

Abstract
Motivation: Leveraging the large compendium of genomic data to predict biomedical pathways and specific mechanisms of protein interactions genome-wide in metazoan organisms has been challenging. In contrast to unicellular organisms, biological and technical variation originating from diverse tissues and cell-lineages is often the largest source of variation in metazoan data compendia. Therefore, a new computational strategy accounting for the tissue heterogeneity in the functional genomic data is needed to accurately translate the vast amount of human genomic data into specific interaction-level hypotheses.
Results: We developed an integrated, scalable strategy for inferring multiple human gene interaction types that takes advantage of data from diverse tissue and cell-lineage origins. Our approach specifically predicts both the presence of a functional association and also the most likely interaction type among human genes or its protein products on a whole-genome scale. We demonstrate that directly incorporating tissue contextual information improves the accuracy of our predictions, and further, that such genome-wide results can be used to significantly refine regulatory interactions from primary experimental datasets (e.g. ChIP-Seq, mass spectrometry).
Availability and implementation: An interactive website hosting all of our interaction predictions is publically available at http://pathwaynet.princeton.edu. Software was implemented using the open-source Sleipnir library, which is available for download at https://bitbucket.org/libsleipnir/libsleipnir.bitbucket.org.
Contact: ogt@cs.princeton.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The molecular activity in a cellular system is maintained by a complex interplay between genes, gene products, metabolites and the environment (Hand and Hardewig, 1996). In particular, intricate biomolecular pathways are formed by a combination of diverse types of mechanistic pairwise interactions, including physical binding in protein–protein complexes (de Lange, 2005), small-molecule-based modifications (Mann and Jensen, 2003) and regulatory actions by activators and repressors (Cowell, 1994). Mapping out these cellular pathways at a whole-genome level is a crucial step for the advancement of human systems biology, aiding at every level from deciphering cellular function to understanding the molecular cause of many complex human diseases.
Functional genomic datasets such as gene expression, cellular localization and DNA/protein binding assays each captures distinct aspects of the cellular activity across multiple cell types and perturbations. However, turning these different instances or ‘views’ of a complex system into an understanding of pathways has proven to be a challenging undertaking. Especially in complex metazoan organisms, such as humans, tissue and cell type-specific expression underlie cellular development, function, and homeostasis (Britten and Davidson, 1969). Consequently, much of the biological and technical variation of the functional genomic data is driven by tissue and cell-lineage heterogeneity. Translating the vast amount of genomic data into specific pathway-level hypotheses thus requires development of algorithmic and statistical approaches that satisfy three requirements: (i) inferring specific individual types of gene interactions, (ii) being scalable to the whole genome and (iii) robust in leveraging biological data from any of the diverse tissue contexts (i.e. experimental results drawn from differing tissues and potentially mixed samples or samples from tissue-culture experiments).
In this work, we developed a tissue-context aggregation-based approach for predicting and studying multiple types of pathway-level gene interactions for metazoan mammalian organisms, specifically applying this approach to the human data compendium. For our approach, we first built a catalog of tissue and interaction-type specific gold standards (e.g. phosphorylation (kinase-substrate) interactions among brain expressed proteins) restricted to gene pairs coexpressed in any of 77 tissues based on curated pathway databases and gene-to-tissue expression profiling. We then utilize a Support Vector Machine (SVM) (Noble, 2006) to generate initial predictions of multiple gene interaction types independently in the context of each tissue (i.e. utilizing the tissue-specific gold standard) by integrating ∼1600 heterogeneous human experimental datasets [e.g. mRNA expression, transcription factor (TF) and kinase motifs and post-translational modifications (PTMs)]. Finally, we aggregate the tissue-context based predictions to obtain the most probable set of interaction labels or each gene pair across the set of pathway-level interaction types (in this study we predict transcriptional regulation, co-complex, phosphorylation and the more general post-translational regulation). Our methodology uniquely allows us to harness the wealth of information in high-throughput genomic data collections by simultaneously separating the heterogeneity originating from diverse tissues to improve signals for predicting different individual interaction types.
To our knowledge, prediction of such genome-wide pathway-level interaction networks in metazoans is an open problem. Many recent studies have begun to address the challenges by inferring or experimentally capturing physical interaction networks (Rhodes et al., 2005; Rual et al., 2005; Schmitt et al., 2014; Stelzl et al., 2005; von Mering et al., 2007), genetic interaction networks (Bassik et al. 2013), Bayesian integration for functional association networks (Date and Stoeckert, 2006; Huttenhower et al., 2009; Lee et al., 2004; Mostafavi et al., 2008; Park et al., 2013; Troyanskaya et al., 2003; Wong et al., 2012) or predicting regulatory networks from specific primary datasets (Margolin et al., 2006; Neph et al., 2012a,b). However, most previous efforts for predicting pathway interactions have been focused on unicellular model organisms (e.g. Escherichia. coli) (Haynes et al., 2013; Marbach et al., 2012), while genome-wide integrated analysis in mammalian organisms have been focused on cross-species integration for inferring multiple types of functional couplings [e.g co-membership to metabolic pathways, signaling pathways or protein–protein interactions (PPI)] (Alexeyenko and Sonnhammer, 2009). No prior integrative method to our knowledge utilizes one of the most significant and important sources of biological variation in human datasets: tissue context. Although several tissue-specific datasets have been generated and analyzed in previous work (Lonsdale et al., 2013; Su et al., 2004), we demonstrate that methodological development is required in addition to the inclusion of tissue-specific data to improve the prediction accuracy of integrated interaction predictions in metazoans. Our work extends the methodological advancements achieved studying pathway interactions among a focused subset of genes or in unicellular model organisms and provides a platform for applying such methods to human data by addressing the challenge of tissue heterogeneity.
Ultimately, we envision that our genome-wide interaction networks cannot only be useful to biology researchers investigating a specific protein or interactions of interest, but also be leveraged to increase the interpretability of new high-throughput studies (ChIP-Seq, proteomics, disease samples, etc.) that capture condition-specific cellular states. As a proof of concept, we demonstrate the utility of our networks by overlaying our interaction networks to identify potential regulatory targets of TFs on 690 ChIP-Seq experimental datasets generated by the ENCODE project (Landt et al., 2012). In addition, we generated the first in vivo derived binding/recognition motifs for cancer-associated TANK-binding kinase 1 (TBK1) by integrating our predicted phosphorylation network with a recent TBK1 knockdown phospho-proteomics study (Kim et al., 2013). Finally, we provide a web-based interface for exploring all our interaction networks and integrative analysis of user data at http://pathwaynet.princeton.edu.
2 Methods
2.1 Tissue-aware integration
The final output of our new prediction pipeline consists of predicted probabilities of gene pair associations for multiple pathway level interaction types (e.g. transcriptional regulation, phosphorylation). Each interaction type network is derived from tissue-aware integration of intermediate per-tissue based SVM classifiers (77 tissues total) that capture tissue-relevant interaction signal while integrating across ∼50 000 genome-scale experiments. Details of the construction of gold standard interaction pairs, tissue context, SVM classifier and input datasets are described in the following sections.
2.1.1 Interaction catalog construction
Unfortunately, there exists no comprehensive curated gold standard repository for known human pathway level interactions. For each interaction type evaluated here, we assembled a gold standard from various sources that have collected experimentally validated interactions. This resulted in 51 525 unique experimentally validated positive interaction labels across four interaction types: transcriptional regulation, phosphorylation, protein co-complexes and post-translational regulation (individual term counts in Supplementary Table S1). Detailed descriptions of interaction type definitions and interaction gold standard construction are provided in Supplementary Information.
All training, evaluation and predictions were limited to the search space of gene pairs in accordance with the protein property that defines each interaction type. Specifically, transcriptional regulation was limited to the subset of gene pairs that included at least one human TF [total 1321 TFs from annotation study (Vaquerizas et al., 2009)]. Likewise, phosphorylation was limited to the gene pairs that included at least one of 514 human kinases (Manning et al., 2002) and post-translational regulation to the subset that included at least 1 of 1881 protein modifying enzymes (e.g. includes the 514 kinases, 217 e3 ligases) (Gene Ontology Consortium, 2004). All genes and its protein products were represented by Entrez gene ids and the final gene-holdout cross-validation background priors used in evaluation were as follows: transcriptional regulation: 0.002; phosphorylation: 0.002; post-translational regulation: 0.0004; co-complex: 0.0005. These priors represent the fraction of known interacting pairs over all positive and negative interactions (negative example construction detailed in Supplementary Information) and were utilized to calculate fold improvement over background for each classification task.
2.1.2 Data sources and preprocessing
We collected a total of 1564 mRNA publically available human expression datasets (Affymetrix U133A and U133 Plus 2.0 platform) from NCBI Gene Expression Omnibus (GEO) (Edgar et al., 2002) (full list of GEO datasets listed in Supplementary Table S2). Expression data were normalized according to the procedure described in (Park et al., 2013).
For non-expression data, data types that closely related to the output interaction type were excluded to avoid any circularity (e.g. no physical interaction data was used in our prediction pipeline as we observe a large overlap of co-complex or phosphorylation positive examples with PPI databases commonly used such as BioGrid (Stark et al., 2011)). Motif information for TFs were obtained from JASPAR (Sandelin et al., 2004), DNaseI profiling (Neph et al., 2012a,b), FastCompare (Elemento and Tavazoie, 2005), CISBP (Ray et al., 2013) and TRANSFAC (Matys et al., 2003), miRNA from MSigDB mir database (Subramanian et al., 2005) and EBI MicroCosm database (Griffiths-Jones et al., 2008) and protein kinases from PhosphoMotif (Amanchy et al., 2007). Protein domain-domain and motif interactions were obtained from PrePPI (Zhang et al., 2013), DOMINE (Yellaboina et al., 2011) and the iELM webserver (Weatheritt et al., 2012). For PrePPI, only the structure-based protein-pair scores were exclusively extracted from PrePPI without including the other types of non-structural data that were later integrated in the study by Zhang et al. (2013). Short linear motifs were identified for all human proteins from iELM. For each occurrence three types of iELM scores were utilized: conservation score (reflecting the overall quality of the motif conservation), relative local conservation score (reflecting the constraint on each residue relative to a window of adjacent residues), and the IUPRED score (indicating the level of protein disorder). PTM potential of proteins was derived by catalogs made from UniProt (Apweiler et al., 2004) and PTMcode (Minguez et al., 2012). To capture the cellular component profile similarity between genes, we took the semantic similarity of Gene Ontology (GO) (Gene Ontology Consortium, 2004) cellular component annotation profile for terms that had more than 100 gene annotations between all gene pairs (each GO term was weighted by the normalized information content,
where i is the gene index and t is the GO term). To capture the phenotype similarity between genes, chemical and genetic perturbation studies curated by the MSigDB (Subramanian et al., 2005) were summarized into gene pairwise similarity phenotype profile scores. Detailed descriptions of how each dataset were used as features provided in Supplementary Information.
2.1.3 Human tissue context construction
In order to capture a wide variety of human tissues in our study, we cataloged genes that are probabilistically identified to be expressed across a set of 77 diverse human tissues utilizing the Gene Expression Barcode methodology (McCall et al., 2011; Zilliox and Irizarry, 2007). This methodology provides a probabilistic framework for determining if an expression value is more likely to come from an expressed or silenced (i.e. unexpressed) distribution, modeled for each gene. Specifically in our study, biologically informative tissue terms were curated from the BRENDA Tissue Ontology (BTO) (Gremse et al., 2011). Next, text-mining of sample descriptions and textual information available in GEO (Edgar et al., 2002) was utilized to annotate expression samples to BTO terms (detailed descriptions of the sample annotation process are provided in the Supplementary Information). Next the Barcode methodology was applied to each expression sample with a tissue BTO term annotation (total 14 092 expression samples) and genes that had an average Barcode probability above 0.7 across tissue annotated expression samples were flagged as transcriptionally active in the tissue (results robust to Barcode cutoff Supplementary Fig. S1).
2.1.4 Tissue-aware data integration
The goal of our integration is to harness the information from the genomic data compendium to predict accurate pathway level interactions. Specifically, the integration is designed to model and exploit the tissue-specific variation across genomic datasets for robust integration in metazoan interactome prediction. Unfortunately, although many diverse types of experimentally validated gene interactions have been curated by multiple databases (Kanehisa and Goto, 2000; Schaefer et al., 2009), the set of tissues in which any given interaction occurs is often not annotated and usually unknown. Thus, we take the approach of generating tissue-specific interaction learning examples by overlaying the Barcode derived tissue contexts onto the known gene interactions. Specifically, for each interaction type, a SVM (Noble, 2006) classifier was trained per tissue context. The training gold standard for each interaction type i and tissue t was define as the following:
where gs is an interaction example for interaction type i, and genes n, m. Tissue contexts t are all genes identified by our Barcode analysis to be transcribed in tissue t and Ubiq (ubiquitous) are genes that are transcribed across all tissues. Thus, a gene pair was considered a tissue-specific interaction example if both genes were expressed in the tissue, while ubiquitous gene interactions (i.e. interactions between genes expressed in all tissues) were treated separately as an independent context to accurately capture tissue-specific variation. Predicting for co-complex, we were unable to separate out ubiquitous gene pair examples due to the high percentage of such pairs ∼84% (transcriptional regulation is ∼35%) in the gold standard.
For each training interaction example a feature vector was constructed from a total of 1590 datasets as described above. Continuous expression features were binned into 0.2 z-score intervals and missing values were set to 0 (Lewis et al., 2006). The set of feature vectors for positive and negative training examples were used to train a linear SVM according to the following formulation:
where n is the training example gene interactions, w is the weight vector for each dataset, yi is the training label of interaction example i and xi is the data vector for all the features for gene pair i. All classifiers were trained using the identical gene-wise 3-fold cross validation split and tissue-contexts with fewer than 30 gene pair cross-validation training examples were dropped. Finally, we merged the tissue-context based intermediate-predictions to obtain the most probable set of labels for each gene pair by assigning the mean predicted score of the top quartile of prediction values across tissue-contexts for each interaction type (performed best compared with other aggregation methods based on our cross-validation results, Supplementary Fig. S2). In addition, one global ‘tissue-unaware’ classifier was trained for each interaction type using the entire gold standard (i.e. non-tissue segmented). Also, we generated bagging-like predictions for each interaction type, where the prediction pipeline (e.g. data) was set constant but each gene tissue expression profile was randomly swapped between genes (resulting in equal number of total tissue contexts and number of gene assignments). All subsequent evaluation analyses were conducted using these gene-wise 3-fold cross-validated networks (i.e. 3-fold cross-validation split of genes with each fold of tissue-aware SVM training were conducted among only gold standard pairs where both genes not part of the held out gene set) to avoid any potential circularity.
2.2 Transcriptional regulation network used for analysis of ChIP-Seq data
Six hundred and ninety ChIP-Seq (Park, 2009) datasets generated by the ENCODE project were used to identify potential TF regulatory targets for a total of 109 TFs. All ChIP-Seq data were handled as ‘Uniform Peaks’ identified based on the ENCODE analysis and normalization pipeline (Landt et al., 2012). A gene was considered a possible target of a TF if the TF’s ChIP-Seq peak overlapped a window surrounding the gene’s transcription start site (TSS). Windows of ±500, 1000 and 2000 bp were used to identify potential regulatory targets. Next, for each TF, specific GO biological process terms (with 5–200 gene annotations) experimentally annotated to that TF was identified. The potential regulatory targets of each TF identified through ChIP-Seq data alone were then tested for enrichment of GO biological processes associated with the TF using a hypergeometric test. Next, our transcriptional regulation network was used to refine ChIP-Seq identified targets by filtering away targets that associated with the TF with a network probability score less than the prior (i.e. no data support). The GO term enrichment analysis was then repeated on the network-refined ChIP-Seq target genes.
2.3 Phosphorylation network used for analysis of phospho-proteomics data
In addition to ChIP-Seq data, we demonstrated the utility of our networks to identify novel phospho-binding or recognition motifs from mass-spectrometry proteomics data. We considered a mass spectrometry experiment that measured altered phosphoproteins following RNAi-mediated knockdown of TBK1 (Kim et al., 2013). In this study, a total of 1154 proteins were identified for a loss of phosphopeptide (PEP score < 0.5 and Mass error < 5 ppm). Protein motif discovery tool FIRE (Elemento et al., 2007; Lieber et al., 2010) was applied to these differentially phosphorylated protein sequences to find enriched motifs that could potentially be TBK1 recognition sites in its targets. Next, similar to the ChIP-Seq example, we refined the mass-spectroscopy targets using our predicted phosphorylation network by filtered out differentially phosphorylated proteins that were linked to TBK1 with a network probability less than the prior. FIRE was re-applied to this filtered set of differentially phosphorylated proteins for discovery of enriched motifs. Identical to the ChIP-Seq analysis, cross-validated networks were used for the analysis.
2.4 Implementation
All software was implemented using the open-source Sleipnir library (Huttenhower et al., 2008), which interfaces with the open-source SVMperf package (Joachims, 2006) for linear kernel SVM classifiers (error parameter C was set to 250 and error-rate loss function was used). All network predictions and evaluations were conducted for the 17 939 genes that were available on the Affymetrix U133A and U133 Plus 2.0 platforms.
3 Results
We demonstrate the advantage of incorporating tissue context for predicting multiple pathway-level interaction types (transcriptional regulation, co-complex, phosphorylation and the more general post-translational regulation). Specifically, we compare our tissue-aware learning approach with a simpler version that does not use information about tissue heterogeneity among human protein coding genes (i.e. tissue-unaware learning). In total, we apply our tissue-aware learning methodology integrating ∼50 000 genome-scale experiments to generate whole-genome networks prioritizing gene/protein interactions with strong supporting evidence for each of the predicted pathway-level interaction types (prediction schematic shown in Fig. 1). We demonstrate the utility of our interaction networks in robustly retrieving pathway members across 447 expert-curated pathways. In addition, we show our interaction networks can be used to accurately identify false positive regulatory targets in primary user datasets generated by ChIP-Seq and Mass spectrometry based phospho-proteomics.
Fig. 1.
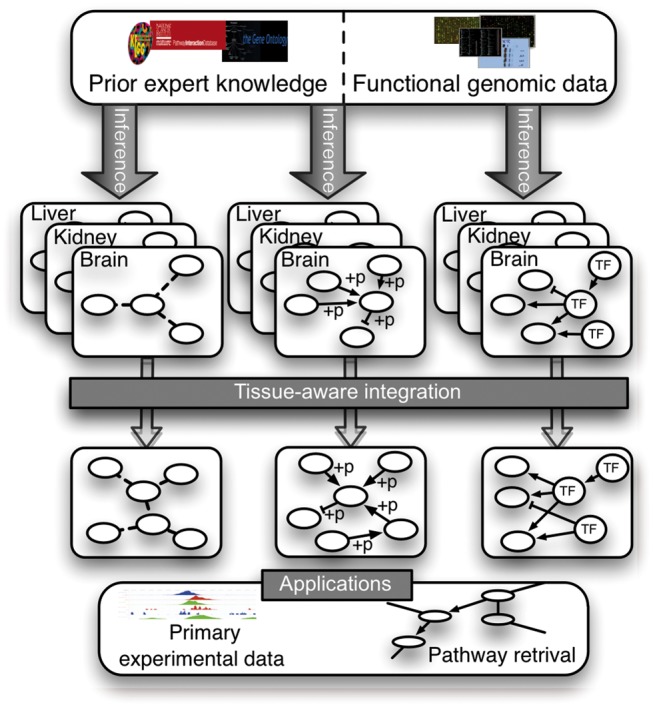
Schematic of our tissue-aware integrative pipeline for inferring metazoan gene interactions. We collect tissue and interaction-type specific gold standards, restricted to protein pairs that are both expressed in the tissue, based on curated databases and expression profiling of total 77 tissues. Next, we infer each interaction type network by integrating the genomic data compendium independently in the context of each tissue (i.e. with tissue-specific learning examples), resulting in multiple intermediate tissue-context networks per interaction-type. Following, we integrate the tissue-context based predictions to obtain the most probable set of labels for each gene pair across the interaction types. Finally, our predicted networks can be used in multiple applications such as pathway retrieval or aiding the interpretation of condition-specific primary datasets
3.1 Tissue-aware learning improves human gene interaction predictions
To address the challenge of predicting pathway-level interactions in metazoans, we ask the question if incorporating gene-level tissue contextual information can improve network prediction. To measure the benefits of this approach, we conducted a 3-fold cross-validation experiment on both our tissue-aware learning method and a simpler tissue-unaware learning method for predicting four interaction types (i.e. transcriptional regulation, phosphorylation, co-complex and post-translational regulation). For each interaction type, we conduct a strict gene-wise holdout evaluation where at each fold the evaluation gold standard pairs include no genes observed during the training stage (i.e. SVM training feature vectors consist of gene-pairs with no overlap with the holdout evaluation set). In addition, identical human data compendium was used for tissue-aware and tissue-unaware learning predictions.
For all four interaction types, there was a significant performance gain when using gene-tissue contextual information [P < 0.01, testing for the difference of area under the curve (Hanley and McNeil, 1982), Fig. 2] with transcriptional regulation showing the largest performance boost in precision over background (over 2-fold) at low recall. Background priors are derived from current knowledge base resources (further detail in Section 2 and Supplementary Information) and provide a baseline performance expected by a random classifier. Our tissue-aware learning method also outperforms a meta-correlation approach, which calculates the average gene pairwise Pearson correlation over the expression datasets (1564 GEO datasets) shown in sky-blue (Fig. 2). We also observe that the performance gain when applying tissue-aware integration is consistent among the subset of tissue-specific interaction examples (i.e. excluding ubiquitously expressed gene pair examples for evaluation) for all four interactions types (Supplementary Fig.re S3). In addition, our tissue-aware learning method outperforms a random bagging-like (Breiman, 1996) approach (i.e. random assignment of tissue profile to gene with number of bags equal to the number of tissue contexts), except in co-complex, which showed comparable performance gains probably due to the significant portion of ubiquitously expressed gene pairs represented in the gold standard ∼84% (Supplementary Fig. S4). The largest performance gain in transcriptional regulation is consistent with our observation (Supplementary Fig. S8) that TFs are generally more tissue specific compared with other classes of genes (e.g. kinases and housekeeping genes). In combination, these evaluations suggest that incorporating tissue-context can significantly improve the accuracy when predicting gene/protein interactions in human, especially when the interaction gold standard is tissue-heterogeneous.
Fig. 2.
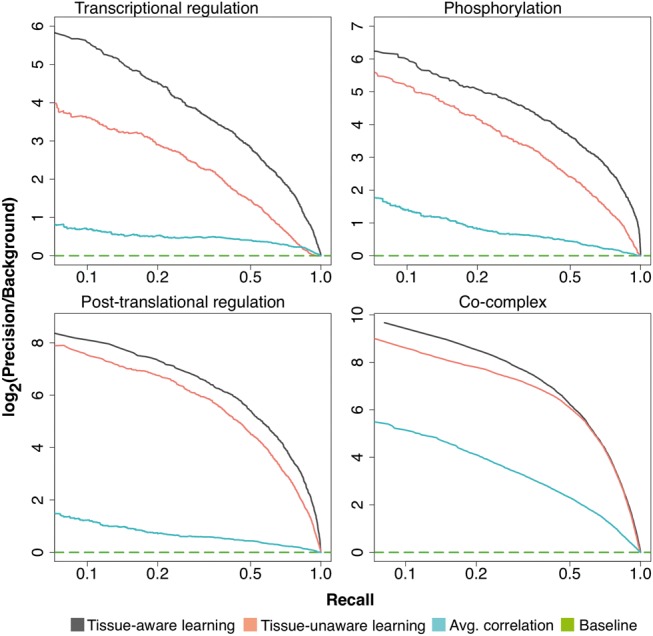
Tissue-aware learning allows improved recovery of pathway interactions. Our tissue-aware learning methodology (black-line) significantly improves prediction accuracy compare to simpler approaches that ignore tissue heterogeneity (labeled tissue-unaware learning and represented as salmon-line) and average correlation across expression datasets (skyblue)
3.2 Enhanced retrieval of cellular pathway components
The concurrent inference of human interaction networks for multiple interaction types allows the generation of specific pathway level hypotheses. For example, often biologists are left with a set of genes that are believed to be functionally associated in a biological process resulting from a high-throughput assay (e.g. differential expression analysis of a multi-condition RNA-Seq experiment). However, understanding the mechanistic connection among a set of functionally related genes has been challenging and requires many experiments that are often extremely time consuming. Thus, it would be of great value if pathway-level predictions inferred from the existing genomic data compendium can be made on any set of genes of interest to an investigator, allowing a systematic prioritization of hypotheses to be experimentally validated.
To address the challenge of pathway component recovery from functionally related genes, we test our ability to prioritize the pathway interactions with known curated human pathways. For example, human FOXO3 is an important TF involved in cell cycle regulation and oxidative stress response along with tumorigenesis and the progression of multiple cancers (Myatt and Lam, 2007). FOXO3 is known to be functionally associated with human kinase AKT1 along with important regulatory genes such as BCL6, GADD45a and YWHAB (Brunet et al., 2001; Fernández de Mattos et al., 2004; Lehtinen et al., 2006). A researcher can investigate the biomolecular interactions among these five clinically important genes, FOXO3, AKT1, BCL6, GADD45a and YWHAB, using our system, which accurately prioritizes many of the interacting pairs with the confirmed interaction type (Fig. 3A, AUC 0.82, full ranked scores in Supplementary Table S3). In addition, such overlay provides a mechanistic hypothesis of the information flow among this set of genes. Starting with kinase AKT1 activating the transcriptional complex FOXO3-YWHAB through phosphorylation, next, the activated FOXO3-YWHAB complex to regulate the expression of targets BCL6 and GADD45A, connecting Akt1 to many downstream cellular processes such as DNA damage and apoptosis.
Fig. 3.
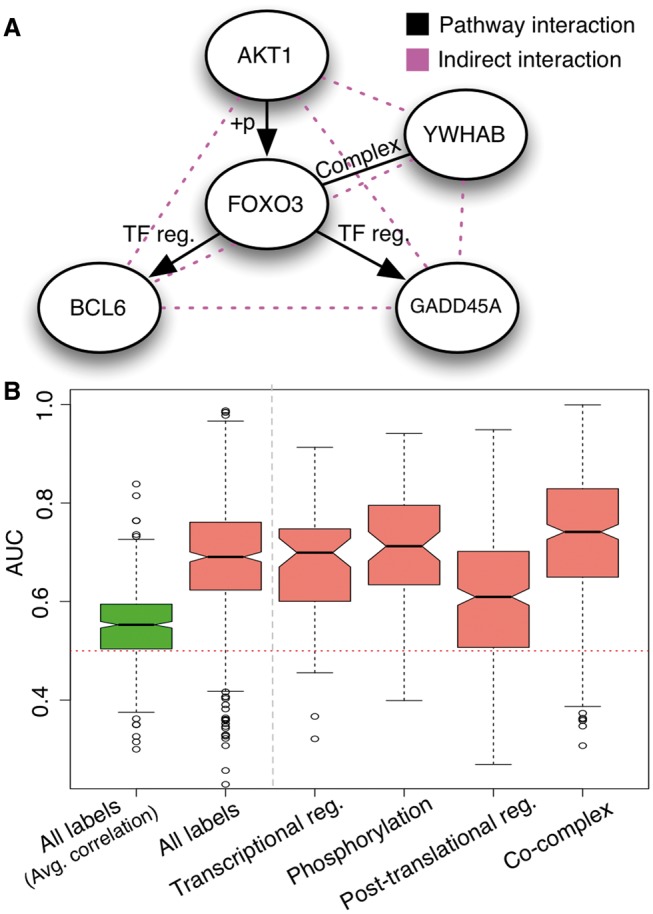
Our multiple human interaction networks allow improved human pathway component retrieval. In panel A, we detail a combination of transcriptional, post-translational and physical interactions that we accurately prioritize surrounding the human tumor suppressor FOXO3. In panel B, we expand our pathway retrieval evaluation analysis to total 447 human curated pathways. Each dot in the box plot represents our accuracy of ranking the pathway interactions above all incorrect interactions between the constitute genes in a pathway. Overall, our predicted networks show significantly improved performance in retrieval of pathway interactions when evaluated for both all interactions types combined (‘all labels’) and also for each individual interaction type (shown in salmon color) compared with average correlation over the expression dataset (‘all labels (Average correlation)’ in lime-green)
To systematically evaluate a broader set of pathways, we assessed our ability to recapitulate the pathway interactions for 447 expert-curated human pathways by Pathway Interaction database (includes all BioCarta pathways and restricted to pathways with minimum 5 genes and 10 interactions) (Schaefer et al., 2009). For each pathway, for the genes constituting the pathway, we assess how accurate our network predictions are in prioritizing the gene pairs with the correct interaction type label compared with all indirect interactions and direct pairs but with incorrect interaction type labels (cross-validated predicted networks were used for the analysis). Our median evaluation performance measures are uniformly well above random (0.5), shown in Figure 3B, with a median AUC of 0.69 across all pathways with comparable performance across different interaction types. Specifically, we observe co-complex interactions to be the top performer, with a median AUC of 0.74 [consistent with previous observations of strong genomic signal of co-complex (Qiu and Noble, 2008)] and post-translational regulation to be the most challenging to predict, with a median AUC of 0.61 (results limited to pathways with at least five gene interactions for the corresponding interaction type, full tabulated results for each pathway in Supplementary Table S4). Also, our results show significant improvement (P < 0.01, Wilcoxon rank test) compared with average gene pairwise correlation over the expression datasets. Finally, our predictions significantly outperform predictions made without incorporating tissue contextual information (i.e. tissue-unaware predictions) and the more general coupling of ‘functional association’ (Huttenhower et al., 2009), thus highlighting the importance of interaction type specific integration of multiple data sources (full interaction type performance comparison for all networks shown in Supplementary Fig. S5). Taken together, these results indicate that our network interactions can generate hypotheses not only about pathway structure from random gene pairs, but can also be used to prioritize the pathway interactions among the more challenging functionally related gene sets.
3.3 Human interaction networks help interpret primary experimental datasets
In addition to pathway component recovery, our predicted interactomes can be used to aid the discovery of regulatory targets in individual investigator generated condition-specific datasets.
3.3.1 Recovery of transcriptional regulatory targets from ChIP-Seq datasets
Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-Seq) has become a prevalent experimental assay that is applied to TFs to measure potential regulatory binding regions across the genome in in vivo settings (Park, 2009). Potential regulatory targets for an immuno-precipitated TF are mainly identified as genes that have a TF ChIP-Seq binding profile proximal to its TSS (Gerstein et al., 2012). However, like many biological assays, ChIP-Seq is known to have a high rate of nonspecific cross-linking due to the usage of formaldehyde (compared with precise UV cross-linking that is specific to <1 angstrom proximity) that can lead to many false positive regulatory candidate targets (Mercer and Mattick, 2013). In addition, even when given a robust TF-binding site, often there can be multiple TSS windows proximal to a binding locus thus obscuring the process of identifying the true regulatory target gene.
With our predicted transcriptional interaction network constructed from over 50 000 genome-scale experiments, we could discriminate false positive regulatory targets identified by a ChIP-Seq experiment either by the result of a non-specific binding or multiple TSS windows proximal to a binding profile. Thus, our approach allows researchers to capture a more accurate binding landscape at the given experimental condition or tissue of the ChIP-Seq experiment. To test this hypothesis, we overlay our transcriptional regulatory network to identify false positive regulatory targets of TFs identified by ChIP-Seq experimental datasets generated for the ENCODE project (total 109 TF, 609 ChIP-Seq experiments) (Landt et al., 2012). Specifically, for each 109 TF, we identify potential regulatory target genes that have binding-profile peaks located within a window surrounding the gene’s TSS. Next, we filter ChIP-Seq identified regulatory target genes that have a predicted probability of less than prior (i.e. no experimental support) in our transcriptional regulatory network. In other words, genes with little experimental evidence in the human genomic data compendium of being transcriptionally regulated by the TF are flagged as false positives.
To evaluate the accuracy of our ability to identify false positive regulatory targets from ChIP-Seq experiments, we test if the regulatory target genes identified for each TF are enriched with genes known to be involved in biological processes associated with the TF. We hypothesized that if false positive TF-target gene pairs are accurately filtered, the enrichment of TF-involved biological processes should improve. Indeed, the evaluation results shown in Figure 4A demonstrate a significant increase in enrichment when filtering based on our transcriptional regulatory network compared with the original regulatory targets identified only from the ChIP-Seq data.
Fig. 4.
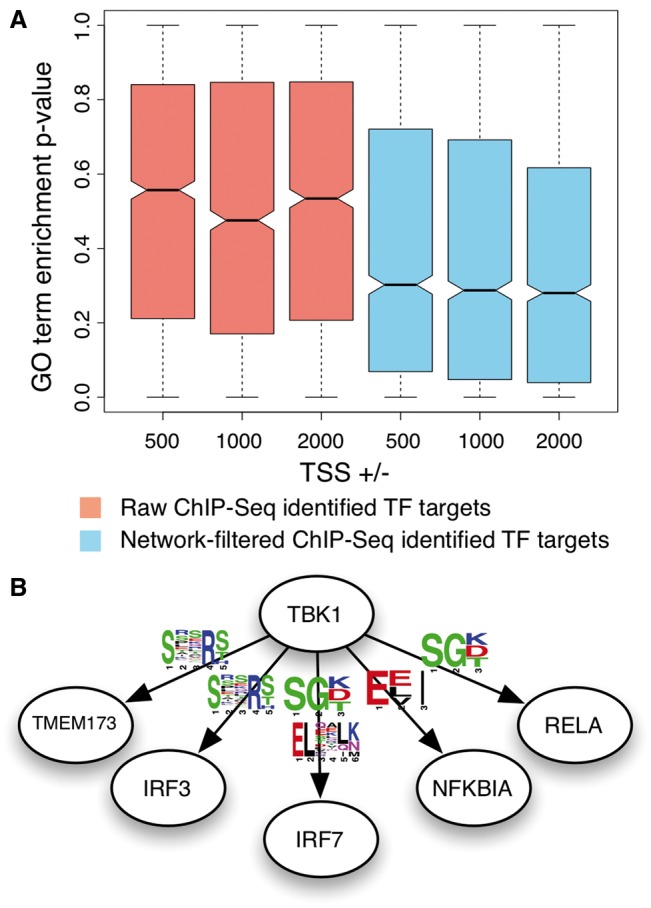
Improving the interpretability of primary experimental data. Our interaction networks can be used to help investigators increase the interpretability of condition-specific primary experimental data. In panel A, ChIP-Seq datasets generated for the ENCODE project (109 TFs) were processed to identify TF and potential regulatory target genes. We observe a significant increase in enrichment of TF associated GO terms among its putative regulator targets, when removing TF-targets pairs that have low predicted probability in our TF regulatory network (skyblue). In panel B, represents our predicted phospho-binding motifs that are present in known TBK1 substrates not identified in the phospho-proteomics study. Such results support the potential biological relevance of these motifs and the accuracy of our phosphorylation network that enabled the analysis
In addition, the improvement in enrichment is consistent across varying windows ± 500, 1000 and 2000 bp) surrounding TSS for identifying regulatory target genes. Interestingly, the TSS window of ±1000 bp that have been used heuristically in a recent study (Tiwari et al., 2012) also showed the best performance in our evaluations. Just based on ChIP-Seq data, the 500 bp window is too restrictive and the 2000 bps window is too promiscuous (i.e. larger increase in false positive regulatory target genes versus true positives). However, our methodology permits a much larger window of ±2000 bp while improving the overall enrichment. Consequently, this allows investigators to identify larger number of functional regulatory targets from the same ChIP-Seq dataset.
3.3.2 Phosphorylation network identifies TBK1 targets from phospho-proteomics data
With the recent advancement of stable isotope labeling techniques (e.g. SILAC), quantitative mass spectrometry has allowed the monitoring of the global alterations of the knock-down or knock-out phenotype of a specific gene at the proteome level (e.g. RNAi targeting a kinase) (Nesvizhskii et al., 2007). However, RNAi/MS studies alone cannot distinguish direct regulatory effects from indirect effects. For example, the collection of differentially phosphorylated proteins after knocking-down a protein kinase will be a mix of direct substrates of the knocked-down kinase and also substrates of other de-activated kinases. This is because often signaling pathways consists of long kinase cascades, complicating any follow-up analysis.
We hypothesized that our phosphorylation interaction network, which summarizes the human data compendium, could be used to prioritize regulatory target proteins from an investigator’s phospho-proteomics study. To test the applicability of our proposed approach, we applied our methodology to a recent phospho-proteomics study (Kim et al., 2013). In this study, loss of phosphorylated proteins was measured using mass spectrometry following the RNAi mediated knock-down of TBK1. TBK1 is an important kinase involved in innate immune response and implicated in multiple human cancers including lung cancer (Guo et al., 2013). Therefore, the identification of regulatory targets of TBK1 and potential binding motifs can provide a great resource for future therapeutic studies.
In the published study, the researchers were not able to report any potential binding/recognition motifs of TBK1, most likely due to the mixture of direct and indirect targets among the differentially phosphorylated proteins. This is especially unfortunate because, although in vitro peptide array studies have been conducted (Hutti et al., 2012; Newman et al., 2013), no known in vivo binding/recognition motif has been identified for kinase TBK1. In fact, when we ran the state-of-art motif discovery tool FIRE (Elemento et al., 2007) on the 2150 differentially phosphorylated protein sequences (1154 unique genes), we retrieved many known binding motifs of other kinases such as ERK1,2 and PKC beta kinase (i.e. not the RNAi knock-down kinase TBK1, motifs shown in Supplementary Figure S6). Interestingly, many of the kinases that recognize the identified motifs were among the differentially phosphorylated proteins (71%). This is consistent with the expectation that many of the substrates of these kinases (and not TBK1) contributing a significant portion to the collection of differentially phosphorylated genes.
To address this challenge, we used our predicted phosphorylation network to refine the identified protein targets by restricting differentially phosphorylated proteins to those that also had a high probability of being regulated by TBK1 in our network. Filtering out proteins that have little experimental evidence of being regulated by TBK1 in the human data compendium could improve subsequent downstream analyses. Thus, we repeated the motif discovery analysis as conducted previously on the sequences of the 258 network-refined protein targets (Supplementary Table S5). As predicted, we no longer retrieved any significant binding motifs of other kinases, compared with our previous analysis, which resulted in multiple known motifs of TBK1 downstream kinase targets (motifs shown in Supplementary Fig. S7).
Due to the lack of proinflammatory stimuli in the experiment (Kim et al., 2013), seven known phosphorylation substrates of TBK1 were not identified to be differentially phosphorylated in this study. Interestingly, as shown in Figure 4B, 4 motifs among the top 10 significant motifs (only identified through our integrated approach, phosphorylation network + FIRE) were present for these known TBK1 substrates, which were not included in the motif discovery analysis (such occurrence or more motif overlap happening by random chance, P-value < 0.05 based on permuting the motif-protein profile). Thus, this result supports the possibility of these motifs being the first in vivo derived motifs identified to be biologically relevant for TBK1 recognizing its phosphorylation substrates.
4 Discussion
Genomic approaches have provided us with great opportunities in unearthing the complexity of human biology and diseases. Although an increasing number of human datasets measuring the molecular changes at the expression, epigenomic and proteomic level has provided us with an invaluable public resource, the efficient integration and identification of regulatory pathways and processes has been challenging. In this work, by integrating ∼1600 human genomic-scale datasets, we provide the means to study human pathway-level interactions at a whole-genome scale. For each pair of genes in the human genome, thousands of experimental data points measuring the behavior of these genes and its protein products were summarized to infer both the presence of a functional association and the most likely pathway interaction type. By applying our interaction networks to experimental datasets, we were able to improve the accuracy of identifying TF regulatory targets from ChIP-Seq data compared with a traditional TSS-proximity method, and also identifying novel kinase substrate recognition motifs from phospho-proteomics data.
This study demonstrates that directly incorporating tissue contextual information in the data integration and inference of gene interactions for metazoan mammalian organisms can significantly improve prediction accuracy. Although we have implemented our system utilizing a maximum-margin hyperplane-based SVM algorithm, we anticipate that the overall approach of directly exploiting tissue and cell-lineage heterogeneity in human datasets can be readily incorporated into many future and existing methods.
Currently, source and target gene information (i.e. directionality) of a predicted regulatory interaction can only be indirectly inferred for gene pairs when there is only one regulator or modifying enzyme protein corresponding to the interaction type (e.g. a predicted phosphorylation interaction that includes one kinase or transcriptional regulation interaction with one TF). Future research will be required to develop methods and incorporate new data sources to unravel the directionality between regulator genes, such as the regulatory directionality between the ∼500 human kinases often chained together in signaling cascades. Furthermore, we expect the explicit prediction of tissue/cell-type specific pathway interaction networks and rewiring to be the next challenge in unraveling human system biology. Major effort will be required to generate a sufficient number of experimentally verified tissue-specific interaction gold standards. To enable such efforts, we have made all of our predicted whole-genome networks publically available at an interactive web-portal, pathwaynet.princeton.edu, for researchers to conduct exploratory analysis for future hypothesis generation.
Supplementary Material
Acknowledgements
The authors acknowledge John Wiggins for technical support.
Funding
This work was supported in part by R01s GM071966 and HG005998 and by P50 GM071508. O.G.T. is a Senior Fellow in the Genetic Networks program at the Canadian Institute for Advanced Research.
Conflict of Interest: none declared.
References
- Alexeyenko A., Sonnhammer E.L.L. (2009) Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Res., 19, 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanchy R., et al. (2007) A curated compendium of phosphorylation motifs. Nat. Biotech., 25, 285–286. [DOI] [PubMed] [Google Scholar]
- Apweiler R., et al. (2004) UniProt: the Universal Protein knowledgebase. Nucleic Acids Res., 32, D115–D119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassik M.C., et al. (2013) A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell, 152, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. (1996) Bagging predictors. Mach. Learn. 24, 123–140. [Google Scholar]
- Britten R.J., Davidson E.H. (1969) Gene regulation for higher cells: a theory. Science, 165, 349–357. [DOI] [PubMed] [Google Scholar]
- Brunet A., et al. (2001) Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol., 21, 952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell I.G. (1994) Repression versus activation in the control of gene transcription. Trends Biochem. Sci., 19, 38–42. [DOI] [PubMed] [Google Scholar]
- Date S.V., Stoeckert C.J. (2006) Computational modeling of the Plasmodium falciparum interactome reveals protein function on a genome-wide scale. Genome Res., 16, 542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Develop., 19, 2100–2110. [DOI] [PubMed] [Google Scholar]
- Edgar R., et al. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res., 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elemento O., et al. (2007) A universal framework for regulatory element discovery across all genomes and data types. Mol. Cell, 28, 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elemento O., Tavazoie S. (2005) Fast and systematic genome-wide discovery of conserved regulatory elements using a non-alignment based approach. Genome Biol., 6, R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández de Mattos S., et al. (2004) FoxO3a and BCR-ABL regulate cyclin D2 transcription through a STAT5/BCL6-dependent mechanism. Mol. Cell. Biol., 24, 10058–10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res., 32, D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M.B., et al. (2012) Architecture of the human regulatory network derived from ENCODE data. Nature, 489, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremse M., et al. (2011) The BRENDA Tissue Ontology (BTO): the first all-integrating ontology of all organisms for enzyme sources. Nucleic Acids Res., 39, D507–D513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., et al. (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res., 36, D154–D158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., et al. (2013) IKBKE is induced by STAT3 and tobacco carcinogen and determines chemosensitivity in non-small cell lung cancer. Oncogene, 32, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hand S.C., Hardewig I. (1996) Downregulation of cellular metabolism during environmental stress: mechanisms and implications. Annu. Rev. Physiol., 58, 539–563. [DOI] [PubMed] [Google Scholar]
- Hanley J.A., McNeil B.J. (1982) The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology, 143, 29–36. [DOI] [PubMed] [Google Scholar]
- Haynes B.C., et al. (2013) Mapping functional transcription factor networks from gene expression data. Genome Res., 23, 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C., et al. (2009) Exploring the human genome with functional maps. Genome Res., 19, 1093–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C., et al. (2008) The Sleipnir library for computational functional genomics. Bioinformatics, 24, 1559–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutti J.E., et al. (2012) Development of a high-throughput assay for identifying inhibitors of TBK1 and IKKε. PLoS One, 7, e41494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachims T. (2006) Training linear SVMs in linear time. In: Proceedings of the 12th ACM SIGKDD international conference on Knowledge discovery and data mining. ACM, Philadelphia, PA, USA, pp. 217–226. [Google Scholar]
- Kanehisa M., Goto S. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res., 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-Y., et al. (2013) Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc. Natl Acad. Sci., 110, 12414–12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt S.G., et al. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res., 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I., et al. (2004) A probabilistic functional network of yeast genes. Science, 306, 1555–1558. [DOI] [PubMed] [Google Scholar]
- Lehtinen M.K., et al. (2006) A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell, 125, 987–1001. [DOI] [PubMed] [Google Scholar]
- Lewis D.P., et al. (2006) Support vector machine learning from heterogeneous data: an empirical analysis using protein sequence and structure. Bioinformatics, 22, 2753–2760. [DOI] [PubMed] [Google Scholar]
- Lieber D.S., et al. (2010) Large-scale discovery and characterization of protein regulatory motifs in eukaryotes. PLoS One, 5, e14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J., et al. (2013) The Genotype-Tissue Expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M., Jensen O.N. (2003) Proteomic analysis of post-translational modifications. Nat. Biotechnol., 21, 255–261. [DOI] [PubMed] [Google Scholar]
- Manning G., et al. (2002) The protein kinase complement of the human genome. Science, 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Marbach D., et al. (2012) Wisdom of crowds for robust gene network inference. Nat. Methods, 9, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin A., et al. (2006) ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics, 7, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V., et al. (2003) TRANSFAC®: transcriptional regulation, from patterns to profiles. Nucleic Acids Res, 31, 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall M.N., et al. (2011) The Gene Expression Barcode: leveraging public data repositories to begin cataloging the human and murine transcriptomes. Nucleic Acids Res, 39, D1011–D1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer T.R., Mattick J.S. (2013) Understanding the regulatory and transcriptional complexity of the genome through structure. Genome Res, 23, 1081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minguez P., et al. (2012) PTMcode: a database of known and predicted functional associations between post-translational modifications in proteins. Nucleic Acids Res., 41, D306–D311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S., et al. (2008) GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol., 9, S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myatt S.S., Lam E.W.F. (2007) The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer, 7, 847–859. [DOI] [PubMed] [Google Scholar]
- Neph S., et al. (2012a) Circuitry and dynamics of human transcription factor regulatory networks. Cell, 150, 1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S., et al. (2012b) An expansive human regulatory lexicon encoded in transcription factor footprints. Nature, 489, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A.I., et al. (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Meth., 4, 787–797. [DOI] [PubMed] [Google Scholar]
- Newman R.H., et al. (2013) Construction of human activity-based phosphorylation networks. Mol. Syst. Biol., 9, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble W.S. (2006) What is a support vector machine? Nat. Biotech., 24, 1565–1567. [DOI] [PubMed] [Google Scholar]
- Park C.Y., et al. (2013) Functional knowledge transfer for high-accuracy prediction of under-studied biological processes. PLoS Comput. Biol., 9, e1002957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P.J. (2009) ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet., 10, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J., Noble W.S. (2008) Predicting co-complexed protein pairs from heterogeneous data. PLoS Comput. Biol., 4, e1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D., et al. (2013) A compendium of RNA-binding motifs for decoding gene regulation. Nature, 499, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D.R., et al. (2005) Probabilistic model of the human protein-protein interaction network. Nat. Biotechnol., 23, 951–959. [DOI] [PubMed] [Google Scholar]
- Rual J.-F., et al. (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature, 437, 1173–1178. [DOI] [PubMed] [Google Scholar]
- Sandelin A., et al. (2004) JASPAR: an open access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res., 32, D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer C.F., et al. (2009) PID: the Pathway Interaction Database. Nucleic Acids Res., 37, D674–D679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt T., et al. (2014) FunCoup 3.0: database of genome-wide functional coupling networks. Nucleic Acids Res., 42, D380–D388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C., et al. (2011) The BioGRID interaction database: 2011 update. Nucleic Acids Res, 39, D698–D704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U., et al. (2005) A human protein-protein interaction network: a resource for annotating the proteome. Cell, 122, 957–968. [DOI] [PubMed] [Google Scholar]
- Su A.I., et al. (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A, 101, 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. U S A, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V.K., et al. (2012) A chromatin-modifying function of JNK during stem cell differentiation. Nat Genet, 44, 94–100. [DOI] [PubMed] [Google Scholar]
- Troyanskaya O.G., et al. (2003) A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc Natl Acad Sci, 100, 8348–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas J.M., et al. (2009) A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet., 10, 252–263. [DOI] [PubMed] [Google Scholar]
- von Mering C., et al. (2007) STRING 7—recent developments in the integration and prediction of protein interactions. Nucleic Acids Res., 35, D358–D362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatheritt R.J., et al. (2012) iELM—a web server to explore short linear motif-mediated interactions. Nucleic Acids Res., 40, W364–W369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A.K., et al. (2012) IMP: a multi-species functional genomics portal for integration, visualization and prediction of protein functions and networks. Nucleic Acids Res., 40, W484–W490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellaboina S., et al. (2011) DOMINE: a comprehensive collection of known and predicted domain-domain interactions. Nucleic Acids Res., 39, D730–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.C., et al. (2013) PrePPI: a structure-informed database of protein–protein interactions. Nucleic Acids Res, 41, D828–D833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilliox M.J., Irizarry R.A. (2007) A gene expression bar code for microarray data. Nat. Meth., 4, 911–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.