the URX neurons in which GPA-8 opposes the functions of GCY-36 and TAX-4 to regulate body fat via the second messenger cGMP.
Together, our data indicate that GPA-8 functions in a discrete signaling pathway with GCY-36 and TAX-4 in the URX neurons to regulate body fat. We find that increased cGMP signaling decreases body fat, whereas blocking cGMP signaling retains or increases body fat. Thus, cGMP functions as an instructive second messenger in the URX neurons for the long-range control of body fat. In these neurons, GCY-36 is the major source of cGMP and TAX-4 is its effector. Because gpa-8 mutants have decreased body fat that is blocked by the absence of gcy-36 and tax-4, our model suggests that GPA-8 must normally oppose the functions of GCY-36 and TAX-4 (Figure 2D).
Oxygen sensing via the body cavity neurons controls the extent of fat mobilization in the periphery
The genetic evidence suggested that gpa-8 opposes the effects of the oxygen sensing genes in URX neurons. In the laboratory, C. elegans feed on living bacteria whose respiration drops the local ambient concentration of oxygen. Under these settings, increasing oxygen concentrations correlate with decreasing food availability. To measure the physiological consequences of food absence and oxygen sensing, we embarked on a series of experiments in which we measured the extent of fat loss upon changing the animals’ exposure to oxygen. We first ascertained that the hypoxia-sensing pathway genes did not alter body fat (Figure S2A). Additionally, evidence from the literature suggests that C. elegans does not experience appreciable hypoxia until ambient oxygen levels reach 3% (Van Voorhies and Ward, 2000). As seen in most organisms, fasting induces a drop in body fat in C. elegans because triglycerides are utilized for energy production in the absence of food supplies (Jo et al., 2009). A fasting time course of wild-type animals revealed that within a 2-3 hour window, adults at atmospheric oxygen (21%) lose nearly 70-80% of their body fat (Figure S2B). To measure the extent to which fat loss is dependent on environmental oxygen exposure, we compared the fasting-induced fat loss of wild-type animals exposed to 21%, versus 10% oxygen (Figure 3A). We chose 10% oxygen because behavioral and imaging studies have shown that URX neurons are not active at this concentration (Zimmer et al., 2009). Well-fed wild-type animals on either live or heat-killed bacterial lawns did not have an appreciable difference in body fat when exposed to 21% and 10% oxygen for the same duration (Figures S2C and S2D). Upon food deprivation and short-term fasting, animals exposed to 10% oxygen over the 2.5-hour window retained significantly greater body fat (~45% of well-fed controls) compared to animals exposed to 21% oxygen (~20% of well-fed controls; Figure 3B). The increase in fat retention at 10% was not accompanied by a change in locomotion or other discernable behavioral differences (E.W. and S.S., unpublished observations). We have previously shown that a substantial loss of body fat requires the transcriptional induction of the atgl-1 lipase (Noble et al., 2013; Srinivasan, 2015). In keeping with the differential effects of oxygen exposure on body fat loss, we found that upon fasting, the extent to which atgl-1 is transcriptionally induced is dependent upon exposure to oxygen. Animals exposed to 21% oxygen showed a significant induction of the atgl-1 reporter, whereas animals exposed to 10% oxygen did not (Figure 3C). Thus, food absence and increased oxygen availability induce peripheral lipid mobilization as judged by body fat levels and atgl-1 induction.
Figure 3. Oxygen-sensing via the body cavity neurons controls the extent of fat mobilization in the periphery.
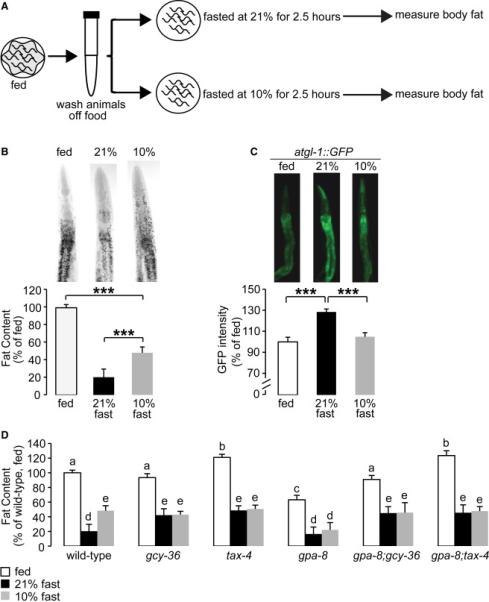
(A) Schematic depiction of the oxygen-dependent fat loss assay. L1 worms were seeded and grown to adulthood on food. Day 1 adults were washed off food over a period of 30 minutes. Worms were then seeded onto plates without food. Worms were then subjected to a fasting period at either 21% or 10% oxygen for an additional 2.5 hours. At the end of the fasting period, worms were fixed and stained with oil Red O.
(B) Wild-type worms were subjected to the fasting assay described in (A). Representative images are shown of fed or fasted wild-type worms, subjected to different O2 concentrations, fixed and stained with oil Red O (upper panels). Data are expressed as a percentage of body fat in wild-type fed controls ± SEM (lower panels; n=20-21). ***, p<0.001 by one-way ANOVA.
(C) atgl-1::GFP worms were subjected to the fasting assay described in (A). The fluorescence intensity of atgl-1 expression was quantified and is expressed as a percentage of wild-type fed controls ± SEM (n=10-15). ***, p<0.001 by one-way ANOVA.
(D) Worms of the indicated genotypes were subjected to the fasting assay described in (A). Fat content was quantified for each genotype and condition. Data are expressed as a percentage of body fat in wild-type fed controls ± SEM (n=12-25). Each genotype was compared to wild-type by one-way ANOVA. Different letters indicate statistical significance. Letters shared in common among groups indicates no significant difference. Groups labeled with “a” were not significantly different from wild-type fed animals. Groups labeled with “b” (p<0.01) and “c” (p<0.001) represent significant differences within the fed condition compared to wild-type. Groups labeled with “d” (p<0.001) and “e” (p<0.01) represent significant differences within the fasted conditions compared to wild-type. See also Figures S2 and S3.
The URX neurons are activated by 21% oxygen via the actions of GCY-36 and TAX-4, and silenced at 10% oxygen. To measure the extent to which the effect of oxygen on fat loss was dependent on neural oxygen sensing via URX, we subjected mutants of the URX-cGMP signaling pathway to the oxygen-dependent fat loss experimental paradigm. Across all genotypes, short-term fasting induced fat loss at both 21% and 10% (Figure 3D). However, the extent to which body fat was mobilized at the two oxygen concentrations varied between mutants. Relative to wild-type animals, gcy-36 and tax-4 mutants showed a significant suppression of fasting-induced fat loss at 21% (Figure 3D). On the other hand, the extent of fat loss at 10% oxygen remained similar to that of wild-type animals in both mutants. Thus, the fat loss elicited by 21% oxygen is abrogated in the gcy-36 and tax-4 mutants, suggesting that activation of URX neurons via these genes is essential for the stimulation of fat loss. Our genetic epistasis experiments had already indicated that GPA-8 opposes the functions of GCY-36 and TAX-4 (Figure 2A). In the oxygen-dependent fasting paradigm, we noted an interesting phenotype in the gpa-8 mutants: although fasting at 21% oxygen elicited fat loss indistinguishably from wild-type animals, they had significantly greater fat loss at 10% oxygen compared to wild-type, gcy-36 and tax-4 animals (Figure 3D). In effect, gpa-8 animals fasted at 10% oxygen resembled those fasted at 21%, again revealing that gpa-8 mutants oppose the gcy-36 and tax-4 mutant phenotypes (Figures 2A, 2D and 3D). The enhanced fasting-induced fat loss of gpa-8 animals at 10% oxygen was fully suppressed in the gpa-8;gcy-36 and gpa-8;tax-4 mutants (Figure 3D), thus the URX responses at 10% and 21% oxygen are both integrated via cGMP signaling. This in turn suggested that in contrast to wild-type animals, gpa-8 mutants raised on heat-killed bacteria would show a differential response at 21% versus 10% oxygen, because in the gpa-8 mutants fasted at 21% oxygen, gcy-36 would be activated in two ways: first, by de-repression via loss of gpa-8 and second, via activation by 21% oxygen. Accordingly, we found that gpa-8 mutants exposed to 21% and 10% oxygen on heat-killed bacterial lawns had decreased body fat at 21% relative to 10% oxygen (Figure S3).
We thus identify two components for the differential modulation of body fat loss by oxygen sensing in URX neurons. One, activation of URX at 21% oxygen via GCY-36 and TAX-4 signaling stimulates fat loss, and gcy-36 and tax-4 mutants retain more body fat at 21% oxygen than wild-type. This is in keeping with the previously observed enhanced behavioral effects of URX at 21% oxygen. Two, silencing of URX at 10% oxygen via GPA-8 signaling serves to minimize fat loss, therefore gpa-8 mutants have enhanced fat loss at 10%. This suggests a role for GPA-8 in keeping URX inactive at 10% oxygen.
Internal fat reservoirs modulate the resting state of URX neurons via GPA-8 signaling
To directly study the effects of GPA-8 signaling on URX neuron function, we turned to Ca2+ imaging in living worms. We used the genetically-encoded calcium indicator GCaMP5k as a reporter of URX activity, because it has been optimized for greater sensitivity and threshold activation properties (Akerboom et al., 2012). Wild-type animals bearing the GCaMP5k transgene expressed under the URX-specific promoter flp-8 were overtly normal and showed robust calcium influx at 21% oxygen (Figures 4A and 4D), as previously described (Schrodel et al., 2013). We observed two properties of URX activation in gpa-8 mutants crossed into the flp-8::GCaMP5k transgenic line. First, at 21% oxygen there was an approximately 30% decrease in maximal activation of URX neurons in the gpa-8 mutants (Figures 4B, 4E and 4G). gcy-36 mutants did not show URX activation at 21% oxygen (Figures 4C and 4F), consistent with published work using GCaMP 3.0 (Zimmer et al., 2009). Second, at 10% oxygen, we observed a nearly two-fold increase in baseline (F0) fluorescence values in gpa-8 mutants relative to wild-type animals (Figure 4H), whereas gcy-36 mutants did not show a difference in baseline (F0) fluorescence (Figure 4H) or flp-8 promoter activity at 10% oxygen (Figure 4I). Importantly, the increase in baseline fluorescence of gpa-8 mutants at 10% oxygen was not a function of a general increase in flp-8 promoter activity at either 21% or 10% oxygen (Figure S4A and Figure 4I). A scatter plot of maximal URX responses (ΔF/F) values, 21% oxygen) versus baseline fluorescence (F0, 10% oxygen) values reveals the effect of loss of GPA-8 on URX function: gpa-8 mutants have increased Ca2+ levels at 10% oxygen, thus elevating the resting state of URX (Figures S4B-S4D). When measuring URX peak responses to the oxygen upshift in absolute GCaMP5k fluorescence levels, no significant difference between wild-type and gpa-8 was observed (Figure 4J); the major effect of GPA-8 therefore lies in controlling Ca2+ levels at 10% oxygen. The URX peak response to the oxygen upshift in absolute GCaMP5k fluorescence levels was decreased in gcy-36 animals compared to wild-type animals (Figure 4J). The consequence of the increased URX basal properties in gpa-8 mutants is to make URX neurons more active at 10%, which in turn dampens maximal responsiveness at 21% oxygen. Because GPA-8 is not involved in the direct sensing of oxygen or in cGMP synthesis, its major effect on URX function is modulatory: it limits baseline Ca2+ and thus ensures that URX is held inactive at 10% oxygen.
Figure 4. GPA-8 controls the resting-state Ca2+ levels in URX neurons.
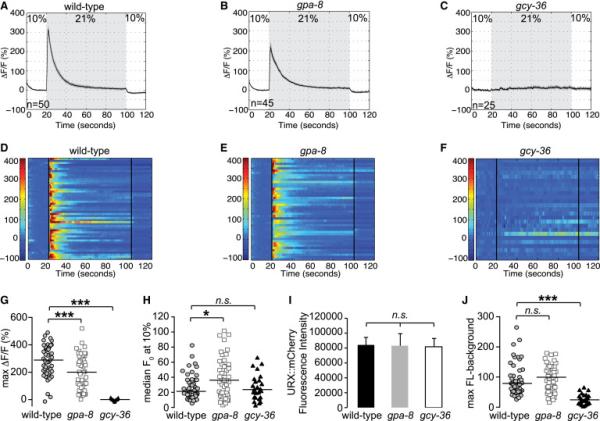
(A-F) Measurements of neuronal activity by Ca2+ imaging of URX neurons for each genotype. The number of animals used for each condition is shown in the figure. We conducted Ca2+ imaging experiments in URX neurons in living wild-type, gpa-8, and gcy-36 mutant animals bearing GCaMP5k under the control of the flp-8 promoter. Oxygen concentrations in the microfluidic chamber were 10% and 21% as indicated. (A-C) For each genotype, black traces show the average percent change of GCaMP5k fluorescence (ΔF/F0) and gray shading indicates SEM. (D-F) Individual URX responses are shown for each genotype; each row represents one animal.
(G) Maximum ΔF/F0 values are shown for individual animals in wild-type, gpa-8, and gcy-36 mutants. Bars indicate the average value within each genotype. ***, p<0.001 by one-way ANOVA.
(H) Individual baseline fluorescence (F0) values at 10% oxygen are shown for individual animals in wild-type, gpa-8, and gcy-36 mutants. Bars indicate the median value within each genotype. *, p<0.05 and n.s., not significant by Kruskal-Wallis test.
(I) We imaged mCherry fluorescence in wild-type, gpa-8, and gcy-36 animals expressing both GCaMP5k and mCherry under the control of the flp-8 promoter. Images were taken in animals exposed to 10% oxygen. For each genotype, the fluorescence intensity was imaged at the same exposure, determined to be within the linear range. Fluorescence intensity was quantified and expressed as an average ± SEM (n=21-27). n.s., not significant by one-way ANOVA.
(J) The background-subtracted maximum fluorescence (max FL) at 21% oxygen is shown for each animal in the wild-type, gpa-8, and gcy-36 backgrounds. Bars indicate the average value within each genotype. ***, p<0.001 and n.s., not significant by one-way ANOVA. See also Figure S4.
In seeking a greater understanding of GPA-8 function, three lines of evidence led us to reason that the URX neurons may detect an internal homeostatic signal. First, resting-state Ca2+ levels in URX neurons were increased in gpa-8 mutants, resembling a tonic increase in neuronal activity. Second, although gpa-8 mutants regulate body fat via resting-state Ca2+ levels in URX at 10% oxygen (Figures 3D and 4H), they do not appreciably alter the physiological response to 21% oxygen (Figure 3D). Third, the positioning of the URX neurons within the body cavity and the coelomic fluid suggests that they may sense the internal milieu (www.wormatlas.org). Thus, we conducted experiments to test whether the role of GPA-8 in keeping URX in a state of diminished activation stems from sensing internal fat reserves.
We decreased body fat levels in the intestine by RNAi-mediated inactivation of each of two genes: Acetyl CoA Carboxylase (ACC/pod-2) and palmitic acid elongase elo-2 in adult C. elegans. ACC/pod-2 is an enzyme that generates malonyl CoA, which is the precursor to fatty acid synthesis in eukaryotes (Salway, 1999). elo-2 encodes an enzyme that converts C16:0 fatty acids to C18:0 fatty acids (Kniazeva et al., 2003). Thus ACC/pod-2 and elo-2 inactivation each inhibit fat synthesis, and pod-2 null mutants arrest at an early larval stage. ACC/pod-2 and elo-2 are only expressed in the C. elegans intestine (Nomura et al., 2010) (Figure S5A), and we found that RNAi-mediated inactivation of pod-2 (post-development; after the L4 stage) or elo-2 leads to a decrease in body fat by greater than 80% compared to control treated worms (Figure S5B). We did not observe any additional physical defects or differences in egg production at the time of imaging in pod-2 or elo-2 inactivated animals (Figures S5C and S5D). We measured the effect of this substantial decrease in body fat in the intestine, on URX activation. In response to 21% oxygen, we observed a decrease in maximal activation (ΔF/F0) of URX neurons (Figures 5A, 5B, 5E, 5F, 5I, 5J, S6A, S6C, S6E and 6A). Next, in response to 10% oxygen, we observed a greater than two-fold increase in baseline (F0) fluorescence values in both ACC/pod-2 and elo-2 inactivated animals relative to wild-type controls (Figures 6B, S6A, S6C and S6E). The increase in resting-state Ca2+ levels at 10% oxygen was not a consequence of increased promoter activity in URX neurons; co-expressed flp-8::mCherry was not appreciably different across the experimental conditions (Figure 6C). Our evidence indicates that a substantial drop in body fat reserves in the intestine has the capacity to modify Ca2+-regulated URX activation properties. The decrease in maximal fluorescence (at 21%) and concomitant increase in baseline fluorescence (at 10%) seen with inactivation of ACC/pod-2 or elo-2 strongly resembled the URX response of gpa-8 mutants (Figures 4B, 4E, 4G and 4H). Therefore we measured URX responses upon fat depletion in the gpa-8 mutants and found that inactivation of either ACC/pod-2 or elo-2 in the gpa-8 mutant background did not lead to an additive effect on URX activation (Figures 5C, 5D, 5G, 5H, 5K, 5L, S6B, S6D and S6F). When measuring the URX response to the oxygen upshift via absolute GCaMP5k fluorescence levels, no significant differences across the experimental conditions were observed (Figure 6D). Together, these experiments indicate that the increased resting-state Ca2+ levels in URX neurons elicited by depleting intestinal fat reserves occurs via GPA-8 signaling. Thus, GPA-8 signaling serves to limit the tonic activity of URX neurons in the ‘off’ state when the animals are exposed to 10% oxygen.
Figure 5. The activation of URX neurons is modulated by internal fat reserves, in a GPA-8-dependent manner.
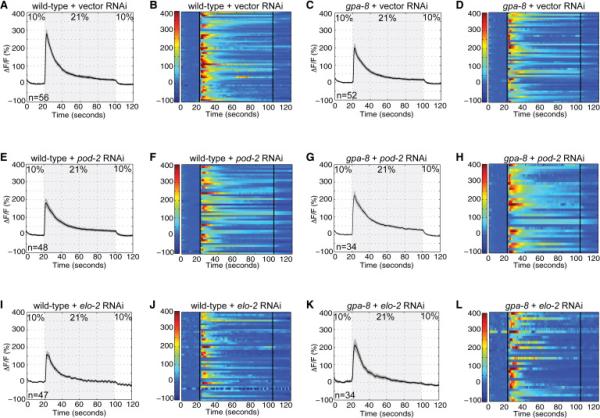
(A, C, E, G, I, K) Measurements of neuronal activity by Ca2+ imaging of URX neurons for each genotype and condition. The number of animals for each condition is given in the figure. We conducted Ca2+ imaging in vector, pod-2, or elo-2 RNAi-treated wild-type and gpa-8 mutant animals bearing GCaMP5k under the control of the flp-8 promoter. Oxygen concentrations in the microfluidic chamber were 10% and 21% as indicated. Black traces show the average percent change of GCaMP5k fluorescence (ΔF/F0) and gray shading indicates SEM. Average ΔF/F0 at the depicted oxygen concentrations in (A, E, I) wild-type animals or (C, G, K) gpa-8 mutants treated with vector control RNAi (top row), pod-2 RNAi (middle row), or elo-2 RNAi (bottom row). (B, D, F, H, J, L) Individual URX responses are shown for each genotype and condition; each row represents one animal. See also Figure S5.
Figure 6. The resting-state Ca2+ levels in URX neurons are modulated by internal fat reserves, in a GPA-8-dependent manner.
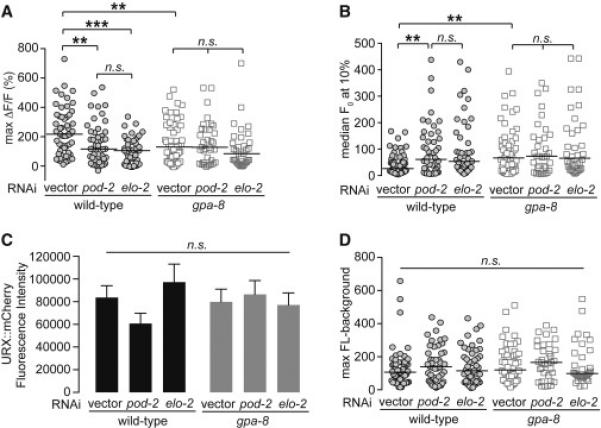
(A) Individual maximum ΔF/F0 values are shown for each genotype and condition, as denoted in the figure. Bars indicate the average value within each genotype. **, p<0.01; ***, p<0.001; and n.s., not significant by two-way ANOVA.
(B) Individual baseline fluorescence (F0) values at 10% oxygen are shown for each genotype and condition. Bars indicate the median value within each genotype. **, p<0.01 and n.s., not significant by Kruskal-Wallis.
(C) mCherry fluorescence intensity in RNAi-treated animals expressing both GCaMP5k and mCherry under the control of the flp-8 promoter. Images were taken in animals exposed to 10% oxygen. For each genotype, the fluorescence intensity was imaged at the same exposure, determined to be within the linear range. Fluorescence intensity was quantified and expressed as an average ± SEM (n=21-27). n.s., not significant by two-way ANOVA.
(D) The background-subtracted maximum fluorescence (max FL) at 21% oxygen is shown for each genotype and condition. Bars indicate the average value within each genotype. n.s., not significant by one-way ANOVA. See also Figure S6.
DISCUSSION
Here, we identify the C. elegans body cavity neurons as homeostatic sensors and integrators of food availability and body fat status (Figure 7). Our evidence points to a model in which oxygen sensation via the URX neurons functions to stimulate body fat loss. Fat stores in turn, regulate the tonic activity of URX neurons. The interoception of fat reserves by the body cavity neurons thus performs a critical function: it ensures that a neural signal to stimulate fat loss is deployed only when there are adequate internal fat reserves (modeled in Figure 7A). Our model predicts that the activity state of URX neurons reflects the net balance between oxygen sensation and internal fat reserves (Figures 7B-7E). In the fat-replete state (Figure 7B), an internal nutrient sufficiency signal activates GPA-8, leading to the inhibition of GCY-36 and cGMP production, which reduces tonic activity of URX. When animals are exposed to low oxygen (~10%) and food is present, there is no evoked response. In this setting, net URX activity is off, and fat loss via ATGL-1 activation is minimal. As food supplies dwindle (Figure 7C), GCY-36 becomes maximally activated by the increase in environmental oxygen to 21%. Active GCY-36 increases cGMP synthesis, TAX-4-mediated Ca2+ influx and URX activity, ultimately stimulating body fat loss via an unknown neuroendocrine signal. While food supplies are still low and as fat stores become depleted (Figure 7D), the internal nutrient sufficiency signal is lost and GPA-8 is no longer kept active. This leads to de-repression of GCY-36, and increased tonic activity of URX. In this physiological setting, although food withdrawal and exposure to 21% oxygen would still activate GCY-36, the net maximal activation of URX is diminished because of its already-increased tonic state. Diminished URX activation would then be predicted to minimize the release of a fat-loss-stimulating signal. Upon re-encountering food (Figure 7E), URX activity is predicted to remain low until fat stores are replete again and the nutrient sufficiency signal is restored. The proposed homeostatic loop defines a novel interoceptive mechanism for the body cavity neurons that integrates external and internal nutrient status.
Figure 7. Schematic depiction of a homeostatic neuroendocrine axis that integrates external and internal nutrient status.
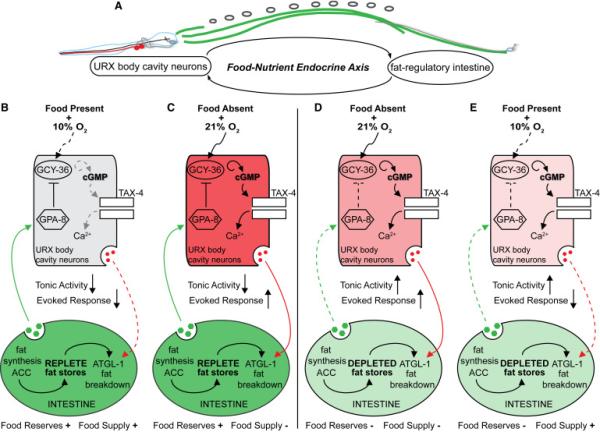
(A) We have identified a neuroendocrine axis which operates to communicate oxygen availability to regulate body fat stores in the intestine, the major metabolic and fat-regulatory organ for C. elegans. A feedback signal from the intestine relays fat status to the URX body cavity neurons by modulating their tonic activity. This homeostatic loop ensures that neural stimulation of fat mobilization only occurs when there are sufficient fat reserves.
(B) In the fat-replete state, an internal nutrient sufficiency signal activates GPA-8 (depicted in green). This leads to the inhibition of GCY-36 and cGMP production, keeping the tonic activity of URX neurons low. In the presence of food, when animals are exposed to low oxygen (~10%), there is no evoked response. In this setting, net URX activity is off, and fat mobilization is minimal (dotted red line).
(C) As food supplies dwindle and ambient oxygen levels rise to 21%, GCY-36 becomes maximally activated and generates cGMP, which in turn activates the cyclic nucleotide gated channel TAX-4. This allows Ca2+ influx and stimulates the evoked response of URX neurons. In this setting, net URX activity is high, leading to the release of a signal that stimulates body fat loss (depicted in red).
(D) As fat stores become depleted in the continued absence of food supplies, at 21% oxygen the internal nutrient sufficiency signal is lost and GPA-8 is no longer kept active (dotted green line). This leads to de-repression of GCY-36, and increases the tonic activity of URX. Although 21% oxygen would still activate GCY-36, the net activity of URX is diminished because of its already-increased tonic state, thus slowly minimizing the release of a fat loss signal.
(E) Upon re-encountering food in the fat-depleted state, net URX activity would be predicted to remain low until fat stores are replete again and the nutrient sufficiency signal is restored.
The utilization of fat for the production of energy employs a cascade of cell-autonomous biochemical reactions that require molecular oxygen in the mitochondria (Salway, 1999). Our data suggest that neural perception of oxygen availability is an additional, previously unknown feature underlying the neural control of body fat. Oxygen sensation under normoxia in the mammalian nervous system has been documented to regulate peripheral metabolism (Frappell et al., 1992), however the mechanisms underlying this effect remain obscure. In C. elegans, the intestine is not directly innervated, thus information relay from the nervous system to metabolic sites must involve endocrine effects. The neuroendocrine mechanisms by which URX neurons communicate information to the intestine are not yet known.
Visualization of URX activity via Ca2+ imaging unexpectedly revealed that the extent of body fat reserves themselves alter URX function in a GPA-8-dependent manner. URX neurons are known to be sensors of environmental oxygen (Persson et al., 2009; Busch et al., 2012) and transducers of 5-HT encoded food presence (Noble et al., 2013). We propose that the direct or indirect perception of internal body fat reserves via GPA-8 signaling is another property of the body cavity neurons. Changes in fat reserves alter the tonic activation state of URX neurons, in keeping with the narrow dynamic range of body fat homeostasis. Thus, our experiments reveal a novel facet of neuronal function in this context: integration between the sensation of oxygen, an external sensory cue, and the perception of body fat, an internal sensory cue. These disparate modalities are integrated via the actions of a cGMP-mediated signal transduction pathway in the URX interoceptive neurons, whose activation status is a measure of the counterbalance between oxygen availability and fat reserves. Although there is currently scant evidence in the literature for the modulation of soluble guanylyl cyclases by G proteins, in C. elegans the ASJ light-sensing neurons utilize a G protein-dependent cGMP transduction pathway that is independent of phosphodiesterase activity (Liu et al., 2010). Our results point to a potential new mode of action for Gustducin/GPA-8 in cellular signal transduction that elicits robust effects on whole body physiology.
There are two possible models for the integration of internal metabolic state information in the nervous system. First, changes in internal metabolic state may alter the ability of neurons to perceive external sensory information by directly modulating the magnitude of the maximal response elicited by a given sensory cue. This model implies that the external sensory receptors overlap with those of internal state sensing, and that each function is dependent on the other. Alternatively, internal state sensing may function to modify the tonic or basal properties of neurons, while retaining their full sensory capacity. A key aspect supporting this alternate model, is that the dynamic range of internal metabolic parameters is much narrower than the many orders of magnitude typically processed by external sensory receptors (Axel, 2005). In this scenario, the cellular machinery used for internal state sensing is distinct from that of external sensing, and integration between external and internal states occurs downstream of receptors. In either model, the result of integration would lead to a metabolic-state-dependent modulation of neuronal activity. Our data favor the second model. We propose that a nutrient sufficiency signal via GPA-8 regulates the tonic activation of the URX neurons, whereas oxygen sensing via GCY-36 regulates the stimulated activation of URX neurons. In support of our model, observations of animal behavior and physiology suggest that sensory functions are enhanced or diminished by, rather than fully dependent upon, internal state.
EXPERIMENTAL PROCEDURES
Animal Maintenance and Strains
All animals were cultured as described (Brenner, 1974). N2 Bristol, obtained from the Caenorhabditis Genetic Center (CGC), was used as the reference wild-type strain. The following mutant strains were used: NL1147 gpa-10(pk36)V, DA1084 egl-30(ad806)I, CX2205 odr-3(n2150)V, NL793 gpa-9(pk438)V, NL1137 gpa-5(pk376)X, NL334 gpa-2(pk16)V, NL2330 gpa-13(pk1270)V, NL795 gpa-7(pk610)IV, NL332 gpa-1(pk15)V, RB1800 gpa-17(ok2334)III, NL797 gpa-15(pk477)I, DG1856 goa-1(sa734)I, NL1146 gpa-6(pk480)X, NL787 gpa-11(pk349)II, NL790 gpa-4(pk381)IV, NL594 gpa-12(pk322)X, NL788 gpa-14(pk342)I, NL335 gpa-3(pk35)V, NL1142 gpa-8(pk345)V, KQ1384 tax-4(p678)III, AX1297 gcy-36(db66)X, SSR866 gpa-8(pk345);tax-4(p678), SSR1047 gpa-8(pk345);gcy-36(db66), FK229 egl-4(ks61)IV, and SSR915 gpa-8(pk345);egl-4(ks61). The following transgenic strains were generated: SSR896 atgl-1::GFP, SSR1080 gpa-8(pk345);atgl-1::GFP, SSR1128 tax-4(p678);Pgcy-36:tax-4::GFP, SSR634 gpa-8(pk345);Pgpa-8:gpa-8::GFP, SSR1008 gpa-8(pk345);Pgpa-8:gpa-8::GFP, SSR1011 gpa-8(pk345);Pgcy-36:gpa-8::GFP, SSR688 Pgcy-36::gpa-8SAS, SSR691 Pflp-8::gpa-8SAS, SSR1070 N2;flp-8::mCherry;flp-8::GCaMP5k, SSR1066 gpa-8(pk345);flp-8::mCherry;flp-8::GCaMP5k, and SSR1218 gcy-36(db66);flp-8::mCherry;flp-8::GCaMP5k. All experiments were performed on day 1 adults.
Cloning and Transgenic Strain Construction
Promoters and genes were cloned using standard PCR techniques from N2 Bristol worm lysates and cloned using Gateway Technology™ (Life Technologies). Promoter lengths were determined based on functional rescue and are available upon request. All rescue plasmids were generated using polycistronic GFP. Transgenic rescue strains were constructed by microinjection into the C. elegans germline followed by visual selection of transgenic animals under fluorescence. For the microinjections, 5-10 ng/μl of the desired plasmid was injected with 25 ng/μl of an unc-122::GFP coinjection marker and 65-70 ng/μl of an empty vector to maintain a total injection mix concentration of 100 ng/μl. In each case, 10–20 stable transgenic lines were generated. Two lines were selected for experimentation based on consistency of expression and transmission rate. For GCaMP5k transgenic animals, 5 ng/μl of Pflp-8::GCaMP5k was injected with 2 ng/μl of a Pflp-8::mCherry coinjection marker.
Triglyceride Extraction and Quantitation
Triglycerides were extracted from wild-type and mutant C. elegans as described (Bligh and Dyer, 1959; Noble et al., 2013). Extracted lipids were quantified using the Enzychrom™ Triglyceride Assay kit (Bioassay Systems) according to the manufacturer's instructions.
Oil Red O Staining
Oil Red O staining was performed as described (Noble et al., 2013) with the following change: animals were fixed for 5 minutes with 4% formaldehyde (Fisher Scientific) and 0.5% β-mercaptoethanol (Acros Organics) before undergoing three freeze-thaw cycles. For oil Red O experiments in which animals were treated with a non-hydrolyzable cGMP analog, animals were seeded on plates containing either 200 μM 8-(4-Chlorophenylthio)-guanosine 3’,5’-cyclic monophosphate (Sigma) or 10% dimethyl sulfoxide (DMSO) vehicle. Within a single experiment, 2000-3000 animals were fixed and stained. All experiments were repeated at least 3 times. Wild-type animals were included as controls in each independent experiment.
RNAi
RNAi experiments were conducted as previously described (Noble et al., 2013). Plates were seeded with HT115 bacteria containing vector or the relevant RNAi clone four days prior to seeding larvae.
Image Acquisition and Quantitation
Black and white images of oil Red O stained worms were captured using a 10× objective on a Zeiss Axio Imager microscope. Images were quantified using ImageJ software (NIH). Lipid droplet staining in the first four pairs of intestinal cells was quantified, as described (Noble et al., 2013). Within each experiment, approximately 10–20 animals from each condition were quantified.
Oxygen-dependent Fat Loss Assay
For each strain, approximately 3000 synchronized L1 larvae were seeded onto each of three plates. Worms were grown at 20° C for 52 h after which all plates were transferred to the bench top. Worms subjected to the fasting protocol were washed off the plates with PBS with 5 sequential washes over a 30-minute period to eliminate residual bacteria, and then seeded onto NGM plates without food. Worms were then subjected to a 2.5-h fasting period at either 21% or 10% oxygen. To establish the time course of fasting, pilot experiments were conducted at atmospheric (21%) oxygen. The ‘21% fasted’ plates were placed in a non-airtight container at room temperature. The ‘fed’ control plates were placed in a similar but separate container. The ‘10% fasted’ plates were placed in a custom-designed sealed acrylic oxygen chamber (TSRI Instrumentation and Design Lab), fitted with inlet and outlet valves. The inlet valve was connected via bubble tubing to a pressurized oxygen and nitrogen pre-mixture containing 10% oxygen (Praxair, Inc), and the outlet valve was exposed to air. All plates were positioned right side up with the lids slightly ajar. The sealed chamber was then perfused for 15 minutes with 10% oxygen. Following perfusion, both valves were closed. During the experiment, pressure inside the chamber was held constant, as judged by a gauge placed inside the oxygen chamber. The chamber was kept at room temperature for an additional 2 h 15 minutes, so that all fasted conditions remained off food for a total of 2.5 h following the washes. At the end of this period, worms from the respective conditions were collected for oil Red O staining.
Calcium Imaging
N2;flp-8::mCherry;flp-8::GCaMP5K, gpa-8(pk345);flp-8::mCherry;flp-8::GCaMP5K, and gcy-36(db66);flp-8::mCherry;flp-8::GCaMP5k transgenic animals were used for GCaMP5k imaging. We used a microfluidic chamber constructed with the oxygen-permeable poly(dimethylsiloxane) (PDMS) as described (Zimmer et al., 2009). A Valvebank II (AutoMate Scientific, Inc.) was used to control input from two pressurized pre-mixtures of oxygen and nitrogen containing either 10% oxygen or 21% oxygen (Praxair, Inc). The gas flow rate was set to 0.26 psi at the outlet of the chamber as judged by a VWR™ traceable pressure meter. Immediately before imaging, individual day 1 adult animals were sequentially transferred to two unseeded plates. Individual C. elegans adults were then transported into the chamber in a drop of S Basal buffer containing 5 mM tetramisole hydrochloride (Sigma) via Tygon tubing (Norton). Animals were constantly submerged in S Basal buffer while inside the chamber. After the animals were immobilized inside the chamber, GCaMP5k fluorescence was visualized at 40x magnification using a spinning disk confocal microscope (Olympus) using MetaMorph™ (version 6.3r7, Molecular Devices). Worms were pre-exposed to 10% oxygen for 5 minutes in the microfluidic chamber as described (Zimmer et al., 2009). GCaMP5k fluorescence was recorded by stream acquisition for two minutes at a rate of 8.34 frames/second with an exposure time of 20 ms using a 12-bit Hamamatsu ORCA-ER digital camera. Each animal was recorded once. GCaMP5k-expressing neurons were marked by a region of interest (ROI). The position of the ROI was tracked using the “Track Objects” function in MetaMorph™. An adjacent ROI was used to subtract background from the total integrated fluorescence intensity of the ROI. Data were analyzed using MATLAB (MathWorks, Inc.). Fluorescence intensity is presented as the percent change in fluorescence relative to the baseline (ΔF/F0). F0 was measured in worms exposed to 10% oxygen during the first 9-13 seconds for each recording and calculated as an average over that period. Maximum ΔF/F0 was measured in worms exposed to 21% oxygen during the first 21-23 seconds for each recording and calculated as an average over that period. All animals were day 1 adults at the time of imaging. The number of animals used for each condition is denoted in the figures.
Statistics
All oil Red O results are presented relative to wild-type unless otherwise noted. Error bars represent SEM. Student's t-test, one-way ANOVA, and two-way ANOVA were used where indicated. Bonferroni's correction for multiple comparisons was used for all ANOVAs. Kruskal-Wallis with Dunn's multiple comparison tests was used where indicated. All experiments were performed at least three times. Wild-type animals were included as controls for each experiment.
Supplementary Material
Highlights.
Oxygen sensing neurons regulate body fat metabolism via a neuroendocrine signal.
Fluctuations in normoxic oxygen determine the magnitude of body fat loss.
An interoceptive fat signal controls the tonic activity of oxygen sensing neurons.
The balance between neuronal oxygen sensing and internal reserves drives fat loss.
ACKNOWLEDGEMENTS
This work was supported by research grants to S. Srinivasan from the NIH/NIDDK (R01 DK095804) and S. Skora from the European Community's Seventh Framework Programme (FP7/2007-2013, ERC grant agreement no. 281869 – C. elegans Neurocircuits). We are grateful to the Knockout Consortium at Tokyo Women's Medical University. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). We thank Dr. Kathryn Spencer, Dorris Neuroscience Center, The Scripps Research Institute for assistance with Ca2+ imaging. We also thank Dr. Rosalind Hussey, Dr. Lavinia Palamiuc, and other members of the Srinivasan Lab for critical comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.S. and E.W. designed the study. E.W. conducted the experiments with critical contributions from C.C., H.R. and S. Skora. S.S. and E.W. analyzed the data. S.S. wrote the manuscript with critical input from E.W. and M.Z. All authors read and approved the manuscript.
REFERENCES
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axel R. Scents and sensibility: a molecular logic of olfactory perception (Nobel lecture). Angewandte Chemie. 2005;44:6110–6127. doi: 10.1002/anie.200501726. [DOI] [PubMed] [Google Scholar]
- Bargmann CI. Chemosensation in C. elegans. WormBook : the online review of C. elegans biology. 2006:1–29. doi: 10.1895/wormbook.1.123.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiani C, Mendel J. Heterotrimeric G proteins in C. elegans. WormBook : the online review of C. elegans biology. 2006:1–25. doi: 10.1895/wormbook.1.75.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR, Morrison C. The brain, appetite, and obesity. Annual review of psychology. 2008;59:55–92. doi: 10.1146/annurev.psych.59.103006.093551. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Canadian journal of biochemistry and physiology. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch KE, Laurent P, Soltesz Z, Murphy RJ, Faivre O, Hedwig B, Thomas M, Smith HL, de Bono M. Tonic signaling from O(2) sensors sets neural circuit activity and behavioral state. Nature neuroscience. 2012;15:581–591. doi: 10.1038/nn.3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon WB. The Wisdom Of The Body. 1932 [Google Scholar]
- Cao L, Choi EY, Liu X, Martin A, Wang C, Xu X, During MJ. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamicadipocyte axis. Cell metabolism. 2011;14:324–338. doi: 10.1016/j.cmet.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AJ, Chronis N, Karow DS, Marletta MA, Bargmann CI. A distributed chemosensory circuit for oxygen preference in C. elegans. PLoS biology. 2006;4:e274. doi: 10.1371/journal.pbio.0040274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung BH, Arellano-Carbajal F, Rybicki I, de Bono M. Soluble guanylate cyclases act in neurons exposed to the body fluid to promote C. elegans aggregation behavior. Current biology : CB. 2004;14:1105–1111. doi: 10.1016/j.cub.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Cheung BH, Cohen M, Rogers C, Albayram O, de Bono M. Experience-dependent modulation of C. elegans behavior by ambient oxygen. Current biology : CB. 2005;15:905–917. doi: 10.1016/j.cub.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Coates JC, de Bono M. Antagonistic pathways in neurons exposed to body fluid regulate social feeding in Caenorhabditis elegans. Nature. 2002;419:925–929. doi: 10.1038/nature01170. [DOI] [PubMed] [Google Scholar]
- Coburn CM, Bargmann CI. A putative cyclic nucleotide-gated channel is required for sensory development and function in C. elegans. Neuron. 1996;17:695–706. doi: 10.1016/s0896-6273(00)80201-9. [DOI] [PubMed] [Google Scholar]
- Craig AD. How do you feel? Interoception: the sense of the physiological condition of the body. Nature reviews. Neuroscience. 2002;3:655–666. doi: 10.1038/nrn894. [DOI] [PubMed] [Google Scholar]
- Davis RE, Swiderski RE, Rahmouni K, Nishimura DY, Mullins RF, Agassandian K, Philp AR, Searby CC, Andrews MP, Thompson S, et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19422–19427. doi: 10.1073/pnas.0708571104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entchev EV, Patel DS, Zhan M, Steele AJ, Lu H, Ch'ng Q. A gene-expression-based neural code for food abundance that modulates lifespan. eLife. 2015;4:e06259. doi: 10.7554/eLife.06259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappell PB, Dotta A, Mortola JP. Metabolism during normoxia, hyperoxia, and recovery in newborn rats. Canadian journal of physiology and pharmacology. 1992;70:408–411. doi: 10.1139/y92-051. [DOI] [PubMed] [Google Scholar]
- Gray JM, Karow DS, Lu H, Chang AJ, Chang JS, Ellis RE, Marletta MA, Bargmann CI. Oxygen sensation and social feeding mediated by a C. elegans guanylate cyclase homologue. Nature. 2004;430:317–322. doi: 10.1038/nature02714. [DOI] [PubMed] [Google Scholar]
- Jansen G, Thijssen KL, Werner P, van der Horst M, Hazendonk E, Plasterk RH. The complete family of genes encoding G proteins of Caenorhabditis elegans. Nature genetics. 1999;21:414–419. doi: 10.1038/7753. [DOI] [PubMed] [Google Scholar]
- Jo H, Shim J, Lee JH, Lee J, Kim JB. IRE-1 and HSP-4 contribute to energy homeostasis via fasting-induced lipases in C. elegans. Cell metabolism. 2009;9:440–448. doi: 10.1016/j.cmet.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Kniazeva M, Crawford QT, Seiber M, Wang CY, Han M. Monomethyl branched-chain fatty acids play an essential role in Caenorhabditis elegans development. PLoS biology. 2004;2:E257. doi: 10.1371/journal.pbio.0020257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeva M, Sieber M, McCauley S, Zhang K, Watts JL, Han M. Suppression of the ELO-2 FA elongation activity results in alterations of the fatty acid composition and multiple physiological defects, including abnormal ultradian rhythms, in Caenorhabditis elegans. Genetics. 2003;163:159–169. doi: 10.1093/genetics/163.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam TK, Pocai A, Gutierrez-Juarez R, Obici S, Bryan J, Aguilar-Bryan L, Schwartz GJ, Rossetti L. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nature medicine. 2005;11:320–327. doi: 10.1038/nm1201. [DOI] [PubMed] [Google Scholar]
- Lee BH, Liu J, Wong D, Srinivasan S, Ashrafi K. Hyperactive neuroendocrine secretion causes size, feeding, and metabolic defects of C. elegans Bardet-Biedl syndrome mutants. PLoS biology. 2011;9:e1001219. doi: 10.1371/journal.pbio.1001219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux GA, Ashrafi K. Neural Regulatory Pathways of Feeding and Fat in Caenorhabditis elegans. Annual review of genetics. 2015;49:413–438. doi: 10.1146/annurev-genet-120213-092244. [DOI] [PubMed] [Google Scholar]
- Libert S, Pletcher SD. Modulation of longevity by environmental sensing. Cell. 2007;131:1231–1234. doi: 10.1016/j.cell.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Liu J, Ward A, Gao J, Dong Y, Nishio N, Inada H, Kang L, Yu Y, Ma D, Xu T, et al. C. elegans phototransduction requires a G protein-dependent cGMP pathway and a taste receptor homolog. Nature neuroscience. 2010;13:715–722. doi: 10.1038/nn.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Cai D. Counterbalance between BAG and URX neurons via guanylate cyclases controls lifespan homeostasis in C. elegans. The EMBO journal. 2013;32:1529–1542. doi: 10.1038/emboj.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mighiu PI, Yue JT, Filippi BM, Abraham MA, Chari M, Lam CK, Yang CS, Christian NR, Charron MJ, Lam TK. Hypothalamic glucagon signaling inhibits hepatic glucose production. Nature medicine. 2013;19:766–772. doi: 10.1038/nm.3115. [DOI] [PubMed] [Google Scholar]
- Mok CA, Healey MP, Shekhar T, Leroux MR, Heon E, Zhen M. Mutations in a guanylate cyclase GCY-35/GCY-36 modify Bardet-Biedl syndrome-associated phenotypes in Caenorhabditis elegans. PLoS genetics. 2011;7:e1002335. doi: 10.1371/journal.pgen.1002335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mykytyn K, Nishimura DY, Searby CC, Shastri M, Yen HJ, Beck JS, Braun T, Streb LM, Cornier AS, Cox GF, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nature genetics. 2002;31:435–438. doi: 10.1038/ng935. [DOI] [PubMed] [Google Scholar]
- Noble T, Stieglitz J, Srinivasan S. An Integrated Neurotransmitter Circuit in C. elegans Controls Body Fat via an Instructive Neuroendocrine Signal to Control C. elegans Body Fat. Cell metabolism. 2013;18:672–684. doi: 10.1016/j.cmet.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Horikawa M, Shimamura S, Hashimoto T, Sakamoto K. Fat accumulation in Caenorhabditis elegans is mediated by SREBP homolog SBP-1. Genes & nutrition. 2010;5:17–27. doi: 10.1007/s12263-009-0157-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson A, Gross E, Laurent P, Busch KE, Bretes H, de Bono M. Natural variation in a neural globin tunes oxygen sensing in wild Caenorhabditis elegans. Nature. 2009;458:1030–1033. doi: 10.1038/nature07820. [DOI] [PubMed] [Google Scholar]
- Salway JG. Metabolism at a glance. Blackwell Science; 1999. [Google Scholar]
- Schrodel T, Prevedel R, Aumayr K, Zimmer M, Vaziri A. Brain-wide 3D imaging of neuronal activity in Caenorhabditis elegans with sculpted light. Nature methods. 2013;10:1013–1020. doi: 10.1038/nmeth.2637. [DOI] [PubMed] [Google Scholar]
- Scott K. Out of thin air: sensory detection of oxygen and carbon dioxide. Neuron. 2011;69:194–202. doi: 10.1016/j.neuron.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S. Regulation of Body Fat in Caenorhabditis elegans. Annual review of physiology. 2015;77:161–178. doi: 10.1146/annurev-physiol-021014-071704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Sadegh L, Elle IC, Christensen AG, Faergeman NJ, Ashrafi K. Serotonin regulates C. elegans fat and feeding through independent molecular mechanisms. Cell metabolism. 2008;7:533–544. doi: 10.1016/j.cmet.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvia DM, Fuhrmann JJ, Hartel PG, Zuberer DA. Principles and Applications of Soil Microbiology. Prentice Hall; Upper Saddle River, New Jersey: 1998. [Google Scholar]
- Van Voorhies WA, Ward S. Broad oxygen tolerance in the nematode Caenorhabditis elegans. The Journal of experimental biology. 2000;203:2467–2478. doi: 10.1242/jeb.203.16.2467. [DOI] [PubMed] [Google Scholar]
- Wang PY, Caspi L, Lam CK, Chari M, Li X, Light PE, Gutierrez-Juarez R, Ang M, Schwartz GJ, Lam TK. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature. 2008;452:1012–1016. doi: 10.1038/nature06852. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Wingrove JA, O'Farrell PH. Nitric oxide contributes to behavioral, cellular, and developmental responses to low oxygen in Drosophila. Cell. 1999;98:105–114. doi: 10.1016/S0092-8674(00)80610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yemini E, Jucikas T, Grundy LJ, Brown AE, Schafer WR. A database of Caenorhabditis elegans behavioral phenotypes. Nature methods. 2013;10:877–879. doi: 10.1038/nmeth.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer M, Gray JM, Pokala N, Chang AJ, Karow DS, Marletta MA, Hudson ML, Morton DB, Chronis N, Bargmann CI. Neurons detect increases and decreases in oxygen levels using distinct guanylate cyclases. Neuron. 2009;61:865–879. doi: 10.1016/j.neuron.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.