

Abstract
African Pygmies practicing a mobile hunter-gatherer lifestyle are phenotypically and genetically diverged from other anatomically modern humans, and they likely experienced strong selective pressures due to their unique lifestyle in the Central African rainforest. To identify genomic targets of adaptation, we sequenced the genomes of four Biaka Pygmies from the Central African Republic and jointly analyzed these data with the genome sequences of three Baka Pygmies from Cameroon and nine Yoruba famers. To account for the complex demographic history of these populations that includes both isolation and gene flow, we fit models using the joint allele frequency spectrum and validated them using independent approaches. Our two best-fit models both suggest ancient divergence between the ancestors of the farmers and Pygmies, 90,000 or 150,000 yr ago. We also find that bidirectional asymmetric gene flow is statistically better supported than a single pulse of unidirectional gene flow from farmers to Pygmies, as previously suggested. We then applied complementary statistics to scan the genome for evidence of selective sweeps and polygenic selection. We found that conventional statistical outlier approaches were biased toward identifying candidates in regions of high mutation or low recombination rate. To avoid this bias, we assigned P-values for candidates using whole-genome simulations incorporating demography and variation in both recombination and mutation rates. We found that genes and gene sets involved in muscle development, bone synthesis, immunity, reproduction, cell signaling and development, and energy metabolism are likely to be targets of positive natural selection in Western African Pygmies or their recent ancestors.
Recent archaeological and genetic studies suggest that anatomically modern humans (AMH) originated in Africa prior to 160–190 thousand yr ago (kya) (Cavalli-Sforza et al. 1994; McDougall et al. 2005; Garrigan and Hammer 2006). Before the invention of agriculture in the Neolithic (∼6–10 kya), hunting and gathering was the subsistence strategy used by early human societies (Cavalli-Sforza 1986; Scheinfeldt et al. 2010; Hill et al. 2011). Among extant African human populations, the Pygmies, commonly identified by their short stature (mean adult height <160 cm), are one of the few that still predominantly practice a hunting and gathering lifestyle. Western Pygmies (e.g., Baka and Biaka) mainly reside in the rainforest west of the Congo Basin, whereas Eastern Pygmies (e.g., Mbuti and Efe) live in and around the Ituri rainforest and further south extending toward Lake Victoria (Cavalli-Sforza et al. 1994). Although still living as mobile hunter-gatherers, Pygmies have established social and economic contacts with nearby settled farmers (Cavalli-Sforza et al. 1994; Joiris 2003). For example, the Efe Pygmies trade forest food to Lese farmers in exchange for cultivated goods (Terashima 1987). Moreover, most Pygmies now speak Niger-Kordofanian (e.g., Bantu) or Nilo-Saharan languages, possibly acquired from neighboring farmers, especially since the expansion of Bantu-speaking agriculturalists beginning ∼5 kya (Blench 2006).
Recent genetic evidence favors a single origin of African Pygmies (Patin et al. 2009; Batini et al. 2011; Veeramah et al. 2012).Western Pygmies have likely experienced greater genetic admixture with neighboring farmer populations than Eastern Pygmies (Patin et al. 2009; Tishkoff et al. 2009; Veeramah et al. 2012; Verdu et al. 2013). Several mitochondrial and multilocus DNA studies estimated that African Pygmies diverged from the ancestors of present-day Niger-Cordofanian agriculturalists ∼60 kya (95% C.I.: 25–130 kya) (Patin et al. 2009), ∼70 kya (95% C.I.: 51–106 kya) (Batini et al. 2011), and ∼49 kya (95% C.I.: 10–105 kya) (Veeramah et al. 2012). However, because each of these studies used <60 loci, they either made strong a priori assumptions to restrict parameter space in their demographic modeling (Patin et al. 2009) or did not have sufficient statistical power to infer gene flow (Batini et al. 2011; Veeramah et al. 2012). Thus, a comprehensive understanding of the demographic prehistory of African Pygmies remains lacking.
Pygmy populations have long been studied because of their distinct phenotypes, particularly short stature. Physiological evidence suggests that short stature is associated with low growth hormone binding protein and insulin-like growth factor-1 (IGF1) levels in Pygmy groups (Baumann et al. 1989; Dávila et al. 2002). Using high-density SNP chip data, several population genetic studies have reported candidates for Pygmy short stature, including genes in the IGF1 pathway (Pickrell et al. 2009; Jarvis et al. 2012; Migliano et al. 2013), the iodine-dependent thyroid hormone pathway (López-Herráez et al. 2009; Migliano et al. 2013), and the bone homeostatsis/skeletal remodeling pathway (Mendizabal et al. 2012). Lachance et al. (2012) searched for signals of positive selection in five high-coverage Western Pygmy genomes and suggested that short stature may be due to selection on genes involved in development of the anterior pituitary, as well as the crosstalk between the adiponectin and insulin-signaling pathways. A more recent study using admixture mapping identified 16 regions associated with height in Batwa Pygmies, which were enriched for SNPs associated with height in Europeans and for genes with growth hormone receptor and regulation functions (Perry et al. 2014).
Several hypotheses have been proposed regarding Pygmy adaptation to the dense, humid forest environment, all of which may influence stature. These include thermoregulatory adaptation to the tropical forest (Cavalli-Sforza 1986), reduction of caloric intake in a food-limited environment (Shea and Bailey 1996), improved mobility in the dense forest (Diamond 1991), and earlier reproduction to compensate for short lifespans (Migliano et al. 2007). In addition, the equatorial rainforest in Central Africa is enriched in pathogens and parasites, such as malaria and haemorrhagic fever (Ohenjo et al. 2006). Loci involved in immunity have thus been suggested to be targets for adaptation (Jarvis et al. 2012; Lachance et al. 2012).
Although previous studies have identified many possible targets of adaptive selection in African Pygmies, challenges remain. First, demographic events and local genomic architecture (e.g., heterogeneity in mutation and recombination rates) can mimic the genetic patterns generated by adaptation (Schaffner et al. 2005; Teshima et al. 2006). High false positive and false negative rates are expected in studies that determine candidates of natural selection based solely on selecting outliers from the distribution of a test statistic (Jeffreys et al. 2005; Schaffner et al. 2005; Teshima et al. 2006; Akey 2009). In addition, the large genomic sizes of candidate regions (on the order of 100 kb), especially for those reported in SNP-microarray studies, make inference of the genetic basis of adaptation difficult.
Understanding genetic adaptation in African Pygmies, therefore, requires not only high-coverage whole-genome data, but also realistic demographic models to assess statistical significance. To provide a genomic perspective on adaptation in Pygmies, we sequenced four Western Biaka Pygmies from the Central African Republic and combined these data with similar data from three Baka Pygmies (Lachance et al. 2012) from Cameroon and nine unrelated Yoruba farmers. We inferred the demographic history of these populations and searched for positive selection using several complementary statistical methods. We assessed statistical significance in our selection scans using genome-scale simulations that incorporated recombination and mutation rate heterogeneity along the genome. Finally, we functionally annotated our candidates, and we discuss their biological impact. Our analysis thus provides unique insights into the complex demographic and adaptive history of Western African Pygmies.
Results
Demographic history inference for Western African Pygmies and farmers
We used the demographic inference tool ∂a∂i (Gutenkunst et al. 2009) to infer the joint demographic history of one farmer (Yoruba) and two Pygmy (Baka and Biaka) populations using our high coverage (median = 60.5×) Complete Genomics (CGI) (Drmanac et al. 2010) whole-genome data. After removing single-nucleotide variants (SNVs) that failed quality control (see Methods), we used 1.58 million intergenic autosomal SNVs to build a three-population unfolded allele frequency spectrum (AFS) (Methods), which we statistically corrected to account for ancestral state misidentification (Hernandez et al. 2007). We chose this statistical approach over obtaining consensus outgroup information from multiple primates for ancestral alleles because the latter causes a substantial reduction in our data and does not completely alleviate the problem of ancestral state misidentification (Hernandez et al. 2007). We also found that removing sites within functional ENCODE elements (Supplemental Material; Gerstein et al. 2012) had little effect on the resulting AFS (Supplemental Fig. S1), so we kept those sites in our analysis.
To guide development of three-population models, we first considered simpler one- and two-population models. These initial models consistently suggested a more recent divergence between the two Pygmy populations than between either of those populations and the farmers. Based on these results and previously published inferences (Patin et al. 2009; Batini et al. 2011; Veeramah et al. 2012; Verdu et al. 2013), we tested multiple three-population models, considering a variety of scenarios for gene flow and population size changes (Supplemental Table S1). The best-fit three-population demographic model, Model-1, had continuous asymmetric gene flow (composite log-likelihood = −6712) (Fig. 1A; Supplemental Table 1). The joint frequency spectra resulting from this model qualitatively reproduce the data (Fig. 1C), although our model does produce an excess of high-frequency shared variants. In Model-1, the ancestors of contemporary farmers and Pygmies diverged ∼156 kya (95% C.I.: 140–164 kya) from an ancestral population that had expanded roughly threefold prior to divergence. The ancestors of the farmers and Pygmies remained isolated until ∼40 kya (95% C.I.: 36–44 kya), at which point bidirectional gene flow began, with the flow from farmers to Pygmies being 10 times greater than from Pygmies to farmers (Table 1). Following the Pygmy-farmer divergence, the effective population size of farmers increased and the effective population size of Pygmies decreased. The Baka and Biaka diverged much more recently, ∼5 kya (95% C.I.: 4.7–5.7 kya). Because our small sample size limits power to infer recent demographic events (Robinson et al. 2014), we assumed that the Baka-Biaka divergence did not change the rates of gene flow with the Yoruba, and our model includes no Baka-Biaka gene flow.
Figure 1.

Best-fit demographic models and observed and predicted frequency spectra for African farmer (Yoruba) and Pygmy (Baka and Biaka) populations. (A) The continuous asymmetric gene flow model (Model-1) with the 10 free parameters labeled. (B) The single-pulse admixture model (Model-2) with the nine free parameters labeled. (C) The marginal spectra for each pair of populations. Row one is data, rows two (Model-1) and four (Model-2) are models, and rows three and five are Anscombe residuals of model minus data for Model-1 and Model-2, respectively.
Table 1.
Parameter estimates and confidence intervals for two best-fit demographic models
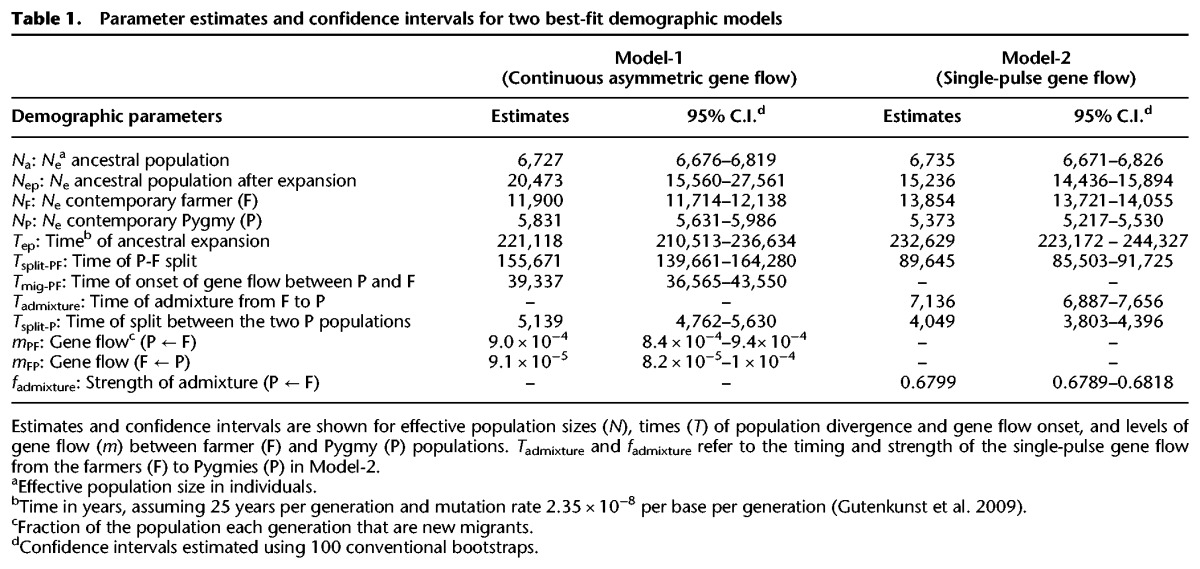
Our second best-fit model involves a recent pulse of unidirectional gene flow from farmers to Pygmies (Model-2) (Fig. 1B,C; Table 1) after the divergence of the two populations. The maximum composite log-likelihood of Model-2 (−7737) is lower than Model-1. In Model-2, we inferred that Pygmies and farmers diverged ∼90 kya (95% C.I.: 85–92 kya). The pulse of gene flow is estimated to have occurred ∼7 kya (95% C.I.: 6.8–7.7 kya), whereas the inferred admixture proportion in our Pygmy sample resulting from the pulse of gene flow from the farmers is ∼68% (C.I.: 67.9%–68.2%).
Model selection and validation of demographic inference
We used three approaches to validate our demographic inference (Methods). First, to remove the effects of linkage, we refit our models to a subset of the data in which variant sites were at least 0.01 centiMorgan (cM) apart. The two best-fitting models remained the same as using the whole data set, and the parameter estimates were compatible (Supplemental Table S2). Under the assumption that the likelihoods calculated using the thinned data set are full likelihoods, we applied the Akaike (AIC) (Akaike 1974) and Bayesian information criteria (BIC) (Schwarz 1978) for model selection. Both AIC and BIC prefer the continuous asymmetric gene-flow model to the single-pulse gene flow model (Supplemental Table S2).
As a second validation, we used patterns of linkage disequilibrium (LD) decay, information not utilized by ∂a∂i. We calculated LD using sliding windows of 0.1 cM in the real data and in simulated whole-genome data, using 100 models drawn from the parameter confidence intervals of our two best-fit demographic models. We found that the patterns of LD decay predicted by the models generally matched the data well for both Pygmies and farmers (Supplemental Fig. S2), but with discrepancies at different distance regimes. This comparison of LD decay suggests that the two best-fit models capture different aspects of the demographic history of our populations, and not perfectly.
As a third validation, we applied the pairwise sequentially Markovian coalescent (PSMC) (Li and Durbin 2011) and multiple sequentially Markovian coalescent (MSMC) (Schiffels and Durbin 2014) as independent means to explore the demographic history of our populations (Methods). As a test of goodness-of-fit, we applied both methods to our intergenic data and simulations under both models (Supplemental Fig. S3). Under Model-1, the PSMC curves of the simulated Pygmy and farmer genomes split at about the same time as in the PSMC analysis of the real data, whereas the two simulated populations of Model-2 do not show clear separation until ∼70 kya (Supplemental Fig. S3A–C). The MSMC curves of Model-1 and those of the real data agree well, but Model-2 seems to fit the MSMC curve from the real data poorly (Supplemental Fig. S3D,E). Together, the PSMC/MSMC results suggest that Model-1 qualitatively fits the data better, and the inferred ancient divergence time in Model-1 is plausible.
In general, these validations suggest that Model-1 is our best estimate of demographic history for these populations, but it is an imperfect model. In order to lessen the impact of model misspecification on our selection inference, we conservatively report candidates under both Model-1 and Model-2.
Prioritizing selection candidates using whole-genome demographic simulations
Because conventional statistical outlier approaches are prone to false positives, we used MaCS (Chen et al. 2009) to perform whole-genome simulations under our realistic demographic models to assign statistical significance (P-values) in our selection scan (Methods). Methods for detecting natural selection often rely on summaries of local genetic variation, and they may be biased by variation in mutation rate across the genome (Reich et al. 2002; Drake et al. 2005; Schaffner et al. 2005; Sainudiin et al. 2007). Indeed, we found that if mutation rate variation is not controlled for, selection scan candidates are highly enriched in genomic regions with greater heterozygosity (Supplemental Material; Supplemental Figs. S4–S7). From here on, we thus used the per-window mutation rate approach (Methods) for all simulations to account for possible biases due to genomic mutation rate heterogeneity. We recognize that this approach may discount some selection signals, yielding a more conservative inference of natural selection. The distribution of P-values was also sensitive to the genetic recombination map used in the simulations (Supplemental Figs. S8, S9). To assess possible biases due to imperfection of the genetic recombination map, we ran two sets of simulations, using two published genetic maps: the African American map (Hinch et al. 2011) and the HapMap Yoruba map (Methods; The International HapMap Consortium 2007). Both these maps likely represent the recombination process better in the Yoruba than in the Pygmies, but no Pygmy-specific map is available.
Our top hits are the top 0.5% of windows in the P-value distribution of each test statistic. To avoid potential biases due to the choice of map and/or null model, we restricted our candidates to those that are top hits using all four combinations of the two genetic maps and the two best-fit demographic models. Unless mentioned otherwise we report P-values and false discovery rates obtained using Model-1 and the African American map, because they are the most conservative (Supplemental Figs. S8, S9).
To illustrate the importance of using P-values to determine candidates, rather than relying on outliers in the distribution of a test statistic, we plotted the P-value based on Model-1 as a function of the G2D statistic for each of the windows (Fig. 2; similar result holds for the iHS analysis, Supplemental Fig. S10). Quadrant I contains windows that have extreme G2D values but are not statistically significant when the confounding effects of demography and genomic architecture are controlled for. Conversely, Quadrant III contains windows that are statistically significant even though their G2D values are not extreme on a genome-wide basis. Because the association between functional elements (e.g., exon and regulatory sequences) and selection is not expected if a large fraction of significant tests are false positives, we validated our P-value approach by comparing the spatial distribution of our candidates for selection with the distribution of known functional sequences in the genome (Voight et al. 2006; Williamson et al. 2007; Mendizabal et al. 2012). As expected, we found that our top hits of the P-value approach were enriched in exons of genes (one-sided Fisher's exact test, P = 0.029) (Supplemental Table S3). Interestingly, we find no enrichment of top hits in regions deemed functional based on five types of ENCODE (Supplemental Material; Gerstein et al. 2012) regulatory elements (Methods; Supplemental Table S3).
Figure 2.
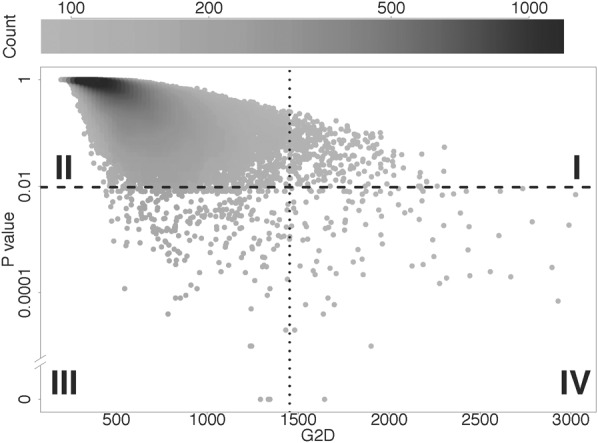
Importance of using P-values to define candidates in the G2D analysis. Each point is a window of 500 single-nucleotide variants, and shading represents the density of points. The vertical dotted line and the horizontal dashed line are the top 0.5% significance cutoffs for the G2D and P-value distributions, respectively. Windows in Quadrant I are outliers in the G2D distribution but are not statistically significant when the effects of demography and genome architecture are controlled for. In Quadrant III are the many windows that are statistically significant even though their G2D values are modest.
Evidence of local adaptation in Western African Pygmies: iHS
To detect recent incomplete selective sweeps, we scanned the genome using the haplotype-based iHS statistic (Voight et al. 2006) for the farmer and Pygmy samples separately (Methods). Using all four simulation sets, we defined Pygmy-specific signals as those windows that were a top hit (the top 0.5% in the P-value distribution) in the Pygmy sample, but not in the Yoruba sample (not within the top 1% in the P-value distribution), yielding 35 distinct genomic regions (Supplemental Table S4). We used a looser P-value cutoff to define Yoruba top hits in order to be more conservative in identifying regions as Pygmy specific. We evaluated the robustness of this prioritization criterion for iHS candidates by repeating the same analysis using four and seven individuals randomly sampled from the nine Yoruba genomes. The same genomic regions were identified as candidates in all experiments, suggesting that our iHS analysis is consistent even when the sample size is small.
Five of our candidate regions contain genes associated with bone synthesis. EPHB1 (locus: Chr 3: 134572433–134716365) (Fig. 3A) is an ephrin receptor at sites of osteogenesis. Interestingly, this region has been previously associated with the short stature in Pygmies (Jarvis et al. 2012). Our candidate region spans ∼140 kb, containing exon 2 and exon 3 of EPHB1 (which has a size of >460 kb and 16 coding exons). Elevated FST has been widely used to infer selection (Nielsen et al. 2009; Pickrell et al. 2009; Jarvis et al. 2012), and FST is elevated in this region, although we found no nonsynonymous variants. To further investigate the signal of selection, we used hierarchical clustering and network analysis of the phased haplotypes (Supplemental Material) for the region around exon 3 (±10 kb). Interestingly, both analyses suggest that Pygmy and farmer groups are almost fixed for different haplotypes (Fig. 3B,C). This is consistent with an incomplete selective sweep (Voight et al. 2006; Pickrell et al. 2009; Pritchard et al. 2010) and indicative of different selective pressures in these two groups. The other four bone synthesis–related candidates are SLCO2A1 (locus: Chr 3: 133506737–133863702), ZBTB38 (locus: Chr 3: 141105569–141333249), TSPAN5 (locus: Chr 4: 99496207–99673561), and GAREM (locus: Chr 18: 29766032–29896024) (Supplemental Material; Supplemental Fig. S11).
Figure 3.
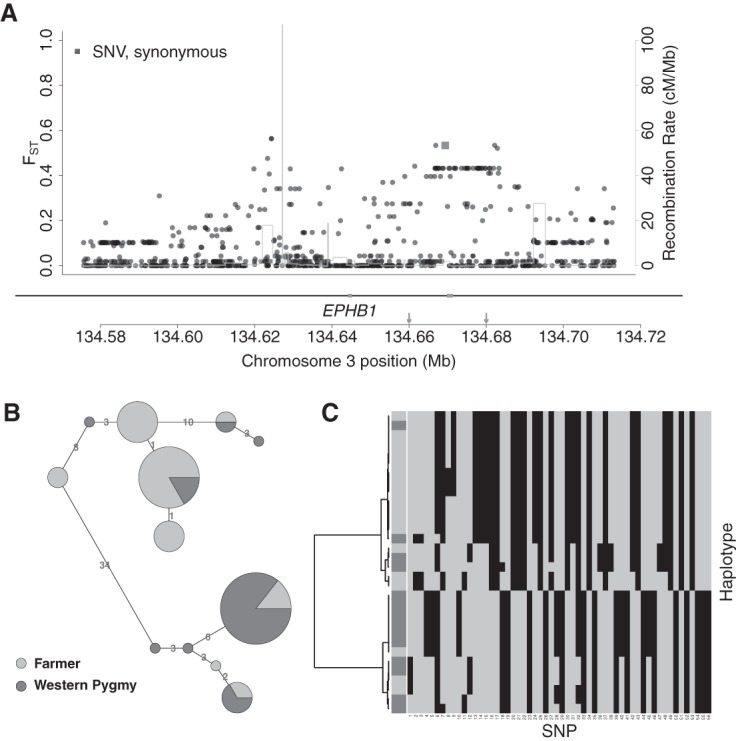
Candidate selection signal in EPHB1. (A) Distribution of FST, functional annotation (RefSeq and ENCODE elements) (Methods), and recombination rate (Hinch et al. 2011) for all variants (dots) in the candidate locus Chr 3: 134572433–134716365. Genes are shown under the plot with black lines (noncoding sequences) and gray boxes (exons). (B) Haplotype network for the region (Chr 3: 134.66–134.68 Mb, the region between the two arrows in A) with elevated FST. Each circle is a haplotype with size proportional to the number of chromosomes compared to the size of a single chromosome shown on the legend. Haplotypes are connected by lines indicating the pairwise nucleotide distance between them. (C) Hierarchical clustering of the haplotypes in B. Rows show individual haplotypes. The first column indicates population membership for haplotypes, using the same shading code as in B. The remaining columns are SNPs, with gray and black for ancestral and derived alleles, respectively.
Consistent with the hypothesis of selection for mobility (Diamond 1991), we found candidate loci in several muscle-related genes (Supplemental Table S4). In particular, OBSCN (spans >150 kb with 81 exons within the candidate locus Chr 1: 228103665–228842760) (Fig. 4A), an obscurin gene, has an important role in the organization of myofibrils during assembly and may mediate interactions between the sarcoplasmic reticulum (striated muscle fibers found in the skeletal system) and myofibrils (Young et al. 2001; Ackermann et al. 2014). Within this gene, 16 of 46 nonsynonymous amino acid variants are predicted as functionally important by either SIFT or PolyPhen-2. The SNV with the largest FST (Chr 1: 228475848, rs437129, FST = 0.54) in this region is nonsynonymous and functionally important (PolyPhen-2 score = 0.968, although SIFT score = 0.43). It is fixed for the ancestral allele (guanine, panTro3, hg19) in our Pygmy sample but is segregating at much lower frequency in our Yoruba farmer sample (allele frequency for G = 0.39 or 7/18) in both homozygote and heterozygote forms. The ancestral allele (G) frequencies of rs437129 in Yoruba, Luhya, and African American based on the 1000 Genomes Project (Phase I) are 0.551, 0.665, and 0.590 (dbSNP 137). Analyses of the haplotypes between the two nonsynonymous sites with FST > 0.5 (Chr 1: 228475848 and Chr 1: 228520973, including the 10-kb flanking region) (Fig. 4B,C) suggest the existence of two major haplotypes in our sample that are relatively population specific. We thus postulate that natural selection might have acted in different directions for this region between these two groups. Other muscle-related genes include COX10 (locus: Chr 17: 13911228–14241158) and LARGE (locus: Chr 22: 34224706–34359718) (Supplemental Material).
Figure 4.
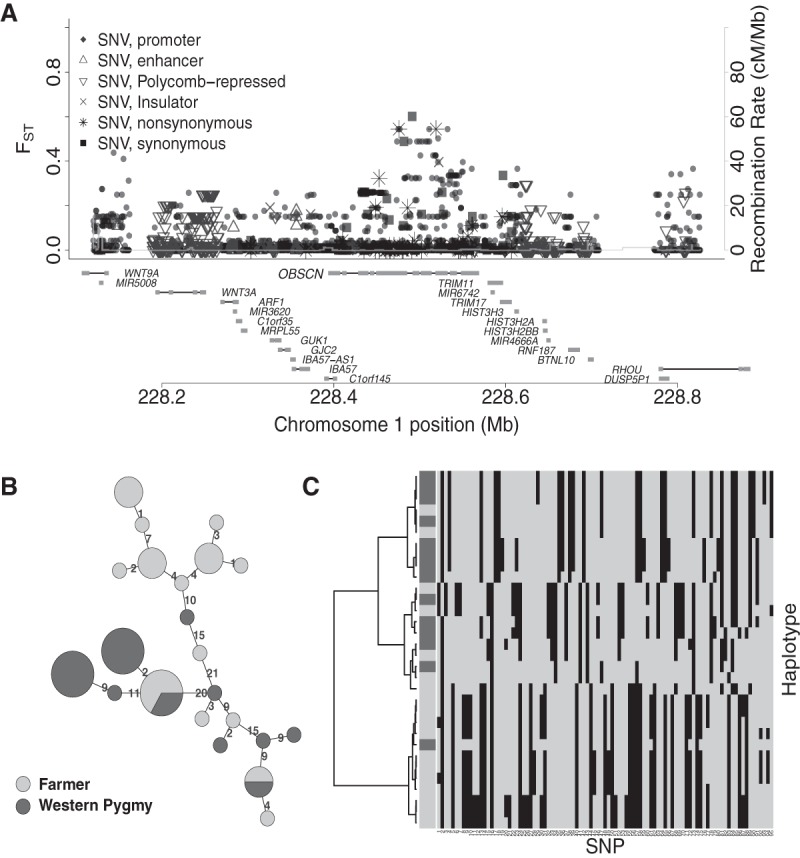
Candidate selection signal near OBSCN. (A) As in Figure 3, but for the region Chr 1: 228103665–228842760. (B,C) As in Figure 3, but for the region Chr 1: 228.46–228.54 Mb with elevated FST.
Our whole-genome selection scan also identified a variety of genes (Supplemental Table S4) involved in immune function, one of the most common targets of adaptive evolution (Williamson et al. 2007; Barreiro and Quintana-Murci 2009), and in reproduction, which is compatible with the life-history tradeoff hypothesis (Migliano et al. 2007). Other functional categories for genes of potential interest within the top hits of our iHS signals (Supplemental Table S4) include energy metabolism, cell signaling, and neural development.
Evidence of local adaptation in Western African Pygmies: G2D
To complement our iHS scan, we performed a scan using the G2D statistic (Nielsen et al. 2009), which measures how different the local farmer-Pygmy 2-D joint allele frequency spectrum is from the genome-wide spectrum. We found low P-value top-hit windows on all 22 chromosomes (Supplemental Fig. S12). To identify Pygmy-specific signals of selection, we used the composite likelihood ratio (CLR) (Nielsen et al. 2005) statistic. Our Pygmy-specific top-hit windows satisfied three conditions for all four simulation sets: (1) They were in the top 0.5% of the P-value distribution of the G2D statistic; (2) they were in the top 0.5% of the P-value distribution of the Pygmy-specific CLR statistic; and (3) they were not within the top 1% of the P-value distribution of the Yoruba-specific CLR statistic. This procedure identified seven distinct Pygmy-specific candidates (Supplemental Table S5), and these candidates do not overlap with those from iHS scan.
Our top candidate region from the G2D scans (locus: Chr 6: 32968692–33049012; P-value = 9.90 × 10−6, FDR = 0.03) (Fig. 5A) includes three members of the Class II Human Leukocyte Antigen (HLA) gene family, HLA-DPB1, HLA-DOA, and HLA-DPA1, which play a critical role in initiating the immune response to invading pathogens (Barreiro and Quintana-Murci 2009; O'Brien et al. 2011). The HLA region has a complex genomic architecture with several recombination hotspots (Fig. 5A; also see Jeffreys et al. 2005). To avoid possible artifacts due to sequencing and genotyping errors, we reanalyzed this region after removing variants violating Hardy-Weinberg equilibrium, an indicator of possible genotyping errors. Fourteen of 1478 SNVs in this region fail the HWE test (cutoff P < 0.05); yet the P-value for this region remains the same after their removal. Eleven nonsynonymous variants were found in this region; of these sites, five are predicted to be deleterious or possibly damaging by SIFT (SIFT score ≤0.05) (Kumar et al. 2009) and/or PolyPhen-2 (PolyPhen-2 score ≥0.995) (Adzhubei et al. 2010). Haplotype analyses (Fig. 5B,C) of the region with elevated FST around the gene HLA-DPA1 show that although the farmer samples possess two major haplotypes, most of the Pygmy samples belong to a single haplogroup. Because of the existence of several recombination hotspots in this locus, we plotted the diplotypes for this region in our sample to avoid possible biases due to phasing error (Supplemental Fig. S13). Consistent with the haplotype analyses, most of the Pygmy samples (five of seven) are homozygous for a single diplotype, whereas the farmers have two diplotypes. We thus hypothesize that a specific immunity-related pressure has driven the evolution of this locus in the Pygmies.
Figure 5.
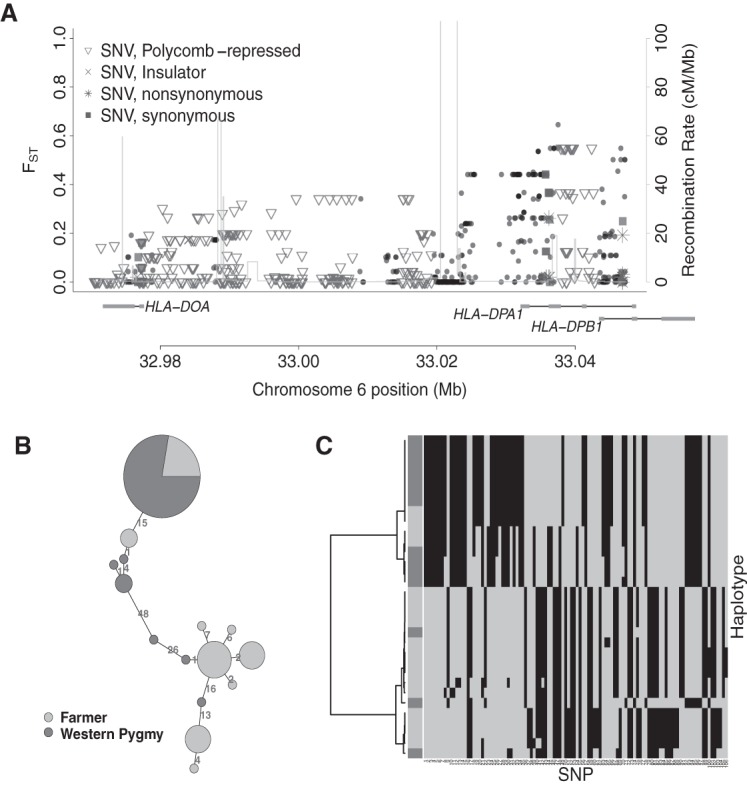
Candidate selection signal near HLA-DPA1. (A) As in Figure 3, but for the candidate locus Chr 6: 32968692–33049012. (B,C) As in Figure 3, but for the region Chr 6: 33.03–33.05 Mb with elevated FST.
This scan also identified two candidate regions that contain genes associated with bone synthesis and development. The gene FLNB in the first region (locus: Chr 3: 57918877–58055004) encodes filamin B, a multifunctional cytoplasmic protein that plays a critical role in skeletal development. Flnb knockout mice are phenotypically similar to individuals with spondylocarpotarsal syndrome as they exhibited short stature and similar skeletal abnormalities (Farrington-Rock et al. 2008). FLNB is known to be associated with height in African Pygmies (Jarvis et al. 2012; Lachance et al. 2012) and has also been reported to be associated with osteoporosis in women (Wilson et al. 2009). Although we did not find any amino acid substitution variants in FLNB in our sample, we did find many variants with large FST that may lie in regulatory elements (Supplemental Fig. S14). The second region (locus: Chr 1: 179361049–179468857) contains the gene AXDND1. Although the function of AXDND1 is unclear, a recent genome-wide association study reported a statistically significant association between this gene and fracture risk. This implies a potential role of AXDND1 in bone synthesis or musculoskeletal traits (Medina-Gomez and Rivadeneira 2014).
Other G2D candidate loci include the reproduction-related gene LAMC1 (locus: Chr 1: 183076845–183184161) and the gene regulation-related gene ZNF (Chr 19: 12386669–12523799) (Supplemental Material).
Inference of polygenic selection in Western African Pygmies
To detect polygenic selection that results in small allele frequency changes at multiple loci involved in a biological function or pathway (Pritchard et al. 2010; Berg and Coop 2014), we used FST (Weir and Cockerham's estimator) (Weir and Cockerham 1984) to estimate the level of population differentiation for each SNV and compared the FST distribution of the SNVs of all genes in a given gene set (a specific biological function or pathway) to that in the rest of our genic sequences. We used 1454 Gene Ontology (GO) gene sets from the Gene Set Enrichment Analysis (GSEA) project (Subramanian et al. 2005).
Using the Mann-Whitney U test, we found 113 gene sets that show significant evidence of having larger FST values compared to the FST distributions of the rest of genic sequences (one-sided test, Bonferroni corrected P < 10−10). Surprisingly, however, we also found as many or more significant gene sets in 72.9% and 48.5% of our neutral whole-genome simulations under Model-1 and Model-2, respectively. This suggests that demographic processes and genomic architecture can mimic the signals of polygenic adaptation, and in turn suggests that many of these 113 significant gene sets are false positives.
Only three of the 113 significant gene sets had significant Mann-Whitney U tests <5% of the time in all of our neutral whole-genome simulation sets, and we consider these sets as true positives (Supplemental Table S6). Among these three gene sets, there were no overlapping genes, nor did any genes overlap with those identified in our G2D and iHS analysis, suggesting that the significance of three gene sets is not due to hitchhiking on selective sweeps. The two strongest signals of polygenic selection are both related to immunity (GO categories: “Antigen binding,” Bonferroni corrected P-value = 2.31 × 10−25; “Pattern recognition receptor activity,” Bonferroni corrected P-value = 5.04 × 10−14). The other candidate is “G1 phase of mitotic cell cycle” (Bonferroni corrected P-value = 1.75 × 10−19). Although the corresponding phenotype for this group is unknown, accurate transition from G1 phase of the cell cycle is crucial for control of eukaryotic cell proliferation (Bertoli et al. 2013).
Discussion
Ancient divergence and more recent gene flow between African farmer and Pygmy populations
Our demographic inference for the farmer (Yoruba) and Western Pygmy hunter-gatherer groups (Baka and Biaka) offers insight into the demographic dynamics of sub-Saharan Africa over the past hundreds of thousands of years. The deep divergence time between the ancestors of the agricultural and Pygmy groups we found in Model-1 (∼155 kya, 95% C.I.: 139–164 kya) is inconsistent with several recent publications (Patin et al. 2009; Batini et al. 2011; Veeramah et al. 2012), whereas the divergence time inferred in Model-2 (∼90 kya, 95% C.I. 85–91 kya) is more consistent with those earlier studies. Our PSMC analysis is consistent with old divergence between the ancestors of the two groups (Supplemental Fig. S3), and our MSMC analysis supports Model-1 over Model-2 (Supplemental Fig. S3); however, these results must be interpreted carefully because these methods do not explicitly model population divergence. Africa experienced dramatic climate fluctuations between dry and wet conditions near the end of Marine Isotope Stage 6 (MIS 6; 190–135 kya) and through the whole MIS 5 (75–135 kya) (Blome et al. 2012; Rito et al. 2013). It is believed that it was about this time period when dramatic climate change caused forest defragmentation in Central Africa. (Blome et al. 2012; Rito et al. 2013; Ziegler et al. 2013). Such environmental changes may give rise to different niches (e.g., savanna versus forest), and in turn promoted population isolation and differentiation (Blome et al. 2012, Ziegler et al. 2013). We thus speculate that environmental change and forest fragmentation may have caused the ancestors of Pygmy rainforest dwellers to diverge from those of agricultural groups within the past 75–190 kya.
Both our best-fit models infer asymmetric gene flow, with greater flow from farmers to Pygmies than vice versa. These inferences are consistent with observed socioeconomic contacts between contemporary Pygmies and farmers (Terashima 1987; Bahuchet 2012), and was also observed previously in Patin et al. (2009). However, Patin et al. (2009) had little power to infer gene flow since divergence (95% C.I. covers 0). Interestingly, our Model-2 suggests a single pulse of gene flow from farmers to Pygmies ∼7 kya, resulting in a ∼68% admixture in our Western Pygmy sample. This admixture proportion is consistent with the recent findings of Verdu et al. (2013), who analyzed autosomal microsatellite data from 23 Central African Pygmy and non-Pygmy populations and inferred admixture proportions of up to 50%–70% in these Pygmy populations; although recent evidence shows that admixture proportions may vary between 0%–90% among individual Pygmies (Patin et al. 2009, 2014; Tishkoff et al. 2009; Jarvis et al. 2012). This substantial agriculturalist genetic ancestry in Pygmies has been hypothesized to be a consequence of the recent expansion of Bantu/Niger-Kordofanian-speaking farmers from West Africa ∼5 kya (Cavalli-Sforza 1986; Tishkoff et al. 2009; Patin et al. 2014). Indeed, our inferred time of admixture coincides with the time of Neolithic agricultural development in Africa ∼5–10 kya (Phillipson 2005), as well as with the estimated times of agriculturalist expansion for both Bantu-speaking (5.6 kya, 95% C.I.: 3.2–8.2 kya) and Niger-Kodorfanian-speaking (7.3 kya, 95% C.I.: 5.7–9.6 kya) people (Li et al. 2014). Many Pygmies today speak languages adopted from neighboring Bantu/Sudanic-speaking farmer groups, with whom they exchange goods (Bahuchet 2012). Because the social–economic relationship between the two groups promotes intermarriage (Terashima 1987, Bahuchet 2012), this symbiotic bond may contribute to the observed substantial admixture in the Pygmy groups, especially since the development and expansion of agriculture in Africa.
There are important differences between the approach used here and those used in earlier demographic studies of African Pygmies (Patin et al. 2009, Batini et al. 2011, Veeramah et al. 2012). First, we jointly estimated all parameters simultaneously for a given model, but some previous studies first estimated effective population sizes and then optimized other model parameters given the pre-estimated population sizes (Patin et al. 2009). They thus explored a smaller region of parameter space, potentially biasing their inferences. Second, our inference was based on whole-genome sequencing data with a relatively small sample size of 16 genomes, whereas these previous studies all used less than 60 loci, but had much larger samples of more than 100 individuals. Two of these studies (Patin et al. 2009; Batini et al. 2011) inferred recent population contraction in the Pygmy groups. Our small sample size limits our power to detect such recent events, but the simulation study of Robinson et al. (2014) suggests that ∂a∂i can confidently infer ancient events in models of similar complexity to those we infer here.
Our inferred dates are based on a phylogeny-based mutation rate of 2.35 × 10−8 per-site per-generation (Gutenkunst et al. 2009; compatible with Nachman and Crowell 2000). This value is compatible to those used in those earlier studies, i.e., an autosomal mutation rate of 2.5 × 10−8 and 2.6 × 10−8 per site per generation for Patin et al. (2009) and Veeramah et al. (2012) and a mitochondrial substitution rate of 27.8 × 10−8 per site per generation in Batini et al. (2011). A generation time of 25 yr was used to convert time estimates to years, although there is some evidence that generation time may differ between the two populations (Migliano et al. 2007). Our date estimates would be two times older if we used the rate of ∼1.2 × 10−8 per-site per-generation estimated by recent pedigree-based whole-genome sequence studies (Conrad et al. 2011; Kong et al. 2012). For example, the split time between the ancestors of Pygmies and farmers would be pushed back further to ∼300 kya, which predates the earliest emergence of AMH in the fossil record ∼200 kya (McDougall et al. 2005; Scheinfeldt et al. 2010). This deep Pygmy-farmer divergence could be in part due to imperfections in the model. For example, our model does not incorporate archaic admixture, which has been reported in Western African Pygmies (Hammer et al. 2011). Such introgression might cause us to overestimate the Pygmy-farmer divergence. Nevertheless, both approaches to estimating the human mutation rate have limitations, including inaccuracy of the human-chimpanzee divergence time in the phylogenetic approach and false negative mutations in the pedigree sequencing approach (Veeramah and Hammer 2014). We used the phylogenetic estimate because of its history in population genetics, but caution is advised when comparing population genetic date estimates with the fossil record.
Importance of prioritizing selection candidates using P-values from whole-genome simulations
Our results highlight the importance of using a model-based approach to assess statistical significance in whole-genome selection scans. Genomic scan studies using the tail of an empirical summary statistic distribution (an “outlier” approach) to define a significance cutoff for positive selection have been highly criticized. Nonselective forces, including demography and local genomic architecture, such as variation in mutation and recombination rates (Reich et al. 2002; Drake et al. 2005; Jeffreys et al. 2005; Schaffner et al. 2005; Sainudiin et al. 2007) across loci, can produce signals similar to positive selection (Tajima 1989; Andolfatto and Przeworski 2000; Wall et al. 2002; Jensen et al. 2005; Schaffner et al. 2005; Teshima et al. 2006). For example, we observed that larger G2D scores are associated with higher heterozygosity (Supplemental Fig. S6), so candidates determined using an empirical outlier approach might be biased toward regions with higher mutation rates. By matching local mutation rate in our simulations to local heterozygosity in the data, we eliminate this bias (Supplemental Fig. S7). Worryingly, the false targets identified by a genomic scan that fails to account for nonselective forces can be misleading because they might still make biological sense a posteriori (Pavlidis et al. 2012).
Prioritizing selection candidates based on P-values identifies candidates that would be missed by an empirical P-value approach and avoids potential false positive candidates caused by demography or recombination and mutation rate variation (Fig. 2). Even more striking is the high proportion of GO gene sets that are identified as significant by a Mann-Whitney U test but that are not significant when compared against our neutral simulations that account for demographic history and genomic architecture. Caution is still advised when interpreting our selection scan results, particularly for the results of iHS scan, because power may be limited due to our relatively small sample of seven Pygmies and nine Yoruba farmers (Pickrell et al. 2009). However, a recent simulation study showed that iHS has up to 80% power with a similar sample size to detect classic hard sweeps (Ferrer-Admetlla et al. 2014).
Candidates of adaptation in Western African Pygmy groups
With our high coverage whole-genome sequencing data, we conducted comprehensive model-based selection scans for Western African Pygmies using a series of complementary statistical approaches. Many loci detected by our approach are involved in muscle development, bone synthesis, immunity, reproduction, cell signaling and development, and energy metabolism (see Results).
Of particular interest are several genomic regions that show signatures of selection in African Pygmies that might contribute to short stature. Seven genes known to be associated with bone synthesis were identified by either iHS or G2D analysis. Among them, FLNB, EPHB1, and TSPAN5 have been functionally shown to affect body size through gene knockout or knockdown experiments in mice (Iwai et al. 2007; Farrington-Rock et al. 2008; Benson et al. 2012; Zhou et al. 2014); and FLNB, AXDND1, ZBTB38, and GAREM have been shown to be associated with human height in multiple populations (Lettre et al. 2008; Weedon et al. 2008; Kim et al. 2012; Wang et al. 2013; Medina-Gomez and Rivadeneira 2014; Wood et al. 2014). EPHB1 was reported to be genetically associated with height in African Pygmies (Jarvis et al. 2012). Interestingly, although we found no nonsynonymous variants in the locus containing EPHB1, the Pygmy and farmer populations are each nearly fixed for a single population-specific haplotype (Fig. 3B,C), a pattern expected under an incomplete selective sweep (Voight et al. 2006; Pickrell et al. 2009; Pritchard et al. 2010). FLNB (locus: Chr 3: 57,918,877–58,055,004) is within the locus Chr 3: 45–60 Mb that was also previously reported to be associated with height in Pygmies (Jarvis et al. 2012; Lachance et al. 2012). Clinically, nonsense mutations in FLNB cause spondylocarpotarsal synostosis syndrome (SCT), a recessive disease characterized by short stature and fusions of the vertebrae and carpal and tarsal bones (Krakow et al. 2004). Our observation of many large FST variants within ENCODE regulatory sequences (Supplemental Fig. S14) around this locus suggests that short stature in Western African Pygmies might arise through cis-regulatory evolution.
Several studies (e.g., Diamond 1991; Venkataraman et al. 2013) have hypothesized that the ability to quickly climb trees and move in dense forest is a potential adaptation of Pygmy hunter-gatherers. One of our candidates, OBSCN, a myofibrils-regulating obscurin gene, harbors several highly differentiated, putatively functionally important SNVs, including rs437129 (see Results). Our haplotype analyses suggest that rs437129 is associated with population-specific haplotypes in the Pygmies and the farmers (Fig. 4B,C), although the signal is noisy. In Pygmies, the fixed allele of rs437129 is consistent with the ancestral state (panTro3, hg19). Under a classic selective sweep model, one might expect a derived beneficial allele to sweep up in frequency, but a nearby ancestral allele could hitchhike with the selected site (Smith and Haigh 1974). However, selection may sometimes favor an ancestral allele that has been segregating in the population (Pritchard et al. 2010). Because accessing essential foods is crucial for hunter-gatherers, mobility-related adaptation to locomotor efficiency amid dense vegetation has been emphasized in several recent studies (Diamond 1991; Bramble and Lieberman 2004; Perry and Dominy 2009). Indeed, Venkataraman et al. (2013) recently presented evidence of a positive correlation between tree-climbing ability and muscle fiber length in African Twa and Asian Agta Pygmies compared to neighboring non-tree-climbing farmers, suggesting that natural selection might have favored anatomical structures (e.g., muscle fiber length) that promote safe vertical climbing (Venkataraman et al. 2013). A plausible evolutionary explanation for our observed selective signal is that natural selection favors the ancestral haplotype of OBSCN possessed in hunter-gatherer Pygmies to adapt specific muscle architecture to locomotor efficiency, whereas local adaptation outside the forest to an alternative allele or relaxation of selection might promote the observed population differentiation around this locus. The signal we found around the gene OBSCN could thus be the first genetic evidence that supports the mobility hypothesis.
We used several complementary statistical tests to detect different modes of adaptation. The haplotype-based iHS test has the greatest power for detecting recent (<30 kya) incomplete sweeps, but the AFS-based G2D test is capable of detecting completed and ongoing sweeps that occurred <300 kya as well as balancing selection (Sabeti et al. 2006; Nielsen et al. 2009). Our gene set enrichment analysis, on the other hand, has little power to detect sweeps but can detect polygenic selection (Daub et al. 2013). It is thus not surprising that there is no overlap among the candidates identified by our different tests. All three tests did, however, detect regions including genes associated with immunity (see Results). The pervasive signals of selection on immune function we found in all three scans are consistent with the view that genes involved in pathogen response are among the most common targets of adaptive evolution (Williamson et al. 2007; Barreiro and Quintana-Murci 2009; Jarvis et al. 2012; Novembre and Han 2012).
We leveraged whole-genome sequence data from African Pygmy and agriculturalist populations to infer their prehistory and search for Pygmy-specific adaptation signals through a carefully designed computational and statistical framework. In doing so, we accounted for many potentially confounding factors, including demography and mutation and recombination rate heterogeneity. Future work may be needed to account for additional confounding factors, but we believe the framework presented here offers great promise for shedding light on the complex demographic and adaptive history of human populations.
Methods
Whole-genome sequencing data and data quality assurance
Our Biaka Pygmy (N = 4) DNA samples were obtained from publicly available cell lines administrated by the Centre d'Etude du Polymorphism Human Genome Diversity Panel (Li et al. 2008). Details regarding the Baka Pygmies (N = 3) samples are in Lachance et al. (2012) and SNP data are available with dbSNP batch IDs: Lachance2012Cell_snp, Lachance2012Cell_deletion, Lachance2012Cell_insertion, and Lachance2012Cell_complex_ substitution. Whole-genome sequencing data for the unrelated Yoruba farmers (N = 9) were downloaded from the CGI data repository (Drmanac et al. 2010). The median coverage across the samples was 60.5× (SD = 8.54×). Genome assembly and variant calling were done using the standard CGI Assembly Pipeline 1.10, CGA Tools 1.4, and NCBI Human Reference Genome build 37 (Supplemental Material). After applying quality control filters (Supplemental Material), we found 10,865,288 autosomal single-nucleotide variants in our samples.
Estimation of demographic parameters using ∂a∂i
We used the allele frequency spectrum (AFS)-based demographic inference tool ∂a∂i (Gutenkunst et al. 2009) to build and fit our demographic models (Supplemental Material). After additional data quality control steps, we built an unfolded AFS using 1,575,394 intergenic SNVs, polarized via human-chimpanzee alignment and statistically corrected to mitigate possible biases due to ancestral state misidentification (Supplemental Material). Because linkage among sites means that ∂a∂i calculates a composite rather than the full likelihood, confidence intervals of model parameters were estimated via 100 nonparametric bootstraps of the intergenic data. These confidence intervals thus account for sampling uncertainty within the data, but not for systematic uncertainties (e.g., the assumed mutation rate).
Assessment of demographic model
The composite likelihood ∂a∂i calculates is not the full likelihood due to the linkage. To minimize linkage in our model selection analysis, we thinned our data by choosing variants at least 0.01 cM apart and refit the candidate models to the resulting sub-data set. We then calculated AIC (Akaike 1974) and BIC (Schwarz 1978) for model selection. In our comparisons of real and simulated LD decay, we estimated LD between pairs of variants by their correlation coefficient (r2) using a genotype code (0, 1, or 2 reference alleles). We performed our PSMC/MSMC analyses (v0.6.3) (Li and Durbin 2011; Schiffels and Durbin 2014) using the default parameters suggested by the authors. To assess variation in the inferred PSMC curves, we analyzed 100 nonparametric bootstraps using the utility provided with the PSMC software. We used MSMC command: msmc --fixedRecombination --skipAmbiguous on the haplotypes of random Pygmy-Yoruba pairs phased using the Python scripts from the authors.
Coalescent whole-genome simulations
We used MaCS (Chen et al. 2009) for our coalescent simulations because of its ability to efficiently perform whole-genome simulations with recombination. To avoid potential underestimation of recombination rates, we removed the first 5 Mb on each chromosome as suggested by the creators of the African American recombination map (Hinch et al. 2011). For consistency, we also did this for the HapMap Yoruba map. To model mutational heterogeneity, we carried out a three-step procedure. First, we divided the genome into 25,000-bp windows and estimated the population genetic mutation parameter using ∂a∂i given a demographic model. Second, we performed each MaCS using a mutation parameter , the largest θ estimated among all of the windows. Third, for each window, we adjusted its mutation rate by dropping a proportion of the simulated variants. All simulations presented here model the effects of demography, recombination heterogeneity, and mutation heterogeneity. For our simulations, we excluded the regions that were excluded in the real data due to our quality control criteria.
Scan for signals of selection
All test statistics were calculated using predefined sliding windows of 500 SNVs, with a step size of 100 SNVs. Windows longer than 1 Mb were dropped to avoid complex genomic regions, such as centromeres or large structure variants. To maximize statistical power and focus on signals of selection in Western Pygmies generally, for all our tests, we combined samples from the two Pygmy populations because they are so recently diverged. We calculated statistical significance of each window using our whole-genome coalescent simulations under the best-fit demographic models. To account for uncertainty in parameters, we drew 1000 parameter sets from the confidence intervals from each model, assuming that they had a multivariate normal distribution. The per-window P-value was defined as the fraction of simulations with statistic values greater than or equal to the observed value of the same window in the real data. Candidates for each neutrality test were defined as the top 0.5% of the P-value distribution. We then ran 100,000 additional local simulations for each candidate to obtain a finer P-value resolution. We estimated false discovery rates using the method of Williamson et al. (2007).
We computed the G2D score defined as in Nielsen et al. (2009) and the integrated haplotype score (iHS) (Voight et al. 2006) using the software selscan (Szpiech and Hernandez 2014). Haplotype phasing was done using BEAGLE v3.1.1 (Supplemental Material; Browning and Browning 2007). To account for possible biases in the iHS analysis due to phasing errors, haplotype phase was estimated using the same procedure used for both the real and simulated data. iHS was calculated with the default parameters in selscan, standardized, and quantified the strength of selection following Voight et al. (2006). To search for evidence of polygenic selection, we downloaded 1454 Gene Ontology gene sets from the Gene Set Enrichment Analysis (GSEA) project at the Broad Institute in January 2014 (Subramanian et al. 2005), discarding 13 gene sets that shared >90% of their genes with another set. One-sided (alternative distribution is greater than the null) Mann-Whitney U tests were performed in R (R Development Core Team 2012). In our simulations, the genic FST distributions were obtained by calculating FST for all SNVs within the same genomic regions that are defined as genes in the real data (RefSeq, downloaded from UCSC Genome Browser in May 2013). The likelihood of a gene set being significant was calculated as
| (1) |
where |S| is the total number of whole-genome simulations; s is a given whole-genome simulation; and I is an indicator function of being significant under s.
Data access
The Biaka sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP067698. The variants for Biaka genomes have been submitted to NCBI dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) under submitter batch ID: HammerLab_Biaka_CGI.
Acknowledgments
Support for this work was provided by the National Institutes of Health (NIH) to J.D.W. and M.F.H. (R01 HG005226). P.H. and R.N.G. were supported by National Science Foundation (NSF) grant DEB-1146074. S.A.T. was supported by NIH grants 1R01GM113657-01 and 8DP1ES022577-04. J.L. is grateful for support from NIH NRSA postdoctoral fellowship F32HG006648.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.192971.115.
References
- Ackermann MA, Shriver M, Perry NA, Hu LY, Kontrogianni-Konstantopoulos A. 2014. Obscurins: Goliaths and Davids take over non-muscle tissues. PLoS One 9: e88162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike H. 1974. A new look at the statistical model identification. IEEE Trans Automat Contr 19: 716–723. [Google Scholar]
- Akey JM. 2009. Constructing genomic maps of positive selection in humans: Where do we go from here? Genome Res 19: 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P, Przeworski M. 2000. A genome-wide departure from the standard neutral model in natural populations of Drosophila. Genetics 156: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahuchet S. 2012. Changing language, remaining Pygmy. Hum Biol 84: 11–43. [DOI] [PubMed] [Google Scholar]
- Barreiro LB, Quintana-Murci L. 2009. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet 11: 17–30. [DOI] [PubMed] [Google Scholar]
- Batini C, Lopes J, Behar DM, Calafell F, Jorde LB, van der Veen L, Quintana-Murci L, Spedini G, Destro-Bisol G, Comas D. 2011. Insights into the demographic history of African Pygmies from complete mitochondrial genomes. Mol Biol Evol 28: 1099–1110. [DOI] [PubMed] [Google Scholar]
- Baumann G, Shaw MA, Merimee TJ. 1989. Low levels of high-affinity growth hormone-binding protein in African Pygmies. N Engl J Med 320: 1705–1709. [DOI] [PubMed] [Google Scholar]
- Benson MD, Opperman LA, Westerlund J, Fernandez CR, San Miguel S, Henkemeyer M, Chenaux G. 2012. Ephrin-B stimulation of calvarial bone formation. Dev Dyn 241: 1901–1910. [DOI] [PubMed] [Google Scholar]
- Berg JJ, Coop G. 2014. A population genetic signal of polygenic adaptation. PLoS Genet 10: e1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, de Bruin RA. 2013. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14: 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blench R. 2006. Archaeology, language, and the African past. Rowman Altamira Press, Lanham, MD. [Google Scholar]
- Blome MW, Cohen AS, Tryon CA, Brooks AS, Russell J. 2012. The environmental context for the origins of modern human diversity: a synthesis of regional variability in African climate 150,000–30,000 years ago. J Hum Evol 62: 563–592. [DOI] [PubMed] [Google Scholar]
- Bramble DM, Lieberman DE. 2004. Endurance running and the evolution of Homo. Nature 432: 345–352. [DOI] [PubMed] [Google Scholar]
- Browning SR, Browning BL. 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81: 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli-Sforza LL. 1986. African pygmies. Academic Press, San Diego, CA. [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza A. 1994. The history and geography of human genes. Princeton University Press, Princeton, NJ. [Google Scholar]
- Chen GK, Marjoram P, Wall JD. 2009. Fast and flexible simulation of DNA sequence data. Genome Res 19: 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang YJ, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub JT, Hofer T, Cutivet E, Dupanloup I, Quintana-Murci L, Robinson-Rechavi M, Excoffier L. 2013. Evidence for polygenic adaptation to pathogens in the human genome. Mol Biol Evol 30: 1544–1558. [DOI] [PubMed] [Google Scholar]
- Dávila N, Shea BT, Omoto K, Mercado M, Misawa S, Baumann G. 2002. Growth hormone binding protein, insulin-like growth factor-I and short stature in two pygmy populations from the Philippines. J Pediatr Endocrinol Metab 15: 269–276. [DOI] [PubMed] [Google Scholar]
- Diamond JM. 1991. Anthropology. Why are pygmies small? Nature 354: 111–112. [DOI] [PubMed] [Google Scholar]
- Drake JA, Bird C, Nemesh J, Thomas DJ, Newton-Cheh C, Reymond A, Excoffier L, Attar H, Antonarakis SE, Dermitzakis ET, et al. 2005. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat Genet 38: 223–227. [DOI] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81. [DOI] [PubMed] [Google Scholar]
- Farrington-Rock C, Kirilova V, Dillard-Telm L, Borowsky AD, Chalk S, Rock MJ, Cohn DH, Krakow D. 2008. Disruption of the Flnb gene in mice phenocopies the human disease spondylocarpotarsal synostosis syndrome. Hum Mol Genet 17: 631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Admetlla A, Liang M, Korneliussen T, Nielsen R. 2014. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol Biol Evol 31: 1275–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan D, Hammer MF. 2006. Reconstructing human origins in the genomic era. Nat Rev Genet 7: 669–680. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. 2012. Architecture of the human regulatory network derived from ENCODE data. Nature 489: 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5: e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. 2011. Genetic evidence for archaic admixture in Africa. Proc Natl Acad Sci 108: 15123–15128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez RD, Williamson SH, Bustamante CD. 2007. Context dependence, ancestral misidentification, and spurious signatures of natural selection. Mol Biol Evol 24: 1792–1800. [DOI] [PubMed] [Google Scholar]
- Hill KR, Walker RS, Bozicevicć M, Eder J, Headland T, Hewlett B, Hurtado AM, Marlowe F, Wiessner P, Wood B. 2011. Co-residence patterns in hunter-gatherer societies show unique human social structure. Science 331: 1286–1289. [DOI] [PubMed] [Google Scholar]
- Hinch AG, Tandon A, Patterson N, Song Y, Rohland N, Palmer CD, Chen GK, Wang K, Buxbaum SG, Akylbekova EL, et al. 2011. The landscape of recombination in African Americans. Nature 476: 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K, Ishii M, Ohshima S, Miyatake K, Saeki Y. 2007. Expression and function of transmembrane-4 superfamily (tetraspanin) proteins in osteoclasts: reciprocal roles of Tspan-5 and NET-6 during osteoclastogenesis. Allergol Int 56: 457–463. [DOI] [PubMed] [Google Scholar]
- Jarvis JP, Scheinfeldt LB, Soi S, Lambert C, Omberg L, Ferwerda B, Froment A, Bodo JM, Beggs W, Hoffman G, et al. 2012. Patterns of ancestry, signatures of natural selection, and genetic association with stature in Western African pygmies. PLoS Genet 8: e1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Neumann R, Panayi M, Myers S, Donnelly P. 2005. Human recombination hot spots hidden in regions of strong marker association. Nat Genet 37: 601–606. [DOI] [PubMed] [Google Scholar]
- Jensen JD, Kim Y, DuMont VB, Aquadro CF, Bustamante CD. 2005. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics 170: 1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiris DV. 2003. The framework of Central African hunter-gatherers and neighbouring societies. Afr Study Monogr 28: 57–79. [Google Scholar]
- Kim JJ, Park YM, Baik KH, Choi HY, Yang GS, Koh I, Hwang JA, Lee J, Lee YS, Rhee H, et al. 2012. Exome sequencing and subsequent association studies identify five amino acid-altering variants influencing human height. Hum Genet 131: 471–478. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakow D, Robertson SP, King LM, Morgan T, Sebald ET, Bertolotto C, Wachsmann-Hogiu S, Acuna D, Shapiro SS, Takafuta T, et al. 2004. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat Genet 36: 405–410. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Lachance J, Vernot B, Elbers CC, Ferwerda B, Froment A, Bodo JM, Lema G, Fu W, Nyambo TB, Rebbeck TR, et al. 2012. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell 150: 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, Sanna S, Eyheramendy S, Voight BF, Butler JL, Guiducci C, et al. 2008. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet 40: 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2011. Inference of human population history from individual whole-genome sequences. Nature 475: 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, Cann HM, Barsh GS, Feldman M, Cavalli-Sforza LL, et al. 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319: 1100–1104. [DOI] [PubMed] [Google Scholar]
- Li S, Schlebusch C, Jakobsson M. 2014. Genetic variation reveals large-scale population expansion and migration during the expansion of Bantu-speaking peoples. Proc Biol Sci 281: 20141448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Herráez DL, Bauchet M, Tang K, Theunert C, Pugach I, Li J, Nandineni MR, Gross A, Scholz M, Stoneking M. 2009. Genetic variation and recent positive selection in worldwide human populations: evidence from nearly 1 million SNPs. PLoS One 4: e7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall I, Brown FH, Fleagle JG. 2005. Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature 433: 733–736. [DOI] [PubMed] [Google Scholar]
- Medina-Gomez C, Rivadeneira F. 2014. Update on the genetic basis of disorders of the musculoskeletal system (ECTS 2013). IBMS BoneKEy 11: 508. [Google Scholar]
- Mendizabal I, Marigorta UM, Lao O, Comas D. 2012. Adaptive evolution of loci covarying with the human African Pygmy phenotype. Hum Genet 131: 1305–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliano AB, Vinicius L, Lahr MM. 2007. Life history trade-offs explain the evolution of human pygmies. Proc Natl Acad Sci 104: 20216–20219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliano AB, Romero IG, Metspalu M, Leavesley M, Pagani L, Antao T, Huang DW, Sherman BT, Siddle K, Scholes C, et al. 2013. Evolution of the pygmy phenotype: evidence of positive selection from genome-wide scans in African, Asian, and Melanesian pygmies. Hum Biol 85: 251–284. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. 2000. Estimate of the mutation rate per nucleotide in humans. Genetics 156: 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. 2005. Genomic scans for selective sweeps using SNP data. Genome Res 15: 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Hubisz MJ, Hellmann I, Torgerson D, Andrés AM, Albrechtsen A, Gutenkunst R, Adams MD, Cargill M, Boyko A, et al. 2009. Darwinian and demographic forces affecting human protein coding genes. Genome Res 19: 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Han E. 2012. Human population structure and the adaptive response to pathogen-induced selection pressures. Philos Trans R Soc Lond B Biol Sci 367: 878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien T, Kohaar I, Pfeiffer R, Maeder D, Yeager M, Schadt E, Prokunina-Olsson L. 2011. Risk alleles for chronic hepatitis B are associated with decreased mRNA expression of HLA-DPA1 and HLA-DPB1 in normal human liver. Genes Immun 12: 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohenjo N, Willis R, Jackson D, Nettleton C, Good K, Mugarura B. 2006. Health of Indigenous people in Africa. Lancet 367: 1937–1946. [DOI] [PubMed] [Google Scholar]
- Patin E, Laval G, Barreiro LB, Salas A, Semino O, Santachiara-Benerecetti S, Kidd KK, Kidd JR, Van der Veen L, Hombert JM, et al. 2009. Inferring the demographic history of African farmers and Pygmy hunter–gatherers using a multilocus resequencing data set. PLoS Genet 5: e1000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patin E, Siddle KJ, Laval G, Quach H, Harmant C, Becker N, Froment A, Régnault B, Lemée L, Gravel S, et al. 2014. The impact of agricultural emergence on the genetic history of African rainforest hunter-gatherers and agriculturalists. Nat Commun 5: 3163. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Jensen JD, Stephan W, Stamatakis A. 2012. A critical assessment of storytelling: gene ontology categories and the importance of validating genomic scans. Mol Biol Evol 29: 3237–3248. [DOI] [PubMed] [Google Scholar]
- Perry GH, Dominy NJ. 2009. Evolution of the human pygmy phenotype. Trends Ecol Evol 24: 218–225. [DOI] [PubMed] [Google Scholar]
- Perry GH, Foll M, Grenier JC, Patin E, Nedelec Y, Pacis A, Barakatt M, Gravel S, Zhou X, Nsobya SL, et al. 2014. Adaptive, convergent origins of the pygmy phenotype in African rainforest hunter-gatherers. Proc Natl Acad Sci 111: E3596–E3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipson DW. 2005. African archaeology. Cambridge University Press, New York. [Google Scholar]
- Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, et al. 2009. Signals of recent positive selection in a worldwide sample of human populations. Genome Res 19: 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Pickrell JK, Coop G. 2010. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol 20: R208–R215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Reich DE, Schaffner SF, Daly MJ, McVean G, Mullikin JC, Higgins JM, Richter DJ, Lander ES, Altshuler D. 2002. Human genome sequence variation and the influence of gene history, mutation and recombination. Nat Genet 32: 135–142. [DOI] [PubMed] [Google Scholar]
- Rito T, Richards MB, Fernandes V, Alshamali F, Cerny V, Pereira L, Soares P. 2013. The first modern human dispersals across Africa. PLoS One 8: e80031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JD, Coffman AJ, Hickerson MJ, Gutenkunst RN. 2014. Sampling strategies for frequency spectrum-based population genomic inference. BMC Evol Biol 14: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti P, Schaffner S, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen T, Altshuler D, Lander E. 2006. Positive natural selection in the human lineage. Science 312: 1614–1620. [DOI] [PubMed] [Google Scholar]
- Sainudiin R, Clark AG, Durrett RT. 2007. Simple models of genomic variation in human SNP density. BMC Genomics 8: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D. 2005. Calibrating a coalescent simulation of human genome sequence variation. Genome Res 15: 1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinfeldt LB, Soi S, Tishkoff SA. 2010. Working toward a synthesis of archaeological, linguistic, and genetic data for inferring African population history. Proc Natl Acad Sci 107Suppl 2: 8931–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffels S, Durbin R. 2014. Inferring human population size and separation history from multiple genome sequences. Nat Genet 46: 919–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G. 1978. Estimating the dimension of a model. Ann Stat 6: 461–464. [Google Scholar]
- Shea BT, Bailey RC. 1996. Allometry and adaptation of body proportions and stature in African pygmies. Am J Phys Anthropol 100: 311–340. [DOI] [PubMed] [Google Scholar]
- Smith JM, Haigh J. 1974. The hitch-hiking effect of a favourable gene. Genet Res 23: 23–35. [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpiech ZA, Hernandez RD. 2014. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol 31: 2824–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima H. 1987. Why Efe girls marry farmers?: socio-ecological backgrounds of inter-ethnic marriage in the Ituri forest of central Africa. Afr Study Monogr 6: 65–83. [Google Scholar]
- Teshima KM, Coop G, Przeworski M. 2006. How reliable are empirical genomic scans for selective sweeps? Genome Res 16: 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, et al. 2009. The genetic structure and history of Africans and African Americans. Science 324: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, Hammer MF. 2014. The impact of whole-genome sequencing on the reconstruction of human population history. Nat Rev Genet 15: 149–162. [DOI] [PubMed] [Google Scholar]
- Veeramah KR, Wegmann D, Woerner A, Mendez FL, Watkins JC, Destro-Bisol G, Soodyall H, Louie L, Hammer MF. 2012. An early divergence of KhoeSan ancestors from those of other modern humans is supported by an ABC-based analysis of autosomal resequencing data. Mol Biol Evol 29: 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman VV, Kraft TS, Dominy NJ. 2013. Tree climbing and human evolution. Proc Natl Acad Sci 110: 1237–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdu P, Becker NS, Froment A, Georges M, Grugni V, Quintana-Murci L, Hombert JM, Van der Veen L, Le Bomin S, Bahuchet S, et al. 2013. Sociocultural behavior, sex-biased admixture, and effective population sizes in Central African Pygmies and non-Pygmies. Mol Biol Evol 30: 918–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. 2006. A map of recent positive selection in the human genome. PLoS Biol 4: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Andolfatto P, Przeworski M. 2002. Testing models of selection and demography in Drosophila simulans. Genetics 162: 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang ZM, Teng YC, Shi JX, Wang HF, Yuan WT, Chu X, Wang DF, Wang W, Huang W. 2013. An SNP of the ZBTB38 gene is associated with idiopathic short stature in the Chinese Han population. Clin Endocrinol 79: 402–408. [DOI] [PubMed] [Google Scholar]
- Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, Freathy RM, Perry JR, Stevens S, Hall AS, et al. 2008. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet 40: 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- Williamson SH, Hubisz MJ, Clark AG, Payseur BA, Bustamante CD, Nielsen R. 2007. Localizing recent adaptive evolution in the human genome. PLoS Genet 3: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SG, Jones MR, Mullin BH, Dick IM, Richards JB, Pastinen TM, Grundberg E, Ljunggren Ö, Surdulescu GL, Dudbridge F, et al. 2009. Common sequence variation in FLNB regulates bone structure in women in the general population and FLNB mRNA expression in osteoblasts in vitro. J Bone Miner Res 24: 1989–1997. [DOI] [PubMed] [Google Scholar]
- Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Luan JA, Kutalik Z, et al. 2014. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 46: 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P, Ehler E, Gautel M. 2001. Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J Cell Biol 154: 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Fujiwara T, Ye S, Li X, Zhao H. 2014. Downregulation of Notch modulators, tetraspanin 5 and 10, inhibits osteoclastogenesis in vitro. Calcif Tissue Int 95: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler M, Simon MH, Hall IR, Barker S, Stringer C, Zahn R. 2013. Development of Middle Stone Age innovation linked to rapid climate change. Nat Commun 4: 1905. [DOI] [PMC free article] [PubMed] [Google Scholar]