


Abstract
Any profound comprehension of gene function requires detailed information about the subcellular localization, molecular interactions and spatio-temporal dynamics of gene products. We developed a multifunctional integrase (MIN) tag for rapid and versatile genome engineering that serves not only as a genetic entry site for the Bxb1 integrase but also as a novel epitope tag for standardized detection and precipitation. For the systematic study of epigenetic factors, including Dnmt1, Dnmt3a, Dnmt3b, Tet1, Tet2, Tet3 and Uhrf1, we generated MIN-tagged embryonic stem cell lines and created a toolbox of prefabricated modules that can be integrated via Bxb1-mediated recombination. We used these functional modules to study protein interactions and their spatio-temporal dynamics as well as gene expression and specific mutations during cellular differentiation and in response to external stimuli. Our genome engineering strategy provides a versatile open platform for efficient generation of multiple isogenic cell lines to study gene function under physiological conditions.
INTRODUCTION
In the last decades targeted gene disruption has been a widely used approach to gain first insights into gene function. However, gene disruption studies are often hampered by high functional redundancy in mammalian systems and yield little information about the subcellular localization, interactions and spatio-temporal dynamics of gene products. In order to gain comprehensive understanding of gene function these studies need to be complemented by more complex genetic manipulations such as fluorophore knockin, specific domain deletions or introduction of point mutations. Additionally, a systematic analysis of gene function requires application of biochemical as well as imaging techniques, which usually rely on the generation of gene specific antibodies, a technically demanding and time-consuming process.
Recently, RNA guided endonucleases (RGENs) derived from the prokaryotic Type II CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR-associated) system have emerged as promising tools for the manipulation and modification of genetic sequences (1–4).
The specificity of RGENs is mediated by small guide RNAs (gRNAs) that bind to 20 bp within the target sequence and recruit the Cas9 nuclease to introduce a double strand break. Although this two-component system has greatly facilitated the generation of gene disruptions in bacteria, plants and mammals, concerns have been raised about considerable off-target effects (5–7). Furthermore, the low frequency of homologous recombination in mammals makes insertion of exogenous components such as fluorophore tags difficult and time-consuming.
In addition to RGENs, phage-derived serine integrases have received considerable attention as novel tools for genome engineering. Recently, Bxb1 was shown to have the highest accuracy and efficiency in a screen of fifteen candidate serine integrases tested in mammalian cells (8). Serine integrases are unidirectional, site-specific recombinases that promote the conservative recombination between phage attachment sites (attP) and bacterial attachment sites (attB) (9) with much higher recombination efficiencies (up to 80%) than the commonly used bidirectional tyrosine integrases, Cre or Flp (9–12).
In this study, we aim to combine the advantages of both RGENS and unidirectional integrases into one fast, widely applicable and flexible method. We developed a novel strategy for genome engineering based on a CRISPR/Cas assisted in-frame insertion of an attP site, which we refer to as the multifunctional integrase (MIN) tag. At the genetic level, the MIN-tag serves as an attachment site for the serine integrase Bxb1 that can be used to introduce a broad range of prefabricated functional cassettes into the genomic locus with high specificity and efficiency. At the protein level, the MIN-tag functions as a novel epitope tag that can be detected with a highly specific monoclonal antibody and used for immunoprecipitation as well as immunofluorescence experiments. To demonstrate the versatility of the strategy, we generated MIN-tagged murine embryonic stem cell (mESC) lines for a variety of major epigenetic factors, including Dnmt1, Dnmt3a, Dnmt3b, Tet1, Tet2, Tet3 and Uhrf1. We created a toolbox of vectors for Bxb1-mediated recombination to generate isogenic cell lines harboring knockout cassettes, fluorescent protein fusions, enzymatic tags and specific mutations; all derived from a single entry cell line ensuring maximal biological comparability. We demonstrate the power of this strategy using proximity-dependent protein labeling to identify novel interactors of TET1 in mESCs as well as to systematically study the subcellular localization, binding kinetics and protein expression dynamics of the de novo methyltransferase DNMT3B during epiblast differentiation.
MATERIALS AND METHODS
Western blotting and immunoprecipitation
Western blot analysis was performed using the following primary antibodies: anti-DNMT1, anti-DNMT3a (Imgenex, 64B1446); anti-DNMT3b (Abcam, 52A1018); anti-UHRF1 (13); anti-TET1, anti-TET2 and anti-TET3 (14); anti-GFP antibody (Roche, 11814460001); anti-β-Actin (Sigma, A5441); anti-SNF2H (Abcam, ab22012). Blots were probed with anti-rat (Jackson ImmunoResearch, 112-035-068), anti-mouse (Sigma, A9044) and anti-rabbit (Biorad, 170–6515) secondary antibodies conjugated to horseradish peroxidase (HRP) and visualized using an ECL detection kit (Pierce). An anti-mouse antibody conjugated to Alexa 488 (Life Technologies, A21202) was used for fluorescence detection of western blots using the Typhoon 9400 (GE Healthcare) imaging system.
For immunoprecipitation, ∼1 × 106 Dnmt1attP/attP, Dnmt3battP/attP or wt cells were harvested in ice cold phosphate buffered saline (PBS), washed twice and subsequently homogenized in 200 μl lysis buffer (20 mM Tris/HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 1 mM PMSF, 0.5% NP40). After centrifugation (10 min, 14 000 g, 4°C) the supernatant was adjusted with dilution buffer (20 mM Tris/HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 1 mM PMSF) to a final volume of 300 μl. A total of 50 μl were mixed with sodium dodecyl sulphate (SDS)-containing sample buffer (referred to as input (I)). For pull-downs, 100 μl (4 μg) of either 5A10 DNMT1 antibody (15) or the newly generated MIN-tag antibody 1E1 was added to the cell lysates and incubated 2 h at 4°C. For pull-down of immunocomplexes, 40 μl of protein G agarose beads (GE Healthcare, Freiburg, Germany) equilibrated in dilution buffer were added and incubation continued for 2 h. After centrifugation (2 min, 5000 × g, 4°C) 50 μl of the supernatant was collected (referred to as flow-through (FT)) while the remaining supernatant was removed. The beads were washed twice with 1 ml dilution buffer containing 300 mM NaCl. After the last washing step, the beads were resuspended in 50 μl Laemmli buffer and boiled for 10 min at 95°C. For immunoblot analysis, 3% of the input and the flow-through as well as 30% of the bound (B) fraction were separated on a 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to western blot analysis.
Immunofluorescence staining and microscopy
Immunostaining was performed as described previously (16). Briefly, cells cultured on coverslips were fixed with 4% paraformaldehyde for 10 min, washed with PBST (PBS, 0.02% Tween20) and permeabilized with PBS supplemented with 0.5% Triton X-100. Both primary and secondary antibody were diluted in blocking solution (PBST, 2% BSA, 0.5% fish skin gelatin). Coverslips with cells were incubated with primary and secondary antibody solutions in dark humid chambers for 1 h at RT; washings after primary and secondary antibodies were done with PBST. Following secondary antibody incubations, cells were post-fixed with 4% paraformaldehyde for 10 min. For DNA counterstaining, coverslips were incubated in a solution of DAPI (2 μg/ml) in PBS. Coverslips were mounted in antifade medium (Vectashield, Vector Laboratories) and sealed with colorless nail polish.
For immunolabeling, the following primary antibodies were used: anti-DNMT1 (15); anti-DNMT3A (Imgenex, 64B1446); anti-DNMT3B (Abcam, 52A1018); anti-UHRF1 (13); anti-TET1, anti-TET2 (14); GFP-Booster_ATTO488 (Chromotek). The secondary antibodies were anti-rabbit conjugated to DyLight fluorophore 594 (Jackson ImmunoResearch, 711-505-152), anti-mouse conjugated to Alexa 488 (Life Technologies, A21202), anti-rat conjugated to Alexa 488 (Life Technologies, A21208) or Alexa 594 (Life Technologies, A21209).
Single optical sections or stacks of optical sections were collected using a Leica TCS SP5 confocal microscope equipped with Plan Apo 63×/1.4 NA oil immersion objective and lasers with excitation lines 405, 488, 561 and 633 nm.
Live cell imaging experiments were performed on an UltraVIEW VoX spinning disc microscope assembled to an Axio Observer D1 inverted stand (Zeiss) and using a 63×/1.4 NA Plan-Apochromat oil immersion objective. The microscope was equipped with a heated environmental chamber set to 37°C and 5% CO2. Fluorophores were excited with 488 nm or 561 nm solid-state diode laser lines. Confocal image series were typically recorded with 14-bit image depth, a frame size of 1024 × 1024 pixels and a pixel size of 110 nm. z-stacks of 12 μm with a step size of 1 μm were recorded every 30 min for about 24 h or for the live cell series of Dnmt3battP/attP every hour for 60 h. To avoid photodamage of the cells, the AOTF of the laser was set to low transmission values of 6–10%. Binning was set to 2×.
Super-resolution microscopy
Super-resolution images were obtained with a DeltaVision OMX V3 3D-SIM microscope (Applied Precision Imaging, GE Healthcare), equipped with a 60×/1.42 NA PlanApo oil objective and sCMOS cameras (Olympus). A z-step size of 125 nm was used during acquisition. SI raw data were reconstructed and deconvolved with the SoftWorX 4.0 software package (Applied Precision). FIJI and Photoshop CS5.1 (Adobe) were used for image processing and assembly.
Antigen preparation, immunization, generation of hybridomas and ELISA screening
For the translated attP peptide, the MIN antigen (attP peptide) was designed with the following sequence SGQPPRSQWCTVQT-Cys. Peptides were synthesized, HPLC purified and coupled to OVA (Peps4LifeSciences-Anette Jacob; Heidelberg). Lou/c rats were immunized subcutaneously and intraperitoneally with a mixture of 50 μg peptide-OVA, 5 nmol CPG oligonucleotide (Tib Molbiol, Berlin), 500 μl PBS and 500 μl incomplete Freund's adjuvant. A boost without adjuvant was given 6 weeks after primary injection. Fusion of the myeloma cell line P3 × 63-Ag8.653 with the rat immune spleen cells was performed using polyethylene glycol 1500 (PEG 1500, Roche, Mannheim, Germany). After fusion, the cells were plated in 96 well plates using RPMI1640 with 20% fetal calf serum, penicillin/streptomycin, pyruvate, non-essential amino acids (Gibco) supplemented by hypoxanthine-aminopterin-thymidine, (HAT) (Sigma, St Louis, MO, USA). Hybridoma supernatants were tested in a solid-phase immunoassay. Microliter plates were coated with avidin (3 μg/ml, Sigma) over night. After blocking with 2% FCS in PBS, plates were incubated with biotinylated MIN peptide at a concentration of 0.2 μg/ml in blocking buffer. After washing the plates, the hybridoma supernatants were added. Bound rat mAbs were detected with a cocktail of HRP-labeled mouse mAbs against the rat IgG heavy chains, thus avoiding IgM mAbs (α-IgG1, α-IgG2a, α-IgG2b (ATCC, Manassas, VA, USA), α-IgG2c (Ascenion, Munich, Germany). HRP substrate conversion was visualized with ready to use TMB (1-Step™ Ultra TMB-ELISA, Thermo). MIN-tag clone 1E1 (rat IgG1) was stably subcloned and further characterized.
A set of 25 rat derived hybridoma supernatants were tested for specificity against an integrated attP peptide in the Dnmt1 locus using both western blot analysis and high content microscopy. Western blots were prepared as mentioned previously. Each supernatant was used in a 1:10 dilution. Blots were probed with an anti-rat secondary antibody conjugated to HRP.
Cells were prepared for immunofluorescence as described above, with the exception that cells were fixed on a 96-well Cell Carrier® plate (Greiner). Cells in individual wells were incubated with the various hybridoma supernatants (1:100) for 1 h. As a secondary antibody, anti-rat conjugated to Alexa 488 (Life Technologies, A21208) was used. Nuclei were counterstained using DAPI. Images of stained cells were acquired automatically with an Operetta high-content imaging system using a 40× air objective (PerkinElmer). DAPI and ATTO488 coupled antibodies were excited and their emissions recorded using standard filter sets. Exposure times were 10 and 400 ms for DAPI and ATTO488, respectively. All monoclonal antibodies described in this study are available upon request.
The MIN antibody are available via http://human.bio.lmu.de/_webtools/MINtool/AB_info.html.
DNA methylation analysis
For the analysis of DNA methylation levels, genomic DNA was isolated using the QIAamp DNA Mini Kit (QIAGEN). Bisulfite treatment was performed using the EZ DNA Methylation-Gold™ Kit (Zymo Research Corporation) according to the manufacturer's protocol. Subsequently, the major satellite repeats sequence was amplified using the primers described in (17). The biotinylated polymerase chain reaction (PCR) products of the second PCR were analyzed by pyrosequencing (Varionostic GmbH, Ulm, Germany).
Targeting donor and plasmid construction
Plasmid sequences can be found in Supplementary Table S6. Targeting donor constructs were either synthesized as ssDNA oligonucleotides (Integrated DNA Technologies) or produced by amplifying 300 to 200 bp long homology arms with the respective external and internal primer sets (Supplementary Table S2). These PCR products of the 5′ and 3′ homology arms were pooled and an overlap extension PCR with the external primers was performed to yield the final targeting fragments. The gRNA vector was synthesized at Eurofins MWG Operon based on the sequences described (3). The subcloning of targeting sequences was performed by circular amplification. The surrogate reporter (pSR) was generated by inserting in vitro annealed DNA oligos via AsiSI and NruI into pCAG-mCh (18). eGFP was amplified using the primers eGFP-F and eGFP-R and sequentially cloned into pCAG-mCh-NruI linker to generate the pSR construct. Reporters were generated by subcloning in vitro annealed DNA oligos containing CRISPR target sites into KpnI and NheI digested pSR. The attB-GFP-knockin construct was generated from R6K-NFLAP (19) by ligation free cloning (20) rearranging the backbone sequences into the artificial intron and introducing the attB site 5′ of the GFP open reading frame (ORF), removing its start codon. The attB-GFP-Poly(A) and attB-mCh-Poly(A) constructs were created by amplifying the GFP ORF including the stop codon and SV40 Poly(A) signal from pCAG-eGFP-IB and inserted into the attB-LAP-tag backbone by ligation free cloning. The attB-mCh-Poly(A)-mPGK-PuroR construct was generated by subcloning the mPGK-PuroR sequence from pPthc-Oct3/4 (21) and ligating it into the EcoRV site of the attB-mCh-Poly(A) construct. The attB-GFP-Poly(A)-mPGK-NeoR was produced by first exchanging the PuroR in pPthc-Oct3/4 with NeoR from pEGFP-C1 (22) using HindIII. The combined mPGK-NeoR was then subcloned into the attB-GFP-Poly(A) vector via the same EcoRV site mentioned previously. The attB-GFP-Dnmt1cDNA-Poly(A), attB-GFP-Tet1cDNA-Poly(A) and attB-GFP-Dnmt3b1cDNA-Poly(A) constructs were generated by inserting the appropriate cDNAs from constructs reported previously (17,23–24) via AsiSI/NotI sites into the attB-GFP-Poly(A) and attB-mCh-Poly(A) vectors respectively. The attB-GFP-Dnmt3b6-Poly(A), attB-GFP-Tet1-d1–1363-Poly(A), attB-GFP-Tet1-d833–1053-Poly(A), attB-GFP-Tet1-d833–1363-Poly(A) vectors were produced via circular amplification with overlap extension primers using the above mentioned attB-GFP-Dnmt1/Dnmt3b1/Tet1cDNA-Poly(A) constructs as templates.
The attB-GFP-Dnmt3b6-Poly(A)-mPGK-NeoR and attB-mCh-Dnmt3b1-Poly(A)-mPGK-PuroR integration constructs were created by inserting the Dnmt3b6 and Dnmt3b1 sequences (from attB-GFP-Dnmt3b6-Poly(A) and attB-GFP-Dnmt3b1-Poly(A)) using AsiSI/NotI sites into attB-GFP-Poly(A)-mPGK-NeoR and attB-mCh-Poly(A)-mPGK-PuroR vectors, respectively.
All constructs described in this study are available via Addgene or via http://human.bio.lmu.de/_webtools/MINtool/.
Cell culture
J1 ESCs were maintained on gelatin-coated dishes in Dulbecco's modified Eagle's medium supplemented with 16% fetal bovine serum (FBS, Biochrom), 0.1 mM ß-mercaptoethanol (Invitrogen), 2 mM L-glutamine, 1× MEM Non-essential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin (PAA Laboratories GmbH), 1000 U/ml recombinant mouse LIF (Millipore) and 2i (1 μM PD032591 and 3 μM CHIR99021 (Axon Medchem, Netherlands), referred to as ESC medium. Differentiation of naive pluripotent stem cells to epiblast-like cells was performed according to the protocol of (25). Briefly, J1 ESCs were maintained in the ground state in Geltrex (Life Technologies) coated flasks and cultured in N2B27 (50% neurobasal medium (Life Technologies), 50% DMEM/F12 (Life Technologies), 2 mM L-glutamine (Life Technologies), 0.1 mM β-mercaptoethanol, N2 supplement (Life Technologies), B27 serum-free supplement (Life Technologies) containing 2i and 1000 U/ml LIF 100 U/ml Penicillin-streptomycin) for at least three passages before differentiation. To differentiate naive ESCs into epiblast-like cells, cells were replated in N2B27 differentiation medium containing 10 ng/ml Fgf2 (R&D), 20 ng/ml Activin A (R6D) and 0.1× Knockout Serum Replacement (KSR)(Life Technologies). Time point 0 h in differentiation time-course experiments corresponds to the time N2B27 differentiation medium was added to cells.
Generation of MIN-tagged and Bxb1-mediated knockin cell lines
To produce MIN-tagged cell lines, 5 × 105 cells were dissociated and seeded in 0.2% gelatin (Sigma-Aldrich) coated p35 plates. After 3 h, cells were transfected with 2 μg of the MIN-tag donor/homology ssDNA oligo or PCR product, 0.5 μg gRNA construct, 0.5 μg surrogate reporter construct and 1 μg Cas9 using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. For Bxb1-mediated recombination of attB constructs, 5 × 105 cells were transfected with 1 μg pCAG-NLS-HA-Bxb1 expression plasmid ((26) addgene 51271), 1 μg of the respective attB construct and 0.5 μg Bxb1 surrogate reporter. For both MIN-Tagging and Bxb1-mediated recombination, cells were dissociated, resuspended in ESC medium 48 h post transfection and then analyzed and sorted with a FACS Aria II (Becton Dickinson). For MIN-tagging, enrichment of cells with RGEN activity was accomplished by single-cell sorting GFP and mCherry positive cells into 96-well plates (Falcon) containing 150 μl of ESC medium. For Bxb1-mediated recombination, cells with Bxb1 activity were enriched for by single-cell sorting GFP positive cells into 96-well plates. Alternatively for Bxb1-mediated integration using antibiotic selection, cells were replated into p150 plates with ESC medium containing G418 (0.5 mg/ml, AppliChem) and puromycin (1 μg/ml, AppliChem) 48 h post transfection.
Identification of MIN-tagged and Bxb1-mediated knockin cell lines with restriction fragment analysis and PCR screening
After ∼7 days (until colonies were readily visible), plates from single-cell sortings were screened for colony growth. Surviving colonies were dissociated and individually replated onto two 96-well plates. Genomic DNA was isolated from one plate after 2–3 days, while the second plate remained in culture. To identify MIN-tagged clones, the region surrounding the ATG (or stop codon in the case of C-terminal tagging) was PCR amplified using the appropriate external and screening primers (Supplementary Table S2). For restriction fragment analysis, 10 μl of these PCR products were digested with either HincII or SacII and then analyzed on 1.5% agarose gels. PCRs of positive clones were confirmed by Sanger sequencing. To screen for Bxb1-mediated recombiation, we employed a three-primer PCR strategy using the respective external primers flanking the MIN-tagged locus and an attL-specific primer (Supplementary Figure S3A, Table S2). For Bxb1-mediated integrations using antibiotic selection, mESC colonies were picked, dissociated using trypsin and plated into individual wells on 96-well plates ∼7 days after starting antibiotic selection. Genomic DNA isolation and screening PCRs were performed as described above. Clones harboring the desired MIN-tag insertion or Bxb1-mediated integration were expanded, frozen and stored in liquid nitrogen.
All cell lines are available at http://human.bio.lmu.de/_webtools/MINtool/cell_lines.html.
Genomic DNA isolation for PCR
Cells were lysed in multi-well plates by the addition of 50 μl lysis buffer (10mM Tris/HCl pH 7.4, 10mM EDTA, 10mM NaCl, 50μg/ml Proteinase K, 1.7 μM SDS) per well. The Plates were subsequently incubated at −80°C for 15 min, followed by 3 h at 56°C. Heat inactivation of Proteinase K was performed by incubation at 85°C for 20 min. The resulting crude DNA lysates were directly subjected to PCR.
BioID
BioID experiments were performed after (27) using extracted crude nuclei (adapted from (28)) as input material. In brief, cells were cultured for 48 h with or without addition of 50 μM biotin. Cell pellets (∼4 ×107 cells) were washed once in buffer A (10 mM HEPES/KOH pH 7.9, 10 mM KCl, 1.5 mM MgCl2) and resuspended in buffer A containing 0.15% NP-40 and 1× protease inhibitor (SERVA). Samples were homogenized using a pellet pestle. After centrifugation, crude nuclei pellets were washed once with PBS. Crude nuclei were resuspended in BioID-lysis buffer (0.2% SDS, 50 mM Tris/HCl pH 7.4, 500 mM NaCl, 1 mM DTT, 1× protease inhibitor), supplemented with 2% Triton X-100 and subjected to sonication twice using a Branson Sonifier 450 (15% amplitude, 0.3 s pulse, 0.6 s pause, total time 30 s). Samples were diluted 1:1 with 50 mM Tris/HCl pH 7.4 after the first sonication step. Pulldown of biotinylated proteins was performed overnight at 4°C with rotation using M-280 Streptavidin Dynabeads (Life Technologies) for subsequent mass spectrometry or Streptactin-Superflow agarose beads (IBA) for SDS-PAGE analysis, respectively. Beads were washed with wash buffer 1 (2% SDS), wash buffer 2 (0.1% desoxycholic acid, 1% Triton X-100, 1 mM EDTA, 500 mM NaCl, 50 mM HEPES/KOH pH 7.5) and wash buffer 3 (0.5% desoxycholic acid, 0.5% NP-40, 1 mM EDTA, 500 mM NaCl, 10 mM Tris/HCl pH 7.4) followed by two washing steps with 50 mM Tris/HCl pH 7.4. For SDS-PAGE analysis, proteins were silverstained after (29).
Digest of proteins and sample preparation for LC-MS/MS
On-beads digest of proteins was performed as described in (28). All steps were carried out at room temperature. Beads were resuspended in 2 M Urea in Tris/HCl pH 7.5, reduced with 10 mM DTT for 20 min and subsequently alkylated with 50 mM chloroacetamide for 20 min. A total of 0.25 μg Pierce Trypsin Protease (Thermo Scientific) was added for 2 h. Beads were collected by centrifugation and the resulting peptide supernatant was further incubated overnight with addition of 0.1 μg trypsin. Peptides were desalted using StageTips (30).
LC-MS/MS and data analysis
Peptides were reconstituted in 20 μl mobile phase A (2% v/v acetonitrile, 0.1% v/v formic acid) and analyzed by tandem mass spectrometry using a EASY-nLC 1000 nano-HPLC system connected to a LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). About 2–4 μl of the peptide mixture were separated onto a PepMap RSLC column (75 μm ID, 150 mm length, C18 stationary phase with 2 μm particle size and 100 Å pore size, Thermo Fisher Scientific) and introduced into the mass spectrometer at a flow rate of 300 nl/min running a gradient from 5 to 35% mobile phase B (98% v/v acetonitrile, 0.1% v/v formic acid). Ion source and transmission parameters of the mass spectrometer were set to spray voltage = 2 kV, capillary temperature = 275°C. The mass spectrometer was operated in data-dependent mode, selecting up to 10 precursors from a MS1 scan (resolution = 60 000) in the range of m/z 250–1800 for collision-induced dissociation (CID). Singly (+1) charged precursor ions and precursors of unknown charge states were rejected. CID was performed for 10 ms using 35% normalized collision energy and the activation q of 0.25. Dynamic exclusion was activated with a repeat count of one, exclusion duration of 30 s, list size of 500 and the mass window of ±10 ppm. Ion target values were 1 000 000 (or maximum 10 ms fill time) for full scans and 10 000 (or maximum 100 ms fill time) for MS/MS scans, respectively. Raw data were analyzed using MaxQuant Version 1.5.2.8 (31) using the MaxLFQ label free quantification algorithm (32) and the match-between-runs functionality. UniprotKB MOUSE.fasta was used as a reference database (33). A maximum of two missed cleavages and a false discovery rate of 1% were set as parameters. Oxidation of methionine and biotinylation were searched as variable modifications and carbamidomethylation of cysteine residues as fixed modification. For statistical analysis, the Perseus software version 1.5.1.6 was used (31). Significance was tested using a two sided Student's t-test and a permutation based FDR calculation. GO enrichment analysis was performed with the Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GOrilla, (34)). A P-value < 0.01 was considered significant.
FRAP
Live cell imaging and FRAP experiments were typically performed on an UltraVIEW VoX spinning disc microscope with integrated FRAP PhotoKinesis accessory (PerkinElmer) assembled to an Axio Observer D1 inverted stand (Zeiss) and using a 63×/1.4 NA Plan-Apochromat oil immersion objective. The microscope was equipped with a heated environmental chamber set to 37°C. Fluorophores were excited with 488 nm (exposure time: 400 ms, laser power: 15%) or 561 nm (exposure time: 450 ms, laser power: 30%) solid-state diode laser lines. Confocal image series were typically recorded with 14-bit image depth, a frame size of 256 × 256 pixels and a pixel size of 110 nm. For photobleaching experiments, the bleach regions, typically with a diameter of 2 μm, were manually chosen to cover the chromocenters. Photobleaching was performed using one iteration with the acousto-optical tunable filter (AOTF) of the 488 nm laser line set to 100% transmission. Typically, 10 pre-bleach images were acquired at a rate of 1 s per timepoint and 60 post-bleach frames were recorded at a rate of 10 s per timepoint. Data correction, normalization and quantitative evaluations were performed by automated processing with ImageJ (http://rsb.info.nih.gov/ij/) using a set of newly developed macros followed by calculations in Excel.
RESULTS
A fast and efficient strategy to generate MIN-tagged genomic loci
Our novel genome engineering strategy relies on the CRISPR/Cas-assisted insertion of the MIN-tag sequence into the open reading frame of a target gene either directly downstream of the start codon or upstream of the stop codon (Figure 1A and Supplementary Figure S2H). Neither regulatory regions nor gene structure are altered, leading to preservation of the endogenous expression pattern and post-transcriptional processing of the gene of interest.
Figure 1.
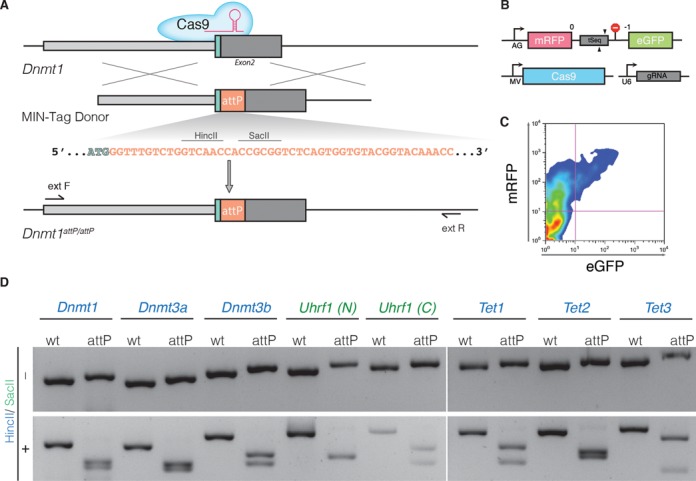
Generation of MIN-tagged cell lines. (A) Schematic overview of MIN-tag insertion into the Dnmt1 locus via CRISPR/Cas assisted gene editing. The MIN-tag donor harbors the attP site and homology to the genomic sequence 5′ and 3′ of the start codon. Integration is facilitated by double strand breaks created by Cas9 directed to the target sequence by a specific gRNA. Restriction enzyme recognition sites used for screening in this study are indicated above the attP sequence. (B) Schematic overview of the surrogate reporter used to enrich for cells expressing a functional Cas9 complex. The respective Cas9 target sequence (tSeq) is placed downstream of mRFP followed by a stop codon and an out-of-frame GFP ORF. This surrogate reporter is transfected into the cells together with a vector expressing Cas9 and a U6 driven gRNA expression cassette. (C) Cells that express a functional Cas9 complex can then be identified by expression of GFP and enriched via FACS. (D) Screening PCRs followed by restriction digest with HincII or SacII of all generated MIN-tagged cell lines. (N) and (C) refer to N- and C-terminal tagging, respectively.
Since epigenetic processes undergo dramatic changes during early embryonic development and are tightly regulated, we tested the efficacy and versatility of our method by targeting the DNA modifying enzymes Dnmt1, Dnmt3a, Dnmt3b, Tet1, Tet2 and Tet3 as well as the chromatin binding protein Uhrf1 in mESCs (Figure 1D). We generated targeting donors containing the 48 bp MIN-tag sequence flanked by short homology arms (200–300 bp for PCR-based donors or 76 bp for single stranded DNA oligos). We next designed specific gRNAs to target sequences located either in close proximity to or overlapping the start or stop codon of the respective genes. As scarless integration of the MIN-tag requires a resistance free selection strategy we used a surrogate reporter assay to enrich for cells that express an active Cas9:gRNA complex by fluorescence-activated cell sorting (FACS) (Figure 1B and C). In this reporter assay, the target sequence is inserted between the ORF of mCherry (mCh) and GFP thereby disrupting the reading frame of the fusion. GFP is expressed only when the target sequence is cleaved by a specific and active Cas9:gRNA complex, which causes small, frameshifting insertions or deletions by non-homologous end joining (NHEJ) restoring the reading frame of the fluorescent protein (35). For each targeting, we co-transfected mESCs with a mixture of surrogate reporter construct, gRNA vector, Cas9 expression plasmid and the specific targeting MIN-tag donor fragment. After single cell sorting of GFP positive cells and expansion of the resulting colonies, we isolated genomic DNA by a fast and simplified in-well lysis protocol to screen for positive clones by PCR and analytical restriction digest. This allows the identification of hetero- and homozygous insertions already at this stage (Supplementary Figure S1D). Combined, all targeting yielded positive clones with an average efficiency of 3% for homozygous and 1% for heterozygous insertions (Supplementary Table S1). All targeted genes were expressed normally and subcellular localization as well as enzymatic activity was not disrupted in comparison to wild-type (wt) cells (Supplementary Figures S1 and S2). In addition, the possibility of C-terminal tagging (see Uhrf1 (C); Figure 1D and Supplementary Figure S2H) allows the MIN-tag to be used in cases where N-terminal targeting disturbs protein function.
Taken together, these results demonstrate that the MIN-tag can efficiently be integrated at precise genomic locations using a CRISPR/Cas assisted, fluorescence based selection strategy.
Generation of a highly specific monoclonal antibody recognizing the MIN epitope
Insertion of the MIN-tag into the ORF of target genes leads to expression of a small peptide that does not occur in the mammalian proteome (Figure 2A). This unique feature allowed us to generate a highly specific monoclonal antibody against MIN-tagged proteins. Immunofluorescence (IF) stainings of a mixed Dnmt1attP/attP and wt culture distinguished single MIN-tagged cells and colonies from wt cells, demonstrating the high specificity of the anti-MIN antibody (Figure 2B). Pull-down experiments in Dnmt1attP/attP cell extracts showed a quantitative enrichment of DNMT1 in the bound fraction (Figure 2C). Furthermore, pull-down of DNMT3B using the anti-MIN antibody efficiently co-precipitated SNF2H, a known interactor of DNMT3B, in protein extracts of Dnmt3battP/attP cells, but not in wt control extracts (Figure 2D) (36).
Figure 2.
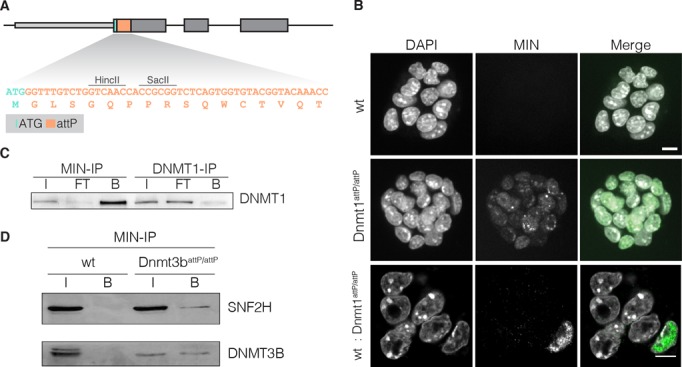
Application of the anti-MIN monoclonal antibody. (A) DNA sequence of the attP site and corresponding translated MIN peptide sequence (orange). (B) Fluorescence micrographs of wt mESCs, Dnmt1attp/attp cells and of a mixed culture (1:10) of wt and Dnmt1attP/attP cells stained with the anti-MIN antibody. DAPI is used as DNA counterstain. Scale bars represent 5 μm. (C) IP experiments performed with anti-MIN and anti-DNMT1 antibody in Dnmt1attP/attP cell extracts (input (I), flow through (FT), bound (B)). (D) Co-IP of DNMT3B in wt and Dnmt3battP/attP cells using the anti-MIN antibody. DNMT3B co-precipitated SNF2H in Dnmt3battP/attP cells as determined by western blot.
Collectively, these data show that the MIN-tag can be utilized as a universal epitope tag for IF and immunoprecipitation (IP), thus allowing the investigation of localization and molecular interactions of MIN-tagged proteins.
Functionalization of MIN-tagged genes by Bxb1-mediated recombination
To demonstrate the versatility of the MIN-tag as a Bxb1 integration site, we constructed a toolbox of functional cassettes, which we recombined into the MIN-tagged locus of the maintenance DNA methyltransferase Dnmt1 (Dnmt1attP/attP). First, we generated a knockout vector carrying the attB site directly in front of the ORF of GFP followed by a stop codon and a polyadenylation signal (attB-GFP-Poly(A), Figure 3A) that we transfected together with a codon-optimized Bxb1 expression construct in the Dnmt1attP/attP cell line. Successful recombination events were identified by GFP expression and single cells sorted by FACS (Figure 3B). We designed a multiplex PCR strategy that takes advantage of the unique attL site generated by successful recombination to facilitate identification of positive clones and their zygosity (Figure 3D and Supplementary Figure S3A). PCR screening of sorted clones revealed that the attB-GFP-Poly(A) construct had been successfully integrated into both alleles in 13 (56.5%) clones (Supplementary Table S3). Of those, we examined three clonal cell lines all of which exhibited no residual expression of DNMT1 by western blot analysis and IF (Figure 3F; Supplementary Figure S3B and C). For functional characterization, we analyzed DNA methylation levels at major satellite repeats, one of the main substrates for DNA methylation activity of DNMT1 during replication (37,38). Due to the loss of the maintenance DNA methyltransferase in the Dnmt1KO/KO clones, a severe hypomethylation was observed at this sequence (Figure 3E). Taken together, our attB-GFP-Poly(A) vector proved to be a valuable tool to generate genetically-defined gene knockouts in MIN-tagged cell lines.
Figure 3.
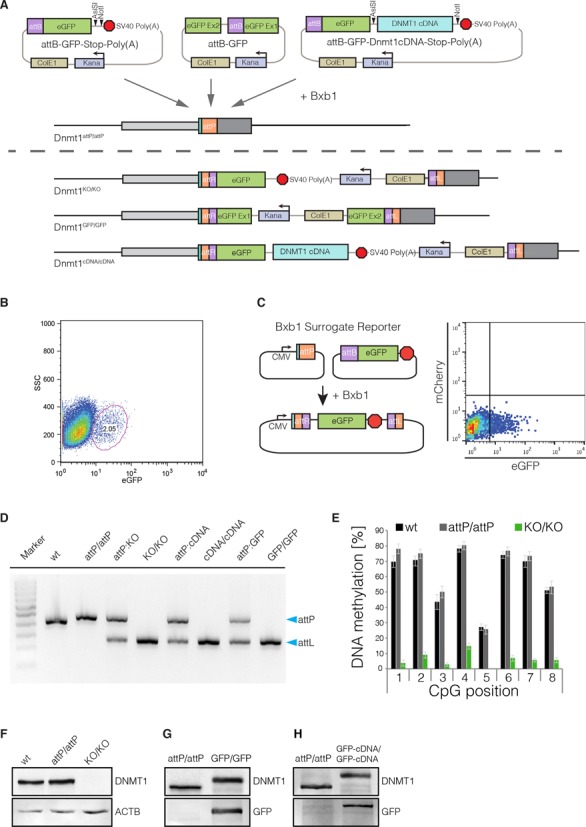
Bxb1-mediated insertion of functional cassettes into the Dnmt1 locus. (A) Schematic outline of the strategy and vectors used to create knockout, GFP knockin and cDNA knockin functionalizations of the Dnmt1attP/attP cell line. cDNAs can be cloned into the attB-GFP-Stop-Poly(A) vector using the 8-cutters AsiSI and NotI. (B) FACS plot depicting the gating and sorting of mESCs to enrich for cells positive for integration of the knockout cassette (2.05% of parent population) based on GFP expression. (C) The Bxb1 surrogate reporter consists of a constitutive CMV promoter followed by an attP site. If Bxb1 and attB donor plasmid containing GFP is present in the cell, recombination of the donor into the reporter leads to expression of GFP. The Bxb1 surrogate reporter can be used to enrich for successful recombination events by FACS. (D) Gel electrophoresis of the multiplex PCR for wt, Dnmt1attP/attP (attP/attP), Dnmt1KO/KO (KO/KO), Dnmt1cDNA/cDNA (cDNA/cDNA) and Dnmt1GFP/GFP (GFP/GFP) as well as 1:1 mixtures with Dnmt1attP/attP genomic DNA, to control for amplification biases. Blue arrows indicate expected sizes of the non-recombined (attP) and recombined allele (attL). (E) DNA methylation levels at the major satellite repeats of Dnmt1KO/KO cells compared to wt and Dnmt1attP/attP cells. (F) Western blot analysis of DNMT1 expression levels in wt, Dnmt1attP/attP and Dnmt1KO/KO cells generated by Bxb1-mediated insertion of a knockout cassette. (G) Western blot analysis of DNMT1 and GFP expression in Dnmt1attP/attP and homozygous GFP-knockin cells (Dnmt1GFP/GFP) generated by Bxb1-mediated insertion. (H) Western blot analysis of DNMT1 and GFP expression in Dnmt1attP/attP and Dnmt1cDNA/cDNA cells expressing a GFP-Dnmt1 minigene from the endogenous promoter.
Second, we designed a GFP knockin construct that can be used to generate in-frame GFP fusions of MIN-tagged genes. To avoid disruption of the gene locus and preserve the endogenous splicing sites, we placed the bacterial backbone sequences into an artificial intron splitting the GFP ORF into two exons (19) (Figure 3A). After recombination and FACS sorting for GFP expressing cells, the GFP knockin construct integrated in both alleles of the Dnmt1 locus in 13 clones (41.9%), without altering physiological DNMT1 expression levels (Figure 3G, Supplementary Figure S3D and Table S3). Live cell imaging of Dnmt1GFP/GFP cells revealed a normal localization of GFP-DNMT1 throughout the cell cycle (15,24)(Supplementary Figure S3E), demonstrating that DNMT1 regulation was not impaired. Albeit only at low frequencies, Bxb1 has been shown to damage recombination sites (8). Therefore, we sought to confirm that the Bxb1-mediated recombination of the GFP cassette at the MIN-tagged locus occurred without error via site-specific recombination. We sequenced the region flanking the attL site in the Dnmt1GFP/GFP cell line (Supplementary Figure S4) and determined that the GFP cassette was accurately integrated in a scarless fashion. In summary, this attB-GFP vector is suited to express GFP fusion proteins from the endogenous promoter preserving physiological regulation and splicing of the target gene.
Finally, we investigated whether the MIN-tag can be used to generate cell lines expressing mutants of the target gene for functional screenings or disease modeling. We cloned the cDNA of Dnmt1 into the attB-GFP-Poly(A) construct in-frame with GFP and performed recombination as described above. We identified 10 (66.6%) clones in which integration had occurred, of which 9 (60%) were homozygous for the Dnmt1 cDNA knockin (Supplementary Table S3). Expression analysis by western blot and live cell imaging revealed that the endogenous DNMT1 protein was completely replaced by the Dnmt1 mini gene product and exhibited normal localization (Figure 3H, Supplementary Figure S3F).
All in all, we show that MIN-tagged entry cell lines can be efficiently functionalized with a flexible toolbox of attB-vectors to generate gene knockouts, N-terminal fusion constructs such as GFP and cDNA knockins. In total, we generated 15 derivatives of our MIN-tagged cell lines so far. The efficiency of Bxb1-mediated recombination ranged from 33 to 67%, with an average of 50% (Supplementary Table S3, Figure S5). This demonstrates the efficacy of our system as well as the simplicity with which MIN-tagged cell lines can be modified and functionalized by prefabricated cassettes. The error-prone step of CRISPR/Cas-mediated insertion of the MIN-tag is necessary only once to generate an entry cell line, which can then be specifically manipulated with a variety of recombination vectors, allowing maximum biological comparability.
Using the MIN-tag strategy to study endogenous protein regulation
As elucidating the function of uncharacterized protein domains requires systematic analysis, we generated a series of deletion constructs covering the N-terminus of TET1, which we aimed to recombine into our Tet1attP/attP cell line (Figure 4A). However, we were unable to identify positive recombination events by FACS due to low expression of this target gene. To circumvent this problem, we developed a surrogate reporter system for Bxb1 mediated recombination that can be used to enrich for positive recombination events (Figure 3C). The Bxb1 surrogate reporter construct consists of a constitutive promoter followed by an attP site and a Poly(A) sequence. Upon transfection, Bxb1 mediates the recombination of a fluorophore (e.g. GFP) containing attB plasmid with the Bxb1 surrogate reporter, which results in the expression of GFP. This allows enrichment of positive recombination events, even when the MIN-tagged gene is not expressed or only at low levels.
Figure 4.
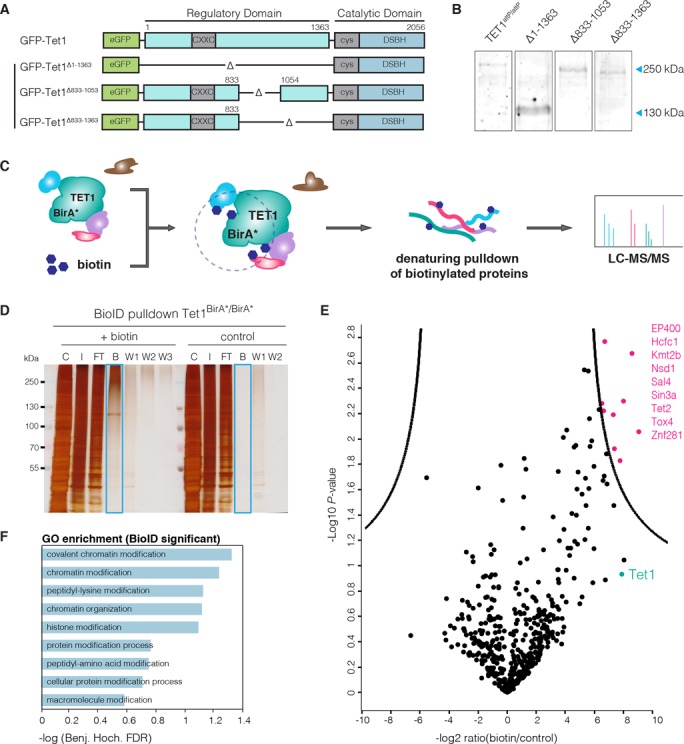
Study of TET1 regulation. (A) Schematic representation of the Tet1 cDNA constructs used for Bxb1-mediated recombination into Tet1attP/attP cells. (B) Western blot analysis of TET1 expression in Tet1attP/attP cell line and its derivatives expressing GFP-TET1Δ1–1363 (Δ1–1363), GFP-TET1Δ833–1053 (1Δ833–1053) and GFP-TET1Δ833–1363 (Δ833–1363). Note that fusion to GFP increases the MW of TET1 constructs by 29 kDa. (C) Schematic representation of the BioID approach as described by Roux etal. (27). (D) SDS-PAGE analysis of a BioID pulldown experiment using the Tet1BirA*/BirA* cell line. Cells were cultured either without (control) or with 50 μM biotin (+biotin). C: Cytoplasm, I: Crude nuclei input, FT: Flowthrough, B: Bound, W1-W3: Wash. (E) Volcano plot of proteins identified in the streptavidin pulldown of the TET1-BioID experiment, quantified with the MaxQuant Label-Free-Quantification algorithm (32). The x-axis reflects the difference in protein abundance in the BioID pull-down compared to the negative control while the y-axis shows the logarithmized P-value of a student's t-test. Significantly enriched proteins are highlighted in pink (FDR = 0.01, S0 = 3, indicated by black line (40)). Experiments were performed in duplicates. (F) GO term enrichment of proteins identified as significant in BioID.
Using the Bxb1 surrogate reporter for enrichment and the above described PCR strategy for screening, we were able to generate four Tet1 knockin cell lines expressing N-terminal deletion constructs from the endogenous promoter. Western blot analysis revealed complete replacement of wt TET1 expression by the knockin constructs (Figure 4B). These cell lines can be used for future systematic studies of the regulatory function of the TET1 N-terminus that is largely unknown so far.
Taking advantage of the MIN-tag strategy to express fusion constructs at endogenous levels, we expanded our toolbox to include a BirA* cassette which we knocked into the Tet1 locus (Supplementary Figure S5G). In contrast to classical IP approaches, proximity-dependent protein labeling by the promiscuous biotin ligase, BirA* (BioID) (27), allows the characterization of the full microenvironment of a protein of interest independent of physical protein–protein interactions. This technique enabled us to pull down proteins within close proximity (∼10 nm radius, (39)) of TET1 that were subsequently identified by LC-MS/MS (Figure 4C). We found nine proteins to be significantly enriched (40) upon addition of exogenous biotin to the culture medium of our Tet1BirA*/BirA* mESC line, including SIN3A, a known interactor of TET1 (41) (Figure 4D and E). Interestingly, these proteins are associated with chromatin modification and organization (Figure 4F). This marks the first time that the BioID method has been used in mESCs and in a non-overexpression context with the BirA* ligase fused to the endogenous protein.
Using the MIN-tag strategy to study dynamic cellular processes
During early embryonic development, the epigenome undergoes massive rearrangements that are precisely regulated. Knockout of the major epigenetic factors is often embryonic lethal (38,42) and over-expression studies frequently fail to reflect the tight regulation of these proteins. Therefore, more flexible and delicate genetic manipulations are needed to study the function of epigenetic factors in vivo. Here, we focus on the de novo DNA methyltransferase 3B (DNMT3B), one of the key factors during epiblast differentiation. While it has been shown that DNMT3B, in concert with DNMT3A and DNMT3L, is responsible for the global wave of de novo DNA methylation occurring during epiblast differentiation (42–44), little is known about its localization and protein kinetics during this developmental time period.
To address this question in a systematic fashion, we generated a homozygous GFP knockin cell line (Dnmt3bGFP/GFP) from the Dnmt3battP/attP cell line by Bxb1-mediated recombination (Figure 5A and 6A). This allowed us to follow expression of DNMT3B under native regulatory conditions and to monitor its localization during the two-day transition from naive pluripotent ESCs to Epiblast-like cells (EpiLCs, (25)) using live cell imaging with high temporal resolution (1 image per hour).
Figure 5.
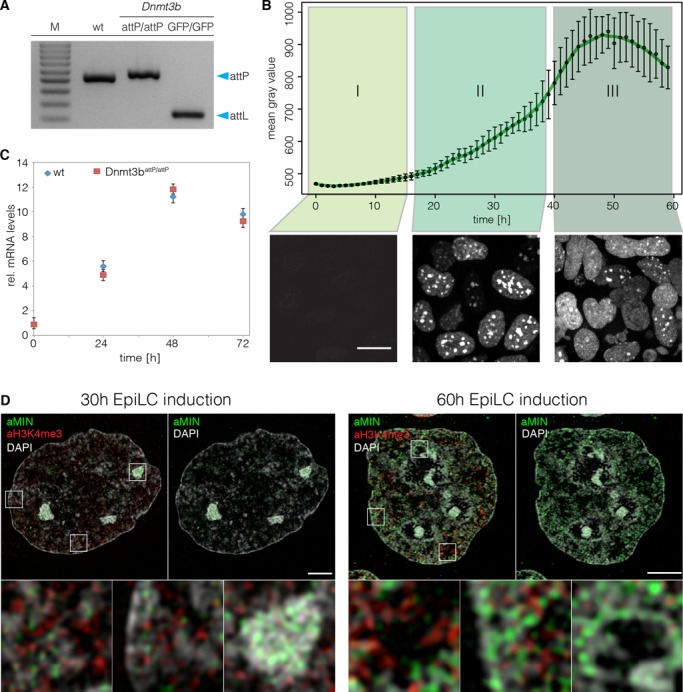
Spatio-temporal dynamics of DNMT3B during epiblast differentiation. (A) Gel electrophoresis of the multiplex screening PCR for wt, Dnmt3battP/attP and Dnmt3bGFP/GFP. Blue arrows indicate expected sizes of the non-recombined (attP) and recombined allele (attL). (B) Evaluation of GFP signals during live cell imaging of Dnmt3bGFP/GFP cells. The graph depicts mean gray values of nuclear GFP signals. Error bars represent standard deviations (n > 81). Lower panels show Z-projections of Dnmt3bGFP/GFP cells representative of the indicated time frame. Scale bar represents 10 μm. (C) Quantitative real-time PCR of Dnmt3b mRNA levels in wt and Dnmt3battP/attP cells during epiblast differentiation. (D) 3D-SIM nuclear mid-sections of anti-MIN (green) and anti-H3K4me3 (red) antibody distributions 30 and 60 h after induction of EpiLC differentiation combined with DAPI counterstaining (gray) in Dnmt3battP/attP cells. Lower panels represent 7× magnifications of selected boxed regions. Scale bars represent 3 μm and 500 nm in insets.
Figure 6.
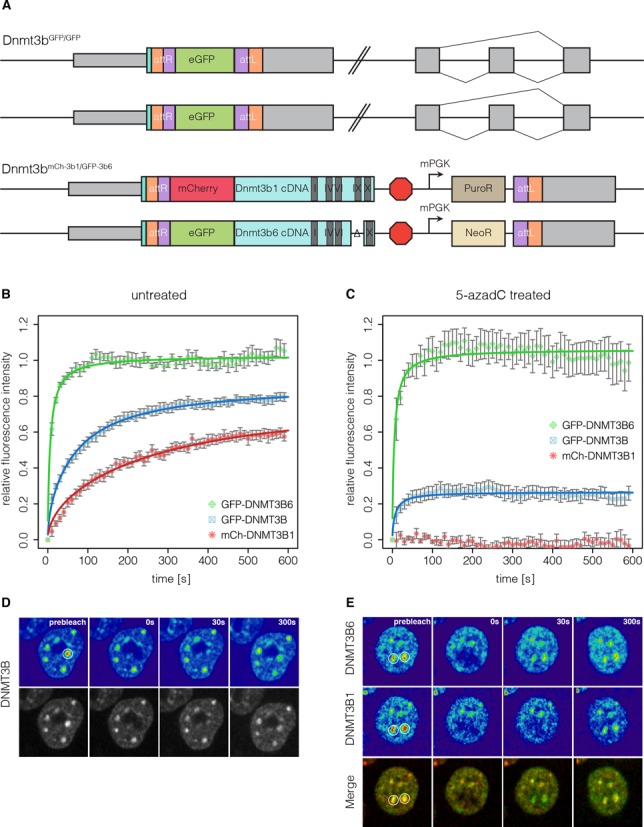
Protein dynamics of DNMT3B and its isoforms during epiblast differentiation. (A) Schematic representation of the Dnmt3b genomic loci in the Dnmt3bGFP/GFP and the Dnmt3bmCh-3b1/GFP-3b6 cell lines. (B) Quantitative evaluation of FRAP experiments (average of 11–14 cells) comparing GFP-DNMT3B with GFP-DNMT3B6 and mCh-DNMT3B1 in Dnmt3bGFP/GFP and the Dnmt3bmCh-3b1/GFP-3b6 cell lines differentiated for 35 h. Error bars represent standard error of the mean. (C) Quantitative evaluation of FRAP experiments (average of 10–12 cells) as in (B) with cells treated with 5-azadC 12 h before imaging. (D and E) Representative images of FRAP experiments performed in (B) and (C), respectively. White circles indicate the bleach ROI with a diameter of 2 μm.
At the naive pluripotent state, we observed very low expression levels of DNMT3B. Upon addition of differentiation medium, protein expression was strongly and uniformly upregulated reaching its maximum at 48–52 h (Figure 5B, Supplementary video 1). Overall, these findings were consistent with Dnmt3b mRNA levels in wt and Dnmt3battP/attP cells (Figure 5C). Interestingly, we observed a highly dynamic subnuclear distribution of DNMT3B during differentiation that can be classified into three patterns (Figure 5B). (i) In the first 14 h of differentiation, DNMT3B is expressed at low levels and no clear enrichment is visible. (ii) Between 14–40 h after initiation of differentiation, DNMT3B expression is upregulated and accumulates at constitutive heterochromatin of chromocenters (CCs). (iii) After 40 h of differentiation, DNMT3B is highly expressed and localization to CCs is diminished. The above-described patterns were not related to specific cell cycle stages, indicating a differentiation stage dependent localization of DNMT3B (Supplementary Figure S6A).
To investigate the specific chromatin distribution of DNMT3B during differentiation in more detail, we performed super-resolution 3D structured illumination microscopy (3D-SIM) with the anti-MIN antibody for protein visualization. DAPI and trimethylated lysine 4 of histone 3 (H3K4me3) were used as markers of heterochromatin and euchromatin (45), respectively. In agreement with the live cell imaging experiments, DNMT3B localizes at CCs, clusters of subcentromeric regions, at the 30 h time point and shows a broader distribution at 60 h after differentiation (Figure 5D). Interestingly, the higher resolution of 3D-SIM revealed an accumulation of the signal in facultative heterochromatin at perinuclear and perinucleolar regions at the 60 h time point (Figure 5D; right panel).
DNMT3B has been shown to be responsible for the methylation of major satellite DNA, a main constituent of CCs (42,46–47). As DNMT3B is enriched at CCs between 14–40 h of differentiation, we investigated whether DNMT3B is actively methylating these sequences during this period. Therefore, we performed fluorescence recovery after photobleaching (FRAP) of GFP-DNMT3B localized at CCs. Using our Dnmt3bGFP/GFP cell line, we performed FRAP experiments at 35 h of differentiation. Using circular regions of interest (ROIs) that encompassed individual CCs, we monitored signal recovery for 10 min after bleaching. We found that the signal exhibited a slow recovery rate (t1/2 = 42 s) and did not recover completely. As DNA methylation has been shown to have a slow turnover rate (48,49), this suggested the immobile fraction (∼20%) of DNMT3B could be catalytically active at CCs (Figure 6B and D, Supplementary Table S4). To test this hypothesis, we performed FRAP experiments on cells treated with the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine (5-azadC), which irreversibly traps DNMTs at their site of action (50). We found that 5-azadC treated CCs exhibited a large immobile fraction (∼80%) suggesting that DNMT3B is actively methylating CCs at this time point. However, we were surprised to find that ∼20% of DNMT3B enzyme still remained mobile (Figure 6C). Considering the long 5-azadC treatment time of 12 h this suggested that a fraction of the enzyme never engaged in catalytic reactions. As our GFP cassette preserves endogenous splicing patterns, the GFP-DNMT3B fusions used in this study represent a mixture of different protein isoforms. This prompted us to investigate the contribution of Dnmt3b splicing isoforms to the observed FRAP kinetics.
For Dnmt3b, nine splicing isoforms, all originating from the same translational start site, have been described (51). Besides the catalytically active isoform DNMT3B1, DNMT3B6 has been shown to be highly expressed in ESCs. This isoform is produced by alternative splicing, skipping exons 23 and 24, resulting in a protein that lacks several highly conserved motifs within the catalytic domain and has therefore been suggested to be inactive (52).
To dissect the contributions of DNMT3B1 and DNMT3B6 to the observed FRAP kinetics of Dnmt3bGFP/GFP cells, we generated a cell line expressing fluorescent fusions of each isoform. For this, we produced cDNA knockin constructs in which DNMT3B1 was fused to a red fluorescent protein mCherry (mCh) and DNMT3B6 was fused to GFP. To facilitate the generation of knockin cell lines expressing each isoform from one allele we equipped the Dnmt3b1 and Dnmt3b6 constructs with a Neomycin and Puromycin resistance cassette, respectively. We successfully established a cell line that simultaneously expressed mCh-DNMT3B1 and GFP-DNMT3B6, both under the control of the endogenous Dnmt3b promoter (Figure 6A, Supplementary Figure S6B), allowing us to directly compare the FRAP kinetics of DNMT3B1 and DNMT3B6 within the same cell. In the absence of 5-azadC, GFP-DNMT3B6 exhibited a fast (t1/2 = 5 s) and complete recovery while mCh-DNMT3B1 recovered slower (t1/2 = 95 s) (Figure 6B, Supplementary Table S4).
Intriguingly, FRAP kinetics of DNMT3B6 were not influenced by the presence of 5-azadC, supporting that it is catalytically inactive. In contrast, DNMT3B1 was completely immobilized by addition of 5-azadC exhibiting virtually no recovery after photobleaching (Figure 6C and E).
Taken together, our MIN-tag strategy enabled us to show that DNMT3B exhibits a dynamic localization to distinct chromatin regions during epiblast differentiation. Super-resolution micrographs of cells stained with anti-MIN antibodies at different time points of epiblast differentiation hint towards progression of de novo DNA methylation in a hierarchical fashion starting at constitutive (CCs) and progressing towards facultative (perinuclear/perinucleolar) heterochromatin. Finally, FRAP experiments revealed that the two isoforms DNMT3B1 and DNMT3B6 exhibit dramatically different DNA binding kinetics.
DISCUSSION
Recent advances in genome engineering technology, based on TALEN and CRISPR/Cas systems, have greatly facilitated the process of manipulating genetic information. Platforms have been established that allow genome-wide gene disruption screenings for factors involved in any biological process (20,53–54). While these methods provide valuable information about the genes and pathways involved, in-depth analysis of target genes is needed to understand their function. This, in turn, requires the implementation of various genetic, cell biological and biochemical techniques. To gain meaningful insights into gene function, these techniques have to be applied under physiological conditions requiring extensive and complex genetic manipulations. Although modern genome engineering tools have made such manipulations possible, a more efficient and universal approach would be highly desirable to implement the above-mentioned techniques in a systematic manner.
The MIN-tag strategy offers a new means of rapid, efficient, yet flexible genetic manipulation of target loci. We show that CRISPR/Cas assisted insertion of the MIN-tag can be performed efficiently with short homology donors. Several studies have shown that CRISPR/Cas mediated gene targeting is associated with a significant risk of off-target cleavage, which can result in indel (insertions or deletions) formation due to NHEJ (5–7,55–56). The MIN-tag strategy requires a single nuclease assisted gene editing event, thereby keeping the likelihood of off-target effects at a minimum. Further modifications are then performed using Bxb1-mediated recombination. In contrast to the phiC31 integrase, Bxb1 has been shown to be highly specific with virtually no unwanted genomic insertions at pseudo attP sites (8–9,57–58). Once a MIN-tagged cell line is established, in-frame fusion of the MIN-tag to the target gene also results in the expression of a novel epitope tag. We show that this epitope tag can be detected by a highly specific antibody, which can be used to screen for positive clones, perform co-immunoprecipitation (co-IP) experiments, as well as conventional and super resolution microscopy.
Using Bxb1 and the MIN-tag toolbox, a MIN-tagged entry cell line can be used to generate multiple isogenic derivatives within 2–3 weeks (Figure 7), without the risk of introducing off-target effects. Our collection of vectors for Bxb1 mediated recombination currently contains over 80 different plasmids (Supplementary Table S5). These prefabricated functional cassettes constitute an expandable toolbox for the simple and flexible genetic alteration of any tagged loci, without the need of locus-specific homology.
Figure 7.
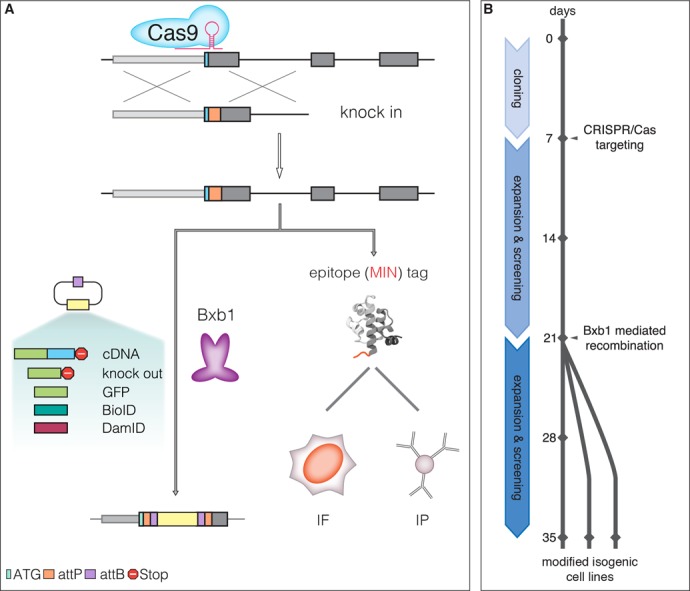
The MIN-tag strategy. (A) Schematic outline of the genome engineering strategy. Small homology donors are used to insert serine integrase (attP) sites in-frame after the ATG codon of target genes via CRISPR/Cas assisted HR. The attP site is translated as a novel epitope tag suitable for IF and IP with the specific monoclonal antibody. The attP site is also recognized by the serine integrase Bxb1 and used for specific and directional integration of attB-carrying functional cassettes into the tagged gene locus. All derivatives are subjected to their endogenous gene regulation ensuring that subsequent studies are performed at physiological expression levels. (B) Timeline for generation of MIN-tagged genes and subsequent modification by Bxb1-mediated recombination. MIN-tagged cell lines can be generated within 2–3 weeks. These cell lines can then be modified within another 2–3 weeks to generate multiple isogenic cell lines with different functional modifications.
Using our stop cassette, we show that the MIN-tag strategy can be used to reliably achieve genetically defined gene disruption of MIN-tagged genes. Harboring a Poly(A) signal, insertion of this cassette efficiently eliminates target gene expression with the added advantage of precluding unwanted downstream initiation. As fluorescent protein reporters are commonly used to study spatio-temporal dynamics and protein kinetics in living cells, we generated a GFP knockin construct (attB-GFP) for Bxb1-mediated integration. GFP knockin cell lines made with this construct retain not only their endogenous expression levels but also their endogenous splicing pattern. Similarly, a BirA* cassette can be introduced at any MIN-tagged locus to allow for proximity-dependent labeling of the microenvironment of a given protein.
Understanding protein function often necessitates the systematic alteration of individual domains through mutations as well as deletions. Equipped with a fluorescent protein and strategic cloning sites, our cDNA knockin cassette is especially tailored for simple and expedient insertion of user-defined cDNAs. PCR-based approaches can be used to easily alter the coding sequence and quickly produce a library of gene specific cDNA mutants. These can then be inserted into target loci by Bxb1-mediated recombination, completely replacing expression of the wt gene while retaining endogenous control. While this strategy does not directly introduce the mutations into the gene locus, it offers a means of inserting and testing multiple mutant constructs in a short time frame without the need to design and perform additional nuclease-assisted targetings. This feature can be used to gain insights into the functional implications of the rapidly growing number of mutations found in cancer and disease. Likewise, the generation of large deletion mutants is easily accomplished facilitating the investigation of protein domain function and interaction mapping. This eliminates the need for excising large genomic regions or cloning long site-specific homology donors.
Obviously, the above mentioned plasmids by no means represent the extent of all possible functional cassettes. For example, MIN-tag toolbox modules allowing inducible protein stabilization or localization (59,60) as well as enzymatic labeling of DNA binding sites (DamID (61)) would greatly assist the elucidation of protein function and protein-chromatin interactions, respectively.
Employing our strategy in mESCs, we inserted the MIN-tag into the genes coding for all mammalian DNA modifying enzymes and a cofactor (Dnmt1, Dnmt3a, Dnmt3b, Tet1, Tet2, Tet3 and Uhrf1). These MIN-tagged cell lines as well as their functional derivatives (Supplementary Table S3) constitute a valuable resource to investigate the role of these proteins during fundamental processes such as pluripotency, cellular reprogramming, embryonic development and disease.
One gold standard method to study protein–protein interactions is co-IP. However, chromatin- or membrane-bound proteins are often barely soluble and consequently difficult to investigate by this approach. Making use of our BirA* cassette, we investigated factors in the microenvironment of TET1, a dioxygenase that oxidizes DNA at methylated cytosines (62). Besides the known interactor SIN3A, we identify eight other proteins in proximity to TET1 that are involved in chromatin modification and organization, including the closely related TET2. This is in accordance with the findings by Costa et al. (63) that TET1 and TET2 have partially overlapping target sites. In conclusion, integration of the BirA* cassette into the endogenous locus is a perfectly suited method to study dynamic protein–protein interactions.
We also applied the MIN-tag strategy to study the de novo DNA methyltransferase DNMT3B during the transition from naive pluripotent ESCs to primed EpiLCs, a period of dramatic epigenetic change. While distinct patterns have been described for ESCs and somatic cells (46,64), the subnuclear distribution of DNMT3B during differentiation remains largely unknown. We discovered that DNMT3B exhibits a highly dynamic subnuclear distribution during epiblast differentiation. Our observations suggest that the global wave of de novo DNA methylation during epiblast differentiation follows a distinct spatio-temporal order, initiating at constitutive pericentromeric heterochromatin followed by transition to facultative heterochromatin.
Exploiting the unique possibilities of our MIN-tag strategy, we furthermore generated a cell line simultaneously expressing differentially tagged splicing isoforms of DNMT3B from different alleles. This approach revealed that the major catalytically active isoform DNMT3B1 was completely immobilized at chromocenters after 5-azadC treatment, while the FRAP kinetics of DNMT3B6 were not affected. This, to our knowledge, is the first time that FRAP has been performed on different isoforms of a protein at endogenous expression levels in the same cell.
While this study was performed using mouse ESCs, our strategy can be applied to any cell type as long as no Bxb1 attP site is present in the respective genomes. The human genome is free of this entry site and introduction of the MIN-tag into cell lines such as human induced pluripotent stem cells should greatly facilitate the generation of clinically relevant disease models. Moreover, MIN-tagged mESCs could be used in blastocyst injections to generate MIN-tagged mice. Different tissues and cells could not only be used for Bxb1-mediated genetic manipulation in vitro, free of the limitation posed by inefficient endogenous homologous recombination, but also to study tissue specific protein regulation with the MIN-tag antibody. Furthermore, widely used cell biological model systems such as HeLa and U2OS cells as well as model organisms such as Caenorhabditis elegans or Drosophila could benefit from the versatility and efficiency of our approach.
In summary, with our combined genome engineering approach, a plethora of functional derivatives can be generated from one entry line with high efficiency. To simplify the distribution of MIN-tagged cell lines and the MIN-tag toolbox as well as to assist with the design of targeting strategies, we have developed a web-tool that is accessible at http://human.bio.lmu.de/_webtools/MINtool/. As entry lines can be shared and the genetic toolbox easily expanded with new functional modules, the MIN-tag strategy represents a dynamic flexible open platform and facilitates systematic functional studies with direct biological comparability.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Ina Poser and Tony Hyman (Max Planck Institute of Molecular Biology & Genetics, Dresden) for providing R6K-NFLAP construct, George Church (Harvard Medical School, Boston) for providing the Cas9 expression construct, Kerry Tucker (Ruprecht- Karls-University, Heidelberg) for providing wt ESCs and Pawel Pelczar (University of Zürich) for providing pCAG-NLS-HA-Bxb1. Furthermore, we thank Robert Engelmeier and Michael Soutschek for help with cell line generation, IF and antibody characterization. We also thank Andy Spiegl, Gregor Jessberger and Jan Langkabel for help with website development. CBM gratefully acknowledges the Life Science Munich Graduate School. ES, CB and KT gratefully acknowledge the International Max Planck Research School for Molecular and Cellular Life Sciences.
FUNDING
Deutsche Forschungsgemeinschaft [SFB 1064 to H.L., E.K. and GRK 1721 to H.L., F.H.]; Funding for open access charge: Deutsche Forschungsgemeinschaft [Collaborative Research Center SFB 1064/A17; GRK 1721].
Conflict of interest statement. None declared.
REFERENCES
- 1.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haurwitz R.E., Jinek M., Wiedenheft B., Zhou K., Doudna J.A. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sampson T.R., Saroj S.D., Llewellyn A.C., Tzeng Y.-L., Weiss D.S. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature. 2013;497:254–257. doi: 10.1038/nature12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuscu C., Arslan S., Singh R., Thorpe J., Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- 6.Wang X., Wang Y., Wu X., Wang J., Wang Y., Qiu Z., Chang T., Huang H., Lin R.-J., Yee J.-K. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015;33:175–178. doi: 10.1038/nbt.3127. [DOI] [PubMed] [Google Scholar]
- 7.Wu X., Scott D.A., Kriz A.J., Chiu A.C., Hsu P.D., Dadon D.B., Cheng A.W., Trevino A.E., Konermann S., Chen S., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Z., Thomas L., Davies B., Chalmers R., Smith M., Brown W. Accuracy and efficiency define Bxb1 integrase as the best of fifteen candidate serine recombinases for the integration of DNA into the human genome. BMC Biotechnol. 2013;13:87–87. doi: 10.1186/1472-6750-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown W.R.A., Lee N.C.O., Xu Z., Smith M.C.M. Serine recombinases as tools for genome engineering. Methods. 2011;53:372–379. doi: 10.1016/j.ymeth.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Bonnet J., Subsoontorn P., Endy D. Rewritable digital data storage in live cells via engineered control of recombination directionality. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8884–8889. doi: 10.1073/pnas.1202344109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang J., Ghosh P., Hatfull G.F., Hong Y. Successive and targeted DNA integrations in the Drosophila genome by Bxb1 and phiC31 integrases. Genetics. 2011;189:391–395. doi: 10.1534/genetics.111.129247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu F., Gamboa M., Farruggio A.P., Hippenmeyer S., Tasic B., Schule B., Chen-Tsai Y., Calos M.P. DICE, an efficient system for iterative genomic editing in human pluripotent stem cells. Nucleic Acids Res. 2014;42:e34. doi: 10.1093/nar/gkt1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Citterio E., Papait R., Nicassio F., Vecchi M., Gomiero P., Mantovani R., Di Fiore P.P., Bonapace I.M. Np95 is a histone-binding protein endowed with ubiquitin ligase activity. Mol. Cell. Biol. 2004;24:2526–2535. doi: 10.1128/MCB.24.6.2526-2535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bauer C., Gobel K., Nagaraj N., Colantuoni C., Wang M., Muller U., Kremmer E., Rottach A., Leonhardt H. Phosphorylation of TET proteins is regulated via O-GlcNAcylation by the O-linked N-acetylglucosamine transferase (OGT) J. Biol. Chem. 2015;290:4801–4812. doi: 10.1074/jbc.M114.605881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider K., Fuchs C., Dobay A., Rottach A., Qin W., Wolf P., Álvarez-Castro J.M., Nalaskowski M.M., Kremmer E., Schmid V., et al. Dissection of cell cycle-dependent dynamics of Dnmt1 by FRAP and diffusion-coupled modeling. Nucleic Acids Res. 2013;41:4860–4876. doi: 10.1093/nar/gkt191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solovei I., Cremer M. 3D-FISH on cultured cells combined with immunostaining. Methods Mol. Biol. 2010;659:117–126. doi: 10.1007/978-1-60761-789-1_8. [DOI] [PubMed] [Google Scholar]
- 17.Meilinger D., Fellinger K., Bultmann S., Rothbauer U., Bonapace I.M., Klinkert W.E.F., Spada F., Leonhardt H. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep. 2009;10:1259–1264. doi: 10.1038/embor.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niwa H., Yamamura K., Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 19.Poser I., Sarov M., Hutchins J.R.A., Heriche J.-K., Toyoda Y., Pozniakovsky A., Weigl D., Nitzsche A., Hegemann B., Bird A.W., et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat. Methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmid-Burgk J.L., Schmidt T., Kaiser V., Honing K., Hornung V. A ligation-independent cloning technique for high-throughput assembly of transcription activator–like effector genes. Nat. Biotechnol. 2013;31:76–81. doi: 10.1038/nbt.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masui S., Shimosato D., Toyooka Y., Yagi R., Takahashi K., Niwa H. An efficient system to establish multiple embryonic stem cell lines carrying an inducible expression unit. Nucleic Acids Res. 2005;33:e43. doi: 10.1093/nar/gni043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Easwaran H.P., Schermelleh L., Leonhardt H., Cardoso M.C. Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep. 2004;5:1181–1186. doi: 10.1038/sj.embor.7400295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frauer C., Rottach A., Meilinger D., Bultmann S., Fellinger K., Hasenoder S., Wang M., Qin W., Soding J., Spada F., et al. Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS One. 2011;6:e16627. doi: 10.1371/journal.pone.0016627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schermelleh L., Haemmer A., Spada F., Rosing N., Meilinger D., Rothbauer U., Cardoso M.C., Leonhardt H. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 2007;35:4301–4312. doi: 10.1093/nar/gkm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayashi K., Saitou M. Generation of eggs from mouse embryonic stem cells and induced pluripotent stem cells. Nat. Protoc. 2013;8:1513–1524. doi: 10.1038/nprot.2013.090. [DOI] [PubMed] [Google Scholar]
- 26.Hermann M., Stillhard P., Wildner H., Seruggia D., Kapp V., Sanchez-Iranzo H., Mercader N., Montoliu L., Zeilhofer H.U., Pelczar P. Binary recombinase systems for high-resolution conditional mutagenesis. Nucleic Acids Res. 2014;42:3894–3907. doi: 10.1093/nar/gkt1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roux K.J., Kim D.I., Raida M., Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012;196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baymaz H.I., Spruijt C.G., Vermeulen M. Identifying nuclear protein-protein interactions using GFP affinity purification and SILAC-based quantitative mass spectrometry. Methods Mol. Biol. 2014;1188:207–226. doi: 10.1007/978-1-4939-1142-4_15. [DOI] [PubMed] [Google Scholar]
- 29.Blum H., Beier H., Gross H.J. Improved silver staining of plant-proteins, RNA and DNA in polyacrylamide gels. Electrophoresis. 1987;8:93–99. [Google Scholar]
- 30.Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 31.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 32.Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48–48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim H., Um E., Cho S.-R., Jung C., Kim H., Kim J.-S. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat. Methods. 2011;8:941–943. doi: 10.1038/nmeth.1733. [DOI] [PubMed] [Google Scholar]
- 36.Geiman T.M., Sankpal U.T., Robertson A.K., Chen Y., Mazumdar M., Heale J.T., Schmiesing J.A., Kim W., Yokomori K., Zhao Y., et al. Isolation and characterization of a novel DNA methyltransferase complex linking DNMT3B with components of the mitotic chromosome condensation machinery. Nucleic Acids Res. 2004;32:2716–2729. doi: 10.1093/nar/gkh589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehnertz B., Ueda Y., Derijck A.A.H.A., Braunschweig U., Perez-Burgos L., Kubicek S., Chen T., Li E., Jenuwein T., Peters A.H.F.M. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003;13:1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 38.Li E., Bestor T.H., Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 39.Kim D.I., Birendra K.C., Zhu W., Motamedchaboki K., Doye V., Roux K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. U.S.A. 2014;111:2453–2461. doi: 10.1073/pnas.1406459111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams K., Christensen J., Pedersen M.T., Johansen J.V., Cloos P.A.C., Rappsilber J., Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 43.Hata K., Okano M., Lei H., Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 44.Okano M., Xie S., Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 45.Bernstein B.E., Kamal M., Lindblad-Toh K., Bekiranov S., Bailey D.K., Huebert D.J., McMahon S., Karlsson E.K., Kulbokas E.J., Gingeras T.R., et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 46.Bachman K.E., Rountree M.R., Baylin S.B. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J. Biol. Chem. 2001;276:32282–32287. doi: 10.1074/jbc.M104661200. [DOI] [PubMed] [Google Scholar]
- 47.Xu G.L., Bestor T.H., Bourc'his D., Hsieh C.L., Tommerup N., Bugge M., Hulten M., Qu X., Russo J.J., Viegas-Pequignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 48.Emperle M., Rajavelu A., Reinhardt R., Jurkowska R.Z., Jeltsch A. Cooperative DNA binding and protein/DNA fiber formation increases the activity of the Dnmt3a DNA methyltransferase. J. Biol. Chem. 2014;289:29602–29613. doi: 10.1074/jbc.M114.572032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gowher H., Jeltsch A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 2001;309:1201–1208. doi: 10.1006/jmbi.2001.4710. [DOI] [PubMed] [Google Scholar]
- 50.Schermelleh L., Spada F., Easwaran H.P., Zolghadr K., Margot J.B., Cardoso M.C., Leonhardt H. Trapped in action: direct visualization of DNA methyltransferase activity in living cells. Nat. Methods. 2005;2:751–756. doi: 10.1038/nmeth794. [DOI] [PubMed] [Google Scholar]
- 51.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M., et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:756–763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weisenberger D.J., Velicescu M., Cheng J.C., Gonzales F.A., Liang G., Jones P.A. Role of the DNA methyltransferase variant DNMT3b3 in DNA methylation. Mol. Cancer Res. 2004;2:62–72. [PubMed] [Google Scholar]
- 53.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G., et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin Y., Cradick T.J., Brown M.T., Deshmukh H., Ranjan P., Sarode N., Wile B.M., Vertino P.M., Stewart F.J., Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42:7473–7485. doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan E.P., Li Y., Velasco-Herrera Mdel C., Yusa K., Bradley A. Off-target assessment of CRISPR-Cas9 guiding RNAs in human iPS and mouse ES cells. Genesis. 2015;53:225–236. doi: 10.1002/dvg.22835. [DOI] [PubMed] [Google Scholar]
- 57.Russell J.P., Chang D.W., Tretiakova A., Padidam M. Phage Bxb1 integrase mediates highly efficient site-specific recombination in mammalian cells. Biotechniques. 2006;40:460–460. doi: 10.2144/000112150. [DOI] [PubMed] [Google Scholar]
- 58.Keravala A., Groth A.C., Jarrahian S., Thyagarajan B., Hoyt J.J., Kirby P.J., Calos M.P. A diversity of serine phage integrases mediate site-specific recombination in mammalian cells. Mol. Genet. Genomics. 2006;276:135–146. doi: 10.1007/s00438-006-0129-5. [DOI] [PubMed] [Google Scholar]
- 59.Banaszynski L.A., Chen L.-C., Maynard-Smith L.A., Ooi A.G.L., Wandless T.J. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kennedy M.J., Hughes R.M., Peteya L.A., Schwartz J.W., Ehlers M.D., Tucker C.L. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Steensel B., Delrow J., Henikoff S. Chromatin profiling using targeted DNA adenine methyltransferase. Nat. Genet. 2001;27:304–308. doi: 10.1038/85871. [DOI] [PubMed] [Google Scholar]
- 62.Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Costa Y., Ding J., Theunissen T.W., Faiola F., Hore T.A., Shliaha P.V., Fidalgo M., Saunders A., Lawrence M., Dietmann S., et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495:370–374. doi: 10.1038/nature11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen T., Tsujimoto N., Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell. Biol. 2004;24:9048–9058. doi: 10.1128/MCB.24.20.9048-9058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.