

ABSTRACT
Pathogenicity islands (PAIs) are mobile integrated genetic elements (MIGEs) that contain a diverse range of virulence factors and are essential in the evolution of pathogenic bacteria. PAIs are widespread among bacteria and integrate into the host genome, commonly at a tRNA locus, via integrase-mediated site-specific recombination. The excision of PAIs is the first step in the horizontal transfer of these elements and is not well understood. In this study, we examined the role of recombination directionality factors (RDFs) and their relationship with integrases in the excision of two PAIs essential for Vibrio cholerae host colonization: Vibrio pathogenicity island 1 (VPI-1) and VPI-2. VPI-1 does not contain an RDF, which allowed us to answer the question of whether RDFs are an absolute requirement for excision. We found that an RDF was required for efficient excision of VPI-2 but not VPI-1 and that RDFs can induce excision of both islands. Expression data revealed that the RDFs act as transcriptional repressors to both VPI-1- and VPI-2-encoded integrases. We demonstrated that the RDFs Vibrio excision factor A (VefA) and VefB bind at the attachment sites (overlapping the int promoter region) of VPI-1 and VPI-2, thus supporting this mode of integrase repression. In addition, V. cholerae RDFs are promiscuous due to their dual functions of promoting excision of both VPI-1 and VPI-2 and acting as negative transcriptional regulators of the integrases. This is the first demonstration of cross talk between PAIs mediated via RDFs which reveals the complex interactions that occur between separately acquired MIGEs.
IMPORTANCE Deciphering the mechanisms of pathogenicity island excision is necessary for understanding the evolution and spread of these elements to their nonpathogenic counterparts. Such mechanistic insight would assist in predicting the mobility of uncharacterized genetic elements. This study identified extensive RDF-mediated cross talk between two nonhomologous VPIs and demonstrated the dual functionality of RDF proteins: (i) inducing PAI excision and (ii) acting as transcriptional regulators. Findings from this study may be implicated in determining the mobilome contribution of other bacteria with multiple MIGEs.
INTRODUCTION
Mobile integrative genetic elements (MIGEs) are the vectors of horizontal gene transfer (HGT) among bacteria via the mechanisms of transformation, transduction, and conjugation (1–3). Members of MIGEs include transposons, integrons, bacteriophages, plasmids, and integrative conjugative elements (ICEs)/conjugative transposons and genomic/pathogenicity islands (GEIs/PAIs). Commensal bacteria can be converted into deadly pathogens through HGT of MIGEs that contain virulence factors, or a pathogen may become more virulent with the acquisition of additional virulence factors (1, 4–6). PAIs were first described for uropathogenic Escherichia coli (UPEC) and have since been described for many pathogenic bacteria (7–17). PAIs range in size from 10 to 200 kb and have a guanine and cytosine (GC) content that differs from that of the core bacterial chromosome. These islands encode virulence factors, are found integrated in the chromosome at the 3′ end of tRNA loci, and are absent from nonpathogenic strains or closely related species (10). PAIs also contain an integrase usually belonging to the tyrosine recombinase (TR) family. Based on phylogenetic analysis of PAI-encoded integrases, it was demonstrated that PAIs are a distinct class of integrative elements and are not degenerate remnants of integrated plasmids, prophages, integrons, or conjugative transposons (2, 18, 19).
Integrases are necessary for both integration and excision of MIGEs by catalyzing site-specific recombination between two DNA substrates through DNA cleavage, formation of a Holliday junction, and rejoining of DNA fragments (20–23). In TR integrase-encoding prophages, it was demonstrated that the excision event also requires an additional protein, an excisionase or recombination directionality factor (RDF) (24–28). In fact, it is believed that all TR integrases require an RDF to perform excision (20, 29). RDFs are small DNA-binding accessory proteins that aid TR integrases in controlling the directionality of their site-specific recombination reactions (29). These proteins govern directionality by favoring one reaction direction and interfering with the other. While the integrases act to catalyze the site-specific recombination reaction necessary for MIGE excision and integration, their cognate RDFs have been described to perform various accessory functions, such as binding and bending DNA near the att sites and acting as positive or negative integrase transcriptional regulators as well as offering stability to their integrase protein partners at the excision site (24, 26, 30–33). Tyrosine recombinase integrases and RDFs have not been studied extensively for PAIs. Phylogenetic analysis of E. coli RDFs and their corresponding integrases from PAIs suggests that they evolved together (18). This coevolution was also observed in the grouping patterns of bacteriophage and transposon integrases with their partner RDFs (29).
Vibrio cholerae O1 serogroup El Tor, the cause of the seventh ongoing cholera pandemic, contains four PAIs that are of particular interest due to their genetic content and unique recombination modules. Vibrio pathogenicity island 1 (VPI-1) and VPI-2 are present in all cholera pandemic strains, both classical and El Tor biotypes (12–14, 17, 34). The presence of two additional PAIs, named Vibrio seventh pandemic island I (VSP-I) and VSP-II, distinguishes biotype El Tor from the classical biotype strains that caused the first six pandemics (12–14, 34). The genes encoding the toxin coregulated pilus (TCP), a type IV bundle-forming pilus, critical for intestinal colonization, are found within VPI-1 (13, 35–37). Vibrio pathogenicity island 2 contains genes for the production of sialidase as well as a sialic acid transport and catabolism gene cluster (12, 38–40). Both VPI-1 and VPI-2 have a TR integrase (intV) located adjacent to a tRNA locus, are flanked by attachment (att) sites, and have a low GC content compared to that of the V. cholerae core genome (8, 12). It was proposed that VPI-1 was actually a filamentous phage named VPIΦ; however, this has proven not to be the case, and the region is indeed a PAI (41, 42). Two RDF genes, vefA and vefB, were identified by bioinformatic analysis in VPI-2, and a third one, vefC, is found in VSP-II; however, no RDF was identified in VPI-1 (43) (Fig. 1A).
FIG 1.

Vibrio pathogenicity islands (VPIs), VPI excision, and Vibrio excision factors (Vefs). (A) VPI-1 inserts at the transfer-messenger RNA (tmRNA) gene and contains genes encoding the toxin-coregulated pilus (TCP) and accessory colonization factor (ACF) as well as an integrase (intV1) and a transposase-like gene (vpiT). VPI-2 inserts into a tRNA-serine locus and contains a restriction modification system (RMS) and a sialic acid scavenging, transport, and catabolism (SAM) region as well as an integrase (intV2) and two recombination directionality factors (RDFs) encoded by vefA and vefB. Vibrio seventh pandemic island II (VSP-II) contains a third RDF encoded by vefC. (B) VPIs can excise from the chromosome and form a circular intermediate containing an attP site and leaving a unique attB site left in the bacterial chromosome. Primers used to detect the attachment (att) sites in two-stage nested PCR assays are represented by arrows on the left and right sides of the attP and attB sites. (C) Amino acid sequence alignment of VefA, VefB, and VefC using ClustalW with default settings in JalView. A 59% identity is shared between VefA and VefC, 46% between VefA and VefB, and 49% between VefB and VefC. Predicted secondary structures of VefA, VefB, and VefC were generated using JNet Secondary Structure Prediction. Arrows represent sheets and cylinders represent helices.
PAI dynamics have been examined in UPEC strain 536, which contains six PAIs (PAI I536 to PAI VI536). All of the PAIs were shown to excise from the chromosome with the exception of PAI IV536 and PAI VI536 (44, 45). RDFs were not identified or examined in these studies. A more recent study, however, found by bioinformatics that each PAI536 contained its own cognate putative RDF (18). In PAIs of Yersinia pseudotuberculosis and Shigella flexneri, excision from the bacterial chromosome was demonstrated and shown to require the cognate island integrase and RDFs named Hef and Rox, respectively (7, 46). In V. cholerae, PAI excision was examined in VPI-1, VPI-2, and VSP-II (8, 42, 43). For VPI-1, it was shown that the cognate TR integrase and the transposase VpiT both contributed to excision, since only in the absence of both intV1 and vpiT was excision abolished (42). For VPI-2, the integrase and an RDF were shown to be required for excision (8, 43). For VSP-II, the cognate integrase was required for excision and formation of a nonreplicative circular intermediate (CI) (8).
In this study, we examined the role of RDFs and their relationship with integrases in the excision of both VPI-1 and VPI-2 to address the question of whether RDFs are an absolute requirement for excision, since the VPI-1 region does not contain an RDF. We have identified extensive RDF-mediated cross talk between VPIs and demonstrated dual functionality of these proteins to aid in PAI excision and act as transcriptional regulators.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
All bacterial strains and plasmids used in this study are listed in Table 1. Bacterial strains were grown overnight aerobically at 225 rpm in Luria broth (LB) (Fisher Scientific, Fair Lawn, NJ) at 37°C unless otherwise stated. The diaminopimelic acid (DAP) auxotroph Escherichia coli β2155 was grown in the presence of 0.3 mM DAP (Sigma, St. Louis, MO). When appropriate, antibiotics were added to media at the following concentrations: 200 μg/ml for streptomycin (Sm), 25 μg/ml for chloramphenicol (Cm), and 100 μg/ml for ampicillin (Amp) (Fisher Scientific). When selecting for the double-crossover event, LB agar was supplemented with 10% sucrose.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| V. cholerae strains | ||
| N16961 | O1 El Tor; VPI-1 VPI-2 VSP-I VSP-II Smr | 57 |
| O395 | O1 classical; VPI-1 VPI-2 | Laboratory strain |
| MO2 | O139; VPI-1 VPI-2 | Laboratory strain |
| SG-7 | Environmental | Laboratory strain |
| MC0817 (ΔvpiT) | N16961 ΔVC0817 Smr | This study |
| MC0847 (ΔintV1) | N16961 ΔVC0847 Smr | This study |
| CN1758 (ΔintV2) | N16961 ΔVC1758 Smr | This study |
| MC4758 (ΔintV1 ΔintV2) | ΔintV1 ΔVC1758 Smr | This study |
| ΔintV1 pintV1 strain | ΔintV1 strain harboring pBAintV1 | This study |
| ΔintV2 pintV2 strain | ΔintV2 strain harboring pBAintV2 | This study |
| SAM1785 (ΔvefA) | N16961 ΔVC1785 Smr | 43 |
| SAM1809 (ΔvefB) | N16961 ΔVC1809 Smr | 43 |
| MC0497 (ΔvefC) | N16961 ΔVC0497 Smr | This study |
| MC8509 (ΔvefA ΔvefB) | ΔvefA ΔVC1809 Smr | This study |
| MC850997 (ΔvefA ΔvefB ΔvefC) | ΔvefA ΔvefB ΔVC0497 Smr | This study |
| MC85099717 (ΔvefA ΔvefB ΔvefC ΔvpiT) | ΔvefA ΔvefB ΔvefC ΔVC0817 Smr | This study |
| ΔvefA ΔvefB ΔvefC pvefA strain | ΔvefA ΔvefB ΔvefC strain harboring pBBvefA | This study |
| ΔvefA vefB ΔvefC pvefB strain | ΔvefA ΔvefB ΔvefC strain harboring pBAvefB | This study |
| ΔvefA ΔvefB ΔvefC pvefC strain | ΔvefA ΔvefB ΔvefC strain harboring pBAvefC | This study |
| ΔvefA ΔvefB ΔvefC pintV2 strain | ΔvefA ΔvefB ΔvefC strain harboring pBAintV2 | This study |
| E. coli strains | ||
| DH5αλpir | Laboratory collection | |
| β2155λpir | Donor for bacterial conjugation, DAP auxotroph | Laboratory collection |
| BL21(DE3) | Expression strain | Laboratory collection |
| Plasmids | ||
| pJET1.2 | Cloning vector; Amr | Fermentas |
| pDS132 | Suicide vector; SacB Cmr | 58 |
| pBAD33 | Arabinose promoter; Cmr | 59 |
| pDSΔvpiT | pDS132 harboring truncated vpiT gene | This study |
| pDSΔintV1 | pDS132 harboring truncated intV1 gene | This study |
| pDSΔintV2 | pDS132 harboring truncated intV2 gene | This study |
| pDSΔvefA | pDS132 harboring truncated vefA gene | 43 |
| pDSΔvefB | pDS132 harboring truncated vefB gene | 43 |
| pDSΔvefC | pDS132 harboring truncated vefC gene | This study |
| pBAintV1 | pBAD33 with full copy of intV1 | This study |
| pBAintV2 | pBAD33 with full copy of intV2 | This study |
| pBAvefA | pBAD33 with full copy of vefA | This study |
| pBAvefB | pBAD33 with full copy of vefB | This study |
| pBAvefC | pBAD33 with full copy of vefC | This study |
| pMALc5x-His6x | Expression vector, TEV site; Amr | 60 |
| pMALc5x-His6x-VefA | pMALc5x-His6x harboring VefA | This study |
| pMALc5x-His6x-VefB | pMALc5x-His6x harboring VefB | This study |
Mutant strain construction.
Primers were designed and purchased from Integrated DNA Technologies (Coralville, IA) in order to create in-frame deletions of genes in V. cholerae N16961 using splicing by overlap extension (SOE) PCR and homologous recombination (47). The mutant strains are listed in Table 1, and primers used for their construction are found in Table S1 in the supplemental material. In order to make the ΔintV1 strain, two sets of primers were designed to amplify a region flanking the integrase gene, VC0847. Primer pairs VC0847A/VC0847B and VC0847C/VC0847D were used to generate PCR fragments of 556 bp and 548 bp, respectively. The purified products from the PCRs with VC0847A/VC0847B and VC0847C/VC0847D were used as the template for PCR with primers VC0847A and VC0847D to form a PCR product containing a truncated intV1 gene of 351 bp. This product was blunt-end cloned into the vector pJET1.2 (Thermo Fisher Scientific, Pittsburgh, PA) and transformed into E. coli DH5α. Following plasmid isolation (Qiagen, Valencia, CA) and restriction digestion with SacI and XbaI (Thermo Fisher Scientific), the fragment was cloned into the suicide vector pDS132. pDSΔintV1 containing the truncated VC0847A/VC0847D insert was then transformed into the DAP auxotroph strain E. coli β2155, which served as the donor for conjugation with V. cholerae N16961. Selective plating without DAP but with Cm allowed for detection of single-crossover events via homologous recombination of pDSΔintV1 into the chromosome of V. cholerae; these events were confirmed by PCR with primer pair VC0847A/VC0847D. The double-crossover event was selected for by plating on LB agar containing 10% sucrose without Cm. Colony PCR using primer pair VC0847A/VC0847D and flanking primers confirmed the double-crossover event. All mutant strains were created using the same method and confirmed by sequencing. Primers used for each mutant construction are listed in Table S1. The ΔintV1 strain (ΔVC0847) has a 918-bp deletion resulting in a 351-bp truncated gene, the ΔvpiT strain (ΔVC0817) has an 816-bp deletion resulting in a 168-bp truncated gene, and the ΔintV2 strain (ΔVC1758) has 1,065 bp deleted and a truncated intV2 gene of 171 bp. The ΔvefC strain (ΔVC0497) has 171 bp deleted and a 30-bp truncated vefC gene.
Excision assays.
In order to observe excision of VPI-1 and VPI-2 from the V. cholerae genome, two excision assays were performed to detect unique sites in the chromosome (attB1 and attB2) and in the circular intermediate (CI) (attP1 and attP2) formed following the excision event (Fig. 1B). Genomic DNA was isolated from overnight liquid incubations using the GNOME kit (MP Biomedicals, Santa Ana, CA) according to the manufacturer's procedure and was used as the template in the attB assays to detect the empty chromosomal site following island excision. Additionally, plasmid DNA was isolated using a Qiagen plasmid isolation kit per the manufacturer's instructions and was used as the template for the attP assay in order to observe the island containing attP site following site-specific recombination from the V. cholerae chromosome. Due to the low rate of island excision under normal laboratory growth conditions, a two-stage nested PCR was performed to observe excision products in the form of attB and attP. Primers were designed based on the V. cholerae N16961 genome in order to amplify the island containing attP site or chromosomal attB site for VPI-1 and VPI-2 (Fig. 1B) and are listed in Table S1 in the supplemental material. PCR was performed in 20-μl reaction mixtures with Taq DNA polymerase (Denville, Holliston, MA) under the following conditions: 95°C for 5 min followed by 30 cycles of 94°C 30 s, 60°C 30 s or 90 s, and 72°C for 30 s and then 72°C for 5 min. Genomic or plasmid DNA (10 ng) was used as the template for the first round of PCR, and 1 μl of PCR product from this reaction was used as the template for the second nested round of PCR. PCR products were analyzed on a 2% agarose gel and stained with ethidium bromide for visualization under UV light. At least two biological replicates and three technical replicates were performed for each assay.
Mutant strain complementation.
Mutant strains which were defective for excision or which showed reduced excision were complemented with expression plasmids containing full intact genes. The integrase gene intV1 and its ribosomal binding site were amplified using primers VC0847F and VC0847R with HotStar HiFidelity polymerase (Qiagen) per the manufacturer's instructions. The PCR product was cloned into pJET1.2 and transformed into E. coli DH5α. Following plasmid isolation and restriction digestion with XbaI and SacI, the fragment was cloned into the arabinose-inducible plasmid pBAD33. The resulting plasmid, pBAintV1, was transformed into E. coli β2155, which was used as the donor strain in a conjugation with the ΔintV1 strain. The resulting ΔintV1 pintV1 strain was used to observe excision. Each complementation strain was created in this manner using primers listed in Table S1 in the supplemental material except pBAVefA, which was cloned from pBBR1MCSVefA into pBAD33 using XbaI and KpnI restriction enzymes. All complementation strains were induced with 0.02% (wt/vol) arabinose unless otherwise stated.
qPCR.
RNA was extracted from wild-type and ΔvefA ΔvefB ΔvefC V. cholerae (N16961) cells grown to mid-logarithmic stage (optical density at 600 nm [OD600] = 0.5 to 0.6) using TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. RNA was subsequently treated with Turbo DNase (Invitrogen) by following the manufacturer's instructions and quantified with a Nanodrop spectrophotometer. cDNA was synthesized starting with 500 ng of DNase-treated RNA as the template using Superscript III reverse transcriptase (Invitrogen). A 1:10 dilution of each sample was used as the template for quantitative real-time PCR (qPCR). PCRs were performed using Fast SYBR green master mix (Applied Biosystems, Carlsbad, CA) and run on an Applied Biosystems 7500 fast real-time PCR system. Cycling conditions were as follows: holding stage at 95°C for 25 s and cycling stage at 95°C for 3 s and 60°C for 30 s. Primers for intV1, intV2, and topI are listed in Table S1 in the supplemental material. Data were analyzed using the Applied Biosystems 7500 software. Relative gene expression was determined using the threshold cycle (ΔΔCT) method (48).
Protein purification of VefA and VefB.
Vibrio cholerae VefA and VefB were separately cloned into the pMALc5x-His6x expression plasmid using primers BamHIVC1785R/NcoIVC1785F and NcoIVefBFwd/BamHIVefBRev, respectively (see Table S1 in the supplemental material), in which a 6×His-tagged maltose-binding protein (MBP) is fused to VefA/VefB, separated by a tobacco etch virus (TEV) protease cleavage site. The Q5 site-directed mutagenesis kit (New England Biolabs, Ipswich, MA) was used to remove a 4-bp sequence in the pMALVefA construct, allowing in-frame translation of the fusion protein, by following the manufacturer's protocol. Primers VefAmutRev/VefAmutFwd used in this reaction are listed in Table S1. Plasmid DNA was confirmed by sequencing and transformed into E. coli BL21(DE3) using a standard CaCl2 method.
pMALc5x-His6x-VefA and pMALc5x-His6x-VefB were expressed in E. coli BL21(DE3). Ten milliliters of overnight culture was inoculated into 1 liter of terrific broth (TB) supplemented with glucose at 37°C and induced with 1 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) at an OD600 of 0.5. Growth continued overnight at 18°C. Cells were harvested by centrifugation (5,000 × g for 20 min at 4°C) and were resuspended in amylose wash buffer (50 mM sodium phosphate, 200 mM NaCl [pH 7.5]) supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF) and 1 mM benzimidine. Bacterial cells were lysed on ice using a high-pressure homogenizer (EmulsiFlex-C5; Avestin, Ottawa, Canada). Cell debris was removed by centrifugation (15,000 × g for 1 h at 4°C). The supernatant was passed through a column containing 30 ml of amylose resin (New England Biolabs). The column was washed with 10 column volumes (CV) of amylose wash buffer. The fusion protein, either MBP-VefA or MBP-VefB, was eluted with 3 CV of amylose elution buffer (50 mM sodium phosphate, 200 mM NaCl, 30 mM maltose [pH 7.5]).
A hexahistidine-tagged TEV protease was added to the eluent in a 1:10 molar ratio (TEV to MBP-VefX), and the cleavage reaction proceeded for 45 min for MBP-VefA and overnight for MBP-VefB, at 4°C. Further TEV cleavage reaction time resulted in significant precipitation and loss of VefA. The cleavage mixture was centrifuged, adjusted to 30 mM imidazole, and subjected to immobilized metal affinity chromatography (IMAC) using nickel resin to remove the His-tagged TEV and MBP as well as any remaining uncleaved fusion protein. The flowthrough and first two CV of wash with IMAC wash buffer (50 mM sodium phosphate, 200 mM NaCl, 30 mM imidazole [pH 7.5]) contained either VefA or VefB. The VefB IMAC flowthrough and wash contained equal parts MBP and VefB and was subjected to further purification. Following a 2-fold dilution, VefB was captured on a heparin column (GE Healthcare Life Sciences, Pittsburgh, PA) using 20 mM Tris, 100 mM NaCl, 1 mM EDTA, and 1 mM dithiothreitol (DTT) (pH 8.0). A linear gradient up to 1 M NaCl was performed to elute VefB from the column. Fractions that contained VefB (56 to 60% NaCl) were pooled. Successive concentrations and dilutions using 3.5-kDa-cutoff concentrators (EMD Millipore, Billerica, MA) with 20 mM Tris (pH 8.0) were performed to reduce the NaCl concentration to 115 mM. Protein molecular weight was confirmed by mass spectrometry, and protein purity was determined to be higher than 95% by a 16% SDS-PAGE Tris-tricine gel.
Electrophoretic mobility shift assays (EMSAs).
DNA fragments attR1 and attL2 were amplified using primers sets attR1Fwd/attR1Rev and attL2Fwd/attL2Rev, respectively (see Table S1 in the supplemental material). Various concentrations of purified VefA or VefB were incubated with 30 ng of target DNA in binding buffer (10 mM Tris, 150 mM KCl, 0.1 mM dithiothreitol, 0.1 mM EDTA, 5% polyethylene glycol [PEG] [pH 7.4]) for 20 min at room temperature, and 10 μl was loaded onto a prerun (200 V for 2 h at 4°C) 8% native acrylamide gel. The gel was run at 200 V for 2.5 h in 1× Tris-acetate-EDTA (TAE) buffer at 4°C. Following electrophoresis, gels were stained in an ethidium bromide bath (0.5 μg/ml) for 20 min, washed with water, and imaged.
RESULTS
All three of the RDFs VefA, VefB, and VefC can induce VPI-2 excision.
VPI-2 contains a TR integrase and two RDFs (Fig. 1A). A two-stage nested PCR approach was used to confirm excision of VPI-2 in several V. cholerae wild-type strains that contained these genes through detection of both the circular intermediate attP2 and the empty attB2 site left in the chromosome following excision (Fig. 1B). Excision was detected through both the attP2 and attB2 PCR assays in the second round of PCR for V. cholerae strainsN16961, O395, and MO2 (see Fig. S1A and B in the supplemental material). In strain SG-7, which is devoid of VPI-2, attB2 is present in every copy of its genome, and thus, the empty site was detected in the first and second rounds of PCR (see Fig. S1B).
Two RDFs are present on VPI-2 (encoded by vefA and vefB), and a third RDF is present in VSP-II (encoded by vefC) (43). These Vef protein sequences are diverse, containing a shared sequence identity of <50% (Fig. 1C); however, their predicted secondary structures are similar (Fig. 1C). Each RDF contains three α-helices, with the second and third separated by a β-sheet (Fig. 1C). This conserved helix-turn-helix motif suggests that the Vefs are capable of binding DNA (49). The RDFs share 21 conserved amino acids, with the majority of them near the winged helix of the C terminus (Fig. 1C).
To determine the role of all three RDFs in excision of VPI-2, the two-stage nested PCR assay was used to detect the empty attB2 site in single, double, and triple RDF mutant strains (Fig. 2A). We constructed in-frame nonpolar deletions of VC0497 (vefC), VC1785 (vefA), and VC1809 (vefB) in V. cholerae N16961 and examined excision using the attB2 excision assay. A wild-type excision product (524 bp) for VPI-2 was observed in the second round of the attB2 PCR assay for each single RDF mutant and a double mutant strain lacking vefA and vefB (Fig. 2A). Only the triple RDF mutant (ΔvefA ΔvefB ΔvefC) gave no excision product, which demonstrates that at least one RDF is required for excision of VPI-2 (Fig. 2A). The triple RDF mutant was complemented with a functional copy of each RDF individually. Each RDF restored the VPI-2 excision phenotype, which was detected in the first round of PCR, evidenced by a 1,449-bp product, indicative of excision above the wild-type level that we termed superexcision (Fig. 2B). These data demonstrate that an RDF is required for VPI-2 excision and that all three RDFs can induce excision of VPI-2.
FIG 2.
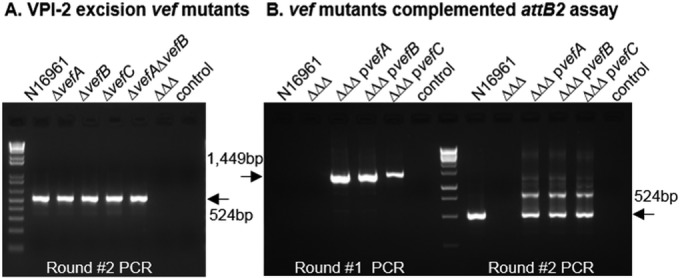
RDFs promote VPI-2 excision. (A) Excision of VPI-2 was examined using the attB2 excision assay in the single, double, and triple (ΔΔΔ) RDF mutant backgrounds. Only results of round 2 are shown, as no excision products were detected in round 1. (B) The triple RDF mutant was complemented with the gene encoding each RDF, vefA, vefB and vefC, individually (pvefA, pvefB, or pvefC) and examined for VPI-2 excision. Excision of the complements was detected in round 1 and is referred to as a superexcision phenotype. “control” indicates sample with no template DNA.
intV2 can mediate VPI-1 excision in the absence of intV1.
VPI-1, which contains a cognate integrase IntV1 but lacks any RDFs, was examined for excision similarly to VPI-2, using attP1 and attB1 two-stage PCR assays (see Fig. S1C and D in the supplemental material). In the attP1 PCR assay, no product was detected in the first round of PCR, indicating a low excision rate as previously suggested (42); however, a 483-bp excision product was detected in the second round of PCR (see Fig. S1C). Similarly, the attB1 assay gave an attB1 PCR product in the second round of PCR for all strains examined (see Fig. S1D), indicating that VPI-1 is capable of excision from these strains.
Next, we reexamined the role of intV1 (VC0847) in excision by constructing an in-frame deletion in VC0847 in V. cholerae N16961. The excision of VPI-1 was nearly abolished in the intV1 mutant, as evidenced by a faint 217-bp PCR band present in the second round of PCR (Fig. 3A). This suggests that there was low-level excision occurring. In the vpiT (VC0817) single mutant strain, VPI-1 excision occurred at a level similar to that seen for the wild type (Fig. 3A). However, no excision was observed in the ΔintV1 ΔvpiT strain using our attB1 assay, consistent with previous findings (Fig. 3A) (42). Interestingly, in the double integrase (ΔintV1 ΔintV2) mutant, excision was abolished, which suggests that IntV2 from VPI-2 plays a role in VPI-1 excision (Fig. 3A). To further investigate this result, we complemented the ΔintV1 ΔintV2 double mutant with a functional intV2 gene and found that VPI-1 excision was restored in the attB1 excision assay (Fig. 3A). These data indicate cross talk between the VPIs mediated by integrases. Analysis of IntV1 and IntV2 amino acid sequences revealed significant homology (53% amino acid identity) displaying a strongly conserved C-terminal domain, typical of TR integrases, which houses the catalytic domain. Both IntV1 and IntV2 contain the essential tyrosine (Y) nucleophile surrounded by the highly conserved “RHR triad” (in tertiary structure) (50), revealing highly similar active sites (see Fig. S2A in the supplemental material). Three conserved domains were identified in both integrases: the N terminus contained a DUF4102 (pfam13356) domain as well as a SAM-like domain (pfam14659) common to a wide variety of phage integrases, and the C-terminal domain contained a P4 catalytic domain (pfam00589) responsible for the breaking and rejoining of DNA fragments during recombination (see Fig. S2A). The data suggest that both integrases can catalyze excision of VPI-1, which suggests that either the integrases have a more promiscuous att site or there is a common att site that both integrases act on.
FIG 3.
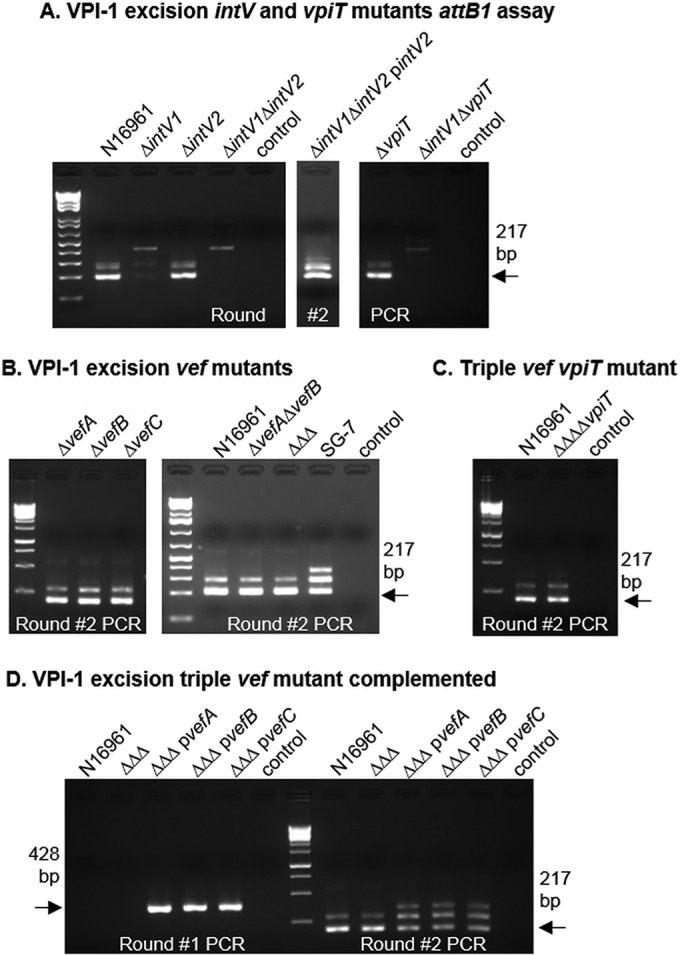
Role of integrases and RDFs in excision of VPI-1. (A) VPI-1 excision in the wild type (N16961) and intV and vpiT single and double mutants. The double intV mutant was complemented with intV2 to confirm the role of intV2 in VPI-1 excision. (B) Excision of VPI-1 using the attB1 excision assay in single, double, and triple (ΔΔΔ) RDF mutant backgrounds. (C) Excision of VPI-1 in the quadruple mutant (ΔΔΔΔvpiT), which lacks all three RDFs and the transposase encoded by vpiT. In panels A to C, the results of only round 2 are shown, as no excision products were detected in round 1. (D) VPI-1 excision in the ΔΔΔ mutant complemented with each RDF individually. Excision of the complements was detected in round 1 and is referred to as a superexcision phenotype. “control” indicates sample with no template DNA.
RDFs are not essential for VPI-1 excision but can promote excision.
Although we were able to observe excision of VPI-1 both in our attP1 and attB1 excision assays, this island contains no annotated RDF (see Fig. S1C and D). To explore this further, VPI-1 excision was examined in the RDF mutant strains using the attB1 excision assay, and excision was observed in the single, double, and triple mutants (Fig. 3B). While no RDF has been annotated within VPI-1, it is possible that one with little sequence homology exists. In order to search for potential new RDFs, we used a method similar to one that Fogg et al. used to identify the E. coli Φ24B Xis (20). The DNA sequence of the entire VPI-1 region and flanking open reading frames (ORFs) (VC0816 to VC0848, genome coordinates 872764 to 915211) was analyzed for all ORFs using the bacterial code which includes start codons GTG, TTG, CTG, and ATT. This analysis identified 35 ORFs between 50 and 110 amino acids in length (typical RDF protein length). These were further analyzed using the HHpred server, a more sensitive analysis tool based on hidden Markov models (HMMs) (51). None of the 35 identified ORFs yielded any similarity to previously described excisionases. However, when the vpiT (VC0817) amino acid sequence was submitted to HHpred, 52 amino acids near the N terminus displayed a distant relationship to TorI, a previously characterized excisionase (25, 31, 52). Although the shared identity in this region was 12%, with an E value of 0.0061, the predicted secondary elements of these regions were nearly identical (see Fig. S3 in the supplemental material). To eliminate the possibility of the VpiT aiding in the excision of VPI-1, a quadruple mutant was constructed in which the three RDFs and vpiT were deleted. The quadruple mutant was examined, and the excision of VPI-1 was still detected (Fig. 3C), suggesting that the VpiT is not involved and that RDFs are not necessary for integrase-mediated VPI-1 excision.
We next examined excision of VPI-1 in the triple RDF mutant complemented strains as described for VPI-2. In this case, we also observed the superexcision phenotype with each RDF (Fig. 3D). Complementation with empty pBAD33 had no effect on excision of either island (data not shown). These data suggest that although the RDFs are not essential for VPI-1 excision, they do function to promote excision of VPI-1.
RDFs are negative regulators of intV1 and intV2.
In order to determine if the RDFs play a role in controlling transcription of the integrase, real-time quantitative PCR was used to determine the transcript levels of intV1 and intV2 in wild-type and ΔvefA ΔvefB ΔvefC cells. We found that intV1 and intV2 levels are 4.5-fold (P < 0.005) and 2.5-fold (P < 0.05) higher in the triple RDF mutant than in the wild type (Fig. 4A), indicating that the RDFs act as negative transcriptional regulators of intV1 and intV2.
FIG 4.
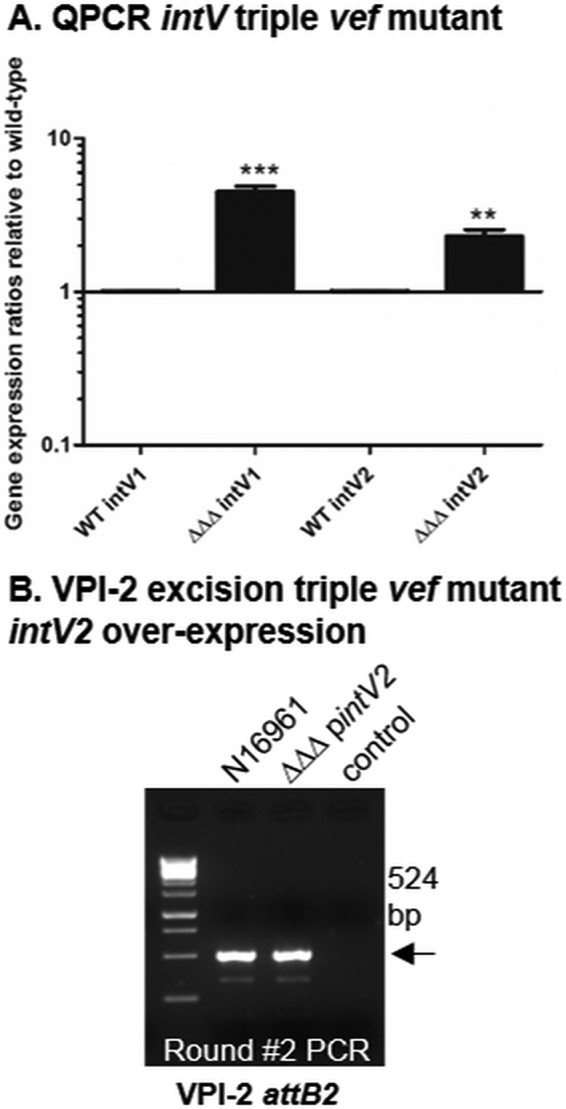
RDFs act as transcriptional repressors of the integrases encoded by intV1 and intV2. Expression of intV1 and intV2 in wild-type (WT) versus ΔvefA ΔvefB ΔvefC logarithmic cells was analyzed using real-time quantitative PCR. Fold change values were extracted using the ΔΔCT method and are represented on a log 10 scale. (A) intV expression in the wild type compared to the triple Vef mutant. (B) VPI-2 attB2 excision assay of intV2 overexpressed in the triple Vef mutant background. Only results of round 2 are shown, as no excision products were detected in round 1. “control” indicates sample with no template DNA.
Overexpression of intV2 can compensate for the absence of RDFs in VPI-2 excision.
The transcriptional data revealed higher levels of intV1 than intV2 in the ΔvefA ΔvefB ΔvefC mutant. This higher level of integrase observed in the triple RDF mutant may explain the VPI-1 excision phenotype still observed in this strain (Fig. 3B and 4A). To determine if increasing the levels of intV2 can restore VPI-2 excision, in the absence of RDFs, the intV2 gene was ectopically expressed under the control of an arabinose promoter in the ΔvefA ΔvefB ΔvefC background. The attB2 assay was used to examine the excision phenotype of this strain. Overexpression of intV2 in the RDF-negative background did indeed result in restoration of VPI-2 excision (Fig. 4B). These data suggest that if integrase levels are high enough, they do not require an RDF to aid in their excision function, which may explain why VPI-1 excision still occurs in an RDF-negative background.
VefA and VefB can bind the att sites of VPI-1 and VPI-2.
The RDF-mediated mechanism of integrase control we observed has also been proposed for the E. coli KpIE1 prophage (31), which contains a recombination module similar to that of VPI-1 and VPI-2, in that the integrase promoter is within the att site (see Fig. S3 in the supplemental material). We next sought to determine whether the V. cholerae RDF VefA can bind at the attR1 and attL2 sites of VPI-1 and VPI-2, respectively. The DNA fragments attR1 and attL2 encompass the region between the att site and translational start site of the integrases IntV1 and IntV2, respectively (Fig. 5A). Electrophoretic mobility shift assays (EMSAs) were used to determine whether VefA can bind these att sites (Fig. 5B). Increasing amounts of VefA were incubated with 30 ng of each DNA fragment. Shifts were observed for attR1 and attL2 DNA fragments near 10 μM and 4.1 μM VefA, respectively (Fig. 5B). No VefA binding was detected when using Vibrio vulnificus DNA amplified from VV1_0809 as a control under the same DAN/protein ratios (see Fig. S4D in the supplemental material). A DNA alignment of the attR1 and attL2 DNA fragments using ClustalW showed 76% conservation, and repeats of up to 14 bp were identified (Fig. 5C). Several shared motifs were also identified in the attR1 and attL2 DNA fragments near the core att sites, suggesting that these may be RDF binding sites (Fig. 5C and D). Additionally, the attL2 sequence possessed five repeated motif sequences in the predicted 5′ untranslated region (UTR) of intV2 (Fig. 5C).
FIG 5.

VefA and VefB bind the att sites of VPI-1 and VPI-2. (A) Representation of the DNA sequences attR1 and attL2, which span from the core att site to the translational start site of intV1 and intV2 of VPI-1 and VPI-2, respectively. (B) Electrophoretic mobility shift assays (EMSAs) of the DNA fragments attR1 (30 ng) and attL2 (30 ng) with various concentrations of VefA (0 to 70 μM). Binding of VefA is observed at 10 μM and 4.1 μM for attR1 and attL2, respectively. (C) Alignment of attR1 and attL2 DNA fragments used in EMSAs. Exact repeats shared between the two sequences are boxed. Core att site sequences are boxed and highlighted. Repeated motif sequences identified by MEME are represented with arrows and their corresponding P values. (D) Logo sequences of the three motifs identified in the att sequences in panel C. (E) EMSA of the DNA fragments attR1 (30 ng) and attL2 (30 ng) with various concentrations of VefB (0 to 20.5 μM). Binding of VefB is observed at 2.5 μM and 1.4 μM for attR1 and attL2, respectively.
We next determined whether VefB can also bind at the attachment sites of VPI-1 and VPI-2. Although sharing only 40% identity at the amino acid level, the predicted secondary elements of VefA and VefB are nearly identical (Fig. 1C). However, the numbers and locations of charged residues are distinctly different; the isoelectric point of VefA is 5.31, while VefB's is 10.44. Binding of VefB to the DNA fragments using EMSAs was observed at 2.5 μM for the att site and 1.4 μM for attR1 and attL2 (Fig. 5E). Both VefA and VefB bind to the VPI-2 att site at a higher affinity than the attR1 site of VPI-1 (Fig. 5B and E). To further narrow down where these RDFs may be binding, two DNA fragments were generated from the attL2 sequence, named 1st half (includes core att site) and 2nd half (includes IntV2 start codon), and were used for EMSAs (see Fig. S4A in the supplemental material). Both VefA and VefB were capable of binding the first half; however, only VefB was able to bind the second half of the attL2 DNA fragment (see Fig. S4B and C).
Distribution of RDFs among vibrios.
BLAST analysis revealed over 3,000 representatives (among 105 genera and 283 species) with equal to or greater than 70% query cover and 40% identity to the V. cholerae RDFs across five bacterial classes: Acidobacteria, Alphaproteobacteria, Betaproteobacteria, Cyanobacteria, and Gammaproteobacteria (see Fig. S5 in the supplemental material). The vast majority were members of the order Enterobacteriales: E. coli (787), Klebsiella (276), Salmonella enterica (166), Yersinia (118), and Enterobacter cloacae (109), bacterial groups that are also disproportionally represented in the genome database. These data demonstrate that the occurrence of these RDFs is widespread among bacteria.
In addition, we examined Vibrio species in detail to identify island regions with IntV1 homologs and associated RDFs. From this analysis, IntV1 was found in association with homologs of VefA, VefB, and VefC in numerous other V. cholerae strains (see Fig. S6 in the supplemental material), indicating that at one time VPI-1 may have contained an RDF. For example, V. cholerae O1 strain 12129 (1) contains VPI-1 with both VefA and VefB and a non-O1/non-O139 strain, V. cholerae HE-09, contains a VPI-1 variant with VefC. Several other examples of PAIs containing IntV1 and Vef homologs exist within V. cholerae as well as in other Vibrionaceae (see Fig. S6). These findings demonstrate the association, at least in proximity, of IntV1 and all 3 RDFs found in V. cholerae N16961, suggesting a shared evolutionary history supporting their ability to cross talk.
DISCUSSION
Pathogenicity islands, which possess a wide range of virulent genes, are widespread among bacteria but are the least examined class of MIGEs (7–17). Excision of PAIs from the bacterial chromosome is thought to be the first step in the transfer and spread of these elements. Thus, determining the excision mechanism is essential in understanding the distribution potential of the mobilome and its role in the evolution of bacteria. Vibrio cholerae was selected as a model organism for this study because it contains two PAIs important for its virulence. Given the ability of VPIs to excise from the bacterial genome (8, 42, 43), we sought to decipher this mechanism, particularly how VPI-1 could excise without containing an RDF. We determined that all three RDFs, corresponding to vefA, vefB, and vefC, have redundant roles in promoting excision of VPI-1 and VPI-2, as overexpression of each individual RDF resulted in superexcision of these islands. These data indicate extensive cross talk between three different islands: vefA and vefB, found on VPI-2, can promote excision of VPI-1, and vefC, found on VSP-II, can aid in excision of both VPI-1 and VPI-2. This is the first report of RDF-mediated cross talk in any MIGE. The cross talk was not limited to the RDFs: we found that the cognate integrase of VPI-2 can also play a role in excision of VPI-1. This is not the first report of integrase-mediated cross talk between pathogenicity islands; in UPEC, the cognate integrase of PAI II536 was capable of mediating excision of PAI V536, an island located elsewhere on the genome, and this, too, was one-directional cross talk (44).
Once the role of RDFs in PAI excision was established, we next examined the molecular mechanism by which these proteins act. We performed real-time quantitative PCR to determine whether the RDFs control integrase transcription and found that the Vefs were negative transcriptional regulators. The unique configuration of the VPI recombination modules (att sites, intV, and vef) can provide insight into this negative regulation. The promoter regions of intV1 and intV2 are located at the attachment sites of the PAI, whereas in bacteriophage λ, with which the majority of RDF studies have been done, the integrase promoter lies within the phage genome (see Fig. S3 in the supplemental material). The position of the PAI integrase promoters allows for potential regulation by proteins that bind at the att sites to aid in formation of the excisive intasome (protein-DNA complex necessary for excision), which we propose are the Vefs. In the E. coli KpIE1 prophage, which has a similar integrase and att site arrangement as the VPIs (see Fig. S3), Panis and colleagues also found that the RDF TorI negatively regulates expression of the intS gene (31). Others have found RDFs to act as integrase repressors, such as the RDF Vis from the P4 bacteriophage, which was found to repress the P4 integrase at transcription initiation as well as posttranscriptionally (53).
Our transcriptional analysis of integrase expression sheds light on the question of why we observe an excision product for VPI-1 and not VPI-2 in the absence of the RDFs. Expression levels of intV1 and intV2 were both increased in the ΔvefA ΔvefB ΔvefC mutant compared to the wild type; however, this effect was more pronounced for intV1, resulting in more cognate integrase to facilitate the site-specific recombination reaction, thus allowing excision of VPI-1. The increased levels of intV2 in this genetic background may also aid in the excision of VPI-1, as we have shown that there is cross talk with this integrase in VPI-1 excision. From this observation, we next determined whether VPI-2 excision could occur in an RDF-negative background if more intV2 was present. Overexpressed intV2 in the ΔvefA ΔvefB ΔvefC background resulted in VPI-2 excision detectable at wild-type levels. These data indicate that if enough integrase is provided, intV1 and intV2 can facilitate site-specific recombination and excision of VPIs without the presence of RDFs. These data highlight the importance of the RDFs in regulating the amount of integrase necessary for controlling VPI excision. The importance of integrase stability has also been demonstrated in the excision of a mycophage (which does not appear to require an RDF for excision) (54).
The majority of RDFs that have been described are from prophages and were shown to control directionality of recombination by playing an architectural role by binding at the att sites, to each other, and/or to the integrase (20, 21, 24, 27–29, 55). We determined that VefA and VefB bound the att sites of both islands, although VefB bound both sites at a higher affinity, causing a shift with four times less protein than VefA. VefA and VefB have similar predicted secondary elements, and their basic residues, which are essential for DNA binding (56), are highly conserved. However, VefA contains additional charged residues, including acidic residues in exposed loops. Furthermore, VefA contains a sole Cys at position 70 (Fig. 1C) whose contribution to function remains unknown and warrants further investigation. Thus, the higher affinity of VefB for the att site DNA may be due to its higher positive charge density, allowing for favorable interaction with DNA. The shift of the protein-bound DNA changes as the amount of protein increases, suggesting multiple Vef binding sites and formation of protein oligomers. Cooperative binding of the lambda Xis protein at the att DNA sites has been reported, and formation of this micronucleoprotein filament has been shown to drive DNA bending (26, 27, 30). Therefore, we speculate that the multiple Vef monomers bind at the att site and act similarly to bend the DNA, thereby driving the excision reaction.
Based on our findings, we have proposed a model for RDF function in the control of PAI excision in V. cholerae. In the absence of the RDFs, intV1 and intV2 expression increases; however, as RDF levels increase, the repression of int is enhanced. We showed that RDFs bind at the att sites of VPI-1 and VPI-2 to aid in excision. It is likely that RDF binding at the attR1 and attL2 sites resulted in decreases of intV1 and intV2 transcription, respectively. The negative regulation of intV transcription may be caused by preventing promoter recognition either by altering DNA topology or by direct RDF binding at the intV promoters. It is possible that the integrase also plays a role in regulating its own transcription and that VefB, whose promoter also overlaps with the attR2 site, may also self-regulate.
This is the first report of RDF-mediated cross talk in PAI excision. We have identified Vef homologs in more than 100 genera and 280 species, confirming their prevalence among bacteria. Due to their low sequence homology, the list of included organisms is most likely vastly underrepresented. Understanding the mechanism of PAI excision is important in the evolution and spread of these elements to their nonpathogenic counterparts. This study reveals extensive cross talk, through RDFs and integrases, between multiple PAIs in a single bacterium, to control the excision and probable spread of these elements to other bacteria. It is likely that other species which contain numerous MIGEs are also capable of cross talk. Thus, it is important to consider the genetic content outside an individual element when determining the inputs necessary for excision and its impact on the bacterial mobilome.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Boyd group, past and present, for their review and helpful discussion on earlier versions of the manuscript, Jun Li and Zhengqi Zhang for their assistance in protein purification, and Melinda Duncan for generously sharing equipment and for thoughtful experimental input.
This work was supported by National Science Foundation CAREER awards MCB-1054447 to S.R. and DEB-0844409 to E.F.B.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00704-15.
REFERENCES
- 1.Boyd EF. 2012. Bacteriophage-encoded bacterial virulence factors and phage-pathogenicity island interactions. Adv Virus Res 82:91–118. doi: 10.1016/B978-0-12-394621-8.00014-5. [DOI] [PubMed] [Google Scholar]
- 2.Boyd EF, Almagro-Moreno S, Parent MA. 2009. Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol 17:47–53. doi: 10.1016/j.tim.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Frost LS, Leplae R, Summers AO, Toussaint A. 2005. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol 3:722–732. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- 4.Almagro-Moreno S, Murphy RA, Boyd EF. 2011. How genomics has shaped our understanding of the evolution and emergence of pathogenic Vibrio cholerae, p 85–99. In Fratamico P, Liu Y, Kathariou S (ed), Genomes of foodborne and waterborne pathogens. ASM Press, Washington, DC. [Google Scholar]
- 5.Boyd EF, Carpenter MR, Chowdhury N. 2012. Mobile effector proteins on phage genomes. Bacteriophage 2:139–148. doi: 10.4161/bact.21658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyd EF. 2012. Mobile and integrative genetic elements encoding cargo genes that are important for bacterial virulence, p 289–304. In Faruque SM. (ed), Foodborne and waterborne bacterial pathogens: epidemiology and molecular biology. Horizon Scientific Press, Norfolk, United Kingdom. [Google Scholar]
- 7.Al-Hasani K, Rajakumar K, Bulach D, Robins-Browne R, Adler B, Sakellaris H. 2001. Genetic organization of the she pathogenicity island in Shigella flexneri 2a. Microb Pathog 30:1–8. doi: 10.1006/mpat.2000.0404. [DOI] [PubMed] [Google Scholar]
- 8.Murphy RA, Boyd EF. 2008. Three pathogenicity islands of Vibrio cholerae can excise from the chromosome and form circular intermediates. J Bacteriol 190:636–647. doi: 10.1128/JB.00562-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carniel E, Guilvout I, Prentice M. 1996. Characterization of a large chromosomal “high-pathogenicity island” in biotype 1B Yersinia enterocolitica. J Bacteriol 178:6743–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hacker J, Kaper JB. 2000. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol 54:641–679. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- 11.Hensel M. 2004. Evolution of pathogenicity islands of Salmonella enterica. Int J Med Microbiol 294:95–102. doi: 10.1016/j.ijmm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 12.Jermyn WS, Boyd EF. 2002. Characterization of a novel Vibrio pathogenicity island (VPI-2) encoding neuraminidase (nanH) among toxigenic Vibrio cholerae isolates. Microbiology 148:3681–3693. doi: 10.1099/00221287-148-11-3681. [DOI] [PubMed] [Google Scholar]
- 13.Karaolis DK, Johnson JA, Bailey CC, Boedeker EC, Kaper JB, Reeves PR. 1998. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc Natl Acad Sci U S A 95:3134–3139. doi: 10.1073/pnas.95.6.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Shea YA, Finnan S, Reen FJ, Morrissey JP, O'Gara F, Boyd EF. 2004. The Vibrio seventh pandemic island-II is a 26.9 kb genomic island present in Vibrio cholerae El Tor and O139 serogroup isolates that shows homology to a 43.4 kb genomic island in V. vulnificus. Microbiology 150:4053–4063. doi: 10.1099/mic.0.27172-0. [DOI] [PubMed] [Google Scholar]
- 15.Quirke AM, Reen FJ, Claesson MJ, Boyd EF. 2006. Genomic island identification in Vibrio vulnificus reveals significant genome plasticity in this human pathogen. Bioinformatics 22:905–910. doi: 10.1093/bioinformatics/btl015. [DOI] [PubMed] [Google Scholar]
- 16.Hurley CC, Quirke A, Reen FJ, Boyd EF. 2006. Four genomic islands that mark post-1995 pandemic Vibrio parahaemolyticus isolates. BMC Genomics 7:104. doi: 10.1186/1471-2164-7-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jermyn WS, Boyd EF. 2005. Molecular evolution of Vibrio pathogenicity island-2 (VPI-2): mosaic structure among Vibrio cholerae and Vibrio mimicus natural isolates. Microbiology 151:311–322. doi: 10.1099/mic.0.27621-0. [DOI] [PubMed] [Google Scholar]
- 18.Napolitano MG, Almagro-Moreno S, Boyd EF. 2011. Dichotomy in the evolution of pathogenicity island and bacteriophage encoded integrases from pathogenic Escherichia coli strains. Infect Genet Evol 11:423–436. [DOI] [PubMed] [Google Scholar]
- 19.Napolitano MG, Boyd EF. 2012. Pathogenicity island evolution: a distinct new class of integrative element or a mosaic of other elements? p 273–287. In Roberts AP, Mullany P (ed), Bacterial integrative mobile genetic elements. Landes Bioscience Press, Austin, TX. [Google Scholar]
- 20.Fogg PC, Rigden DJ, Saunders JR, McCarthy AJ, Allison HE. 2011. Characterization of the relationship between integrase, excisionase and antirepressor activities associated with a superinfecting Shiga toxin encoding bacteriophage. Nucleic Acids Res 39:2116–2129. doi: 10.1093/nar/gkq923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flanigan A, Gardner J. 2008. Structural prediction and mutational analysis of the Gifsy-I Xis protein. BMC Microbiol 8:199. doi: 10.1186/1471-2180-8-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramsay JP, Sullivan JT, Stuart GS, Lamont IL, Ronson CW. 2006. Excision and transfer of the Mesorhizobium loti R7A symbiosis island requires an integrase IntS, a novel recombination directionality factor RdfS, and a putative relaxase RlxS. Mol Microbiol 62:723–734. doi: 10.1111/j.1365-2958.2006.05396.x. [DOI] [PubMed] [Google Scholar]
- 23.Lee CA, Auchtung JM, Monson RE, Grossman AD. 2007. Identification and characterization of int (integrase), xis (excisionase) and chromosomal attachment sites of the integrative and conjugative element ICEBs1 of Bacillus subtilis. Mol Microbiol 66:1356–1369. [DOI] [PubMed] [Google Scholar]
- 24.Coddeville M, Ritzenthaler P. 2010. Control of directionality in bacteriophage mv4 site-specific recombination: functional analysis of the Xis factor. J Bacteriol 192:624–635. doi: 10.1128/JB.00986-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panis G, Mejean V, Ansaldi M. 2007. Control and regulation of KplE1 prophage site-specific recombination: a new recombination module analyzed. J Biol Chem 282:21798–21809. doi: 10.1074/jbc.M701827200. [DOI] [PubMed] [Google Scholar]
- 26.Sam MD, Papagiannis CV, Connolly KM, Corselli L, Iwahara J, Lee J, Phillips M, Wojciak JM, Johnson RC, Clubb RT. 2002. Regulation of directionality in bacteriophage lambda site-specific recombination: structure of the Xis protein. J Mol Biol 324:791–805. doi: 10.1016/S0022-2836(02)01150-6. [DOI] [PubMed] [Google Scholar]
- 27.Singh S, Plaks JG, Homa NJ, Amrich CG, Heroux A, Hatfull GF, VanDemark AP. 2014. The structure of Xis reveals the basis for filament formation and insight into DNA bending within a mycobacteriophage intasome. J Mol Biol 426:412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warren D, Sam MD, Manley K, Sarkar D, Lee SY, Abbani M, Wojciak JM, Clubb RT, Landy A. 2003. Identification of the lambda integrase surface that interacts with Xis reveals a residue that is also critical for Int dimer formation. Proc Natl Acad Sci U S A 100:8176–8181. doi: 10.1073/pnas.1033041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis JA, Hatfull GF. 2001. Control of directionality in integrase-mediated recombination: examination of recombination directionality factors (RDFs) including Xis and Cox proteins. Nucleic Acids Res 29:2205–2216. doi: 10.1093/nar/29.11.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abbani MA, Papagiannis CV, Sam MD, Cascio D, Johnson RC, Clubb RT. 2007. Structure of the cooperative Xis-DNA complex reveals a micronucleoprotein filament that regulates phage lambda intasome assembly. Proc Natl Acad Sci U S A 104:2109–2114. doi: 10.1073/pnas.0607820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panis G, Duverger Y, Courvoisier-Dezord E, Champ S, Talla E, Ansaldi M. 2010. Tight regulation of the intS gene of the KplE1 prophage: a new paradigm for integrase gene regulation. PLoS Genet 6(10):e1001149. doi: 10.1371/journal.pgen.1001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Numrych TE, Gumport RI, Gardner JF. 1992. Characterization of the bacteriophage lambda excisionase (Xis) protein: the C-terminus is required for Xis-integrase cooperativity but not for DNA binding. EMBO J 11:3797–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swalla BM, Cho EH, Gumport RI, Gardner JF. 2003. The molecular basis of co-operative DNA binding between lambda integrase and excisionase. Mol Microbiol 50:89–99. doi: 10.1046/j.1365-2958.2003.03687.x. [DOI] [PubMed] [Google Scholar]
- 34.Dziejman M, Balon E, Boyd D, Fraser CM, Heidelberg JF, Mekalanos JJ. 2002. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc Natl Acad Sci U S A 99:1556–1561. doi: 10.1073/pnas.042667999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. 1988. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manning PA. 1997. The tcp gene cluster of Vibrio cholerae. Gene 192:63–70. doi: 10.1016/S0378-1119(97)00036-X. [DOI] [PubMed] [Google Scholar]
- 37.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A 84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Almagro-Moreno S, Boyd EF. 2009. Sialic acid catabolism confers a competitive advantage to pathogenic Vibrio cholerae in the mouse intestine. Infect Immun 77:3807–3816. doi: 10.1128/IAI.00279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Almagro-Moreno S, Boyd EF. 2009. Insights into the evolution of sialic acid catabolism among bacteria. BMC Evol Biol 9:118. doi: 10.1186/1471-2148-9-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chowdhury N, Norris J, McAlister E, Lau SY, Thomas GH, Boyd EF. 2012. The VC1777-VC1779 proteins are members of a sialic acid-specific subfamily of TRAP transporters (SiaPQM) and constitute the sole route of sialic acid uptake in the human pathogen Vibrio cholerae. Microbiology 158:2158–2167. doi: 10.1099/mic.0.059659-0. [DOI] [PubMed] [Google Scholar]
- 41.Faruque SM, Zhu J, Asadulghani Kamruzzaman M, Mekalanos JJ. 2003. Examination of diverse toxin-coregulated pilus-positive Vibrio cholerae strains fails to demonstrate evidence for Vibrio pathogenicity island phage. Infect Immun 71:2993–2999. doi: 10.1128/IAI.71.6.2993-2999.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajanna C, Wang J, Zhang D, Xu Z, Ali A, Hou YM, Karaolis DK. 2003. The vibrio pathogenicity island of epidemic Vibrio cholerae forms precise extrachromosomal circular excision products. J Bacteriol 185:6893–6901. doi: 10.1128/JB.185.23.6893-6901.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Almagro-Moreno S, Napolitano MG, Boyd EF. 2010. Excision dynamics of Vibrio pathogenicity island-2 from Vibrio cholerae: role of a recombination directionality factor VefA. BMC Microbiol 10:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hochhut B, Wilde C, Balling G, Middendorf B, Dobrindt U, Brzuszkiewicz E, Gottschalk G, Carniel E, Hacker J. 2006. Role of pathogenicity island-associated integrases in the genome plasticity of uropathogenic Escherichia coli strain 536. Mol Microbiol 61:584–595. doi: 10.1111/j.1365-2958.2006.05255.x. [DOI] [PubMed] [Google Scholar]
- 45.Dobrindt U, Chowdary MG, Krumbholz G, Hacker J. 2010. Genome dynamics and its impact on evolution of Escherichia coli. Med Microbiol Immunol 199:145–154. doi: 10.1007/s00430-010-0161-2. [DOI] [PubMed] [Google Scholar]
- 46.Lesic B, Bach S, Ghigo JM, Dobrindt U, Hacker J, Carniel E. 2004. Excision of the high-pathogenicity island of Yersinia pseudotuberculosis requires the combined actions of its cognate integrase and Hef, a new recombination directionality factor. Mol Microbiol 52:1337–1348. doi: 10.1111/j.1365-2958.2004.04073.x. [DOI] [PubMed] [Google Scholar]
- 47.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 49.Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. 2005. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol Rev 29:231–262. doi: 10.1016/j.fmrre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 50.Grainge I, Jayaram M. 1999. The integrase family of recombinases: organization and function of the active site. Mol Microbiol 33:449–456. doi: 10.1046/j.1365-2958.1999.01493.x. [DOI] [PubMed] [Google Scholar]
- 51.Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Panis G, Franche N, Mejean V, Ansaldi M. 2012. Insights into the functions of a prophage recombination directionality factor. Viruses 4:2417–2431. doi: 10.3390/v4112417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piazzolla D, Cali S, Spoldi E, Forti F, Sala C, Magnoni F, Deho G, Ghisotti D. 2006. Expression of phage P4 integrase is regulated negatively by both Int and Vis. J Gen Virol 87:2423–2431. doi: 10.1099/vir.0.81875-0. [DOI] [PubMed] [Google Scholar]
- 54.Broussard GW, Oldfield LM, Villanueva VM, Lunt BL, Shine EE, Hatfull GF. 2013. Integration-dependent bacteriophage immunity provides insights into the evolution of genetic switches. Mol Cell 49:237–248. doi: 10.1016/j.molcel.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abbani M, Iwahara M, Clubb RT. 2005. The structure of the excisionase (Xis) protein from conjugative transposon Tn916 provides insights into the regulation of heterobivalent tyrosine recombinases. J Mol Biol 347:11–25. doi: 10.1016/j.jmb.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 56.Jones S, van Heyningen P, Berman HM, Thornton JM. 1999. Protein-DNA interactions: a structural analysis. J Mol Biol 287:877–896. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- 57.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Philippe N, Alcaraz JP, Coursange E, Geiselmann J, Schneider D. 2004. Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51:246–255. doi: 10.1016/j.plasmid.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 59.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu J, Srinivasan P, Pham DN, Rozovsky S. 2012. Expression and purification of the membrane enzyme selenoprotein K. Protein Expr Purif 86:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.