

Abstract
Comparisons of whole-genome sequences from ancient and contemporary samples have pointed to several instances of archaic admixture through interbreeding between the ancestors of modern non-Africans and now extinct hominids such as Neanderthals and Denisovans. One implication of these findings is that some adaptive features in contemporary humans may have entered the population via gene flow with archaic forms in Eurasia. Within Africa, fossil evidence suggests that anatomically modern humans (AMH) and various archaic forms coexisted for much of the last 200,000 yr; however, the absence of ancient DNA in Africa has limited our ability to make a direct comparison between archaic and modern human genomes. Here, we use statistical inference based on high coverage whole-genome data (greater than 60×) from contemporary African Pygmy hunter-gatherers as an alternative means to study the evolutionary history of the genus Homo. Using whole-genome simulations that consider demographic histories that include both isolation and gene flow with neighboring farming populations, our inference method rejects the hypothesis that the ancestors of AMH were genetically isolated in Africa, thus providing the first whole genome-level evidence of African archaic admixture. Our inferences also suggest a complex human evolutionary history in Africa, which involves at least a single admixture event from an unknown archaic population into the ancestors of AMH, likely within the last 30,000 yr.
Introgression, the transfer of genetic material between closely related species through hybridization, is an important and ubiquitous evolutionary force in both plants and animals (Mallet 2007). Although hybrids are often nonviable or infertile, hybridization can be an important driving force for the origin of novel traits and new species (Mallet 2007; Zinner et al. 2011). Within our genus, Homo, there is strong evidence for multiple introgression events between our own species, H. sapiens, and now extinct sister taxa outside Africa (Pääbo 2014). Neanderthal whole-genome sequencing (Green et al. 2010; Prüfer et al. 2014) revealed that Neanderthals contributed an average of ∼2% of the genetic variation of present-day humans living outside of sub-Saharan Africa. This gene flow likely took place 37–86 thousand yr ago (kya), after early modern humans emigrated from Africa and before archaic forms went extinct in Eurasia (Sankararaman et al. 2012), and it may have occurred multiple times (Vernot and Akey 2014, 2015; Kim and Lohmueller 2015). Analyses of genome sequences from another extinct archaic human species, known as Denisovan, found in a cave in Siberia suggest that this archaic form or its closely related species contributed ∼5% of genetic variation to present-day Melanesians (Reich et al. 2010; Pääbo 2014). Furthermore, studies also suggest that the Denisovan genome has sequences that came from admixture with an unknown extinct hominin (Reich et al. 2010) and with Neanderthals (Prüfer et al. 2014). An important implication of these findings is that interbreeding among archaic humans may have promoted adaptation through the transfer of advantageous introgressive alleles (Hardy et al. 2005; Evans et al. 2006; Mendez et al. 2012; Huerta-Sánchez et al. 2014; Sankararaman et al. 2014; Vernot and Akey 2014; Racimo et al. 2015).
Although it is becoming clear that modern humans interbred with archaic humans outside of Africa, it is not clear to what extent similar interbreeding took place in the history of anatomically modern humans (AMH) in Africa, the continent on which AMH originated ∼200 kya (Cavalli-Sforza et al. 1994). Recent fossil evidence suggests that Homo emerged ∼2.8 million yr ago (Mya) in Eastern Africa (Villmoare et al. 2015). Several morphologically mosaic forms of Homo coexisted until ∼35 kya, well after the first appearance of AMH (Bräuer 2008; Rightmire 2009). The persistent coexistence of a variety of transitional forms of Homo with a mosaic of archaic and modern traits throughout Africa during the Pleistocene (Bräuer 2008; Rightmire 2009; Harvati et al. 2011) suggests ample opportunity for interbreeding among the ancestors of modern and archaic humans (Hammer et al. 2011). Unfortunately, owing to the tropical environment over most of sub-Sahara Africa, to date efforts to obtain DNA from archaic hominin fossil bones and teeth have not been successful (Campana et al. 2013; Veeramah and Hammer 2014). Archaic introgression in Africa can thus not be studied by directly comparing DNA sequences from archaic and modern populations.
Using a more indirect approach to infer archaic admixture, Plagnol and Wall (2006) recently analyzed patterns of divergence and linkage disequilibrium (LD) in DNA sequence polymorphism data from extant modern humans. Their summary statistic, S*, exploits the fact that recently introgressed lineages from long-isolated archaic humans show increased divergence and extensive LD (Plagnol and Wall 2006). S* has been widely used to identify ancient admixture in modern humans both inside and outside Africa (Plagnol and Wall 2006; Wall et al. 2009; Hammer et al. 2011; Lachance et al. 2012; Vernot and Akey 2014). Plagnol and Wall (2006) and Wall et al. (2009) inferred a 5% genetic contribution from a now-extinct taxon to the Niger-Kordofanian Yoruba farmers or their ancestors. Hammer et al. (2011) analyzed DNA sequence data from 61 noncoding loci in three contemporary sub-Saharan African populations. They found that hunter-gatherer African populations, including the Biaka Pygmy, Mbuti Pygmy, and San, contain ∼2% genetic material likely introgressed ∼35 kya from an archaic population that split from the ancestors of modern humans ∼700 kya. Recently, Lachance et al. (2012) applied S* to whole-genome data of three African hunter-gatherer populations and showed that their top-ranked S* loci are enriched for long-isolated lineages. These studies provide strong evidence for archaic admixture in Africa, but their inferences are limited because (1) they used genic sequences, in which recent natural selection may complicate inference (Plagnol and Wall 2006; Wall et al. 2009); (2) they surveyed only a small sample of genomic loci (Hammer et al. 2011); or (3) they did not control for confounding effects such as the demographic history of the interrogated populations (Lachance et al. 2012), which can bias S* scores.
Here, we search for whole genome–level evidence of archaic admixture by analyzing high coverage whole-genome sequence data from two Western African Pygmy populations, Biaka and Baka. We chose these African Pygmies to study the evolutionary history of modern humans in Africa because of prior evidence of archaic introgression in these two groups (Garrigan et al. 2005; Hayakawa et al. 2006; Hammer et al. 2011; Lachance et al. 2012) and because these populations are one of the basal groups on the extant human phylogeny, implying that they harbor some of the most ancient genetic lineages in humans (Tishkoff et al. 2009; Pickrell et al. 2012). We apply the S* statistic to detect putatively introgressive genomic loci in the Pygmy populations. We assess the statistical significance of candidates through sophisticated whole-genome simulations that incorporate demography and variation in both recombination and mutation rates. We then test the hypothesis of no archaic introgression in Africa at the whole-genome level by comparing these null simulations with the data. To understand the demographic dynamics between the ancestors of archaic and modern humans, for our candidate introgressive loci, we investigate the joint distribution of time to the most recent common ancestor (TMRCA) (Thomson et al. 2000) and genetic length, which represent the divergence between candidate archaic introgressive and modern human lineages as well as the time of introgression, respectively. Lastly, we discuss the number and timing of archaic admixture events in Africa by analyzing the pattern of LD and the distribution of genetic length using our candidate loci. Together, our results provide the first model-based whole-genome perspective on archaic introgression in Africa.
Results
Whole genome evidence of archaic introgression in Western African Pygmies
We analyzed high coverage (greater than 60×) whole-genome sequencing data from two Western Pygmy populations, Biaka (N = 4) (Hsieh et al. 2016) and Baka (N = 3) (Lachance et al. 2012). To maximize statistical power for all our analyses, we combined the samples of the two populations because they are very recently diverged (Hsieh et al. 2016). After a series of data quality control steps (Methods) to identify candidate archaic sequences, we calculated S* in overlapping 200-SNP (single-nucleotide polymorphism) windows across the entire genome. Because S* is sensitive to local recombination and mutation rate heterogeneity (Supplemental Fig. S1), we assessed the significance of S* for each window using whole-genome coalescent simulations that account for demographic history and this heterogeneity. We used two demographic models recently inferred from the same whole-genome samples (Hsieh et al. 2016) that incorporate both isolation and gene flow with neighboring farming populations (Supplemental Fig. S2; Supplemental Table S1). Because these two models do not include archaic admixture, they serve as the best demographic null models for our sample. To account for uncertainty in the recombination map, we simulated both models using the Yoruba HapMap (The International HapMap Consortium 2007) and African American (Hinch et al. 2011) genetic maps, for a total of four simulation sets. The S* P-value distributions for these simulation sets were highly correlated (Supplemental Fig. S3).
To formally test for evidence of archaic admixture in Africa, we compared the observed S* P-value distribution from our data with our demographic null S* P-value distributions based on the four whole-genome simulation sets. As expected, demographic null S* P-values are uniformly distributed between 0 and 1 (Fig. 1, solid line). More importantly, there is a strong excess of low S* P-values in the real data compared to our demographic null models (one-sided Mann-Whitney U test, P < 2.2 × 10−16) (Fig. 1). This is consistent with the alternative hypothesis of archaic introgressive sequences in our Pygmy whole-genome sample, and we thus reject the demographic null models of no archaic introgression.
Figure 1.
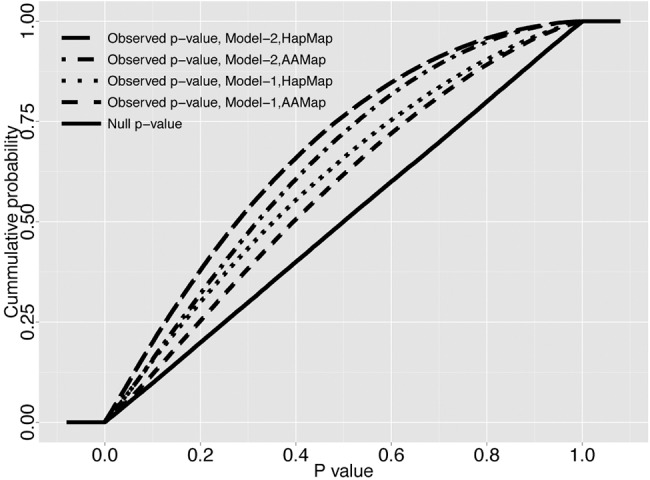
Significant excess of tests with very low P-values in the observed S* P-value distribution. Plotted is the observed (dashed lines) S* P-value distribution for the real data, calculated based on each of the four sets of whole-genome simulations. The four sets arise from the combination of the two demographic null models (Supplemental Fig. S2): Model-1 (the continuous asymmetric gene flow model) and Model-2 (the single-pulse admixture model); and the two genetic recombination maps: HapMap Yoruba map (HapMap) and African American map (AAMap). The solid line represents the null S* P-value distributions of the four whole-genome null simulation sets, derived by calculating P-values using a single randomly chosen simulation from each set. All four are uniformly distributed between 0 and 1 as expected. For the real data (dashed lines), all four analyses show a significant shift to small P-values in the observed S* P-value distribution (one-sided Mann-Whitney U test, P < 2.2 × 10−16), thus rejecting our demographic null hypotheses including no archaic admixture.
The significant difference in S* P-value distribution between the real data and demographic null simulations suggests that our approach can identify sequences with signatures of archaic introgression. The P-value approach identifies different sequences from the conventional statistical outlier approach (Supplemental Fig. S4). Calling the top 1% of windows as significant for both P-value and S*, we find many windows that have extreme S* values are not statistically significant when controlling for the confounding effects of demography and genomic heterogeneity in mutation and recombination rates (Supplemental Fig. S4, quadrant I). Conversely, we also find many windows that are statistically significant although their S* values are not extreme on a genome-wide basis (Supplemental Fig. S4, quadrant III). We chose the significance threshold of the top 1% S* P-values through whole-genome simulations with various plausible archaic admixture scenarios (Methods; Supplemental Fig. S10). We found that with this threshold, our downstream analyses provided consistent inferences with those based on simulations (Supplemental Tables S2–S5). We estimated that the false discovery rate (FDR) in our top 1% of windows ranges from ∼19% to ∼68% across the four neutral whole-genome simulation sets (Supplemental Table S6) using the approach of Williamson et al. (2007) (Methods). To minimize the impact of demographic and recombination model misspecification on our results, we chose candidate introgressive loci only if they were significant (top 1% in P-value distribution) using all four simulation sets. Our inference of candidate introgressive loci is thus more conservative than suggested by the single-simulation FDR estimates. This procedure yielded 265 distinct candidate introgressive loci (∼20 Mb in total length) (Methods) that were spread across the entire genome (Supplemental Fig. S5). Interestingly, there was a marked depletion of candidate archaic lineages in genic regions (one-sided Fisher's exact test, P ∼ 0.01). To illustrate the typical genomic characteristics of our candidate introgressive loci, we generated a network plot of computationally phased haplotypes (Bandelt et al. 1999) for one of the top candidate loci, Chr 16:8702222-8747116 (Fig. 2; Supplemental Fig. S6). The estimated TMRCA for this locus is ∼2.9 million yr ago. For comparison, we also included haplotypes from nine publicly available Yoruba farmer genomes (Drmanac et al. 2010) and rooted the network using the chimpanzee sequence (PanTro3). The network shows a long branch separating four Pygmy haplotypes from a cluster with all remaining Pygmy and Yoruba haplotypes (Fig. 2). The absence of reticulation among the four haplotypes on the basal branch is consistent with recent introgression into modern humans (Mendez et al. 2013).
Figure 2.
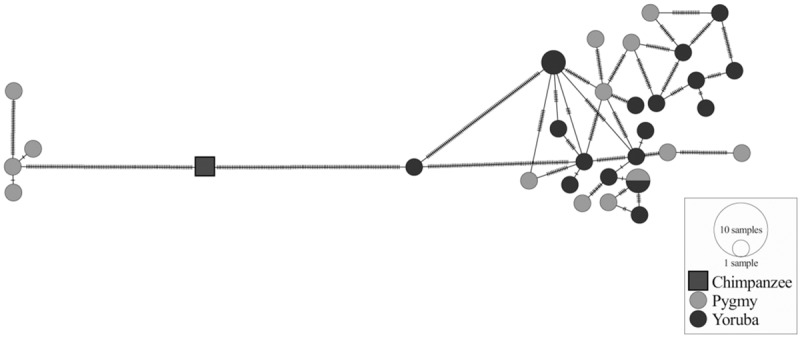
Haplotype network for the candidate introgressive locus Chr 16:8702222-8747116. Each circle is a haplotype with size proportional to the haplotype frequency, and the shade of gray indicates the haplotype frequency in the Pygmy (lighter gray) and Yoruba (darker gray) samples. Vertical bars along each branch indicate the number of mutations separating the haplotypes.
Inference of demographic dynamics using candidate loci for archaic admixture
We further analyzed our 265 candidate introgressive loci to infer the dynamics of admixture in Africa. First, for each candidate locus, we calculated the TMRCA and genetic length. The divergence time between the sequences of two hybridizing species can be inferred from the TMRCA (Lachance et al. 2012), and the time of introgression can be inferred through the genetic length. Both the TMRCA (median: 1.08 Mya; range: 0.53–6.43 Mya) and genetic length (median: 0.157 cM; range: 0.003–0.626 cM) of our candidate introgressive loci have wide distributions (Fig. 3; Supplemental Figs. S7, S8). Assuming that all archaic variants on an introgressive chromosome were in complete linkage disequilibrium (LD) at the time of introgression, we used the inverse of the genetic length observed at present as an estimator for the time when introgression occurred. This analysis suggests that archaic sequences might have introgressed as long ago as 0.97 Mya and as recently as ∼4.6 kya. This implies that interbreeding among archaic and modern humans occurred multiple times or in a continuous fashion. For comparison, we calculated both TMRCA and genetic length for three alternative sets of 265 loci that were randomly drawn from (1) the top 1% loci in the S* distribution; (2) the bottom 1% loci in the S* P-value distribution; and (3) the whole genome. The TMRCA distribution of our candidate introgressive loci is significantly older than those of the other three sub-data sets (one-sided Mann-Whitney U test, P < 3.1 × 10−15 in all three tests) (Fig. 3; Supplemental Fig. S7), suggesting that our P-value approach has enriched for potentially introgressive sequences. On the other hand, the genetic length distribution of our S* P-value candidate loci is also significantly different from those of the other sub-data sets (two-sided Mann-Whitney U test, P < 1.06 × 10−5 in all three tests) (Fig. 3; Supplemental Fig. S8). Together, these results suggest that our S* P-value candidate introgressive loci contain different demographic information from the conventional outliers in the empirical S* distribution.
Figure 3.
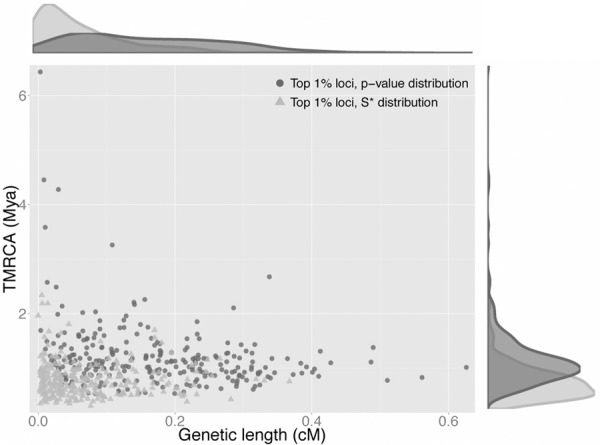
Wide joint distribution of TMRCA and genetic length for the top 1% S* P-value candidate loci. Darker gray dots and lighter gray triangles are, respectively, the two candidate sets from the top 1% candidate loci from the observed S* P-value and empirical S* distributions. The top and right plots show the marginal density of genetic length and TMRCA, respectively, for both candidate sets.
To further investigate the observed pattern of TMRCA and genetic length for our candidate introgressive loci, we used a variant of the D3 statistic (Hammer et al. 2011), , to estimate the minimum of the sizes of the two most basal lineages for a given locus; thus, is sensitive to the admixture proportion (Methods). Interestingly, ∼92% of our candidate introgressive loci (244 of 265) have frequencies <10% (i.e., only contain a single copy of the putative archaic chromosome, ), suggesting a relatively low admixture proportion from archaic to modern humans (Supplemental Fig. S9). Moreover, we found that is positively correlated with TMRCA (Pearson's correlation = 0.52, P < 2.2 × 10−16), but negatively correlated with genetic length (Pearson's correlation = −0.16, P = 0.008). The introgressive archaic lineages that rose to higher frequency (i.e., large ) may result from a strong admixture event or stochastic genetic drift in the past, but we cannot rule out the possible effect of positive natural selection.
To infer the number and the dates of admixture events, we modeled the decay of admixture-induced LD as an exponential function of genetic distance. The number of exponential decays represents the number of admixture events, and the rates of decay can be used to estimate times of admixture (Methods). This approach has been applied to study admixture in many human populations, including cases involving archaic hominins (Moorjani et al. 2011; Sankararaman et al. 2012; Loh et al. 2013; Fu et al. 2014; Pickrell et al. 2014). To assess the efficacy of this LD approach under a complex demographic model, we applied this method to simulated whole-genome data with archaic admixture. Simulations were generated by incorporating various scenarios of archaic admixture into the best-fit demographic model for Western African Pygmies described in Hsieh et al. (2016), which includes population divergence, population isolation, and recent asymmetric gene flow (Methods; Supplemental Figs. S2, S10). The top 1% S* P-value candidate loci for each whole-genome simulation set were determined using the same analysis pipeline as described above for the real data. We found that this approach can correctly infer single-wave admixture when we fit LD curves using a distance range of 0.02 to 1 cM, except for simulations in which admixture occurred 7800 generations ago (Supplemental Table S2). This is expected because this distance range, which is also suggested in Sankararaman et al. (2012), focuses the inference on admixture events up to 5000 generations ago. Indeed, inferences with this distance range do not have the power to detect the older admixture events in simulated two-wave archaic admixture scenarios (7800 and 300 generations ago) (Supplemental Table S4), which results in falsely accepting the single-wave archaic admixture model. This approach does tend to substantially underestimate times since admixture (Supplemental Tables S2–S5), suggesting that these estimated archaic admixture times should be considered as lower bounds, at least when the underlying population history is as complex as the models simulated here. For example, the LD-based method predicts an admixture time of 212 generations ago, which is ∼1/6 of the true time (1200 generations ago), from the simulation of a single 2% archaic admixture event (Supplemental Table S2).
To reduce possible confounding effects due to natural selection, we fit these models to the 244 low-frequency candidate archaic introgressive sequences () (Supplemental Fig. S9). Note that these 244 low-frequency putative archaic lineages have similarly wide distributions in both TMRCA (median: 1.04 Mya; range: 0.53–3.58 Mya) and genetic length (median: 0.16 cM; range: 0.004–0.625 cM) to those of the original 265 candidate loci (Supplemental Fig. S9). Using the single-wave model with the genetic distance range 0.02–1 cM, we inferred that admixture between the ancestors of modern Africans and putative archaic humans occurred ∼312 (95% C.I.: 45–975) generations ago or 9048 (95% C.I.: 1305–28,275) yr ago (Fig. 4A; also see Supplemental Fig. S11A; Supplemental Table S7), assuming a generation time of 29 yr (Methods). We were not able to obtain a stable fit under a two-wave model (a sum of two exponentials) with this genetic distance range. Our inference thus suggests that there was a single archaic admixture event within the past 5000 generations (or ∼150,000 yr).
Figure 4.

Decay of pairwise LD with respect to genetic distance for SNPs ascertained from the top 1% candidate introgressive loci. Black and blue dots are the average estimated LD among pairs of SNPs binned using genetic distance (in 0.001 cM increments) using real data and 100 bootstraps, respectively. The genetic distance is calculated based on the HapMap Yoruba map (The International HapMap Consortium 2007). For the cases of using the African American map (Hinch et al. 2011), see Supplemental Figure S10. The green curve is the fit of a single exponential using the data, while the red and orange curves are the fits of two exponentials using the real data and 100 bootstraps, respectively. (A) Fitting LD decay within genetic distance 0.02–1 cM. (B) Fitting LD decay within genetic distance 0.002–1 cM.
In order to further investigate the pattern of LD decay, we performed parametric bootstraps, in which we simulated whole-genome data under various plausible demographic models with archaic admixture (Supplemental Fig. S10). Interestingly, we found that the LD decay of the top 1% S* P-value loci from the data is not compatible with that from any of the single- and two-pulse archaic admixture simulations (two-sided Mann-Whitney U test, P < 2.2 × 10−16) (Supplemental Fig. S12). Nor are the distributions of genetic length of the top 1% S* P-value loci compatible between the data and the simulations (two-sided Mann-Whitney U test, P < 2.2 × 10−16) (Supplemental Fig. S13). To test whether these observations may result from more than a single pulse of introgression, we carried out LD fits using the wider distance range 0.002–1 cM to search for evidence of possible admixture events that occurred deeper in time. Under the two-wave model and with the same generation time, we inferred admixture waves occurring ∼19,342 (95% C.I.: 2438–44,772) and ∼312 (95% C.I.: 106–1376) generations ago (Fig. 4B; also see Supplemental Fig. S11B; Supplemental Table S7). The two-wave model fits significantly better than the single-wave model (likelihood ratio test, P-value <2.2 × 10−16, χ2 with d.f. = 2), and therefore, we rejected the model of single-wave admixture from archaic to modern humans in Africa. We also evaluated this analysis framework with this genetic distance range in our archaic whole-genome simulations. We found that although this analysis can correctly infer a true two-wave model (Supplemental Table S5), it also tends to falsely reject the single-wave archaic admixture model for simulations of single-wave archaic admixture (Supplemental Table S3). This suggests that at the genetic distance range of 0.002–1 cM, this method does not have statistical specificity to distinguish complex from simpler admixture models. However, the joint distribution of TMRCA and genetic length for the top 1% S* P-value candidate loci from the data is qualitatively more similar to those from the two-wave archaic admixture simulations than to those from the single-wave simulations (Fig. 5). Although none of the archaic admixture simulations statistically agree with the data in the joint distribution (MANOVA test, P < 2.8 × 10−12 for any pair of data and archaic admixture simulations), this suggests that models with two-wave archaic admixture do indeed reproduce the observed S* signal (sequence divergence and LD decay) better than those with single-wave admixture. Although caution is required in interpreting the result of the likelihood-ratio test for distinguishing single-wave from multiple-wave archaic admixture, together our inferences suggest recurrent archaic admixture in AMH evolution in Africa, with evidence that at least one such event occurred as recently as ∼9000 yr ago.
Figure 5.

Comparison of the joint distributions of TMRCA and genetic length for the top 1% S* P-value candidate loci from the data and whole-genome archaic admixture simulations. In each panel, the joint and marginal distributions of TMRCA (million yr ago) and genetic length (cM) of our candidate loci from the data (black cross and solid line in scatter and density plots, respectively) are compared with those from archaic admixture simulations (symbols and dashed lines in scatter and density plots, respectively): (A) two-wave archaic admixture model; (B) single-wave, 2% archaic admixture; (C) single-wave, 5% archaic admixture. TMRCA estimates for archaic simulation candidates were obtained from simulated coalescent trees in MaCS (Chen et al. 2009).
Discussion
The limited fossil record and the absence of ancient DNA within Africa have hampered our understanding of the processes that gave rise to AMH. Thus, here we have taken an indirect, model-based inference approach to test the hypothesis of archaic admixture in Africa using high coverage whole-genome sequence data of two African Pygmy populations, which were previously suggested to possess signals of archaic admixture (Garrigan et al. 2005; Hayakawa et al. 2006; Hammer et al. 2011; Lachance et al. 2012). We reject the hypothesis of no archaic admixture (P < 2.2 × 10−16) (Fig. 1) by comparing patterns of linkage disequilibrium quantified by S* in data with whole-genome simulations. It is important to note that our inference approach incorporates the best-fit demographic null models for our Pygmy sample (Hsieh et al. 2016) and accounts for genomic recombination and mutation rate heterogeneity, which mitigates the effects of these confounding factors. Although previous studies have found evidence for archaic admixture in these populations (Hammer et al. 2011; Lachance et al. 2012), our in-depth analyses reveal more about the timing and dynamics of the admixture process.
We observed our 265 candidate introgressive loci from the top 1% S* P-value distribution across the entire genome; however, there is a strong depletion of candidate introgressive lineages in genic regions (P ∼ 0.01). This is consistent with the recent observation of Neanderthal ancestry “deserts” in Eurasian genomes (Sankararaman et al. 2014; Vernot and Akey 2014), possibly due to genetic incompatibility in hybrids (Zinner et al. 2011; Twyford and Ennos 2012; Sankararaman et al. 2014; Vernot and Akey 2014). Our results also support the hypothesis of complex demographic dynamics between archaic humans and the ancestral population that gave rise to modern humans (Hammer et al. 2011; Hammer 2013). The wide distributions in both TMRCA and genetic length for our candidate introgressive loci (Fig. 3) provide the first model-based genome-level evidence that admixture between archaic humans and the ancestors of anatomically modern humans might be a common feature of human evolution in Africa. The TMRCA distribution of the candidate introgressive loci presented here is compatible with results from a recent study of three African hunter-gatherer populations (Lachance et al. 2012). The genetic length distribution of our putative introgressive loci can be considered as a proxy for time of introgression, which ranges from 0.97 Mya to ∼4.6 kya. However, caution must be exercised as direct inference of admixture time based on tract length distribution can be misleading due to violation of underlying model assumptions (Liang and Nielsen 2014). Moreover, gene flow from archaic humans was relatively weak, with ∼92% of these candidate loci having frequencies of the putative archaic haplotype <10% (Supplemental Fig. S9). Together, our results imply that frequent but low-level interbreeding between archaic and modern humans or their ancestors might have occurred in the past in Africa.
Modeling the decay of archaic-admixture-induced LD in the genetic distance range 0.02–1 cM among our candidate introgressive loci, we found evidence of at least one African archaic admixture event within the last ∼150,000 yr. From our simulation study, this inferred admixture date of ∼9000 (95% C.I.: 1305–28,275) yr ago should be treated as a lower bound because the LD-based method used here indeed tends to substantially underestimate the actual date of admixture (Results; Supplemental Table S2). In addition, by comparing the joint and marginal distributions of TMRCA and genetic length among the inferred candidate loci and whole-genome archaic admixture simulations, we found that two-wave archaic admixture models reproduce the S* signals of the data qualitatively better than single-wave models, although the two-wave models are still an incomplete description of the data (Fig. 5; Supplemental Figs. S12, S13). When using genetic distances in the range of 0.002–1 cM, we rejected a single-wave admixture model. Our simulation study, however, shows that this method also has low statistical specificity (i.e., high false rejection rate) in this genetic distance regime (Supplemental Tables S3, S5). We cannot rule out that processes other than archaic introgression may have produced the signals we observed. Thus, although our inferences indeed reject the null model of no archaic admixture and find modest evidence for recurrent archaic admixture, further work is needed to better characterize the nature of the admixture process in Africa. Indeed, recent studies favor models featuring recurrent archaic admixture outside Africa (Kim and Lohmueller 2015; Vernot and Akey 2015). Neanderthals coexisted and interbred with modern humans as well as Denisovans in Eurasia from at least 100 kya until ∼30 kya before they disappeared from the fossil record (Wall and Hammer 2006; Reich et al. 2011; Pääbo 2014; Prüfer et al. 2014; Veeramah and Hammer 2014). Inside the African continent, although the putative source population(s) of archaic admixture is unclear, hominin fossils indicate the coexistence of several morphologically distinct Homo lineages in the past 2.8 million yr (Potts 2013). Some Middle-Later Pleistocene hominin fossils show intermediate forms with both modern and archaic features (Bräuer 2008; Rightmire 2009; Harvati et al. 2011). Furthermore, recent studies show evidence for Neanderthal-like lineages in several sub-Saharan African populations, possibly resulting from back migration of admixed non-African populations (Wang et al. 2013; Gallego Llorente et al. 2015). Together, these results suggest there was ample opportunity for admixture to occur among different hominin forms in Africa.
We note that the confidence interval for the date of our inferred single-pulse admixture event (9048 kya, 95% C.I.: 1.3–28.2 kya) encompasses the estimated age of fossils (∼13 kya) at the Iwo Elero site in Nigeria that exhibit cranial features intermediate between those of archaic and modern humans (Harvati et al. 2011). However, it is important to point out that archaic introgression need not have been directly into the ancestors of modern Pygmies; rather, it may have resulted from recent gene flow from one or more modern human populations that themselves were recently admixed or that shared recent common ancestry with some unknown archaic hominin(s). The date of the inferred admixture is coincident with the development of agriculture in Africa ∼5–10 kya (Phillipson 2005) and the estimated time of agriculture expansion for Niger-Kodorfanian-speaking farmers ∼7 kya (95% C.I.: 5.7–9.6 kya) (Li et al. 2014). African Pygmies have undergone extensive gene flow with neighboring farmers (Patin et al. 2009; Tishkoff et al. 2009; Jarvis et al. 2012; Hsieh et al. 2016), and recent studies suggest that some Western African populations, including the Niger-Kodorfanian Yoruba farmers from Nigeria, show strong signals of ancient admixture (Plagnol and Wall 2006; Wall et al. 2009). Thus, it is plausible that archaic lineages associated with this inferred admixture event introgressed recently into one or more non-Pygmy African populations, such as the ancestors of African farmers, and subsequently entered the Pygmy population through recent gene flow from these non-Pygmy neighboring groups. Nevertheless, because our simulation study shows the tendency of underestimation of the archaic admixture date using the LD-based approach, caution is warranted while interpreting the inferred admixture time.
Inferring more descriptive demographic models for archaic-AMH divergence time, introgression time, and admixture proportion will require a much larger sample of modern human genome sequences. It will also require more sophisticated demographic models that incorporate not only archaic admixture, but recent changes in the size and structure of AMH populations. Although our inferences are limited as a result of a relatively small sample size, an advantage of our whole-genome inference framework is that it controls for confounding variables through the use of whole-genome simulations and realistic demographic null models that account for variation in genomic mutation and recombination rates. Future work may be needed to control for other sources of uncertainty, such as model misspecification; yet we believe the inference approach presented here offers an improved framework for shedding light on the question of archaic admixture in Africa.
Our results suggest that gene flow between archaic forms and the ancestors of modern humans in Africa may have occurred multiple times or continuously at low levels over evolutionary time. Throughout the entire Pleistocene, the African environment underwent a series of wet-dry episodes (Potts 2013), many of which coincide with the first and last appearances of hominin fossils and major stone tool transitions in Africa. This suggests a scenario characterized by pressure to adapt to a variety of environmental conditions over the course of the Pleistocene (deMenocal 2011; Potts 2013). This may account for the fossil evidence of a variety of forms exhibiting different combinations of archaic and anatomically modern features (Bräuer 2008; Rightmire 2009; Harvati et al. 2011). It is tempting to speculate that recurrent interbreeding among diverse hominins may have played a key role in producing novel genotypes, which in turn facilitated the process of adaptation to changing environmental conditions. Thus, our own species may have emerged as the sole surviving member of the genus Homo as a result of the acquisition of genes that descend from divergent ancestors that occupied different ecological niches over a wider temporal and spatial range (Stringer 2012). The ultimate resolution of this question, and in particular for regions of the genome that code for anatomically modern traits, has important implications for human origin models and the evolutionary processes that took place during the archaic-to-modern transition (Hammer 2013).
Methods
Whole-genome sequencing data and data filtering
DNA of the four Biaka Pygmies were obtained from publicly available cell lines administrated by the Centre d'Etude du Polymorphism Human Genome Diversity Panel (Li et al. 2008). Sequencing of the three Baka Pygmies has been previously described (Lachance et al. 2012) (NCBI dbSNP submitter batch IDs: Lachance2012Cell_snp, Lachance2012Cell_deletion, Lachance2012Cell_insertion, and Lachance2012Cell_complex_substitution.). Nine Yoruba genomes were downloaded from the CGI public data repository (Drmanac et al. 2010). The median coverage across these samples in this study is 60.5×. The genome assembly and variant calling were generated using the standard CGI Assembly Pipeline 1.10, CGA Tools 1.4, and NCBI Human Reference Genome build 37. Because CGI is currently not supporting GRCh38, our data were aligned according to GRCh37/hg19. Given the relatively small sample size of our data, to assure genotyping quality, we included only variants that were (1) fully called across all samples, (2) not in any known/called indels, (3) not in any known/called copy number variants, (4) not in any known segmental duplication regions, and (5) had valid corresponding human-chimpanzee alignment (PanTro3, hg19). Databases that were used for the steps 3, 4, and 5 were downloaded from the UCSC Genome Browser as of May 2013. This filtering process resulted in a total of 10,865,288 autosomal SNVs. According to the Genome Reference Consortium (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/info/index.shtml), the major improvements in GRCh38, compared to GRCh37/hg19, are in (1) providing alternative scaffolds to better represent complex regions (e.g., centromeres) in the genome; (2) closing/reducing the known gaps in the previous releases (sizes of gaps: ∼234 Mb or 7.6% in GRCh37/hg19 and ∼151 Mb or 4.9% in GRCh38); and (3) correcting assembly errors, particularly for complex regions, in the previous releases. Although realigning our data to GRCh38 might yield more data in complex regions, we do not expect this will affect our conclusions, because we excluded most complex regions and known gaps from all of our analyses.
Genetic recombination maps
For our analyses of the data and our simulations, we used two recently published African genetic maps: the Yoruba HapMap recombination map (based on linkage disequilibrium patterns) (The International HapMap Consortium 2007) and the African American recombination map (based on breakpoints of admixture tracts) (Hinch et al. 2011). Because these maps are built using different populations and different inference approaches, they are expected to be sensitive to different time periods of human history. Therefore, to avoid possible biases in our inferences, we chose to repeat all of our analyses with these two maps. Markers within the first 5 Mb on each chromosome were removed due to possible underestimation of rates of recombination in the African American recombination map as suggested by Hinch et al. (2011). For consistency, this filter was also applied to data generated using the HapMap recombination map. Note that because our candidate loci were determined using both the HapMap and African American genetic maps and the recombination rates per megabase (cM/Mb) of these loci are highly correlated between these two maps (Pearson's correlation = 0.97, P < 2.2 × 10−16), we only reported values for both recombination rate and genetic distance using HapMap unless stated otherwise.
Coalescent whole-genome simulations
We performed whole-genome coalescent simulations using MaCS (Chen et al. 2009), which simulates two demographic models that were previously inferred using these samples (Hsieh et al. 2016). To account for mutational heterogeneity, we first divided the whole genome into windows of 25,000 nucleotides. For the jth window, the population genetic mutation parameter was estimated using ∂a∂i (Gutenkunst et al. 2009) given a demographic model. Then each MaCS simulation was conducted using the mutation parameter, , the largest θ estimated among all of the windows. Lastly, for the jth window, we adjusted its mutation rate by dropping the proportion of the simulated variants. We also incorporated the two genetic recombination maps that are available for Africans to capture recombination hotspots in our whole-genome simulations. For sequences between markers in the map, including hotspots and coldspots, a uniform interpolated recombination rate was assumed. For all of our simulated sequences, we excluded the same variant sites that were removed in the real data due to our quality filtering criteria (Methods). To account for the uncertainty of the demographic parameter estimates, we simulated 1000 models drawn from the confidence intervals of the parameter estimates for each of the two best-fit demographic models.
We used the same whole-genome simulation framework to simulate whole-genome data of archaic introgression under various plausible archaic admixture scenarios (Supplemental Fig. S10; Supplemental Tables S2–S5). Models underlying these simulations were based on Hammer et al. (2011), and the best-fit demographic model (Model-1) (Supplemental Fig. S2A) was taken from Hsieh et al. (2016). Because we are interested in parameters related to archaic admixture, for simplicity we fixed other parameters, including the effective population sizes for the ancestors of modern humans, Pygmies, and farmer populations, times of divergence between populations, as well as the asymmetric gene-flow between Pygmy and farmer populations, as described in Hammer et al. (2011) and Hsieh et al. (2016). In addition, we assumed the same population size (6700 individuals) for both the common ancestor of archaic/modern humans and the archaic human population. For single-wave archaic admixture simulations, the time of admixture into modern human lineages was set at 1200, 2700, 5500, or 7800 generations ago (Supplemental Fig. S10). For the two-wave archaic admixture simulations, we set the two events to be 7800 and 300 generations ago in order to investigate extreme cases. The admixture proportion was set to be 2%, 5%, or 10% in each of the simulations (Supplemental Tables S2–S5).
Identification of archaic introgressive sequences and hypothesis testing of no archaic admixture
To detect archaic introgressive sequences, we calculated the summary statistic S*, which is known to be sensitive to archaic admixture without using an archaic reference genome (Plagnol and Wall 2006). S* searches for sequences in which SNPs likely originated in an archaic population (deep TMRCA of individual loci) and are still in strong LD (i.e., these sites are congruent), and it returns variant sites that are congruent. We implemented the dynamic programming approach for calculating S* using the same scoring function (Plagnol and Wall 2006), except that for any given SNP, we chose to only tolerate at most two mismatches among chromosomes because our sample size is smaller (N = 7) than the original study of S* (N = 12, Yoruba population) (Plagnol and Wall 2006). We calculated S* for regions defined by a sliding window of 200 SNPs with a step size of 50 SNPs. Windows longer than 1 Mb were dropped to avoid complex genomic regions, such as centromeres or large structural variants. We determined the significance of S* values from these windows by comparing with our whole-genome simulations. A window was deemed significant if its corresponding P-value was within the top 1% in the P-value distribution. We estimated false discovery rates by adopting the method of Storey and Tibshirani (2003), but estimated the tuning parameter λ using the procedure and parameter settings suggested in Williamson et al. (2007). Together, we generated four sets of whole-genome simulations based on the combination of two models and two genetic recombination maps. Finally, we defined candidate archaic introgression loci to be those windows that were significant in all four whole-genome simulation data sets. The bounds of an introgressive candidate locus are the leftmost and rightmost congruent sites selected by S*; this length thus represents a lower bound in length for each candidate.
To formally test the hypothesis of no admixture, we performed a goodness-of-fit test with a significance level of 1% to compare the P-value distribution from observed data to the expected P-value distribution from a simulation under the best-fit demographic null model. The expected null P-value distribution was constructed by treating a randomly drawn whole-genome simulation as the real data. The goodness-of-fit test was performed using a one-sided Mann-Whitney U test implemented in R (R Core Team 2015).
The haplotype network plot was generated using PopART v1.7 (http://popart.otago.ac.nz). Hierarchical clustering for haplotype data was performed using the R function “hclust” in the stats package (R Core Team 2015) with pairwise nucleotide differences as the distance matrix.
Haplotypes were computationally phased using BEAGLE v3.1.1 (Browning and Browning 2007). To enhance phasing accuracy, we included additional public genotype data: a Bakola and a Bedzen genome (CGI Assembly Pipeline 1.10, CGA Tools 1.4) from Lachance et al. (2012); 16 Biaka Pygmies genotyped by the Human Genome Diversity Project (HGDP; Illumina 650k) (Li et al. 2008). The nine Yoruba genomes were phased separately using the same approach, with an additional four Luhya genomes from the CGI public data repository, genotype data of 81 Yoruba and 86 Luhya samples from the 1000 Genomes Project, and 21 Yoruba and 10 Luhya samples from the HGDP. All positions were converted into hg19 coordinates using UCSC liftOver utility, if necessary.
Statistical inference on archaic admixture in Western African Pygmies
Under the Wright-Fisher evolution, the TMRCA is an estimator for the time of divergence between the archaic and modern human sequences (Wall 2000; Lachance et al. 2012), and the genetic length of a putative archaic sequence represents a proxy of the time of sequence introgression (Pool and Nielsen 2009; Gravel 2012). We used the method of Thomson et al. (2000) to estimate TMRCA for each candidate archaic introgressive locus, assuming a divergence of 6 My between human and chimpanzee (Glazko and Nei 2003) and a generation time of 29 yr (Fenner 2005; Langergraber et al. 2012). For each candidate locus, we estimated the genetic length by taking the difference between the two ends of the locus in terms of their genetic positions according to the two genetic recombination maps (The International HapMap Consortium 2007; Hinch et al. 2011). To further investigate our candidate archaic sequences, for each of these candidate sequences, we calculated the D3 statistic, an estimator for the amount of archaic admixture (Hammer et al. 2011). D3 was originally defined as min(|G1|, |G2|), where G1 and G2 are the two most diverged basal haplotypes of congruent sites identified by S*, identified by clustering all sequences. To avoid potential biases in D3 due to haplotype phasing errors, we instead constructed an estimator of D3, , based on the genotypes of the unphased congruent sites. Under the Wright-Fisher model, the expected frequency for a given allele does not change over time (Hartl et al. 1997). Thus, given that a sequence was truly introgressed from an archaic population into modern humans, the derived allele counts of the congruent sites across this locus would reflect the strength of admixture. Thus, we defined as the mode of the derived allele frequency of the congruent variant sites for each individual locus. Loci that have more than one mode were excluded because they might add noise to downstream analyses.
Parametric coalescent simulations and inference on admixture events using LD information
Following recent studies (Sankararaman et al. 2012; Loh et al. 2013; Moorjani et al. 2013; Pickrell et al. 2014), for any pair of alleles that arose on the archaic lineage and introgressed into modern humans at time tadmix generations ago, the survival probability is defined as , where d is the genetic distance between the two alleles. The two-locus Wright-Fisher diffusion (Ohta and Kimura 1969) predicts that
where is the expected LD at present; n is the number of independent admixture events; tadmix,i is the time of the ith admixture events in generations; and ai is the intercept of the exponential curve. We calculated the expected LD for any pair of congruent sites selected by S* at a genetic distance d as
where S(x) is the set of all pairs of congruent SNPs that are at a genetic distance d. is the covariance between the unphased genotypes observed at SNP i and j, a robust estimator for the measurement of LD (Rogers and Huff 2009; Sankararaman et al. 2012). The R function nls was used for curve-fitting, which maximizes the following log-likelihood function of each model using numerical optimization to obtain the maximum likelihood estimates for both the intercepts ai and rate of decay tadmix,i because there are no close-form solutions for the parameters in nonlinear least squares.
where σ is the variance of residual between Dobserved and Dfitted; and i indexes over the N genetic distance bins. The date estimates of gene flow events were then inferred based on the rate of decay of the best-fit exponential curve. Model selection was performed through a likelihood ratio test in order to determine the best-fit exponential model.
To assess the performance of this approach for distinguishing between single (simple) and two-pulse (complex) models, we simulated whole-genome data of archaic admixture as described in the previous section. Based on our simulation study, we chose to fit LD decay curves to the observed mean LD estimates for d in the range 0.02 cM to 1 cM in increments of 0.001 cM, which resulted in model inferences consistent with the underlying models in our simulations. To test for possible older admixture events, we also fit LD decay curves for d in the range 0.002 cM to 1 cM, although caution is required in interpretation, because LD within a short distance may be confounded by background LD. To ensure the convergence of the best-fit curve for each exponential model, we generated 100 random initial sets as start parameters and performed optimization using a Gauss-Newton algorithm. Finally, the confidence intervals of the parameters (intercepts and rates of decay) for the best-fit exponential curve were estimated based on 100 conventional nonparametric bootstraps. We assumed a generation time of 29 yr (Fenner 2005; Langergraber et al. 2012) to convert the date estimates to physical years.
Data access
The Biaka sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP067698. The variants for Biaka genomes have been submitted to NCBI dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) under submitter batch ID: HammerLab_Biaka_CGI.
Acknowledgments
Support for this work was provided by the National Institutes of Health to J.D.W. and M.F.H. (R01 HG005226). P.H. and R.N.G. were supported by National Science Foundation (NSF) grant DEB-1146074. S.A.T. was supported by National Institutes of Health (NIH) grants 1R01GM113657-01 and 8DP1ES022577-04. J.L. was supported by an NIH NRSA postdoctoral fellowship F32HG006648.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.196634.115.
References
- Bandelt HJ, Forster P, Rohl A. 1999. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48. [DOI] [PubMed] [Google Scholar]
- Bräuer G. 2008. The origin of modern anatomy: by speciation or intraspecific evolution? Evol Anthropol 17: 22–37. [Google Scholar]
- Browning SR, Browning BL. 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81: 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana MG, Bower MA, Crabtree PJ. 2013. Ancient DNA for the archaeologist: the future of African research. Afr Archaeol Rev 30: 21–37. [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza A. 1994. The history and geography of human genes. Princeton University Press, Princeton, NJ. [Google Scholar]
- Chen GK, Marjoram P, Wall JD. 2009. Fast and flexible simulation of DNA sequence data. Genome Res 19: 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deMenocal PB. 2011. Anthropology. Climate and human evolution. Science 331: 540–542. [DOI] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81. [DOI] [PubMed] [Google Scholar]
- Evans PD, Mekel-Bobrov N, Vallender EJ, Hudson RR, Lahn BT. 2006. Evidence that the adaptive allele of the brain size gene microcephalin introgressed into Homo sapiens from an archaic Homo lineage. Proc Natl Acad Sci 103: 18178–18183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner JN. 2005. Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. Am J Phys Anthropol 128: 415–423. [DOI] [PubMed] [Google Scholar]
- Fu Q, Li H, Moorjani P, Jay F, Slepchenko SM, Bondarev AA, Johnson PL, Aximu-Petri A, Prüfer K, de Filippo C, et al. 2014. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514: 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego Llorente M, Jones ER, Eriksson A, Siska V, Arthur KW, Arthur JW, Curtis MC, Stock JT, Coltorti M, Pieruccini P, et al. 2015. Ancient Ethiopian genome reveals extensive Eurasian admixture throughout the African continent. Science 350: 820–822. [DOI] [PubMed] [Google Scholar]
- Garrigan D, Mobasher Z, Kingan SB, Wilder JA, Hammer MF. 2005. Deep haplotype divergence and long-range linkage disequilibrium at Xp21.1 provide evidence that humans descend from a structured ancestral population. Genetics 170: 1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazko GV, Nei M. 2003. Estimation of divergence times for major lineages of primate species. Mol Biol Evol 20: 424–434. [DOI] [PubMed] [Google Scholar]
- Gravel S. 2012. Population genetics models of local ancestry. Genetics 191: 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, et al. 2010. A draft sequence of the Neandertal genome. Science 328: 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5: e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF. 2013. Human hybrids. Sci Am 308: 66–71. [DOI] [PubMed] [Google Scholar]
- Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. 2011. Genetic evidence for archaic admixture in Africa. Proc Natl Acad Sci 108: 15123–15128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Pittman A, Myers A, Gwinn-Hardy K, Fung HC, de Silva R, Hutton M, Duckworth J. 2005. Evidence suggesting that Homo neanderthalensis contributed the H2 MAPT haplotype to Homo sapiens. Biochem Soc Transac 33(Pt 4): 582–585. [DOI] [PubMed] [Google Scholar]
- Hartl DL, Clark AG, Clark AG. 1997. Principles of population genetics. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Harvati K, Stringer C, Grün R, Aubert M, Allsworth-Jones P, Folorunso CA. 2011. The Later Stone Age calvaria from Iwo Eleru, Nigeria: morphology and chronology. PLoS One 6: e24024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa T, Aki I, Varki A, Satta Y, Takahata N. 2006. Fixation of the human-specific CMP-N-acetylneuraminic acid hydroxylase pseudogene and implications of haplotype diversity for human evolution. Genetics 172: 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinch AG, Tandon A, Patterson N, Song Y, Rohland N, Palmer CD, Chen GK, Wang K, Buxbaum SG, Akylbekova EL, et al. 2011. The landscape of recombination in African Americans. Nature 476: 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh P, Veeramah KR, Lachance J, Tishkoff SA, Wall JD, Hammer MF, Gutenkunst RN. 2016. Whole-genome sequence analyses of Western Central African Pygmy hunter-gatherers reveal a complex demographic history and identify candidate genes under positive natural selection. Genome Res (this issue) 26: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Sánchez E, Jin X, Asan, Bianba Z, Peter BM, Vinckenbosch N, Liang Y, Yi X, He M, Somel M, et al. 2014. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512: 194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis JP, Scheinfeldt LB, Soi S, Lambert C, Omberg L, Ferwerda B, Froment A, Bodo JM, Beggs W, Hoffman G, et al. 2012. Patterns of ancestry, signatures of natural selection, and genetic association with stature in Western African Pygmies. PLoS Genet 8: e1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BY, Lohmueller KE. 2015. Selection and reduced population size cannot explain higher amounts of Neandertal ancestry in East Asian than in European human populations. Am J Hum Genet 96: 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance J, Vernot B, Elbers CC, Ferwerda B, Froment A, Bodo JM, Lema G, Fu W, Nyambo TB, Rebbeck TR, et al. 2012. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell 150: 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langergraber KE, Prüfer K, Rowney C, Boesch C, Crockford C, Fawcett K, Inoue E, Inoue-Muruyama M, Mitani JC, Muller MN, et al. 2012. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proc Natl Acad Sci 109: 15716–15721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, Cann HM, Barsh GS, Feldman M, Cavalli-Sforza LL, et al. 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319: 1100–1104. [DOI] [PubMed] [Google Scholar]
- Li S, Schlebusch C, Jakobsson M. 2014. Genetic variation reveals large-scale population expansion and migration during the expansion of Bantu-speaking peoples. Proc Biol Sci 281: 20141448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M, Nielsen R. 2014. The lengths of admixture tracts. Genetics 197: 953–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh PR, Lipson M, Patterson N, Moorjani P, Pickrell JK, Reich D, Berger B. 2013. Inferring admixture histories of human populations using linkage disequilibrium. Genetics 193: 1233–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J. 2007. Hybrid speciation. Nature 446: 279–283. [DOI] [PubMed] [Google Scholar]
- Mendez FL, Watkins JC, Hammer MF. 2012. A haplotype at STAT2 introgressed from Neanderthals and serves as a candidate of positive selection in Papua New Guinea. Am J Hum Genet 91: 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez FL, Watkins JC, Hammer MF. 2013. Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol Biol Evol 30: 798–801. [DOI] [PubMed] [Google Scholar]
- Moorjani P, Patterson N, Hirschhorn JN, Keinan A, Hao L, Atzmon G, Burns E, Ostrer H, Price AL, Reich D. 2011. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet 7: e1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorjani P, Thangaraj K, Patterson N, Lipson M, Loh PR, Govindaraj P, Berger B, Reich D, Singh L. 2013. Genetic evidence for recent population mixture in India. Am J Hum Genet 93: 422–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, Kimura M. 1969. Linkage disequilibrium at steady state determined by random genetic drift and recurrent mutation. Genetics 63: 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pääbo S. 2014. The human condition—a molecular approach. Cell 157: 216–226. [DOI] [PubMed] [Google Scholar]
- Patin E, Laval G, Barreiro LB, Salas A, Semino O, Santachiara-Benerecetti S, Kidd KK, Kidd JR, Van der Veen L, Hombert JM, et al. 2009. Inferring the demographic history of African farmers and Pygmy hunter–gatherers using a multilocus resequencing data set. PLoS Genet 5: e1000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipson DW. 2005. African archaeology. Cambridge University Press, New York. [Google Scholar]
- Pickrell JK, Patterson N, Barbieri C, Berthold F, Gerlach L, Guldemann T, Kure B, Mpoloka SW, Nakagawa H, Naumann C, et al. 2012. The genetic prehistory of southern Africa. Nat Commun 3: 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Patterson N, Loh PR, Lipson M, Berger B, Stoneking M, Pakendorf B, Reich D. 2014. Ancient west Eurasian ancestry in southern and eastern Africa. Proc Natl Acad Sci 111: 2632–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagnol V, Wall JD. 2006. Possible ancestral structure in human populations. PLoS Genet 2: e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool JE, Nielsen R. 2009. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics 181: 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts R. 2013. Hominin evolution in settings of strong environmental variability. Quat Sci Rev 73: 1–13. [Google Scholar]
- Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Racimo F, Sankararaman S, Nielsen R, Huerta-Sánchez E. 2015. Evidence for archaic adaptive introgression in humans. Nat Rev Genet 16: 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PL, et al. 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468: 1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Patterson N, Kircher M, Delfin F, Nandineni MR, Pugach I, Ko AM, Ko YC, Jinam TA, Phipps ME, et al. 2011. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet 89: 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rightmire GP. 2009. Out of Africa: modern human origins special feature: middle and later Pleistocene hominins in Africa and Southwest Asia. Proc Natl Acad Sci 106: 16046–16050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers AR, Huff C. 2009. Linkage disequilibrium between loci with unknown phase. Genetics 182: 839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Patterson N, Li H, Pääbo S, Reich D. 2012. The date of interbreeding between Neandertals and modern humans. PLoS Genet 8: e1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, Patterson N, Reich D. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507: 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci 100: 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer C. 2012. Lone survivors: how we came to be the only humans on earth. Macmillan, New York. [Google Scholar]
- Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW. 2000. Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc Natl Acad Sci 97: 7360–7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, et al. 2009. The genetic structure and history of Africans and African Americans. Science 324: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyford AD, Ennos RA. 2012. Next-generation hybridization and introgression. Heredity 108: 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, Hammer MF. 2014. The impact of whole-genome sequencing on the reconstruction of human population history. Nat Rev Genet 15: 149–162. [DOI] [PubMed] [Google Scholar]
- Vernot B, Akey JM. 2014. Resurrecting surviving Neandertal lineages from modern human genomes. Science 343: 1017–1021. [DOI] [PubMed] [Google Scholar]
- Vernot B, Akey JM. 2015. Complex history of admixture between modern humans and Neandertals. Am J Hum Genet 96: 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villmoare B, Kimbel WH, Seyoum C, Campisano CJ, DiMaggio EN, Rowan J, Braun DR, Arrowsmith JR, Reed KE. 2015. Paleoanthropology. Early Homo at 2.8 Ma from Ledi-Geraru, Afar, Ethiopia. Science 347: 1352–1355. [DOI] [PubMed] [Google Scholar]
- Wall JD. 2000. Detecting ancient admixture in humans using sequence polymorphism data. Genetics 154: 1271–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Hammer MF. 2006. Archaic admixture in the human genome. Curr Opin Genet Dev 16: 606–610. [DOI] [PubMed] [Google Scholar]
- Wall JD, Lohmueller KE, Plagnol V. 2009. Detecting ancient admixture and estimating demographic parameters in multiple human populations. Mol Biol Evol 26: 1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lachance J, Tishkoff SA, Hey J, Xing J. 2013. Apparent variation in Neanderthal admixture among African populations is consistent with gene flow from non-African populations. Genome Biol Evol 5: 2075–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson SH, Hubisz MJ, Clark AG, Payseur BA, Bustamante CD, Nielsen R. 2007. Localizing recent adaptive evolution in the human genome. PLoS Genet 3: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinner D, Arnold ML, Roos C. 2011. The strange blood: natural hybridization in primates. Evol Anthropol 20: 96–103. [DOI] [PubMed] [Google Scholar]