

Abstract
Objective:
To investigate the genetic contributors to cerebrovascular disease and variation in biomarkers of ischemic stroke.
Methods:
The Vitamin Intervention for Stroke Prevention Trial (VISP) was a randomized, controlled clinical trial of B vitamin supplementation to prevent recurrent stroke, myocardial infarction, or death. VISP collected baseline measures of C-reactive protein (CRP), fibrinogen, creatinine, prothrombin fragments F1+2, thrombin-antithrombin complex, and thrombomodulin prior to treatment initiation. Genome-wide association scans were conducted for these traits and follow-up replication analyses were performed.
Results:
We detected an association between CRP single nucleotide polymorphisms (SNPs) and circulating CRP levels (most associated SNP, rs2592902, p = 1.14 × 10−9) in 2,100 VISP participants. We discovered a novel association for CRP level in the AKR1D1 locus (rs2589998, p = 7.3 × 10−8, approaching genome-wide significance) that also is an expression quantitative trait locus for CRP gene expression. We replicated previously identified associations of fibrinogen with SNPs in the FGB and LEPR loci. CRP-associated SNPs and CRP levels were significantly associated with risk of ischemic stroke and recurrent stroke in VISP as well as specific stroke subtypes in METASTROKE. Fibrinogen levels but not fibrinogen-associated SNPs were also found to be associated with recurrent stroke in VISP.
Conclusions:
Our data identify a genetic contribution to inflammatory and hemostatic biomarkers in a stroke population. Additionally, our results suggest shared genetic contributions to circulating CRP levels measured poststroke and risk for incident and recurrent ischemic stroke. These data broaden our understanding of genetic contributors to biomarker variation and ischemic stroke risk, which should be useful in clinical risk evaluation.
Prior data support the heritability of biomarker concentrations involved with inflammatory processes1,2 and hemostasis.3 Genome-wide association studies (GWAS) have identified genetic contributors to circulating levels of these traits in population studies. Investigating biomarkers in populations potentially enriched for genetic determinants can complement studies in the general population, facilitate the identification of genetic risk factors, and address whether the genetic factors influencing the biomarker also influence risk of the disease.4
Because of the known associations between biomarker status and disease risk, GWAS in human biomarker variability can contribute understanding of biomarker pathophysiology5 and development of treatment strategies.6 We hypothesize an association between genetic variability and circulating levels of inflammatory biomarkers and risk for stroke. In a retrospective analysis of prospectively collected data, we sought to test this hypothesis in the Vitamin Intervention for Stroke Prevention (VISP) trial, an ischemic stroke population, for the genetic contributors of 6 biomarkers associated with stroke risk: C-reactive protein (CRP), fibrinogen, creatinine, prothrombin fragments F1+2 (F1+2), thrombin-antithrombin complex (TAT), and thrombomodulin (TM). We next sought to replicate previous associations from biomarker GWAS in participants from the VISP trial. Leveraging these data, we then went on to investigate the relationship between the genetic determinants of these biomarkers with incident and recurrent ischemic stroke in both VISP and METASTROKE.7
The VISP population used for the primary analyses had available genotypic and phenotypic information for both biomarkers and recurrent stroke, with the latter a relatively infrequent phenotype in the stroke literature. The VISP population represents a unique resource since all participants had an ischemic stroke prior to study entry, and entry criteria—including the requirement that participants have total homocysteine levels in the top quartile—are anticipated to have selected a more homogenous sample.
METHODS
A workflow of all analyses can be seen in figure e-1 on the Neurology® Web site at Neurology.org.
VISP participants.
A detailed description of the VISP study design and participants can be found in the e-Methods.
GWAS.
In the VISP population, GWAS analyses were conducted using PLINK v1.0.7.8 We used multivariable linear regression modeling to test correlation of quantitative traits and single nucleotide polymorphism (SNP) markers. We derived the first 10 principal components of ancestry from genotype data9 using the KING software9 and adjusted for population heterogeneity. We included age and sex as covariates in the regression model. No relatives were detected in the VISP population and no adjustments were made to the analysis for relatedness. We analyzed the following biomarkers: creatinine, F1+2, CRP, TAT, TM, and fibrinogen. We used the same regression model to perform association tests between phenotype and expected allele counts. The α was set at p = 5 × 10−8 for the genome-wide significance threshold for the primary analyses. Using standard settings, we generated Manhattan plots (figures 1, e-2) using the R package qqman (https://github.com/stephenturner/qqman). Regional association plots were created using the LocusZoom Plot with Your Data function (https://statgen.sph.umich.edu/locuszoom/genform.php?type=yourdata).10
Figure 1. C-reactive protein (CRP): Manhattan and regional association plots of Vitamin Intervention for Stroke Prevention biomarker genome-wide association studies (GWAS).

(A) GWAS of CRP. (B) Regional association plot of chromosome 1 CRP locus. (C) Regional association plot of chromosome 7 CRP locus. In Manhattan plots, the red line represents a −log10(p) of 5 × 10−8 and the blue line represents a −log10(p) of 1 × 10−6. In regional association plots (B, C), the X-axis represents the base pair position according to the GRCh37/hg19 build of the human genome. Manhattan plots were created using the qqman R package. Regional association plots were created utilizing the genome build/linkage disequilibrium population hg19/1000 Genomes March 2012 EUR.
We accessed publicly available data for these biomarkers (table 1) to establish evidence of replication for the GWAS results. p Value thresholds were based on a Bonferroni-corrected value of p = 2.10 × 10−3 (0.05/24) for CRP and p = 0.025 (0.05/2) for fibrinogen. We interrogated our significant SNPs in a GWAS analysis of CRP in a general population cohort from the Framingham Heart Study.11,12 We assessed significantly associated SNPs from previously published GWAS of the same biomarkers and related phenotypes (i.e., biomarker levels or vascular disease) in the VISP sample using SNPs that were genotyped, imputed, or had appropriate surrogate SNP data by linkage disequilibrium (LD) r2 > 0.9. We investigated the single most significant SNPs from these publications applying appropriate Bonferroni corrections.
Table 1.
Primary findings and replication analyses

Stroke analyses.
Survival analyses of recurrent stroke.
We used Cox proportional hazards models to investigate the associations between standardized CRP levels and SNPs with time until the first recurrent stroke measured from the time of randomization. During the trial, 182 individuals had recurrent stroke. Censoring occurred due to loss to follow-up, death, and early termination. We coded SNPs using additive genetic models, both a basic model (adjusting only for the intervention group) and a full model (adjusting for the intervention group, age, race, sex, smoking, body mass index, diabetes status, hypertension, and low-density lipoprotein).
Replication of associated variants in case-control analysis of ischemic stroke risk.
We determined all the most significantly associated SNPs (or an appropriate surrogate) in the GWAS results from METASTROKE,7 a study of 12,389 individuals with ischemic stroke and 62,004 controls with imputation to the Hapmap2 reference panel.13 We obtained the results of association with all ischemic stroke and the 3 major stroke subtypes (large vessel [atherosclerotic], cardioembolic, and small vessel [lacunar] from the Trial of Org 10172 in Acute Stroke Treatment [TOAST] classification).
Case-control analysis of ischemic stroke risk.
We investigated whether SNPs associated with the biomarker traits also contributed to ischemic stroke risk. For ischemic stroke case-control analyses, we compared VISP study participants (ischemic stroke cases) to publicly available controls from the Gene-Environment Association Studies (GENEVA) Melanoma Study (dbGAP phs000187), restricting the population to individuals of European descent only. This control population was selected because GENEVA controls were genotyped on the same array as VISP (HumanOmni1-Quad_v1-0_B), and this case-control strategy has been published previously.14
Standard protocol approvals, registrations, and patient consents.
All studies received approval from an ethical standards committee on human experimentation and had written informed consent obtained from all participants (or surrogates). The VISP clinical trial identifier is NCT00004734. The FHS clinical trial identifier is NCT00005121. METASTROKE is not a clinical trial but has received all necessary approvals from the applicable regional and institutional review boards.
RESULTS
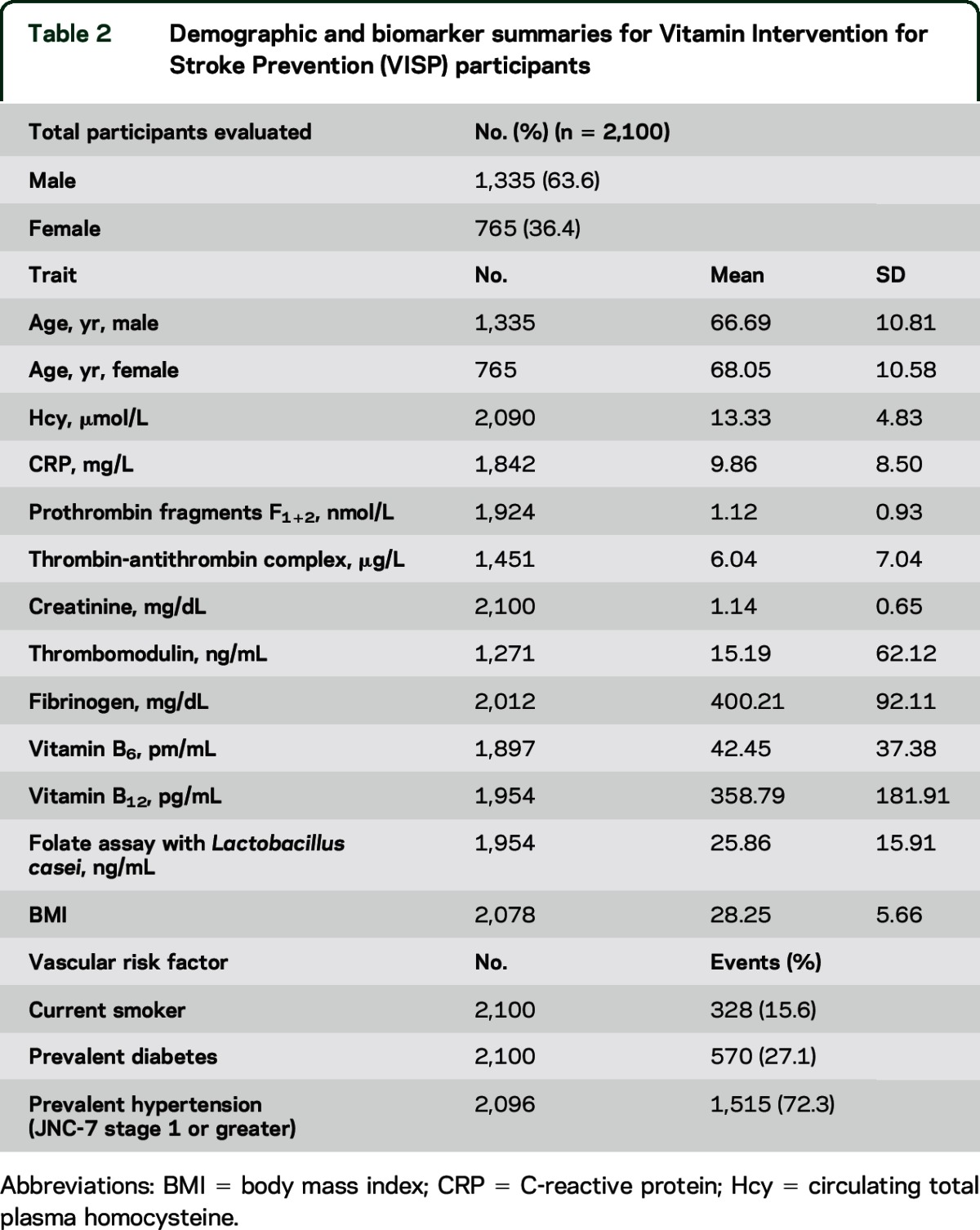
A total of 2,100 VISP participant samples were genotyped using the Illumina (San Diego, CA) HumanOmni1-Quad-v1 array with additional imputation of SNPs using 1000 Genomes Project (1000 G) data, resulting in a total coverage of 7,500,450 SNPs. Table 2 summarizes demographic information.
Table 2.
Demographic and biomarker summaries for Vitamin Intervention for Stroke Prevention (VISP) participants
Primary biomarker results.
CRP.
SNP rs2592902 had the strongest association (p = 1.14 × 10−9) with baseline CRP level. This SNP is 26.4 kb upstream from the CRP structural gene (table 1). Twenty-four significantly associated variants from our GWAS span the 5′ to the 3′ regions of the CRP gene. None of these SNPs is predicted to be damaging coding variants according to the human genome reference assembly GRCh37/hg19. Thus, these SNPs may be regulatory in their effect on CRP variation. A full functional analysis using HaploReg v2 can be found in table e-1.
Two SNPs located on chromosome 7q33, rs2589998 (p = 7.3 × 10−8) and rs2465086 (p = 8.5 × 10−8), approached genome-wide significance with CRP level (table 1). These SNPs are located near the Aldo-Keto Reductase Family 1, Member D1 gene (AKR1D1), and represent novel associations with CRP level. Both SNPs are expression quantitative trait loci (eQTLs) for CRP gene expression in liver (figure e-3).15 We assessed association between these SNPs and CRP levels in the longitudinal, population-based Framingham Heart Study (FHS).11,12 There was no statistically significant evidence supporting the association between rs2589998 and rs2465086 with CRP in FHS (data not shown), indicating a possible stroke population–specific effect.
Fibrinogen.
SNP rs2801231, the most strongly associated with fibrinogen levels, approached genome-wide significance (p = 5.0 × 10−7). This SNP is located on Xq25, with the closest gene, glutamate dehydrogenase 2 mitochondrial precursor (GLUD2), positioned 1.03 Mb downstream.
Creatinine, prothrombin fragments F1+2, TAT, and TM.
Results from the GWAS of creatinine, prothrombin fragments F1+2, TAT, and TM did not yield genome-wide significant associations (figure e-3). We did not identify significant associations in our data for previously published associations of these biomarkers. Table e-2 includes the most significant SNPs from each of these analyses.
Replication of SNPs associated with each biomarker in prior GWAS.
CRP.
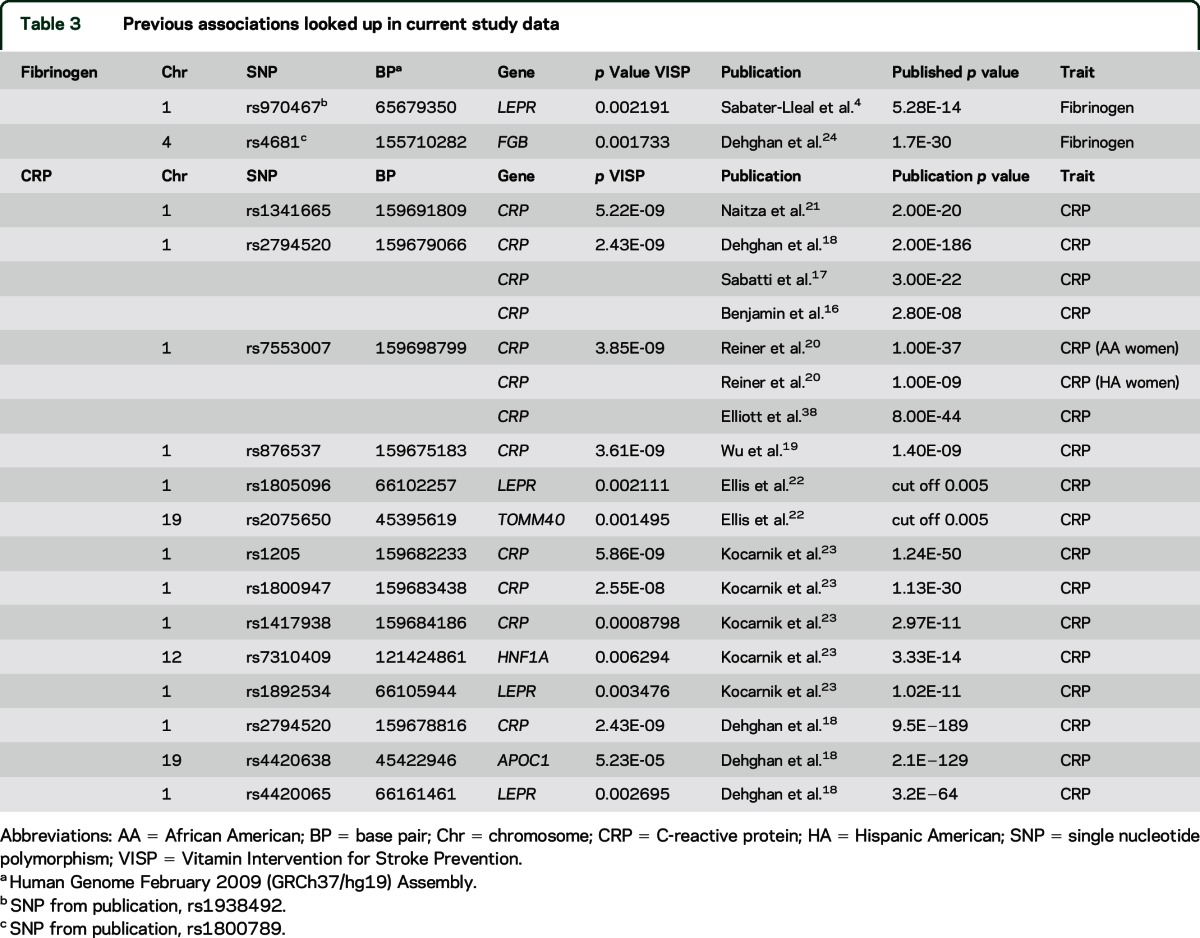
We replicated prior GWAS results including rs2794520 (p = 2.4 × 10−9), previously associated with CRP levels in FHS (p = 3.0 × 10−8),16 an analysis of metabolic traits in a birth cohort from a founder population (p = 3.0 × 10−22),17 and a meta-analysis of CRP levels in 80,000 participants (p = 2.0 × 10−186)18 (table 3). Other replicated variants associated with CRP include rs876537 (p = 3.6 × 10−9),19 rs7553007 (p = 3.8 × 10−9),20 and rs1341665 (p = 5.2 × 10−9).21 In addition to SNPs within or near the CRP gene, we replicated associations between SNPs in TOMM40,22 HNF1A,23 LEPR,22 and APOC118 and CRP levels (table 3).
Table 3.
Previous associations looked up in current study data
Fibrinogen.
The most significantly associated SNPs in prior GWAS of fibrinogen were not genotyped or imputed in VISP. We therefore assessed genic-level replication across LEPR and FGB, which contained the top loci from 2 prior GWAS of fibrinogen levels.4,24 SNPs within these genes showed association with fibrinogen in the VISP analysis (table 3).
CRP levels and CRP SNPs are associated with recurrent stroke in VISP.
Using a basic model adjusting for interventional group only, we found that circulating CRP levels were associated with recurrent stroke in VISP (p = 0.0007). These findings remained significant in the adjusted model (p = 0.0027). Individuals with CRP levels 1 SD above the population mean, 9.86 mg/L, had an approximate 1.25-fold increased risk of recurrent stroke in both models.
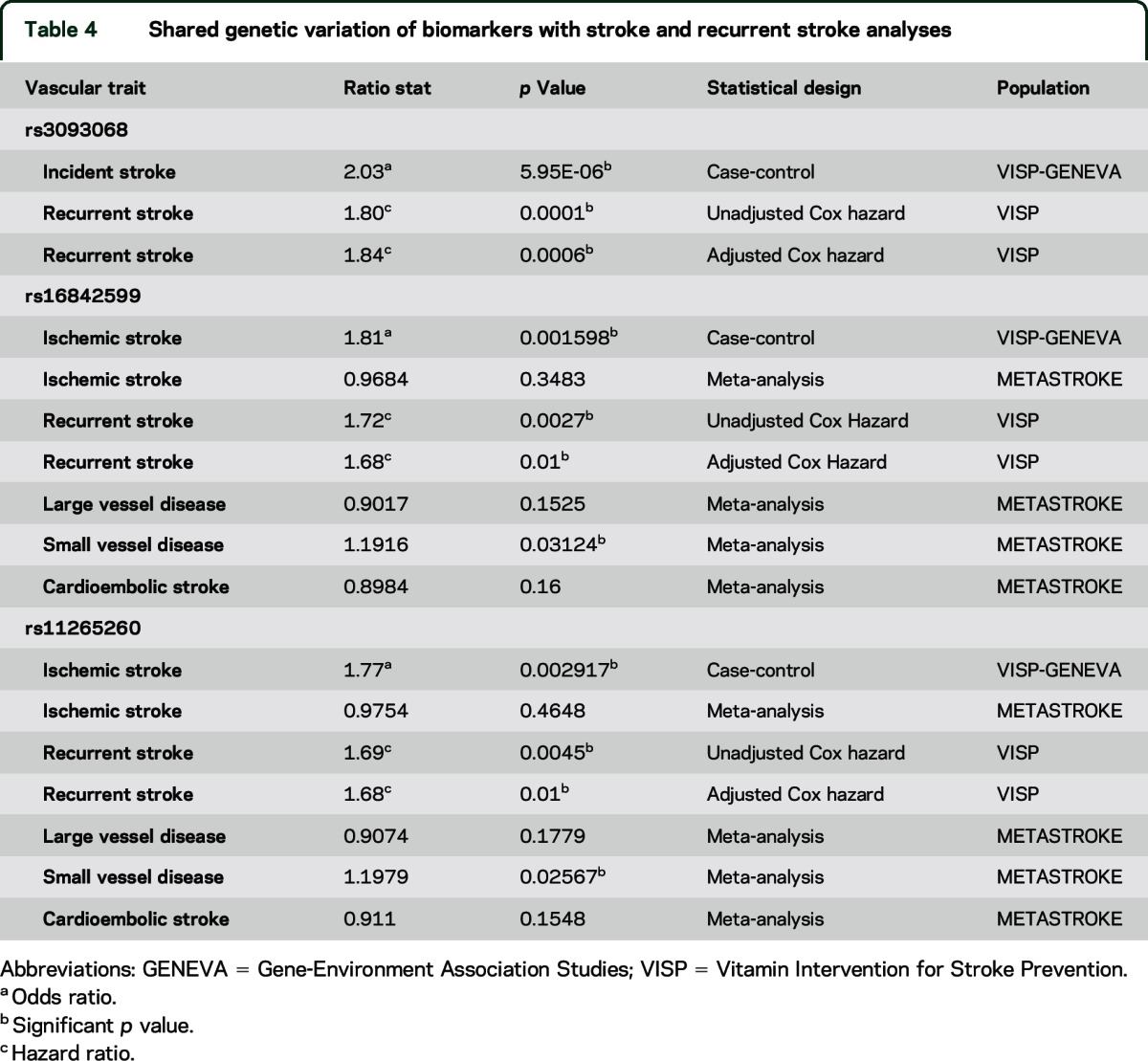
In a fully adjusted model, we found that CRP-associated SNPs, rs3093068, rs16842599, and rs11265260, were also associated with recurrent stroke in VISP (table 4) and confer a 1.8-fold increased risk for recurrent stroke. rs16842599 and rs11265260 are in complete LD with rs3093068 (r2 = 1.0, D' = 1.0, 1000 G phase 1 EUR population).
Table 4.
Shared genetic variation of biomarkers with stroke and recurrent stroke analyses
Fibrinogen levels are associated with recurrent stroke in VISP.
Similar to the CRP analyses, we found that circulating fibrinogen levels were associated with recurrent stroke in VISP (p = 0.001, hazard ratio 1.26). These findings remained significant in the adjusted model (p = 0.0129, hazard ratio 1.22). Individuals with fibrinogen levels 1 SD above the population mean, 400.21 mg/dL, had an approximate 1.24-fold increased risk of recurrent stroke in both models. These data are consistent with previous findings that show increased risk for cerebrovascular events and fibrinogen levels but lack a genetic association.4
Shared genetic susceptibility and replication of incident ischemic stroke and CRP.
All 3 SNPs found to be significantly associated with recurrent stroke were assed for association with incident ischemic stroke in a case-control analysis utilizing VISP (case) and High Density SNP Association Analysis of Melanoma (phs000187) (control). Associations were detected for all 3 SNPs and incident ischemic stroke, with rs3093068 showing the strongest association (p = 5.95 × 10−6) (table 4). We sought to replicate these findings in METASTROKE.7 The METASTROKE data imputed to HapMap2 do not contain rs3093068, thus, we used proxy SNPs. We found that rs12068753 (r2 = 0.98, 1000 G Pilot, EUR) was significantly associated with small vessel disease in METASTROKE (p = 0.027). In fact, 5 of 11 proxy SNPs interrogated were found to be significantly associated with small vessel disease in METASTROKE (corrected p < 0.05) (table e-3). Another SNP rs16842559, in perfect LD with rs3093068, was associated with large vessel disease (p = 0.049). We found no associations between CRP SNPs and all ischemic stroke or cardioembolic stroke in METASTROKE (table e-3).
DISCUSSION
Searching for genetic determinants of human variation in biomarker levels in ischemic stroke cases, we detected associations between variants within the CRP locus and CRP levels at a genome-wide significance level, and replicated prior GWAS associations for CRP and fibrinogen. Additionally, we identified a novel association between 2 SNPs at Ch7q33 and serum CRP measures that approached genome-wide significance; however, this association was not replicated.
CRP binds to the surface of dead or dying cells to activate the complement system via the C1Q complex and initiate the innate immune response.25 CRP is also known to play a fundamental role in the pathogenesis of atherosclerosis.26–28 Elevated high-sensitivity CRP level is a known risk factor for stroke and cardiovascular disease.29–31 We show that elevated CRP levels were associated with recurrent stroke risk. Three CRP-associated SNPs, rs3093068, rs16842599, and rs11265260, were also found to be associated with recurrent stroke in VISP, indicating a new and potentially clinically relevant connection between genetic determinants of CRP and recurrent stroke risk. In case-control analyses, we also found that these same SNPs were associated with overall ischemic stroke risk. Replication attempts in METASTROKE yielded encouraging results and refined potential mechanisms since associations were restricted to small vessel disease (rs12068753) and large vessel disease (rs16842559). CRP regulatory region variants have been associated with ischemic stroke in Han Chinese32 and reside in known regions of significance in African Americans and Hispanic Americans.20 The VISP trial's entry criteria excluded the majority of cardioembolic strokes and operable large vessel carotid disease, so our association with predominantly small vessel disease strokes in VISP contrasts with the EuroCLOT study results that identified this same SNP as associated with large vessel, cardioembolic, and overall stroke.33 Acute elevation of CRP levels is associated with plaque rupture. However, an association between CRP SNPs and small vessel disease in METASTROKE suggests that the association is not driven by acute plaque rupture.
Our association between SNP rs2592902, in the CRP gene, and CRP levels is novel. This SNP is an eQTL for CRP mRNA levels in whole blood as per the Genotype-Tissue Expression project (GTEx) (figure e-3).15 The other 5 CRP GWAS-associated SNPs are located in a region not previously associated with circulating CRP levels, potentially indicating a novel region of CRP regulation. We replicated other loci previously associated with CRP levels, including TOMM40, HNF1A, LEPR, and APOC1 (table 3). The recapitulation of these previously reported associations in our poststroke population provides a measure of confidence in our novel results and extends our understanding of genetic determinants of CRP levels during the subacute phase after ischemic stroke.
We identified a novel association approaching significance between 7p33 (the closest gene, AKR1D1, is 62.5 kb upstream) and CRP levels. AKR1D1 is responsible for catalyzing 5-β-reduction of bile acid intermediates and steroid hormones carrying a δ(4)-3-1 structure.34 Homozygous mutations in AKR1D1 cause bile acid synthesis defect, congenital, 2 (MIM 235555),35 and AKR1D1 is also associated with hepatic dysfunction.36 Functional evaluation using the GTEx dataset shows that in liver the top 2 SNPs at the AKR1D1 locus, rs2465086 and rs2589998, are eQTLs for CRP gene expression (p = 0.02 and p = 0.03, respectively), indicating a novel link between these genes and the biology of CRP function (figure e-3). Whole blood data for AKR1D1 are not available through GTEx. However, we were unable to replicate our genetic findings, indicating that further genetic studies will be necessary to confirm this finding.
The potential clinical importance of our CRP findings is further enhanced by the possibility of shared genetic susceptibility with vascular disease. We identified 3 SNPs—rs3093068, rs16842599, and rs11265260—associated with incident ischemic stroke, recurrent stroke, and stroke subtypes. These findings are critical to our understanding of genetic risk for vascular disease and could prove useful in risk stratification for prevention of both incident and recurrent stroke.
We continued to investigate how vascular biomarkers may be influenced by genetic drivers in VISP by performing a genome-wide analysis of fibrinogen. Fibrinogen is an essential part of the blood coagulation process and a known marker of inflammation. Elevated fibrinogen levels have consistently been associated with an increased risk for both stroke and cardiovascular disease.37 We confirmed the association of genetic variability in the leptin receptor (LEPR) and the fibrinogen β chain (FGB) genes with fibrinogen levels. The LEPR gene protein product is functional mainly within adipose tissue and helps to regulate the hunger/satiation balance. The FGB gene is a component of the active fibrinogen molecule. Both of these genes warrant follow-up as their potential impact on vascular risk and repair could be essential.
We found that fibrinogen levels are associated with recurrent stroke in both unadjusted and adjusted models. In contrast to CRP, no SNPs reached genome-wide significance for fibrinogen and thus were not assessed for association with ischemic stroke or recurrent stroke risk. This has been seen in a previous GWAS of fibrinogen.4
The GWAS for creatinine, prothrombin fragments F1+2, TAT, and TM failed to identify significant genetic associations. We were unable to replicate prior GWAS associations for plasma creatinine measures. Our analyses represent unique GWAS of F1+2, TAT, and TM that have no prior GWAS to attempt replication.
Limitations of our study include the low proportion of individuals from non-European descent, potentially limiting generalizability. Generally, GWAS are limited because of their lacking predictive power for the phenotype of interest, ability to singularly identify causative variants because of linkage disequilibrium especially in the case of rare SNPs, and inability to be reproduced across different populations. Additionally, stroke subtype information is not available for VISP. METASTROKE results suggest that subtype data may be relevant to explain observed associations.
Strengths include an extremely well-phenotyped study population in VISP, although stroke subtype information is lacking, paired with 1000 G imputed genetic data. These data enabled us to take a broad approach to investigating how known biomarkers of vascular disease may be functioning at both the genetic and biological levels of disease in stroke populations (VISP, METASTROKE) and the general population (FHS). Using a comprehensive approach, we generated our primary data, reviewed previously reported analyses, cross-referenced these studies, and investigated how this information may lead to a better understanding of clinical vascular risk. This study also buffers many of the general concerns of GWAS because of our foundation in known biological mechanisms of stroke risk, replication across different studies, and the functional evidence present in our eQTL analyses.
Biomarkers are important in prediction of disease susceptibility, outcomes, treatment response, and recovery. Our data provide confirmation of genetic associations for CRP and fibrinogen in the setting of ischemic stroke. We also show a novel putative association of variants on 7q33 and CRP levels, which requires independent replication, possibly in populations like VISP as FHS is distinctly different. Our results demonstrate that CRP-associated variants across multiple loci influence CRP levels in the acute phase following ischemic stroke, and that there are shared genetic factors between CRP and ischemic stroke risk and recurrent stroke risk. Confirming previous work, we show that elevated fibrinogen levels are associated with ischemic stroke. Evaluation of replication pointed to specific subtypes at highest risk. In total, these analyses demonstrate that known and novel variants contribute to biomarker levels following ischemic stroke, and knowledge of these factors can provide important new information in evaluating risk for cerebrovascular disease.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all the individuals who volunteered and participated in the VISP studies; the physicians involved in VISP; Daniel Gallo and Emily Farber at the University of Virginia's Genome Sciences Laboratory for technical support; and Fang Chen at the University of Virginia.
GLOSSARY
- 1000 G
1000 Genomes Project
- CRP
C-reactive protein
- eQTL
expression quantitative trait locus
- FGB
fibrinogen β chain gene
- FHS
Framingham Heart Study
- GENEVA
Gene-Environment Association Studies
- GTEx
Genotype-Tissue Expression project
- GWAS
genome-wide association studies
- LD
linkage disequilibrium
- LEPR
leptin receptor gene
- SNP
single nucleotide polymorphism
- TAT
thrombin-antithrombin complex
- TM
thrombomodulin
- VISP
Vitamin Intervention for Stroke Prevention
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: On behalf of METASTROKE, The Genomics and Randomized Trials Network (GARNET) Collaborative Research Group, Bruce Weir, Patrick Heagerty, Alex Reiner, Charles Kooperberg, Michele Sale, Bradford Worrall, Richard Weinshilboum, Peter Fasching, Ebony B. Madden, M.G.C. David Valle, Kimberly F. Doheny, Elizabeth Pugh, Stacey Gabriel, Daniel Mirel, Martin Dichgans, Hugh S Markus, Sudha Seshadri, Giorgio B Boncoraglio, Jonathan Rosand, Pankaj Sharma, Peter M Rothwell, Alex P Reiner, Martin Farrall, Jane Maguire, Braxton D Mitchell, Cathie M Sudlow, Robert Clarke, Myriam Fornage, James F Meschia, Unnur Thorsteinsdottir, Anita L DeStefano, Christopher Levi, and Solveig Gretarsdottir
AUTHOR CONTRIBUTIONS
Stephen R. Williams, Fang-Chi Hsu, Keith L. Keene, Wei-Min Chen, Sarah Nelson, Godfrey Dzhivhuho, Joe L. Rowles, Rainer Malik, Josée Dupuis, and Honghuang Lin performed analyses. All authors participated in the acquisition of the data and read and approved the final manuscript.
STUDY FUNDING
Study recruitment and collection of datasets for the VISP trial were supported by a grant (R01 NS34447; PI James Toole) from the National Institute of Neurological Disorders and Stroke (NINDS). GWAS genotyping performed at the Center for Inherited Disease Research (CIDR) (U01 HG004438l; PI David Valle) was funded by the National Human Genome Research Institute and the Genomics and Randomized Trials (GARNET) Network (U01HG00516-03; co-PI Michèle M. Sale and Bradford B. Worrall) and genetic data cleaning was provided by the GARNET Coordinating Center (U01HG005157; PI Bruce S. Weir). Given that all VISP participants were stroke cases, we obtained GWAS data (dbGAP) for external controls from the High Density SNP Association Analysis of Melanoma: Case-Control and Outcomes Investigation (study accession: phs000187.v1.p1); they were also genotyped on the Illumina HumanOmni1-Quad by CIDR. The Framingham Heart Study is funded by the National Heart, Lung and Blood Institute (NHLBI) (contract no. N01-HC-25195; http://www.framinghamheartstudy.org/) and by grants from NINDS (NS17950, www.ninds.nih.gov/), the NHLBI (U01HL 096917; www.nhlbi.nih.gov), and the National Institute on Aging (NIA) (AG08122, AG033193; www.nia.nih.gov). All links are current as of August 4, 2015. METASTROKE is an unfunded consortium of large case-control and cohort GWAS studies of stroke.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Fox ER, Benjamin EJ, Sarpong DF, et al. Epidemiology, heritability, and genetic linkage of C-reactive protein in African Americans (from the Jackson Heart Study). Am J Cardiol 2008;102:835–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vickers MA, Green FR, Terry C, et al. Genotype at a promoter polymorphism of the interleukin-6 gene is associated with baseline levels of plasma C-reactive protein. Cardiovasc Res 2002;53:1029–1034. [DOI] [PubMed] [Google Scholar]
- 3.de Lange M, Snieder H, Ariens RA, Spector TD, Grant PJ. The genetics of haemostasis: a twin study. Lancet 2001;357:101–105. [DOI] [PubMed] [Google Scholar]
- 4.Sabater-Lleal M, Huang J, Chasman D, et al. Multiethnic meta-analysis of genome-wide association studies in >100 000 subjects identifies 23 fibrinogen-associated loci but no strong evidence of a causal association between circulating fibrinogen and cardiovascular disease. Circulation 2013;128:1310–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarwar N, Sandhu MS, Ricketts SL, et al. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet 2010;375:1634–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mora S, Ridker PM. Justification for the Use of Statins in Primary Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER): can C-reactive protein be used to target statin therapy in primary prevention? Am J Cardiol 2006;97:33A–41A. [DOI] [PubMed] [Google Scholar]
- 7.Traylor M, Farrall M, Holliday EG, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol 2012;11:951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics 2010;26:2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010;26:2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families: The Framingham Offspring Study. Am J Epidemiol 1979;110:281–290. [DOI] [PubMed] [Google Scholar]
- 12.Splansky GL, Corey D, Yang Q, et al. The third generation cohort of the National Heart, Lung, and Blood Institute's Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol 2007;165:1328–1335. [DOI] [PubMed] [Google Scholar]
- 13.Frazer KA, Ballinger DG, Cox DR, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007;449:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng YC, Anderson CD, Bione S, et al. Are myocardial infarction–associated single-nucleotide polymorphisms associated with ischemic stroke? Stroke 2012;43:980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lonsdale J, Thomas J, Salvatore M, et al. The genotype-tissue expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benjamin EJ, Dupuis J, Larson MG, et al. Genome-wide association with select biomarker traits in the Framingham Heart Study. BMC medical genetics 2007;8(suppl 1):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabatti C, Service SK, Hartikainen AL, et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet 2009;41:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dehghan A, Dupuis J, Barbalic M, et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation 2011;123:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y, McDade TW, Kuzawa CW, et al. Genome-wide association with C-reactive protein levels in CLHNS: evidence for the CRP and HNF1A loci and their interaction with exposure to a pathogenic environment. Inflammation 2012;35:574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiner AP, Beleza S, Franceschini N, et al. Genome-wide association and population genetic analysis of C-reactive protein in African American and Hispanic American women. Am J Hum Genet 2012;91:502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naitza S, Porcu E, Steri M, et al. A genome-wide association scan on the levels of markers of inflammation in Sardinians reveals associations that underpin its complex regulation. PLoS Genet 2012;8:e1002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellis J, Lange EM, Li J, et al. Large multiethnic Candidate Gene Study for C-reactive protein levels: identification of a novel association at CD36 in African Americans. Hum Genet 2014;133:985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kocarnik JM, Pendergrass SA, Carty CL, et al. Multiancestral analysis of inflammation-related genetic variants and C-reactive protein in the population architecture using genomics and epidemiology study. Circ Cardiovasc Genet 2014;7:178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dehghan A, Yang Q, Peters A, et al. Association of novel genetic Loci with circulating fibrinogen levels: a genome-wide association study in 6 population-based cohorts. Circ Cardiovasc Genet 2009;2:125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure 1999;7:169–177. [DOI] [PubMed] [Google Scholar]
- 26.Libby P. Inflammation in atherosclerosis. Nature 2002;420:868–874. [DOI] [PubMed] [Google Scholar]
- 27.Chamorro A. Role of inflammation in stroke and atherothrombosis. Cerebrovasc Dis 2004;17(suppl 3):1–5. [DOI] [PubMed] [Google Scholar]
- 28.Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest 2003;111:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shoki AH, Mayer-Hamblett N, Wilcox PG, Sin DD, Quon BS. Systematic review of blood biomarkers in cystic fibrosis pulmonary exacerbations. Chest 2013;144:1659–1670. [DOI] [PubMed] [Google Scholar]
- 30.Kuo HK, Yen CJ, Chang CH, Kuo CK, Chen JH, Sorond F. Relation of C-reactive protein to stroke, cognitive disorders, and depression in the general population: systematic review and meta-analysis. Lancet Neurol 2005;4:371–380. [DOI] [PubMed] [Google Scholar]
- 31.Abraham J, Campbell CY, Cheema A, Gluckman TJ, Blumenthal RS, Danyi P. C-reactive protein in cardiovascular risk assessment: a review of the evidence. J Cardiometab Syndr 2007;2:119–123. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, Ding H, Tang JR, et al. C-reactive protein polymorphisms and genetic susceptibility to ischemic stroke and hemorrhagic stroke in the Chinese Han population. Acta Pharmacol Sin 2009;30:291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams FM, Carter AM, Hysi PG, et al. Ischemic stroke is associated with the ABO locus: the EuroCLOT study. Ann Neurol 2013;73:16–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondo KH, Kai MH, Setoguchi Y, et al. Cloning and expression of cDNA of human delta 4-3-oxosteroid 5 beta-reductase and substrate specificity of the expressed enzyme. Eur J Biochem 1994;219:357–363. [DOI] [PubMed] [Google Scholar]
- 35.Lemonde HA, Custard EJ, Bouquet J, et al. Mutations in SRD5B1 (AKR1D1), the gene encoding delta(4)-3-oxosteroid 5beta-reductase, in hepatitis and liver failure in infancy. Gut 2003;52:1494–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzales E, Cresteil D, Baussan C, Dabadie A, Gerhardt MF, Jacquemin E. SRD5B1 (AKR1D1) gene analysis in delta(4)-3-oxosteroid 5beta-reductase deficiency: evidence for primary genetic defect. J Hepatol 2004;40:716–718. [DOI] [PubMed] [Google Scholar]
- 37.Eidelman RS, Hennekens CH. Fibrinogen: a predictor of stroke and marker of atherosclerosis. Eur Heart J 2003;24:499–500. [DOI] [PubMed] [Google Scholar]
- 38.Elliott P, Chambers JC, Zhang W, et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA 2009;302:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.