

Measurements of mRNA ribosome loading in wild-type and clock-deficient plants show that the circadian clock regulates the translation of Arabidopsis mRNAs in concert with diurnal light-dark changes.
Abstract
Circadian control of gene expression is well characterized at the transcriptional level, but little is known about diel or circadian control of translation. Genome-wide translation state profiling of mRNAs in Arabidopsis thaliana seedlings grown in long day was performed to estimate ribosome loading per mRNA. The experiments revealed extensive translational regulation of key biological processes. Notably, translation of mRNAs for ribosomal proteins and mitochondrial respiration peaked at night. Central clock mRNAs are among those subject to fluctuations in ribosome loading. There was no consistent phase relationship between peak translation states and peak transcript levels. The overlay of distinct transcriptional and translational cycles can be expected to alter the waveform of the protein synthesis rate. Plants that constitutively overexpress the clock gene CCA1 showed phase shifts in peak translation, with a 6-h delay from midnight to dawn or from noon to evening being particularly common. Moreover, cycles of ribosome loading that were detected under continuous light in the wild type collapsed in the CCA1 overexpressor. Finally, at the transcript level, the CCA1-ox strain adopted a global pattern of transcript abundance that was broadly correlated with the light-dark environment. Altogether, these data demonstrate that gene-specific diel cycles of ribosome loading are controlled in part by the circadian clock.
INTRODUCTION
To adjust to a changing environment over the course of a day, plant cells regulate gene expression in a diel context. In Arabidopsis thaliana, for example, about one-third of all genes are transcribed under the direction of the circadian clock (Covington et al., 2008). Since most of the energy required for gene expression is spent on translation, it is plausible that translation itself may also be dielly regulated in order for plants to respond to the environment in an energy-efficient manner. Indeed, Arabidopsis plants undergo global cycles of ribosome loading (Pal et al., 2013) but not ribosome abundance (Piques et al., 2009) over the course of the light-dark cycle.
The translation state (TL) of an mRNA is often estimated from its ribosome loading. The more ribosomes bind to an mRNA, the more efficiently it is translated (Mathews et al., 2007). mRNA-ribosome complexes (polysomes) can be fractionated according to the number of ribosomes, and the proportion of the mRNA pool that resides in the different fractions can be used to determine TL. As defined here, TL is independent of the mRNA transcript level. That is, an mRNA’s TL value will be the same between two different samples, as long as the average number of ribosomes per mRNA molecule is the same, even if the total amount of the mRNA differs between the two samples. TL for many Arabidopsis mRNAs is sensitive to a variety of environmental conditions, including hypoxia, heavy metal, drought, sugar, virus, heat, and light exposure, as well as various genetic backgrounds (Kawaguchi et al., 2004; Nicolaï et al., 2006; Kim et al., 2007; Branco-Price et al., 2008; Sormani et al., 2011; Juntawong and Bailey-Serres, 2012; Liu et al., 2012; Moeller et al., 2012; Tiruneh et al., 2013; Yángüez et al., 2013). Extensive coregulation of TL has been reported, for example, for ribosomal protein mRNAs (Kawaguchi et al., 2004; Kim et al., 2007; Juntawong and Bailey-Serres, 2012; Tiruneh et al., 2013), suggesting that the transcriptome is organized into regulons of translational control.
The central oscillator of the circadian clock in Arabidopsis is based on a group of interlocked transcriptional feedback loops. At the core of the oscillator are three groups of genes that regulate gene expression in a cyclical fashion throughout the day by mutual transcriptional repression (Nagel and Kay, 2012; Pokhilko et al., 2012). Transcripts for evening genes (e.g., TIMING OF CAB EXPRESSION1 [TOC1], the transcription factor gene LUX, EARLY FLOWERING3 [ELF3], and ELF4) peak late in the day and repress day genes (e.g., PSEUDO-RESPONSE REGULATOR5 [PRR5], PRR7, and PRR9). Day genes repress morning genes, and morning genes (e.g., CIRCADIAN CLOCK ASSOCIATED1 [CCA1] and LATE ELONGATED HYPOCOTYL [LHY]) repress evening genes. Constitutive overexpression of the morning gene CCA1 disrupts normal clock function (Wang and Tobin, 1998; Green et al., 2002). Under continuous light, the clock of the CCA1-overexpressor strain (CCA1-ox) is disordered and arrhythmic, as many central clock genes and clock output mRNAs are continuously expressed, while the partner of CCA1, LHY, is continuously repressed (Wang and Tobin, 1998; Matsushika et al., 2002). In contrast, under light-dark cycle conditions, mRNAs for several central clock genes and clock outputs continue to cycle in the CCA1-ox strain (Matsushika et al., 2002). Clock genes such as LHY and CCR2/GRP7 still respond to light in CCA1-ox but typically do not anticipate the dark-to-light transition, in keeping with the defect in the clock (Green et al., 2002).
Early studies established that translation of new proteins plays a fundamental role in the operation of the circadian clock (Jacklet, 1977; Nakashima et al., 1981). However, subsequent investigations identified a robust mechanism of transcriptional control at the core of several circadian clocks (Hardin et al., 1992; Aronson et al., 1994; Sehgal et al., 1995; Schaffer et al., 1998; Wang and Tobin, 1998; Strayer et al., 2000). Thus, the role of translational control in circadian clock function has not been studied in detail at the genome level. In recent years, posttranscriptional control of diel and circadian gene expression has attracted significant attention. In particular, many clock mRNAs are alternatively spliced (Staiger et al., 2003; Staiger and Green, 2011; Filichkin and Mockler, 2012; Park et al., 2012), and this must be regulated for proper clock function (Sanchez et al., 2010; Jones et al., 2012). Alternative splicing has also been implicated in temperature compensation of the clock (James et al., 2012; Seo et al., 2012; Kwon et al., 2014). By comparison, control of diel gene expression at the translational level has received comparatively little attention (Kim et al., 2003).
Here, we characterized translational control over the course of the diel light-dark cycle by measuring the ribosome loading of mRNAs in 10-d-old Arabidopsis seedlings grown in a long day. Approximately one in seven mRNAs are subject to robust diel cycles of ribosome loading. These cycles are partially controlled by the circadian clock, given that the translation cycles are substantially remodeled in the CCA1-ox strain. Diel and circadian translational control are particularly common among mRNAs for ribosome biogenesis, the inner mitochondrial membrane, and the photosynthetic apparatus. In summary, we provide a genome-wide characterization of circadian control of gene-specific translation in plants.
RESULTS
Polysome Loading over a Diel Cycle
We monitored polysome loading in 10-d-old wild-type Arabidopsis seedlings over a 16-h-light/8-h-dark cycle at 6 am (Zeitgeber time ZT0), 12 pm (ZT6), 6 pm (ZT12), 12 am (ZT18), and again at 6 am (ZT24). RNA was fractionated into nonpolysomal (NP), small polysomal (SP), and large polysomal (LP) fractions using sucrose density centrifugation. We quantified polysome loading as the fraction of RNA found in SP and LP fractions relative to the total, which also includes NP RNA: (SP+LP)/(NP+SP+LP). In the wild type, polysome loading began at its lowest level at dawn, 6 am (ZT0), peaked during the day, remained elevated through 12 am (ZT18), and returned to low levels again the next dawn (Figures 1A and 1B). These data, obtained in seedlings grown in long day on artificial medium with 1% sucrose, follow a similar pattern as those from vegetative rosettes grown in a 12-h light-dark cycle on soil (Pal et al., 2013), although the drop in translation toward dawn was less pronounced in our experiments. In the CCA1-ox strain, which has a disrupted circadian clock due to constitutive overexpression of CCA1, the pattern was similar to the wild type, but less dramatic (Figures 1A and 1C). Invoking the well-supported notion that ribosome loading reflects the rate of translation initiation (Mathews et al., 2007), these data suggested that diel control of translation may depend on a functional clock.
Figure 1.

Polysome Loading over a Diel Cycle.
Ten-day-old wild-type and CCA1-ox seedlings were grown in a 16-h-light/8-h-dark cycle. Tissue extracts from five time points harvested every 6 h were individually subjected to polysome density gradient centrifugation. The fraction of total RNA recovered in polysomal fractions (small and large polysomes) is plotted in (A) as a percentage of total RNA (nonpolysomes and small and large polysomes shown in [B] and [C]) at each ZT time. Error bars show standard deviations from three biological replicates. The difference in polysome loading between the wild type and CCA1-ox was significant by unpaired two-tailed t test for ZT6 (P = 0.0058) and ZT18 (P = 0.0041). The elevated polysome loading compared with ZT0 was significant in the wild type for ZT6, ZT12, and ZT18 and in CCA1-ox for ZT6.
In order to obtain gene-specific ribosome loading data over the diel cycle, the mRNAs in the NP, SP, LP, and total (TX) RNA fractions were quantified by microarray hybridization at 6 am (ZT0), 12 pm (ZT6), 6 pm (ZT12), and 12 am (ZT18). A TL was calculated for each mRNA: TL = (2xSP+7xLP)/(NP+SP+LP). SP and LP fractions were weighted by 2 and 7, respectively, because mRNA molecules are estimated to be bound by two and seven ribosomes in these fractions, on average (Supplemental Figure 1). TL values were calculated for those 12,342 nuclear-encoded genes that were reliably detected in the SP and LP fractions at all four time points in all three replicates (Supplemental Data Set 1). ΔTL is defined as the TL at the peak minus the TL at the trough. Genes with varying TL across the diel cycle were first identified by significance analysis of microarrays (SAM; Tusher et al., 2001). Using SAM, 1825 mRNAs (15% of 12,342) varied in their TL value across time points at a collective false discovery rate (FDR) of 10% (Figures 2A and 2C). In lieu of a gene-wise FDR, which SAM does not provide, we calculated an empirical permutation-based P value, which confirmed that, besides a large fraction of strong translation cycles, many of the moderate translation cycles (0.3 < ΔTL < 0.7) were statistically significant (Figure 2E). The TL of the majority of mRNAs peaked at noon (ZT6) or midnight (ZT18); peaks at dawn (ZT0) or in the evening (ZT12) were less common. More frequently than not, peak and trough were offset by 12 h (Figure 2A).
Figure 2.

Diel Changes in Ribosome Loading of Arabidopsis mRNAs.
TL is an estimate of ribosome numbers per mRNA, and ΔTL is the difference between the highest and lowest TL for a gene, averaged over replicate samples. Lights-on was at ZT0 and lights-off was at ZT16. (A), (C), (E), (G), and (I) are the wild type. (B), (D), (F), (H), and (J) are CCA1-ox.
(A) and (B) mRNAs were filtered for a translational cycle using SAM. The mRNAs were first sorted into four predefined clusters according to the time of peak TL. Each cluster was then subdivided according to the time of the TL trough. Data were displayed using the heatmap.2 function from the gplots package in R. The number of mRNAs per cluster is given on the right.
(C) and (D) Distribution of all ΔTL values (black trace). ΔTL values were binned in increments of 0.1. The subset of cycling genes that were selected by ANOVA (AOV P < 0.05) (red trace) or SAM (10% FDR) (blue trace) is also illustrated.
(E) and (F) Relationship between ΔTL and SAM P value. We used the test statistic computed by SAM and its permutation-based null distribution to compute an empirical P value for each prefiltered gene. Here, we plotted the negative log of this P value versus the ΔTL for each gene. The 2437 genes with P < 0.05 lie to the right of the broken line.
(G) and (H) Venn diagram showing the overlap among the four classes of differentially translated genes.
(I) and (J) Relationship between raw ANOVA P values from comparing average TL across time points and the corresponding adjusted P values using the Benjamini-Hochberg method. For the wild type and CCA1-ox, a raw P value of 0.05 (red vertical line) corresponded to a FDR of 0.25 and 0.15, respectively. Fifty-four and 1254 genes passed the FDR < 0.05 threshold, respectively (red horizontal line).
Translation states of six representative genes were also analyzed by qRT-PCR (Supplemental Figure 2) as an independent technique, by calculating TL from the levels of transcripts present in the NP, SP, and LP fractions. Evidently, qRT-PCR and microarray results showed similar trends. The qRT-PCR data also confirmed that the changes in TL reflect diel cycles rather than monotonic trends over developmental time.
Because the SAM filter will miss valid cycling genes because of the arbitrary FDR cutoff of 10%, and to capture the majority of true positives while balancing the risk of false positives, we applied two other filters to the raw data. One-way ANOVA, to identify significant variation in TL across time points, yielded 2503 genes (20%) with an uncorrected P value below 0.05 and an FDR of below 0.25 (Figures 2C, 2G, and 2I), indicating that ∼2000 are true positives. As an alternative to ANOVA, we simply applied a moderately stringent threshold (ΔTL > 0.7) or a lenient threshold (ΔTL > 0.3) to the data (Figure 2G; Supplemental Data Set 1). Based on SAM, ANOVA, and the ΔTL > 0.3 threshold, ∼4000 mRNAs were translationally invariant. Taken together, ∼2000 mRNAs have statistically robust translational changes over the diel cycle, ∼4000 mRNAs are invariant, and the remaining 6300 may have statistically marginal changes in their translation. Evidently, a large fraction of the seedling transcriptome is affected.
We modeled diel variation in TL and TX as sine waves with a 24-h period (see Methods). Figure 3A shows four genes whose large R2 values document a good fit to a sine model and whose translation states peak around ZT0, ZT6, ZT12, and ZT18. Figure 3B shows distributions of R2 values for TX and TL in the wild type and CCA1-ox (see below). We used an R2 value of 0.6 as an arbitrary threshold to identify genes with sinusoidal expression, and we identified a peak time as the time at which the sine function reached its maximum. The distribution of peak times is shown in Figure 3C for wild-type TX and TL, as well as for CCA1-ox. While the sine model makes an additional assumption, it has merit because it uses information that we did not previously take into account, namely, the wave form of the data. The sine-modeled phase of our wild-type transcript cycles (Figure 3C) matched those of eleven published experiments (Mockler et al., 2007) with R2 values of up to 0.91, confirming the accuracy of our transcript data and the value of the sine modeling approach. In general, the patterns were consistent with our original analysis but provided additional information. At the TL level, in the wild type there is a strong preference for genes to peak at ZT19, with a secondary preference around ZT7 to ZT10. It appeared from our original analysis that many mRNAs peaked at ZT18 (12 am), while others peaked at ZT0 (6 am). This alternate perspective, made possible by the sine modeling approach, suggests that these two groups may actually behave as one larger group, whose TL peaks are centered about ZT19.
Figure 3.

Diel Cycles of Transcript Levels and Translation States Were Modeled as Sine Waves.
(A) Examples of the fit of representative translation state data to sine waves. The R2 indicates the fraction of the variation in TL that is explained by the sine model.
(B) Distributions of R2 values for TX and TL in the wild type and CCA1-ox. Genes in CCA1-ox have a greater tendency to behave like sine waves in terms of their TX and TL.
(C) Genes with an R2 >0.6 were selected and were binned according to their estimated peak TX and peak TL under the assumption of a sine model. For wild-type TL, the median confidence interval for peak TL was estimated by bootstrapping to be ±1.8 h (5th to 95th percentile range 1.0 to 2.6 h). In (A), PGR5 is photosystem I protein PROTON GRADIENT REGULATION5. LHCB3 is light-harvesting chlorophyll a/b binding protein 3 of photosystem II. At5g08570 is annotated as a pyruvate kinase family protein. MRPL11 is protein 11 of the mitochondrial ribosomal large subunit.
Comparison of Transcript Levels and Translation State over a Diel Cycle
In the wild type, the majority of genes had phase offsets between the peak in transcript abundance (TX) and the TL peak (Figure 4A). After calculating an odds ratio (Figure 4B), coincidence between the TX and TL peak was slightly overrepresented compared with the three other phase relationships. Similar results were obtained when genes were classified by their trough times. In summary, phase shifts between TX and TL cycles are common and gene specific.
Figure 4.

Phase Diagram Showing the Relation between Diel Cycles of mRNA TX Levels and mRNA TL in the Wild Type and CCA1-ox.
Genes were classified as cycling at the TX or TL level, as identified by SAM with a 10% FDR.
(A) and (C) Gene counts. Genes sharing the same phase relationship between peak TX and peak TL were binned together in the indicated cells and counted. Each cell contains the number of genes that peaked at the indicated times based on TX and TL. Invariant genes (Inv) were those that passed our prefiltering step but whose TX or TL did not vary significantly across time points. Coloring indicates the (log2) values in each cell, with yellow indicating lower values, orange indicating medium values, and red indicating higher values. A heat map of genes clustered based on TX cycles is shown in Supplemental Figure 7. (A) is the wild type, and (C) is CCA1-ox.
(B) and (D) Odds ratios for the data shown on the left. Odds ratios were calculated by (1) calculating the odds of a transcript with TX peak at time x having a TL peak at time y, (2) calculating the odds of a transcript not peaking at time x having its TL peak at time y, and (3) dividing the odds from step 1 by the odds from step 2. (B) is the wild type, and (D) is CCA1-ox.
Gene Ontology Enrichment in Coregulated Genes over a Diel Cycle
To identify functional processes that are under diel regulation at the translational level, we searched for enriched functional terms among genes with translation peaks at each time point using Gene Ontology analysis (Figure 5, Table 1). At dawn (ZT0), photosystem proteins were enriched, as well as biosynthesis of sulfur compounds such as glucosinolates and sulfur amino acids. At noon (ZT6), cell growth and division processes such as microtubules were slightly enriched, and these terms were strongly depleted in the evening (ZT12). Of note, hypocotyl growth peaks around noon (Nozue et al., 2007). In the wild type, there was no enrichment of any terms in the evening. The most striking enrichment was observed at night (ZT18). Ribosomal protein translation was highly enriched at this time, together with RNA methylation, nucleolar proteins, and small nuclear ribonucleoproteins. The coordinate diel translation of ribosomal protein mRNAs is displayed in Supplemental Figure 3. Evidently, the ribosome loading of the majority of these mRNAs was high at midnight (ZT18) to dawn (ZT0) and low around noon (ZT6) and evening (ZT12). The small minority of mRNAs that bucked the trend generally represented a less expressed paralog within their gene family; some of them had either very low or very high ribosome loading. Taken together, the results suggest that many ribosomal proteins belong to one regulon of translational control whose diel ribosome loading peaks at night. Besides the ribosomal proteins, several other small functional categories were preferentially translated at night, in particular mitochondrial proteins, the prefoldin complex, a protein that aids in co- or posttranslational protein folding, V-type proton-ATPase, and the DNA-directed RNA polymerase IV and V complexes (Table 1, Figure 5).
Figure 5.

Functional Enrichment among Groups of mRNAs with Common Peaks in Their Ribosome Loading Cycle (Wild Type).
Genes identified as cycling by either SAM at 10% FDR, ΔTL > 0.7, or ANOVA were searched for enrichment of functional gene annotations. The topGO R package was used with default settings and with all 12,342 reliably expressed genes as a background set to identify enriched biological processes (BP) and cellular components (CC). Significantly enriched functional categories are presented separately in a nested fashion. The table presents the number of genes within the background set that are annotated with the given term, the number whose TL peaks at the given time, the enrichment factor, and its FDR-corrected P value. FDR values below 1E-10 are shaded dark green, those below 1E-05 are medium green, and those below 5E-02 are light green. FDRs above 0.05 are not listed. E, to the power of 10; bios, biosynthesis; PS, photosystem; NDH, NADH dehydrogenase; Pol, polymerase; snRNP, small nucleolar ribonucleoprotein particle.
Table 1. Changes in Peak Translation upon Disruption of the Circadian Clock in CCA1-ox.
| Peak in Wild Type | Peak in CCA1-ox | |||||||
|---|---|---|---|---|---|---|---|---|
| Time | 6 am | 12 pm | 6 pm | 12 am | 6 am | 12 pm | 6 pm | 12 am |
| ZT | 0 | 6 | 12 | 18 | 0 | 6 | 12 | 18 |
| GO Enrichment Term | ||||||||
| Translation | ||||||||
| Cytosolic ribosome | – | – | – | X | X | – | – | – |
| RNA methylation | – | – | – | X | X | – | – | – |
| Prefoldin complex | – | – | – | X | X | – | – | – |
| Signal recognition particle | – | – | – | – | – | X | – | – |
| tRNA metabolism | – | – | – | – | – | X | – | – |
| tRNA aminoacylation | – | – | – | – | – | X | X | – |
| rRNA processing | – | – | – | – | – | X | – | – |
| mRNA catabolism | – | – | – | – | – | – | – | X |
| Mitochondrion | ||||||||
| Inner membrane | – | – | – | X | X | – | – | – |
| Envelope | – | – | – | X | X | – | – | – |
| Photorespiration | – | – | – | X | X | – | – | – |
| Plastid | ||||||||
| Thylakoid | X | – | – | – | X | X | – | – |
| Photosystem I | X | – | – | X | X | – | – | – |
| Photosystem II | X | – | – | – | X | – | – | – |
| Photosystem II assembly | – | – | – | X | – | X | – | – |
| Stroma | – | – | – | – | – | X | – | – |
| Plastid nucleoid | – | – | – | – | – | X | – | – |
| Plastid envelope | – | – | – | – | – | X | – | – |
| Protein targeting to plastid | – | – | – | – | – | X | – | – |
| Carbohydrate synthesis | – | – | – | – | – | – | X | – |
| Carbohydrate catabolism | – | – | – | – | X | – | – | – |
| Carotenoid synthesis | – | – | – | – | – | X | X | – |
| Sulfur compound synthesis | X | – | – | – | – | X | – | – |
| Amino acid synthesis | – | – | – | – | X | X | – | – |
| Nucleus | ||||||||
| Nuclear chromatin | – | – | – | – | – | X | X | – |
| Chromatin silencing | – | – | – | – | – | – | – | X |
| DNA methylation | – | X | – | – | – | – | – | – |
| Histone lysine methylation | – | – | – | – | – | – | X | – |
| RNA Pol IV/V | – | – | – | X | – | – | – | – |
| Cell division | ||||||||
| Cytokinesis or cell cycle | – | X | – | – | – | – | X | X |
| Microtubule | – | X | – | – | – | – | X | – |
| Other | ||||||||
| V-type H+ ATPase | – | – | – | X | X | – | – | – |
| Proteasome | – | – | – | X | – | X | – | – |
| Phosphatidylinositol synthesis | – | – | – | X | X | – | – | – |
| ATP binding (kinase, helicase) | – | – | – | – | – | – | X | X |
| Protein phosphorylation | – | – | – | – | – | – | – | X |
| Protein dephosphorylation | X | – | – | – | – | – | – | – |
| Divalent metal ion transport | X | – | – | – | X | – | – | – |
Cohorts of mRNAs with similar diel ribosome loading cycles were searched for enriched functional categories using Gene Ontology (GO). An X indicates that the term is enriched among the mRNAs whose translation peaks at the given time; a dash indicates absence of enrichment. Detailed data are in Figure 5 (wild type) and Supplemental Figure 10 (CCA1-ox).
A more detailed view of ribosome loading dynamics in central energy metabolism is displayed in Supplemental Figure 4. Of those mRNAs that had significant cycles, photosystem I mRNAs were well coordinated (night/dawn peak), as were most of the translocators in the plastid envelope (night peak) and a majority of the light harvesting proteins (dawn/noon peak). Photosystem II proteins yielded less information and Calvin cycle proteins had essentially no TL cycles. Overall, different functional groups of chloroplast proteins have different patterns of ribosome loading. Among mitochondrial proteins with translation cycles, most of which function in oxidative phosphorylation in the inner membrane, most peaked at night/dawn (pattern I), while a smaller subset peaked during the day (pattern II). In glycolysis and associated enzymes, ribosome loading tended to peak during the day. Of note, most glycolytic enzymes did not have translation cycles. Of those three that did, phosphofructokinase and pyruvate kinase both catalyze energetically downhill reactions and are considered to be highly regulated. Regulation of ribosome loading may add another layer of regulation to these enzymes. Also, of the multiple paralogous genes that encode several of these enzymes, not all undergo changes in ribosome loading, and the phase of the ribosome loading cycle can differ between paralogs (e.g., invertase and phosphoglycerate mutase). This finding suggests that translational regulation is one more way for duplicated genes to evolve new and distinct patterns of regulation.
Two other functional groups of mRNAs with noteworthy translation dynamics are shown in Supplemental Figures 5 and 6. Many redox-related enzymes had translation cycles, but the timing of peak TL differed among individual mRNAs (Supplemental Figure 5A). These enzymes also had a tendency for relatively high absolute ribosome loading (Supplemental Figure 5B). Proteins that function in protein turnover also revealed biases in absolute translation states, with proteases and proteasome subunits scoring high and E3 ligases scoring low, on average (Supplemental Figure 6). Taken together, these data suggest that besides the ribosomal protein mRNAs, several other functional groups of mRNAs also form translational regulons.
Global Transcript Profile in the Clock-Deficient CCA1-ox Strain
To distinguish whether the diel cycles of translation are driven by the circadian clock or by diel light-dark changes, we then prepared to compare the wild type and a strain overexpressing the central oscillator gene, CCA1 (CCA1-ox). As a first step, we determined transcript cycles of clock genes because these had not been described at a global level (Supplemental Figure 7). Compared with the wild type, the majority of transcript profiles in CCA1-ox correlated with light and darkness indicating that, as expected, the plants’ ability to anticipate the lights-on signal was severely curtailed. In contrast, the wild type had a variety of diel TX patterns, including many genes that appear to anticipate light changes. Assuming that transcript cycles conform to a sine model, in CCA1-ox, the distribution of TX peak times was relatively narrow, restricted around ZT9 (3 pm) and ZT21 (3 am) (Figure 3C). In the wild type, many more mRNAs peaked between ZT12-ZT16 (evening) and between ZT0-ZT6 (morning).
The broad correlation between light phase and transcript phase is reminiscent of the hypocotyl growth in CCA1-ox (Nozue et al., 2007), which, having escaped from control by the clock, is also strongly driven by light and darkness. As expected (Wang and Tobin, 1998; Green et al., 2002; Matsushika et al., 2002), cycling of key clock-regulated transcripts was muted in CCA1-ox (Figure 6A; Supplemental Figure 8A), e.g., for ELF4, PRR5, PRR7, and PRR9. Additionally, LHY was transcriptionally repressed at all time points. Although diel cycling of LHY mRNA was clearly disrupted in CCA1-ox, in our hands, LHY retained a small peak at the end of night at ZT0. Therefore, we cannot rule out that the CCA1-ox plants may retain weak residual clock activity in long day.
Figure 6.

Diel Cycles of Translation States and mRNA Transcript Levels for Clock-Associated Genes.
Diel cycles of translation states and mRNA transcript levels for clock-associated genes. Previously described clock-associated genes were hand-selected, focusing on the central oscillator, the light input pathways, and selected outputs. EF1A was included for comparison as a weakly cycling mRNA. For better comparability between genes, the signals are row scaled. In detail, for each gene, the mean signal was scaled to 0 (gray), and an average sd was calculated. The signal value for each time point was row-scaled so as to indicate the number of standard deviations that separate each value from the mean for that gene (unitless Z-score, yellow = high). Black indicates that the gene did not pass our prefilter for that time point. The original transcript abundances and translation states are displayed in Supplemental Figure 8.
(A) Transcript abundance. Left: the wild type. Genes were clustered according to their original mRNA expression values using hierarchical clustering based on Pearson coefficients, with replicates averaged. Four major clusters are boxed to aid in interpretation. Middle: CCA1-ox. The genes are ordered according to the wild-type clustering tree. Gene names are shown between the left and middle panels and apply to both panels. Right: CCA1-ox data were reclustered on their own; gene names are shown on the right.
(B) Translation patterns of clock-associated genes are displayed as in (A). TL states were calculated as described in Methods. Note the large cluster of mRNAs whose TL peak shifted from morning/noon (ZT0/ZT6) in the wild type to evening/night (ZT12/ZT18) in CCA1-ox.
In CCA1-ox, the following broad functional annotation patterns were observed (Supplemental Figure 9). At dawn (ZT0), the cohort of peak transcripts was enriched for RNA-biology processes and protein localization. At noon (ZT6) and in the evening (ZT12), the majority of enriched processes were chloroplast and photosynthesis associated. Remarkably, of 309 light reaction mRNAs, 229 peaked during the light period, either at ZT6 or ZT12, a stronger enrichment than in the wild type (note bold, red values for chloroplast [ZT6] and photosystem light reaction [ZT12] in Supplemental Figure 9, column WT). Similar trends were seen for many functional groups responsible for carbohydrate metabolism. In the evening (ZT12), categories such as apoplast, glucose catabolism, and glucosinolates became prominent as well. Finally, at night (ZT18), ion transport and defense responses became predominant. Notably, the majority of these patterns in CCA1-ox were similar, but more accentuated, compared with the wild type. However, for other functional terms, the transcript patterns in CCA1-ox were muted (e.g., defense response, cell wall at ZT18; underlined green values indicate stronger enrichment in the wild type). The term cytokinesis/cell division was biased toward noon (ZT6) in the wild type, yet only weakly enriched, at ZT12, in CCA1-ox.
In summary, the defect of the clock in the CCA1-ox strain disturbs the coordinate transcription of certain functional classes of mRNAs, but also opens the door to a more tightly coordinated transcription for other classes of mRNAs, especially photosynthesis-related mRNAs. In wild-type plants, the functional clock uncouples large numbers of transcripts from the tight control exerted by the overt light environment.
Translational Cycling in Plants with a Disrupted Circadian Clock
We initially hypothesized that translational regulation by the diel cycle would be dampened in CCA1-ox plants. However, polysome microarray analysis of the CCA1-ox strain revealed robust translation cycles (Figures 2B and 2D). The clusters with dawn and evening peaks were enlarged at the expense of the day and night peaks. Thanks in part to a lower variation between replicate experiments, the CCA1-ox TL data yielded a larger number of genes (5521 versus 2780 in the wild type) that were scored statistically significant in their translation cycle by either the SAM or ANOVA methods (Figures 2D, 2F, 2H, and 2J). In CCA1-ox, only 2533 genes were not identified as cycling by any method and were therefore classified as surely invariant.
In CCA1-ox, TX and TL cycles were more highly coordinated than in the wild type (Figure 4C). This was particularly striking at dawn (6 am, ZT0), as evident from the high odds ratios along the diagonal in Figure 4D. Taken together with the previous data from wild-type plants (Figures 4A and 4B), this observation suggests that the clock does not just regulate translation. The fully functional clock in the wild type may also work to separate transcriptional control from translational control, making them more independent of each other. By contrast, in the clock-defective CCA1-ox strain, transcription and translation cycles may be coming under an alternative form of joint control, possibly light-dark transitions and the associated shift in the cellular energy balance.
Diel Cycles of Translation Are Disturbed by Malfunction of the Clock
Next, we examined how the clock defect in the CCA1-ox strain affected the phase of the diel translation cycles (Figures 7C and 7D). Among the mRNAs with robust translation cycles in the wild type, about one-third lost their cycle in CCA1-ox (Figure 7C). Of those that maintained a cycle in CCA1-ox, the minority maintained their peak at the same time as the wild type, while the majority shifted their translation peak to a different time. A 6-h delay was most common. In particular, the vast majority of mRNAs with a wild-type TL peak at 12 am (ZT18) peaked 6 h later in CCA1-ox. These mRNAs preferentially encode ribosomal proteins and mitochondrial proteins (Table 1). Finally, we draw attention to more than 3000 mRNAs that had no robust cycle in the wild type, yet started to cycle in CCA1-ox, thus revealing translation cycles that may be suppressed in the wild type by the fully functional clock (Figure 7C). Overall, the shift in TL in the CCA1-ox strain was also evident when TL was modeled as a sine wave (Figure 3C). Taken together, these data indicate that a functional clock is important for maintaining translation cycles for certain mRNAs and suppressing the cycles of others.
Figure 7.

Phase Diagram Comparing the Expression Cycles between the Wild Type and CCA1-ox.
Genes with significant TX cycles ([A] and [B]) or translation cycles ([C] and [D]) were identified using SAM with the 10% FDR cutoff. All other genes were called invariant (Inv). The genes were binned according to the peak in the wild type and the peak in CCA1-ox.
(A) and (C) Numbers of genes with a given phase relationship are indicated as a heat map.
(B) and (D) Odds ratios for the data shown on the left. For details, see legend to Figure 4.
For comparison, at the transcript level, it was more common for genes to peak at the same time in the wild type and CCA1-ox (Figures 7A and 7B). Only a small fraction lost their cycle. However, like at the translation level, many mRNAs that had no consistent transcript cycle in the wild type did cycle in CCA1-ox.
The shifts in translation cycles in the CCA1-ox strain stood out in starker contrast after Gene Ontology analysis (Figure 5 for wild type, Supplemental Figure 10 for CCA1-ox, and Table 1 for a summary). The translation phase of the following major processes was delayed by 6 h: cytosolic ribosome (Supplemental Figure 3) and RNA methylation, mitochondrial inner membrane and envelope, photorespiration, sulfur compound synthesis, and cytokinesis. For some of the smaller categories, such as the prefoldin complex, V-type proton ATPase, and microtubules, the 6-h delay was very striking, given that a sizable fraction of genes in each group was translationally regulated in a coordinated fashion. Second, certain functional classes maintained a peak in TL at a given time or showed only a slight shift, for example, photosystem proteins and metal ion transport. Third, several functional categories only revealed coordinate translation in CCA1-ox, after disruption of the clock, but not in the wild type. For example, mRNAs for tRNA metabolism, specifically aminoacyl-tRNA synthetases, typically did not cycle in the wild type but did cycle in CCA1-ox with a peak at ZT6 or ZT12. The opposite pattern, loss of coordinate translation, also occurred occasionally (e.g., RNA polymerase IV/V).
It should be understood that not all mRNAs within one larger category follow the same dynamic. For example, in the wild type, the mRNAs for amino acid biosynthesis had gene-specific translation peaks at each time point but appeared to coalesce into a broader peak with enrichment at ZT0 and ZT6 in CCA1-ox. These data again underscore coregulation of both large and small, functionally related, groups of mRNAs and suggest that, besides the well-known ribosomal protein mRNAs, many other mRNAs are targets of translational control.
The changes in ribosome loading over the diel cycle differ dramatically from changes previously described to occur in response to shorter, unexpected, dark or light treatments (Supplemental Figure 11). Using k-means clustering, it is evident that three different shifts from light to darkness (e.g., ZT12 evening to ZT18 night) did not resemble the response that occurs when light-grown seedlings are exposed to one hour of darkness in midday at ZT8 (L→1 hD; Juntawong et al., 2012). Instead, it resembled more closely the response of dark-exposed seedlings to reillumination (Juntawong et al., 2012) and the response of dark-grown seedlings to 4 h of white light, which stimulates their deetiolation (Liu et al., 2012). Vice versa, the diel response to a dark-to-light shift (ZT0 to ZT6) resembled the response to a 1-h exposure to darkness. Of the six clusters defined by k-means, clusters 1, 2, 3, and 4 all supported this anticorrelated pattern. In fact, in previous studies, ribosomal proteins were among the most highly repressed mRNAs in response to darkness and induced by light, whereas here, ribosomal proteins (clusters 1 and 2) were translationally stimulated during the night (Table 1). Evidently, many other mRNAs follow a similar pattern, including mitochondrial (cluster 1) and cytoskeletal proteins (cluster 3) and polysaccharide synthesis (cluster 4). Only clusters 5 and 6 (enriched for chloroplast, Golgi, cell wall, and glucose catabolism) followed the more intuitive expectation of a correlated behavior. These data suggest that mRNAs belonging to the night cluster under diel conditions also tend to be translationally inhibited when darkness is experienced by the plant as a stress condition.
Translational Control of Circadian Clock mRNAs
Next, we addressed whether clock mRNAs were subject to translational control. Because the levels of many central clock mRNAs drop below the reliably detectable limit at one time point and were therefore filtered out in our previous analysis, we refiltered the raw array data such that genes must be present in all three replicates of SP and LP fractions for at least two time points, but not all four time points (14,397 genes). In CCA1-ox, the transcript cycles of many clock-associated genes became muted, and the peaks and troughs often coincided with darkness (ZT0 and ZT18) or light (ZT6 and ZT12; see Figure 6A and Supplemental Figure 8A, middle and right panels), not unlike the transcriptome as a whole (Supplemental Figure 7).
At the translation level, clock-associated transcripts (Figure 6B; Supplemental Figure 8B, left panels) fell into two broad groups in the wild type, one with peaks at dawn or noon (ZT0 or ZT6; e.g., GRP8, E3 ubiquitin-protein ligase COP1, and phytochrome D [PHYD]), and a second with peaks in the night (ZT18; e.g., LUX and UVR8). Upon disruption of the clock in CCA1-ox, the translation patterns of both groups changed dramatically. The late night group tended to be delayed to dawn or noon (e.g., LUX and UVR8), while the dawn-noon group was delayed until noon (GRP8 and COP1) or evening (e.g., photoreceptors including PHYE, PHYD, CRY2, and PHOT2) or beyond (Figure 6B), roughly consistent with the pattern across the entire translatome (Figures 7C and 7D).
Figure 8 displays the same cycles of transcript levels and translation states in the context of a clock wiring diagram (Pokhilko et al., 2012). For the wild type, several evening genes showed a 6-h phase delay between peak transcript level and peak translation (e.g., TOC1 and LUX). Among genes with a transcript peak around noon, PRR5 and GIGANTEA (GI) also had an offset in their translation peak. In contrast, among genes with a dawn transcript peak, coincidence of TX and TL was more common (e.g., LHY and CCA1, also see CRY1 and the bZIP transcription factor gene HYH in Figure 6). While inferences about the behavior of single genes are sensitive to noise, in the aggregate it seems clear that translation states are a way for plants to fine-tune the expression of clock regulatory mRNAs. These data suggest that the clock relies not only on transcriptional control but also on translational control at the level of ribosome loading for proper functionality.
Figure 8.

Translation Cycles Affecting the Arabidopsis Circadian Clock.
A wiring diagram for the Arabidopsis clock (Pokhilko et al., 2012) was amended to illustrate cycles of TX levels and TLs. Arrows and T-bars indicate stimulation and repression, respectively. Yellow suns indicate light-regulated transcription, and blue suns indicate light-dependent posttranscriptional effects. TX levels and TLs are displayed in the form of heat maps. The upper row of each set of rectangles indicates the TX and the lower row indicates the TL. The left set indicates the wild type and the right set CCA1-ox. Data are from Supplemental Figure 8; n = 3. For ELF4, TL data are omitted because of poor expression data. Black symbolizes mRNA expression below threshold.
Given that the amplitude of TL cycles is generally small, we wanted to simulate how the waveform of the protein synthesis rate might be affected by phase offsets between a TX cycle and a TL cycle (Figure 9). Figure 9A assumes that TX and TL both follow plain sinusoidal patterns. The rate of protein synthesis was calculated by multiplying TX by TL. A 9-h offset between TX and TL causes an asymmetric peak in the synthesis rate, and a 12-h offset can produce a plateau-like pattern. It is very challenging to measure protein synthesis rates empirically (Schwanhäusser et al., 2011), especially with enough precision to distinguish relatively small differences, and especially in plants, where the key technique, stable isotope labeling with amino acids, is just barely becoming more common (Lewandowska et al., 2013). This is why a simulation is useful. Data from selected Arabidopsis mRNAs are shown to exemplify how TL may modulate the synthesis rate (Figures 9B to 9I). The photoreceptors PHOT2 and UVR8 (AT5G63860, UV-B receptor) predict that the translation dynamic amplifies the TX dynamic or extends the protein synthesis into a plateau-like pattern, respectively. For data as those from LP1 (AT2G38540, lipid transfer protein 1) and RCE1 (AT4G36800, Rub1 conjugating enzyme) the offset between TX and TL cycles causes a delay or advance, respectively, in the protein synthesis rate by several hours, which is not expected from the TX level alone. For MRPL11 (AT4G35490, mitochondrial ribosomal protein L11) and RPL P1 (ribosomal protein P1), the protein synthesis rate is driven primarily by the TL pattern, yet phase-shifted slightly by the weaker TX cycle. Finally, PFK1 and PFK3 are isoforms of phosphofructokinase (At4g29220 and At4g26270; Supplemental Figure 4) that differ in their phase offsets between TX and TL. The PFK data demonstrate that genes related by an ancient gene duplication could evolve different patterns of translational regulation. Taken together, it is plausible that differences in the waveform of protein synthesis rates may help to fine-tune gene function. For example, high translation of the TOC1 and LUX mRNAs at night (Figure 8) may allow these proteins to continue to repress transcription of morning genes, CCA1 and LHY, and of day genes such as GI and PRR9, respectively. In conclusion, relatively shallow cycles of ribosome loading that affect a large number of mRNAs may contribute in a nonlinear fashion to the functioning of complex cellular networks.
Figure 9.

Simulation of Protein Synthesis Rates from mRNA Transcript Level Data and TL Data.
mRNA levels were multiplied with the corresponding translation states in order to simulate the protein synthesis rate from the given mRNA. The results were extrapolated to 42 h to better visualize the cycling behavior. The mRNA levels and simulated protein synthesis rates are mean-centered (mean = 100) and are displayed on the left y axis, while the TL is displayed as is on the right y axis.
(A) A mathematical simulation of TX level (stippled trace), TL in three possible phases (pale traces), and the three calculated protein synthesis rates (dark traces).
(B) to (H) Actual TX and TL data and the protein synthesis rates calculated from them. The difference in the shape of the transcript level trace and the protein synthesis trace is accentuated with dark shading. ZT, zeitgeber time.
Translation Cycles in Constant Light
Finally, we examined the role of the circadian clock in the cycle of translation by measuring TLs in continuous light. CCA1-ox plants grown in a light-dark cycle still exhibit circadian rhythmicity in transcriptional behavior, but CCA1-ox plants grown in constant light do not (Green et al., 2002). We examined polysome loading during and after a shift from long day to constant light conditions (LD>LL) in both the wild type and CCA1-ox. On a global level (Figure 10A), the fraction of wild-type RNA recovered in polysomes rose during the day, as expected (Figure 1), dropped during the subjective night despite continuous illumination, then rose and fell again during day 2. This pattern was clearly evident in the individual dynamics of NP and LP. In CCA1-ox, in contrast, polysomes stayed high during the first subjective night and then fell during day 2. These data further indicate that global polysome loading is controlled by the circadian clock.
Figure 10.

TLs under Continuous Light Conditions.
Plants were grown for 10 d under long-day conditions (LD) and shifted to continuous light (LL) at ZT0. Plant samples were harvested at 6-h intervals for 48 h and fractionated by sucrose density gradient into NP, SP, and LP fractions. The left column contains data from the wild type and the right column is from CCA1-ox. The subjective night is indicated by gray shading.
(A) The percentage of RNA measured by UV absorption that was detected in NP, SP, and LP portions of the gradient. The trace labeled PL (polysomes) is the sum of SP and LP.
(B) The abundances of individual mRNAs for selected genes were quantified by qRT-PCR. Translation states were calculated. The traces from the three biological replicates were centered on their average TL. The error bars indicate the range of the data from n = 3 biological replicates (2 for PRR3). For LHCA1 as a representative mRNA, the traces of the three individual replicates are also shown with thin lines. Letters above each data point indicate which data points are significantly different by ANOVA with Tukey post-hoc test (α = 0.05). Data points that share the same letter are not different from each other. Data sets that lack lettering had few or no significantly different pairs.
We then analyzed the translation states of specific mRNAs using qRT-PCR (Figure 10B). The mRNAs for two chlorophyll binding proteins (CAB4 and LHCA1) and one ribosomal protein (RPL26B) experienced dynamic fluctuations in ribosome loading in the wild type, whereas in CCA1-ox these fluctuations were suppressed. Two central clock mRNAs, PRR3 and possibly CCA1, also displayed cyclical changes in the wild type but not in CCA1-ox. In contrast, the EF1A mRNA maintained an even ribosome loading throughout. Although there was considerable variation between replicates, as illustrated in detail for LHCA1 in the wild type, the general trend for an amplified dynamic on day 2 of the time course was consistently observed. It is noteworthy that the dynamics in the wild type often did not conform to a strict 24-h period (see trough-to-trough period for LHCA1, PRR3, and RPL26B). Together, these observations suggest that the wild type has a trend for cyclical ribosome loading of various mRNAs, even under continuous light, in keeping with the global dynamic (Figure 10A). Moreover, a functional clock appears to be required for these ribosome loading cycles. However, the erratic patterns seen under continuous light suggest that regular light-dark changes assist the clock in organizing the ribosome loading of various mRNAs.
DISCUSSION
In this study, we document diel and circadian regulation of ribosome loading of mRNA in Arabidopsis seedlings. These fluctuations are extensive, affecting ∼15% of transcripts in wild-type seedlings, and affect different classes of genes differently. The specific diel patterns of ribosome loading that are observed in wild-type plants require a functional circadian clock. Here, we provide information about diel effects on translation across the entire transcriptome, gene by gene. Clock-controlled diel translation must be integrated with other internal and external cues that are known to affect translation.
Diel Regulation of Ribosome Loading Is Extensive
Of ∼12,000 mRNAs that were reliably detected in Arabidopsis seedlings, about one-fifth (2503) passed ANOVA with a standard significance threshold (P < 0.05), far more than the 600 expected to pass this threshold by chance alone if data were randomly distributed. More than 7% (890) of genes were scored as dielly fluctuating by SAM at an FDR of 0.05. These are respectable yields of statistically validated genes given that our measure of TL is calculated from three independent data points, i.e., NP, SP, and LP mRNA, that each come with their own margin of error. To optimize biological inference, we applied different filters to our raw translation data, depending on the question at hand. For example, to search for functional enrichment using Gene Ontology, we started with a lenient filter that selected genes through any of three different measures of significance. This approach yielded substantial insight into translationally coregulated mRNAs by uncovering enrichment of numerous functional categories, which were evaluated rigorously for statistical significance by guarding against multiple comparisons (Figure 5). Other analyses, such as comparisons between TX levels and TLs, as well as phase shift diagrams between the wild type and CCA1-ox, were conducted with a more rigorously preselected set of genes (SAM with FDR < 10%), again while performing downstream statistical tests (Figures 4 and 7).
Here, the diel fluctuations in ribosome loading of Arabidopsis mRNAs were revealed in seedling shoots growing on defined medium with 1% sucrose and entrained by a 16-h day (Figure 1). When Pal and coworkers performed global analysis of ribosome loading over the diel cycle in vegetative rosettes in a 12-h day (Piques et al., 2009; Pal et al., 2013), their global drop in ribosome loading was deeper than ours, suggesting that diel translation under natural conditions, i.e., with fluctuating levels of photoassimilate, may be more pronounced than seen here.
Diel Phase Affects Translation in More Than One Way
We found peaks in ribosome loading for mRNAs at all four time points that we surveyed: dawn, noon, evening, and night. Together with the large cohort of translationally flat genes, there appear to be five groups of mRNAs. Of these five, the groups peaking at night, at dawn, and at noon had distinct functional enrichment, underscoring that they are regulated differently. The “night” cohort had the most striking bias, being enriched for mRNAs for cytosolic ribosomal proteins, mitochondrial proteins, and several smaller protein assemblies. Significant functional biases were also evident in the dawn (ZT0) and noon (ZT6) cohorts. Of the photosynthetic apparatus, photosystem I mRNAs were translationally stimulated at night and dawn, photosystem II was biased more toward dawn, and most light-harvesting proteins were stimulated well into the light period, possibly shedding light on translational control of assembly of the photosynthetic apparatus. No positive enrichment was detected among mRNAs whose ribosome loading is flat, nor in the smaller evening cohort (peak at ZT12), although the evening group was strongly depleted for terms that peak at noon and at night, i.e., cytoskeleton and ribosome.
Ribosomal protein mRNAs are the best defined translational regulon in plants, being coregulated under essentially all experimental conditions that have been examined. Their coregulation here further validates the quality of the data. These mRNAs are preferentially repressed in their translation after abiotic stress such as heat (Yángüez et al., 2013), drought (Kawaguchi et al., 2004), and hypoxia (Branco-Price et al., 2008). They are translationally stimulated by light (Liu et al., 2012) and in mutants defective in the translation apparatus (Kim et al., 2007; Tiruneh et al., 2013). These mRNAs are translationally repressed by 1 h of unanticipated darkness in the middle of the day (Juntawong and Bailey-Serres, 2012), while they are translationally stimulated after 2 h of anticipated darkness at night, in our data. These findings suggest that ribosomal protein mRNAs are regulated by darkness in a circadian context.
Diel Translation Is a Function of the Circadian Clock
Three pieces of evidence indicate that the diel regulation of ribosome loading is an output of the circadian clock. First, the clock-compromised CCA1-ox strain has a very distinct translational profile compared with the wild type. Translation cycles continued to be very evident in CCA1-ox, but the dawn and evening peaks were much more pronounced while the night peak and noon peak were relatively small. For several specific functional categories, the translation cycles in CCA1-ox appear to be delayed by about 6 h, for example, ribosomal proteins, indicating that the wild-type clock works to advance the time of peak translation. One caveat when using the CCA1-ox strain is that CCA1 may have additional functions that are entirely independent of the clock. For example, by exposing the cell to CCA1 protein in the evening, when its level is usually low, CCA1 might interact spuriously with partners that have no relation to the clock and that CCA1 does not normally encounter. For this reason, experiments were also performed under continuous light conditions. Second, cycles of diel translation were also evident in the wild type under free-running continuous light conditions (Figure 10), suggesting that they are governed by the circadian clock. Third, these fluctuations of translation under continuous light were broadly disrupted in the CCA1-ox strain (Figure 10). Given that CCA1-ox is substantially clock-deficient under continuous light (Wang and Tobin, 1998), we conclude that cycles of diel translation are driven by the circadian clock.
The clock’s output pathways may well affect translation indirectly because none of the core oscillator components are known translational regulators. Most direct outputs of the clock are transcriptional, and these may well mediate the translational effects seen here. For example, in CCA1-ox plants, many RNA-biology transcripts peak around ZT0, whereas in the wild type, fewer of them do (Supplemental Figure 9). This result adds credence to the hypothesis that the clock may regulate translation via the primary layer of clock output genes. Should one consider whether CCA1-dependent changes in translation may be the far-downstream consequence of physiological or developmental alterations in the CCA1-ox strain? CCA1-ox does have a clearly elongated hypocotyl (Wang and Tobin, 1998). In addition, our wild-type seedlings expressed the floral inducer FT, while CCA1-ox plants did not. Overall, however, CCA1-ox is morphologically quite normal and was physiologically vigorous at the time of our experiments. Therefore, we doubt that the translational changes in CCA1-ox can be attributed solely to mechanisms that are several degrees of separation away from the clock itself.
We collected microarray data of the global transcriptome in CCA1-ox over the diel cycle. Many transcripts assume a cycle that mirrors the light and dark conditions, in keeping with the clock defect. However, the LHY transcript, while expectedly repressed, rose slightly at ZT0 (6 am), an effect we interpret as a residual anticipation of dawn and, thus, residual clock activity (Figure 6). Even though CCA1-ox is not completely clock deficient in the light-dark cycle as previously noted (Green et al., 2002; Matsushika et al., 2002), the strain was broadly affected in its translation, indicating that CCA1 cycling is critical for dielly regulated translation.
The clock-deficient CCA1-ox strain revealed translational cycling of several hundred new mRNAs, which did not cycle in the wild type (Figure 7). With the caveat that lack of statistical significance does not prove absence of a cycle in the wild type, this finding suggests that the clock helps to suppress fluctuations in ribosome loading driven by diel light-dark cycle conditions. Likewise, derepression of cycles in the CCA1-ox strain was also evident at the transcript level.
Global Clock Control of Ribosome Loading
While alternative splicing has been implicated repeatedly as a clock output and as a regulator of circadian clock function, clock control of translation or ribosome loading has rarely been examined. Aside from the global cycles of ribosome loading (Piques et al., 2009; Pal et al., 2013) in Arabidopsis, translation of the LHY mRNA was shown to be stimulated by light, a phenomenon thought to sharpen the peak of LHY protein abundance in the morning (Kim et al., 2003). Our results also showed higher ribosome loading of LHY in the morning, when LHY mRNA peaks, than at the end of the day, when the mRNA is low. This pattern of translational regulation may keep LHY protein levels from rising too early in the night.
Translational control of a clock output has been described in the dinoflagellate Gonyaulax and in the green alga Chlamydomonas reinhardtii (Morse et al., 1989; Mittag et al., 1994). More recently, translational regulation of ribosomal protein mRNAs in mouse was shown to peak at night (Jouffe et al., 2013), which is at first glance similar to the situation in Arabidopsis. However, mice are nocturnal animals, feeding at night and living off their fat deposits during the day. In Arabidopsis, in contrast, energy is harvested and thus more abundant during the day, while the plant lives off its starch deposits at night. Thus, considering the overall energy infrastructure of the mouse and Arabidopsis, the resemblance in the nocturnal peaks of ribosomal protein translation is probably coincidental.
Synthesis of ribosomal proteins occupies a substantial fraction of translational capacity, especially in growing cells (Warner, 1999; Piques et al., 2009). The question arises why the plant would translate ribosomal proteins preferentially at night rather than during the day when energy is more directly available, which would circumvent losses during starch deposition and conversion. The reason is unknown but may be associated with the following. Translational capacity is finite. Ribosomes lying idle in the cell may be a sign of poor stewardship of growth-limiting resources, especially nitrogen. During the light period, the cell may use most of its translational capacity for bulk maintenance, including maintenance of the photosynthetic apparatus, leaving little capacity for ribosomal protein translation. Thus, the finding that ribosomal protein translation preferentially occurs at night may suggest that the cell has spare capacity at that time.
Integration of Clock-Controlled Diel Translation with Other Signals
Most signals known to regulate translation are exogenous environmental cues, including light, darkness, drought, and temperature. In contrast, only a few endogenous cues are known to regulate translation, e.g., sucrose and amino acids (Nicolaï et al., 2006; Lageix et al., 2008; Zhang et al., 2008; Roy and von Arnim, 2013). The clock is another endogenous mechanism now known to affect translation. Clock-controlled and diel ribosome loading must be integrated with other signals that affect ribosome loading simultaneously. These signals can be expected to play a relatively minor role under constant light conditions on medium containing sucrose, but will affect what happens under light-dark cycle conditions. For example, polysome loading rises rapidly upon lights-on, peaking after 1 to 4 h, and then slowly declining toward the end of the day. Conversely, a transient drop in polysome loading occurs around 15 to 30 min after lights-off (Pal et al., 2013). How the effects of light-dark transitions, daylength, photosynthate, and other signals are integrated with signals from the clock will be an important area of future research.
METHODS
Plant Material and Polysome Gradient Fractionation
Arabidopsis thaliana ecotype Columbia was grown on agar plates containing full-strength Murashige and Skoog salts, pH 5.7, and 1% sucrose for 10 d in a 16-h-light (∼80 μmol/m2/s; Philips F17T18/TL741 fluorescent bulbs) and 8-h-dark cycle at 22°C. The CCA1-ox strain overexpresses the CCA1 protein under the control of the cauliflower mosaic virus 35S promoter (35S:CCA1; Wang and Tobin, 1998) and was grown likewise. Three biological replicates were collected. RNA extraction and microarray hybridization closely followed an earlier procedure (Kim et al., 2007), with a few modifications described earlier (Missra and von Arnim, 2014). After sucrose gradient fractionation, we generated three fractions of mRNAs: the NP fraction, the SPs (one to three ribosomes per mRNA), and the LPs (four and more ribosomes per mRNA) (Supplemental Figure 1). We measured the RNA abundance after fractionation in order to reveal the global shift in ribosome loading over the course of the day. Samples for total transcripts were also collected alongside. Following the manufacturer's protocols, LP, SP, NP, and TX RNA fractions were converted to cDNA and hybridized to GeneChip Arabidopsis ATH1 Genome Arrays, which contain 22,746 probe sets representing ∼24,000 genes.
If a given experimental treatment causes a global reduction in polysome loading, the global shift becomes masked during the standardized experimental procedure. The global shift is measured from RNA abundance data after fractionation (Kawaguchi et al., 2004). A small global shift was detected with a peak at ZT6 (noon) and trough at ZT0 (end of night) (Figure 1). However, no global adjustment of our array data was performed. Therefore, a gene that displays a TL cycle identical to the global shift will be regarded as noncycling. The TL cycles described in Figure 2 are cycles beyond the global cycle.
Microarray Data Analysis
Statistical and bioinformatic analyses were performed using R version 3.1.1 (R Core Team, 2014) and Bioconductor version 2.14. Raw signal intensities for each probe set were extracted from CEL files, the Affymetrix proprietary data format, using the affy package version 1.44.0 (Gautier et al., 2004) and normalized using the gcrma package version 2.38 (Wu et al., 2014), all with default settings. Normalization using the rma method from the affy package yielded similar results on this data set. Hybridization signals were classified as present (P), marginal (M), or absent (A), using the mas5calls function from the affy package. qRT-PCR was performed on a Bio-Rad iQ5 instrument using Bio-Rad EvaGreen enzyme and dye reagents with three technical replicates per plate and three biological replications. Primer sequences for qRT-PCR are listed in Supplemental Table 1.
Calculation of Translation States
TLs were calculated for every time point for genes with P calls in all four SP and LP samples, while A calls were permitted in the NP samples. Genes with a variance below 0.001 in hybridization signals for any time point were discarded as artifactual, resulting in 12,342 genes in the wild type. Signals from the gcrma output were unlogged and TL was calculated according to the following formula:
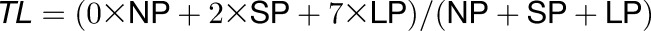 |
This calculation is based on the estimate that mRNAs in the NP, SP, and LP fractions are bound by an average of zero, two, and seven ribosomes, respectively. If all ribosomes were equally active in translation, then TL would indicate a translation rate of proteins produced per mRNA per unit time. We cannot rule out that the average number of ribosomes per mRNA varies with time; for example, it may be lower than seven in the LP at ZT0, when the global polysome loading is lowest. Because we were not able to settle on more precise estimates for each time point, the given values of two and seven were applied to all samples equally. The translation profiles may be skewed slightly as a result. The abundance of total mRNAs (TX) was displayed on a log2 scale, as usual.
Identification of Genes with Diel Fluctuation of Their Translation State
For each gene, the difference between the peak TL and trough TL is the ΔTL value. Differentially translated mRNAs were identified by one of four criteria, ΔTL > 0.3, ΔTL > 0.7, ANOVA with P < 0.05, and SAM (Tusher, 2001) with a collective FDR < 0.10. The lenient cutoff of 0.3 was only used to identify those genes that clearly lack a translation cycle. For SAM (Tusher et al., 2001), we ran the R function “samr” with response type “Multiclass” to identify genes whose TL varied significantly across time points. We used 1000 permutations to compute a test statistic (T) for each gene, as well as a null distribution of 1000 Ts for each gene obtained from random shuffling of the class labels. We then computed an empirical P value for each gene as the number of randomized Ts that were greater than the original, divided by 1000. As an additional check, we permutated the time point labels of our wild-type TL data and then used SAM to search for false-positive TL cycles. Such permutations yielded an average of only 46 translationally cycling genes (range: 0 to 312), compared with 1825 from the original data. For ANOVA with a raw P value <0.05, we also calculated the FDR per gene using the Benjamini-Hochberg method.
Modeling Diel Cycles as Sine Waves
We modeled diel variation in TX and TL as sine waves using a linear model approach to precisely estimate the phase and amplitude, as described elsewhere in more detail (Stolwijk et al., 1999). A sine wave can be described by the following function: 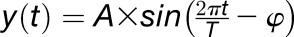 , where
, where  is time,
is time,  is the amplitude,
is the amplitude, 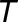 is the period, and
is the period, and 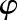 is the phase. This sine function can be transformed into a linear regression formula,
is the phase. This sine function can be transformed into a linear regression formula, 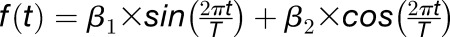 , and fitting a linear model with this formula yields the coefficients
, and fitting a linear model with this formula yields the coefficients 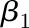 and
and 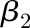 . The amplitude can then be calculated as
. The amplitude can then be calculated as 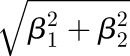 , and the phase as
, and the phase as  . For each gene, we first subtracted the mean TX or TL from the data series and then followed this approach to estimate the phase and amplitude with the “lm” function from the standard R “stats” package, assuming a period of 24 h. The Pearson coefficient (R2) between empirical data and predicted data indicates the percent of the variation in TX or TL for a gene that is explained by the sine wave. This approach is convenient due to its simplicity and computational speed, and it has the advantage that the phase can be any value over a continuous range from 0 to 2π (ZT0 to ZT24), so the peak and trough of TX or TL can occur between the times at which data were collected. A confidence interval was calculated for each TL peak time using bootstrapping.
. For each gene, we first subtracted the mean TX or TL from the data series and then followed this approach to estimate the phase and amplitude with the “lm” function from the standard R “stats” package, assuming a period of 24 h. The Pearson coefficient (R2) between empirical data and predicted data indicates the percent of the variation in TX or TL for a gene that is explained by the sine wave. This approach is convenient due to its simplicity and computational speed, and it has the advantage that the phase can be any value over a continuous range from 0 to 2π (ZT0 to ZT24), so the peak and trough of TX or TL can occur between the times at which data were collected. A confidence interval was calculated for each TL peak time using bootstrapping.
Clustering and Higher Level Analyses
mRNAs with diel fluctuations in TL were clustered using R according to the time of peak TL and, secondarily, the time of trough TL. Hierarchical clustering was performed using the Pearson coefficient as the similarity metric. This and all overlap plots were made using the heatmap.2 function from the gplots package in R package version 2.14.2 (Warnes et al., 2014). Where indicated, the data from individual genes were mean-centered and row-scaled by their Z-score (distance from the mean as multiples of their sd) to better display trends in translation over time. When individual expression values in a time series did not pass our data quality filter, the expression value was replaced with NA. Enrichment of functional annotations was determined using the topGO R package version 2.18 (Alexa and Rahnenführer, 2010) with all 12,342 expressed genes as the reference set. For Gene Ontology analysis, genes were preselected using SAM or ANOVA, or a ΔTL value above 0.7. Functional terms had to be significantly enriched with an FDR of 0.05 or less in either the wild type or CCA1-ox to be considered for presentation. Terms that were substantially overlapping with other terms were omitted.
Accession Numbers
The original microarray hybridization data, metadata, and extracted and normalized data are accessible in NCBI-GEO under superseries GSE61899: Polysome profiling in wild type and CIRCADIAN CLOCK ASSOCIATED1-overexpressing (CCA1-ox) Arabidopsis thaliana over a 24-h diel cycle. Four data sets are grouped together under GSE61899: GSE61895, transcript levels in the wild type; GSE61896, transcript levels in CCA1-ox; GSE61897, polysome wild type; GSE61898, polysome CCA1-ox. A list of Arabidopsis Genome Initiative numbers is provided in Supplemental Table 2.
Supplemental Data
Supplemental Figure 1. Representative polysome gradient UV absorption profiles.
Supplemental Figure 2. Microarray data of mRNA TL are reproducible by qRT-PCR.
Supplemental Figure 3. Heat map of TLs for ribosomal protein mRNAs.
Supplemental Figure 4. Heat map of TLs for mRNAs in central energy metabolism.
Supplemental Figure 5. Translation states of redox-related mRNAs.
Supplemental Figure 6. Translation states of mRNAs related to protein turnover.
Supplemental Figure 7. Diel transcript levels in the wild type and CCA1-ox.
Supplemental Figure 8. Diel cycles of translation states and mRNA transcript levels for clock-associated genes.
Supplemental Figure 9. Gene Ontology analysis of diel transcript levels.
Supplemental Figure 10. Gene Ontology analysis of diel changes in TL in CCA1-ox.
Supplemental Figure 11. Comparison of diel changes in TL with short-term light-dark transitions.
Supplemental Table 1. List of primers.
Supplemental Table 2. List of Arabidopsis Genome Identifier (AGI) numbers of selected genes.
Supplemental Data Set 1. Translation states and transcript levels for all reliably expressed transcripts.
Supplementary Material
Acknowledgments
We thank Elaine Tobin for the kind gift of 35S:CCA1. Bayu Tiruneh performed preliminary meta-analyses of translation. We thank Julia Gouffon for performing microarray hybridizations and Arnold Saxton for comments and help with statistical analysis. Karen Browning and Julia Bailey-Serres kindly provided data before publication. We gratefully acknowledge support from National Science Foundation grants DBI-0820047 and IOS-1456988 and DOE Energy Biosciences Grant DE-FG02-96ER20223. J.S. and K.K. were supported by an NSF-REU grant (Research Experiences for Undergraduates).
AUTHOR CONTRIBUTIONS
A.M., T.L., J.S., and A.G.v.A. performed experiments. B.E., Q.J., and K.K. conducted bioinformatic analyses. A.M., B.E., and A.G.v.A. designed the research. A.M., B.E., and A.G.v.A. wrote the article.
Glossary
- TL
translation
- NP
nonpolysomal
- SP
small polysomal
- LP
large polysomal
- SAM
significance analysis of microarrays
- TX
total
- FDR
false discovery rate
Footnotes
Articles can be viewed online without a subscription.
References
- Alexa A., Rahnenführer J. (2010). topGO: Enrichment analysis for Gene Ontology. R package version 2.18.0, https://www.bioconductor.org.
- Aronson B.D., Johnson K.A., Loros J.J., Dunlap J.C. (1994). Negative feedback defining a circadian clock: autoregulation of the clock gene frequency. Science 263: 1578–1584. [DOI] [PubMed] [Google Scholar]
- Branco-Price C., Kaiser K.A., Jang C.J., Larive C.K., Bailey-Serres J. (2008). Selective mRNA translation coordinates energetic and metabolic adjustments to cellular oxygen deprivation and reoxygenation in Arabidopsis thaliana. Plant J. 56: 743–755. [DOI] [PubMed] [Google Scholar]
- Covington M.F., Maloof J.N., Straume M., Kay S.A., Harmer S.L. (2008). Global transcriptome analysis reveals circadian regulation of key pathways in plant growth and development. Genome Biol. 9: R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filichkin S.A., Mockler T.C. (2012). Unproductive alternative splicing and nonsense mRNAs: a widespread phenomenon among plant circadian clock genes. Biol. Direct 7: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L., Cope L., Bolstad B.M., Irizarry R.A. (2004). affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315. [DOI] [PubMed] [Google Scholar]
- Green R.M., Tingay S., Wang Z.Y., Tobin E.M. (2002). Circadian rhythms confer a higher level of fitness to Arabidopsis plants. Plant Physiol. 129: 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin P.E., Hall J.C., Rosbash M. (1992). Circadian oscillations in period gene mRNA levels are transcriptionally regulated. Proc. Natl. Acad. Sci. USA 89: 11711–11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacklet J.W. (1977). Neuronal circadian rhythm: phase shifting by a protein synthesis inhibitor. Science 198: 69–71. [DOI] [PubMed] [Google Scholar]
- James A.B., Syed N.H., Bordage S., Marshall J., Nimmo G.A., Jenkins G.I., Herzyk P., Brown J.W., Nimmo H.G. (2012). Alternative splicing mediates responses of the Arabidopsis circadian clock to temperature changes. Plant Cell 24: 961–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M.A., Williams B.A., McNicol J., Simpson C.G., Brown J.W., Harmer S.L. (2012). Mutation of Arabidopsis spliceosomal timekeeper locus1 causes circadian clock defects. Plant Cell 24: 4066–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouffe C., Cretenet G., Symul L., Martin E., Atger F., Naef F., Gachon F. (2013). The circadian clock coordinates ribosome biogenesis. PLoS Biol. 11: e1001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juntawong P., Bailey-Serres J. (2012). Dynamic light regulation of translation status in Arabidopsis thaliana. Front. Plant Sci. 3: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi R., Girke T., Bray E.A., Bailey-Serres J. (2004). Differential mRNA translation contributes to gene regulation under non-stress and dehydration stress conditions in Arabidopsis thaliana. Plant J. 38: 823–839. [DOI] [PubMed] [Google Scholar]
- Kim B.H., Cai X., Vaughn J.N., von Arnim A.G. (2007). On the functions of the h subunit of eukaryotic initiation factor 3 in late stages of translation initiation. Genome Biol. 8: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.Y., Song H.R., Taylor B.L., Carré I.A. (2003). Light-regulated translation mediates gated induction of the Arabidopsis clock protein LHY. EMBO J. 22: 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y.J., Park M.J., Kim S.G., Baldwin I.T., Park C.M. (2014). Alternative splicing and nonsense-mediated decay of circadian clock genes under environmental stress conditions in Arabidopsis. BMC Plant Biol. 14: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lageix S., Lanet E., Pouch-Pélissier M.N., Espagnol M.C., Robaglia C., Deragon J.M., Pélissier T. (2008). Arabidopsis eIF2α kinase GCN2 is essential for growth in stress conditions and is activated by wounding. BMC Plant Biol. 8: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowska D., ten Have S., Hodge K., Tillemans V., Lamond A.I., Brown J.W. (2013). Plant SILAC: stable-isotope labelling with amino acids of Arabidopsis seedlings for quantitative proteomics. PLoS One 8: e72207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.J., Wu S.H., Chen H.M., Wu S.H. (2012). Widespread translational control contributes to the regulation of Arabidopsis photomorphogenesis. Mol. Syst. Biol. 8: 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews M.B., Sonenberg N., Hershey J.W.B. (2007). Origins and principles of translational control. In Translational Control in Biology and Medicine, Mathews M.B., Sonenberg N., Hershey J.W.B., eds (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; ), pp. 1–40. [Google Scholar]
- Matsushika A., Makino S., Kojima M., Yamashino T., Mizuno T. (2002). The APRR1/TOC1 quintet implicated in circadian rhythms of Arabidopsis thaliana: II. Characterization with CCA1-overexpressing plants. Plant Cell Physiol. 43: 118–122. [DOI] [PubMed] [Google Scholar]
- Missra A., von Arnim A.G. (2014). Analysis of mRNA translation states in Arabidopsis over the diurnal cycle by polysome microarray. Methods Mol. Biol. 1158: 157–174. [DOI] [PubMed] [Google Scholar]
- Mittag M., Lee D.H., Hastings J.W. (1994). Circadian expression of the luciferin-binding protein correlates with the binding of a protein to the 3′ untranslated region of its mRNA. Proc. Natl. Acad. Sci. USA 91: 5257–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockler T.C., Michael T.P., Priest H.D., Shen R., Sullivan C.M., Givan S.A., McEntee C., Kay S.A., Chory J. (2007). The DIURNAL project: DIURNAL and circadian expression profiling, model-based pattern matching, and promoter analysis. Cold Spring Harb. Symp. Quant. Biol. 72: 353–363. [DOI] [PubMed] [Google Scholar]
- Moeller J.R., Moscou M.J., Bancroft T., Skadsen R.W., Wise R.P., Whitham S.A. (2012). Differential accumulation of host mRNAs on polyribosomes during obligate pathogen-plant interactions. Mol. Biosyst. 8: 2153–2165. [DOI] [PubMed] [Google Scholar]
- Morse D., Milos P.M., Roux E., Hastings J.W. (1989). Circadian regulation of bioluminescence in Gonyaulax involves translational control. Proc. Natl. Acad. Sci. USA 86: 172–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel D.H., Kay S.A. (2012). Complexity in the wiring and regulation of plant circadian networks. Curr. Biol. 22: R648–R657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima H., Perlman J., Feldman J.F. (1981). Cycloheximide-induced phase shifting of circadian clock of Neurospora. Am. J. Physiol. 241: R31–R35. [DOI] [PubMed] [Google Scholar]
- Nicolaï M., Roncato M.A., Canoy A.S., Rouquié D., Sarda X., Freyssinet G., Robaglia C. (2006). Large-scale analysis of mRNA translation states during sucrose starvation in arabidopsis cells identifies cell proliferation and chromatin structure as targets of translational control. Plant Physiol. 141: 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozue K., Covington M.F., Duek P.D., Lorrain S., Fankhauser C., Harmer S.L., Maloof J.N. (2007). Rhythmic growth explained by coincidence between internal and external cues. Nature 448: 358–361. [DOI] [PubMed] [Google Scholar]
- Pal S.K., et al. (2013). Diurnal changes of polysome loading track sucrose content in the rosette of wild-type arabidopsis and the starchless pgm mutant. Plant Physiol. 162: 1246–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M.J., Seo P.J., Park C.M. (2012). CCA1 alternative splicing as a way of linking the circadian clock to temperature response in Arabidopsis. Plant Signal. Behav. 7: 1194–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piques M., Schulze W.X., Höhne M., Usadel B., Gibon Y., Rohwer J., Stitt M. (2009). Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol. Syst. Biol. 5: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokhilko A., Fernández A.P., Edwards K.D., Southern M.M., Halliday K.J., Millar A.J. (2012). The clock gene circuit in Arabidopsis includes a repressilator with additional feedback loops. Mol. Syst. Biol. 8: 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2014). R: A Language and Environment for Statistical Computing. (Vienna, Austria: R Foundation for Statistical Computing).
- Roy B., von Arnim A.G. (2013). Translational regulation of cytoplasmic mRNAs. Arabidopsis Book 11: e0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez S.E., et al. (2010). A methyl transferase links the circadian clock to the regulation of alternative splicing. Nature 468: 112–116. [DOI] [PubMed] [Google Scholar]
- Schaffer R., Ramsay N., Samach A., Corden S., Putterill J., Carré I.A., Coupland G. (1998). The late elongated hypocotyl mutation of Arabidopsis disrupts circadian rhythms and the photoperiodic control of flowering. Cell 93: 1219–1229. [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011). Global quantification of mammalian gene expression control. Nature 473: 337–342. [DOI] [PubMed] [Google Scholar]
- Sehgal A., Rothenfluh-Hilfiker A., Hunter-Ensor M., Chen Y., Myers M.P., Young M.W. (1995). Rhythmic expression of timeless: a basis for promoting circadian cycles in period gene autoregulation. Science 270: 808–810. [DOI] [PubMed] [Google Scholar]
- Seo P.J., Park M.J., Lim M.H., Kim S.G., Lee M., Baldwin I.T., Park C.M. (2012). A self-regulatory circuit of CIRCADIAN CLOCK-ASSOCIATED1 underlies the circadian clock regulation of temperature responses in Arabidopsis. Plant Cell 24: 2427–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sormani R., Delannoy E., Lageix S., Bitton F., Lanet E., Saez-Vasquez J., Deragon J.M., Renou J.P., Robaglia C. (2011). Sublethal cadmium intoxication in Arabidopsis thaliana impacts translation at multiple levels. Plant Cell Physiol. 52: 436–447. [DOI] [PubMed] [Google Scholar]
- Staiger D., Green R. (2011). RNA-based regulation in the plant circadian clock. Trends Plant Sci. 16: 517–523. [DOI] [PubMed] [Google Scholar]
- Staiger D., Zecca L., Wieczorek Kirk D.A., Apel K., Eckstein L. (2003). The circadian clock regulated RNA-binding protein AtGRP7 autoregulates its expression by influencing alternative splicing of its own pre-mRNA. Plant J. 33: 361–371. [DOI] [PubMed] [Google Scholar]
- Stolwijk A.M., Straatman H., Zielhuis G.A. (1999). Studying seasonality by using sine and cosine functions in regression analysis. J. Epidemiol. Community Health 53: 235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strayer C., Oyama T., Schultz T.F., Raman R., Somers D.E., Más P., Panda S., Kreps J.A., Kay S.A. (2000). Cloning of the Arabidopsis clock gene TOC1, an autoregulatory response regulator homolog. Science 289: 768–771. [DOI] [PubMed] [Google Scholar]
- Tiruneh B.S., Kim B.H., Gallie D.R., Roy B., von Arnim A.G. (2013). The global translation profile in a ribosomal protein mutant resembles that of an eIF3 mutant. BMC Biol. 11: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher V.G., Tibshirani R., Chu G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98: 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.Y., Tobin E.M. (1998). Constitutive expression of the CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) gene disrupts circadian rhythms and suppresses its own expression. Cell 93: 1207–1217. [DOI] [PubMed] [Google Scholar]
- Warner J.R. (1999). The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24: 437–440. [DOI] [PubMed] [Google Scholar]
- Warnes G.R., Bolker B., Bonebakker L., Gentleman R., Huber W., Liaw A.,.Lumley T., Maechler M., Magnusson A., Moeller S., Schwartz M., Venables B. (2014). gplots: Various R programming tools for plotting data. R package version 2.14.2. http://CRAN.R-project.org/package=gplots.
- Wu J., MacDonald J., Gentry J., Irizarry R. (2014). gcrma: Background adjustment using sequence information. R package version 2.38.0, http://www.bioconductor.org/packages/3.0/bioc/html/gcrma.html.
- Yángüez E., Castro-Sanz A.B., Fernández-Bautista N., Oliveros J.C., Castellano M.M. (2013). Analysis of genome-wide changes in the translatome of Arabidopsis seedlings subjected to heat stress. PLoS One 8: e71425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wang Y., Kanyuka K., Parry M.A., Powers S.J., Halford N.G. (2008). GCN2-dependent phosphorylation of eukaryotic translation initiation factor-2alpha in Arabidopsis. J. Exp. Bot. 59: 3131–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.