

Abstract
Multiple myeloma (MM) remains an incurable malignancy due, in part, to the influence of the bone marrow microenvironment on survival and drug response. Identification of microenvironment-specific survival signaling determinants is critical for the rational design of therapy and elimination of MM. Previously, we have shown that collaborative signaling between β1 integrin-mediated adhesion to fibronectin and interleukin-6 confers a more malignant phenotype via amplification of signal transducer and activator of transcription 3 (STAT3) activation. Further characterization of the events modulated under these conditions with quantitative phosphotyrosine profiling identified 193 differentially phosphorylated peptides. Seventy-seven phosphorylations were upregulated upon adhesion, including PYK2/FAK2, Paxillin, CASL and p130CAS consistent with focal adhesion (FA) formation. We hypothesized that the collaborative signaling between β1 integrin and gp130 (IL-6 beta receptor, IL-6 signal transducer) was mediated by FA formation and proline-rich tyrosine kinase 2 (PYK2) activity. Both pharmacological and molecular targeting of PYK2 attenuated the amplification of STAT3 phosphorylation under co-stimulatory conditions. Co-culture of MM cells with patient bone marrow stromal cells (BMSC) showed similar β1 integrin-specific enhancement of PYK2 and STAT3 signaling. Molecular and pharmacological targeting of PYK2 specifically induced cell death and reduced clonogenic growth in BMSC-adherent myeloma cell lines, aldehyde dehydrogenase-positive MM cancer stem cells and patient specimens. Finally, PYK2 inhibition similarly attenuated MM progression in vivo. These data identify a novel PYK2-mediated survival pathway in MM cells and MM cancer stem cells within the context of microenvironmental cues, providing preclinical support for the use of the clinical stage FAK/PYK2 inhibitors for treatment of MM, especially in a minimal residual disease setting.
Introduction
Multiple myeloma (MM) remains a mortal disease of bone marrow resident malignant plasma cells characterized by classical hallmarks of end organ damage.1–3 Nearly all myeloma patients succumb to the disease due to the existence of a population of tumor cells that evade initial therapy, contributing to minimal residual disease (MRD), and subsequent relapse of drug-resistant tumor cells.4,5 MRD results, in part, from the survival signaling afforded adherent myeloma cells by complex cues from soluble and physical determinants of the bone marrow microenvironment. This hypothesis has identified mechanisms of environment-mediated drug resistance and survival signaling as critical considerations in drug discovery and therapeutic design in myeloma and other hematologic malignancies.6–10
The successful development of therapeutics targeting environment-mediated survival signaling (and environment-mediated drug resistance) requires preclinical models that account for the tumor and the tumor microenvironment (TME). We, and others, have demonstrated that (1) myeloma cell survival and drug resistance imparted by cellular adhesion is dynamic and environment dependent; (2) altered drug response is dominated by post-translational events; and (3) novel signaling networks and survival pathways are elicited under co-stimulatory conditions (soluble and physical components) that would not be identified in models without TME cues.2,11–13 These data reveal inherent limitations in cell culture models of drug discovery that do not account for multiple stimuli from the TME. Within the bone marrow microenvironment, myeloma survival and resistance to therapy likely involves the ‘collaboration’ between dynamic soluble and physical effectors of the TME,2,12,14 providing a differential selective advantage.
To investigate signaling modulated by the TME, we utilized a reductionist approach, reconstructing the complexity of the bone marrow microenvironment from a model of adhesion to fibronectin (FN) or exposure to interleukin-6 (IL-6) as separate representative physical and soluble microenvironment determinants to a combined model examining the intracellular signaling and biological sequelae following exposure of FN-adhered myeloma cells to IL-6.2 With this model, we previously demonstrated that IL-6 and FN-adhesion collaborated via post-translational alterations of the IL-6 receptor signaling complex to enhance signal transducer and activator of transcription 3 (STAT3) activity and a more malignant phenotype.2 β1 integrin-mediated adhesion to FN altered phosphorylation events from gp130 (IL-6 beta receptor, IL-6 signal transducer). Together, these data suggested that FN-adhesion modulates signaling from gp130, accounting for the subsequent phenotype. Therefore, we wanted to further investigate signaling events induced by FN-adhesion combined with IL-6.
To address this question, we surveyed the intracellular signaling induced by FN/IL-6 collaborative signaling by combining phosphotyrosine immunoprecipitation with liquid chromatography– tandem mass spectrometry to examine the tyrosine phos-phoproteome in the FN/IL-6-model of the TME.15–17 From these data, we hypothesized that focal adhesion formation and proline-rich tyrosine kinase 2 (PYK2), a kinase recently linked with myeloma progression,18 is critical for the enhanced JAK1/STAT3 activation observed under co-stimulatory conditions. Within this report, we demonstrate that PYK2 is positioned upstream of JAK1/STAT3 signaling and is a critical mediator of a novel myeloma cell survival pathway activated in the context of co-stimulation within multiple models of the TME and in vivo; as such, PYK2 represents a viable anti-myeloma target.
Results
β1 integrin preferentially amplifies JAK and STAT3 phosphorylation following IL-6 ligation in myeloma cell lines and patient specimens We have previously demonstrated that stimulation of FN-adherent myeloma cells with IL-6 facilitated a β1 integrin-dependent enhancement of STAT3 activation.2 Further characterization of the signaling in co-stimulated cells demonstrated a preferential phosphorylation of both Janus kinase 1 (JAK1) and STAT3 in myeloma cell lines and patient specimens (Figure 1a and b). Quantification of STAT3 phosphorylation in patient specimens using flow cytometry method (FCM) confirmed the western blot data in six of seven CD138-selected specimens examined by FCM and seven of eight samples including western blot analysis (Figure 1c), or 87.5% of patient samples. Interestingly, FCM data suggest that adhesion to FN may enhance STAT3 phosphorylation by lowering the threshold of its activation, increasing the number of responding cells. In characterizing other signaling effectors of IL-6, we demonstrated that the co-stimulatory phosphorylation of JAK1 and STAT3 was specific. Both AKT and extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation were not increased under the same conditions (Figure 1d and e). To confirm β1 integrin involvement, we demonstrated that enhanced JAK1 and STAT3 phosphorylation was abrogated following knockdown of β1 integrin in myeloma cell lines with RNA interference (RNAi; Figure 1f and g).2 Together, these data demonstrated that cellular adhesion to FN acutely alters the signaling dynamics elicited by IL-6, suggestive of larger scale alterations in the phosphoproteome under adherent conditions.
Figure 1.
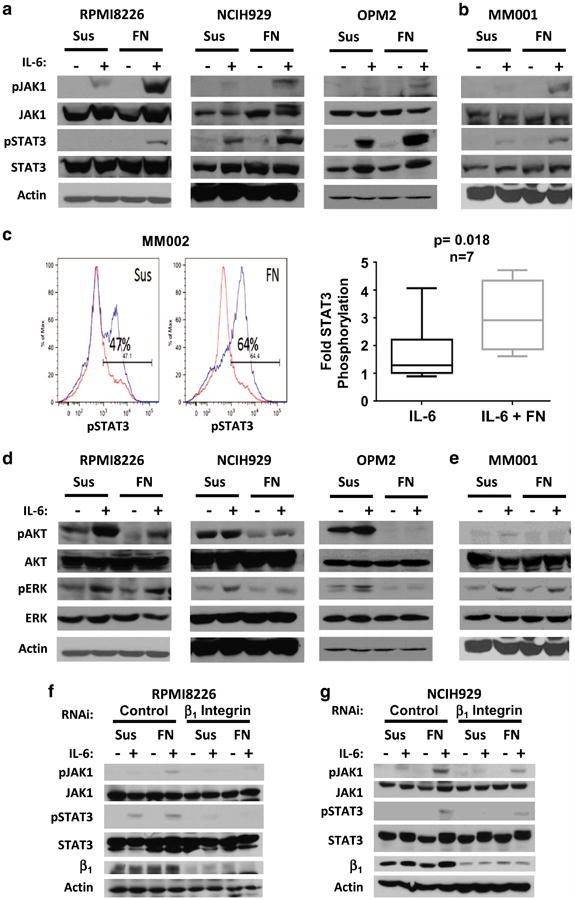
IL-6 induced JAK1 and STAT3 phosphorylation in myeloma cells is selectively increased by β1 integrin-mediated adhesion. Myeloma cell lines (a) and patient specimens (b, c) display amplified JAK1 and STAT3 phosphorylation in response to 1 ng/ml IL-6 when adhered to the extracellular matrix component fibronectin (FN) compared with cells grown in suspension (Sus). IL-6 induced STAT3 phosphorylation at tyrosine 705 was measured in seven myeloma patient specimens ex vivo by FCM (c, IL-6 stimulated cells are indicated by blue tracing). In contrast, activation of other downstream IL-6 signaling pathways, such as AKT and ERK, is not enhanced by adhesion (d, e). Knockdown of β1 integrin by RNA interference abolished adhesion-mediated enhancement of JAK1 and STAT3 phosphorylation in IL-6 treated cells (f, g). Cell lines (a, d, f, g) or primary cells (b, c, e) were grown in Sus or adhered to FN-coated plates (FN) for 1 h prior to stimulation with 1 ng/ml IL-6 for 30 min. Protein phosphorylation was assessed by western blot (a, b, d-g) or flow cytometry (FCM, c). s.d. are indicated by boxes and data range are indicated by whiskers. The mean is represented by a line within boxes. P-value was determined using Student's T-test, using a 95% confidence interval. The P-value <0.05 was considered to be significant. All immunoblots are representative of at least three independent experiments. RNA interference data were repeated using three unique constructs per target.
Characterization of the tyrosine phosphoproteome induced by adhesion to fibronectin and identification of PYK2 phosphorylation in adhered myeloma cells
To characterize downstream signaling events induced by FN adhesion or IL-6 alone, as well as collaborative signaling between FN and IL-6, phosphotyrosine proteomic profiling was used to compare cells in suspension (Sus), Sus+IL-6, FN-adhesion and FN+IL-6. Relative quantification identified 193 unique phospho-peptides across four conditions that demonstrated consistency in magnitude and direction of phosphorylation between duplicate samples. Hierarchical cluster analysis of the phosphopeptide heat map revealed four unique groups of phosphorylation events correlating with the four experimental conditions. Of these, 152 phosphorylation events were specific to adhesion (Figure 2a–d). These phosphopeptides corresponded to proteins participating in signal transduction, cellular adhesion/cytoskeletal assembly, RNA processing, transcriptional regulation, metabolism, cell cycle and vesicle trafficking (Supplementary Table 1). Because our previous results demonstrated that FN-adhesion-specific events facilitated STAT3 recruitment to gp130 and enhanced STAT3 activation,2 we initially focused on FN-adhesion-specific phosphorylation events. Enhanced STAT3 phosphorylation (8.7-fold increased phosphorylation in co-stimulated cells vs IL-6 alone) and decreased ERK1 (MK03) phosphorylation (0.43-fold in co-stimulated cells vs IL-6 alone) was observed within the phosphoproteomic data set providing further validation of this methodology consistent with our western blot results (Figure 1a, b, d and e).2
Figure 2.

Adhesion to fibronectin alters the IL-6-induced phosphotyrosine proteome of 8226 myeloma cells. 8226 myeloma cells were grown in suspension (Sus) or adhered to FN plates (FN) for 1 h prior to stimulation with 1 ng/ml IL-6 for 30 min. Lysates from 108 cells per condition were digested with trypsin, and phosphotyrosine peptides were enriched by immunoprecipitation. Peptides are grouped according to changes in relative intensity between sample groups: phosphorylation induced by (a) suspension, (b) adhesion to FN, (c) Sus+IL-6 or (d) adhesion to FN+IL-6. Peptides with increasing relative abundance are indicated by blue shading; decreasing phosphorylation events are indicated by yellow shading. Peptides are identified by gene symbols.
Examination of the list of tyrosine phosphorylation events positively regulated by adhesion to FN-identified phosphorylation events consistent with focal adhesion formation including paxillin (pY118), BCAR1/p130CAS (pY128 and pY249), CASL/NEDD9 (pY166), β-actin (pY166), tubulin 1α (pY357), as well as the focal adhesion kinase (FAK) FAK2/PYK2 (pY580). Quantification of peptide phosphorylation relative to myeloma cells in Sus demonstrated the greatest increase in pY580 of PYK2 compared with all other peptides, suggesting that PYK2 may be a central component in FA signaling in myeloma cells (Supplementary Table 2). PYK2 autophosphorylation (pY402) following adhesion to FN was validated by western blot analysis (Figure 3a and b) because sequences around this residue are not amenable to analysis by tryptic digestion. In addition to 77 upregulated FN-specific phosphorylation events, levels of 75 phosphorylated peptides were also negatively regulated upon FN adhesion, 20 phosphorylation sites were specifically modulated by IL-6 stimulation alone and 21 were unique to co-stimulation conditions (Figure 2a–d; Supplementary Figure 1A–D with peptide sequences). These data suggest that FN adhesion stimulates novel signaling events linking focal adhesions and cytokine signaling in myeloma cells. Our phosphotyrosine proteomic screen also suggests that focal adhesion formation and activation of PYK2 may be a critical step in the unique and complex signaling events observed in the context of the TME.
Figure 3.

PYK2 mediates the amplification of IL-6-induced STAT3 phosphorylation and concomitant gene expression in adhered myeloma cells. Adhesion of myeloma cell lines (a) or primary myeloma specimens (b) to the ECM component fibronectin (FN) stimulates PYK2 autophosphorylation at tyrosine 402. Adhesion-mediated amplification of STAT3 phosphorylation at tyrosine 705 is abrogated by blockade of PYK2 protein expression (c, d) or inhibition of PYK2 kinase activity (e, f). DEPTOR expression in myeloma cells is induced by FN-mediated adhesion and IL-6 stimulation (g) and inhibition of STAT3 or PYK2 expression by ASO treatment reduced DEPTOR expression. PYK2 kinase activity was assessed by monitoring PYK2 autophosphorylation at tyrosine 402. Cells were either treated with siRNA or ASO 48 h prior to stimulation with IL-6. Cells were grown in suspension (Sus) or adhered to FN-coated plates (FN) for 1 h prior to stimulation with 1 ng/ml IL-6 for 30 min. VS-6062 was added just before plating. All immunoblots are representative of at least three independent experiments. RNA interference data were repeated using three unique constructs per target.
PYK2 is an upstream mediator of the enhanced STAT3 phosphorylation observed under co-stimulatory conditions
PYK2 is a FAK family protein19,20 previously identified in myeloma cells and has been linked to MM progression.18,21–23 Our phosphoproteomic analysis suggests PYK2 is well suited to modulate cross talk between soluble and physical effectors (activated focal adhesions) in the bone marrow niche. In support of this, PYK2 signaling has been linked to cell surface receptor signaling.24–28 In Figure 3a and b, we validated that PYK2 is phosphorylated upon adhesion to FN in all myeloma cell lines and CD138-selected patient myeloma cells examined. We hypothesized that adhesion-induced PYK2 activation contributes to enhanced STAT3 phosphorylation. Treatment of RPMI8226 or NCIH929 myeloma cells with PYK2 RNAi attenuated the enhanced STAT3 phosphorylation induced under collaborative conditions (Figure 3c and d). This treatment did not affect the low levels of STAT3 phosphorylation induced by IL-6 alone. PYK2 RNAi did not affect the expression of other IL-6 signaling complex proteins or β1 integrin expression (data not shown). Similar results are demonstrated with the FAK/PYK2 kinase inhibitor VS-6062 (Figure 3e and f). Molecular targeting of PYK2 did not significantly attenuate adhesion of RPMI8226 or NCIH929 cells to FN (data not shown; P > 0.05). These results indicate that PYK2 is a key upstream determinant in the enhanced STAT3 signaling linking β1 integrin-mediated adhesion and gp130.
DEP domain-containing mTOR-interacting protein (DEPTOR, DEPDC6) is a negative regulator of the mTOR pathway, causing reduced cell growth and proliferation. DEPTOR is overexpressed in myeloma with increased c-maf expression and reduced expression of DEPTOR in myeloma cells leads to apoptosis.29 We show for the first time that DEPTOR protein (Figure 3g) and RNA (data not shown) expression is induced by FN-mediated adhesion and IL-6 stimulation. Moreover, pretreatment of myeloma cells with STAT3 or PYK2 RNAi attenuated co-stimulation induced DEPTOR expression. These data suggest that DEPTOR represents a novel downstream effector of PYK2 and STAT3 signaling under co-stimulatory conditions.
PYK2 modulates STAT3 phosphorylation in myeloma cells upon adhesion to patient BMSCs
We next wanted to determine whether PYK2 and subsequent signaling translated to more complex and more biologically relevant models of the TME. Myeloma cells were examined under three conditions: cells incubated in (1) monoculture (M, myeloma cells alone), (2) co-culture with patient bone marrow stromal cells (BMSCs) separated by a transwell membrane (Tw; providing only soluble factors from the TME) and (3) co-culture with patient BMSCs with direct adhesion (Cx; both physical and soluble components; Figure 4a). Within this more biologically complex model, we demonstrate that PYK2, JAK1 and STAT3 phosphorylation were enhanced in only myeloma cells co-cultured under adherent conditions in all cell lines examined (Figure 4b and c). Increased PYK2, JAK1 and STAT3 phosphorylation was observed in RPMI8226 cells upon adhesion to all patient BMSCs utilized (Supplementary Figure 2A; three individual patient BMSCs). Similar to the FN/IL-6 model, STAT3 phosphorylation was preferential, occurring at the exclusion of AKT and ERK1/2 phosphorylation (Supplementary Figure 2B). Preferential PYK2, JAK1 and STAT3 phosphorylation is similarly observed in patient myeloma cells upon adhesion to BMSCs, but not in conditions without direct contact (Figure 4d and e).
Figure 4.
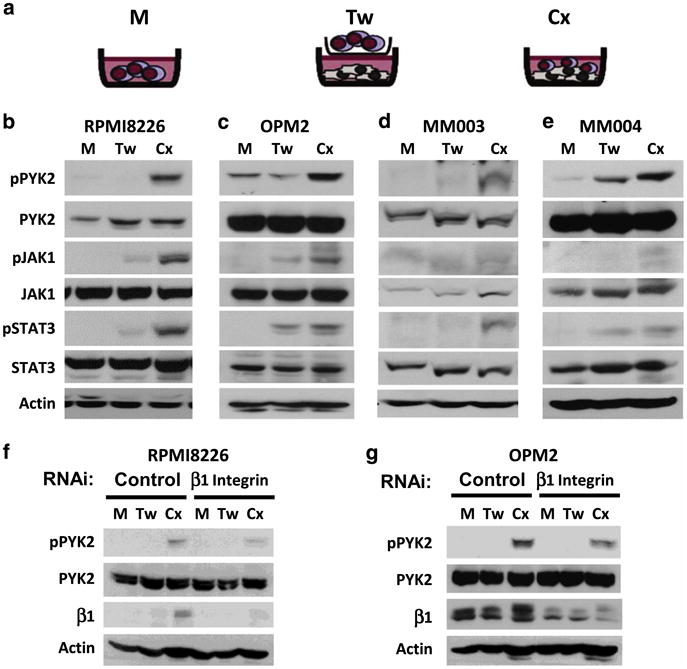
Adhesion-mediated amplification of STAT3 phosphorylation in a complex model of the bone marrow microenvironment requires β1 integrin. Myeloma cells were either grown in monoculture (M) or in co-culture with patient-derived bone marrow stromal cells (BMSCs). Co-cultured myeloma cells were either separated from BMSCs by transwell inserts that allow soluble factor diffusion (Tw) or adhered to BMSC monolayers (Cx, a). Adhesion to patient-derived BMSCs enhances PYK2, JAK1 and STAT3 phosphorylation in myeloma cells lines (b, c) and patient specimens (d, e). Knockdown of β1 integrin diminishes the activation of PYK2 induced by adhesion to BMSCs (f, g). All immunoblots are representative of at least three independent experiments. RNA interference data were repeated using three unique constructs per target.
We examined the role of β1 integrin and the IL-6 signal transducer, gp130, in the amplification of STAT3 phosphorylation in myeloma cells adhered to BMSCs. The activation of PYK2 under co-culture conditions was dependent upon β1 integrin-mediated adhesion to BMSCs, as incubation of RPMI8226 and OPM2 myeloma cell lines with β1 integrin small interfering RNA attenuated co-culture-associated PYK2 phosphorylation (Figure 4f and g). Of note, increased β1 integrin expression was seen in myeloma cells under co-culture conditions. BMSC-induced STAT3 phosphorylation in myeloma cells was also decreased by gp130 knockdown (Supplementary Figure 2C). Taken together, these data indicate that β1 integrin/gp130 cross talk is responsible, at least in part, for enhanced STAT3 signaling observed in myeloma cells adhered to BMSC. Importantly, STAT3 and PYK2 phosphorylation was not induced in the BMSCs under these co-cultured conditions, demonstrating myeloma cell-specific phosphorylation of PYK2 and STAT3 under co-cultured conditions (Supplementary Figure 3). Furthermore, treatment of RPMI8226 and OPM2 myeloma cells with the FAK/PYK2 tyrosine kinase inhibitor (TKI) VS-6062 decreased PYK2 and STAT3 phosphorylation induced by co-stimulatory conditions (Figure 5a and b). These data further demonstrate a novel β1 integrin-dependent PYK2/STAT3 signaling pathway under collaborative signaling between soluble and physical effectors of the TME.
Figure 5.

PYK2 inhibition or knockdown induces apoptosis in myeloma cells adhered to patient-derived bone marrow stroma. Myeloma cells were treated with increasing amounts of PYK2 inhibitor VS-6062 (vehicle, 200 nM or 400 nM) and incubated for 24 h in monoculture (M) or co-culture. Treatment of myeloma cells with VS-6062 blocked PYK2 autophosphorylation at Y402 and STAT3 phosphorylation at Y705 (a, b). Co-cultured myeloma cells were either separated from BMSCs by transwell inserts that allow soluble factor diffusion (Tw) or adhered to BMSC monolayers (Cx, c–f). In a separate experiment, myeloma cell lines (g, h) or patient specimens (i) were treated with antisense oligonucleotides (ASOs) 48 h prior to co-culture for 24 h with patient BMSCs. Cells were treated with control, STAT3 (AZD9150), PYK2 or STAT3 (AZD9150)/PYK2 ASO in combination. Knockdown efficiency was assessed by immunoblot 48 h after ASO treatment (j). Apoptosis was measured by monitoring Annexin V surface expression (c, e, g–i) and/or Caspase3 activation (d, f). Error bars indicate s.d., n = 4. Indicated P-values were generated using T-test with a 95% confidence interval. P-values <0.05 were considered to be significant.
PYK2 regulates myeloma cell survival under adherent co-culture conditions
We demonstrated that targeting PYK2 with VS-6062 or RNAi attenuated STAT3 signaling, placing PYK2 upstream of STAT3 signaling under co-stimulatory conditions. Therefore, we next determined the consequences of targeting PYK2 on myeloma survival. Incubation of myeloma cells with nanomolar concentrations of VS-6062 induced co-culture-specific cell death as quantified by annexin V surface expression and caspase-3 activation by FCM in RPMI8226 (Figure 5c and d) and OPM2 (Figure 5e and f) myeloma cell lines. To address the possibility that VS-6062 might be toxic to BMSC through inhibition of FAK/PYK2 signaling, we demonstrated that the TKI does not impact the viability of BMSCs at concentrations up to 10 μM, several-fold higher than concentrations needed to induce myeloma cell death (data not shown).
Even with highly specific pharmacologic kinase inhibitors, TKIs can have off-target effects. Furthermore, under co-culture conditions, we cannot state that VS-6062 is targeting only myeloma cells. To address these issues, we utilized clinically relevant antisense oligonucleotides (ASOs). Myeloma cell lines and patient specimens were treated with ASOs targeting STAT3 (AZD9150), PYK2, as well as the combination for 48 h prior to incubation in the three culture conditions. Targeting this pathway with ASO similarly induced apoptosis only in myeloma cells incubated under co-stimulatory conditions as quantified by FCM (Figure 5g–j). STAT3 ASO (AZD9150)-induced Annexin V-positive cells in RPMI8226 cells (P = 0.02) and trended toward increased apoptosis of OPM2 cells (P = 0.061). PYK2 ASO-induced statistically significant Annexin V staining in both RPMI8226 and OPM2 myeloma cell lines (P = 0.006 and 0.001, respectively), as well as a patient specimen (P = 0.02). Consistent with our data placing PYK2 upstream of JAK/STAT3 signaling, the combined knockdown of PYK2 and STAT3 did not afford a statistically significant increase in myeloma cell death above that of PYK2 alone. Background apoptosis observed in control cells adhered to BMSCs (Cx) did not impact the significance of apoptosis measured in experimental groups and is likely an artifact resulting from myeloma-BMSC separation just prior to staining. These results provide evidence that the collaboration of BMSC adhesion and soluble determinants modulate PYK2-dependent myeloma cell survival.
To further assess the impact of PYK2 inhibition on myeloma cell survival, we employed the clinically relevant FAK/PYK2 inhibitor, VS-6063 (defactinib, Verastem, Inc., Cambridge, MA, USA) currently being evaluated in phase I and II clinical trials (NCT01870609, NCT01778803, NCT01951690 and NCT02004028). Phosphorylation of PYK2 in myeloma cell lines (Figure 6a) and myeloma patient specimens (Figure 6b) adhered to BMSCs could be blocked using nanomolar concentrations of inhibitor (Figure 6a and b), replicating results observed with VS-6062. To assess the effects of VS-6063 on survival of both myeloma cell lines and patient specimens, we employed colony formation assays. Consistent with an increased survival advantage in the context of BMSC, this assay demonstrated increased MM cell clonogenic survival in the presence of BMSCs (Figure 6c and d). Inhibition of PYK2 with VS-6063 preferentially inhibited clonogenic growth of RPMI8226, NCIH929 and patient MM cells grown in co-culture relative to cells grown in the absence of BMSCs (Figure 6e and f).
Figure 6.

PYK2 inhibition reduces long-term viability and clonogenic growth of myeloma cells co-cultured with patient BMSCs. Myeloma cell lines (a, c, e) and patient specimens (b, d) were cultured in monoculture (light bars) or co-cultured with patient BMSCs (dark bars) in the presence of indicated concentrations of the clinically relevant FAK/PYK2 inhibitor VS-6063 (defactinib). PYK2 activation in myeloma cell lines (a) or patient specimens (b) grown in monoculture (M) or co-cultured with BMSCs (Cx) was assessed by measuring pY402 via immunoblot. Tumor cell colony formation in methylcellulose was used to quantify clonogenic growth as previously described (c–f).31,33 Cell lines and patient specimens shown in panels c and d match those displayed directly below in panels e and f. Clonogenic growth of an ALDH-positive myeloma cell subpopulation was also measured (g, h). Error bars are represented as s.e.
MRD has also been attributed to a small population of putative cancer stem cells with self-renewal properties and the ability to propagate disease in myeloma and other malignancies.30–32 To further investigate the potential of targeting PYK2 in other aspects of MRD, we examined the effect of the TME on myeloma progenitor cells defined as aldehyde dehydrogenase (ALDH +)-positive myeloma cells.31,33 Within this context, the percentage of ALDH+ myeloma cells increased in the presence of BMSCs (Figure 6g), suggesting that this population of cells is also influenced by the TME. Importantly, ALDH+ myeloma cancer stem cells co-cultured with BMSCs also demonstrated increased sensitivity to treatment with the PYK2 inhibitor VS-6063 relative to ALDH+ cells cultured without BMSC (Figure 6h). Together, these data further affirm the role of PYK2 in TME-mediated MM cell survival and that targeting PYK2 represents a therapeutic strategy to overcome the protective effects of the TME and its potential contribution to multiple aspects of MRD.
Finally, we wanted to examine the role of PYK2 in an in vivo model of the TME. As in human myeloma cells, PYK2 TKI attenuated PYK2 and STAT3 phosphorylation and induced apoptosis in 5TGM1 murine myeloma cells cultured in direct contact with M2-10B4 murine BMSCs (Figure 7a and b). Employing the immune competent 5TGM1-C57BL/KaLwRijHsd murine model, we demonstrated that PYK2 TKI activity was effective in an in vivo model of the myeloma TME. One million luciferase-expressing 5TGM1 cells were tail-vein injected into six mice per condition. Oral administration of the VS-4718 PYK2/FAK TKI twice daily significantly reduced murine myeloma disease burden as quantified by immunoglobulin G2b levels and luciferase activity (Figure 7c and d; P = 0.03 and 0.02, respectively). Furthermore, PYK2 inhibition with VS-4718 also afforded a statistically significant overall survival advantage to treated mice relative to vehicle control (Figure 7e, P = 0.018). Collectively, our results demonstrate that PYK2 signaling is critical for myeloma cell survival in the context on the TME.
Figure 7.
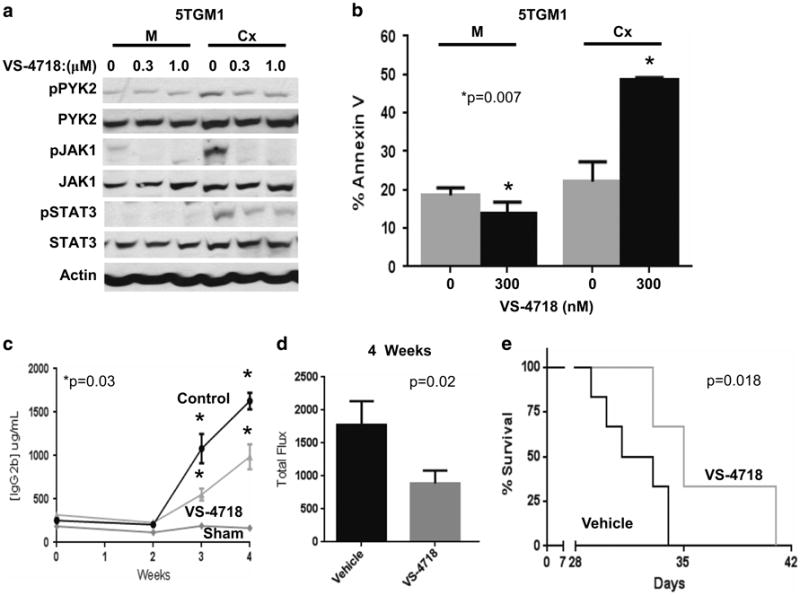
PYK2 inhibition reduces tumor burden and increases overall survival in vivo. 5TGM1 murine myeloma cells were cultured in the presence of murine M2-10B4 bone marrow stoma for 2 h. Myeloma cells were either adhered to stroma (Cx) or grown in monoculture (M) and PYK2 activation was assessed by measuring pY402 via immunoblot (a). 5TGM1 myeloma cells were treated with indicated concentrations of Pyk2 inhibitor VS-4718 for 24 h in monoculture or in co-culture with M2-10B4 stroma, and apoptosis was measured by measuring surface Annexin V expression via FCM (b). One million 5TGM1-luc cells were injected into 6- to 8-week-old C57BL/KaLwRijHsd mice. Tumor burden was assessed by measuring levels of IgG2b paraprotein by ELISA weekly (c) or luciferase activity at 4 weeks (d). Survival was assessed until the onset of hind-limb paralysis (e). Error bars are represented as s.e.m. Indicated P-values were generated using T-test with a 95% confidence interval in c and d. The significance of Kaplan–Meyer survival curves was determined by the log-rank test. P-values <0.05 were considered to be significant.
Discussion
The high mortality observed in MM is, in part, the consequence of MRD afforded by complex signaling events in the TME and putative MM cancer stem cells. We and others have previously demonstrated myeloma cells co-cultured in contact with BMSCs were afforded a survival advantage compared with cells co-cultured without contact.12,13,34 This experience demonstrates that the combination of soluble and physical effectors provides a selective advantage, suggesting microenvironment-specific heterogeneity in survival signaling conferred by the TME. Therefore, it is critical to identify microenvironment-specific survival pathways and drug targets.
We have previously shown that when myeloma cells are adhered to fibronectin, STAT3 phosphorylation is enhanced in response to IL-6 stimulation, leading to a more aggressive tumor phenotype.2 In the present work, we have extended those findings by identifying a novel molecular intermediate of gp130 and integrin cooperative signaling using phosphoproteomic screening of a preclinical TME model. Specifically, we identified the FAK PYK2 as the central figure in the unique signaling events modulated by the collaborative signaling between the soluble and physical effectors of the TME. Enhanced JAK/STAT3 phosphorylation was observed under cooperative signaling between receptors of soluble factors and adhesion in both the reductionist (FN/IL-6) and more clinically relevant patient BMSC co-stimulatory models of the TME. We demonstrate that this enhanced signaling is dependent on upstream activation of the FAK, PYK2. Critically, targeting PYK2 with pharmacologic and molecular techniques preferentially induces death and attenuates clonogenic growth in myeloma cells under co-stimulatory conditions. Attenuation of PYK2 also had disease-modifying effects in the 5TGM1 murine model. These results indicate that under co-stimulatory conditions, myeloma cells and myeloma stem cells require PYK2 for survival. As such, these data illustrate the potential of PYK2 as a novel TME-specific target to combat myeloma and MRD by disrupting the survival pathways resulting from the complex network of environmental cues in the bone marrow microenvironment.
PYK2 expression has been shown to correlate with myeloma disease progression.18 Our results indicate that adhesion-dependent PYK2 activation modulates myeloma cell survival, at least in part, via enhanced JAK/STAT signaling. This conclusion is consistent with previous findings linking PYK2 to JAK-dependent Type I and Type II cytokine signaling.25–28 This work is novel because it is the first description of a downstream PYK2-mediated STAT3 activation mechanism associated with these receptor families. The involvement of PYK2 in JAK-dependent receptor signaling is interesting because PYK2 and FAK1 are one of only two families of tyrosine kinases to contain FERM (4.1, ezrin, radixin and moesin) protein-binding domains.20 The other group of kinases is the JAK family that imparts kinase activity to gp130 and other cytokine receptors. These facts suggest that PYK2 is well suited to modulate cross talk between gp130 and β1 integrin-activated focal adhesions via direct contact. We anticipate that it will be critical to further examine the biochemical relationship between JAK and PYK2 under co-stimulatory conditions.
We also identified DEPTOR as a downstream effector of PYK2 and STAT3 signaling in our model system. DEPTOR is an inhibitor of TORC1 signaling linked to myeloma survival29 and its role in survival in this system will be an active component of future studies. Furthermore, our work does not exclude the influence of additional PYK2-modulated signaling pathways induced by BMSCs on myeloma cell survival. Our ASO data provide evidence that PYK2-specific, yet STAT3-independent events, may contribute to myeloma cell survival upon interacting with the TME. PYK2 has also been shown to negatively regulate osteoblast differentiation and activity in mice,35 suggesting that PYK2 may negatively influence WNT/β-catenin, osterix and RUNX2 signaling.23,36 Furthermore, adhesion-mediated PYK2 activity may also influence signaling from additional upstream receptors. PYK2 has been linked to STAT3 signal amplification through the receptor tyrosine kinase, epidermal growth factor receptor.24 Furthermore, PYK2 also associates with the receptors FGFR3 and c-MET.37,38 These results indicate that adhesion-mediated activation of PYK2 may modulate signaling events from multiple soluble receptors critical to myeloma survival in the TME, thus, making PYK2 a more attractive therapeutic target.
Our examination of the tyrosine phosphoproteome initially identified PYK2 as a candidate intermediate of FN/IL-6 cooperative signaling. In addition, these data highlight other important signaling cross talk downstream of IL-6 receptor and adhesion receptors. We identified 193 differentially modified tyrosine phosphopeptides in myeloma cells. Phosphopeptide heat map analysis revealed four unique groupings of phosphorylation events correlating with the experimental conditions. These phosphopeptides corresponded to proteins participating in numerous cellular functions including cytoskeletal/adhesion and signal transduction, as well as cellular metabolism, RNA processing and cell cycle (Supplementary Table 1). Further investigation is needed into the potential role of TME-regulated RNA processing and metabolism intermediates as alternate/additional anti-myeloma targets. In addition to the 152 differentially regulated peptides identified following adhesion alone, 21 phosphopeptides were demonstrated to be specific to co-stimulation (FN/IL-6), including the phosphatases SHIP1 and SHP2. Fuhler et al. have demonstrated a role of phosphatases in myeloma survival and drug resistance, suggesting that they too may be critical mediators of the survival signaling induced under co-stimulatory conditions.39,40
This work is significant because it is the first report linking PYK2 to the amplification of STAT3 signaling in the context of TME-specific myeloma survival.41 We identify PYK2 as a novel TME-specific target to potentially combat MRD by disrupting the unique survival pathways resulting from the complex network of environmental cues of the TME in myeloma cells, in putative myeloma cancer stem cells, and in vivo. As such, we provide preclinical evidence and rationale supporting the clinical use of FAK/PYK2 inhibitors, such as VS-6063, for the treatment of MM, ideally in the setting of MRD. Finally, the TME has been linked to survival and drug response in diverse tumors42–47 Therefore, PYK2 or other signaling factors identified in our phosphoproteomic analysis may have broad application for survival of a spectrum of malignancies in the context of the TME.
Materials and Methods
Cell culture
All cell lines were grown as previously published,2 and are routinely tested for mycoplasma contamination and κ/λ expression. Cell lines were authenticated using short tandem repeat DNA typing according to ATCC guidelines using the GenePrint 10 System (Promega, Madison, WI, USA).48
Primary tumor specimens were harvested from bone marrow by aspiration and purified by positive selection versus an anti-CD138 affinity column (Miltenyi, Auburn, CA, USA). Primary patient BMSCs were obtained from the flow-through from CD138-selected marrow samples. Nonadherent cells were aspirated for 3–4 weeks until a morphologically similar monolayer of bone marrow fibroblasts cells remained. Specimens were obtained from myeloma patients enrolled in Moffitt Cancer Center's Total Cancer Care program after obtaining informed consent. This program is approved by the institutional review board at the University of South Florida.
RNAi and ASOs
ASO to STAT3 (AZD9150),49 PYK2 and non-specific controls were from Isis Pharmaceuticals (Carlsbad, CA, USA). Twenty-four to 48 h prior to experiments, ASOs were added directly to tissue culture medium of cell lines or primary tumor cells at concentrations of 2.5 μm or 10.0 μm, respectively. Non-silencing control and small interfering RNA targeting three sequences each of PYK2, β1 integrin and gp130 was purchased from Thermo Scientific (Waltham, MA, USA) and transfected into cell lines by electroporation as previously described.50
Apoptosis assays
Apoptosis was quantified 48 h after treatment with VS-6062, VS-4718 (Verastem, Inc.) or ASO by FCM using either antibodies directed to activated caspase-3 or annexin surface expression per manufacturer's directions (BD Pharmingen, San Jose, CA, USA).
Tumor cell colony formation
Tumor cell colony formation in methylcellulose was used to quantify in vitro clonogenic growth according to our previously published methods.31,33 Myeloma cells (1000 cells/ml) were washed twice with phosphate-buffered saline following treatment, then plated in triplicate into 35 mm2 tissue culture dishes containing 1.2% methylcellulose, 30% fetal bovine serum, 1% bovine serum albumin, 10-4 m 2-mercaptoethanol and 2 mm L-glutamine. For clinical specimens, mononuclear cells were isolated from primary clinical bone marrow aspirates. Following 10–21 days of culture at 37 °C and 5% CO2, tumor colonies consisting of more than 40 cells were quantified using an inverted microscope. Drug treatments were carried out using VS-6063 for 96 h in complete media at the indicated concentrations.
FC and cell isolation
Cells were stained with fluorescein isothiocyanate–conjugated mouse anti-human CD138 or isotype control antibody (BD Pharmingen, San Diego, CA, USA) for 30 min at 4 °C, then stained for ALDH activity using the Aldefluor reagent (Stem Cell Technologies, Vancouver, BC, Canada) according to the manufacturer's instructions. Cells were subsequently washed in phosphate-buffered saline containing 5 μm propidium iodide (Sigma, St Louis, MO, USA) and analyzed on a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA, USA) or isolated using a FACSAria II cell sorter. Cells were initially gated to exclude propidium iodide positive cells then analyzed for CD138 or ALDH expression.
Immunoblotting
Cell extract, preparation and immunoblotting were performed as previously described.2,51 PYK2, STAT3 and JAK1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). AKT and pSTAT3 (pY 705), and anti-phosphotyrosine antibodies were from Cell Signaling (Boston, MA, USA). Anti-pJAK1 (pYpY 1022/1023) was from Invitrogen (Grand Island, NY, USA). pERK1/2 (pT 202/pY 204), ERK1 and β1 integrin antisera was from BD Biosciences (San Jose, CA, USA). pAKT (pS 473) antibody was from Imgenex (San Diego, CA, USA). Anti-pPYK2 (pY402) was from R&D Systems (Minneapolis, MN, USA). Actin antibody was from Sigma and horseradish peroxidase-conjugated secondary antibody was from Jackson ImmunoResearch (West Grove, PA, USA). STAT3 phosphorylation in primary cells was examined by FC. Cells were fixed in 1.6% paraformaldehyde, then permeabilized in 95% methanol for 10 min, and incubated in 1% fetal bovine serum overnight prior to staining with Alexa647-conjugated anti-pSTAT3 (pY 705; BD Pharmingen) for 2 h at 4 °C. Fibronectin was purchased from Invitrogen.
Liquid chromatography–tandem mass spectrometry phosphoproteomics
After phosphotyrosine immunoprecipitation at the peptide level according to the manufacturer's instructions, tryptic phosphopeptides were eluted with 0.15% trifluoroacetic acid and concentrated to 20 μl using vacuum centrifugation (Speedvac, Thermo, San Jose, CA, USA). A nanoflow liquid chromatograph (U3000, Dionex, Sunnyvale, CA, USA) coupled to an electrospray ion trap mass spectrometer (LTQ-Orbitrap, Thermo) was used for tandem mass spectrometry peptide sequencing experiments for identification and relative quantification. Each sample was analyzed in duplicate, as previously described.52
5TGM1 myeloma mouse model
A total of 1×106 5TGM1 cells were injected into 6- to 8-week-old female C57BL/KaLwRijHsd mice via tail vein. Beginning at day 14, mice were treated with VS-4718 in 0.5% carboxymethyl cellulose (Sigma-Aldrich; St Louis, MO, USA) and 0.1% Tween80 (Sigma-Aldrich) at 50 mg/kg twice daily by oral gavage. Immunoglobulin G2b serum levels were measured by ELISA once a week for 4 weeks per the manufacturer's instructions (Bethyl, Montgomery, TX, USA). Tumor burden was also measured by luciferase assay using an IVES-200 imaging system. Mice were monitored daily and euthanized at the onset of hind-limb paralysis. All procedures were carried out in compliance with the laws, regulations and guidelines of the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and with the approval of the University of South Florida's Animal Care and Use Committee. Sample size was predetermined using power analysis and mice were sorted by simple randomization without blinding prior to the experiment.
Supplementary Material
Acknowledgments
We thank Jianguo Tao and William S Dalton for helpful discussions and critical reading of the manuscript. This work was supported by Florida Department of Health Bankhead-Coley Team Science Program grant 2BT03 (to KSH and LHA). The Moffitt Proteomics Facility is supported by the US Army Medical Research and Materiel Command under Award W81XWH-08-2-0101 for a National Functional Genomics Center, the National Cancer Institute under Award P30-CA076292 as a Cancer Center Support Grant and the Moffitt Foundation. Patient specimens were obtained from the Total Cancer Care program at Moffitt Cancer Center. Patient specimen collection, phosphoproteome mapping and flow cytometry were performed by the Translational Research, Proteomics and Flow Cytometry Core facilities at Moffitt Cancer Center. Antisense oligonucleotides were provided by Isis Pharmaceuticals (Carlsbad, CA, USA). The focal adhesion kinase inhibitors VS-6062,VS-6063 (defactinib), and VS-4718 were provided by Verastem, Inc. (Needham, MA, USA).
Abbreviations
- ASO
anti-sense oligonucleotide
- BMSC
bone marrow stromal cell
- ERK
extracellular signal-regulated kinase
- FAK
focal adhesion kinase
- FCM
flow cytometry method
- FERM
4.1 protein, ezrin, radixin, moesin domain
- FN
fibronectin
- gp130
IL-6 beta receptor, IL-6 signal transducer
- IL-6
interleukin-6
- JAK
Janus kinase
- MRD
minimal residual disease
- PYK2
proline-rich tyrosine kinase 2
- STAT
signal transducer and activator of transcription
- Sus
suspension
- TME
tumor microenvironment
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Kesanakurti D, Chetty C, Dinh DH, Gujrati M, Rao JS. Role of MMP-2 in the regulation of IL-6/Stat3 survival signaling via interaction with alpha5beta1 integrin in glioma. Oncogene. 2013;32:327–340. doi: 10.1038/onc.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shain KH, Yarde DN, Meads MB, Huang M, Jove R, Hazlehurst LA, et al. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009;69:1009–1015. doi: 10.1158/0008-5472.CAN-08-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajkumar SV. Multiple myeloma: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013;88:226–235. doi: 10.1002/ajh.23390. [DOI] [PubMed] [Google Scholar]
- 4.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 5.Rajkumar SV. Multiple myeloma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:57–65. doi: 10.1002/ajh.21913. [DOI] [PubMed] [Google Scholar]
- 6.Hazlehurst LA, Bewry NN, Nair RR, Pinilla-Ibarz J. Signaling networks associated with BCR-ABL-dependent transformation. Cancer Control. 2009;16:100–107. doi: 10.1177/107327480901600202. [DOI] [PubMed] [Google Scholar]
- 7.Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–4351. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia. 2009;23:10–24. doi: 10.1038/leu.2008.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shain KH, Dalton WS. Environmental-mediated drug resistance: a target for multiple myeloma therapy. Expert Rev Hematol. 2009;2:649–662. doi: 10.1586/ehm.09.55. [DOI] [PubMed] [Google Scholar]
- 10.Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9:665–674. doi: 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- 11.Landowski TH, Olashaw NE, Agrawal D, Dalton WS. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-kappa B (RelB/p50) in myeloma cells. Oncogene. 2003;22:2417–2421. doi: 10.1038/sj.onc.1206315. [DOI] [PubMed] [Google Scholar]
- 12.Nefedova Y, Landowski TH, Dalton WS. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia. 2003;17:1175–1182. doi: 10.1038/sj.leu.2402924. [DOI] [PubMed] [Google Scholar]
- 13.Nefedova Y, Cheng P, Alsina M, Dalton WS, Gabrilovich DI. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004;103:3503–3510. doi: 10.1182/blood-2003-07-2340. [DOI] [PubMed] [Google Scholar]
- 14.McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to antic-ancer drug activity. Nat Med. 2010;16:483–489. doi: 10.1038/nm.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J, et al. A chemical and phospho-proteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6:291–299. doi: 10.1038/nchembio.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23:94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- 17.St-Germain JR, Taylor P, Tong J, Jin LL, Nikolic A, Stewart II, et al. Multiple myeloma phosphotyrosine proteomic profile associated with FGFR3 expression, ligand activation, and drug inhibition. Proc Natl Acad Sci USA. 2009;106:20127–20132. doi: 10.1073/pnas.0910957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Moschetta M, Huynh D, Tai YT, Zhang Y, Zhang W, et al. Pyk2 promotes tumor progression in multiple myeloma. Blood. 2014;124:2675–2686. doi: 10.1182/blood-2014-03-563981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butler B, Blystone SD. Tyrosine phosphorylation of beta3 integrin provides a binding site for Pyk2. J Biol Chem. 2005;280:14556–14562. doi: 10.1074/jbc.M411765200. [DOI] [PubMed] [Google Scholar]
- 20.Golubovskaya VM. Targeting focal adhesion kinase in cancer-part I. Anticancer Agents Med Chem. 2010;10:713. doi: 10.2174/187152010794728693. [DOI] [PubMed] [Google Scholar]
- 21.Chauhan D, Hideshima T, Pandey P, Treon S, Teoh G, Raje N, et al. RAFTK/PYK2-dependent and -independent apoptosis in multiple myeloma cells. Oncogene. 1999;18:6733–6740. doi: 10.1038/sj.onc.1203082. [DOI] [PubMed] [Google Scholar]
- 22.Chauhan D, Pandey P, Hideshima T, Treon S, Raje N, Davies FE, et al. SHP2 mediates the protective effect of interleukin-6 against dexamethasone-induced apoptosis in multiple myeloma cells. J Biol Chem. 2000;275:27845–27850. doi: 10.1074/jbc.M003428200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Moschetta M, Huynh D, Tai YT, Zhang Y, Zhang W, et al. Pyk2 promotes tumor progression in multiple myeloma. Blood. 2014;124:2675–2686. doi: 10.1182/blood-2014-03-563981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi CS, Kehrl JH. Pyk2 amplifies epidermal growth factor and c-Src-induced Stat3 activation. J Biol Chem. 2004;279:17224–17231. doi: 10.1074/jbc.M311875200. [DOI] [PubMed] [Google Scholar]
- 25.Benbernou N, Muegge K, Durum SK. Interleukin (IL)-7 induces rapid activation of Pyk2, which is bound to Janus kinase 1 and IL-7Ralpha. J Biol Chem. 2000;275:7060–7065. doi: 10.1074/jbc.275.10.7060. [DOI] [PubMed] [Google Scholar]
- 26.Miyazaki T, Takaoka A, Nogueira L, Dikic I, Fujii H, Tsujino S, et al. Pyk2 is a downstream mediator of the IL-2 receptor-coupled Jak signaling pathway. Genes Dev. 1998;12:770–775. doi: 10.1101/gad.12.6.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takaoka A, Tanaka N, Mitani Y, Miyazaki T, Fujii H, Sato M, et al. Protein tyrosine kinase Pyk2 mediates the Jak-dependent activation of MAPK and Stat1 in IFN-gamma, but not IFN-alpha, signaling. EMBO J. 1999;18:2480–2488. doi: 10.1093/emboj/18.9.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Tassiulas I, Park-Min KH, Reid AC, Gil-Henn H, Schlessinger J, et al. ‘Tuning’ of type I interferon-induced Jak-STAT1 signaling by calcium-dependent kinases in macrophages. Nat Immunol. 2008;9:186–193. doi: 10.1038/ni1548. [DOI] [PubMed] [Google Scholar]
- 29.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boucher K, Parquet N, Widen R, Shain K, Baz R, Alsina M, et al. Stemness of B-cell progenitors in multiple myeloma bone marrow. Clin Cancer Res. 2012;18:6155–6168. doi: 10.1158/1078-0432.CCR-12-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68:190–197. doi: 10.1158/0008-5472.CAN-07-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y, Shi J, Gu Z, Salama ME, Das S, Wendlandt E, et al. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res. 2015;75:594–604. doi: 10.1158/0008-5472.CAN-14-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103:2332–2336. doi: 10.1182/blood-2003-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirshner J, Thulien KJ, Martin LD, Debes Marun C, Reiman T, Belch AR, et al. A unique three-dimensional model for evaluating the impact of therapy on multiple myeloma. Blood. 2008;112:2935–2945. doi: 10.1182/blood-2008-02-142430. [DOI] [PubMed] [Google Scholar]
- 35.Buckbinder L, Crawford DT, Qi H, Ke HZ, Olson LM, Long KR, et al. Proline-rich tyrosine kinase 2 regulates osteoprogenitor cells and bone formation, and offers an anabolic treatment approach for osteoporosis. Proc Natl Acad Sci USA. 2007;104:10619–10624. doi: 10.1073/pnas.0701421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kingsley LA, Chirgwin JM, Guise TA. Breaking new ground to build bone. Proc Natl Acad Sci USA. 2007;104:10753–10754. doi: 10.1073/pnas.0704357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guessous F, Yang Y, Johnson E, Marcinkiewicz L, Smith M, Zhang Y, et al. Cooperation between c-Met and focal adhesion kinase family members in medulloblastoma and implications for therapy. Mol Cancer Ther. 2012;11:288–297. doi: 10.1158/1535-7163.MCT-11-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer AN, Gastwirt RF, Schlaepfer DD, Donoghue DJ. The cytoplasmic tyrosine kinase Pyk2 as a novel effector of fibroblast growth factor receptor 3 activation. J Biol Chem. 2004;279:28450–28457. doi: 10.1074/jbc.M403335200. [DOI] [PubMed] [Google Scholar]
- 39.Fuhler GM, Brooks R, Toms B, Iyer S, Gengo EA, Park MY, et al. Therapeutic potential of SH2 domain-containing inositol-5′-phosphatase 1 (SHIP1) and SHIP2 inhibition in cancer. Mol Med. 2012;18:65–75. doi: 10.2119/molmed.2011.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kerr WG. Inhibitor and activator: dual functions for SHIP in immunity and cancer. Ann NY Acad Sci. 2011;1217:1–17. doi: 10.1111/j.1749-6632.2010.05869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–2526. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 42.Ara T, Nakata R, Sheard MA, Shimada H, Buettner R, Groshen SG, et al. Critical role of STAT3 in IL-6-mediated drug resistance in human neuroblastoma. Cancer Res. 2013;73:3852–3864. doi: 10.1158/0008-5472.CAN-12-2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Codony-Servat J, Marin-Aguilera M, Visa L, Garcia-Albeniz X, Pineda E, Fernandez PL, et al. Nuclear factor-kappa B and interleukin-6 related docetaxel resistance in castration-resistant prostate cancer. Prostate. 2013;73:512–521. doi: 10.1002/pros.22591. [DOI] [PubMed] [Google Scholar]
- 44.Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F, Ha SJ, et al. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancer Ther. 2012;11:2254–2264. doi: 10.1158/1535-7163.MCT-12-0311. [DOI] [PubMed] [Google Scholar]
- 45.Lin HY, Hou SC, Chen SC, Kao MC, Yu CC, Funayama S, et al. (-)-Epigallocatechin gallate induces Fas/CD95-mediated apoptosis through inhibiting constitutive and IL-6-induced JAK/STAT3 signaling in head and neck squamous cell carcinoma cells. J Agric Food Chem. 2012;60:2480–2489. doi: 10.1021/jf204362n. [DOI] [PubMed] [Google Scholar]
- 46.Shi Z, Yang WM, Chen LP, Yang DH, Zhou Q, Zhu J, et al. Enhanced chemo-sensitization in multidrug-resistant human breast cancer cells by inhibition of IL-6 and IL-8 production. Breast Cancer Res Treat. 2012;135:737–747. doi: 10.1007/s10549-012-2196-0. [DOI] [PubMed] [Google Scholar]
- 47.Voorhees PM, Chen Q, Small GW, Kuhn DJ, Hunsucker SA, Nemeth JA, et al. Targeted inhibition of interleukin-6 with CNTO 328 sensitizes pre-clinical models of multiple myeloma to dexamethasone-mediated cell death. Br J Haematol. 2009;145:481–490. doi: 10.1111/j.1365-2141.2009.07647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Authentication of Human Cell Lines: Standardization of STR Profiling ATCC Standards Development Organization, pp Designation: ASN-0002. Publication No ANSI/ATCC ASN-0002-2011. 2012 [Google Scholar]
- 49.Burel SA, Han SR, Lee HS, Norris DA, Lee BS, Machemer T, et al. Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucleic Acid Ther. 2013;23:213–227. doi: 10.1089/nat.2013.0422. [DOI] [PubMed] [Google Scholar]
- 50.Yarde DN, Oliveira V, Mathews L, Wang X, Villagra A, Boulware D, et al. Targeting the Fanconi anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res. 2009;69:9367–9375. doi: 10.1158/0008-5472.CAN-09-2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meads MB, Li ZW, Dalton WS. A novel TNF receptor-associated factor 6 binding domain mediates NF-kappaB signaling by the common cytokine receptor beta subunit. J Immunol. 2010;185:1606–1615. doi: 10.4049/jimmunol.0902026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang G, Fang B, Liu RZ, Lin H, Kinose F, Bai Y, et al. Mass spectrometry mapping of epidermal growth factor receptor phosphorylation related to oncogenic mutations and tyrosine kinase inhibitor sensitivity. J Proteome Res. 2011;10:305–319. doi: 10.1021/pr1006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.