

ABSTRACT
Myxobacteria form complex social communities that elicit multicellular behaviors. One such behavior is kin recognition, in which cells identify siblings via their polymorphic TraA cell surface receptor, to transiently fuse outer membranes and exchange their contents. In addition, outer membrane exchange (OME) regulates behaviors, such as inhibition of wild-type Myxococcus xanthus (DK1622) from swarming. Here we monitored the fate of motile cells and surprisingly found they were killed by nonmotile siblings. The kill phenotype required OME (i.e., was TraA dependent). The genetic basis of killing was traced to ancestral strains used to construct DK1622. Specifically, the kill phenotype mapped to a large “polyploid prophage,” Mx alpha. Sensitive strains contained a 200-kb deletion that removed two of three Mx alpha units. To explain these results, we suggest that Mx alpha expresses a toxin-antitoxin cassette that uses the OME machinery of M. xanthus to transfer a toxin that makes the population “addicted” to Mx alpha. Thus, siblings that lost Mx alpha units (no immunity) are killed by cells that harbor the element. To test this, an Mx alpha-harboring laboratory strain was engineered (by traA allele swap) to recognize a closely related species, Myxococcus fulvus. As a result, M. fulvus, which lacks Mx alpha, was killed. These TraA-mediated antagonisms provide an explanation for how kin recognition specificity might have evolved in myxobacteria. That is, recognition specificity is determined by polymorphisms in traA, which we hypothesize were selected for because OME with non-kin leads to lethal outcomes.
IMPORTANCE The transition from single cell to multicellular life is considered a major evolutionary event. Myxobacteria have successfully made this transition. For example, in response to starvation, individual cells aggregate into multicellular fruiting bodies wherein cells differentiate into spores. To build fruits, cells need to recognize their siblings, and in part, this is mediated by the TraA cell surface receptor. Surprisingly, we report that TraA recognition can also involve sibling killing. We show that killing originates from a prophage-like element that has apparently hijacked the TraA system to deliver a toxin to kin. We hypothesize that this killing system has imposed selective pressures on kin recognition, which in turn has resulted in TraA polymorphisms and hence many different recognition groups.
INTRODUCTION
Myxobacteria inhabit the soil and, as such, live in taxonomically diverse environments in which thousands of microbial species and subspecies compete for scarce resources (1). Remarkably, from these heterogeneous populations, myxobacteria assemble collectives that function like tissues. These multicellular behaviors include rhythmic and synchronized movements that culminate in fruiting body formation. To accomplish this, myxobacteria must recognize their neighbors to determine if they are friend, foe, or food. How myxobacteria recognize kin and assemble homogenous populations is an emerging field of study.
The most thoroughly described cell-cell recognition system in myxobacteria is mediated by the TraA polymorphic cell surface receptor. This receptor, with its partner protein, TraB, controls the fusion and exchange of outer membrane (OM) material between cells (2). To engage in OM exchange (OME), the partnering cells must express traA alleles that belong to the same recognition group (3). Because OME leads to the transfer of many different proteins and lipids, it can, in principle, result in cooperative or antagonist interactions. In other bacterial transport systems, cargo transfer is typically unidirectional and the outcomes are usually antagonistic to the recipient. For example, the type III, IV, V, and VI secretion systems transfer effectors to target cells that act as toxins or as virulence or selfish elements (4–7). Cooperative bacterial transfer systems have rarely been described. In contrast, OME involves bidirectional cargo transfer, in which both cells must express compatible TraA/B machinery, suggesting that these interactions are mutually sought.
Myxobacteria are gliding bacteria that translocate in a smooth motion on solid surfaces along their long axis (8). The movement of cell groups is called swarming and is a cooperative behavior, because their expansion rate increases with cell density (8). In OME, gliding is indirectly required to facilitate membrane fusion and fission (9). Gliding is powered by two separate engines, referred to as A (adventurous) and S (social) motility (8). The S-engine consists of type IV pili, and the A-engine is a multiprotein complex that includes mobile cell surface adhesins (10). S-motility is proficient at swarming on soft, moist agar and requires cell-cell contact. In contrast, A-motility is adapted for hard and drier surfaces, on which individual or small groups of cells move (11). A nonmotile mutant (A− S−) therefore typically requires two mutations, one in each system.
Because Myxococcus xanthus is both a social and predatory species, it is a good model system to study the interplay between cooperative and competitive interactions. Its extensive social behaviors suggest that M. xanthus has evolved a means to regulate these interactions. One example is fruiting body development in which a subpopulation develops into environmentally resilient spores in response to starvation, while other cells lyse or form persister-like cells (12). How cell fates are determined is poorly understood but may involve competitive interactions interwoven within cooperative behaviors. Likewise, OME appears to involve both cooperative and competitive interactions. Cooperative interactions are suggested by sharing of cellular resources and, in some cases, the ability of cells to repair their damaged sibling cells (13). In contrast, the swarming and developmental behaviors of some motile strains can be antagonized by OME with some nonmotile strains (2). This antagonistic response is potent, as a ratio of 1 nonmotile cell to 50 motile cells blocks the latter from swarming (14). The nature of swarm inhibition is the focus of this study, in which we found that ancestral strains kill siblings that were derived from them. The kill phenotype required OME and was engineered into a laboratory strain to antagonize an environmental isolate. We suggest that the kill phenotype arises from a toxin-antitoxin system that maps to a large polyploid prophage-like element that was fortuitously deleted in laboratory strains. We discuss social and evolutionary implications of these findings.
MATERIALS AND METHODS
Growth conditions.
The bacterial strains used in this study are listed in Table 1. M. xanthus was grown in the dark at 33°C in CTT medium (1% Casitone, 10 mM Tris-HCl [pH 7.6], 8 mM MgSO4, 1 mM KH2PO4) with or without kanamycin (Km; 50 μg ml−1), zeocin (Zm; 50 μg ml−1), oxytetracycline (Tc; 10 μg ml−1), or galactose (Gal, 2%), as needed. For swarm inhibition assays, agar (1.5%) plates consisted of 0.5× CTT (0.5% Casitone) with 2 mM CaCl2 added after autoclaving or TPM (10 mM Tris-HCl [pH 7.6], 8 mM MgSO4, 1 mM KH2PO4) agar was used. Standard competition assays were done on 1.5% agar plates with 0.25× CTT (0.25% Casitone). For competition assays on semisolid agar, CTT with 0.5% agar was used. To generate micrographs of mixed swarms, 0.25× CTT–0.8% agarose pads were made on glass microscope slides. To determine the numbers of CFU of mixed cultures, CTT agar plates were supplemented with antibiotics to select for a particular strain, and colonies were counted after 6 days of incubation. tdTomato expression was induced in liquid and on agar plates with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). To grow Myxococcus fulvus, 0.5× CTT was supplemented with 0.5% yeast extract. For routine cloning, Escherichia coli DH5α pir+ was grown at 37°C in LB, and tetracycline (10 μg ml−1) or Km (50 μg ml−1) was used for selection as needed.
TABLE 1.
Strains used in this study
| Strain | Relevant propertiese | Source and/or reference(s) |
|---|---|---|
| DH5α pir+ | E. coli cloning strain | Lab collection |
| DK101b | A+ S− pilQ1; M. xanthus, also known as FB or DZF1 | 18, 27 |
| DK1622 | A+ S+; WTd M. xanthus, ΔMx alpha | 18, 28 |
| HW-1 | A+ S+; WT M. fulvus | ATCC; 48 |
| YS (DK1600)b | A+ S− pilG or pilH1; derived from FB | 18, 47 |
| DZ1 | A− S−; ΔMx alpha, derived from FB, multiple mutations | 30, 31 |
| DZ2 | A+ S+; WT M. xanthus, contains Mx alpha repeats | 20, 21 |
| DK360b | A− S− cglE1 pilQ1 | 15, 49 |
| DK391b | A− S− cglD1 pilQ1 | 15 |
| DK1217b | A− S+ aglB1 (aglQ1); parent strain to DK1622 | 18 |
| DK1633b | A− S− cglC1 pilQ1633 | 18 |
| DK8601b | A− S− aglB1 ΔpilA::Tc Tcr | 17 |
| DK8605b | A− S+ aglB1 PpilA-gfp Kmr | 17 |
| DK8606b | A− S− aglB1 ΔpilA PpilA-gfp Kmr | 17 |
| DK8615a | A+ S− ΔpilQ | 18 |
| DK8616b | A− S− aglB1 ΔpilQ | 18 |
| DK10410a | A+ S− ΔpilA | 50 |
| DW703a | A+ S+ ΔpilS PpilA-gfp Kmr | This study |
| DW704b | A− S− cglF1 ΔpilA::Tc Tcr | 2 |
| DW709a | A+ S− ΔpilA PpilA-gfp Kmr | 51 |
| DW1048b | A− S− aglB1 ΔpilA::Tc PpilA-SSIM-mCherry(pDP1) Tcr Kmr | 9 |
| DW1411b | A− S− aglB1 ΔpilA::Tc PpilA-SSOM-mCherry(pXW6) Tcr Smr | 2 |
| DW1415a | A+ S− ΔpilQ traA::pDP2 Kmr | 2 |
| DW1438a | A− S− Ω1903 (Tn5) cglC2 ΔpilQ Kmr | 15 |
| DW1443a | A− S− Ω1931 (Tn5) cglE1 ΔpilQ Kmr | 15 |
| DW1445a | A− S− Ω1919 (Tn5) cglF1 ΔpilQ Kmr | 15 |
| DW1467b | A− S− aglB1 ΔpilA::Tc ΔtraA Tcr | 3 |
| DW1470b | A− S− DW1467 PpilA-traAM .fulvus (pDP25) Kmr Tcr | 3 |
| DW1482a | A+ S− ΔpilQ ΔtraA | This study |
| DW1613a | A+ S− DW709 traA::pAD4 Kmr Zmr | This study |
| DW1614b | A− S− DW1470 PpilA-SSOM-mCherry(pXW6) Kmr Smr Tcr | This study |
| DW1615a | A+ S− ΔpilQ ΔtraA PpilA-traAM .fulvus (pDP25) PpilA-SSOM-mCherry(pXW6) Kmr Smr | This study |
| DW1616a | A+ S+ PpilA-SSIM-mCherry(pDP1) Kmr | This study |
| DW1617a | A+ S− ΔpilQ omrA::mini-Tn5 pTdTomato Kmr Tcr | This study |
| DW1618a | A+ S− ΔpilQ omrB::pAD3 pTdTomato Kmr Tcr | This study |
| DW1619a | A+ S− ΔpilQ PpilA-SSIM-mCherry(pDP1) Kmr | This study |
| DW1620b | A+ S− pilQ1 pTdTomato Tcr | This study |
| DW2401b | A+ S+ (DK1217/pDP110) Kmr | This study |
| DW2402b | A+ S− (DK8601/pDP110) Kmr | This study |
| DW2403b,c | A− S− aglB1 ΔpilQ ΔMx alpha-29 (markerless) | This study |
| DW2404a | A+ S− ΔpilQ pTdTomato Tcr | This study |
Derived from DK1622 (ΔMx alpha).
Derived from DK101 (FB) and contains three Mx alpha units.
See Materials and Methods for details.
WT, wild type.
Unless otherwise noted, strains listed are M. xanthus strains.
Strain constructions.
Gene disruptions were made by amplifying internal gene fragments by PCR that were then cloned into pCR-XL-TOPO or pCR2.1-TOPO (see Table S1 in the supplemental material). The Tn5-Ω2213 insertion site was identified by a PCR-based method as previously described (15). For aglB1 (aglQ1) rescue, a plasmid was made by amplifying the aglRQS operon and cloning it into pCR2.1-TOPO, generating pDP110. The markerless ΔMx alpha deletion cassette was made by PCR amplification of the corresponding upstream and downstream DNA fragments, and these fragments were placed in pBJ114 by three-piece Gibson cloning (New England BioLabs) to create pCV101. Primers used for PCR are listed in Table S2 in the supplemental material. Colony PCR and restriction analyses were used to confirm clone construction. Verified plasmids (see Table S1) were electroporated into M. xanthus strains, and recombinants were selected on CTT agar with the appropriate antibiotics. M. xanthus transformants were then isolated and verified by diagnostic PCR and/or phenotypic analysis. For DW2403 (ΔMx alpha-29) strain verification, we used diagnostic PCR with primers against MXF1DRAFT_07228 and confirmed that a deletion had occurred in Mx alpha. Additional diagnostic PCRs confirmed that the Mx alpha region corresponding to the end of contig 48 was also absent; however, a region corresponding to contig 58 was unexpectedly present. From the counterselection step, seven additional Galr Kms clones that showed no antagonistic phenotype were tested by PCR and were all found to contain different types of deletions in Mx alpha, but none of them contained the full deletion as planned. We concluded that the large Mx alpha repeats were inherently unstable and that deletions spontaneously occurred at different positions within Mx alpha. See Discussion for further details.
Swarm inhibition.
Experiments were typically done by mixing motile and nonmotile strains at a 1:1 ratio (∼3 × 109 CFU ml−1) and pipetting the mixtures onto the described plates. Unless stated otherwise, the plates were incubated for 72 h at 33°C, and micrographs were taken with a stereomicroscope at 3.2× magnification or with a 10× phase-contrast objective on a compound microscope (2). Time-lapse microscopy was done as described previously (2).
Competition experiments.
Myxococcus strains were grown in CTT overnight, and cells were harvested at mid-log growth (∼3 × 108 CFU ml−1). For fluorescent labeling experiments, either one or both strains were labeled with green fluorescent protein (GFP), tdTomato, or mCherry and mixed at the indicated ratios. Strain mixtures were transferred to agarose pads (5 μl, for direct microscopy) or agar plates (30 μl, to harvest cells) and incubated in a humid chamber. At the indicated times, either the colony edge was observed on agarose pads or cells were collected and washed twice in TPM and observed on a glass slide by phase-contrast and fluorescence microscopies with a fluorescein isothiocyanate (FITC) or Texas Red filter set. At least 200 cells were counted for each replicate to determine the strain ratios. Micrographs were obtained as described previously (2). To determine the numbers of CFU from competition experiments, cells with Tc or Km markers were similarly mixed and collected, and viable cells were enumerated by serial dilution onto selective plates.
RESULTS
Swarm inhibition is caused by sibling killing.
In earlier work, we found that nonmotile strains of M. xanthus inhibit the ability of apparent isogenic motile strains to swarm (Fig. 1A) (2). Swarm inhibition is not caused by physical obstruction of nonmotile cells but instead is TraA/B dependent. To investigate this phenomenon further, the edge of mixed inoculums was observed at a high resolution. After 24 h of incubation, motile cells had migrated beyond the inoculum spot (Fig. 1B). However, by 48 h, those individual cells or small groups of cells seen at 24 h had mostly disappeared, although their phase-bright “slime trails” remained (Fig. 1B). The disappearance of cells from the swarm edge raised the possibility that cells either had returned to the colony or had lysed. To track the fate of such cells, time-lapse microscopy was used 24 h after the cell mixture was plated. As previously reported (2, 14), many of the cells at the swarm edge moved slowly or not at all (compare Movies S1 and S2 in the supplemental material), suggesting that those cells were sick or dead. In addition, in two cases, isolated cells lysed (see Movie S1).
FIG 1.
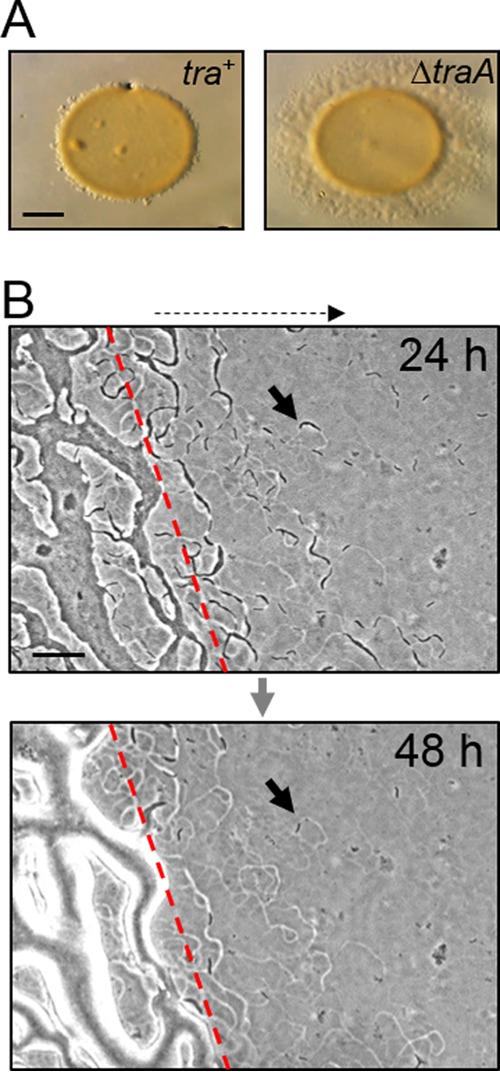
A nonmotile strain inhibits A-motility swarm expansion of a related strain and depletes motile cells from the swarm edge by a Tra-dependent mechanism. (A) Nonmotile strain DK8601 was mixed 1:1 with the indicated isogenic A-motile strains DK8615 (ΔpilQ) and DW1415 (ΔpilQ traA::km) and incubated for 72 h. Bar, 1 mm. (B) Phase-contrast micrographs of the same tra+ strain mixture 24 h after mixing. Top panel, cells (arrow) that have migrated out from the inoculum edge (red dashed line); bottom panel, the identical field 24 h later revealing that most motile cells at the swarm fringe have disappeared (arrow), although slime trails remain. Bar, 100 μm. The dashed arrow at the top of panel B shows the direction of swarm expansion.
Our results suggested that motile cells at the swarm periphery died and lysed following contact with their nonmotile siblings. To investigate the fate of motile cells within the colony center, strains were differentially labeled with fluorescent proteins. Here, motile and nonmotile strains were labeled with GFP (cytoplasm) and mCherry (cytoplasmic membrane), respectively, and their fitness was assessed. As expected, shortly after mixing and plating, there were a substantial number of green- and red-labeled cells (Fig. 2A, first row). Over time, however, the number of GFP-labeled cells decreased, and by 48 h the green motile cells were rarely detected (Fig. 2A, second row). To clearly delineate individual cells, the colony was collected in buffer and cells were viewed on glass microscope slides. Again, the green motile cells were rarely seen by 72 h (see Fig. S1 in the supplemental material, top row, tra+). These results suggest that the motile GFP-labeled cells had lysed after extended contact with nonmotile siblings.
FIG 2.

Nonmotile cells kill A-motile cells by a Tra-dependent mechanism. (A) The nonmotile (NM) strain DW1048 labeled with mCherry was mixed at a 10:1 ratio with an A-motile strain labeled with GFP (neither reporter can be exchanged [9]). Top and bottom panels (DW709 and DW1613, respectively) are identical, except for the traA allele in the motile strain. Micrographs of the swarm edge were taken at early and late times. Note the difference in green fluorescence and swarm flares at 48 h between strain mixtures. Bar, 100 μm. (B) Numbers of CFU were determined between 1:1 mixtures of an NM strain (DK8601; Tcr) mixed with Kmr motile strains that were either tra+ (DW1619) or a traA mutant (DW1415). (C) Susceptible cells become filamentous when mixed with aggressor cells. GFP-labeled strains with different traA alleles (DW709 and DW1613) were mixed at a 10:1 ratio with an aggressor strain (DW1411; mCherry) and incubated for 24 h on agarose pads. Bar, 10 μm.
To quantify population dynamics, strain mixtures were collected at various times, washed, and microscopically examined on glass slides. The ratio of motile to nonmotile cells was then determined by fluorescence microscopy. As found above, the ratio dramatically decreased over time. In this assay, by 72 h, the motile cell population was ∼100-fold lower (limit of assay) than the nonmotile cell population (Fig. 3A). To assess a wider dynamic range, the cell populations were enumerated by viable CFU. In this assay, the motile and nonmotile cells were differentiated by Km- or Tc-resistant markers that the strains carried. After 72 h, no viable motile cells were detected (≥104-fold decrease) (Fig. 2B). In contrast, the nonmotile population grew. We conclude that the swarm inhibition was caused by nonmotile cells killing their motile siblings.
FIG 3.

Antagonism depends on a hard surface and OmrA. (A) An A+ S− strain labeled with GFP (DW709, tra+) was mixed at a 1:1 ratio with a nonmotile aggressor strain (DW1048; mCherry) and placed on hard agar (HA; 1.5%) or soft agar (SA; 0.5%). As a control, a traA::km mutant (A+ S−, DW1613) was mixed with DW1048 on HA. (B) An omrA mutation confers resistance. Indicated A-motile strains (omrA, DW1617; omrB, DW1618; control, DW1619) were incubated with a nonmotile aggressor strain (DK8606; GFP labeled). Three independent experiments were carried out, and the data are plotted as the means ± standard errors. All strain ratios were determined by fluorescence microscopy.
Sibling killing is Tra dependent.
We previously showed that swarm inhibition is Tra dependent (Fig. 1A) (2, 14). That is, when either strain in the mixture contains a traA or traB mutation, swarm inhibition is abolished. We tested whether swarm relief correlated with motile cell survival when OME was blocked. As expected, when the motile cells contained a traA mutation, they swarmed out from the inoculum (Fig. 2A, compare the second and fourth rows). In addition, and in contrast to Tra+ strain mixtures, the isogenic TraA mutant flourished when mixed with the same nonmotile strain (Fig. 2A, compare the second and fourth rows). To quantify this, the number of CFU in each population was determined. The motile strain with the traA mutation survived as well as the nonmotile strain (Fig. 2B), indicating that the kill phenotype was Tra dependent.
Target cells become filamentous.
The morphological fate of motile cells was tracked during swarm inhibition at high magnification. Interestingly, by 24 h, the surviving GFP-labeled motile cells became filamentous, ranging in length from 12 to 20 μm (Fig. 2C). Filamentation was Tra dependent, as a traA mutant did not elongate (length ∼7 μm) (Fig. 2C). Attempts to transfer filamentous cells to glass slides for a detailed inspection were unsuccessful, suggesting that filamentous cells had lysed following physical manipulation. Because filamentation is a response to stress, including exposure to poisons (16), we hypothesized that a toxin was delivered by a Tra-dependent mechanism from nonmotile to motile cells, which then led to filamentation and death.
Semisolid agar abolishes killing.
OME requires TraA/B function on a hard agar surface; it occurs neither in liquid nor on semisolid agar (9, 17). Susceptibility of the killing effect was thus tested, and consistent with prior findings, there was no killing on semisolid agar, whereas killing occurred on hard agar (Fig. 3A). This finding suggests that killing, like OME, requires sustained cell-cell contacts on a hard surface and that it is not mediated by a diffusible factor.
An omrA mutation confers resistance.
omrA was identified from a forward screen for factors required for swarm inhibition. In contrast to TraA/B, OmrA and the codiscovered OmrB proteins are not required for OME but instead are specifically involved in how cells respond to OME (14). Here, OmrA and OmrB were tested for involvement in killing. Importantly, the omrA mutant was not killed and actually outcompeted the nonmotile strain by about 5-fold (Fig. 3B). This indicates that the omrA mutation confers resistance to killing and explains how it was discovered in the screen (14). In contrast, a strain containing a mutation in omrB (identified by bioinformatics methods to function in the OmrA pathway) was killed, although there was a modest delay (Fig. 3B). This result correlates with the partial swarm relief phenotype that is conferred by an omrB mutation (14).
Sibling antagonism is not correlated with motility phenotypes.
We sought to identify the genetic determinant(s) that caused killing; however a feasible forward screen was not apparent to us. As an alternative approach, we surveyed interactions between different laboratory strains in order to obtain clues about the genetic basis of killing. Because our initial observation was swarm inhibition (2, 14), we tested whether motility phenotypes might be involved in killing. However, through a series of experiments, we determined that phenotypic differences in A- and S-motility were not the cause of the kill phenotype (for details, see Fig. S2, S3, and S4 and the text in the supplemental material).
DK1622-derived strains are sensitive to killing by ancestral strains.
To continue the search for genetic factors involved in sibling rivalry, we expanded the panel of strains surveyed. From these studies, we discovered that swarm inhibition was correlated with an ancestral strain background. Specifically, in a nonmotile DK101 (A− S−) strain background, there was swarm inhibition, whereas in a nonmotile DK1622 background, there was no swarm inhibition. As shown in Fig. 4A, this result was repeatable between three different sets of nonmotile strains. Here each strain set contained a different A-motility mutation. As outlined in Fig. 5, DK1622 was derived from DK101 via two intermediate strains. The construction of DK1622, which occurred 4 decades ago, was necessitated because the predecessor strains, including DK101, lacked S-motility (18).
FIG 4.

Antagonistic behavior is correlated with ancestral strains. (A) Swarm inhibition assay at 72 h of an A+ S− strain (DK8615) mixed with nonmotile strains that contain three different A-motility mutations placed in either DK1622 (DW1438, DW1443, DW1445) or DK101 (DK1633, DK360, DW704) backgrounds. Bar, 1 mm. (B) A susceptible strain labeled with GFP (DW709) was mixed 1:1 with the indicated unlabeled ancestral strains and a nonaggressive control (DK8615). All strains were A-motile. After a 48-h incubation, the ratio of cells was determined. Experiments were done in triplicate, and the means ± standard errors are shown. (C) Same as described for panel B, except the competitor was a nonmotile aggressor strain labeled with mCherry (DW1048). (D) The nonmotile strain DZ1 was mixed 1:1 with the indicated strains, and no swarm inhibition was observed. Bar, 1 mm. (E) DNA agarose gel of diagnostic PCRs with primers that are specific to the Mx alpha region absent from DK1622. The locus tag was MXF1DRAFT_07228 from DZF1 (contig 40), and the product size was 441 bp. See Table 1 for strain details.
FIG 5.
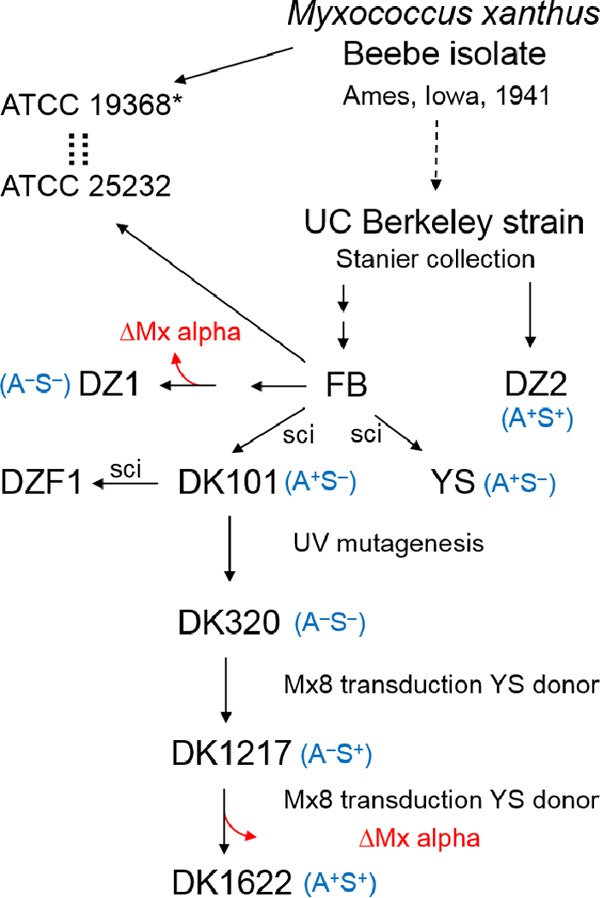
Flowchart and historical information for the derivation of M. xanthus laboratory strains. The first isolation and description of the species M. xanthus were reported by Beebe in 1941 (44). Although the origin of currently used M. xanthus laboratory strains is murky, Wu and colleagues (45) indicated that the Beebe strain was transferred to UC Berkeley, where it was maintained in Roger Stanier's strain collection. Both FB and DZ2 were obtained from the Berkeley collection (20, 21, 46). The claim by Wu et al. is supported by the fact that ATCC strains 19368 and 25232 are cross-listed in the ATCC database. The Beebe isolate was indeed deposited in ATCC as strain 19368. However, in the early 1960s, ATCC personnel were no longer able to revive this strain (denoted by an asterisk) (ATCC technical support staff, personal communication). The ATCC consequently requested that Marty Dworkin (University of Minnesota) deposit his M. xanthus FB strain (ATCC 25232), with the understanding that it was the same strain as 19368; hence the strains were cross-listed. It should be further noted that FB was not maintained as a pure culture; it was a mixture of related strains that had evolved from a common ancestor during prolonged laboratory propagation (47). Additional strain details can be found in Table 1, the text, and the supplemental material. Dashed arrows/lines, presumed relationships; solid arrows, known relationships; two arrows, multiple steps; sci, single-colony isolate.
To test the hypothesis that strain background has a role in antagonism, three different ancestral strains were investigated. Two of these strains, DK101 and YS, were directly derived from strain FB (18, 19) and were the parental strains used to create DK1622 (18) (Fig. 5). The third strain, DZ2, shares a common ancestor with DK1622 but was stored independently by the Zusman lab (20, 21). When any of these ancestral strains was mixed with a DK1622-derived strain, the former strains readily outcompeted the latter strain (Fig. 4B). Similarly, DK1217, which is the direct parent of DK1622 (Fig. 5), readily outcompeted a DK1622-derived strain (see Fig. S4 in the supplemental material). In addition, when these motile ancestral strains were mixed with a labeled DK8601 aggressor strain, they were not killed, whereas the control strain was killed (Fig. 4C). Taken together, we conclude that ancestral strains (DK101, DZ2, YS, and DK1217) antagonize their DK1622 sibling.
Ancestral strains, including DK1217, contain Mx alpha.
A notable difference between DK1622 and strains YS and DK101 is that the former strain has an ∼200-kb deletion (22, 23). The deleted region contains a defective prophage-like element called Mx alpha (22, 24). Work by Zissler and colleagues showed that Mx alpha particles contained only a small portion (∼35 kb) of the prophage-like region (300 kb) and were not lytic. In our studies, we also found no evidence that Mx alpha particles kill. However, because prophages can contain toxins (25, 26), a possible explanation for killing is that a toxin-antitoxin gene cassette resides in the region missing from DK1622 (note that Table 1 indicates the Mx alpha genotype of all M. xanthus strains in footnotes or the table body). There was, however, an inconsistency with the hypothesis that Mx alpha was the genetic determinant for killing. Namely, the Mx alpha deletion was presumed to have occurred following UV mutagenesis of DK101 to create DK320 and prior to construction of DK1217 (Fig. 5) (18, 23). To investigate whether the Mx alpha region was correlated with the killing phenotype, the ancestral strains were screened for this deletion by PCR. Here, a DNA marker that corresponded to the deleted Mx alpha region was identified by comparing the draft genome sequence of DK101 (also known as DZF1) (27) to the complete DK1622 genome (28) (Fig. 6). Importantly, all of the aggressor strains, including DK1217, contained the Mx alpha region that was absent from DK1622 (Fig. 4E). Therefore, there was a correlation between the aggressor phenotype and the presence of the complete Mx alpha region.
FIG 6.

Genomic organization of the Mx alpha units in DK1622 and DZF1 (DK101). (A) Organization of ORFs found in DK1622 from MXAN_1800 to MXAN_1900. Predicted gene functions are color coded. S/T, serine/threonine. (B) The same ORF map as that in panel A with corresponding map position of seven contigs from the DZF1 that perfectly map to this region (top). The DZF1 draft genome has 75 total contigs (27). Regions in nine DZF1 contigs that are homologous to MXAN_1800 to MXAN_1900 and are absent from the DK1622 genome are shown at the bottom. The green and blue contig bars presumably represent two different Mx alpha units. Contig numbers are given on the left. Note that there are gaps in and between some contigs in relation to the DK1622 region. In addition, contig regions that are not homologous to the DK1622 region (insertions) are not shown. In total, these nine contigs contain 200 kb of DNA. (C) Simplified Mx alpha map illustrating the deleted region in DK1622. See the text for additional details.
A report by Magrini and colleagues suggested that the nonmotile strain DZ1 also contains a deletion in Mx alpha (29). As outlined (Fig. 5), DZ1 was derived from FB independently of DK1622 (30, 31). To test the proposed correlation, DZ1 was screened and found to lack the Mx alpha diagnostic marker (Fig. 4E) and exhibited no antagonism toward DK1622 (Fig. 4D). These findings support the idea that Mx alpha contains a genetic determinant involved in killing. In addition, the finding that DK1622 and DZ1 independently and spontaneously deleted part or all of Mx alpha suggests that this element is unstable during laboratory growth.
Mx alpha is necessary for the kill phenotype and resistance.
To directly test whether Mx alpha is involved in killing, a ΔMx alpha mutation was created in a nonmotile aggressor strain that contained Mx alpha by use of plasmid pCV101 (which contains ΔMx alpha and a counterselectable cassette). Importantly, this strain (DW2403, ΔMx alpha-29) no longer killed nor caused swarm inhibition (Fig. 7A). In addition, DW2403 was susceptible to being killed (Fig. 7B). We conclude that Mx alpha is necessary for the kill phenotype and for resistance to killing. To explain these results, we suggest that Mx alpha contains a toxin that kills siblings mediated by OME delivery and a cognate antitoxin that confers immunity.
FIG 7.
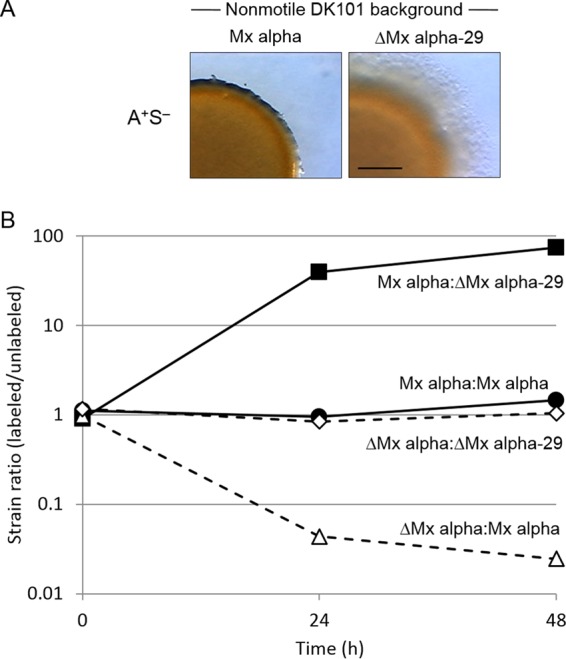
Deletion of Mx alpha region prevents antagonism. (A) Swarm inhibition at 48 h. A-motile strain DW2404 (ΔMx alpha) was mixed 1:1 with isogenic nonmotile strain DK8616 (Mx alpha) or DW2403 (ΔMx alpha-29). Bar, 1 mm. (B) Fitness experiments in which either an Mx alpha aggressor strain (DK101 labeled with tdTomato [DW1620]; solid lines) or a ΔMx alpha nonaggressor strain (DW2404 tdTomato labeled; dashed lines) was mixed 1:1 with an isogenic Mx alpha strain (DK8616) or a ΔMx alpha-29 strain (DW2403). Strain fitness was microscopically determined by counting labeled and unlabeled cells.
Mx alpha is polyploid.
Starich and Zissler showed by Southern blotting with DNA from purified particles that Mx alpha consists of three large repeat units, two of which are absent from DK1622 (22). Genome comparisons indeed showed that DK1622 contains a single Mx alpha unit that spans a 100-kb region (MXAN_1801 to MXAN_1900) (Fig. 6). The DZF1 draft genome, which consists of 75 contigs (27), contains seven contigs that perfectly match MXAN_1801 to MXAN_1900 and nine other contigs that are unique to DZF1 yet share homology to the aforementioned DK1622 region (Fig. 6). These 16 contigs from DZF1 span 300 kb and constitute three imperfect repeats. That is, alleles of some genes are present in all repeats and other genes are unique to a particular copy. In total, 84 open reading frames (ORFs) between MXAN_1801 and MXAN_1900 have alternative alleles in DZF1 that were absent from DK1622 (see Table S2 in the supplemental material). These alternative alleles typically share 50 to 99% identity at the amino acid level. Last, we note that the Mx alpha region contains several candidate toxin and antitoxin ORFs.
Engineered laboratory strain kills environmental isolate.
Previously, we showed that when a laboratory strain heterologously expresses a traA allele from M. fulvus (traAM. fulvus allele), it empowers OME with the corresponding M. fulvus HW-1 environmental isolate (3), which was otherwise unable to engage in OME with DK1622. In addition, there is a fitness gain for the laboratory strain in competition experiments with M. fulvus (3). Conversely, when the traADK1622 gene is deleted from a laboratory strain (DK1622 related), which prevents OME with environmental isolates belonging to the TraADK1622 recognition group, its fitness markedly decreases against those isolates (3). Our current findings suggest an explanation for these results. To investigate this, we tested whether the M. fulvus strain was killed in a manner that depended on OME and Mx alpha. Here, two different DK1217-derived laboratory strains that contained either the traADK1622 or traAM. fulvus allele were mixed with M. fulvus. Based on the resulting number of CFU, the M. fulvus strain outcompeted the traADK1622 strain by 1,000-fold after 24 h (Fig. 8B). In contrast, there was nearly a 100-fold decrease in the relative fitness of M. fulvus when the laboratory strain contained the traAM. fulvus allele. In fact, the survival ratios of M. fulvus and of the engineered M. xanthus strain with traAM. fulvus were nearly equal, although for both strains their CFU numbers were lower than the CFU number in the starting inoculum (Fig. 8B). In contrast, M. fulvus outcompeted DK1622 (i.e., ΔMx alpha) expressing TraAM. fulvus (Fig. 8B). The magnitude of the antagonistic interactions was also evident by visual and microscopic inspection of inoculum mixtures (Fig. 8A). Robust colony growth was observed when M. fulvus dominated the laboratory strains (Fig. 8A, left and right colonies). However, when the DK1217 traAM. fulvus strain containing the entire Mx alpha region was mixed with M. fulvus, the inoculum remained translucent after 48 h, indicating intense bidirectional antagonism that blocked either strain from swarming or growing (Fig. 8A, middle panels). From these results, we conclude that a kill phenotype can be activated toward siblings and nonsiblings, including between different species, by engineering compatible traA alleles for OME and hence cargo (toxin) delivery. We also note that M. fulvus has an uncharacterized mechanism(s) to kill M. xanthus that does not depend on OME.
FIG 8.
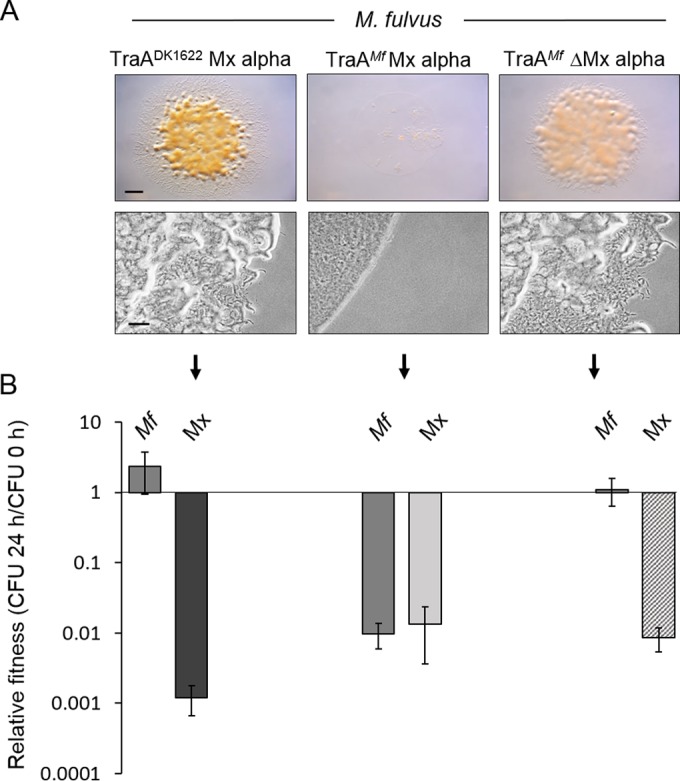
Interspecies antagonism is mediated by traA allele-specific interactions and Mx alpha. (A) M. fulvus (Mf) and M. xanthus (Mx) strains with the indicated properties (DW1048, DW1614, and DW1615 [left to right]) were mixed at 1:1 ratios, and after 24 h, phase-contrast micrographs were taken (bottom), or after 48 h, stereomicrographs were taken (top). Note that the middle top panel was translucent. Bars, 1 mm (top) and 100 μm (bottom). (B) The relative fitness of the same strain mixtures as described for panel A was determined by dividing the number of CFU from 24 h by the number of CFU from 0 h for each strain.
DISCUSSION
Here, we surprisingly discovered that M. xanthus cells kill siblings derived from the same parental lineage by a mechanism that involves OME. This finding provides evolutionary insight into why OME requires kin recognition (3). Namely, OME among neighboring cells can lead to beneficial outcomes; however, it can also have lethal consequences. Therefore, kin recognition provides a mechanism by which cell-cell selectivity reduces the chance of lethal encounters between isolates. Selectivity is derived from polymorphisms within the TraA variable domain; OME occurs only between isolates that have identical or nearly identical traA alleles (3). The kill phenotype also explains why our previous screen to isolate swarm relief mutants was so powerful (14). Indeed, instead of a screen, as originally conceived, a genetic selection was imposed. Thus, in mixed cultures, motile cells were annihilated by their nonmotile siblings unless they contained a mutation that blocked killing. The >50 mutations isolated all mapped to traAB or omrA (14).
A working model for killing is outlined in Fig. 9. In this model, the Mx alpha cell is hypothesized to produce toxin-antitoxin factors. Toxin delivery is mediated by OME, because when OME is blocked by a tra mutation, incompatible traA alleles, or environmental conditions, antagonism is abolished. The finding that an omrA mutation confers resistance provides clues about the toxicity mechanism. Based on sequence similarity to MprF from Staphylococcus aureus, OmrA is predicted to function as an aminoacyl phospholipid flippase (14). In S. aureus, altered MprF function confers resistance to cationic antibiotics, such as daptomycin, that act on the cytoplasmic membrane (32). Thus, by analogy, an omrA mutation will alter the homeostasis of the cytoplasmic membrane and, in turn, may block how a toxin interacts and/or traverses the cytoplasmic membrane (Fig. 9). Alternatively, as was recently described for contact-dependent inhibition (CDI) and type VI secretion (5, 33), OmrA may serve as a receptor to facilitate toxin delivery across the cytoplasmic membrane. Another clue in support of a toxin-mediated interaction is the filamentation response of susceptible cells. Filamentation is a morphological marker of cells stressed by an insult, such as a poison that blocks a core metabolic function (16). In this model, aggressor cells are resistant to toxin-mediated sibling attack and thus do not show a filamentation phenotype, because they express an antitoxin. Finally, we note that in ongoing work, the toxin-antitoxin genes in Mx alpha have been identified, and we are currently characterizing them.
FIG 9.
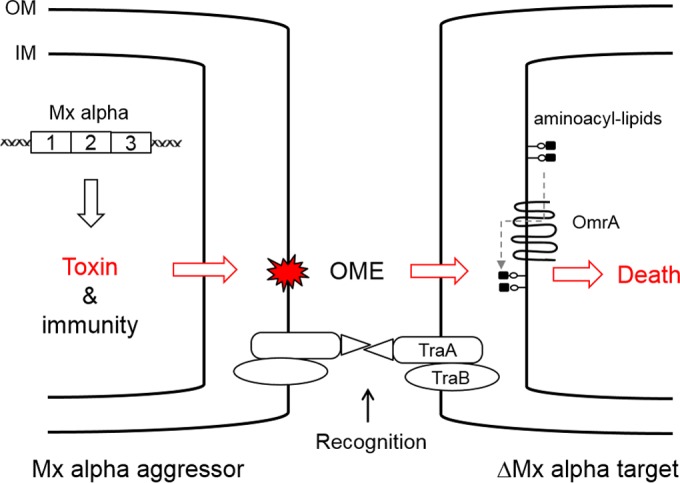
Working model for how OME and Mx alpha mediate killing. The Mx alpha units that are absent from DK1622 are proposed to contain a toxin-antitoxin system. The toxin is transferred to a target cell by an OME-mediated process. For the toxin to kill, the target cell must express OmrA and lack the antitoxin. Lollipops represent phospholipid molecules; OM, outer membrane; IM, inner membrane. See the text for additional details.
Mx alpha was discovered as a latent prophage involved in specialized transduction of Tn5-marked Mx alpha DNA from strain YS (22, 24). From culture supernatants, low levels of transducing particles were isolated and observed by electron microscopy. When incubated with other Myxococcus strains, these particles do not form plaques and thus are likely to represent defective phage. In support of this, the particles have a small diameter (35 nm) and can package only ∼35 kb of DNA, which is insufficient to contain a single Mx alpha unit (22). Mx alpha has similarities to other phage-like elements called genetic transfer agents (GTAs), which package and exchange genomic DNA between cells but do not infect recipients (34). The primary difference between GTAs and Mx alpha is that the latter transfers its own DNA, whereas GTAs conduct generalized transduction.
Mx alpha contains 84 ORFs (e.g., see Table S2 in the supplemental material) that are present in multiple alleles. Consequently, Mx alpha has polyploid qualities. To our knowledge, this is the first example of a large region in a bacterial genome that is polyploid—a chromosomal segment with allelic variation for a large set of genes. These features imply that allelic differences in Mx alpha provide selective advantages that allow their retention. Given that prophages confer immunity to infection against phage, one possible role for being polyploid is to provide a broad spectrum of phage resistance. Bioinformatically, this hypothesis is difficult to assess because many of the Mx alpha ORFs, like other phage genes, contain no predicted functions (see Table S2).
Our results shed light on how large tandem repeats might have remained stable in M. xanthus. Typically, large DNA repeats are unstable in bacterial genomes because homologous recombination leads to their removal (35). In addition, the Mx alpha units represent >3% of the M. xanthus genome and, consequently, are a burden as selective pressures strive to minimize bacterial genome size. This puzzle is highlighted by the homologs of Mx alpha that are sometimes present in other environmental isolates (22), including in the sequenced genomes of Myxococcus virescens DSM 2260 and distantly related Stigmatella aurantiaca DW4/3-1 and Haliangium ochraceum DSM 14365 species (36). A plausible explanation for their presence comes from the discovery of their role in fratricide behavior. That is, cells that lose Mx alpha, or portions of it, become susceptible to killing by siblings that still harbor an intact Mx alpha. Last, it should be noted that in the generation of DK1622 and DZ1, the parental strains were grown in liquid medium, an unnatural environment for this terrestrial bacterium and a condition where OME-dependent killing cannot occur. Thus, cells that spontaneously delete Mx alpha or portions thereof in liquid medium would escape lethal encounters. Once cured of Mx alpha, isolated DK1622 and DZ1 strains could be propagated on agar.
During fruiting body formation, ∼80% of the cells lyse (12, 37). Lysis has generally been assumed to be the result of a poorly defined programmed cell death pathway. However, our finding of sibling rivalry raises the possibility that cell-cell competition during development plays a role in determining cell fate. Although this idea is speculative, cell competition does lead to sibling killing during Bacillus subtilis development, in a process called cannibalism (38). Like M. xanthus, surviving B. subtilis cells benefit from sibling lysis by the release of their nutrients. Similarly, individual Dictyostelium discoideum amoebae coalesce into fruiting bodies in response to starvation, and those cells compete to become a spore or to terminally differentiate into a stalk cell (39). In M. xanthus, monocultures of traA mutants develop (2, 13), indicating that under laboratory conditions, OME is not required. Future studies in M. xanthus will need to test whether developmental lysis is a result of a toxin-antitoxin system, cell competition, and/or OME function.
Previously, it was shown that OME leads to beneficial outcomes (13, 40). Here, OME was found to lead to adversarial interactions, which is a typical response for bacterial cell-cell transfer systems. For instance, CDI (5) and type VI secretion (7) mediate bacterial competition. Interestingly, a fratricide behavior also arises from monocultures of Salmonella. In those cultures, a subpopulation of cells undergo a DNA rearrangement that results in the expression of an otherwise silent toxin-antitoxin gene cassette, which in turn blocks sibling growth by a CDI mechanism (41). In addition, clonemate killing was described in Paenibacillus dendritiformis and Streptococcus pneumoniae (42, 43). These findings show that bacteria have evolved systems to compete with not only related strains but also their own progeny.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jehee Moon for technical assistance.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Funding Statement
This work was supported in part by the USDA National Institute of Food and Agriculture Hatch project 227896 subaward WYO-472-12 to D.W.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00964-15.
REFERENCES
- 1.Vos M, Velicer GJ. 2009. Social conflict in centimeter-and global-scale populations of the bacterium Myxococcus xanthus. Curr Biol 19:1763–1767. doi: 10.1016/j.cub.2009.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pathak DT, Wei X, Bucuvalas A, Haft DH, Gerloff DL, Wall D. 2012. Cell contact-dependent outer membrane exchange in myxobacteria: genetic determinants and mechanism. PLoS Genet 8:e1002626. doi: 10.1371/journal.pgen.1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pathak DT, Wei X, Dey A, Wall D. 2013. Molecular recognition by a polymorphic cell surface receptor governs cooperative behaviors in bacteria. PLoS Genet 9:e1003891. doi: 10.1371/journal.pgen.1003891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christie PJ, Cascales E. 2005. Structural and dynamic properties of bacterial type IV secretion systems (review). Mol Membr Biol 22:51–61. doi: 10.1080/09687860500063316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willett JL, Gucinski GC, Fatherree JP, Low DA, Hayes CS. 2015. Contact-dependent growth inhibition toxins exploit multiple independent cell-entry pathways. Proc Natl Acad Sci U S A 112:11341–11346. doi: 10.1073/pnas.1512124112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mota LJ, Cornelis GR. 2005. The bacterial injection kit: type III secretion systems. Ann Med 37:234–249. doi: 10.1080/07853890510037329. [DOI] [PubMed] [Google Scholar]
- 7.Russell AB, Peterson SB, Mougous JD. 2014. Type VI secretion system effectors: poisons with a purpose. Nat Rev Microbiol 12:137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spormann AM. 1999. Gliding motility in bacteria: insights from studies of Myxococcus xanthus. Microbiol Mol Biol Rev 63:621–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei X, Pathak DT, Wall D. 2011. Heterologous protein transfer within structured myxobacteria biofilms. Mol Microbiol 81:315–326. doi: 10.1111/j.1365-2958.2011.07710.x. [DOI] [PubMed] [Google Scholar]
- 10.Jarrell KF, McBride MJ. 2008. The surprisingly diverse ways that prokaryotes move. Nat Rev Microbiol 6:466–476. doi: 10.1038/nrmicro1900. [DOI] [PubMed] [Google Scholar]
- 11.Shi W, Zusman DR. 1993. The two motility systems of Myxococcus xanthus show different selective advantages on various surfaces. Proc Natl Acad Sci U S A 90:3378–3382. doi: 10.1073/pnas.90.8.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee B, Holkenbrink C, Treuner-Lange A, Higgs PI. 2012. Myxococcus xanthus developmental cell fate production: heterogeneous accumulation of developmental regulatory proteins and reexamination of the role of MazF in developmental lysis. J Bacteriol 194:3058–3068. doi: 10.1128/JB.06756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vassallo C, Pathak DT, Cao P, Zuckerman DM, Hoiczyk E, Wall D. 2015. Cell rejuvenation and social behaviors promoted by LPS exchange in myxobacteria. Proc Natl Acad Sci U S A 112:E2939–E2946. doi: 10.1073/pnas.1503553112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dey A, Wall D. 2014. A genetic screen in Myxococcus xanthus identifies mutants that uncouple outer membrane exchange from a downstream cellular response. J Bacteriol 196:4324–4332. doi: 10.1128/JB.02217-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pathak DT, Wall D. 2012. Identification of the cglC, cglD, cglE, and cglF genes and their role in cell contact-dependent gliding motility in Myxococcus xanthus. J Bacteriol 194:1940–1949. doi: 10.1128/JB.00055-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Justice SS, Hunstad DA, Cegelski L, Hultgren SJ. 2008. Morphological plasticity as a bacterial survival strategy. Nat Rev Microbiol 6:162–168. doi: 10.1038/nrmicro1820. [DOI] [PubMed] [Google Scholar]
- 17.Wall D, Kaiser D. 1998. Alignment enhances the cell-to-cell transfer of pilus phenotype. Proc Natl Acad Sci U S A 95:3054–3058. doi: 10.1073/pnas.95.6.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wall D, Kolenbrander PE, Kaiser D. 1999. The Myxococcus xanthus pilQ (sglA) gene encodes a secretin homolog required for type IV pilus biogenesis, social motility, and development. J Bacteriol 181:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodgkin J, Kaiser D. 1977. Cell-to-cell stimulation of movement in nonmotile mutants of Myxococcus. Proc Natl Acad Sci U S A 74:2938–2942. doi: 10.1073/pnas.74.7.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller S, Willett JW, Bahr SM, Darnell CL, Hummels KR, Dong CK, Vlamakis HC, Kirby JR. 2013. Draft genome sequence of Myxococcus xanthus wild-type strain DZ2, a model organism for predation and development. Genome Announc 1:e00217–13. doi: 10.1128/genomeA.00217-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campos JM, Zusman DR. 1975. Regulation of development in Myxococcus xanthus: effect of 3′:5′-cyclic AMP, ADP, and nutrition. Proc Natl Acad Sci U S A 72:518–522. doi: 10.1073/pnas.72.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starich T, Zissler J. 1989. Movement of multiple DNA units between Myxococcus xanthus cells. J Bacteriol 171:2323–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Keseler IM, Shimkets LJ. 1990. Genome size of Myxococcus xanthus determined by pulsed-field gel electrophoresis. J Bacteriol 172:4206–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starich T, Cordes P, Zissler J. 1985. Transposon tagging to detect a latent virus in Myxococcus xanthus. Science 230:541–543. doi: 10.1126/science.2996138. [DOI] [PubMed] [Google Scholar]
- 25.Short FL, Blower TR, Salmond GP. 2012. A promiscuous antitoxin of bacteriophage T4 ensures successful viral replication. Mol Microbiol 83:665–668. doi: 10.1111/j.1365-2958.2012.07974.x. [DOI] [PubMed] [Google Scholar]
- 26.Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H. 2003. Prophage genomics. Microbiol Mol Biol Rev 67:238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller S, Willett JW, Bahr SM, Scott JC, Wilson JM, Darnell CL, Vlamakis HC, Kirby JR. 2013. Draft genome of a type 4 pilus defective Myxococcus xanthus strain, DZF1. Genome Announc 1:e00392-13. doi: 10.1128/genomeA.00392-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen JA, Ronning CM, Barbazuk WB, Blanchard M, Field C, Halling C, Hinkle G, Iartchuk O, Kim HS, Mackenzie C, Madupu R, Miller N, Shvartsbeyn A, Sullivan SA, Vaudin M, Wiegand R, Kaplan HB. 2006. Evolution of sensory complexity recorded in a myxobacterial genome. Proc Natl Acad Sci U S A 103:15200–15205. doi: 10.1073/pnas.0607335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magrini V, Storms ML, Youderian P. 1999. Site-specific recombination of temperate Myxococcus xanthus phage Mx8: regulation of integrase activity by reversible, covalent modification. J Bacteriol 181:4062–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zusman DR, Krotoski DM, Cumsky M. 1978. Chromosome replication in Myxococcus xanthus. J Bacteriol 133:122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zusman D, Rosenberg E. 1970. DNA cycle of Myxococcus xanthus. J Mol Biol 49:609–619. doi: 10.1016/0022-2836(70)90285-8. [DOI] [PubMed] [Google Scholar]
- 32.Ernst CM, Peschel A. 2011. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol Microbiol 80:290–299. doi: 10.1111/j.1365-2958.2011.07576.x. [DOI] [PubMed] [Google Scholar]
- 33.Whitney JC, Quentin D, Sawai S, LeRoux M, Harding BN, Ledvina HE, Tran BQ, Robinson H, Goo YA, Goodlett DR, Raunser S, Mougous JD. 2015. An interbacterial NAD(P)(+) glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell 163:607–619. doi: 10.1016/j.cell.2015.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lang AS, Zhaxybayeva O, Beatty JT. 2012. Gene transfer agents: phage-like elements of genetic exchange. Nat Rev Microbiol 10:472–482. doi: 10.1038/nrmicro2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth JR, Benson N, Galitski T, Haack K, Lawrence JG, Miesel L. 1996. Rearrangements of the bacterial chromosome: formation and applications, p 2256–2276. In Neidhardt FC, Curtis R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC. [Google Scholar]
- 36.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Pillay M, Ratner A, Huang J, Woyke T, Huntemann M, Anderson I, Billis K, Varghese N, Mavromatis K, Pati A, Ivanova NN, Kyrpides NC. 2014. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res 42:D560–567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boynton TO, McMurry JL, Shimkets LJ. 2013. Characterization of Myxococcus xanthus MazF and implications for a new point of regulation. Mol Microbiol 87:1267–1276. doi: 10.1111/mmi.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez-Pastor JE, Hobbs EC, Losick R. 2003. Cannibalism by sporulating bacteria. Science 301:510–513. doi: 10.1126/science.1086462. [DOI] [PubMed] [Google Scholar]
- 39.Ho HI, Hirose S, Kuspa A, Shaulsky G. 2013. Kin recognition protects cooperators against cheaters. Curr Biol 23:1590–1595. doi: 10.1016/j.cub.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao P, Dey A, Vassallo CN, Wall D. 2015. How myxobacteria cooperate. J Mol Biol 427:3709–3721. doi: 10.1016/j.jmb.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koskiniemi S, Garza-Sanchez F, Sandegren L, Webb JS, Braaten BA, Poole SJ, Andersson DI, Hayes CS, Low DA. 2014. Selection of orphan Rhs toxin expression in evolved Salmonella enterica serovar Typhimurium. PLoS Genet 10:e1004255. doi: 10.1371/journal.pgen.1004255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Be'er A, Florin EL, Fisher CR, Swinney HL, Payne SM. 2011. Surviving bacterial sibling rivalry: inducible and reversible phenotypic switching in Paenibacillus dendritiformis. mBio 2:e00069. doi: 10.1128/mBio.00069-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guiral S, Mitchell TJ, Martin B, Claverys JP. 2005. Competence-programmed predation of noncompetent cells in the human pathogen Streptococcus pneumoniae: genetic requirements. Proc Natl Acad Sci U S A 102:8710–8715. doi: 10.1073/pnas.0500879102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beebe JM. 1941. The morphology and cytology of Myxococcus xanthus, n. sp. J Bacteriol 42:193–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu Y, Kaiser AD, Jiang Y, Alber MS. 2009. Periodic reversal of direction allows myxobacteria to swarm. Proc Natl Acad Sci U S A 106:1222–1227. doi: 10.1073/pnas.0811662106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dworkin M. 1962. Nutritional requirements for vegetative growth of Myxococcus xanthus. J Bacteriol 84:250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wireman JW, Dworkin M. 1975. Morphogenesis and developmental interactions in myxobacteria. Science 189:516–523. doi: 10.1126/science.806967. [DOI] [PubMed] [Google Scholar]
- 48.Li ZF, Li X, Liu H, Liu X, Han K, Wu ZH, Hu W, Li FF, Li YZ. 2011. Genome sequence of the halotolerant marine bacterium Myxococcus fulvus HW-1. J Bacteriol 193:5015–5016. doi: 10.1128/JB.05516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hodgkin J, Kaiser D. 1979. Genetics of gliding motility in Myxococcus xanthus (Myxobacterales): genes controlling movement of single cells. Mol Gen Genet 171:167–176. doi: 10.1007/BF00270003. [DOI] [Google Scholar]
- 50.Wu SS, Kaiser D. 1997. Regulation of expression of the pilA gene in Myxococcus xanthus. J Bacteriol 179:7748–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei X, Vassallo CN, Pathak DT, Wall D. 2014. Myxobacteria produce outer membrane-enclosed tubes in unstructured environments. J Bacteriol 196:1807–1814. doi: 10.1128/JB.00850-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.