

Summary
Aging is associated with the decline of protein, cell, and organ function. Here, we use an integrated approach to characterize gene expression, bulk translation, and cell biology in the brains and livers of young and old rats. We identify 468 differences in protein abundance between young and old animals. The majority are a consequence of altered translation output, that is, the combined effect of changes in transcript abundance and translation efficiency. In addition, we identify 130 proteins whose overall abundance remains unchanged but whose sub-cellular localization, phosphorylation state, or splice-form varies. While some protein-level differences appear to be a generic property of the rats’ chronological age, the majority are specific to one organ. These may be a consequence of the organ’s physiology or the chronological age of the cells within the tissue. Taken together, our study provides an initial view of the proteome at the molecular, sub-cellular, and organ level in young and old rats.
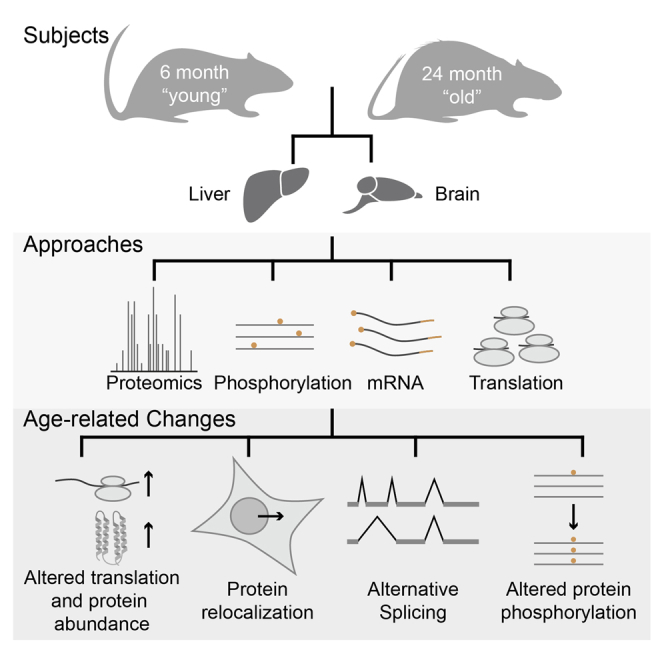
Graphical Abstract
Highlights
-
•
An integrated approach identifies molecular alterations between young and old rats
-
•
Changes in translation output explain the majority of the altered protein abundances
-
•
Key protein complexes are altered in abundance and composition
-
•
We provide a rich data resource to stimulate further studies of aging
Ori et al. quantified the molecular alterations that occur between young and old rats in two organs: brain and liver. By integrating genomic and proteomic measurements, the authors were able to reveal that changes in translation are the primary cause of protein level alterations during aging. However, they also identified other levels of regulation such as protein localization and phosphorylation that co-participate in modifying the proteome in old animals.
Introduction
Aging is a multifactorial process that is associated with a progressive loss of physiological integrity, resulting in functional decline and increased morbidity. A number of studies have monitored system-wide changes in gene expression associated with aging in mammals (de Magalhães et al., 2009) and identified common and organ-specific transcriptional changes (Schumacher et al., 2008). For example, changes in transcript identity and abundance have been studied in brain across various species including mice, rats, and humans (Jiang et al., 2001, Lee et al., 2000, Lu et al., 2004, Wood et al., 2013). These studies underline that on a global scale, aging organs are not subject to massive alterations; rather, alterations are subtle. Consistently, changes in age-related expression account for only a small fraction of the genes monitored in all species analyzed, generally <5%. The regulatory changes that are conserved across species are reduced even further and restricted to very few genes involved in calcium signaling and other synaptic functions (Loerch et al., 2008). Similarly, ribosomal profiling has revealed that the vast majority of genes were changed <2-fold in their translation levels in young and old animals (98% of 11,000 quantified genes) (Toyama et al., 2013). Furthermore, a previous large-scale study that used state-of-the-art shotgun proteomics to investigate various tissues of aging mice concluded that the vast majority of proteins are unchanged in abundance (Walther and Mann, 2011). Therefore, it remains unclear whether the described changes in transcriptional programs are at all reflected at the proteome level and, if so, whether other types of regulation contribute to the deterioration of the proteome during aging.
There are many ways to alter a protein’s function without changing its abundance. Function can be modified by altering protein structure, post-translational modifications, or localization within a cell. For example, tissues with low cell proliferation rates and thus low regenerative capacity, such as brain, contain a subset of proteins with extremely long half-lives (Savas et al., 2012, Toyama et al., 2013) that are more vulnerable to damage accumulation and loss of function, phenomena that might not be observable by measuring protein abundance. Degenerated proteins might also be unable to maintain their proper subcellular localization in old cells (David et al., 2010, Kaganovich et al., 2008), thus perturbing signaling pathways and gene expression programs that alter post-translational modifications and mRNA levels as a secondary response. To study molecular-level changes like these and to identify correlations between them, it is necessary to combine several system-wide technologies, thereby simultaneously monitoring levels of protein, mRNA, and post-translational modifications, as well as translation rates, all within a single biological specimen.
In this study, we combined shotgun mass spectrometry with subcellular fractionation, RNA sequencing, and ribosome profiling to investigate physiological changes in the liver and brain of young and old rats (Figure 1A). We demonstrate that this integrative analysis consistently identifies subtle alterations within old cells that would not become apparent using any of the single techniques alone. The system-wide comparison of the brain, which has a limited capacity for cell renewal, and the liver, which has more robust renewal capabilities, allowed us to identify specific alterations (in protein kinases and mediators of signaling in brain, and metabolic processes in liver) as well as common responses (inflammation and activation of immune response) that differentiate organs from young and old animals. Our work captures unappreciated differences in the biology of young and old cells. We identify changes in the abundance and phosphorylation patterns of individual protein species with subcellular resolution. We show that these differences are driven by changes in translation, perturbed subcellular protein localization, protein phosphorylation and alternative splicing.
Figure 1.

Integrated Genomics and Proteomics Analysis of Aging Brain and Liver
(A) Brain and liver samples obtained from three “young” (6-month-old) and three “old” (24-month-old) rats were compared by next-generation RNA sequencing, ribosomal profiling and shotgun mass spectrometry. Prior to mass spectrometry, organ homogenates were fractionated in four subcellular fractions: nuclei (nuc), post-nuclear fraction 1 (pn1; enriched for mitochondria), post-nuclear fraction 2 (pn2; enriched for cytosolic membranes) and soluble cytosolic proteins (sol). For proteomics measurements, each sample was analyzed in technical duplicate (repeated injection of the same sample).
(B) Reproducibility of protein abundance measurements. The histogram shows the distribution of pairwise correlations between all technical replicates for brain and liver.
(C) The boxplots depict all the pairwise correlations between samples from all the subcellular fractions.
(D and E) Less than 10% of the quantified transcript and protein groups were affected in young versus old rats, both in brain (D) and liver (E). Differential protein expression was assessed by label-free quantification and significantly affected proteins were defined using a q value cut-off of 0.1. Additionally, proteins that were identified exclusively and consistently in one age group but not the other were considered as age-affected (dark orange). Translation output was measured by ribosome footprints. Significantly affected transcripts were defined using an adjusted p value cut-off of 0.01.
(F) Significantly affected transcripts were grouped according to whether they were affected by a change in transcript abundance (as quantified by RNA-seq), translation efficiency or by a combined effect.
(G) Established aging markers were recovered at the protein level. Histones H3.1 and H3.3 were quantified using proteotypic peptides measured by targeted proteomics (see Supplemental Experimental Procedures). The ratio between the two proteins is displayed relative to its value in young animals (set to 1) and it represents the average value ± SEM (n = 3 animals per age group). For beta-galactosidase and alpha-synuclein, protein abundances are indicated as average abundance score (sum of peptide intensities normalized by protein molecular weight) ± SEM (n = 3 animals per age group).
See also Figures S1 and S2 and Tables S1 and S2.
Results
To identify age-related molecular changes in the liver and brain, we sacrificed three “young” (6-month-old) and three “old” (24-month-old) rats from multiple litters, the latter of which represent old, but not dying, animals at the age of 50% expected survival. Brain and liver were harvested and identical samples were split for analysis at the transcription, translation and proteome levels by next-generation RNA sequencing, ribosome profiling and tandem shotgun mass spectrometry, respectively (Figure 1A). The mRNA abundance and translation output (total ribosome footprint reads) of 8,975 and 6,490 transcripts were compared between young and old animals in brain and liver, respectively, and changes in translation efficiency were inferred by comparing these measured values (Table S1). To increase proteomic coverage and obtain insights into subcellular localization, brain and liver samples were fractionated into nuclei, post-nuclear fraction 1 (pn1; enriched for mitochondria), post-nuclear fraction 2 (pn2; enriched for cytoplasmic membranes) and soluble cytosolic proteins (sol) according to a previously established procedure (Blobel and Potter, 1966, Lovtrup-Rein and McEwen, 1966). All samples were analyzed by shotgun mass spectrometry, enabling us to perform 14,131 comparisons of protein abundances across two age groups and four subcellular fractions, covering 4,714 protein groups (i.e., collections of alternative protein isoforms containing shared peptides) mapping to 4,697 unique gene identifiers (Table S2).
The proteomic measurements were highly reproducible, as indicated by the high correlation between technical replicates (on average Pearson’s r = 0.974; Figure 1B). More importantly, correlation values across biological replicates of different age groups were significantly lower as compared to samples from the same age group (Wilcoxon rank sum test p value 2.5e-5 and 8.1e-5 for brain and liver, respectively; Figure 1C), demonstrating that the variation of protein abundance across age groups is more pronounced than the variation of protein levels across individuals of the same age. At the same time, the observed coefficients of variation among both, young and old animals were very low (median coefficient of variation ∼25%; Figure S1A). Similarly, both ribosome profiling and RNA sequencing (RNA-seq) were highly reproducible, as indicated by high correlation between replicates (Figures S2A, S2B, S2D, and S2E). We thus conclude that the measured changes in protein and transcript abundance, and ribosome occupancy significantly discriminate samples obtained from the two age groups with the number of analyzed animals (n = 3 for each age group) using rigorous statistics (Figures S1B, S1C, S2C, and S2F).
In agreement with previous studies (Jiang et al., 2001, Lee et al., 2000, Lu et al., 2004, Toyama et al., 2013, Walther and Mann, 2011, Wood et al., 2013), our data underline that age-specific variations of the transcriptome and proteome are much less pronounced than tissue-specific differences (Figure S1D). The majority of genes and proteins (>90%) are stably expressed and maintained in both the brains and livers of old animals (Figures 1D and 1E). Ribosome profiling provides an inclusive measure of “translation output” that reflects the net effect of changes in mRNA levels and ribosome occupancy. We identified 658 and 490 transcripts, in brain and liver respectively, with changes in translation output (adjusted p value < 0.01, Table S1). Of these, 168 (brain) and 283 (liver) were caused exclusively by changes in mRNA abundance. The “translation efficiency” of an mRNA is the overall translation output from ribosome profiling normalized over the transcript abundance from RNA-seq. Among differentially expressed transcripts, 96 (brain) and 9 (liver) displayed exclusively a change in translation efficiency but not in transcript abundance (Figure 1F). In order to directly relate changes in protein production to the observed alterations of protein abundance, we compared our translation output values from ribosome profiling to our proteomic measurements. At the proteomic level, significant abundance changes for 204 protein groups in young versus old organs (q value < 0.1, Table S2) were detected. A total of 264 proteins were identified exclusively and consistently in all three replicates of one age group but in no replicates of the other age group; these were considered as potentially affected (Table S2). We conclude that hundreds of genes and proteins were identified that differentiate young and old organs and constitute a comprehensive resource for the scientific community to query molecular alterations of cells in their physiological ground state during aging in mammals.
Age Markers Were Consistently Identified
The sensitivity of our approach recovers alterations of previously established age markers. A global decrease in the abundance ratio between histone H3.1 and H3.3 was observed in both old organs (Figure 1G). Histone H3.1 is incorporated in nucleosomes only during mitosis (Wu et al., 1982) and is replaced by the H3.3 variant outside of mitosis, typically during transcription (Ahmad and Henikoff, 2002, Schwartz and Ahmad, 2005). Thus, in a tissue that is largely post-mitotic such as the brain, the H3.1/H3.3 ratio is expected to decrease with age (Maze et al., 2015); this is what we observe. We also observed a 2-fold increase in the abundance of the enzyme beta-galactosidase in old brain (Figure 1G), a widely used marker of cellular senescence (Dimri et al., 1995). Concurrently, we detected beta-galactosidase in old liver but not young (Table S2). A decreased level of alpha-synuclein, a protein involved in synaptic plasticity and neurodegenerative disorders, was also detected in brain (Figure 1G), corroborating a previous study in mice (Mak et al., 2009). Additionally, we detected significant changes at the level of protein abundance or translation output for 97 age-related factors (Hühne et al., 2014) (Tables S1 and S2). We conclude that our integrative analysis recapitulates trends of several known age markers but also identifies a large and comprehensive set of transcripts and proteins that are linked to age-specific alterations for the first time.
Identification of Common and Organ-Specific Alterations
Liver and brain have different regenerative capacities. Most neurons in the adult brain are non-dividing cells that must survive for an organism’s lifespan (Spalding et al., 2005). In contrast, liver cells, such as hepatocytes, are replaced every few months throughout adult life in rodents (Arber et al., 1988, Toyama et al., 2013). One would thus predict that age-related effects onto the proteome are organ-specific. A comparison of proteomic changes across the different biochemical fractions supports this hypothesis: we identified the highest number of differentially expressed proteins in the nuclear fraction of the brain (7% of nuclear proteins affected: Figure 1D), whereas in the liver the most changes were found in the cytosolic fraction (5% of cytosolic proteins affected: Figure 1E). Overall, a larger fraction of the proteome was affected in brain, the tissue with lower regenerative capacity: 8% of all quantified proteins versus 5% in liver (Figures 1D and 1E).
The identities of the transcripts and proteins affected were also indicative of organ-specific differences between young and old animals. Out of 1,099 transcripts abundances that were altered, only 48, considerably less than expected (Fisher’s exact test: p value < 0.0001, odds ratio 0.04), were common to brain and liver and 37 of them (77%) changed concordantly (Figure 2A). Most of these transcripts are involved in the regulation of the immune system and stress response (Table S1). The protein abundance data showed a similar trend: only eight out of the 447 affected protein groups are common to brain and liver (Fisher’s exact test: p value < 0.001, odds ratio 0.02; Figure 2B).
Figure 2.

Common and Organ-Specific Alterations of the Proteome in Old Rats
(A and B) We identified only 48 transcripts affected at the level of translation output (A) and eight proteins affected at the level of protein abundance (B) that were altered in both brain and liver. The most represented gene ontology (GO) terms among consistently affected transcripts are shown in (A) while the five consistently affected proteins are indicated in (B).
(C–F) Functional enrichment was performed on the list of quantified transcripts and proteins that were ranked according to the level of differential expression (fold change) using GOrilla (Eden et al., 2009). Displayed GO terms are representative cases selected from among those significantly enriched (cut-off for transcripts: q value < 0.05, minimum number of transcript >4, fold enrichment ≥2; for proteins the same criteria were applied with the exception of q value < 0.2). The fold enrichments are plotted using positive values for terms enriched in transcripts/proteins that are increased in old animals, or using negative values for terms enriched in transcripts/proteins that are decreased in old animals.
Gene ontology analysis confirmed that different functional modules are affected in brain and liver, but also underlined the existence of common aspects of altered protein function in old tissues. Commonalities include the enrichment of altered genes encoding for factors involved in cell communication, hormone response, immune response/inflammation, and the depletion of respiratory chain components (Figures 2C–2F; Table S3). The latter, previously described at the mRNA level across multiple organs and species (Zahn et al., 2006), likely arises from mitochondrial dysfunction and might be accompanied by an increase of reactive oxygen species and generation of pro-inflammatory signals (Green et al., 2011). Another common feature is the regulation of proteins involved in post-translational modifications, although different pathways are affected in the two organs. Whereas several protein kinases (Figure S3A) and methyltransferases are altered in brain (Figure 2D), it is largely acetyltransferases that are affected in liver (Figure 2F). We speculate that some of these alterations might be responsible for the changes in the epigenetic landscape described in aged cells (Rhie et al., 2013, Sun et al., 2014).
Organ-specific alterations appear to be linked to their physiology. In brain, alterations of neuronal communication and synaptic transmission occur at multiple levels: we observed depletion of intracellular mediators of signaling such as protein kinases, multiple ion channels and G protein-coupled receptors, in the brains of old rats (Figure 2C). In liver, several metabolic networks are altered (Figures 2E and 2F). In particular, we observed an increased abundance of enzymes involved in pyruvate metabolism and the tricarboxylic acid cycle, lipid catabolism (in particular fatty acid oxidation), and maintenance of redox homeostasis (Figure 2F). These alterations are consistent and might underlie the metabolic changes that have been observed in old liver in mice (Houtkooper et al., 2011).
In summary, by providing a detailed picture of the molecular alterations that distinguish young and old organs, our data suggest a molecular basis for several hallmarks of aging including mitochondrial dysfunction, increased inflammation, and changes in regulators of the epigenetic landscape (López-Otín et al., 2013).
Protein Complexes Are Affected at Multiple Levels
We have previously shown that the composition of the nuclear pore complex (NPC) is altered during aging through the loss of long-lived scaffold components (D’Angelo et al., 2009, Toyama et al., 2013), affecting its permeability barrier. We wanted to investigate the maintenance of protein complexes more generally and found that the alterations of the proteome that occur between young and old animals affect protein complexes in two different ways.
First, the overall abundance of some protein complexes is different in young and old rat. Consistent with a previous study in mice and worms (Houtkooper et al., 2013), we observed that the abundance of components of the mitochondrial ribosome was lower in old versus young brain (Figure 3A). This phenomenon was not observed in the liver. However, other protein complexes in liver were similarly affected by age: the abundance of cytosolic proteasomes was higher in old liver versus young (Figure 3B) and the abundance of NPCs was lower (Figure 3C). Such changes might result in a reduced functional output and longevity, as in the case of the mitochondrial ribosome (Houtkooper et al., 2013).
Figure 3.

Proteomic Changes Affect the Abundance and Composition of Protein Complexes
(A–C) We analyzed changes in protein complex abundance using a gene-set enrichment approach (see Supplemental Experimental Procedures). Definitions covering 270 large protein complexes curated from different resources and the literature (A.O., M.I., et al., unpublished data) were used. Only protein complexes that had at least five members quantified were considered. A q value cut-off of 0.1 was used to determine significantly affected protein complexes. All proteins identified in the respective sample are plotted according to their average abundance score and average fold change between young and old animals (both log2-transformed). Positive values indicate higher expression in old animals and negative values indicate higher expression in young animals. Orange dots indicate the identified members of the affected complex. Boxplots show the distribution of abundances of complex members in young and old animals.
(D) Changes in protein abundance affect the composition of several protein complexes in old rats. For each selected example, the quantified complex members are displayed and the significantly affected cases are highlighted in red (indicating increased abundance in old animals) or blue (indicating decreased abundance in old animals). Compositional changes were inferred from protein abundance scores obtained by tandem mass spectrometry as previously described (Ori et al., 2013). A q value cut-off of 0.2 was used to determine significantly affected protein complex members.
See also Table S4.
Second, a subset of protein complexes undergoes compositional changes in young versus old animals. Using algorithms that we have previously developed (Ori et al., 2013), we identified changes in protein abundance for specific members of complexes involved in chromatin regulation (e.g., polycomb repressive complex I), RNA processing and transport (e.g., TREX and exon junction complex), and complexes involved in vesicular transport (e.g., COPI, COPII, retromer complex: Figure 3D; Table S4). Our data suggests that changes of composition that occur in old animals might cause loss of protein complex function due to complex misassembly, as described for the nuclear pore (D’Angelo et al., 2012, Lessard et al., 2007), or might mediate adaptation of its functionality, for example, under increased stress conditions.
Translation Output Contributes Significantly to Proteomic Alterations
The combination of ribosome profiling and shotgun proteomics data allowed us to determine the contribution of translation output, and thus protein synthesis, to alteration of protein abundances (Battle et al., 2015, Guo et al., 2010). Globally, we found that the changes in translation output between young and old animals are reflected by consistent changes in protein level. We categorized all transcripts into three groups: significantly increased, significantly decreased, and not affected at the level of translation output. The corresponding proteomic fold changes of altered transcripts were shifted substantially and significantly relative to transcripts without evidence for significant translational change (Figures 4A and 4B; Table S5).
Figure 4.

Changes in Translation Correspond to Changes in Protein Abundance
(A and B) The distribution of the corresponding protein changes for transcripts showing increased (red) or decreased (blue) translation output in old animals is significantly and consistently shifted relative to that of unaffected transcripts (gray).
(C and D) Changes in translation output and protein abundance are positively correlated despite the limited overlap in cases that rise to significance using both methods. We assessed the correlation between translation and protein abundance by calculating the Spearman’s rank correlation coefficient between the respective fold changes: 0.13 and 0.25 for brain and liver, respectively. The correlation coefficients increased to 0.37 for brain and 0.42 for liver when only significant cases (either at the translation or protein level) were taken into account.
(E and F) The distribution of the corresponding changes in translation for proteins increased (red) or decreased (blue) in old animals is significantly and consistently shifted relative to the distribution of unaffected proteins (gray).
(G) The chaperonin alpha-crystallin B is shown as an example of a protein that is affected at the level of translation output, and it is consistently changed at the protein level in multiple subcellular fractions.
See also Table S5.
Notably, for ∼75% of these transcripts, the changes in translation output and protein abundance are concordant in directionality (Figures 4A and 4B, dashed lines). Also, the observed fold-changes in translation output and protein abundances in both brain and liver were positively correlated, meaning that we see a correlation between the changes in the abundance of a protein, as assessed by proteomics, and the abundance and/or translation of the mRNA that encodes it in our ribosome profiling experiments (Spearman’s rank correlation coefficient between translation and protein abundance fold changes: 0.13 and 0.25 for brain and liver, respectively: Figures 4C and 4D). Conversely, if proteins are categorized into groups that are significantly increased, significantly decreased, and not affected in their abundance, their respective fold changes in translation output are similarly shifted, although with smaller effect size (compare Figures 4E and 4F to 4A and 4B).
Taken together, this analysis suggests that differences in protein synthesis (translation output) give rise to an appreciable fraction of the observed changes in protein abundance that discriminate young and old organs (Figures 4E and 4F). This overall correspondence emerges despite limited overlap between datasets (Figures 4C and 4D), suggesting that many genes might be showing subtle changes in synthesis and abundance that do not rise to statistical significance when studied with only one of the techniques. We show that on a global scale, changes in translation output—although small—impact on protein abundance between young and old animals, as exemplified by the increase of chaperonin alpha-crystallin B in old brains (Figure 4G).
Altered Protein Localization
The data integration also revealed exceptions from the overall trend, in which significant changes in protein abundance could not be explained by changes in translation output, particularly in brain (Figures 4G and 5A). These discrepancies point to alternative mechanisms that differentially control protein abundance across age groups, such as protein degradation, or changes in protein localization. In the latter case, the increased abundance of a protein in one compartment might be counterbalanced by a decrease in another, leading to a signal we can observe in our subcellular fractions that would not be detectable by proteomics on total lysates or measurements of translation output. Indeed, we observed opposite abundance changes in different subcellular fractions for nine proteins—seven in brain and two in liver. We interpreted these data as indicative of change in subcellular localization. The potentially relocalized proteins include RNA and protein-modifying enzymes, proteins involved in translation, and nuclear transport factors (Figure 5B). For example, exportin 5, which interacts with NPCs during the export of miRNAs (Lund et al., 2004) and tRNAs (Calado et al., 2002), shows an increased abundance in the cytoplasm of cells from old brains, implying a potential alteration of nuclear transport activity. Two protein kinases—TRAF2- and NCK-interacting kinase (TNIK) and brain-specific kinase (BRSK1, also referred to as SAD-B)—also show proteomic alterations suggestive of subcellular redistribution with no changes in translation output. Whereas TNIK showed an increased abundance at cytosolic membranes, BRSK1 was less abundant there and more in soluble cytosol of old brains (Figure 5A). These potential relocalization events highlight an additional level of proteome alteration between young and old animals. Our data, along with the previously described age-related deterioration of the NPC (D’Angelo et al., 2009), suggest a remodeling of the protein and RNA transport machineries that is associated with animal age.
Figure 5.

Changes in Protein Localization and Phosphorylation
(A) The intracellular distribution of the kinase BRSK1 changes between young and old brain. BRSK1 shows significant changes in protein abundance with opposite signs in two distinct subcellular fractions, whereas it is not affected at the level of translation output.
(B) Another eight proteins display behavior similar to BRSK1, suggesting that their intracellular distribution might change in young versus old animals.
(C and D) Changes in phosphopeptide abundance were assessed by label-free quantification using the same procedure used for protein quantification (q value < 0.1). In addition, phosphopeptides identified exclusively and consistently in one age group but not the other were considered as age-affected (dark orange).
(E) Comparison of functional annotations between affected phosphosites in brain and liver. Affected phosphosites were annotated using the GO slim annotation associated to the corresponding protein group using QuickGO (Binns et al., 2009). Annotations were compared by selecting terms that were at least 1.5 times more frequent among affected phosphosites in one of the two organs. Only the 15 most represented categories per organ were considered.
(F) We identified a decreased level of phosphorylation of several microtubule-associated proteins (MAPS) in membrane fractions that mimics the relocalization of BRSK1 (A).
Changes in Protein Phosphorylation
Changes in post-translational modifications, such as phosphorylation, have been shown to drive cancer (Krueger and Srivastava, 2006) and a series of developmental processes (Huang and Reichardt, 2001, Sancho et al., 2004). However, in the context of aging, evidence for alterations of post-translational modifications remain largely anecdotal. We identified several kinases that are differentially expressed between young and old animals, particularly in the brain (Figure S3A). In total, we found perturbations in the levels of 12 protein kinases belonging to different families. These include the major kinases that control cell growth, neuronal morphogenesis and plasticity such as the beta-adrenergic receptor kinase 1, the calcium/calmodulin-dependent protein kinases (CAMK) types I and IV, the cyclin-dependent kinases 5 and 19, and the serine/threonine-protein kinase mTOR (Figure S3A).
We next tested whether these changes in kinase levels impact downstream target phosphorylation levels by performing a phospho-proteomic analysis of all subcellular fractions, tissues, and age groups described above. Similarly to the protein abundance measurements, we observed high reproducibility between technical replicates (on average Pearson’s r = 0.957; Figure S4A), lower correlation values across age groups as compared to samples from the same age group (Figure S4B), and median coefficient of variation ∼25% (Figure S4C). Across all four subcellular fractions and two organs, we made 2,497 comparisons of phosphosite abundance, covering 1,437 unique phosphosites, of which 75 (occurring on 68 proteins) were found to change significantly (q value < 0.1; Figures 5C and 5D; Table S6). In addition, we found 168 phosphosites (on 160 proteins) that were uniquely and consistently identified in all the replicates of one age group but not the other (Table S6). For 136 affected phosphosites, we covered both changes in protein abundance and phosphorylation state on the same set of samples and thus could infer changes in the fraction of protein molecules phosphorylated (Figure S4D). In 19 out of the 136 phosphosites both the protein and phosphosite level changed in the same organ and subcellular fraction. In 17 of these 19 cases the fold changes were consistent (Figure S4E), implying that the phosphorylation state of the protein is not changed. In contrast, 2 of the 19 phosphosites had opposing fold changes, and the remaining 117 phosphosites had not-affected protein abundances while phosphosite level changed. The latter two scenarios are indicative of an alteration of the fraction of protein molecules phosphorylated (Figure S4D).
In liver, we found effects on the phosphorylation of proteins involved in metabolic processes and energy production, similarly to the biological processes impacted at the protein abundance level (Figure 5E). However, we also identified several altered phosphosites in transcriptional regulators, including c-JUN and FOXA1, as well as proteins involved in stress responses and homeostatic processes. Similarly to the observed protein abundances, a larger fraction of phosphosites changed in brain as compared to the liver, which is consistent with the large number of protein kinases affected in brain. In addition to those kinases affected at the protein level, we also found changes in phosphorylation of a different subset of kinases (Table S6), possibly because their activity is regulated through their phosphorylation status. Interestingly, the affected phosphosites were not equally distributed between cell compartments of brain, but primarily identified in membrane (pn2) and nuclear fractions (Figures 5C and 5D). A very prominent fraction of altered phosphosites was detected in cytoskeletal proteins (mostly in pn2), in particular in multiple microtubule-associated proteins (MAPS) (Figure 5F). We speculate that this observation is related to the above-discussed redistribution of BRSK1 kinase away from membranes in old brain (Figure 5A). BRSK1 is known to associate with synaptic vesicles (Inoue et al., 2006) and it is required to control the polarization of neurons (Kishi et al., 2005) through a mechanism that ultimately leads to the phosphorylation of MAPS (Barnes et al., 2007). While this specific hypothesis remains to be tested, the overall dataset suggests that altered protein phosphorylation and localization might have important physiological outcomes in old animals.
Multiple Levels of Regulation Modulate Functional Networks between Young and Old Animals
The integration of multiple measurements on the same sample provides a powerful approach to capture functional modules that discriminate between different cellular states. To take advantage of this, we combined genes affected at the level of translation, protein abundance and protein phosphorylation and used them to reconstruct functional networks (see Supplemental Experimental Procedures) that are altered between young and old animals (Figure 6). Specifically in brain, we identified three related networks that are involved in signal transduction. The majority of the nodes in these networks displayed reduced translation output or protein abundance in old animals. Among these, several mediators of the calcium signaling pathway, such as the kinases CAMK1D and CAMK4, were reduced in their protein abundance, while the kinases CAMK2D and CAMK2G were affected in their phosphorylation state (Figure 6A). We found up-stream mediators of calcium signaling to be affected, including several voltage-gated channels and calcium transporters, as well as down-stream effectors such as the CREB-regulated transcription coactivator 1 (CRTC1; Figure 6A). Taken together, our data suggests that the response to stimuli might be modified at multiple levels in the brain of old animals, mainly through the depletion of several factors involved in mediating calcium signaling.
Figure 6.

The Impact of Age on Functional Networks Occurs at Multiple Levels of Regulation
(A and B) Selected functional networks that are altered between young and old rats are displayed. The node fill color indicates significant fold change in protein abundance and the node border color indicates significant fold change in translation output. Changes in phosphorylation status are represented by the circled letter “P.” Increased abundance in old animals is indicated in red and decreased abundance is indicated in blue.
See also Figures S5 and S7.
We also found two related functional networks that are common to both brain and liver. These clusters comprise multiple mediators of the immune response such as proteins involved in antigen processing and presentation, including all the three components of the C1q complex that are involved in the first step of the classical complement activation pathway by direct recognition of pathogens or antibody:antigen complexes (Figures 6A and 6B). As many of these proteins are generally not expressed in hepatocytes and neuronal cells, such as CD4, we speculate that the observed abundance increase derives from an increased number of immune cells that are recruited into the old organ by pro-inflammatory stimuli.
Alternative Splicing
Another potential source of age-related alterations of the cellular proteome is the process of alternative splicing, which results in a single gene coding for multiple proteins (Wood et al., 2013). Inspired by the fact that multiple factors involved in the regulation of alternative splicing and RNA processing were affected in old animals (Figure 6A), we analyzed potential differences in mRNA splicing between young and old animals (see Supplemental Experimental Procedures). We found significant differences in the expression of 41 and 61 transcript isoforms from 24 and 39 genes in brain and liver, respectively (Figure S6; Table S7). Among these were two isoforms in the brain of Sgk1, a serine/threonine protein kinase that plays an important role in cellular stress response and two isoforms in the liver of Whsc1, a histone H3K36 N-methyltransferase (Figure 7A). A longer isoform (ENSRNOT00000016121) of Sgk1 predominates in old brain, whereas a shorter isoform (ENSRNOT00000040736) is equally prevalent in young animals (Figure 7B). Comparison of the number of isoform-specific reads suggests that this change is primarily due to a large increase in the levels of the long isoform with age (Figure 7C). In liver, the short isoform (ENSRNOT00000050238) of Whsc1 is more prevalent in young animals compared to the long isoform (ENSRNOT00000021952), whereas in old animals the two isoforms are expressed at similar levels (Figure 7B). For Whsc1, this is due primarily to an increase in expression of the longer isoform with age, bringing it to a similar expression level as the short isoform (Figure 7C). Our analysis of alternative splicing demonstrates that there are age-related changes in the abundance of specific transcript isoforms with distinct coding potential, which might result in functional proteome diversification in old organs.
Figure 7.

Alternative Expression of Splicing Isoforms
(A) Sgk1 and Whsc1 each have two isoforms with substantial expression in brain and liver, respectively. The lengths of Whsc1 introns have been scaled down by a factor of 2 for display purposes.
(B) A comparison of percent spliced-in (PSI) values generated by MISO (Katz et al., 2010) shows a significant difference in isoform expression levels between young and old animals (for each isoform, Bayes factor ≥1012). Error bars represent 95% confidence intervals.
(C) Isoform-specific expression (reads per kb, normalized for overall sequencing depth) shows the underlying changes in isoform abundances responsible for differences in PSI values.
Discussion
Here, we present an integrated comparison of gene expression, translation, protein abundance, and phosphorylation in organs from young and old rats. Our work expands the list of proteins that are affected by chronological age in mammals. Although some of the functional modules discussed above were previously identified as hallmarks of aging (López-Otín et al., 2013), we identified hundreds of molecular events underlying these processes that were previously unknown to be affected by age. We thus provide a rich resource that should stimulate the generation of new, experimentally testable hypotheses, leading to a better understanding of aging on the organism level.
The comparison of two organs with different physiology and regenerative capacity enabled us to distinguish organ-specific effects from more systemic effects of aging. Intuitively, our results suggest that organ-specific effects of age are tightly linked to the organ function. For example, in brain, multiple alterations of key signaling mediators are observed. We speculate that these alterations might be part of a progressive functional deterioration that affect the maintenance of neuronal plasticity in old brains (Bading, 2013) and other phenotypes observed the aging brain (Burke and Barnes, 2006). Notably, 45 of the changes that we identified in old rat brains are consistent with a previous transcriptomics study of aging human brains (Lu et al., 2004) (Figure S7), suggesting that age-related changes in the proteome and transcriptome are to some extent conserved from rat to humans.
The systemic impact of chronological age on proteome homeostasis manifests on many levels. In the liver, the majority of age-dependent changes are driven by alteration of transcript abundance (58% of the affected transcripts versus only 25% in brain; Figure 1F), suggesting the occurrence of age-related changes in transcriptional regulation. In contrast, the brain appeared to be affected by age largely at the translational level. For example, specifically in aged brain, we observed that the translational output of multiple ribosomal subunits increased (Figure 6A); these subunits were also more abundant on the protein level (Figure S5). A similar effect of orchestrated ribosomal gene expression was described throughout the lifespan of the short-lived fish Nothobranchius furzeri (Baumgart et al., 2014), suggesting that this phenomenon might represent a conserved feature of the aging brain. Conversely, a decreased protein level of multiple factors involved in translation initiation accompanied the increased abundance of ribosomal subunits (Figure 6A). Taken together, our data suggest that an age-associated remodeling of the translation machinery in the brain may ultimately lead to alterations of the translation efficiency of a subset of transcripts in old animals. Specifically, we identified 15% of the brain transcripts to be affected by a change in translation (versus only 2% in liver).
Despite the correlation between translation output and protein abundances, not all the observed changes of protein abundance could be explained by changes in translation output, particularly in brain (Figure 4E). This phenomenon strongly indicates a higher degree of post-translational control in the brain as compared to the liver. Indeed, our proteomic analysis revealed that key regulators of protein homeostasis were altered in aged brain, including several components of the ubiquitin-proteasome and autophagy systems (Figure S3B). These findings imply that altered protein homeostasis, which has been shown to affect organism longevity under stress-response conditions (Kevei and Hoppe, 2014), also leads to detectable proteomic alterations that occur between young and old animals. The exact consequences and targets of such alterations are likely complex and remain to be explored in detail. In addition, more fine-grained genomic and proteomic investigations at multiple time points across organism life-span are required to shed light on the dynamics and interplay between systemic and organ-specific effects of aging and thus reveal which alterations are causative of, or a result of, aging.
From our measurements performed on the bulk organ lysate, it is impossible to estimate whether the observed changes affect most of the cells within an organ or whether they originate from a subpopulation of cells that is more prone to the effects of aging or from a change in the proportion between cell types in an organ. Most of the proteomics changes that we describe at the level of both protein abundance and phosphorylation level are quite large (typically >2-fold). It is therefore reasonable to assume that they might affect a major proportion of cells. However, we cannot exclude that events occurring in a minor fraction of cells will be missed by our approach. Higher resolution studies focusing on specific anatomical regions or cell subpopulations will be required to detect more subtle alterations.
Experimental Procedures
Tissue Fractionation
Liver and brain were harvested from three Fischer 344 rats for each age group. Nuclei were purified according to previously described protocols (Blobel and Potter, 1966, Lovtrup-Rein and McEwen, 1966). Further fractionation was based on previous descriptions (Toyama et al., 2013).
Determination of Transcription and Translation Changes
RNA-seq and ribosome profiling libraries were prepared using an Illumina TruSeq kit and standard ribosome profiling protocols (Ingolia et al., 2009, Ingolia et al., 2012), and sequenced using the Illumina HiSeq platform. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO: GSE66715 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66715).
Determination of Protein and Phosphosite Abundance Changes
Proteins from subcellular fractions were solubilized and digested into peptides as described in Ori et al. (2014). For phosphoproteome analysis, phosphopeptides were isolated from peptide mixtures by TiO2-based affinity enrichment using the Titansphere Phos-TiO Kit (MZ Analysentechnik) as described in Bui et al. (2013). All samples were measured by shotgun mass spectrometry (see Supplemental Experimental Procedures). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) (Vizcaíno et al., 2014) via the PRIDE partner repository (Vizcaíno et al., 2013) with the dataset identifier PXD002467.
Author Contributions
A.O., B.H.T., M.S.H., N.T.I., M.W.H., and M.B. designed experiments and wrote the manuscript. A.O., B.H.T., M.S.H., and T.B. performed experiments and analyzed data. M.I. and P.B. designed algorithms for protein complex analysis. M.W.H. and M.B. oversaw the project.
Acknowledgments
We gratefully acknowledge support from EMBL’s Proteomics Core Facility, Centre for Statistical Data Analysis, in particular Dr. Bernd Klaus, and the Centre for Biomolecular Network Analysis, in particular Dr. Matt Rogon. We thank Drs. Oliver Rinner and Lukas Reiter for access to the software SpectroDive (Biognosys AG). A.O. was supported by postdoctoral fellowships from the Alexander von Humboldt foundation and Marie Curie Actions. T.B. was supported by EMBL Interdisciplinary Postdoc Programme (EIPOD) under Marie Curie Actions COFUND. N.T.I. is supported from the Searle Scholars Program (11-SSP-229) and the Paul F. Glenn Center for Aging Research. M.B. acknowledges funding by EMBL.
Published: September 17, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.cels.2015.08.012.
Contributor Information
Nicholas T. Ingolia, Email: ingolia@berkeley.edu.
Martin W. Hetzer, Email: hetzer@salk.edu.
Martin Beck, Email: mbeck@embl.de.
Accession Numbers
The accession number for the data reported in this paper is GEO: GSE66715 and ProteomeXchange: PXD002467.
Supplemental Information
References
- Ahmad K., Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell. 2002;9:1191–1200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- Arber N., Zajicek G., Ariel I. The streaming liver. II. Hepatocyte life history. Liver. 1988;8:80–87. doi: 10.1111/j.1600-0676.1988.tb00972.x. [DOI] [PubMed] [Google Scholar]
- Bading H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013;14:593–608. doi: 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- Barnes A.P., Lilley B.N., Pan Y.A., Plummer L.J., Powell A.W., Raines A.N., Sanes J.R., Polleux F. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–563. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Battle A., Khan Z., Wang S.H., Mitrano A., Ford M.J., Pritchard J.K., Gilad Y. Genomic variation. Impact of regulatory variation from RNA to protein. Science. 2015;347:664–667. doi: 10.1126/science.1260793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart M., Groth M., Priebe S., Savino A., Testa G., Dix A., Ripa R., Spallotta F., Gaetano C., Ori M. RNA-seq of the aging brain in the short-lived fish N. furzeri - conserved pathways and novel genes associated with neurogenesis. Aging Cell. 2014;13:965–974. doi: 10.1111/acel.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns D., Dimmer E., Huntley R., Barrell D., O’Donovan C., Apweiler R. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics. 2009;25:3045–3046. doi: 10.1093/bioinformatics/btp536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel G., Potter V.R. Nuclei from rat liver: isolation method that combines purity with high yield. Science. 1966;154:1662–1665. doi: 10.1126/science.154.3757.1662. [DOI] [PubMed] [Google Scholar]
- Bui K.H., von Appen A., DiGuilio A.L., Ori A., Sparks L., Mackmull M.T., Bock T., Hagen W., Andrés-Pons A., Glavy J.S., Beck M. Integrated structural analysis of the human nuclear pore complex scaffold. Cell. 2013;155:1233–1243. doi: 10.1016/j.cell.2013.10.055. [DOI] [PubMed] [Google Scholar]
- Burke S.N., Barnes C.A. Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 2006;7:30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- Calado A., Treichel N., Müller E.C., Otto A., Kutay U. Exportin-5-mediated nuclear export of eukaryotic elongation factor 1A and tRNA. EMBO J. 2002;21:6216–6224. doi: 10.1093/emboj/cdf620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo M.A., Raices M., Panowski S.H., Hetzer M.W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–295. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo M.A., Gomez-Cavazos J.S., Mei A., Lackner D.H., Hetzer M.W. A change in nuclear pore complex composition regulates cell differentiation. Dev. Cell. 2012;22:446–458. doi: 10.1016/j.devcel.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David D.C., Ollikainen N., Trinidad J.C., Cary M.P., Burlingame A.L., Kenyon C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhães J.P., Curado J., Church G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green D.R., Galluzzi L., Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H., Ingolia N.T., Weissman J.S., Bartel D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper R.H., Argmann C., Houten S.M., Cantó C., Jeninga E.H., Andreux P.A., Thomas C., Doenlen R., Schoonjans K., Auwerx J. The metabolic footprint of aging in mice. Sci. Rep. 2011;1:134. doi: 10.1038/srep00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper R.H., Mouchiroud L., Ryu D., Moullan N., Katsyuba E., Knott G., Williams R.W., Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E.J., Reichardt L.F. Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hühne R., Thalheim T., Sühnel J. AgeFactDB--the JenAge Ageing Factor Database--towards data integration in ageing research. Nucleic Acids Res. 2014;42:D892–D896. doi: 10.1093/nar/gkt1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue E., Mochida S., Takagi H., Higa S., Deguchi-Tawarada M., Takao-Rikitsu E., Inoue M., Yao I., Takeuchi K., Kitajima I. SAD: a presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron. 2006;50:261–275. doi: 10.1016/j.neuron.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Jiang C.H., Tsien J.Z., Schultz P.G., Hu Y. The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc. Natl. Acad. Sci. USA. 2001;98:1930–1934. doi: 10.1073/pnas.98.4.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D., Kopito R., Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y., Wang E.T., Airoldi E.M., Burge C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevei É., Hoppe T. Ubiquitin sets the timer: impacts on aging and longevity. Nat. Struct. Mol. Biol. 2014;21:290–292. doi: 10.1038/nsmb.2806. [DOI] [PubMed] [Google Scholar]
- Kishi M., Pan Y.A., Crump J.G., Sanes J.R. Mammalian SAD kinases are required for neuronal polarization. Science. 2005;307:929–932. doi: 10.1126/science.1107403. [DOI] [PubMed] [Google Scholar]
- Krueger K.E., Srivastava S. Posttranslational protein modifications: current implications for cancer detection, prevention, and therapeutics. Mol. Cell. Proteomics. 2006;5:1799–1810. doi: 10.1074/mcp.R600009-MCP200. [DOI] [PubMed] [Google Scholar]
- Lee C.K., Weindruch R., Prolla T.A. Gene-expression profile of the ageing brain in mice. Nat. Genet. 2000;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- Lessard J., Wu J.I., Ranish J.A., Wan M., Winslow M.M., Staahl B.T., Wu H., Aebersold R., Graef I.A., Crabtree G.R. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerch P.M., Lu T., Dakin K.A., Vann J.M., Isaacs A., Geula C., Wang J., Pan Y., Gabuzda D.H., Li C. Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE. 2008;3:e3329. doi: 10.1371/journal.pone.0003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovtrup-Rein H., McEwen B.S. Isolation and fractionation of rat brain nuclei. J. Cell Biol. 1966;30:405–415. doi: 10.1083/jcb.30.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T., Pan Y., Kao S.Y., Li C., Kohane I., Chan J., Yankner B.A. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Lund E., Güttinger S., Calado A., Dahlberg J.E., Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- Mak S.K., McCormack A.L., Langston J.W., Kordower J.H., Di Monte D.A. Decreased alpha-synuclein expression in the aging mouse substantia nigra. Exp. Neurol. 2009;220:359–365. doi: 10.1016/j.expneurol.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Maze I., Wenderski W., Noh K.M., Bagot R.C., Tzavaras N., Purushothaman I., Elsässer S.J., Guo Y., Ionete C., Hurd Y.L. Critical role of histone turnover in neuronal transcription and plasticity. Neuron. 2015;87:77–94. doi: 10.1016/j.neuron.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A., Banterle N., Iskar M., Andrés-Pons A., Escher C., Khanh Bui H., Sparks L., Solis-Mezarino V., Rinner O., Bork P. Cell type-specific nuclear pores: a case in point for context-dependent stoichiometry of molecular machines. Mol. Syst. Biol. 2013;9:648. doi: 10.1038/msb.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A., Andrés-Pons A., Beck M. The use of targeted proteomics to determine the stoichiometry of large macromolecular assemblies. Methods Cell Biol. 2014;122:117–146. doi: 10.1016/B978-0-12-417160-2.00006-0. [DOI] [PubMed] [Google Scholar]
- Rhie B.H., Song Y.H., Ryu H.Y., Ahn S.H. Cellular aging is associated with increased ubiquitylation of histone H2B in yeast telomeric heterochromatin. Biochem. Biophys. Res. Commun. 2013;439:570–575. doi: 10.1016/j.bbrc.2013.09.017. [DOI] [PubMed] [Google Scholar]
- Sancho E., Batlle E., Clevers H. Signaling pathways in intestinal development and cancer. Annu. Rev. Cell Dev. Biol. 2004;20:695–723. doi: 10.1146/annurev.cellbio.20.010403.092805. [DOI] [PubMed] [Google Scholar]
- Savas J.N., Toyama B.H., Xu T., Yates J.R., 3rd, Hetzer M.W. Extremely long-lived nuclear pore proteins in the rat brain. Science. 2012;335:942. doi: 10.1126/science.1217421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B., van der Pluijm I., Moorhouse M.J., Kosteas T., Robinson A.R., Suh Y., Breit T.M., van Steeg H., Niedernhofer L.J., van Ijcken W. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 2008;4:e1000161. doi: 10.1371/journal.pgen.1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz B.E., Ahmad K. Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 2005;19:804–814. doi: 10.1101/gad.1259805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding K.L., Bhardwaj R.D., Buchholz B.A., Druid H., Frisén J. Retrospective birth dating of cells in humans. Cell. 2005;122:133–143. doi: 10.1016/j.cell.2005.04.028. [DOI] [PubMed] [Google Scholar]
- Sun D., Luo M., Jeong M., Rodriguez B., Xia Z., Hannah R., Wang H., Le T., Faull K.F., Chen R. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama B.H., Savas J.N., Park S.K., Harris M.S., Ingolia N.T., Yates J.R., 3rd, Hetzer M.W. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell. 2013;154:971–982. doi: 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J.A., Côté R.G., Csordas A., Dianes J.A., Fabregat A., Foster J.M., Griss J., Alpi E., Birim M., Contell J. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J.A., Deutsch E.W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J.A., Sun Z., Farrah T., Bandeira N. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther D.M., Mann M. Accurate quantification of more than 4000 mouse tissue proteins reveals minimal proteome changes during aging. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M110.004523. M110.004523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S.H., Craig T., Li Y., Merry B., de Magalhães J.P. Whole transcriptome sequencing of the aging rat brain reveals dynamic RNA changes in the dark matter of the genome. Age (Dordr.) 2013;35:763–776. doi: 10.1007/s11357-012-9410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.S., Tsai S., Bonner W.M. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell. 1982;31:367–374. doi: 10.1016/0092-8674(82)90130-1. [DOI] [PubMed] [Google Scholar]
- Zahn J.M., Sonu R., Vogel H., Crane E., Mazan-Mamczarz K., Rabkin R., Davis R.W., Becker K.G., Owen A.B., Kim S.K. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006;2:e115. doi: 10.1371/journal.pgen.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.