

A novel imaging-based method is introduced to quantitatively localize Golgi proteins at nanometer resolution. The method reveals different intra-Golgi trafficking of secretory cargoes.
Abstract
Cellular functions of the Golgi are determined by the unique distribution of its resident proteins. Currently, electron microscopy is required for the localization of a Golgi protein at the sub-Golgi level. We developed a quantitative sub-Golgi localization method based on centers of fluorescence masses of nocodazole-induced Golgi ministacks under conventional optical microscopy. Our method is rapid, convenient, and quantitative, and it yields a practical localization resolution of ∼30 nm. The method was validated by the previous electron microscopy data. We quantitatively studied the intra-Golgi trafficking of synchronized secretory membrane cargoes and directly demonstrated the cisternal progression of cargoes from the cis- to the trans-Golgi. Our data suggest that the constitutive efflux of secretory cargoes could be restricted at the Golgi stack, and the entry of the trans-Golgi network in secretory pathway could be signal dependent.
INTRODUCTION
The mammalian Golgi complex plays essential roles in posttranslational modifications and sorting of secretory/endocytic proteins and lipids (cargoes; Glick and Luini, 2011; Klumperman, 2011; Lu and Hong, 2014). The basic structural unit of the Golgi is the Golgi stack, which comprises 4–11 tightly adjacent and flattened membrane sacs called cisternae. The mammalian Golgi ribbon is a compound organization of Golgi stacks that are laterally connected by reticular and tubular membrane networks (Klumperman, 2011). The Golgi stack is further divided into three regions: the cis-, medial-, and trans-Golgi. At the trans side, the trans-cisterna further develops into an anastomosing membrane compartment referred to as the trans-Golgi network (TGN). Although the elaborate structure and organization of the Golgi has attracted much attention, the Golgi remains one of the most mysterious and controversial organelles. For example, there are still heated debates on how the Golgi maintains its cisternal organization and works in membrane trafficking (Glick and Luini, 2011). One of the major challenges in studying the Golgi is to resolve the cisternal or sub-Golgi localizations of its resident proteins or transiting cargoes. There is a lack of a method to rapidly and quantitatively achieve nanometer localization accuracy of Golgi proteins. Electron microscopy (EM), especially immunogold EM, has been the technique of choice, but it requires an expensive instrument and is technically demanding and time consuming. Various superresolution light microscopy techniques are available to achieve a resolution of 20–110 nm at optimal conditions, but they require special microscope configurations and/or fluorophore labeling. Of great importance, both EM and superresolution techniques provide qualitative instead of quantitative localization data.
The resolution of conventional light microscopy (∼250 nm) is too low to resolve Golgi cisternae, which are <100 nm in thickness (Klumperman, 2011). Even under a high-end fluorescence microscope, the mammalian Golgi appears as a bright perinuclear lump with little resolvable structural features. The difficulty is partially due to the dense congregation of randomly oriented Golgi stacks. When single ribbons of Golgi stacks are well spread, as in certain cells, it is possible to vaguely discern the cis- from the trans-region of the Golgi(Antony et al., 1992; Nilsson et al., 1993). Based on single Golgi ribbons, an intensity line profile approach has been proposed to quantify the localization of a Golgi protein (Dejgaard et al., 2007; Starr et al., 2007). However, Golgi ribbons suitable for line profile analysis are not available for every cells, and it is subjective to judge and select Golgi ribbons. Golgi stacks could also be spatially dispersed by the treatment of the microtubule-depolymerizing drug nocodazole. Under prolonged nocodazole treatment (>2 h), the Golgi disassembles and reemerges as several hundred ministacks at the endoplasmic reticulum (ER) exit sites (ERES; Cole et al., 1996). Extensive studies have demonstrated that Golgi ministacks are valid models of native Golgi stacks, as they have similar structures (Trucco et al., 2004) and cellular functions, such as membrane trafficking and posttranslational modifications (Rogalski et al., 1984; Van De Moortele et al., 1993; Cole et al., 1996; Trucco et al., 2004). Of note, the Golgi is naturally dispersed as ministacks in plant and insect cells (Klumperman, 2011).
Taking advantage of the simplified organization of the nocodazole-induced Golgi ministack, Dejgaard et al. (2007) reported a method to calculate the relative distance ratio among three maximum intensity positions (peaks) of the test protein and cis- and trans-Golgi markers from triple-fluorescence-labeled Golgi. Their method was unable to achieve subpixel accuracy, however, which greatly limited its resolution. Here we introduce a novel imaging method named Golgi protein localization by imaging centers of mass (GLIM) to rapidly, quantitatively, and systematically study the localization of Golgi proteins by conventional light microscopy. Using GLIM, we quantitatively mapped many Golgi proteins and studied the intra-Golgi trafficking of synchronized secretory membrane cargoes. Our quantitative data demonstrate the differential cisternal progression of cargoes from the cis- to the trans-Golgi and suggest that the entry of the TGN into the secretory pathway could be signal dependent.
RESULTS
Development of GLIM
In our method, HeLa cells (hereafter cells) triply labeled by either endogenous or ectopically expressed GM130, the test protein, and β1,4-galactosyltransferase–mCherry (GalT-mCherry) are treated with nocodazole and imaged by a spinning-disk confocal or wide-field microscope as three-dimensional (3D) image stacks. GM130 (Nakamura et al., 1995) and GalT-mCherry (Roth and Berger, 1982) serve as references to mark the cis- and trans-Golgi. GM130, GalT-mCherry, and the test protein normally appear as tightly adjacent single fuzzy spots in a ministack, which can be occasionally resolved, as exemplified by the TGN marker Vamp4-GFP (Steegmaier et al., 1999; Figure 1, A–D). Golgi ministacks are visually inspected, and those that display only one object in each channel are masked by regions of interest (ROIs).
FIGURE 1:
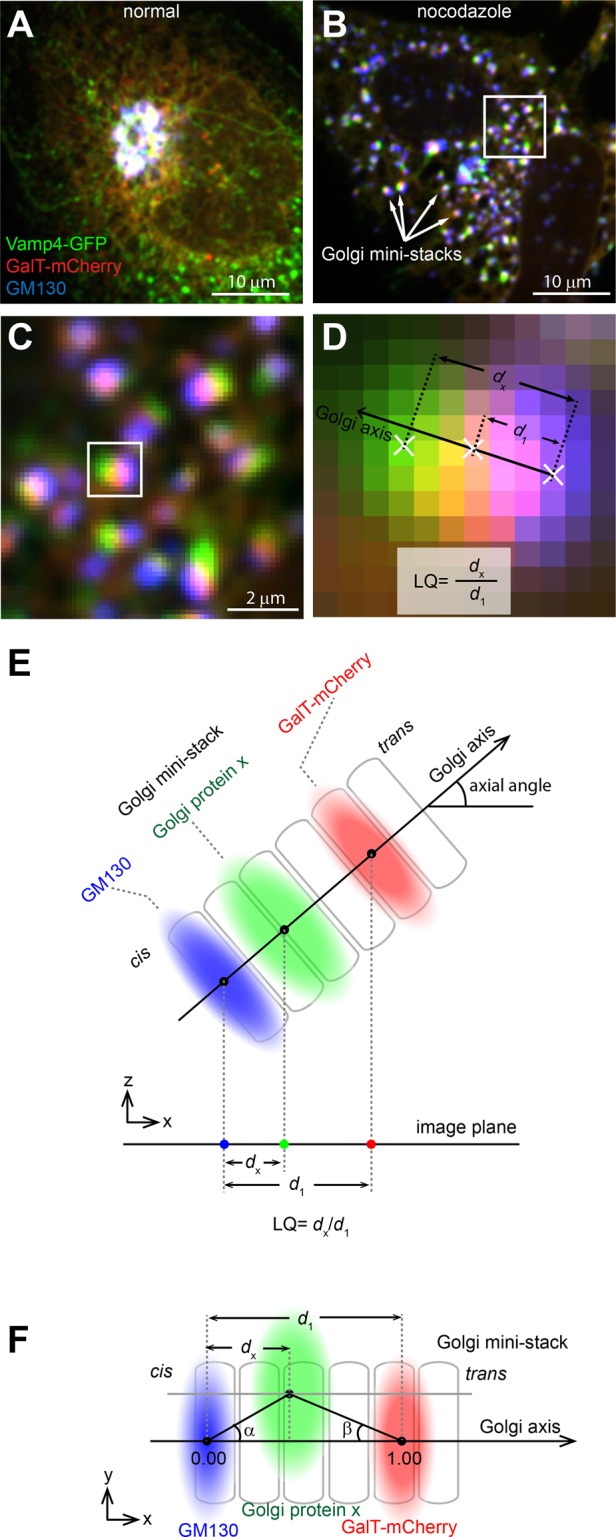
Overview of GLIM. HeLa cells transiently expressing Vamp4-GFP (green) and GalT-mCherry (red) were treated without (A) or with (B) nocodazole and processed for immunofluorescence labeling of endogenous GM130 (artificially colored as blue). Arrows indicate examples of Golgi ministacks. Scale bars, 10 μm. The boxed region in B is enlarged in C to show Golgi ministacks, which display visible separation of the three Golgi proteins. Scale bar, 2 μm. The boxed Golgi ministack is further enlarged in D to schematically illustrate centers of mass (denoted as white crosses) and the calculation of the LQ. (E) Schematic diagram illustrating the Golgi axis, axial angle, d1, dx, and the calculation of the LQ. During imaging, the Golgi ministack in 3D space is projected onto the image plane, and the diagram shows the xz-view. (F) Schematic diagram illustrating angle α, angle β, and the projected distance dx. The Golgi ministack image is in the xy-plane or the image plane and the center of the protein x is off-axis.
To localize the test protein at subpixel accuracy, we calculate the center of fluorescence mass (hereafter center) of the Golgi ministack within each ROI (Figure 1D). We define the Golgi ministack axis (hereafter Golgi axis) as a vector from the center of GM130 to that of GalT-mCherry in the image plane or 3D space (Figure 1, E and F) and the axial angle as the angle between the 3D Golgi axis and the image plane (Figure 1E). The ministack is assumed to have an infinite order of rotational symmetry around its axis and further modeled as a linear or one-dimensional structure.
Correction of the chromatic shift.
To assess and correct chromatic shift aberration in light microscopy, we image 110-nm beads emitting three-color fluorescence. The shift in z-axis is not considered in this study due to the z-average projection. The shift in the xy-plane is subpixel for our microscopes (Figure 2, A–G). Throughout this work, the red channel is fiducially defined as free of chromatic shift aberration, and therefore centers of red beads are true positions of beads and denoted as (0, 0) (Figure 2C). Polynomial fitting is used to correct the chromatic shift in green and far-red channels. Judging by the mean magnitude of shifts, we found that the first-order polynomial or linear fitting yielded the best performance from zeroth to fourth order (Supplemental Figure S1). We routinely imaged hundreds of beads to calibrate the linear fitting tool, which was subsequently used to correct chromatic shifts of centers acquired in the same imaging session. A significant improvement in localization is achieved after such correction (Figure 2, D–G). The localization uncertainty of shift-corrected centers of beads was consistently ∼8 nm in both green and far-red channels (Figure 2, F and G), which was determined as (SDx + SDy)/2, where SDx and SDy are SDs of centers in the x- and y-axes, respectively. We evaluated the accuracy of our localization method in vivo under a typical experimental condition, in which Golgi ministacks were stained by an anti-GM130 antibody followed by a mixture of secondary antibodies in three colors (Figure 2H). After chromatic shift correction, the localization uncertainty was ∼30 nm (Figure 2, I and J), indicating that our method was able to achieve nanometer accuracy. Despite microscopic thermal and mechanical drifts, such a high accuracy in positioning was made possible by the use of red channel objects as fiduciary markers.
FIGURE 2:

Correction of the chromatic shift. (A) The three-color merged image of 110-nm beads acquired by the spinning-disk confocal microscope. Each bead emits green, red, and far-red (artificially colored as blue) fluorescence. Scale bar, 10 μm. (B) Enlarged view of a merged single-bead image with barely visible chromatic shift. Scale bar, 200 nm. (C) Diagram showing that shifts of green and far-red bead images can be represented by vectors from the center of the red bead image (0,0) to centers of green (green vector) and far-red (blue vector) bead images, respectively. (D, E) The chromatic shift before and after shift correction. The boxed areas correspond to the image in A. The green and blue vectors were plotted before and after shift correction. To visualize the tiny shift, the magnitude of each vector is amplified 200-fold. (F, G) Scatter plots showing centers of green (green dots) and far-red (blue dots) beads before and after shift correction. (H) Evaluation of our localization method in vivo under a typical experimental condition. Golgi ministacks were stained by anti-GM130 antibody, followed by a mixture of three secondary antibodies conjugated with Alexa Fluor 488, 594, or 647. Scale bar, 10 μm. (I, J) The localization analysis of images described in H by scatter plots, which show green (green dots) and far-red (blue dots) centers of Golgi mini-stacks before and after shift-correction.
Localization quotient.
Three criteria were developed to ensure data quality. 1) The signal-to-noise ratio criterion, for which the ratio of the total intensity of a ministack to the SD of the background must be ≥30 in each channel. The criterion is important for the positioning accuracy of the center of mass, which depends on the signal-to-noise ratios of Golgi ministacks. 2) The axial angle or distance criterion, for which d1, or the distance between centers of GM130 and GalT-mCherry (Figure 1E), must be ≥70 nm. When the axial angle approaches 90°, the ministack becomes nonresolvable, as d1 approaches 0. 3) The collinearity criterion, in which either |tanα| or |tanβ| must be ≤0.3 (see Figure 1F for the definition of angles α and β). This criterion ensures that the three centers of a ministack are sufficiently collinear for our one-dimensional model of the Golgi ministack. Ministacks that fulfill these criteria are referred to as analyzable ministacks.
To describe quantitatively the localization of a test Golgi protein (x) and normalize random axial angles and inhomogeneous axial lengths of Golgi ministacks, we introduced the localization quotient (LQ), defined as dx/d1, where dx is the distance from the center of x to that of GM130 (Figure 1, D–F). The LQs of GM130 and GalT-mCherry are 0.00 and 1.00, respectively. If the center of x is off-axis, its projection axial distance is used for the calculation (Figure 1F). The biological explanation of off-axis could be a gradual detachment of trans-Golgi cisternae (Mogelsvang et al., 2004; De Matteis and Luini, 2008) or the lateral budding/tethering of transport carriers at the time of fixation. We observed that LQs seemed insignificantly affected by image background subtraction values and expression levels of GalT-mCherry (Supplemental Figure S2, A–C). Using GS15 as an example, the selection of ROIs and the analysis of LQs in one and multiple cells are shown in Figure 3. LQs of a Golgi protein varied among ministacks within a cell (Figure 3B), and different cells had slightly different mean LQs (Figure 3C), which could be attributed to their distinct cellular statuses, such as cell cycle stage. However, a statistical analysis of many analyzable ministacks (typically >50) from multiple cells (typically 5–10) generally yielded convergent LQs. The LQ of a Golgi protein could be robustly reproduced from independently conducted experiments (Supplemental Table S1) or with different types of microscopes (confocal or wide-field; Supplemental Table S2).
FIGURE 3:
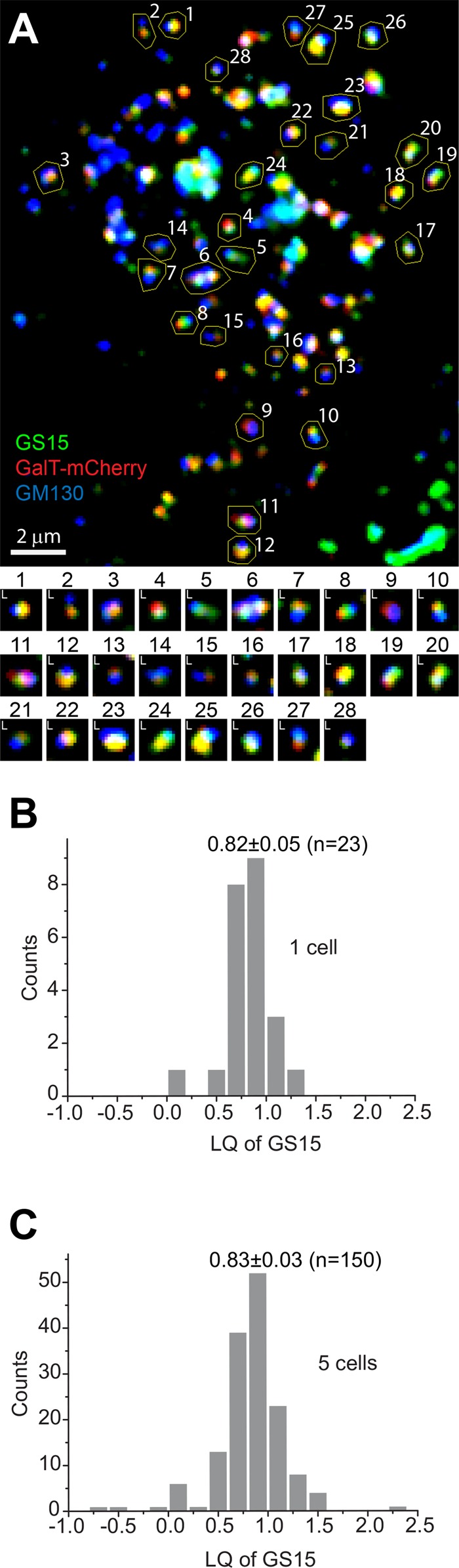
A typical example of GLIM, here acquiring the LQ of GS15. (A) Nocodazole-treated cells expressing GalT-mCherry (red) were stained for endogenous GM130 (blue) and GS15 (green). Golgi ministacks were selected by manually drawn ROIs and labeled by identification numbers. Images of these masked Golgi ministacks are shown below with their identification numbers. L, the Golgi mini-stack is valid for the calculation of the LQ (analyzable Golgi ministacks). Scale bar, 2 μm. (B) Histogram of LQs of 23 ministacks (denoted by L) from the cell in A. (C) Histogram of LQs of 150 ministacks from five cells. The value shown in each histogram is mean ± SEM.
Except for the manual selection of ministacks, data analysis was aided by software tools. The LQs of a Golgi protein could be acquired within hours, including imaging and postacquisition analysis. Thus, we have introduced the concept of GLIM as a novel imaging method for quantitative study of localizations of Golgi proteins.
Validation of GLIM and a quantitative map of the Golgi
Using GLIM, we mapped various green fluorescent protein (GFP)–tagged and endogenous Golgi proteins in HeLa cells (Table 1, Figure 4A, and Supplemental Figure S3A). GFP-tagged proteins include Sec31a (ERES; Tang et al., 2000), ERGIC53 (the ER–Golgi intermediate compartment [ERGIC]; cis; Schweizer et al., 1988), Golgin84 (cis; Diao et al., 2003), GPP130 (cis/medial; Linstedt et al., 1997), TPST1/2 (tyrosylprotein sulfotransferase 1/2; trans; Baeuerle and Huttner, 1987), sialyltransferase (SialT; trans/TGN; Rabouille et al., 1995), Rab6 (trans/TGN; Antony et al., 1992), GCC88 (TGN; Luke et al., 2003)), and Vamp4 (TGN; Steegmaier et al., 1999); endogenous proteins include GS27/membrin (ERGIC/cis; Hay et al., 1998), KDEL receptor (KDELR; ERGIC/cis; Griffiths et al., 1994), β-COP (ERGIC/cis; Oprins et al., 1993), GS28/GOS-28 (cis/medial/trans; Volchuk et al., 2004), acyl-CoA binding domain–containing 3 (ACBD3; sub-Golgi localization unknown; Sohda et al., 2001), Giantin (medial; Martinez-Menarguez et al., 2001), GS15 (trans/TGN; Volchuk et al., 2004), Arl1 (trans/TGN; Lu et al., 2001), Golgin97 (TGN; Lu and Hong, 2003), Golgin245 (TGN; Brown et al., 2001), Syntaxin6 (TGN; Bock et al., 1997), Vti1a (TGN; Kreykenbohm et al., 2002), TGN46 (TGN; Luzio et al., 1990), furin (TGN; Molloy et al., 1994), cation-independent mannose 6-phosphate receptor (CI-M6PR; TGN; Brown, 1990), and GGA2 (TGN; Dell’Angelica et al., 2000; Hirst et al., 2000). In these experiments, endogenous GM130 was labeled by either mouse or rabbit antibody, as they had almost identical LQs (Table 1 and Figure 4A). Regions of the Golgi were operationally defined according to LQ: the ERES/ERGIC, <−0.25; cis, 0.00 ± 0.25; medial, 0.50 ± 0.25; trans-Golgi, 1.00 ± 0.25; and TGN, 1.25–2.00. This definition is consistent with qualitative sub-Golgi localization data from immunogold EM or, in limited cases, fluorescence microscopy in the literature, therefore validating GLIM (Table 1). In cell lines of different tissue origins, LQs of Golgi proteins have very similar values (Supplemental Figure S4A), in agreement with a conserved molecular organization of the Golgi complex among mammalian cells.
TABLE 1:
LQ of Golgi proteins.
| Golgi protein | LQ (mean) | SEM | n | Localization regiona | Reported localization |
|---|---|---|---|---|---|
| Sec31a-GFP | −0.65 | 0.06 | 67 | ERES/ERGIC | ERES/ERGIC ( Tang et al., 2000) |
| GS27 | −0.22 | 0.03 | 101 | cis | ERGIC/cis ( Hay et al., 1998) |
| GFP-ERGIC53 | −0.16 | 0.02 | 198 | cis | ERGIC/cis ( Hauri et al., 2000) |
| KDELR | −0.11 | 0.03 | 130 | cis | ERGIC/cis ( Griffiths et al., 1994) |
| GM130 | 0.00a | − | − | cis | cis ( Nakamura et al., 1995) |
| GM130 (r)b | 0.03 | 0.01 | 284 | cis | cis ( Nakamura et al., 1995) |
| β-COP | 0.04 | 0.09 | 57 | cis | ERGIC/cis ( Oprins et al., 1993) |
| GFP-Golgin84 | 0.26 | 0.03 | 108 | Medial | cis ( Diao et al., 2003) |
| ACBD3 | 0.27 | 0.08 | 44 | Medial | Golgi stack ( Sohda et al., 2001) |
| GS28 | 0.53 | 0.08 | 125 | Medial | cis/medial/trans (Volchuk et al., 2004) |
| Giantin | 0.59 | 0.11 | 32 | Medial | medial ( Martinez-Menarguez et al., 2001) |
| TPST2-GFP | 0.64 | 0.02 | 154 | Medial | trans ( Baeuerle and Huttner, 1987) |
| TPST1-GFP | 0.76 | 0.04 | 111 | trans | trans ( Baeuerle and Huttner, 1987) |
| GS15 | 0.82 | 0.03 | 97 | trans | trans/TGN ( Volchuk et al., 2004) |
| SialT-AcGFP | 0.82 | 0.04 | 41 | trans | trans/TGN ( Rabouille et al., 1995) |
| GFP-GPP130 | 0.84 | 0.02 | 168 | trans | cis/medial ( Linstedt et al., 1997) |
| GalT-mCherry | 1.00a | − | − | trans | trans ( Roth and Berger, 1982) |
| GFP-Rab6 | 1.04 | 0.04 | 262 | trans | trans/TGN ( Antony et al., 1992) |
| Arl1 | 1.20 | 0.05 | 26 | trans | trans/TGN ( Lu et al., 2001) |
| Vti1a | 1.26 | 0.02 | 162 | TGN | TGN ( Kreykenbohm et al., 2002) |
| GFP-GCC88 | 1.31 | 0.04 | 62 | TGN | TGN ( Luke et al., 2003) |
| Golgin97 | 1.32 | 0.03 | 174 | TGN | TGN ( Lu and Hong, 2003) |
| Golgin245 | 1.42 | 0.05 | 126 | TGN | TGN ( Brown et al., 2001) |
| CI-M6PR | 1.46 | 0.24 | 42 | TGN | TGN ( Brown, 1990) |
| TGN46 | 1.47 | 0.02 | 203 | TGN | TGN ( Luzio et al., 1990) |
| Syntaxin6 | 1.56 | 0.11 | 84 | TGN | TGN ( Bock et al., 1997) |
| Vamp4-GFP | 1.57 | 0.04 | 157 | TGN | TGN ( Steegmaier et al., 1999) |
| Furin | 1.62 | 0.11 | 43 | TGN | TGN ( Molloy et al., 1994) |
| GGA2 | 1.96 | 0.23 | 33 | TGN | TGN ( Dell’Angelica et al., 2000; Hirst et al., 2000) |
The table corresponds to Figure 4A.
aAs defined in Results.
bGM130 (r) denotes data acquired by GM130 rabbit antibody when the LQ of costained GM130 mouse antibody was defined as 0.00.
FIGURE 4:

Mapping multiple Golgi proteins via GLIM. (A) A localization map of 25 Golgi proteins in HeLa cells (see also Table 1). Regions including the ERES/ERGIC, cis-, medial-, and trans-Golgi, and TGN are quantitatively defined (see Results), corresponding to the schematic Golgi ministack model on the right. (B) LQs of endogenous and their corresponding tagged proteins. Error bars, SEMs. In B, n ≥ 42 Golgi ministacks were analyzed for each LQ.
The mean axial length of Golgi ministacks in three dimensions can be estimated by multiplying the corresponding mean two-dimensional (2D) projected or imaged values by π/2 (see Materials and Methods, Projection of unit-length 3D line segment). To obtain the mean 2D projected axial length, the axial angle criterion was dropped to allow random orientation of ministacks. The 3D axial length of the stacked region, defined by the distance from GM130 to GalT-mCherry, and that of the Golgi complex, defined by the distance from GM130 to TGN46, were measured to be 344 ± 6 nm (n = 401) and 501 ± 8 nm (n = 401; mean ± SEM; Supplemental Figure S4, B and C), respectively. Our estimated values are consistent with the EM tomography data (Ladinsky et al., 1999, 2002).
Assessment of the effect of tagging on the Golgi localization
Tagged fusion proteins are widely used for subcellular localization study. Taking advantage of GLIM, we compared LQs of tagged versus endogenous Golgi proteins to assess the effect of tagging. Whereas tagging did not significantly change the LQs of GM130, ACBD3, Golgin97, and CI-M6PR (Figure 4B), it dramatically shifted the sub-Golgi localization of KDELR from cis- to trans-Golgi, as noted previously (Dejgaard et al., 2007). For GalT, tagging mCherry, GFP, and hemagglutinin (HA) at its C-terminus quantitatively affected its localization. The mislocalization of KDELR and GalT by GFP tagging could be due to the fact that mCherry (Shaner et al., 2004) and HA are monomers, whereas GFP (enhanced GFP) is prone to dimerization (kd = 0.11 mM; Zacharias et al., 2002) at high local concentration. In summary, GLIM is able to assess quantitatively the effect of tagging on a protein’s sub-Golgi localization.
The organization of a Golgi ministack changes dynamically
The concept of GLIM is not limited to fixed cells. With the availability of fluorescence proteins at the blue and far-red ends of the spectrum, it is possible to perform three-color live-cell imaging. We tagged the far-red fluorescence protein iRFP670 (Shcherbakova and Verkhusha, 2013) to GalT and coexpressed it together with GFP-Golgin84 and mCherry-GM130 for live-cell imaging (Supplemental Video S1). For a given ministack, LQ data can be missing for certain frames since the ministack could be either nonanalyzable or lost to tracking. Figure 5A shows a histogram of LQs of all ministacks through all time points. The mean LQ of GFP-Golgin84, 0.24 ± 0.02 (n = 295), agrees with the value obtained from our fixed cells (Table 1), demonstrating the validity of GLIM in live-cell imaging. Examining the plot of LQ versus time, we observe heterogeneous Golgin84 localization at any time point (Figure 5B), consistent with what we found in fixed cells (Figure 3B). Intriguingly, as shown by a typical trace in Figure 5C, Golgin84 LQ of a ministack appeared to oscillate randomly around the population mean on a time scale of minutes, suggesting that the organization of a Golgi ministack can change dynamically. Further study along this line would help us to understand the dynamic organization of the Golgi.
FIGURE 5:
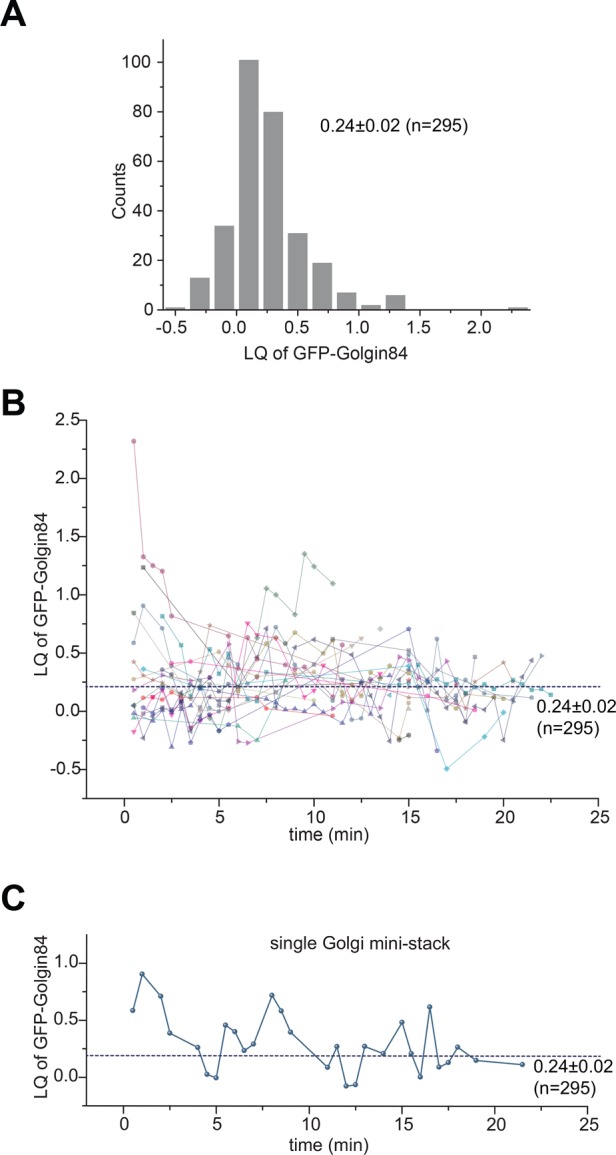
The dynamic fluctuation of the LQ of Golgin84 during live-cell imaging. Cells transiently expressing GalT-iRFP670, GFP-Golgin84, and mCherry-GM130 were imaged live for 24 min with a time interval of 30 s (Supplemental Video S1). (A) Histogram of LQs of GFP-Golgin84. We plotted together 295 LQs from 29 Golgi ministacks at various time points. (B) The LQ of GFP-Golgin84 for each Golgi ministack, represented by different shapes and colors, plotted against time (minutes). Nearest available data points are connected by lines of the same color. Note that some Golgi ministacks have only single data points. (C) Typical LQ trace showing dynamic changes of the organization of the Golgi ministack. The dotted horizontal line indicates the population mean. The value indicated is mean ± SEM.
Quantitative visualization of the intra-Golgi transport of vesicular stomatitis virus protein G–GFP
With the aid of GLIM, we attempted to study quantitatively intra-Golgi transport probed by GFP-tagged temperature-sensitive vesicular stomatitis virus protein G (VSVGtsO45-GFP; hereafter VSVG-GFP), a classic secretory membrane cargo (Kreis and Lodish, 1986; Beckers et al., 1987). When expressed at 40°C, VSVG-GFP misfolded and was arrested in the ER (Figure 6A; 0 min). At 32°C, it rapidly refolded and entered the secretory pathway by sequentially transiting the ERES, ERGIC, Golgi, and plasma membrane (PM) in a synchronous trafficking wave (chase; Figure 6A; 10 and 120 min). We quantified the Golgi-ministack localized VSVG-GFP and found that it rapidly increased at the beginning of the chase, peaked at 25 min, and decreased to an almost undetectable level after 200 min (Figure 6B) as previously reported (Hirschberg et al., 1998). The LQ of VSVG-GFP was also observed to increase rapidly and almost linearly up to ∼0.8 (trans-Golgi) within 25 min after the chase, in agreement with the cisternal progression model (Trucco et al., 2004; Figure 6C). The LQ subsequently advanced slowly and eventually plateaued at ∼1.1 (Figure 6C). Because the Golgi pool of VSVG-GFP was observed to decline rapidly after 25 min of chase (Figure 6B), the rate of Golgi exit toward the PM (anterograde trafficking) and the ER (retrograde trafficking) must exceed that of secretory entry of VSVG-GFP, suggesting that cisternal progression and the Golgi exit of a cargo can take place simultaneously during intra-Golgi trafficking. Of note, the LQ of VSVG-GFP did not go beyond 1.25 even after 4 h of chase, when a majority of it had already exited the Golgi, suggesting that VSVG-GFP can exit the Golgi at the trans-Golgi instead of the TGN. A similar result was obtained when the chase was conducted in the presence of protein synthesis inhibitor cycloheximide (Figure 6D), ruling out the effect of the newly synthesized VSVG-GFP at the early secretory pathway. Superresolution 3D-structured illumination microscopy (3D-SIM) was also used to reveal qualitatively the exit site of VSVG-GFP in the native Golgi complex (without nocodazole treatment; Figure 6E). After 40 min of chase, the line profile of a typical Golgi ribbon showed that the peak of VSVG-GFP did not reach that of GalT-mCherry, consistent with data obtained from GLIM.
FIGURE 6:

Quantitative study of the intra-Golgi trafficking of VSVG-GFP via GLIM. Cells transiently expressing VSVG-GFP and GalT-mCherry were incubated and treated with nocodazole (except as indicated otherwise) at 40°C, followed by the chase at 32°C for various lengths of time. (A) Cells were fixed after the chase at 32°C for 0 (left), 10 (middle) and 120 (right) min. Endogenous GM130 (blue) was immunostained. VSVG-GFP and GalT-mCherry are shown as green and red, respectively. Scale bar, 10 μm. (B) The fraction of Golgi-localized VSVG-GFP during the chase. More than 20 cells were analyzed at each time. (C) The LQ of VSVG-GFP during the chase. (D) Cells at 40°C were chased at 32°C in the presence of 10 μg/ml cycloheximide. (E) Cells were incubated at 40°C and subsequently chased for 40 min at 32°C in the absence of nocodazole. Left, image from superresolution 3D-SIM. The boxed Golgi ribbon is magnified and shown in the upper right corner. The arrow indicates the intensity line profile, which is plotted on the right. Scale bar, 5 μm. (F, G) Cells at 40°C were subjected to (F) incubation at 15°C for 1 h before the chase at 40°C or (G) incubation at 20°C for 2 h before the chase at 32°C. Error bars, SEMs. In C, D, F, and G, the number of ministacks analyzed at each time point ranges from 12 to 216.
The intra-Golgi trafficking of VSVG-GFP was further characterized by GLIM after 15 or 20°C temperature block, which is known to arrest secretory cargoes at the ERGIC/cis-Golgi or trans-Golgi/TGN, respectively (Matlin and Simons, 1983; Saraste and Kuismanen, 1984; Griffiths et al., 1985; Figure 6, F and G). At 15 and 20°C, the LQ of VSVG-GFP was arrested at −0.06 ± 0.03 (n = 97) and 0.56 ± 0.03 (n = 124), corresponding to the cis- and medial-Golgi localization, respectively. It seems surprising that the LQ of VSVG at 20°C did not correspond to the trans-Golgi/TGN. However, our finding is consistent with the fact that VSVG at 20°C is only partially resistant to endoglycosidase H digestion and can be found at other cisternae in addition to the trans-side of the Golgi (Griffiths et al., 1985). The subsequent chase after 15°C block was performed at 40°C to inhibit the ER pool of VSVG-GFP from entering the secretory pathway (Figure 6F), since VSVGtsO45 is no longer thermosensitive outside the ER (de Silva et al., 1990), whereas the chase after 20°C was conducted at 32°C (Figure 6G). Both experiments further demonstrated that synchronized VSVG-GFP cargoes gradually stop and disappear at the trans-Golgi instead of the TGN, as both LQs never exceeded 1.25. It seems highly unlikely that VSVG-GFP could transit the TGN, whereas the trans-Golgi could impose a rate-limiting step, since we have never observed the LQs of VSVG-GFP and other PM targeted cargoes (described later) to be >1.25 under various chase conditions and sampling frequencies. Therefore our experiments clearly demonstrated the power of GLIM for studying intra-Golgi trafficking, and our data suggested that VSVG-GFP can exit at the trans-Golgi instead of the TGN.
A caveat in interpreting LQs acquired in temperature-shift experiments is that the Golgi organization could change accordingly. Indeed, we found that, whereas the LQs of the trans-Golgi marker GS15 at 32 and 37°C are similar, that of the TGN marker Golgin245 increased from 1.42 ± 0.05 (n = 126) at 37°C to 1.62 ± 0.03 (n = 210) at 32°C. The result demonstrates a possible “distortion” of the TGN and suggests that the TGN can be redefined as the region with LQ > 1.25 at 32°C. Nonetheless, this temperature effect should not compromise our conclusion that secretory VSVG-GFP cannot reach the TGN.
Signal-dependent entry of the TGN during the secretory pathway
During endocytic trafficking, only cargoes with TGN localization/sorting signals can enter the TGN, whereas the rest are either recycled via the early or recycling endosome to the PM or degraded in the lysosome (Lu and Hong, 2014). In the secretory pathway, the Golgi complex has been conventionally modeled as a linear pipe with cargoes entering at the cis-Golgi and exiting at the TGN. However, our high-resolution localization data from GLIM revealed that VSVG can transit and exit at the Golgi stack but never reach the TGN. To further explore whether the new finding applies to other secretory cargoes, we used the newly established retention using selective hooks (RUSH) system to synchronize the secretion of cargoes under a more physiological condition than temperature block (Boncompain et al., 2012). In the RUSH system, streptavidin-binding peptide (SBP)–fused reporters are retained in the ER due to the interaction of SBP with streptavidin-fused ER hooks. On the addition of biotin and subsequent biotin–streptavidin interaction, reporters are released from ER hooks and synchronously enter the secretory pathway (chase). We found that our PM-targeted transmembrane reporters (cargoes; Figure 7A), including chimera TNFα-SBP-GFP (type II transmembrane protein; Figure 7B), signal sequence (ss)-SBP-GFP-E-cadherin (type I transmembrane protein; Figure 7C), and ss-SBP-GFP-CD59 (glycosylphosphatidylinositol [GPI]-anchored protein; Figure 7D), transited the ministack from the cis- to the trans-Golgi as a synchronized traffic wave during the chase (Supplemental Figure S3B), similar to VSVG-GFP. Fitting their traces by a single-exponential function yielded LQ plateau of 1.03 (n = 1), 1.05 ± 0.08 (mean ± SD; n = 3), and 0.98 (n = 1) for TNFα-SBP-GFP, ss-SBP-GFP-E-cadherin, and ss-SBP-GFP-CD59, respectively, suggesting that they could exit the Golgi at the trans-Golgi but not the TGN. In contrast, a TGN-targeted reporter, ss-SBP-GFP-CD8a-furin (type I transmembrane protein), which contains the CD8a transmembrane domain and the furin cytosolic domain (Figure 7A), was observed to reach the TGN via the secretory pathway, as its LQ plateaued at 1.59 ± 0.03 (mean ± SD; n = 3), which is significantly different from ss-SBP-GFP-E-cadherin (p = 4 × 10−4; Figure 7I).
FIGURE 7:

The secretory targeting of the TGN is signal dependent. (A) Schematic diagram showing ER hooks and secretory membrane reporters used in the RUSH system: 1) Ii-Strep (hook), 2) ss-Strep-KDEL (hook), 3) ss-SBP-GFP-E-cadherin, 4) TNFα-SBP-GFP, 5) ss-SBP-GFP-CD59, 6) ss-SBP-GFP-CD8a-furin (wild type [WT]), 7) ss-SBP-GFP-CD8a-furin (Y mutation), 8) ss-SBP-GFP-CD8a-furin (AC mutation), and 9) ss-SBP-GFP-CD8a-furin (Y+AC mutation). memb., membrane; -N and -C, the N- and C-termini of proteins, respectively; ss, signal sequence; Strep., streptavidin; SBP, streptavidin-binding peptide; furin(c), furin cytosolic domain. (B–H) During intra-Golgi transport assays, reporters were coexpressed with an ER hook, synchronously released into the secretory pathway by biotin treatment at 0 min, and subsequently monitored at different chase times (minutes). LQ plots were fitted by a single-exponential function, and the resulting adjusted R2 (adj. R2) and plateau are shown in each plot. Error bars, SEMs. (I) Statistical comparison of LQ plateaus of reporters. n, number of independent experiments. For reporters with n = 1, the data are from B and C. For reporters with n ≥ 3, adjusted R2 values of all curve fittings are ≥0.85. The number on the top of each column is mean ± SD. Error bars, SDs. In B–I, the number of ministacks analyzed at each time point ranges from 12 to 131.
It was previously demonstrated that furin cytosolic domain possesses two TGN targeting signals—tyrosine-based Y762KGL and acidic cluster S776DSEEDE (Figure 7A), either of which is sufficient for the TGN localization after endocytosis (Schafer et al., 1995). Consistent with the redundancy of the two signals in the endocytic pathway, which we independently verified (Supplemental Figure S5), mutation of either the tyrosine motif from YKGL to AKGA (Y mutation) or the acidic cluster motif from SDSEEDE to ADAAAAA (AC mutation) did not abolish their entry of the TGN via the secretory pathway, as their LQs plateaued at 1.49 ± 0.04 (mean ± SD; n = 3) and 1.62 ± 0.09 (mean ± SD; n = 4), respectively (Figure 7, F, G, and I). Only when both tyrosine and acidic cluster motifs were mutated (Y+AC mutation) was the plateau of furin chimera significantly reduced to 0.87 ± 0.11 (mean ± SD; n = 3) in comparison to the wild type (p = 4 × 10−4; Figure 7, H–I), suggesting that the entry of the TGN via the secretory pathway could be dependent on the same TGN targeting signals as the endocytic trafficking pathway. It is possible that, similar to PM targeted cargoes, furin chimera could also exit at the trans-Golgi and target to the PM, and, subsequently, the TGN localization of furin could indirectly result from the endosome-to-TGN retrograde trafficking. However, under the same experimental conditions, we did not observe a synchronized appearance of wild-type furin chimera at the PM at any time during the chase (up to 360 min), whereas Y+AC mutant could be easily detected there after 40 min of chase (Supplemental Figure S6). Together with results of VSVG-GFP, our finding suggests a novel model for intra-Golgi transport. The cisternal progression and efflux of PM-targeted constitutive secretory cargoes, including GPI-anchored proteins, could be restricted by default to the Golgi stack; only cargoes with TGN-targeting signals could enter and exit the TGN during the secretory pathway.
DISCUSSION
Conventional light microscopic studies on the Golgi structure are qualitative and hampered by a limited resolution. The arrival of various superresolution microscopy techniques such as 3D-SIM, stimulated emission depletion, photoactivated localization microscopy, and stochastic optical reconstruction microscopy makes it possible to resolve sub-Golgi structures under the light microscope (Schermelleh et al., 2010). However, their application to the field of the Golgi is limited by the following disadvantages. First, superresolution techniques require either special instruments or hardware modifications that are beyond most cell biology labs. Second, conventional fluorescence labeling protocols are incompatible with most superresolution methods. Third, although some superresolution methods claim to achieve a resolution of 20 nm, the practical resolution of these techniques is much lower (>50 nm). Fourth, it is still very technically challenging for these techniques to achieve multicolor 3D or live-cell imaging, either singly or in combination, which is routine work for conventional light microscopy. Here we developed GLIM as a quantitative method to systematically localize Golgi proteins under conventional optical microscopy. The practical localization accuracy of GLIM, which is ∼30 nm, approaches that of the immunogold EM, which is ∼20 nm, considering the size of the antibody and gold particle complex. However, GLIM is much more rapid, convenient, and quantitative. We believe that it is also superior to current superresolution techniques in localizing Golgi proteins because, in addition to its high accuracy and reproducibility, it is compatible with conventional microscopes and fluorescence labeling protocols. Of importance, we demonstrated that GLIM also works for live-cell imaging.
We validated GLIM by mapping >20 Golgi proteins, which were found to localize randomly from the cis-Golgi to the TGN. LQs of these proteins are consistent with their reported sub-Golgi localizations. The mechanism governing the localization of a Golgi protein is elusive, but it could be determined by the dynamic equilibrium of two opposite movement within the Golgi—anterograde and retrograde trafficking. GLIM can help greatly in elucidating this mechanism. When GLIM was applied to live-cell imaging, we found that the organization of a Golgi ministack randomly oscillates around the population mean on a time scale of minutes. The observation is consistent with our results from fixed cells, which demonstrated a wide distribution of LQs. Although the molecular mechanism behind such findings remains to be explored, it is tempting to speculate that the periodic fluctuation of the LQ could be the result of discontinuous retrograde trafficking, such as by vesicles, to the cis side of the Golgi after the resident protein is carried to the trans side by cisternal progression.
We successfully applied GLIM to monitor the intra-Golgi trafficking of PM-targeted secretory membrane cargoes, such as VSVG, tumor necrosis factor α (TNFα), E-cadherin, and CD59, synchronized by either temperature shift or biotin treatment. We observed the successive transition of these cargoes through the ERES/ERGIC and from the cis- to the trans-Golgi cisternae. Their axial movement, as shown in plots of the LQ versus time, slowed gradually and appeared to be stagnant at LQ ≈ 1, which corresponds to the trans-Golgi, for a prolonged time, during which they are known to be packed into membrane carriers destined for the PM. In contrast, the secretory membrane cargo with furin cytosolic tail transited the Golgi ministack, and its LQ plateaued at the TGN, where it is known to be subsequently sorted to endosomes or the PM (Lu and Hong, 2014). The secretory TGN targeting of furin chimera could be due to its retention at the TGN by either AP1 clathrin coat complex or Mint3, which specifically interacts with furin tyrosine or acidic cluster motif, respectively (Teuchert et al., 1999; Han et al., 2008). The secretory TGN targeting seems to adopt the same types of signals as the endocytic TGN targeting—tyrosine and acidic cluster motif—although mechanisms of the two targeting pathways could be distinct, as the former involves retention during cisternal progression, whereas the latter requires retrieval by vesicular trafficking.
A possible model to account for our intra-Golgi trafficking kinetic data is that the anterograde membrane flow from the Golgi stack to the TGN could be selective. Whereas PM targeted cargoes could exit before and at the trans-Golgi, cargoes possessing TGN targeting signals could enter and exit at the TGN. Our model is in contrast to the conventional view that the TGN is the Golgi exit site at which secretory cargoes are sorted to various post-Golgi compartments (Griffiths and Simons, 1986). This view was mainly developed from immuno-EM studies of virus spike proteins such as VSVG in the 1980s (Griffiths et al., 1985). However, we argue here that the precise Golgi exit site of cargoes has not been clearly resolved, and the question is open. The TGN was morphologically defined as the trans-most Golgi cisterna together with its attached tubular and reticular network of membrane (Griffiths and Simons, 1986; De Matteis and Luini, 2008). This definition could be inconsistent with the modern view of the TGN, which is marked by clathrin and accessory proteins (Klumperman, 2011). Owing to technical limitations, that early EM work was conducted using single thin sections and lacked immunogold EM colocalization within the same cells. It is now known from the EM tomography of the Golgi that trans-Golgi cisternae can also have budding profiles (Ladinsky et al., 2002). Therefore it might have been difficult in that early work to determine from the morphology whether the cisterna plus tubular profile of VSVG staining is the TGN or the trans-Golgi with its associated budding profile. In contrast, the definition of the TGN in GLIM is based on the LQs of molecular markers. Consistent with our model, the EM tomography data of the Golgi stack at 20°C revealed that the trans-Golgi instead of the TGN could be the cargo exit site of the Golgi (Ladinsky et al., 2002). Recently, based on EM tomography data, it has been proposed that the trans-Golgi could be the exit site for constitutively secreted cargoes such as VSVG, whereas the TGN could serve as the exit site for endosome/lysosome- or secretory granule–targeted proteins (Mogelsvang et al., 2004; Klumperman, 2011).
An alternative interpretation of our intra-Golgi trafficking kinetic data is that both constitutive and TGN cargoes can transit the TGN but with distinct residence times. Constitutive cargoes can be rapidly packed into transport carriers destined to the PM, so that they have a much shorter residence time at the TGN than the Golgi stack. Therefore constitutive cargoes appear depleted at the TGN. On the other hand, TGN cargoes can have a longer residence time at the TGN due to the retention by their TGN targeting signals. With our current technology, it seems impossible to completely rule out this alternative model. However, GLIM clearly revealed the differential anterograde trafficking of PM targeted from TGN-targeted cargoes, an observation not easily produced by available alternative approaches.
Our TGN model predicts that posttranslational modification reactions for constitutive cargoes, such as sialylation, fucosylation, sulfation, and endoproteolytic cleavage, could take place in the Golgi stack instead of the TGN. Although some early studies localized these reactions at the TGN, we argue that precise subcellular locations of those posttranslational modifications should be reexamined, and some of them could take place in the trans-Golgi instead of the previously reported TGN. We are in the process of defining the precise locations of these reactions by GLIM. Indeed, LQs of TPST1/2 and SialT suggest that protein sulfation and sialylation could take place in the Golgi stack. The new intra-Golgi transport data generated by GLIM could also be used to assess or test previously proposed Golgi models. In a recently proposed rapid partitioning model, a secretory cargo is expected to evenly distribute throughout the entire Golgi complex upon its entry of the Golgi, whereas the cisternal progression model predicts that a cargo wave propagates from the cis- to the trans-Golgi (Patterson et al., 2008). Our intra-Golgi trafficking data on synchronized membrane cargoes provide direct evidence of progressive cargo waves moving from the cis- to the trans-Golgi, therefore the cisternal progression model. To our knowledge, this is the first study to resolve quantitatively intra-Golgi transport under a conventional light microscope. We believe that GLIM can help to pave the way to quantitative study of the Golgi and resolve contested views on the intra-Golgi transport.
MATERIALS AND METHODS
DNA plasmids
The following mammalian expression DNA plasmids were generous gifts: Sec31a-GFP and Vamp4-GFP (W. Hong; Tran et al., 2007), GFP-ERGIC53 (H. Hauri; Ben-Tekaya et al., 2005), GFP-GM130 (M. De Matties; Marra et al., 2001), GFP-Golgin84 (M. Lowe; Diao et al., 2003), GFP-GPP130 (A. Linstedt; Linstedt et al., 1997), GFP-KDELR and VSVGtsO45-GFP (J. Lippincott-Schwartz; Presley et al., 1997), piRFP670-N1 (V. Verkhusha; Addgene [Cambridge, United Kingdom] plasmid #45457; Shcherbakova and Verkhusha, 2013), pME-Flag-CD59-GPI (R. Watanabe; Addgene plasmid #50378; Rivier et al., 2010), TPST1/2-GFP (D. Stephens; Addgene plasmid #66617/66618), and ss-Strep-KDEL_ss-SBP-GFP-E-cadherin, and Ii-Strep_TNFα-SBP-GFP (F. Perez; Boncompain et al., 2012). CD8a-CI-M6PR (Mahajan et al., 2013) and GFP-Golgin97 (Lu et al., 2006) were previously described. To clone GalT-mCherry, an insert containing the coding sequence of amino acids 1–81 of β1,4-galactosyltransferase (GalT) was released by digesting GalT-tdTomato (Lu et al., 2013) using NheI/BamHI and subsequently ligated into pmCherry-N1 vector (Takara Bio, Shiga, Japan) at the same restriction sites. To clone GalT-HA, a duplex DNA oligonucleotide that encodes a HA tag and possesses BamHI and NotI overhangs was made by annealing two synthetic oligonucleotides. GalT-mCherry was digested by BamHI/NotI to remove mCherry and subsequently ligated with the duplex DNA oligonucleotide to replace mCherry with a HA tag. To clone GalT-iRFP670, the coding sequence of amino acids 1–81 of GalT was amplified by the PCR and ligated into piRFP670-N1 vector (Addgene plasmid #45457) at EcoRI/BamHI sites. To clone GFP-ACBD3, the coding sequence of ACBD3 was released from an IMAGE clone (GenBank Accession No. BC045533) by SpeI, end-blunted, digested by EcoRI, and ligated to pEGFP-C2 (Takara Bio), which was prepared by BamHI digestion, end-blunting, and subsequent EcoRI digestion. To clone SialT-AcGFP, the coding sequence of β-galactosamide α-2,6-sialyltransferase (GenBank Accession No. BC040009) was amplified by the PCR and ligated into pAcGFP1-N1 (Takara Bio) vector at XhoI/BamHI sites. To clone GFP-Rab6, human Rab6 coding sequence was amplified by the PCR and cloned into pEGFP-C2 at EcoRI/BamHI. To clone GFP-GCC88, the 5’ fragment of GCC88 coding sequence was PCR amplified, and the 3’ fragment was released by enzyme digestion of an IMAGE clone (GenBank Accession No. BC014100). The two fragments were connected by a native BamHI site and cloned into pEGFP-C3 vector (Takara Bio) at XhoI/EcoRI sites. To clone pmCherry-GM130, the N-terminal fragment of GM130 was amplified by PCR, and the C-terminal fragment of GM130 was released by digesting GFP-GM130 using BamHI, and the two fragments were ligated into pmCherry-C2 vector (Takara Bio) at XhoI/BamHI sites. To clone RUSH system bicistronic expression construct ss-Strep-KDEL_ss-SBP-GFP-CD8a-furin, the coding sequence for CD8a-furin chimera was amplified by PCR using pCI-neo-CD8a-furin (Mahajan et al., 2013) as the template and ligated into FseI/XhoI-digested ss-Strep-KDEL_ss-SBP-GFP-E-cadherin at FseI/XhoI sites. The Y762KGL-to-A762KGA mutation (tyrosine motif or Y mutation), S776DSEEDE-to-A776DAAAAA mutation (acidic cluster or AC mutation) or double mutation (Y+AC mutation) in ss-SBP-GFP-CD8a-furin was introduced by standard overlapping PCRs. To clone bicistronic expression construct ss-Strep-KDEL_ss-SBP-GFP-CD59, the coding sequence of mature CD59 was amplified by PCR using pME-Flag-CD59-GPI (Addgene plasmid #50378) and ligated into FseI/XbaI-digested ss-Strep-KDEL_ss-SBP-GFP-E-cadherin at FseI/XbaI sites. All PCRs were conducted using Pfu DNA polymerase. All plasmids constructed in this work were confirmed by sequencing.
Antibodies
The following primary antibodies were purchased: mouse monoclonal antibodies (mAbs) against GM130, GS15, GS27, GS28, Vti1a, Syntaxin 6, GGA2 and Golgin245 (BD Bioscience, San Jose, CA); CI-M6PR mouse mAb, furin rabbit polyclonal antibody (pAb), and β-COP rabbit pAb (Thermo Scientific, Waltham, MA); Golgin97 mouse mAb (Invitrogen, Carlsbad, CA); KDELR mouse mAb (StressGen Biotechnologies, San Diego, CA); GM130 rabbit mAb, TGN46 rabbit pAb, and Giantin rabbit pAb (Abcam); ACBD3 rabbit pAb (Sigma-Aldrich, St. Louis, MO); and HA mouse mAb (Santa Cruz Biotechnology, Dallas, TX). CD8a mouse mAb was from OKT8 hybridoma culture supernatant. Arl1 rabbit pAb was previously described (Lu et al., 2001).
Cell culture, immunofluorescence, and transfection
HeLa, BSC1, C2C12, and NRK fibroblast cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). hTERT RPE1 cells were cultured in DMEM/F-12 supplemented with 10% FBS. For immunofluorescence, cells were grown on No. 1.5 12-mm-diameter glass coverslips. Nocodazole 33 μM (Sigma-Aldrich) was used to induce the formation of Golgi ministacks. Cells were fixed by 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). After washing by PBS, the residual amount of PFA left within cells was blocked by NH4Cl. Cells were subsequently processed for immunofluorescence labeling by primary antibodies, followed by Alexa Fluor 488–, 594–, and/or 647–conjugated goat anti-mouse or anti-rabbit secondary antibodies (Invitrogen). Both primary and secondary antibodies were diluted in PBS containing 5% FBS, 2% bovine serum albumin, and 0.1% Saponin (Sigma-Aldrich). Labeled cells were mounted in Mowiol mounting medium containing 12% Mowiol 4-88 (EMD Millipore, Billerica, MA), 30% glycerol, and 100 mM Tris, pH 8.5. After drying of the mounting medium, coverslips were sealed in nail polish and stored at −20°C. Cell transfection was conducted using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s manual.
Imaging
Cells were imaged under a spinning-disk confocal microscope system comprising an Olympus IX81 microscope (Olympus, Tokyo, Japan) equipped with an oil objective lens (100×, 1.45 numerical aperture [NA]), a piezo z-stage, Yokogawa CSU-X1 spinning head, and an electron- multiplying charge coupled device (EMCCD) camera (Evolve; Photometrics, Tucson, AZ). Fluorophores were excited by three lines of 50-mW solid-state lasers: 488, 561 and 640 nm (Sapphire; Coherent, Santa Clara, CA). Emission lights were sequentially collected through a quad-band dichroic mirror and filters (mounted in the filter wheel of CSU-X1) optimized for GFP/Alexa Fluor 488, mCherry/Alexa Fluor 594, and Alexa Fluor 647. The microscope system was controlled by MetaMorph software (Molecular Devices, Sunnyvale, CA). A microruler (Geller MicroAnalytical Laboratory, Topsfield, MA) measured the pixel size to be 89 or 67 nm, depending on the extra magnification placed between the EMCCD camera and the spinning head. The z-step of image stacks was 130 nm. The gain of the EMCCD camera was 1×. The digitizer used was either 5 MHz with EM gain 20–400 or 1.25 MHz standard.
Cells were also imaged under a wide-field microscope system comprising an Olympus IX83 equipped with an oil objective lens (100×, NA 1.40), a motorized stage, motorized filter cubes, an scientific complementary metal oxide semiconductor (sCMOS ) camera (Neo; Andor, Belfast, United Kingdom), and a 200-W metal-halide excitation light source (Lumen Pro 200; Prior Scientific, Rockland, MA). Dichroic mirrors and filters in filter turrets were optimized for GFP/Alexa Fluor 488, mCherry/Alexa Fluor 594, and Alexa Fluor 647. The microscope system was controlled by MetaMorph software and only center quadrant of the sCMOS sensor was used for imaging. The pixel size was measured by the microruler to be 64 nm. The z-step of image stacks was 200 nm. Camera settings were rolling shutter, 200-MHz digitizer, and 16 bit (low noise, high well capacity).
Maximum excitation illumination power was used for both spinning-disk confocal and wide-field microscopy. Settings of camera and exposure time were chosen according to the brightness of samples to ensure that the resulting image had a large dynamic range. Typically, exposure time of 100–400 ms or 1–5 s was used for spinning-disk confocal or wide-field imaging, respectively.
The superresolution 3D-SIM was conducted using an Elyra PS.1 (Zeiss, Jena, Germany) equipped with an oil Plan-Apochromat objective lens (100×, NA 1.46), EMCCD camera, and three lines of excitation lasers (488, 561, and 642 nm) under the control of Zen 2012 software (Zeiss). The chromatic shift aberration of the system was calibrated and corrected by TetraSpeck beads (110 nm in diameter; Invitrogen). Image stacks were acquired. The pixel size was 25 nm. The z-step of image stacks was 100 nm. The intensity line profile of the Golgi ribbon imaged by superresolution 3D-SIM was generated using ImageJ (National Institutes of Health, Bethesda, MD).
Live-cell imaging
HeLa cells grown on a glass-bottom Petri dish (MatTek, Ashland, MA) were cotransfected with DNA plasmids encoding mCherry-GM130, GalT-iRFP670, and GFP-Golgin84 using Lipofectamine 2000. After ∼20 h, cells were treated with nocodazole for 2.5 h and imaged in phenol red–free Leibovitz-15 medium (Invitrogen) supplemented with 10% FBS and nocodazole at 34°C. Two-dimensional time-lapse images were acquired under the spinning-disk confocal microscope. The chromatic shift was corrected as described later (Microscope chromatic shift calibration).
VSVG trafficking assays
HeLa cells were cotransfected using DNA plasmids encoding VSVGtsO45-GFP and GalT-mCherry and incubated at 40°C overnight. After nocodazole treatment for 3 h at 40°C, four types of VSVG transport assays were subsequently conducted in the continuous presence of nocodazole. 1) Cells were transferred to 32°C for various lengths of time (chase). 2) To inhibit the synthesis of new VSVG-GFP, cells were transferred to 32°C to chase in the presence of 10 μg/ml cycloheximide (Sigma-Aldrich). 3) For the 15°C temperature-block experiment, cells were incubated at 15°C for 1 h to accumulate VSVG-GFP at the ERGIC and subsequently shifted to 40°C to chase. 4) For the 20°C temperature-block experiment, cells were incubated at 20°C for 2 h and subsequently shifted to 32°C to chase. The chase was conducted until VSVG-GFP that remained in the Golgi was too weak to be reliably analyzed. Cells were fixed and processed for immunofluorescence labeling of GM130.
To quantify the fraction of VSVG-GFP in the Golgi in Figure 6B, cells were imaged under the wide-field microscope. ROIs of the Golgi were segmented using GM130 intensity in ImageJ. VSVG-GFP–positive cells were identified, and, in each cell, the fraction of VSVG-GFP within the Golgi ROI versus the total cellular VSVG-GFP was calculated at each time point of the chase.
Synchronization of protein secretion by the RUSH system
HeLa cells were cotransfected with a DNA plasmid encoding GalT-mCherry and a RUSH system bicistronic expression plasmid—Ii-Strep_TNFα-SBP-GFP, ss-Strep-KDEL_ss-SBP-GFP-E-cadherin, ss-Strep-KDEL_ss-SBP-GFP-CD59, ss-Strep-KDEL_ss-SBP-GFP-CD8a-furin, ss-Strep-KDEL_ss-SBP-GFP-CD8a-furin (Y mutation), ss-Strep-KDEL_ss-SBP-GFP-CD8a-furin (AC mutation), or ss-Strep-KDEL_ss-SBP-GFP-CD8a-furin (Y+AC mutation)—using Lipofectamine2000. Streptavidin, 50 ng/ml, was added to the culture medium to neutralize free biotin. After 20 h, cells were treated with nocodazole in the presence of 50 ng/ml streptavidin for 3 h, followed by 40 μM biotin in the presence of nocodazole and 10 μg/ml cycloheximide to release the reporter into the secretory pathway. Cells were fixed after chase and processed for GM130 immunofluorescence labeling and imaging.
Image analysis for Golgi ministacks
Three-color image (GM130, GalT-mCherry, and test protein x) stacks were visually examined in ImageJ to find the plane that contained ministacks of interest. Three consecutive z-sections centering this plane were averaged (average z-projection). The image in each channel was subsequently subjected to global background subtraction, so that only ministacks of interest had nonzero values, whereas pixels of background were all zero. The color-merged image was generated, and Golgi ministacks were visually inspected. Those that displayed only one object in each channel were manually masked by ROIs. Golgi ministacks are inhomogeneous, and a significant number of them were not selected: 1) large, bright Golgi chunks; 2) those displaying ring or crescent shapes; 3) those with tightly adjacent fluorescence structures, such as neighbor ministacks or endosomes, as many Golgi proteins also localize to endosomes; and 4) those missing one of the three fluorescence signals. Typically, ∼10 Golgi ministacks could be selected from each cell. For our live-cell imaging analysis, Golgi ministacks in the 2D time- lapse images were manually tracked.
The center of mass and the total intensity of the Golgi ministack within each ROI were calculated in ImageJ. The xy-coordinates of centers were subjected to a calibrated first-order polynomial function (Supplemental Note S1) to correct the chromatic shift (see later description, Microscope chromatic shift calibration). Shift-corrected coordinates of three channels were exported to OriginPro8.5 (OriginLab, Northampton, MA) for further analysis. We calculated dx, d1, and tan α or tan β (Figure 1, E and F) using shift-corrected x- and y-coordinates. Analyzable Golgi ministacks were selected if they fulfill the following three empirical criteria described in Results: 1) the signal-to-noise ratio criterion, in which the ratio of the total intensity of a ministack to the SD of the background must be ≥30 in each channel; 2) the axial angle or distance criterion, in which d1 must be ≥70 nm; and 3) the collinearity criterion, in which either |tanα| or |tanβ| must be ≤0.3. Typically, 50–70% ROI–masked Golgi ministacks fulfilled these criteria. The LQ of test protein x in an analyzable ministack was calculated as dx/d1, where dx is the projected distance from the center of x to that of GM130 and d1 is the distance from the center of GM130 to that of GalT-mCherry (Figure 1, D–F). LQs were further statistically analyzed for their means, SEMs, and histograms in OriginPro8.5. In the intra-Golgi transport assay using the RUSH system, the LQ versus time plot was fitted in OriginPro8.5 by a single-exponential function, y = y0 + A1 exp[−(x − x0)/t1], where y0 is the LQ plateau.
Microscope chromatic shift calibration
TetraSpeck beads (110 nm in diameter; Invitrogen) were spread onto the surface of a glass coverslip in Mowiol mounting medium. Three-color image stacks of beads were acquired before and after each Golgi imaging session, and these images were combined for analysis. Similar to the method described earlier for image analysis of Golgi ministacks, bead images were sequentially subjected to z-projection, background subtraction, merge, and single-bead ROI selection to obtain xy-coordinates of centers of mass in three channels. These xy-coordinates were fitted by a first-order polynomial function in MATLAB (MathWorks; Supplemental Note S1) for calibration, with the assumption that xy-coordinates of red fluorescence are true positions of beads. In practice, this was proposed as a least square–fitting problem and solved using QR matrix decomposition (Golub and Van Loan, 1996). After calibration, the resulting function was used to correct the chromatic shift of xy-coordinates obtained in the same imaging session.
Projection of unit-length 3D line segment
Assume that an infinite number of randomly oriented unit-length 3D line segments are projected into a single 2D plane. The mean 2D length after the projection is computed as
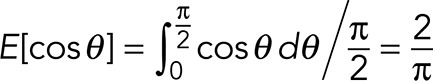 |
where θ is its orientation with respect to the 2D plane. As a result, for a projected 2D line segment, its length in original 3D space is calculated by multiplying with π/2.
Supplementary Material
Acknowledgments
We thank W. Hong (Institute of Molecular and Cell Biology, Singapore) for sharing the spinning-disk confocal microscope; W. Hong and H. Hauri (University of Basel, Basel, Switzerland), M. De Matties (Consorzio Mario Negri Sud, Santa Maria Imbaro, Italy), M. Lowe (University of Manchester, Manchester, United Kingdom), A. Linstedt (Carnegie Mellon University, Pittsburgh, PA), J. Lippincott-Schwartz (National Institutes of Health, Bethesda, MD), V. Verkhusha (Albert Einstein College of Medicine, New York, NY), R. Watanabe (University of Geneva, Geneva, Switzerland), D. Stephens (University of Bristol, Bristol, United Kingdom), and F. Perez (Institut Curie, Paris, France) for providing DNA plasmids; and T. Wohland (National University of Singapore, Singapore) and F. Bard (Institute of Molecular and Cell Biology, Singapore) for helpful discussions. This work was supported by grants from the National Medical Research Council (NMRC/CBRG/007/2012) and Ministry of Education (AcRF Tier1 RG 18/11 and RG 48/13) to L.L.
Abbreviations used:
- CI-M6PR
cation-independent mannose 6-phosphate receptor
- ERES
endoplasmic reticulum exit site
- ERGIC
endoplasmic reticulum–Golgi intermediate compartment
- GalT
β1,4-galactosyltransferase
- GLIM
Golgi protein localization by imaging centers of mass
- GPI
glycosylphosphatidylinositol
- KDELR
KDEL receptor
- LQ
localization quotient
- PM
plasma membrane
- ROI
region of interest
- RUSH
retention using selective hooks
- SBP
streptavidin-binding peptide
- ss
signal sequence
- TGN
trans-Golgi network
- VSVG
vesicular stomatitis virus protein G.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-09-0664) on January 13, 2016.
REFERENCES
- Antony C, Cibert C, Geraud G, Santa Maria A, Maro B, Mayau V, Goud B. The small GTP-binding protein rab6p is distributed from medial Golgi to the trans-Golgi network as determined by a confocal microscopic approach. J Cell Sci. 1992;103:785–796. doi: 10.1242/jcs.103.3.785. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Huttner WB. Tyrosine sulfation is a trans-Golgi-specific protein modification. J Cell Biol. 1987;105:2655–2664. doi: 10.1083/jcb.105.6.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers CJ, Keller DS, Balch WE. Semi-intact cells permeable to macromolecules: use in reconstitution of protein transport from the endoplasmic reticulum to the Golgi complex. Cell. 1987;50:523–534. doi: 10.1016/0092-8674(87)90025-0. [DOI] [PubMed] [Google Scholar]
- Ben-Tekaya H, Miura K, Pepperkok R, Hauri HP. Live imaging of bidirectional traffic from the ERGIC. J Cell Sci. 2005;118:357–367. doi: 10.1242/jcs.01615. [DOI] [PubMed] [Google Scholar]
- Bock JB, Klumperman J, Davanger S, Scheller RH. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol Biol Cell. 1997;8:1261–1271. doi: 10.1091/mbc.8.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, Mercanti V, Jollivet F, Raposo G, Perez F. Synchronization of secretory protein traffic in populations of cells. Nat Methods. 2012;9:493–498. doi: 10.1038/nmeth.1928. [DOI] [PubMed] [Google Scholar]
- Brown DL, Heimann K, Lock J, Kjer-Nielsen L, van Vliet C, Stow JL, Gleeson PA. The GRIP domain is a specific targeting sequence for a population of trans-Golgi network derived tubulo-vesicular carriers. Traffic. 2001;2:336–344. doi: 10.1034/j.1600-0854.2001.002005336.x. [DOI] [PubMed] [Google Scholar]
- Brown WJ. Cation-independent mannose 6-phosphate receptors are concentrated in trans Golgi elements in normal human and I-cell disease fibroblasts. Eur J Cell Biol. 1990;51:201–210. [PubMed] [Google Scholar]
- Cole NB, Sciaky N, Marotta A, Song J, Lippincott-Schwartz J. Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol Biol Cell. 1996;7:631–650. doi: 10.1091/mbc.7.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejgaard SY, Murshid A, Dee KM, Presley JF. Confocal microscopy-based linescan methodologies for intra-Golgi localization of proteins. J Histochem Cytochem. 2007;55:709–719. doi: 10.1369/jhc.6A7090.2007. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC, Puertollano R, Mullins C, Aguilar RC, Vargas JD, Hartnell LM, Bonifacino JS. GGAs: a family of ADP ribosylation factor-binding proteins related to adaptors and associated with the Golgi complex. J Cell Biol. 2000;149:81–94. doi: 10.1083/jcb.149.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis MA, Luini A. Exiting the Golgi complex. Nat Rev Mol Cell Biol. 2008;9:273–284. doi: 10.1038/nrm2378. [DOI] [PubMed] [Google Scholar]
- de Silva AM, Balch WE, Helenius A. Quality control in the endoplasmic reticulum: folding and misfolding of vesicular stomatitis virus G protein in cells and in vitro. J Cell Biol. 1990;111:857–866. doi: 10.1083/jcb.111.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao A, Rahman D, Pappin DJ, Lucocq J, Lowe M. The coiled-coil membrane protein golgin-84 is a novel rab effector required for Golgi ribbon formation. J Cell Biol. 2003;160:201–212. doi: 10.1083/jcb.200207045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BS, Luini A. Models for Golgi traffic: a critical assessment. Cold Spring Harb Perspect Biol. 2011;3:a005215. doi: 10.1101/cshperspect.a005215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub GH, Van Loan CF. Matrix Computations. Baltimore, MD: Johns Hopkins University Press; 1996. [Google Scholar]
- Griffiths G, Ericsson M, Krijnse-Locker J, Nilsson T, Goud B, Soling HD, Tang BL, Wong SH, Hong W. Localization of the Lys, Asp, Glu, Leu tetrapeptide receptor to the Golgi complex and the intermediate compartment in mammalian cells. J Cell Biol. 1994;127:1557–1574. doi: 10.1083/jcb.127.6.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, Pfeiffer S, Simons K, Matlin K. Exit of newly synthesized membrane proteins from the trans cisterna of the Golgi complex to the plasma membrane. J Cell Biol. 1985;101:949–964. doi: 10.1083/jcb.101.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, Simons K. The trans Golgi network: sorting at the exit site of the Golgi complex. Science. 1986;234:438–443. doi: 10.1126/science.2945253. [DOI] [PubMed] [Google Scholar]
- Han J, Wang Y, Wang S, Chi C. Interaction of Mint3 with Furin regulates the localization of Furin in the trans-Golgi network. J Cell Sci. 2008;121:2217–2223. doi: 10.1242/jcs.019745. [DOI] [PubMed] [Google Scholar]
- Hauri HP, Kappeler F, Andersson H, Appenzeller C. ERGIC-53 and traffic in the secretory pathway. J Cell Sci. 2000;113:587–596. doi: 10.1242/jcs.113.4.587. [DOI] [PubMed] [Google Scholar]
- Hay JC, Klumperman J, Oorschot V, Steegmaier M, Kuo CS, Scheller RH. Localization, dynamics, and protein interactions reveal distinct roles for ER and Golgi SNAREs. J Cell Biol. 1998;141:1489–1502. doi: 10.1083/jcb.141.7.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Lui WW, Bright NA, Totty N, Seaman MN, Robinson MS. A family of proteins with gamma-adaptin and VHS domains that facilitate trafficking between the trans-Golgi network and the vacuole/lysosome. J Cell Biol. 2000;149:67–80. doi: 10.1083/jcb.149.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J. Architecture of the mammalian Golgi. Cold Spring Harb Perspect Biol. 2011;3:a005181. doi: 10.1101/cshperspect.a005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis TE, Lodish HF. Oligomerization is essential for transport of vesicular stomatitis viral glycoprotein to the cell surface. Cell. 1986;46:929–937. doi: 10.1016/0092-8674(86)90075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreykenbohm V, Wenzel D, Antonin W, Atlachkine V, von Mollard GF. The SNAREs vti1a and vti1b have distinct localization and SNARE complex partners. Eur J Cell Biol. 2002;81:273–280. doi: 10.1078/0171-9335-00247. [DOI] [PubMed] [Google Scholar]
- Ladinsky MS, Mastronarde DN, McIntosh JR, Howell KE, Staehelin LA. Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J Cell Biol. 1999;144:1135–1149. doi: 10.1083/jcb.144.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky MS, Wu CC, McIntosh S, McIntosh JR, Howell KE. Structure of the Golgi and distribution of reporter molecules at 20 degrees C reveals the complexity of the exit compartments. Mol Biol Cell. 2002;13:2810–2825. doi: 10.1091/mbc.01-12-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt AD, Mehta A, Suhan J, Reggio H, Hauri HP. Sequence and overexpression of GPP130/GIMPc: evidence for saturable pH-sensitive targeting of a type II early Golgi membrane protein. Mol Biol Cell. 1997;8:1073–1087. doi: 10.1091/mbc.8.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Hannoush RN, Goess BC, Varadarajan S, Shair MD, Kirchhausen T. The small molecule dispergo tubulates the endoplasmic reticulum and inhibits export. Mol Biol Cell. 2013;24:1020–1029. doi: 10.1091/mbc.E12-08-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Hong W. Interaction of Arl1-GTP with GRIP domains recruits autoantigens Golgin-97 and Golgin-245/p230 onto the Golgi. Mol Biol Cell. 2003;14:3767–3781. doi: 10.1091/mbc.E03-01-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Hong W. From endosomes to the trans-Golgi network. Semin Cell Dev Biol. 2014;31:30–39. doi: 10.1016/j.semcdb.2014.04.024. [DOI] [PubMed] [Google Scholar]
- Lu L, Horstmann H, Ng C, Hong W. Regulation of Golgi structure and function by ARF-like protein 1 (Arl1) J Cell Sci. 2001;114:4543–4555. doi: 10.1242/jcs.114.24.4543. [DOI] [PubMed] [Google Scholar]
- Lu L, Tai G, Wu M, Song H, Hong W. Multilayer interactions determine the Golgi localization of GRIP golgins. Traffic. 2006;7:1399–1407. doi: 10.1111/j.1600-0854.2006.00473.x. [DOI] [PubMed] [Google Scholar]
- Luke MR, Kjer-Nielsen L, Brown DL, Stow JL, Gleeson PA. GRIP domain-mediated targeting of two new coiled-coil proteins, GCC88 and GCC185, to subcompartments of the trans-Golgi network. J Biol Chem. 2003;278:4216–4226. doi: 10.1074/jbc.M210387200. [DOI] [PubMed] [Google Scholar]
- Luzio JP, Brake B, Banting G, Howell KE, Braghetta P, Stanley KK. Identification, sequencing and expression of an integral membrane protein of the trans-Golgi network (TGN38) Biochem J. 1990;270:97–102. doi: 10.1042/bj2700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan D, Boh BK, Zhou Y, Chen L, Cornvik TC, Hong W, Lu L. Mammalian Mon2/Ysl2 regulates endosome-to-Golgi trafficking but possesses no guanine nucleotide exchange activity toward Arl1 GTPase. Sci Rep. 2013;3:3362. doi: 10.1038/srep03362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra P, Maffucci T, Daniele T, Tullio GD, Ikehara Y, Chan EK, Luini A, Beznoussenko G, Mironov A, De Matteis MA. The GM130 and GRASP65 Golgi proteins cycle through and define a subdomain of the intermediate compartment. Nat Cell Biol. 2001;3:1101–1113. doi: 10.1038/ncb1201-1101. [DOI] [PubMed] [Google Scholar]
- Martinez-Menarguez JA, Prekeris R, Oorschot VM, Scheller R, Slot JW, Geuze HJ, Klumperman J. Peri-Golgi vesicles contain retrograde but not anterograde proteins consistent with the cisternal progression model of intra-Golgi transport. J Cell Biol. 2001;155:1213–1224. doi: 10.1083/jcb.200108029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin KS, Simons K. Reduced temperature prevents transfer of a membrane glycoprotein to the cell surface but does not prevent terminal glycosylation. Cell. 1983;34:233–243. doi: 10.1016/0092-8674(83)90154-x. [DOI] [PubMed] [Google Scholar]
- Mogelsvang S, Marsh BJ, Ladinsky MS, Howell KE. Predicting function from structure: 3D structure studies of the mammalian Golgi complex. Traffic. 2004;5:338–345. doi: 10.1111/j.1398-9219.2004.00186.x. [DOI] [PubMed] [Google Scholar]
- Molloy SS, Thomas L, VanSlyke JK, Stenberg PE, Thomas G. Intracellular trafficking and activation of the furin proprotein convertase: localization to the TGN and recycling from the cell surface. EMBO J. 1994;13:18–33. doi: 10.1002/j.1460-2075.1994.tb06231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T, Pypaert M, Hoe MH, Slusarewicz P, Berger EG, Warren G. Overlapping distribution of two glycosyltransferases in the Golgi apparatus of HeLa cells. J Cell Biol. 1993;120:5–13. doi: 10.1083/jcb.120.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprins A, Duden R, Kreis TE, Geuze HJ, Slot JW. Beta-COP localizes mainly to the cis-Golgi side in exocrine pancreas. J Cell Biol. 1993;121:49–59. doi: 10.1083/jcb.121.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Hirschberg K, Polishchuk RS, Gerlich D, Phair RD, Lippincott-Schwartz J. Transport through the Golgi apparatus by rapid partitioning within a two-phase membrane system. Cell. 2008;133:1055–1067. doi: 10.1016/j.cell.2008.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Hui N, Hunte F, Kieckbusch R, Berger EG, Warren G, Nilsson T. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides. J Cell Sci. 1995;108:1617–1627. doi: 10.1242/jcs.108.4.1617. [DOI] [PubMed] [Google Scholar]
- Rivier AS, Castillon GA, Michon L, Fukasawa M, Romanova-Michaelides M, Jaensch N, Hanada K, Watanabe R. Exit of GPI-anchored proteins from the ER differs in yeast and mammalian cells. Traffic. 2010;11:1017–1033. doi: 10.1111/j.1600-0854.2010.01081.x. [DOI] [PubMed] [Google Scholar]
- Rogalski AA, Bergmann JE, Singer SJ. Effect of microtubule assembly status on the intracellular processing and surface expression of an integral protein of the plasma membrane. J Cell Biol. 1984;99:1101–1109. doi: 10.1083/jcb.99.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J, Berger EG. Immunocytochemical localization of galactosyltransferase in HeLa cells: codistribution with thiamine pyrophosphatase in trans-Golgi cisternae. J Cell Biol. 1982;93:223–229. doi: 10.1083/jcb.93.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste J, Kuismanen E. Pre- and post-Golgi vacuoles operate in the transport of Semliki Forest virus membrane glycoproteins to the cell surface. Cell. 1984;38:535–549. doi: 10.1016/0092-8674(84)90508-7. [DOI] [PubMed] [Google Scholar]
- Schafer W, Stroh A, Berghofer S, Seiler J, Vey M, Kruse ML, Kern HF, Klenk HD, Garten W. Two independent targeting signals in the cytoplasmic domain determine trans-Golgi network localization and endosomal trafficking of the proprotein convertase furin. EMBO J. 1995;14:2424–2435. doi: 10.1002/j.1460-2075.1995.tb07240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermelleh L, Heintzmann R, Leonhardt H. A guide to super-resolution fluorescence microscopy. J Cell Biol. 2010;190:165–175. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A, Fransen JA, Bachi T, Ginsel L, Hauri HP. Identification, by a monoclonal antibody, of a 53-kD protein associated with a tubulo-vesicular compartment at the cis-side of the Golgi apparatus. J Cell Biol. 1988;107:1643–1653. doi: 10.1083/jcb.107.5.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shcherbakova DM, Verkhusha VV. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat Methods. 2013;10:751–754. doi: 10.1038/nmeth.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohda M, Misumi Y, Yamamoto A, Yano A, Nakamura N, Ikehara Y. Identification and characterization of a novel Golgi protein, GCP60, that interacts with the integral membrane protein giantin. J Biol Chem. 2001;276:45298–45306. doi: 10.1074/jbc.M108961200. [DOI] [PubMed] [Google Scholar]
- Starr T, Forsten-Williams K, Storrie B. Both post-Golgi and intra-Golgi cycling affect the distribution of the Golgi phosphoprotein GPP130. Traffic. 2007;8:1265–1279. doi: 10.1111/j.1600-0854.2007.00607.x. [DOI] [PubMed] [Google Scholar]
- Steegmaier M, Klumperman J, Foletti DL, Yoo JS, Scheller RH. Vesicle-associated membrane protein 4 is implicated in trans-Golgi network vesicle trafficking. Mol Biol Cell. 1999;10:1957–1972. doi: 10.1091/mbc.10.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang BL, Zhang T, Low DY, Wong ET, Horstmann H, Hong W. Mammalian homologues of yeast sec31p. An ubiquitously expressed form is localized to endoplasmic reticulum (ER) exit sites and is essential for ER-Golgi transport. J Biol Chem. 2000;275:13597–13604. doi: 10.1074/jbc.275.18.13597. [DOI] [PubMed] [Google Scholar]
- Teuchert M, Schafer W, Berghofer S, Hoflack B, Klenk HD, Garten W. Sorting of furin at the trans-Golgi network. Interaction of the cytoplasmic tail sorting signals with AP-1 Golgi-specific assembly proteins. J Biol Chem. 1999;274:8199–8207. doi: 10.1074/jbc.274.12.8199. [DOI] [PubMed] [Google Scholar]
- Tran TH, Zeng Q, Hong W. VAMP4 cycles from the cell surface to the trans-Golgi network via sorting and recycling endosomes. J Cell Sci. 2007;120:1028–1041. doi: 10.1242/jcs.03387. [DOI] [PubMed] [Google Scholar]
- Trucco A, Polishchuk RS, Martella O, Di Pentima A, Fusella A, Di Giandomenico D, San Pietro E, Beznoussenko GV, Polishchuk EV, Baldassarre M, et al. Secretory traffic triggers the formation of tubular continuities across Golgi sub-compartments. Nat Cell Biol. 2004;6:1071–1081. doi: 10.1038/ncb1180. [DOI] [PubMed] [Google Scholar]
- Van De Moortele S, Picart R, Tixier-Vidal A, Tougard C. Nocodazole and taxol affect subcellular compartments but not secretory activity of GH3B6 prolactin cells. Eur J Cell Biol. 1993;60:217–227. [PubMed] [Google Scholar]
- Volchuk A, Ravazzola M, Perrelet A, Eng WS, Di Liberto M, Varlamov O, Fukasawa M, Engel T, Sollner TH, Rothman JE, Orci L. Countercurrent distribution of two distinct SNARE complexes mediating transport within the Golgi stack. Mol Biol Cell. 2004;15:1506–1518. doi: 10.1091/mbc.E03-08-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.