

Abstract
Localized aggressive periodontitis (LAP) is a distinct form of early-onset periodontitis linked to periodontal infection with uncontrolled inflammation and leukocyte-mediated tissue destruction. The resolution of inflammation is an active process orchestrated by specialized proresolving lipid mediators (SPMs). Since the level of the Maresin pathway marker 14-hydroxy-docosahexaenoic acid (14-HDHA) was lower in activated peripheral blood from LAP patients, we investigated the Maresin 1 (MaR1) biosynthetic pathway in these subjects and its role in regulating phagocyte functions. Macrophages from LAP patients had a lower level of expression of 12-lipoxygenase (∼30%) and reduced MaR1 (LAP versus healthy controls [HC], 87.8 ± 50 pg/106 cells versus 239.1 ± 32 pg/106 cells). Phagocytosis by LAP macrophages was reduced ∼40% compared to that of HC, and killing of periodontal pathogens, including Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans, were similarly reduced. LAP neutrophils also displayed slower kinetics (∼30%) and decreased maximal phagocytosis (∼20% lower) with these pathogens than those of HC. The administration of MaR1 at 1 nM enhanced phagocytosis (31 to 65% increase), intracellular antimicrobial reactive oxygen species production (26 to 71% increase), bacterial killing of these periodontal pathogens (22 to 38% reduction of bacterial titers), and restored impairment of LAP phagocytes. Together, these results suggest that therapeutics targeting the Maresin pathway have clinical utility in treating LAP and other oral diseases associated with infection, inflammation, and altered phagocyte functions.
INTRODUCTION
Resolution of inflammation is an active process orchestrated by specialized proresolving lipid mediators (SPMs) derived from essential fatty acids (1). During self-resolving inflammation, there is a temporal lipid mediator (LM) class switch from classic proinflammatory lipid mediators, such as leukotrienes (LT) and prostaglandins (PG), to SPMs that include lipoxins (LX), resolvins (Rv), protectins (PD), and maresins (MaR) (2, 3). SPMs counterregulate acute inflammation via separate but related anti-inflammatory and proresolving actions in diverse experimental animal disease models in vivo (4). Failure to resolve acute inflammation may result in chronic inflammation, fibrosis, tissue damage, and loss of organ function (1, 5). It is well appreciated that uncontrolled inflammation is a unifying factor in the pathogenesis of several chronic inflammatory diseases, including cardiovascular, lung, joint (3–5), and periodontal diseases (6). Thus, dysregulated biosynthesis of proresolving lipid mediators may underlie part of the pathogenesis of uncontrolled inflammation (1) and oral inflammation (6).
Localized aggressive periodontitis (LAP) is a clinically distinct form of rapidly progressing periodontal disease linked to abnormal leukocyte-mediated tissue destruction with infection and uncontrolled inflammation (7). The sites with periodontal pockets in LAP patients harbor higher levels of periodontal pathogens, including Porphyromonas gingivalis (8) and Aggregatibacter actinomycetemcomitans (9, 10). Earlier results indicate that activated peripheral blood from LAP patients exhibits an imbalanced lipid mediator profile with higher production of proinflammatory mediators, including leukotriene B4 (LTB4; 5S,12R-dihydroxy-6Z,8E,10E,14Z-eicosatetraenoic acid), and lower levels of proresolving mediators, including lipoxin A4 (LXA4; 5S,6R,15S-trihydroxyeicosa-7E,9E,11Z,13E-tetraenoic acid). Of note, levels of 14-hydroxy-docosahexaenoic acid (14-HDHA; 14S-hydroxy-4Z,7Z,10Z,12E,16Z,19Z-DHA), a marker of the Maresin 1 biosynthetic pathway, are reduced in activated peripheral blood from LAP patients compared to those from healthy control donors (11, 12). Therefore, LAP may serve as a model human disease to investigate the biosynthesis and actions of Maresin 1 in resolving inflammation and infection.
Maresin 1 (MaR1) was first identified in macrophages as a potent proresolving lipid mediator (12) and recently was identified in several human tissues (13). Total organic synthesis was achieved, and the complete stereochemistry of bioactive MaR1 was established as 7R,14S-dihydroxydocosa-4Z,8E,10E,12Z,16Z,19Z-hexaenoic acid (14). In addition to its proresolving actions, MaR1 also promotes tissue regeneration and relieves inflammatory neuropathic pain (14). Along these lines, we recently cloned human macrophage 12-lipoxygenase (12-LOX) that proved identical to the human platelet 12-LOX in nucleotide sequence and demonstrated its initiating role in MaR1 biosynthesis (15, 16). In the present study, we investigated the MaR1 biosynthetic pathway in leukocytes from LAP patients and its role in regulating phagocyte functions in the containment and killing of periodontal pathogens.
MATERIALS AND METHODS
Human samples.
Human samples were obtained following informed consent approved by the Forsyth Institute Review Board (FIRB) (protocol number 11-05). Venous blood (∼60 ml; 25 U/ml heparin) was collected from patients with a diagnosis of localized aggressive periodontitis (LAP) according to the American Academy of Periodontology (AAP) guidelines (17) and from age- and gender-matched healthy volunteers (HC) with no signs of periodontal disease. All blood donors otherwise were healthy nonsmokers and had denied taking any nonsteroidal anti-inflammatory drugs (NSAIDs) for at least 2 weeks prior to the experiment. LAP subjects usually aggregate in families and present with severe, early-onset bone loss around first molars and incisor teeth (17, 18). The LAP patient cohort was characterized by a hyperresponsive neutrophil phenotype (elevated LTB4 and IL-8-induced superoxide generation) (18). The patients were diagnosed by a licensed periodontist (H. Hasturk) in the Center for Clinical and Translational Research at the Forsyth Institute. In addition, macrophages were prepared from peripheral blood monocytes donated from healthy human volunteers at the Children's Hospital, Boston, for independent experiments, including the dose response of MaR1 in enhancing macrophage phagocytosis and intracellular reactive oxygen species (ROS) generation as described below.
Human neutrophil and monocyte isolation and macrophage culture.
Neutrophils and peripheral blood mononuclear cells (PBMCs) were separated from human whole blood by Ficoll-Histopaque density gradient centrifugation (Histopaque 1077 and 1119; Sigma-Aldrich). Neutrophils were isolated after isotonic lysis of red blood cells followed by two washes in phosphate-buffered saline (Sigma-Aldrich). Monocytes were obtained after adhesion purification and incubated with RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% (vol/vol) fetal bovine serum (FBS) (Life Technologies) in the presence of human recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF; R&D Systems) (20 ng/ml, 7 days, 37°C, pH 7.4) for macrophage differentiation, which were used throughout the experiments unless otherwise specified. For lipid mediator metabololipidomics (13), monocytes were cultured in phenol red-free RPMI 1640 medium supplemented with 10% human serum (Lonza) as described previously (14).
Oral bacterial preparation.
Porphyromonas gingivalis (A7436) was cultured on 2% Trypticase soy agar (TSA) supplemented with 2.6% brain heart infusion agar (BD BBL), 1% (wt/vol) yeast extract (BD Bacto), 5% defibrinated sheep red blood cells (Northeast Laboratory Services), 5 μg/ml of hemin, and 0.5 μg/ml vitamin K (Sigma-Aldrich). Aggregatibacter actinomycetemcomitans (ATCC Y4), Fusobacterium nucleatum subsp. nucleatum (ATCC 25586), and Streptococcus intermedius (ATCC 27335) were grown on TSA plates (BD Diagnostic Systems). All cultures were placed in an anaerobic chamber (85% N2, 10% CO2, 5% H2 at 37°C). Colonies were transferred from the plate to respective broth media, namely, P. gingivalis into Wilkin's broth (Oxoid) and the others into brain heart infusion broth (BD BBL), and grown for 4 days. Bacterial titers were determined at 600 nm using a spectrometer (SmartSpec 3000; Bio-Rad) and adjusted to an OD of 1.0 (approximately 109 CFU/ml) prior to experiments.
Expression of surface markers and 12-LOX in human macrophages.
Macrophages from LAP patients and HC were harvested, fixed, and stained with CD54, CD206, and HLA-DR antibodies (Ab) (clones HCD54, 19.2, and G46-6; BD Biosciences). For 12-lipoxygenase (12-LOX) expression, intracellular staining was carried out using rabbit anti-human 12-LOX Ab (Novus) and Alexa Fluor-conjugated goat anti-rabbit Ab (Jackson) after cell permeabilization (Cytofix/Cytoperm; BD Biosciences) as described previously (16). Expression levels of the proteins were monitored by flow cytometry (FACSCanto II; BD Biosciences) and analyzed with FlowJo (TreeStar).
LC-MS/MS-based LM metabololipidomics.
All samples for liquid chromatography-tandem mass spectroscopy (LC-MS/MS) analysis were subjected to solid-phase extraction (13). Briefly, 1-ml macrophage incubations were stopped by adding two volumes of ice-cold methanol with deuterium-labeled internal standards d8-5S-hydroxyeicosatetraenoic acid, d4-leukotriene B4, d5-lipoxin A4, d5-resolvin D2, and d4-prostaglandin E2 (500 pg each) to facilitate quantification and sample recovery. Supernatants were loaded onto a C18 column following centrifugation and protein precipitation. LMs were eluted with the methyl formate fraction, dried, and resuspended in methanol-water (50:50, vol/vol) prior to injection into the LC-MS/MS system QTrap 6500 (ABSciex), as detailed previously (13).
Maresin 1 preparation.
MaR1 was prepared by total organic synthesis as described in reference 14 as part of the NIH Program Project P01-GM095467 (C. N. Serhan), and additional MaR1 for these experiments was purchased from Cayman Chemical. The compound was validated with LC-MS/MS and the concentration calibrated using UV absorption at a maximum wavelength of 271 nm before experiments.
Macrophage and neutrophil phagocytosis in vitro.
GM-CSF-differentiated human macrophages (5 ×104 cells/well) or neutrophils (105 cells/well) were adhered onto 96-well plates and incubated with vehicle (phosphate-buffered saline containing Ca2+ and Mg2+, termed PBS+/+; Sigma-Aldrich) or the indicated concentration of MaR1 for 15 min, followed by incubation with fluorescence-labeled bacteria (BacLight Green; Molecular Probes) (phagocyte-to-bacterium ratio, 1:50) or fluorescent microspheres (FluoSpheres; Molecular Probes) (phagocyte-to-particle ratio, 1:10) for the indicated time (15 min to 3 h). Plates were washed 3 times with PBS+/+, extracellular fluorescence was quenched by Trypan blue (1:15 dilution), and phagocytosis was determined by a SpectraMax M3 fluorescent plate reader (Molecular Devices). For direct comparison of macrophage phagocytosis between LAP patients and HC, macrophages (2 × 104 cells/well) were used under essentially the same conditions as those described above and subjected to flow cytometry to determine the mean florescence intensity (MFI).
Macrophage and neutrophil intracellular reactive oxygen species generation.
Macrophages (5 ×104 cells/well) and neutrophils (105 cells/well) were preincubated with the fluorescent intracellular ROS indicator carboxy-H2DCF-DA (reactive oxygen species detection reagents; Invitrogen) for 30 min and washed. The cells then were incubated with vehicle (PBS+/+; pH 7.4) or the indicated concentration of MaR1 for 15 min, followed by the addition of bacteria (phagocyte-to-bacterium ratio, 1:50) for 1 h. Intracellular ROS generation was determined using a SpectraMax M3 fluorescent plate reader (Molecular Devices).
Transmission electron microscopy (TEM).
Freshly isolated neutrophils (106 cells) from LAP patients and HC were incubated with P. gingivalis (polymorphonuclear leukocyte [PMN]-to-bacterium ratio of 1:50) for 1 h; cells were fixed with formaldehyde-glutaraldehyde-picric acid (FGP) and submitted to the Electron Microscopy Core Facility (Harvard Medical School) for further processing and analysis.
Bacterial killing by macrophages.
Macrophages (5 × 104 cells/well) from HC and LAP patients were incubated with the indicated concentration of MaR1 for 15 min, followed by the addition of bacteria (macrophage-to-bacterium ratio of 1:50) for 60 min. Supernatant was gently mixed, diluted with PBS, and plated (50 μl) onto respective agar plates to determine bacterial titers. Photos were taken after 7 days, and CFU were counted using ImageJ (NIH).
Statistical analysis.
Results are expressed as means ± standard errors of the means (SEM). Statistical analysis was performed using Student's t test (two independent groups) and one-way analysis of variance (ANOVA) (multiple groups). A P value of ≤0.05 was considered significant.
RESULTS
Macrophages from LAP patients display lower 12-lipoxygenase and reduced endogenous MaR1.
Since human 12-LOX is the key initiating enzyme in the biosynthesis of MaR1 (15, 16) (Fig. 1A), we first investigated the levels of 12-LOX in macrophages from LAP patients. These LAP macrophages exhibited significantly lower 12-LOX levels (∼30% reduction) than healthy control (HC) subjects (Fig. 1B). We next identified and quantified the endogenous levels of MaR1 and its biosynthetic pathway marker, 14-HDHA, in LAP and HC macrophage incubations using LC-MS/MS-based LM-metabololipidomics. Representative multiple reaction monitoring (MRM) chromatograms and MS/MS spectra for the identification of MaR1 are shown in Fig. 1C and D. MaR1 levels were significantly lower in the LAP macrophages than in HC (87.8 ± 50 pg/106 cells versus 239.1 ± 32 pg/106 cells; ∼65% reduction), with a reduction in its pathway marker, 14-HDHA (625 ± 251 pg/106 cells versus 935 ± 217 pg/106 cells; ∼30% reduction), that was not statistically significant (Fig. 1E). Taken together, these findings suggested that in LAP macrophages MaR1 biosynthesis is dysregulated.
FIG 1.

LAP macrophages have lower 12-LOX and reduced MaR1 levels. (A) Maresin 1 biosynthetic pathway (12, 15, 16). (B) Human 12-LOX was determined using flow cytometry in human macrophages from LAP patients (black bar) and matched healthy controls (HC; open bar) (see Materials and Methods for details). (Left) Results are expressed as percentages normalized to values for HC and are means ± SEM (n = 7 paired donors). **, P < 0.01 for LAP versus HC. (Right) Representative histograms. (C to E) 14-HDHA and MaR1 levels in HC and LAP macrophage incubations (RPMI with 20 ng/ml GM-CSF, 7 days, 37°C, pH 7.4) determined using LC-MS/MS-based LM-metabololipidomics (see Materials and Methods for details). (C) MRM chromatograms (MaR1, m/z 359/177; 14-HDHA, m/z 343/205). (D) Representative MS/MS spectrum from LAP macrophage incubations used for the identification of MaR1. (E) MaR1 and 14-HDHA levels are expressed as means ± SEM (n = 6 paired donors). *, P < 0.05 for LAP versus HC.
MaR1 restores impaired phagocytosis of P. gingivalis and A. actinomycetemcomitans in LAP patient macrophages.
Because macrophage clearance of bacteria is critical to control infection and resolution (2, 3) and MaR1 enhances macrophage phagocytosis of zymosan and apoptotic PMN (12, 14), we questioned whether MaR1 could enhance the phagocytosis of two periodontal pathogens, namely, P. gingivalis and A. actinomycetemcomitans. Indeed, MaR1 dose dependently (10 pM to 100 nM) enhanced the phagocytosis of both bacteria (∼10 to 35% above vehicle) by macrophages from healthy donors (Fig. 2A). Since macrophages from LAP patients had lower MaR1 levels, we next investigated their ability to phagocytize these periodontal pathogens. Macrophages from LAP patients exhibited an approximately 40% reduction in phagocytosis of both P. gingivalis and A. actinomycetemcomitans compared to the levels for HC at 1 h and a similar ratio at 3 h (Fig. 2B). When MaR1 (0.1 to 10 nM) was incubated with macrophages from LAP patients, a statistically significant increase in phagocytosis of P. gingivalis (∼37% at 1 nM) and A. actinomycetemcomitans (∼65% at 1 nM) was observed, reaching levels that were comparable to those obtained with macrophages from HC (Fig. 2C). These results demonstrate that MaR1 enhances macrophage phagocytosis of P. gingivalis and A. actinomycetemcomitans, rescuing the phagocytic function that also was observed to be impaired in macrophages from LAP patients.
FIG 2.
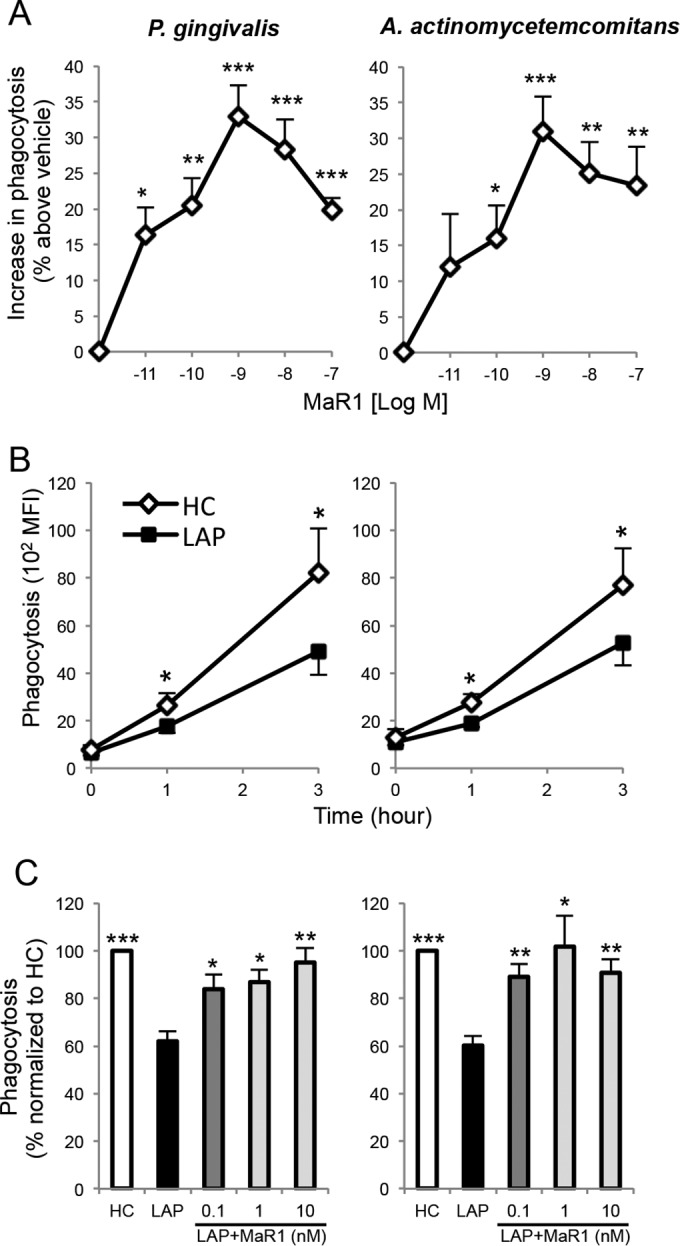
MaR1 enhances phagocytosis of P. gingivalis and A. actinomycetemcomitans and restores diminished LAP macrophage phagocytosis. (A) Macrophages from healthy donors (5 × 104 cells/well) were incubated (37°C) with vehicle (PBS+/+; pH 7.4) or MaR1 (10 pM to 100 nM) for 15 min prior to addition of fluorescent-labeled bacteria (Mϕ-to-bacterium ratio of 1:50) for 60 min. Phagocytosis was assessed using a fluorescence plate reader. Results are expressed as percent increase above the level for the vehicle; results are means ± SEM from n = 6 to 7 healthy donors. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) versus vehicle alone. (B and C) Macrophages (peripheral monocytes were collected and incubated with RPMI and 20 ng/ml GM-CSF; see Materials and Methods for details) obtained from LAP patients and matched HC (2 × 104 cells/well) were incubated with the fluorescence-labeled bacteria (1:50) for the indicated time intervals, and phagocytosis was assessed using flow cytometry. (B) Results are expressed as mean fluorescence intensity (MFI); results are means ± SEM from n = 4 to 6 paired donors. *, P < 0.05 for HC versus LAP. (C) Macrophages were incubated (37°C) with vehicle (PBS+/+; pH 7.4) or MaR1 (0.1 to 10 nM) for 15 min, followed by incubation with the fluorescence-labeled bacteria for 60 min. Results are expressed as percentages normalized to values for HC; results are means ± SEM from n = 3 to 6 paired donors (n = 6 for vehicle alone and n = 3 to 4 for MaR1 dose response with LAP macrophages). P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) versus LAP plus vehicle.
MaR1 rescues impaired LAP macrophage clearance of both P. gingivalis and A. actinomycetemcomitans by enhancing intracellular antimicrobial ROS.
We next determined if macrophages from LAP patients have diminished bacterial killing. For these experiments, live bacteria were incubated with macrophages from HC and LAP patients, and the supernatants were collected and cultured to determine bacterial titers. Supernatants from LAP macrophages gave higher bacterial titers of both P. gingivalis (26% higher) and A. actinomycetemcomitans (14% higher) than macrophages from HC (Fig. 3). The incubation of macrophages from HC and LAP patients with MaR1 (1 nM) significantly reduced bacterial titers of both of P. gingivalis and A. actinomycetemcomitans. Since MaR1 enhanced macrophage uptake and clearance of these pathogens, we next questioned whether LAP macrophages produce less intracellular ROS following the engulfment of these bacteria (18, 19) and whether MaR1 could enhance ROS production. To address this, macrophages were incubated with a fluorescent intracellular ROS indicator followed by P. gingivalis and A. actinomycetemcomitans in the presence of either MaR1 or vehicle (see Materials and Methods for details). MaR1 dose dependently enhanced macrophage intracellular ROS generation with both pathogens (see Fig. S1A in the supplemental material). Intracellular ROS levels in macrophages from HC and LAP patients without bacteria were not statistically different (see Fig. S1B, inset). The addition of bacteria resulted in an ∼20 to 30% increase in intracellular ROS. MaR1 (1 nM) enhanced intracellular ROS production in both HC and LAP macrophages with greater increases in incubations with A. actinomycetemcomitans (33% increase above vehicle) than with P. gingivalis (see Fig. S1B). These results indicated that intracellular ROS production is not reduced in LAP macrophage and that MaR1 further increases ROS in both LAP and HC macrophage when incubated with oral pathogens (P. gingivalis and A. actinomycetemcomitans).
FIG 3.
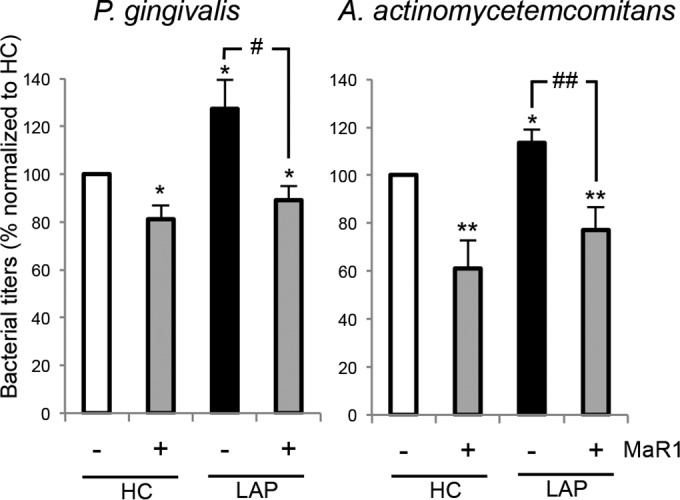
MaR1 rescues diminished LAP macrophage killing of P. gingivalis and A. actinomycetemcomitans. Macrophages (5 × 104 cells/well) from HC and LAP patients were incubated with vehicle (PBS+/+) or MaR1 (1 nM) for 15 min, followed by addition of bacteria (Mϕ-to-bacterium ratio, 1:50) for 60 min. Serial dilutions of the incubations were plated onto agar plates for 7 days. Bacterial titers were counted using ImageJ. Results are expressed as percentages normalized to values for HC (100%); results are means ± SEM for n = 6 paired donors. P < 0.05 (*) and P < 0.01 (**) versus HC plus vehicle. P < 0.05 (#) and P < 0.01 (##) for MaR versus vehicle with LAP.
Neutrophils from LAP patients display generalized impairment in phagocytosis.
Since neutrophils play an important role in the pathogenesis of LAP (7, 18), we next investigated whether neutrophils from the LAP patients also display impaired phagocytosis of P. gingivalis and A. actinomycetemcomitans. By incubating fluorescence-labeled bacteria with the neutrophils, it was apparent that LAP neutrophils exhibited impaired phagocytosis, with slower kinetics from 0 to 2 h and reduced maximal phagocytosis (Fig. 4A). We next questioned whether this abnormality is specific to certain bacteria by testing F. nucleatum, S. intermedius, and latex particles. LAP neutrophils all gave reduced phagocytosis (Fig. 4B). We next determined whether LAP neutrophils exhibited different levels of impairment with different oral bacteria. A comparison of the kinetics of bacterial phagocytosis demonstrated that LAP neutrophils exhibited a 51% reduction in the rate of phagocytosis with F. nucleatum, 32% reduction with P. gingivalis, 31% with A. actinomycetemcomitans, and 23% with S. intermedius compared to levels for HC neutrophils (Fig. 4C). Similar results were obtained when we assessed the extent of phagocytosis at 60 min, revealing that LAP neutrophils phagocytized significantly less F. nucleatum than S. intermedius compared to phagocytosis by HC (Fig. 4D). We also found reduced maximal phagocytosis with LAP neutrophils, with a 46% reduction in F. nucleatum and ∼20% reduction in the extent of phagocytosis for other bacteria assessed compared to values obtained with HC neutrophils (Fig. 4D). These results indicate that LAP neutrophils, compared to HC, display generalized impairment in phagocytosis, yet a differential response may exist for F. nucleatum.
FIG 4.

LAP neutrophils exhibit generalized diminished phagocytosis. (A to D) Isolated human PMNs (105 cells/well) were incubated with fluorescence-labeled P. gingivalis, A. actinomycetemcomitans, F. nucleatum, S. intermedius (PMN-to-bacterium ratio, 1:50), or latex particles (PMN-to-particle ratio, 1:10) for the indicated time intervals. Phagocytosis was determined using a fluorescence plate reader. (A and B) Results are expressed as relative fluorescence units (RFU) and are means ± SEM for n = 3 to 6 paired donors. *, P < 0.05 for HC versus LAP. (C) The rate of phagocytosis (from 0 h to maximal phagocytosis) was calculated for each experiment and is represented as the rate of phagocytosis for each species of bacterium. Results are expressed as percent reduction in level for LAP compared to that for HC; results are means ± SEM for n = 3 to 4 paired donors. P < 0.05 (*) and P < 0.01 (**) for LAP versus HC. (D) The extent of phagocytosis after 1 h and the maximal values from both LAP and HC were obtained. Results are expressed as percent reduction of the level for LAP compared to that for HC; results are means ± SEM from n = 3 to 6 paired donors. P < 0.05 (*) and P < 0.01 (**) for LAP versus HC; #, P < 0.05 for HC versus LAP plus F. nucleatum. Abbreviations: P. g, P. gingivalis; A. a, A. actinomycetemcomitans; F. n, F. nucleatum; S. i, S. intermedius.
MaR1 partially restores LAP neutrophil phagocytosis of P. gingivalis and A. actinomycetemcomitans.
We next questioned whether MaR1 could act on neutrophils to rescue phagocytic functions in neutrophils from LAP patients. The incubation of neutrophils with MaR1 dose dependently enhanced neutrophil phagocytosis of P. gingivalis (∼42% increase above vehicle at 1 nM) and A. actinomycetemcomitans (∼46% increase above vehicle at 1 nM) with both LAP and HC neutrophils (Fig. 5) and partially restores the defect in LAP neutrophils above 1 pM concentrations. In addition, MaR1 dose dependently enhanced both LAP and HC neutrophil intracellular ROS generation with both pathogens (see Fig. S3 in the supplemental material). These results indicated that MaR1 enhances neutrophil intracellular ROS generation and partially restores LAP neutrophil-impaired phagocytosis of P. gingivalis and A. actinomycetemcomitans.
FIG 5.

MaR1 partially restores LAP neutrophil-impaired phagocytosis of P. gingivalis and A. actinomycetemcomitans. (A) Isolated PMNs (105 cells/well) from LAP patients and HC were incubated (37°C) with MaR1 (0.1 pM to 100 nM) or vehicle (PBS+/+; pH 7.4) alone for 15 min prior to the addition of fluorescence-labeled bacteria (PMN-to-bacterium ratio, 1:50) for 60 min. Phagocytosis was determined using a fluorescence plate reader. Results are expressed as percent changes normalized to levels for HC; results are means ± SEM from n = 5 to 6 donors. P < 0.05 (#), P < 0.01 (##), and P < 0.001 (###) for HC plus MaR1 versus HC plus vehicle; P < 0.05 (*) and P < 0.01 (**) for LAP plus MaR1 versus LAP plus vehicle; P < 0.05 (§), P < 0.01 (§§), and P < 0.001 (§§§) for LAP versus HC plus vehicle. (B) Representative TEM images of LAP neutrophils with P. gingivalis. (Top) Phagocytosis of P. gingivalis. (Bottom) P. gingivalis within the phagolysosome.
Given the impaired phagocytosis of neutrophils from LAP patients, samples were taken for inspection using TEM. No apparent structural differences were observed between LAP and HC neutrophils or their phagocytic apparatus. Representative TEM images of LAP neutrophils after incubation with P. gingivalis are shown (Fig. 5).
DISCUSSION
In the present study, we investigated macrophage biosynthesis of MaR1 in cells from LAP patients using LM metabololipidomics and assessed the action of MaR1 in regulating phagocyte functions. We found that in cells from LAP patients, MaR1 biosynthesis is dysregulated and that their phagocytes (neutrophils and macrophages) display an impaired ability to phagocytize and kill the periodontal pathogens P. gingivalis and A. actinomycetemcomitans. Exogenous MaR1 partially restored this impairment in LAP neutrophils and macrophages, increasing intracellular ROS production, phagocyte ingestion, and clearance of the periodontal pathogens. In parallel incubations, MaR1 also enhanced these functions in phagocytes from healthy controls.
Maresin 1 biosynthesis in humans was established recently using recombinant human macrophage 12-LOX enzyme and synthetic intermediates (15, 16). It was demonstrated that 12-LOX is the key initiating enzyme that produces 14S-hydroperoxy-docosahexaenoic acid (14S-HpDHA) from the precursor docosahexaenoic acid (DHA) (15), which is known to be selectively stored in human and other mammalian tissues, including neural and lymphoid tissues (20, 21). The intermediate 14S-HpDHA then is converted via 12-LOX-mediated epoxidation to 13S,14S-epoxy-docosahexaenoic acid (13,14-epoxy-maresin; 13,14-eMaR), which is further converted to MaR1 by human macrophages (16) (Fig. 1A). 14-HpDHA also could be enzymatically reduced to 14-HDHA, the corresponding alcohol, which can be used as a marker of the Maresin biosynthetic pathway (12) and was reported to be lower in activated LAP whole blood (11). In the present experiments, we found that macrophages from LAP patients have lower 12-LOX (∼70%) and reduced levels of 14-HDHA (∼70%), as well as MaR1 (∼35%), compared to those of cells from healthy control donors (Fig. 1B and E). These results suggest that the MaR1 biosynthetic pathway is diminished in leukocytes in patients with localized aggressive periodontitis.
Bacterial clearance is critical for containing infection and its resolution, and MaR1 enhances macrophage phagocytosis of both zymosan and apoptotic PMN (12, 14); therefore, in the present study we assessed whether MaR1 could specifically enhance phagocytosis and clearance of the periodontal pathogens P. gingivalis and A. actinomycetemcomitans. MaR1 enhanced the phagocytosis of both bacteria in the concentrations identified from endogenous production (Fig. 1E). This observation led us to question whether LAP macrophages with lower levels of MaR1 exhibit impaired phagocytosis of these pathogens. Earlier, it was discovered that LAP neutrophils display defective phagocytosis of Staphylococcus aureus (22, 23). Recently, macrophages from LAP patients also were found to display impaired phagocytosis of zymosan that was rescued with Resolvin E1 (11). It was unknown whether these phagocytes also display reduced phagocytosis of periodontal pathogens. In the present report, we found impaired phagocytosis of both P. gingivalis and A. actinomycetemcomitans by phagocytes (neutrophils and macrophages) from LAP patients (Fig. 2B and 4A). Of note, LAP macrophages did not express different levels of surface markers for polarized phenotypes (CD54, CD2016, and HLA-DR; n = 4 paired donors; data not shown). The incubation of HC and LAP macrophages with periodontal pathogens (cell-to-bacterium ratio of 1:50) for 1 h did not initiate higher levels of MaR1. Instead, the levels of MaR1 were reduced similarly in both HC and LAP macrophages, indicating the pathogens did not alter the biosynthesis differently between the two groups, and MaR1 may be rapidly metabolized or its biosynthesis inhibited under these conditions (see Fig. S5 in the supplemental material). Importantly, the addition of exogenous MaR1 normalized the impaired phagocytosis in LAP phagocytes and enhanced the clearance ability of LAP macrophages (Fig. 2C, 3, and 5).
It is now widely appreciated that periodontal disease is a polymicrobial infection modulated by host response (24). Besides P. gingivalis and A. actinomycetemcomitans, there is a growing body of evidence implicating the potential role of other commensal oral bacteria in the biofilm. S. intermedius is a Gram-positive, facultative anaerobic commensal oral bacterium that has an antagonistic relationship with periodontal pathogens like P. gingivalis and A. actinomycetemcomitans (previously known as Bacteroides gingivalis and Actinobacillus actinomycetemcomitans in the literature [25]). Additionally, F. nucleatum recently has been implicated as a potential target pathogen (26). F. nucleatum also is prevalent within deep periodontal pockets of LAP patients (27), and its abundance in the biofilm is associated with the severity of periodontal inflammation in adolescents (28). In addition, F. nucleatum is essential for the growth of P. gingivalis and A. actinomycetemcomitans (29), and it can synergistically create a stronger inflammatory response with more tissue destruction in coinfections with P. gingivalis in a mouse periodontitis model (30). In the current study, we investigated LAP neutrophil phagocytosis of four oral bacteria and compared results to those for HC. The LAP neutrophils exhibited generalized impairment in the phagocytosis of each of the four bacteria, giving lower rates of phagocytosis and reduced maximal phagocytosis. The most prominent deficiency was in the extent of phagocytosis of F. nucleatum (42% reduction), followed by A. actinomycetemcomitans (27% reduction), P. gingivalis (24% reduction), and S. intermedius (18% reduction), compared to that of HC after 1 h of incubation (Fig. 4C). It was reported earlier that LAP neutrophils exhibit 10 to 20% reduced phagocytosis of Staphylococcus aureus at 1 h, which also coincided with the reduced rate of phagocytosis (23). In addition, the reduction in maximal phagocytosis with LAP neutrophils is also about 20% for the oral bacteria tested here, except for F. nucleatum at 46%. The results suggest that F. nucleatum plays an important role in dysbiosis (31) in patients with localized aggressive periodontitis, and the imbalanced kinetics suggest a mechanism for host-mediated disruption of biofilm homeostasis. Of note, in our experiments MaR1 enhanced the macrophage phagocytosis of F. nucleatum (∼30% increase at 1 nM) with cells from healthy donors (see Fig. S4 in the supplemental material).
Following phagocytosis, intracellular ROS generation is critical for oxidative bacterial killing within the phagocytes (19). Earlier, our group demonstrated that resolvins D1 and D2 (RvD1 and RvD2) and Protectin (PD1) increase intracellular ROS generation during human PMN ingestion of Escherichia coli (32, 33). In the present study, we report that MaR1 enhanced the phagocytosis of P. gingivalis and A. actinomycetemcomitans and also increased intracellular ROS generation within phagocytes from both HC and LAP (see Fig. S1 to S3 in the supplemental material). In addition, LAP neutrophils and macrophages produced levels of intracellular ROS similar to those of HC when incubated with both pathogens. However, under the same conditions, fewer bacteria were ingested by the LAP phagocytes, indicating that LAP phagocytes present with hyperresponsive intracellular ROS generation. It was reported earlier that LAP neutrophils release higher levels of extracellular ROS after activation (17), which is counterregulated and reduced by Resolvin E1 (RvE1) and aspirin-triggered lipoxin analog, members of other families of proresolving lipid mediators biosynthesized from eicosapentaenoic acid (EPA) and arachidonic acid (AA) (34). Topical RvE1 resolves inflammation and restores tissue homeostasis in a rabbit P. gingivalis-infected periodontitis model, demonstrating its protective role (35).
Taking our results together, we identified for the first time several functional defects in phagocytes obtained from LAP patients. They displayed impaired bacterial containment and killing along with reduced MaR1 production in the macrophages. The exogenous administration of MaR1 partially corrected this defect, enhancing the phagocytosis and killing of periodontal pathogens as well as stimulating intracellular ROS generation. Moreover, these results suggest that the MaR1 biosynthetic pathway underlies some of the pathogenesis in LAP and that new therapeutics targeting this Maresin biosynthetic and related pathways may have clinical utility in treating localized aggressive periodontitis and other oral diseases associated with bacterial infection, uncontrolled inflammation, and failed resolution.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yannis Koroneos (Forsyth Institute) for technical assistance in cell isolation, Maria Ericsson (Harvard Medical School core facility) for guidance in electron microscopy analysis, and Mary Halm Small for expert assistance in manuscript preparation.
This work was supported by National Institute of Dental and Craniofacial Research grants NIH/NIDCR DE018917 (to H.H.) and NIH/NIDCR DE015566 (to T.E.V.D.) as well as National Institute of General Medical Sciences grant NIH/NIGMS GM038765 (to C.N.S.).
The funders had no role in study design, data collection and interpretation, or decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01131-15.
REFERENCES
- 1.Serhan CN. 2014. Pro-resolving lipid mediators are leads for resolution physiology. Nature 510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. 2001. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol 2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 3.Serhan CN, Savill J. 2005. Resolution of inflammation: the beginning programs the end. Nat Immunol 6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 4.Serhan CN, Chiang N, Van Dyke TE. 2008. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathan C, Ding A. 2010. Nonresolving inflammation. Cell 140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 6.Van Dyke TE, Serhan CN. 2003. Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases. J Dent Res 82:82–90. doi: 10.1177/154405910308200202. [DOI] [PubMed] [Google Scholar]
- 7.Kantarci A, Oyaizu K, Van Dyke TE. 2003. Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. J Periodontol 74:66–75. doi: 10.1902/jop.2003.74.1.66. [DOI] [PubMed] [Google Scholar]
- 8.Lopez NJ, Mellado JC, Leighton GX. 1996. Occurrence of Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis and Prevotella intermedia in juvenile periodontitis. J Clin Periodontol 23:101–105. doi: 10.1111/j.1600-051X.1996.tb00541.x. [DOI] [PubMed] [Google Scholar]
- 9.Slots J. 1976. The predominant cultivable organisms in juvenile periodontitis. Scand J Dent Res 84:1–10. [DOI] [PubMed] [Google Scholar]
- 10.Mandell RL, Socransky SS. 1981. A selective medium for Actinobacillus actinomycetemcomitans and the incidence of the organism in juvenile periodontitis. J Periodontol 52:593–598. doi: 10.1902/jop.1981.52.10.593. [DOI] [PubMed] [Google Scholar]
- 11.Fredman G, Oh SF, Ayilavarapu S, Hasturk H, Serhan CN, Van Dyke TE. 2011. Impaired phagocytosis in localized aggressive periodontitis: rescue by resolvin E1. PLoS One 6:e24422. doi: 10.1371/journal.pone.0024422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, Oh SF, Spite M. 2009. Maresins: novel macrophage mediators with potent anti-inflammatory and pro-resolving actions. J Exp Med 206:15–23. doi: 10.1084/jem.20081880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. 2014. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol 307:C39–C54. doi: 10.1152/ajpcell.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serhan CN, Dalli J, Karamnov S, Choi A, Park CK, Xu ZZ, Ji RR, Zhu M, Petasis NA. 2012. Macrophage pro-resolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J 26:1755–1765. doi: 10.1096/fj.11-201442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng B, Wang CW, Arnardottir HH, Li Y, Cheng CY, Dalli J, Serhan CN. 2014. Maresin biosynthesis and identification of maresin 2, a new anti-inflammatory and pro-resolving mediator from human macrophages. PLoS One 9:e102362. doi: 10.1371/journal.pone.0102362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggstrom JZ, Petasis NA, Serhan CN. 2013. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J 27:2573–2583. doi: 10.1096/fj.13-227728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armitage GC. 1999. Development of a classification system for periodontal diseases and conditions. Ann Periodontol 4:1–6. doi: 10.1902/annals.1999.4.1.1. [DOI] [PubMed] [Google Scholar]
- 18.Gronert K, Kantarci A, Levy BD, Clish CB, Odparlik S, Hasturk H, Badwey JA, Colgan SP, Van Dyke TE, Serhan CN. 2004. A molecular defect in intracellular lipid signaling in human neutrophils in localized aggressive periodontal tissue damage. J Immunol 172:1856–1861. doi: 10.4049/jimmunol.172.3.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majno G, Joris I. 2004. Bacterial killing: metabolic aspect of phagocytosis, p 419–422. In Majno G, Joris I (ed), Cells, tissues, and disease: principles of general pathology, 2nd ed Oxford University Press, New York, NY. [Google Scholar]
- 20.Bazan NG. 1990. Supply of n-3 polyunsaturated fatty acids and their significance in the central nervous system, p 1–22. In Wurtman RJ, Wurtman JJ (ed), Nutrition and the brain, vol 8 Raven Press, New York, NY. [Google Scholar]
- 21.Calder PC. 2008. The relationship between the fatty acid composition of immune cells and their function. Prostaglandins Leukot Essent Fatty Acids 79:101–108. doi: 10.1016/j.plefa.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 22.Cainciola LJ, Genco RJ, Patters MR, McKenna J, van Oss CJ. 1977. Defective polymorphonuclear leukocyte function in a human periodontal disease. Nature 265:445–447. doi: 10.1038/265445a0. [DOI] [PubMed] [Google Scholar]
- 23.Van Dyke TE, Zinney W, Winkel K, Taufiq A, Offenbacher S, Arnold RR. 1986. Neutrophil function in localized juvenile periodontitis. Phagocytosis, superoxide production and specific granule release. J Periodontol 57:703–708. [DOI] [PubMed] [Google Scholar]
- 24.Darveau RP. 2010. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 25.Hillman JD, Socransky SS, Shivers M. 1985. The relationships between streptococcal species and periodontopathic bacteria in human dental plaque. Arch Oral Biol 30:791–795. doi: 10.1016/0003-9969(85)90133-5. [DOI] [PubMed] [Google Scholar]
- 26.Han YW. 2015. Fusobacterium nucleatum: a commensal-turned pathogen. Curr Opin Microbiol 23:141–147. doi: 10.1016/j.mib.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oettinger-Barak O, Sela MN, Sprecher H, Machtei EE. 2014. Clinical and microbiological characterization of localized aggressive periodontitis: a cohort study. Aust Dent J 59:165–171. doi: 10.1111/adj.12165. [DOI] [PubMed] [Google Scholar]
- 28.Yang NY, Zhang Q, Li JL, Yang SH, Shi Q. 2014. Progression of periodontal inflammation in adolescents is associated with increased number of Porphyromonas gingivalis, Prevotella intermedia, Tannerella forsythensis, and Fusobacterium nucleatum. Int J Paediatr Dent 24:226–233. doi: 10.1111/ipd.12065. [DOI] [PubMed] [Google Scholar]
- 29.Bradshaw DJ, Marsh PD, Watson GK, Allison C. 1998. Role of Fusobacterium nucleatum and coaggregation in anaerobe survival in planktonic and biofilm oral microbial communities during aeration. Infect Immun 66:4729–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polak D, Wilensky A, Shapira L, Halabi A, Goldstein D, Weiss EI, Houri-Haddad Y. 2009. Mouse model of experimental periodontitis induced by Porphyromonas gingivalis/Fusobacterium nucleatum infection: bone loss and host response. J Clin Periodontol 36:406–410. doi: 10.1111/j.1600-051X.2009.01393.x. [DOI] [PubMed] [Google Scholar]
- 31.Hajishengallis G. 2015. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 15:30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery TW, Schmidt BA, Serhan CN. 2012. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN. 2009. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, Petasis NA, Levy BD, Serhan CN, Van Dyke TE. 2006. RvE1 protects from local inflammation and osteoclast mediated bone destruction in periodontitis. FASEB J 20:401–403. [DOI] [PubMed] [Google Scholar]
- 35.Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, Van Dyke TE. 2007. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol 179:7021–7029. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.