

Abstract
Climatic adaptation is an example of a genotype-by-environment interaction (G×E) of fitness. Selection upon gene expression regulatory variation can contribute to adaptive phenotypic diversity; however, surprisingly few studies have examined how genome-wide patterns of gene expression G×E are manifested in response to environmental stress and other selective agents that cause climatic adaptation. Here, we characterize drought-responsive expression divergence between upland (drought-adapted) and lowland (mesic) ecotypes of the perennial C4 grass, Panicum hallii, in natural field conditions. Overall, we find that cis-regulatory elements contributed to gene expression divergence across 47% of genes, 7.2% of which exhibit drought-responsive G×E. While less well-represented, we observe 1294 genes (7.8%) with trans effects. Trans-by-environment interactions are weaker and much less common than cis G×E, occurring in only 0.7% of trans-regulated genes. Finally, gene expression heterosis is highly enriched in expression phenotypes with significant G×E. As such, modes of inheritance that drive heterosis, such as dominance or overdominance, may be common among G×E genes. Interestingly, motifs specific to drought-responsive transcription factors are highly enriched in the promoters of genes exhibiting G×E and trans regulation, indicating that expression G×E and heterosis may result from the evolution of transcription factors or their binding sites. P. hallii serves as the genomic model for its close relative and emerging biofuel crop, switchgrass (Panicum virgatum). Accordingly, the results here not only aid in the discovery of the genetic mechanisms that underlie local adaptation but also provide a foundation to improve switchgrass yield under water-limited conditions.
Locally adapted populations outperform foreign populations in their native site but are less fit at other sites. There is a growing appreciation that this reciprocal home site advantage, which characterizes local adaptation, is driven by selection not only on coding sequence polymorphisms but also on gene expression regulatory variants (King and Wilson 1975; Hoekstra and Coyne 2007; Prud'homme et al. 2007; Stapley et al. 2010; Fraser 2011). Genotype-by-environment interactions (G×E), which underlie adaptive physiological variation and cause adaptation, have been partially resolved in a variety of species (e.g., Angert and Schemske 2005; Ågren and Schemske 2012; Anderson et al. 2012); however, the patterns of G×E at the gene expression level are less well understood (but see Hannah et al. 2006; Fraser 2011; Des Marais et al. 2013; Lasky et al. 2014).
Several studies have suggested that gene expression regulatory elements may be particularly likely to be involved in adaptive evolution (Wray et al. 2003; Wittkopp and Kalay 2011; Fraser 2013). This conclusion is supported by the observation that expression regulation can be tissue and environment specific while nonsynonymous mutations may alter protein function regardless of environmental cues (Wagner and Lynch 2008). Gene expression regulatory elements underlying adaptation may exist in close physical proximity to the target gene (cis) or in distant and often physically unlinked regions (trans). While cis-acting elements typically cause expression variation in a single gene, trans-acting modifiers may affect expression of several or, in the case of global transcription factors, thousands of genes. Mutations at loci with wide-ranging pleiotropic effects are typically deleterious (Keightley and Hill 1990; Kondrashov and Turelli 1992), leading to the hypothesis that trans-acting expression regulation may be subjected to purifying selection (Emerson et al. 2010), while cis-acting regulatory elements may respond to directional selection and contribute to adaptive differentiation (Schaefke et al. 2013). Indeed, many regulatory factors underlying physiological responses to drought and other stresses are conserved across broad phylogenetic distances (Matsukura et al. 2010; Des Marais et al. 2012; Mizoi et al. 2012).
Differentiation among plant populations across abiotic stress gradients provides some of the most compelling and best understood examples of adaptation in nature (e.g., Clausen et al. 1948; Stebbins 1950; McMillan 1959; Rieseberg and Willis 2007). In particular, natural selection imposed by differences in soil water availability is one of the most common and strongest drivers of local adaptation among plant populations (Stebbins 1952; Juenger 2013). To survive and reproduce in regions with a threat of periodic droughts, plants must alter their physiology quickly and effectively (Bohnert et al. 1995; Tisné et al. 2010). Drought responses at both the gene expression and whole-plant levels are implicated as major forces underlying local adaptation in a variety of plant species (Ramanjulu and Bartels 2002; Chaves et al. 2003). Given these strong selective forces, it is not surprising that many plant species have evolved “upland” and “lowland” ecotypes adapted to xeric and mesic environments, respectively (Porter 1966; Yu and Nguyen 1994; Kumar et al. 2008).
Panicum hallii variety hallii and var. filipes represent an ideal system to study the genetics of adaptation to drought. P. hallii is a genetically tractable diploid model system for C4 perennial grasses with complex genomes (e.g., P. virgatum/Switchgrass). Accordingly, a variety of genomic tools have been recently developed in P. hallii (Lowry et al. 2012, 2013). Importantly, the two varieties display striking ecological divergence, where the lowland var. filipes is primarily found in mesic regions of the Gulf Coast and Rio Grande Valley of Texas and northern Mexico, while the upland var. hallii’s range extends into dry regions of the Chihuahuan and Sonoran deserts (Waller 1976; Lowry et al. 2013).
Here, we define the extent and direction of drought-responsive gene expression at both whole-transcriptome and allele-specific expression (ASE) levels. Specifically, we tested three hypotheses: (1) divergence between upland and lowland ecotypes is characterized by drought-responsive differential gene expression; (2) such gene expression G×E is regulated primarily by cis factors; and (3) trans-regulated genes will be associated with regulatory elements that are known to respond to abiotic stress. To assess these hypotheses, we conducted a combined analysis of parental genotypes of each variety and their hybrid progeny across drought and drought recovery conditions. In doing so, we characterize the effects of cis-, trans-, and drought-responsive gene expression regulation that may underlie physiological divergence between the upland and lowland ecotypes of P. hallii.
Results and Discussion
Climatic context of the experimental drought
The two parental genotypes used in this experiment, HAL2 (var. hallii) and FIL2 (var. filipes) (Fig. 1A), represent the reference genomes for each variety. The HAL2 accession was collected from a population in xeric oak-shrub savanna at the Ladybird Johnson Wildflower Center (Austin, TX, USA 97.87°W, 30.18°N), while the FIL2 accession was collected from the Corpus Christi Botanical Gardens (Corpus Christi, TX, USA, 97.40°W, 27.65°N). These sites are representative of xeric hill country savanna and mesic coastal plains habitats.
Figure 1.
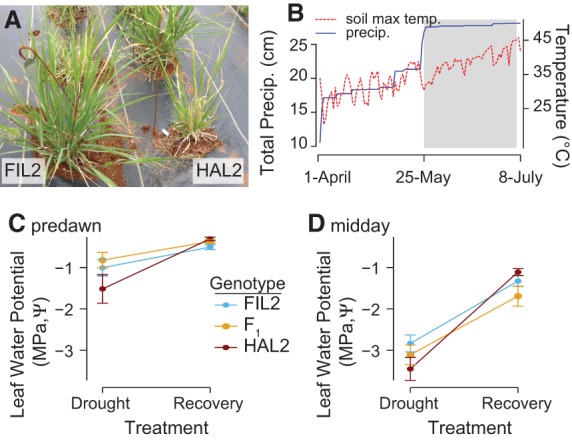
Climatic conditions and leaf-level responses across a natural drought and recovery. (A) An image of FIL2 (left) and HAL2 (right) that was taken prior to the onset of drought demonstrates the reduced growth rate of HAL2 compared to its lowland relative. From May 25 through July 5, 2013, <1 cm of rain fell at the site of the experiment. (B) Cumulative rainfall is presented along with daily maximum soil temperature. Leaf water potential (MPa, Ψ) was measured on July 5 (drought) and again on July 8 after irrigation and natural rainfall (recovery); means ± SE are presented for Ψ at predawn (C) and midday (D).
We grew the two ecotypes and their F1 hybrid progeny (“F1”) in a replicated common garden. The genotypes were vegetatively cloned (HAL2: n = 28, FIL2: n = 35, and F1: n = 29), planted in the field in October 2012, and subjected to a 41-d natural drought at the Ladybird Johnson Wildflower Center in 2013 (Fig. 1B). The 2013 drought represented the driest period from May 25 to July 5 since 1946 in Austin, TX (Supplemental Fig. S1A,B). We utilized this natural drought to test for responses to soil moisture variation by comparing droughted plants to those subjected to an experimental rewatering treatment. On July 5, 2013, after 41 d of drought, we collected leaf tissue and measured leaf water potential. On July 7, 2013, each plant was irrigated with 4 L of water. On July 8, 2013, leaf water potential was again assayed, and leaf tissue was collected on an independent subset of plants. Samples collected from the first harvest constituted the “drought” treatment, while the subsequent harvest, which followed watering, represented the “recovery” experimental treatment.
Leaf water potential (Ψ) was 2.2× lower in the drought treatment (F(df=1) = 78.7, P < 0.001) (Fig. 1C,D) regardless of genotype, indicating that our recovery treatment sufficiently relaxed the leaf-level physiological stresses of drought. While there was little evidence of an additive effect of genotype on Ψ (F(df=2) = 0.8, P > 0.1), there was a significant genotype-by-environment interaction (G×E) (F(df=2) = 3.43, P < 0.05) (Fig. 1C,D). This G×E was driven by strong drought responses of the HAL2 genotype in both predawn and midday leaf water potential measurements. Increased phenotypic plasticity to drought may contribute to the whole-plant drought adaptation characteristic of var. hallii (Lowry et al. 2015).
Drought-responsive gene expression divergence between upland and lowland ecotypes
Leaf tissue was harvested and RNA was extracted from the most recently expanded leaf from 17 FIL2, 12 HAL2, and 14 F1 clones in the drought treatment and 17 FIL2, 13 HAL2, and 15 F1 clones in the recovery treatment. Whole RNA sequencing yielded 22,256 genes with mean counts >5 (Supplemental Fig. S2). These expressed genes constituted 59.3% of the total gene models, indicating that a majority of genes were expressed in the most recently expanded leaves under natural field conditions. We fit a generalized linear model via DESeq2 (Love et al. 2014) to the transcript count data and calculated Wald test contrast P-values for genotype, treatment, and G×E model terms. For each contrast, we applied multiple testing corrections via Q-values calculated from the P-value distributions (Storey 2002). Genes with Q-values ≤0.05 were determined to have a significant effect. We also estimated the proportion of true null hypotheses (π0) (Storey et al. 2004) as an alternative approach to count the number of genes that were affected by an experimental factor.
Across all genes, 15,332 (π0 = 0.264) and 16,766 (π0 = 0.224) genes were affected by treatment and genotype, respectively (Fig. 2A). While the number of genes affected by each factor was relatively similar, the effect size (calculated as the mean absolute value of the log2 fold change [LFC]) due to the genotype term (1.59) was >2.5× that of treatment (0.63). Consistent with this observation, the most common expression profile was found among genes regulated in the same direction across both treatments between HAL2 and FIL2 (Fig. 2A). However, 5471 genes were differentially expressed exclusively in the drought or drought-recovery treatment. Interestingly, these drought-responsive genes tended to be up-regulated (HAL2 > FIL2) in drought (52%, binomial P = 0.01) but down-regulated in the recovery treatment (58%, binomial P < 0.0001) (Fig. 2A).
Figure 2.
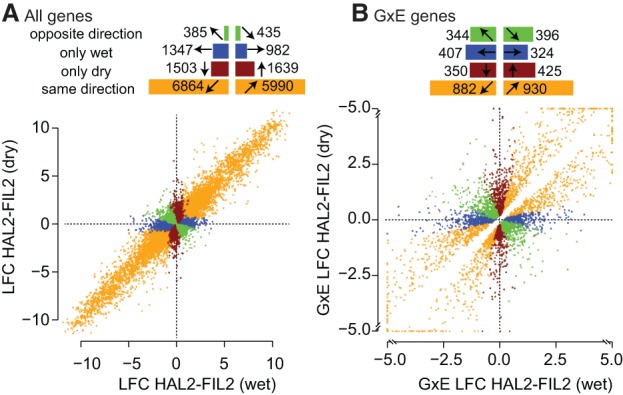
Distribution of differential expression of total counts between HAL2 and FIL2 parents in drought and recovery treatments. Log2 fold changes (LFC) between HAL2 and FIL2 were calculated for all genes independently in each experimental treatment. (A) All genes with significant genotype effects are presented. (B) Only those genes with significant G×E effects are presented; those G×E genes with log2 fold changes greater than 5 or less than −5 are shrunk to 5 (−5) for plotting purposes. In each panel, genes were binned by the direction and significance of the LFC. The numbers of genes belonging to each bin are displayed as horizontal bar plots at the top of each panel.
We also observed significant (q ≤ 0.05) patterns of genotype-by-environment interactions (G×E) among the expression of 3907 genes (Fig. 2B). Nearly half of the G×E genes (1795, 46%) were driven by differential expression in both treatments (orange points, Fig. 2B). However, the experimental treatment drove differential expression across many of the remaining genes: 33.6% of the genes declared to have significant G×E effects were differentially expressed in one treatment but not the other. Finally, 20.5% of G×E genes had opposite response directions (green points, Fig. 2B). These rank- and sign-changing G×E genes demonstrate phenotypic trade-offs between environments and represent an interesting and potentially evolutionarily important subset of loci that may underlie adaptive differences in response to drought between var. hallii and var. filipes (Des Marais et al. 2013; Juenger 2013; Lasky et al. 2014).
To determine the identity and characteristics of the significantly differentially expressed genes, we calculated the overrepresentation of gene ontology (GO) terms and sequence motifs in the promoter (Supplemental Tables S1, S2). Across all G×E genes that were differentially expressed in drought (red points, Fig. 1A), there was a significant enrichment of several hundred GO terms (Supplemental Table S1). Some of the most interesting were those that were responsive to heat, drought, and reactive-oxygen species (Supplemental Table S1). Additionally, GO terms enriched among the opposite direction G×E genes (green points, Fig. 2B) included responses to heat, flowering, and guard-cell development. Physiological divergence of these traits has been observed among differentially drought-adapted genotypes in other systems (Lebaudy et al. 2008; Wilczek et al. 2009). Interestingly, the most highly enriched motif in HAL2 up-regulated genes is specific to an abscisic acid (ABA) responsive transcription factor (ABRE) (ABADESI2, P < 1 × 10−21). ABREs are known to increase expression in drought and elevated ABA concentrations (Lam and Chua 1991; Busk and Pagès 1998; Narusaka et al. 2003), driving adaptive drought responses (Des Marais and Juenger 2010).
Impact of drought on gene expression heterosis
The cis- and trans-acting regulatory elements that cause environmental responses are thought to contribute to patterns of heterosis at both the gene expression and whole-plant levels (Hochholdinger and Hoecker 2007; Chen 2013). We explored patterns of gene expression heterosis between var. hallii and var. filipes by contrasting total transcript abundance of each gene among HAL2, FIL2, and their F1 hybrid. Each gene was classified into one of seven expression categories based on Q-values (α = 0.05) for three separate contrasts (Table 1; Stupar and Springer 2006; Paschold et al. 2012): (1) no differential expression; (2) additive effects; (3) high-parent heterosis (hp); (4) low-parent heterosis (lp); (5) above high-parent heterosis (>hp); (6) below low-parent heterosis (<lp); and (7) ambiguous expression patterns (Table 1). It is important to note that these categorizations are conducted on the log2 scale, so genes with “additive” effects may not appear to be linear on an untransformed scale.
Table 1.
Heterosis categorization of all expressed genes
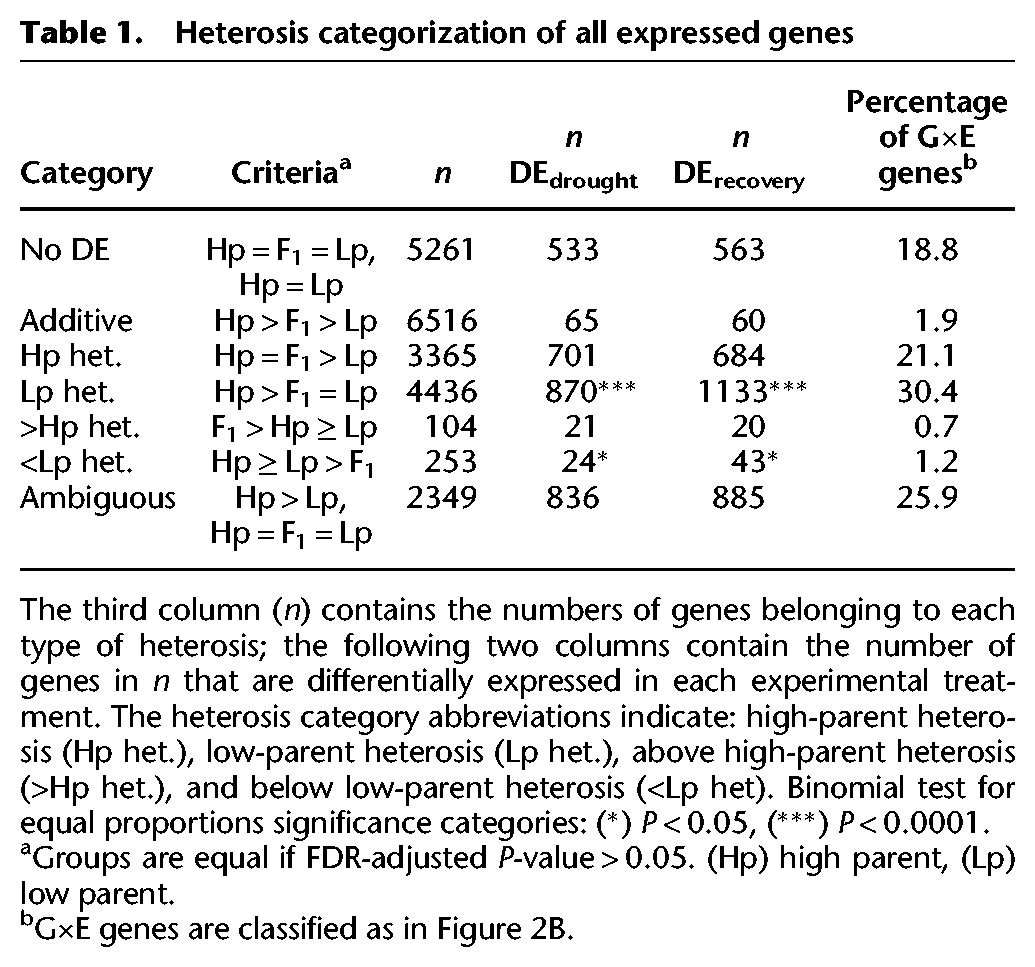
Across environmental treatments, a slight majority of differentially expressed genes showed additive gene expression patterns (Table 1). Additive expression patterns were found almost exclusively among the genes exhibiting significant differential expression in both environments. In fact, only 125 (1.9%) of the additive genes were differentially expressed in a single environment, and four (0.08%) additive genes were differentially expressed in opposite directions across environments (Supplemental Fig. S3).
The extreme scarcity of additive genes that were differentially expressed in only a single environment was balanced by a significant overrepresentation of genes displaying both G×E and heterosis. Hybrids exhibited the expression values of the high parent or low parent across 3365 (19.8%) and 4436 (26.1%) genes, respectively (Table 1; Supplemental Fig. S3). This bias toward below-mid-parent heterosis was highly significant (binomial test P < 0.0001). Among these 7801 genes, 3431 (44.0%) displayed significant differential expression in one environment but not the other. As such, genes with high- or low-parent heterosis represented 93% of all genes with G×E (Supplemental Fig. S3). Physiological G×E effects often underlie the fitness G×E that constitute local adaptation (Clausen et al. 1948; Ågren and Schemske 2012; Juenger 2013) and may be a result of G×E at specific loci (Hall et al. 2010; Des Marais et al. 2012; Ågren et al. 2013). Therefore, it is plausible that such heterotic G×E genes contribute to the adaptive divergence observed between these upland and lowland ecotypes.
Heterotic genes that significantly exceeded the range of the parents comprised an extreme minority. Only 104 (0.4%) and 253 (1.1%) genes were found to have above high-parent and below low-parent heterosis, respectively. Many of the heterotic genes were initially classified as conserved (Fig. 2, gray points). However, genetic variation was present for these genes between HAL2 and FIL2 but only was revealed within the F1 hybrid, implicating multiple antagonistic loci, epistatic interactions, or other forms of trans-acting gene expression regulation (Rieseberg et al. 1999; Li et al. 2008; Chen 2010).
Inference of cis- and cis-treatment gene expression G×E via a linear model
We quantified allele-specific expression for 16,465 genes by the presence of HAL2- or FIL2-specific single nucleotide polymorphisms. We then partitioned the variance of ASE into that attributable to allelic imbalance in the F1 generation (cis) and differential allelic imbalance across parental (F0) and F1 generations (trans) using a linear modeling approach (Figs. 3, 4A,B). This methodology is analogous to the binomial and χ2 tests (Supplemental Figs. S4, S5) that have been traditionally used to assess cis- and trans-acting gene expression regulation (Wittkopp et al. 2004; McManus et al. 2010; Bader et al. 2015); however, linear modeling affords several benefits, including the capability to incorporate biological replicates and to control for experimental design variables (i.e., blocking, treatment, and other covariates). We tested the effect of cis, trans, experimental treatments, and all additive and interactive combinations therein using a linear model specification within the DESeq2 negative binomial framework (Bader et al. 2015) (see the Supplemental Material for comparisons between the DESeq2 model specification, mixed models, and traditional cis-trans test methods).
Figure 3.
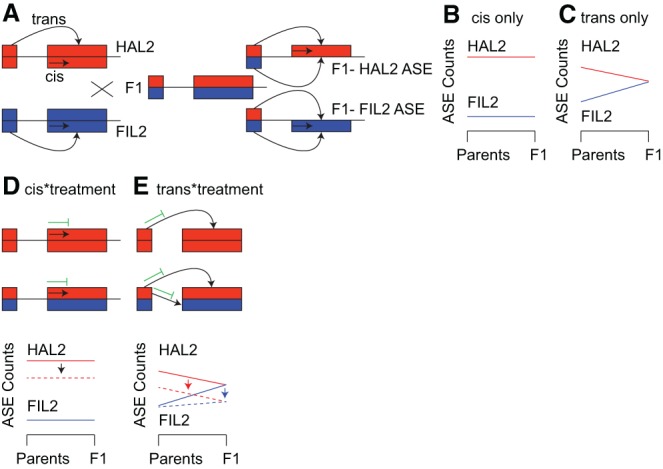
Description of the F0/F1 ASE test for cis- and trans-acting gene expression regulation. (A) Each inbred parent possesses only the cis and trans factors associated with that genotype, while the F1 hybrid possesses both. (B) In the simplest case where ASE patterns are affected by a single cis element, allelic imbalance ratios are identical across generations. (C) In the case where a single trans factor causes ASE variation, the F1 ASE counts should be identical for both alleles since both trans factors are acting on each allele. To illustrate the effects of cis- and trans-by-treatment interactions, take the case where an experimental treatment induces the expression of a HAL2-specific cis- or trans-acting repressor (green symbol, dashed lines). The resultant expression patterns would shift so that the difference between alleles (cis-by-treatment, D) or the slope of the genotype*generation interaction depends on the treatment (trans-by-treatment, E). The scheme presented here is a simplification, and all combinations of cis, trans additive effects and interactions with the environment are possible.
Figure 4.
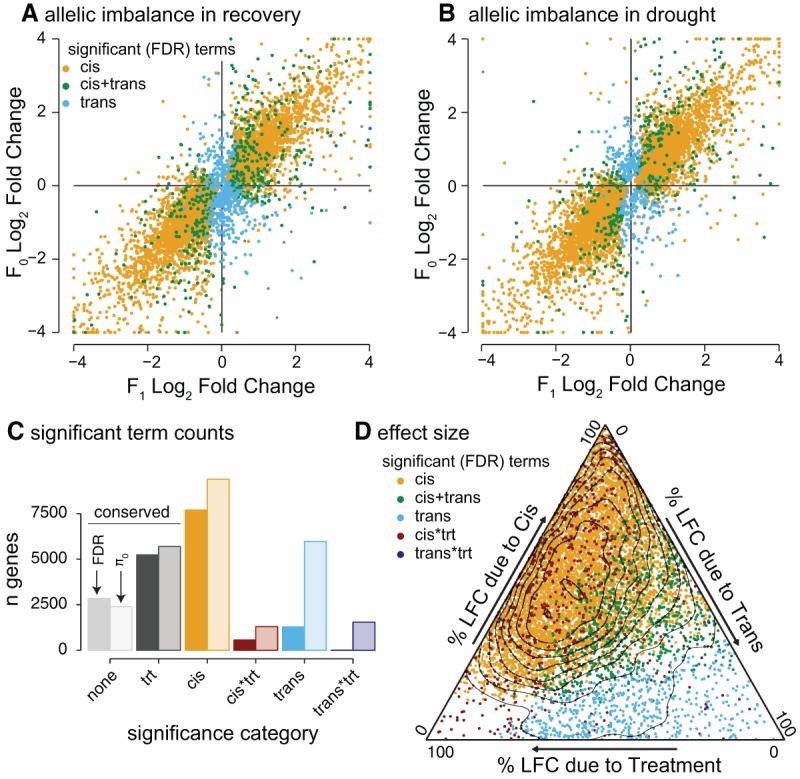
Distribution of cis, trans, and environmental interaction effects. The log2 fold change of the FIL2 vs. the HAL2 allele in the F0 and F1 generations is plotted for the recovery (A) and drought treatments (B). Coloration is derived from the significance of terms (allele—cis, allele*generation—trans). (C) All ASE genes were categorized by the model terms that caused significant differential expression. The number of genes in each bin was determined by a strict FDR threshold (left, darker bars) and the estimated number of true null hypotheses (π0, right, transparent bars) methods. Where both additive and interactive terms were significant for cis or trans (e.g., best model = y ∼ cis + cis*treatment), the model was binned into the interactive effect category. (D) For those genes with significant genetic effects (cis and/or trans), the proportions of log2 fold change due to cis, trans, and treatment are plotted in the ternary diagram. Total variance is the sum of the three components and variance attributable to interaction terms is ignored (but see Fig. 5).
We found 17,524 (78.7%) genes that were differentially expressed when contrasting the parents; however, only 8086 (49.8%) of the 16,465 genes that had detectable ASE had significant cis- or trans-regulated allele-specific expression (Fig. 4A–C). This discrepancy was primarily a result of larger allelic imbalance effect sizes in the F0 compared to the F1 generation (Fig. 4A,B; Supplemental Fig. S6; Supplemental Material). Nonetheless, our experiment had surprisingly strong power to detect cis effects. We found that >47% (7699) of gene expression patterns were affected by cis factors, a finding that exceeded other published studies using linear modeling to test for cis effects, which ranged from 15% to 30% (Cubillos et al. 2014; Bader et al. 2015). The increased power is likely attributable to significant genomic divergence between the parents and individual, rather than pooled, sequencing of each biological replicate (Liu et al. 2014).
Combined, the additive effects of cis factors and treatment explained the bulk of differential ASE (Fig. 4D); however, many genes also displayed complex G×E patterns (Supplemental Fig. S6). Across the 8379 genes with genotype-driven ASE, we estimated that expression was affected by a cis-by-treatment term across 2030 genes (π0 = 0.76); however, more stringent methods revealed many fewer genes (562, Q-value ≤0.05) (Fig. 4C,D). As in the G×E allelic imbalance tests, the most common patterns of cis-by-treatment effects were found in cases where both expression responses had the same sign and the alleles retained their relative ranks across treatments but the difference between means shifted by treatment (e.g., HAL2 >> FIL2 in wet, HAL2 > FIL2 in dry) (Supplemental Fig. S6).
Trans and trans-by-treatment expression regulatory divergence
In the linear model, inference of the trans effect is captured by an allele-by-generation interaction (Fig. 3; Bader et al. 2015). We estimated that the expression of 5969 (π0 = 0.63) of the 16,465 genes with quantifiable ASE were affected by trans factors; however, in many cases the trans effect size was relatively small (Fig. 4D). Therefore, it is not surprising that more stringent Q-value thresholding found only 1285 trans-regulated genes (19.8%) (Fig. 4C), most of which were also regulated by cis factors (Fig. 4D). Of these genes, 154 (Supplemental Fig. S7) showed significant homogenization in the F1 generation, a pattern potentially driven by the complementation of nonfunctional trans factors in the F1 (Supplemental Fig. S7; Paschold et al. 2012).
Among the trans-regulated genes, we observed patterns indicative of compensatory evolution in the expression of 312 genes (Supplemental Fig. S7). Expression of these genes was conserved in the parents but divergent in the F1. Such compensatory evolution is likely a result of co-evolution among cis and trans factors (Landry et al. 2005). In this case, selection may have favored consistent expression among genotypes, but the trans-acting factors producing such consistency were not identical-by-state in each genotype.
Transcription factors and other regulatory elements are expected to drive the expression of trans-regulated genes. As such, we expected that many transcription-factor target motifs would be highly enriched among compensatory and other trans-regulated genes. Since differential drought adaptation is one of the primary factors characterizing HAL2-FIL2 divergence, we hypothesized that genes that have diverged due to trans regulation would tend to contain drought-responsive element binding factor target motifs. The data strongly support this hypothesis: Three of the four most enriched motifs among all compensatory trans-regulated gene promoters corresponded to the binding sites of ABA-responsive transcription factors (Supplemental Table S2), including the above mentioned ABADESI2 and two other ABRE members.
We found that many genes (1287, π0 = 0.812) were marginally affected by trans-by-treatment interactions (Fig. 4C). However, FDR thresholding reduced this number to only nine significant genes (Fig. 4C,D). This discrepancy was driven by the relatively small effects of trans-by-treatment interactions (Fig. 4D; Supplemental Fig. S8) and decreased power to detect interaction terms relative to main effects in linear models. The nine genes with expression affected trans-by-treatment interactions (Table 2; Fig. 5; Supplemental Fig. S8) had the strongest effects and spanned a diverse array of possible G×E expression patterns (Fig. 5; Supplemental Fig. S9). Due to the small number of genes affected by trans-treatment interaction, neither GO nor motif enrichment analyses can be reasonably applied. We therefore examined the Arabidopsis annotations of these genes directly. Interestingly, five of these nine were orthologs of Arabidopsis or rice genes with annotations related to drought or other stress responses: (1) UTG85A2 (Fig. 5A)—such glycosyltransferase family genes have been shown to disrupt the abscisic acid responsive pathway (Priest et al. 2006), affecting drought responses (Tognetti et al. 2010); (2) TINY2, a DREB family transcription factor (Fig. 5B)—DREBs are the most extensively studied drought-responsive group of genes in Arabidopsis (Sakuma et al. 2002; Agarwal et al. 2006; Xianjun et al. 2011); (3) FT, a central gene in the flowering pathway (Fig. 5C)—several recent studies have implicated a physiological and genetic tie between drought-responsive and flowering pathways (McKay et al. 2003; Lovell et al. 2013, 2015; Riboni et al. 2013; Kimura et al. 2015); (4) RLK1, the rice ortholog (OsLecRK4) which improves resistance to insect herbivory (Liu et al. 2015), and (5) a rice ortholog (OSMate2) of the MATE efflux family gene AT1G71140, which, when overexpressed in A. thaliana, alters a variety of abiotic and biotic stress responses (Tiwari et al. 2014).
Table 2.
Description of the nine genes with significant trans-by-treatment interactions
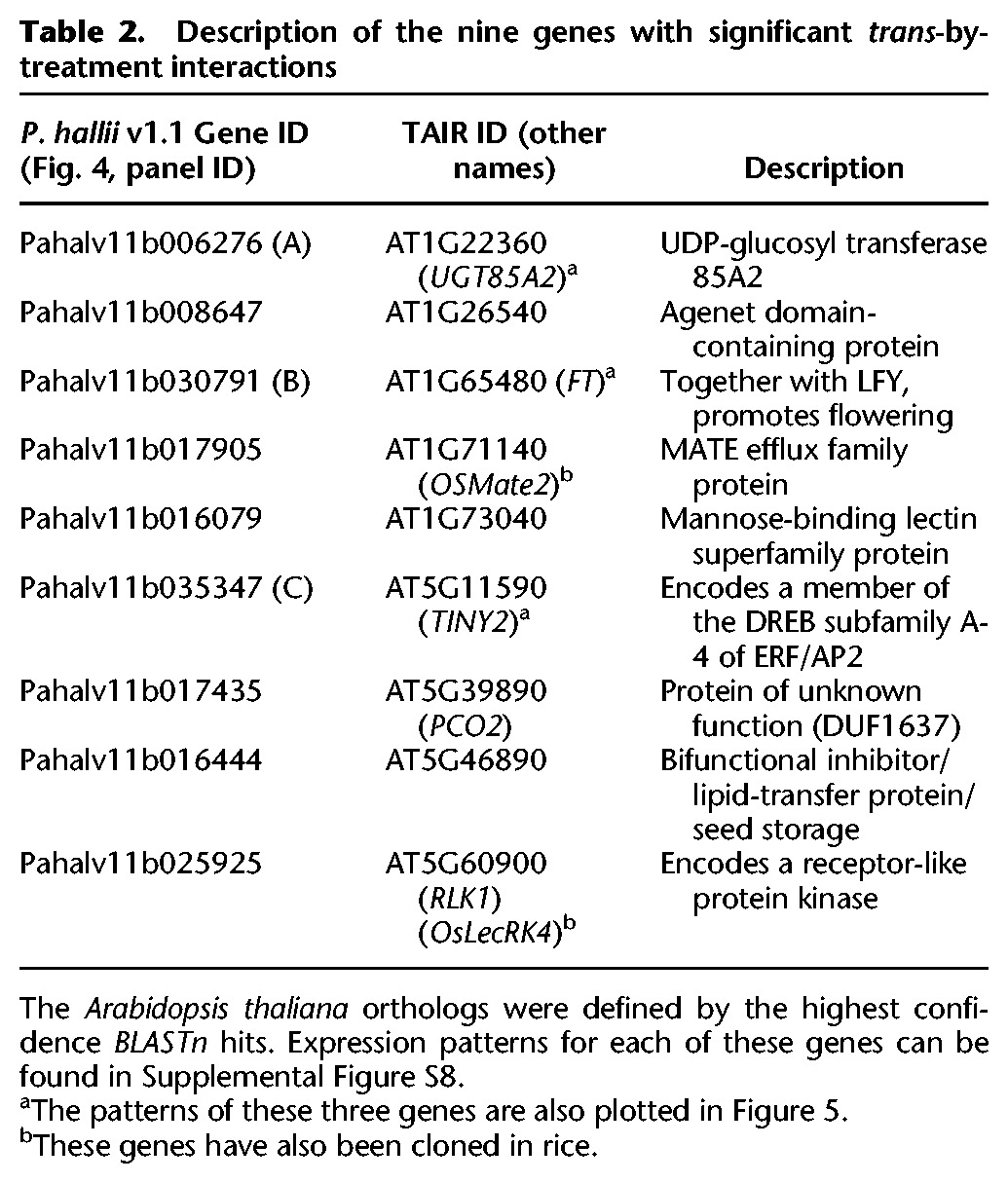
Figure 5.
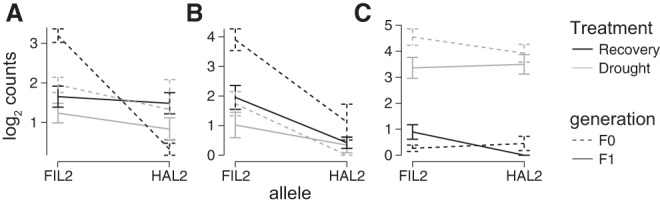
Expression patterns of three genes with significant trans-treatment interactions. Mean log2 transformed and library size corrected counts are plotted for Pahalv11b006276 (A), Pahalv11b030791 (B), and Pahalv11b035347 (C). The Arabidopsis TAIR gene identifiers and pseudonyms for each gene can be found in Table 2.
We found support for similar physiological roles of these genes in Arabidopsis and P. hallii. For example, in the drought treatment, alleles of the P. hallii TINY2 ortholog were strongly diverged in the F0 generation but had identical expression in the F1. However, in the recovery treatment, the transcripts were barely quantifiable, regardless of the genotype or generation. This pattern, which yielded a strong trans-by-treatment effect in our experiment, is consistent with the documented expression patterns of TINY2 in A. thaliana (Wei et al. 2005). Finally, ABRE cis elements are enriched within promoters of DREB2A downstream genes (Sakuma et al. 2006). Since TINY2 is a member of the DREB family of transcription factors and ABRE motifs are significantly overrepresented in the promoters of trans-regulated genes observed here, it is plausible that the P. hallii ortholog of TINY2 may be one of the transcription factors causing such trans regulatory divergence. These observations may provide a fertile avenue for future research about the genetic networks that have diverged to cause differential drought adaptation of upland and lowland ecotypes of P. hallii.
Conclusions
Across all genes, treatment and cis effects accounted for the bulk of variation attributable to experimental factors (Figs. 2A, 4D). This extreme prevalence of cis-acting variation over trans has been observed in several recent analyses using similar approaches (Wittkopp et al. 2004; McManus et al. 2010; Cubillos et al. 2014; Bader et al. 2015). Nonetheless, we estimated that an additional 1468 cis-by-treatment, 2246 trans, and 1436 trans-by-treatment effects existed but were not of sufficient strength to be detected by FDR thresholding methods (Fig. 4C). These results indicated that many genes were influenced primarily by cis factors and secondarily by trans factors and by both cis and trans interactions with the environment. Importantly, several drought-responsive transcription factor-binding sites were highly enriched among genes that responded to drought or were trans regulated, indicating that drought adaptation in P. hallii may be due to similar genetic networks found in model species. Combined, these observations provide a strong foundation for further inquiry into the evolution of drought tolerance in this important genomic model for the emerging biofuel crop, switchgrass (P. virgatum).
Methods
Germplasm and experimental design
The parents, HAL2 (var. hallii; Austin, TX; 30.19° N, 97.87° W) and FIL2 (var. filipes; Corpus Christi, TX; 27.65° N, 97.40° W), were originally germinated from seeds at the University of Texas at Austin. The F1 hybrid was made through a controlled cross, as described in Lowry et al. (2015). The parents and the F1 hybrid were cloned through vegetative propagation to produce replicate plants for the experiment.
We conducted sampling of the plants in the drought treatment on July 5, 2013. Half of the parental and F1 plants were assigned randomly to the drought treatment prior to leaf collections. For each plant, we measured predawn leaf water potential (LWP) between 4:30 a.m. and 6:40 a.m. We then collected leaf tissue for RNA simultaneously with leaf tissue collection for mid-day LWP using a Scholander-type pressure bomb (PMS Instruments, model 1000) between 11:00 a.m. and 1:30 p.m. RNA was extracted from the most recently expanded leaf on a tiller that was representative of the majority of tillers at the time of sampling. The time of leaf collection was recorded to the nearest minute. Leaves for RNA extractions were flash-frozen with liquid nitrogen and packed immediately in dry ice. To simulate a large rainfall event, all plants were rewatered on July 7, 2013. Each plant received 4 L of water, applied by hand to the base of the plant. On July 8, we conducted sampling from the parental and F1 plants that had not been sampled on July 5. These rewatered plants were sampled for predawn and midday LWP and leaf tissue following the same sampling protocol as on July 5.
RNA extraction
P. hallii leaf samples (50–200 mg) were homogenized in Eppendorf tubes with steel beads on a Geno/Grinder 2000 (Spex SamplePrep). Total RNA was extracted with RNeasy Plant Mini kits (Qiagen) and treated with DNase I to remove contaminating genomic DNA. RNA concentration was quantified with Qubit (Invitrogen). Three micrograms of each RNA sample passing quality control (RIN of 5 or greater) were sequenced.
Sequencing effort
Plate-based RNA sample prep was performed on the PerkinElmer SciClone NGS robotic liquid handling system using Illumina's TruSeq Stranded mRNA HT Sample Prep kit following the protocol outlined by Illumina in their user guide and with the following conditions: total RNA starting material was 1 µg per sample, and 10 cycles of PCR was used for library amplification. The prepared libraries were then quantified using KAPA Biosystem's next-generation sequencing library qPCR kit and run on a Roche LightCycler 480 real-time PCR instrument. The quantified libraries were multiplexed into pools of four libraries each, and the pool was prepared for sequencing on the Illumina HiSeq sequencing platform utilizing a TruSeq Paired-End Cluster kit v3 and Illumina's cBot instrument to generate a clustered flowcell for sequencing. Sequencing of the flowcell was performed on the Illumina HiSeq 2000 sequencer using a TruSeq SBS sequencing kit (200 cycles, v3, following a 2 × 150 indexed run recipe).
Raw reads were trimmed, then mapped to the draft v1.1 P. hallii var. filipes genome (which is from the FIL2 accession), and counts were called for each of the 37,638 annotated gene models. There was no evidence of mapping bias toward the FIL2 allele (binomial test P > 0.1) (Supplemental Fig. S2).
Generation of counts and ASE data
Histograms of 50mer frequency were generated for the HAL2 and FIL2 parents, and 50mers were extracted within a frequency range bounded by full width at the half maximum of the genome peak of the individual parents. HAL2/FIL2-shared 50mers were excluded, and the 50mers for the HAL2/FIL2-specific parents were aligned to the FIL2 (v1.1) reference genome using the BWA (Li and Durbin 2009) short read aligner. Uniquely aligning HAL2 and FIL2 50mers (FIL2 aligning perfectly and HAL2 aligning with one mismatch) that aligned to the same location in the FIL2 reference were used to define a set of markers that discriminated between the parental lines. The 50mer marker pairs were then used to classify reads as either HAL2, FIL2, or undetermined. The final set of classified reads were aligned to the P. hallii FIL2 reference genome (v1.1) using short read aligning program GSNAP (Wu and Nacu 2010). HTSeq v0.6.1 (Anders et al. 2015), a Python package, was used to count the reads mapped to annotated genes in the reference genome. Outliers among the biological replicates were identified using Pearson's correlation coefficient, R2 ≥ 0.95 and multidimensional scaling and were not considered for further analysis.
Modeling differential expression
Differential expression was inferred by analyzing raw counts using the DESeq2 package (Love et al. 2014) in the R Environment for Statistical Computing (R Core Team 2012), which infers differential expression after accounting for library size variation and mean-variance structure. Details about the model specification can be found in the Supplemental Material.
Gene ontology and motif enrichment analyses
To qualify the identity and characteristics of the significantly differentially expressed genes, we utilized de novo InterPro (Mitchell et al. 2015), Arabidopsis thaliana, and Oryza sativa ortholog GO terms. We then tested for enrichment of terms in each of the direction and significance groupings (bar plot categories in Fig. 2A) using the classic Fisher's exact test available in the topGO package (http://bioconductor.org/packages/release/bioc/html/topGO.html). To assess patterns of transcription factor binding motifs, promoter sequences (2 kb upstream) for each gene were downloaded from the Phytozome online portal (http://phytozome.jgi.doe.gov/pz/portal.html). We then calculated the statistical enrichment of a set of 485 motifs found in plant species from the newPLACE (http://www.dna.affrc.go.jp/PLACE/) database using the R package PWMEnrich (Frith et al. 2004).
Data access
The sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under BioProject PRJNA306692. Sample information and accession numbers can be found in Supplemental Table S3.
Supplementary Material
Acknowledgments
J. Heiling and B. Whitaker assisted in propagating plants and planting the experiment. Many members of the Juenger laboratory assisted in harvesting leaf tissue and measuring leaf water potentials. We thank M. Simmons, M. Bertelsen, and the Ladybird Johnson Wildflower Center for facilitating our field experiment. Computational analyses were completed on the Stampede system with allocations from the Texas Advance Computing Center. Earlier versions of this manuscript were greatly improved following comments from J.R. Lasky, D. Bolnick, and three anonymous reviewers. J.T.L. was supported by a National Science Foundation IOS fellowship (IOS-1402393). D.B.L. was supported by a Department of Agriculture National Institute of Food and Agriculture (USDA NIFA) fellowship (2011-67012-309969). E.V.S. was supported in part by the Russian Government Program of Competitive Growth of Kazan Federal University. Funding for this project came from grants to T.E.J. from the National Science Foundation (IOS-0922457) and the Department of Energy (DOE) (DE-SC0008451). The work conducted by the DOE Joint Genome Institute was supported by the Office of Science of the DOE under contract DE-AC02-05CH11231.
Author contributions: All authors contributed significantly to this work. J.T.L., S.S., D.B.L., X.W., J.S., and T.E.J. wrote the paper and conducted the statistical analysis. D.B.L. and T.E.J. designed the experiment. D.B.L. and J.E.B. conducted the fieldwork. E.V.S., J. Johnson, and M.W. conducted RNA sequencing. J. Jenkins, C.P., and A.S. generated annotations and processed raw data.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.198135.115.
References
- Agarwal PK, Agarwal P, Reddy MK, Sopory SK. 2006. Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep 25: 1263–1274. [DOI] [PubMed] [Google Scholar]
- Ågren J, Schemske DW. 2012. Reciprocal transplants demonstrate strong adaptive differentiation of the model organism Arabidopsis thaliana in its native range. New Phytol 194: 1112–1122. [DOI] [PubMed] [Google Scholar]
- Ågren J, Oakley CG, McKay JK, Lovell JT, Schemske DW. 2013. Genetic mapping of adaptation reveals fitness tradeoffs in Arabidopsis thaliana. Proc Natl Acad Sci 110: 21077–21082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JT, Lee C-R, Rushworth CA, Colautti RI, Mitchell-Olds T. 2012. Genetic trade-offs and conditional neutrality contribute to local adaptation. Mol Ecol 22: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angert AL, Schemske DW. 2005. The evolution of species’ distributions: reciprocal transplants across the elevation ranges of Mimulus cardinalis and M. lewisii. Evolution 59: 1671–1684. [PubMed] [Google Scholar]
- Bader DM, Wilkening S, Lin G, Tekkedil MM, Dietrich K, Steinmetz LM, Gagneur J. 2015. Negative feedback buffers effects of regulatory variants. Mol Syst Biol 11: 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert HJ, Nelson DE, Jensen RG. 1995. Adaptations to environmental stresses. Plant Cell 7: 1099–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busk PK, Pagès M. 1998. Regulation of abscisic acid-induced transcription. Plant Mol Biol 37: 425–435. [DOI] [PubMed] [Google Scholar]
- Chaves MM, Maroco JP, Pereira JS. 2003. Understanding plant responses to drought—from genes to the whole plant. Funct Plant Biol 30: 239–264. [DOI] [PubMed] [Google Scholar]
- Chen ZJ. 2010. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci 15: 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ. 2013. Genomic and epigenetic insights into the molecular bases of heterosis. Nat Rev Genet 14: 471–482. [DOI] [PubMed] [Google Scholar]
- Clausen J, Keck DD, Hiesey WM. 1948. Experimental studies on the nature of species. III. Environresponses of climatic races of Achillea. Carnegie Institute of Washington, Washington, DC. [Google Scholar]
- Cubillos FA, Stegle O, Grondin C, Canut M, Tisné S, Gy I, Loudet O. 2014. Extensive cis-regulatory variation robust to environmental perturbation in Arabidopsis. Plant Cell 26: 4298–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Des Marais DL, Juenger TE. 2010. Pleiotropy, plasticity, and the evolution of plant abiotic stress tolerance. Ann NY Acad Sci 1206: 56–79. [DOI] [PubMed] [Google Scholar]
- Des Marais DL, McKay JK, Richards JH, Sen S, Wayne T, Juenger TE. 2012. Physiological genomics of response to soil drying in diverse Arabidopsis accessions. Plant Cell 24: 893–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Des Marais DL, Hernandez KM, Juenger TE. 2013. Genotype-by-environment interaction and plasticity: exploring genomic responses of plants to the abiotic environment. Annu Rev Ecol Evol Syst 44: 5–29. [Google Scholar]
- Emerson JJ, Hsieh LC, Sung HM, Wang TY, Huang CJ, Lu HHS, Lu MYJ, Wu SH, Li WH. 2010. Natural selection on cis and trans regulation in yeasts. Genome Res 20: 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB. 2011. Genome-wide approaches to the study of adaptive gene expression evolution: Systematic studies of evolutionary adaptations involving gene expression will allow many fundamental questions in evolutionary biology to be addressed. Bioessays 33: 469–477. [DOI] [PubMed] [Google Scholar]
- Fraser HB. 2013. Gene expression drives local adaptation in humans. Genome Res 23: 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Fu Y, Yu L, Chen J-F, Hansen U, Weng Z. 2004. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res 32: 1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MC, Lowry DB, Willis JH. 2010. Is local adaptation in Mimulus guttatus caused by trade-offs at individual loci? Mol Ecol 19: 2739–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah MA, Wiese D, Freund S, Fiehn O, Heyer AG, Hincha DK. 2006. Natural genetic variation of freezing tolerance in Arabidopsis. Plant Physiol 142: 98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochholdinger F, Hoecker N. 2007. Towards the molecular basis of heterosis. Trends Plant Sci 12: 427–432. [DOI] [PubMed] [Google Scholar]
- Hoekstra HE, Coyne JA. 2007. The locus of evolution: evo devo and the genetics of adaptation. Evolution 61: 995–1016. [DOI] [PubMed] [Google Scholar]
- Juenger TE. 2013. Natural variation and genetic constraints on drought tolerance. Curr Opin Plant Biol 16: 274–281. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Hill WG. 1990. Variation maintained in quantitative traits with mutation-selection balance: pleiotropic side-effects on fitness traits. Proc R Soc Lond B 242: 95–100. [Google Scholar]
- Kimura Y, Aoki S, Ando E, Kitatsuji A, Watanabe A, Ohnishi M, Takahashi K, Inoue S-I, Nakamichi N, Tamada Y, et al. 2015. A flowering integrator, SOC1, affects stomatal opening in Arabidopsis thaliana. Plant Cell Physiol 56: 640–649. [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC. 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–116. [DOI] [PubMed] [Google Scholar]
- Kondrashov AS, Turelli M. 1992. Deleterious mutations, apparent stabilizing selection and the maintenance of quantitative variation. Genetics 132: 603–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Bernier J, Verulkar S, Lafitte HR, Atlin GN. 2008. Breeding for drought tolerance: direct selection for yield, response to selection and use of drought-tolerant donors in upland and lowland-adapted populations. Field Crops Res 107: 221–231. [Google Scholar]
- Lam E, Chua NH. 1991. Tetramer of a 21-base pair synthetic element confers seed expression and transcriptional enhancement in response to water stress and abscisic acid. J Biol Chem 266: 17131–17135. [PubMed] [Google Scholar]
- Landry CR, Wittkopp PJ, Taubes CH, Ranz JM, Clark AG, Hartl DL. 2005. Compensatory cis-trans evolution and the dysregulation of gene expression in interspecific hybrids of Drosophila. Genetics 171: 1813–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasky JR, Des Marais DL, Lowry DB, Povolotskaya I, McKay JK, Richards JH, Keitt TH, Juenger TE. 2014. Natural variation in abiotic stress responsive gene expression and local adaptation to climate in Arabidopsis thaliana. Mol Biol Evol 31: 2283–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebaudy A, Vavasseur A, Hosy E, Dreyer I, Leonhardt N, Thibaud J-B, Véry A-A, Simonneau T, Sentenac H. 2008. Plant adaptation to fluctuating environment and biomass production are strongly dependent on guard cell potassium channels. Proc Natl Acad Sci 105: 5271–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lu K, Chen Z, Mu T, Hu Z, Li X. 2008. Dominance, overdominance and epistasis condition the heterosis in two heterotic rice hybrids. Genetics 180: 1725–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou J, White KP. 2014. RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30: 301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wu H, Chen H, Liu Y, He J, Kang H, Sun Z, Pan G, Wang Q, Hu J, et al. 2015. A gene cluster encoding lectin receptor kinases confers broad-spectrum and durable insect resistance in rice. Nat Biotechnol 33: 301–305. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JT, Juenger TE, Michaels SD, Lasky JR, Platt A, Richards JH, Yu X, Easlon HM, Sen S, McKay JK. 2013. Pleiotropy of FRIGIDA enhances the potential for multivariate adaptation. Proc Biol Sci 280: 20131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JT, Mullen JL, Lowry DB, Awole K, Richards JH, Sen S, Verslues PE, Juenger TE, McKay JK. 2015. Exploiting differential gene expression and epistasis to discover candidate genes for drought-associated QTLs in Arabidopsis thaliana. Plant Cell 27: 969–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB, Purmal CT, Meyer E, Juenger TE. 2012. Microsatellite markers for the native Texas perennial grass, Panicum hallii (Poaceae). Am J Bot 99: e114–e116. [DOI] [PubMed] [Google Scholar]
- Lowry DB, Purmal CT, Juenger TE. 2013. A population genetic transect of Panicum hallii (Poaceae). Am J Bot 100: 592–601. [DOI] [PubMed] [Google Scholar]
- Lowry DB, Hernandez K, Taylor SH, Meyer E, Logan TL, Barry KW, Chapman JA, Rokhsar DS, Schmutz J, Juenger TE. 2015. The genetics of divergence and reproductive isolation between ecotypes of Panicum hallii. New Phytol 205: 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukura S, Mizoi J, Yoshida T, Todaka D, Ito Y, Maruyama K, Shinozaki K, Yamaguchi-Shinozaki K. 2010. Comprehensive analysis of rice DREB2-type genes that encode transcription factors involved in the expression of abiotic stress-responsive genes. Mol Genet Genomics 283: 185–196. [DOI] [PubMed] [Google Scholar]
- McKay JK, Richards JH, Mitchell-Olds T. 2003. Genetics of drought adaptation in Arabidopsis thaliana: I. Pleiotropy contributes to genetic correlations among ecological traits. Mol Ecol 12: 1137–1151. [DOI] [PubMed] [Google Scholar]
- McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, Wittkopp PJ. 2010. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20: 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan C. 1959. The role of ecotypic variation in the distribution of the central grassland of North America. Ecol Monogr 29: 285–308. [Google Scholar]
- Mitchell A, Chang H-Y, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, et al. 2015. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res 43: D213–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. 2012. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim Biophys Acta 1819: 86–96. [DOI] [PubMed] [Google Scholar]
- Narusaka Y, Nakashima K, Shinwari ZK, Sakuma Y, Furihata T, Abe H, Narusaka M, Shinozaki K, Yamaguchi-Shinozaki K. 2003. Interaction between two cis-acting elements, ABRE and DRE, in ABA-dependent expression of Arabidopsis rd29A gene in response to dehydration and high-salinity stresses. Plant J 34: 137–148. [DOI] [PubMed] [Google Scholar]
- Paschold A, Jia Y, Marcon C, Lund S, Larson NB, Yeh C-T, Ossowski S, Lanz C, Nettleton D, Schnable PS, et al. 2012. Complementation contributes to transcriptome complexity in maize (Zea mays L.) hybrids relative to their inbred parents. Genome Res 22: 2445–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter CL. 1966. An analysis of variation between upland and lowland switchgrass Panicum virgatum L in central Oklahoma. Ecology 47: 980–992. [Google Scholar]
- Priest DM, Ambrose SJ, Vaistij FE, Elias L, Higgins GS, Ross ARS, Abrams SR, Bowles DJ. 2006. Use of the glucosyltransferase UGT71B6 to disturb abscisic acid homeostasis in Arabidopsis thaliana. Plant J 46: 492–502. [DOI] [PubMed] [Google Scholar]
- Prud'homme B, Gompel N, Carroll SB. 2007. Emerging principles of regulatory evolution. Proc Natl Acad Sci 104: 8605–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanjulu S, Bartels D. 2002. Drought- and desiccation-induced modulation of gene expression in plants. Plant Cell Environ 25: 141–151. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Riboni M, Galbiati M, Tonelli C, Conti L. 2013. GIGANTEA enables drought escape response via abscisic acid-dependent activation of the florigens and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1. Plant Physiol 162: 1706–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, Willis JH. 2007. Plant speciation. Science 317: 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, Archer MA, Wayne RK. 1999. Transgressive segregation, adaptation and speciation. Heredity 83: 363–372. [DOI] [PubMed] [Google Scholar]
- Sakuma Y, Liu Q, Dubouzet JG, Abe H, Shinozaki K, Yamaguchi-Shinozaki K. 2002. DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem Biophys Res Commun 290: 998–1009. [DOI] [PubMed] [Google Scholar]
- Sakuma Y, Maruyama K, Osakabe Y, Qin F, Seki M, Shinozaki K, Yamaguchi-Shinozaki K. 2006. Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. Plant Cell 18: 1292–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefke B, Emerson JJ, Wang TY, Lu MYJ, Hsieh LC, Li WH. 2013. Inheritance of gene expression level and selective constraints on trans- and cis-regulatory changes in yeast. Mol Biol Evol 30: 2121–2133. [DOI] [PubMed] [Google Scholar]
- Stapley J, Reger J, Feulner PGD, Smadja C, Galindo J, Ekblom R, Bennison C, Ball AD, Beckerman AP, Slate J. 2010. Adaptation genomics: The next generation. Trends Ecol Evol 25: 705–712. [DOI] [PubMed] [Google Scholar]
- Stebbins GL. 1950. Variation and evolution in flowering plants. Columbia University Press, New York. [Google Scholar]
- Stebbins GL Jr. 1952. Aridity as a stimulus to plant evolution. Am Nat 86: 33–44. [Google Scholar]
- Storey JD. 2002. A direct approach to false discovery rates. J R Stat Soc Ser B Stat Methodol 64: 479–498. [Google Scholar]
- Storey JD, Taylor JE, Siegmund D. 2004. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: A unified approach. J R Stat Soc Ser B Stat Methodol 66: 187–205. [Google Scholar]
- Stupar RM, Springer NM. 2006. Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 173: 2199–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisné S, Schmalenbach I, Reymond M, Dauzat M, Pervent M, Vile D, Granier C. 2010. Keep on growing under drought: genetic and developmental bases of the response of rosette area using a recombinant inbred line population. Plant Cell Environ 33: 1875–1887. [DOI] [PubMed] [Google Scholar]
- Tiwari M, Sharma D, Singh M, Tripathi RD, Trivedi PK. 2014. Expression of OsMATE1 and OsMATE2 alters development, stress responses and pathogen susceptibility in Arabidopsis. Sci Rep 4 3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognetti VB, Van Aken O, Morreel K, Vandenbroucke K, van de Cotte B, De Clercq I, Chiwocha S, Fenske R, Prinsen E, Boerjan W, et al. 2010. Perturbation of indole-3-butyric acid homeostasis by the UDP-glucosyltransferase UGT74E2 modulates Arabidopsis architecture and water stress tolerance. Plant Cell 22: 2660–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GP, Lynch VJ. 2008. The gene regulatory logic of transcription factor evolution. Trends Ecol Evol 23: 377–385. [DOI] [PubMed] [Google Scholar]
- Waller FR. 1976. “A biosystematic study of Panicum section Diffusa (Poaceae) in North America.” PhD thesis, Texas A&M University. [Google Scholar]
- Wei G, Pan Y, Lei J, Zhu YX. 2005. Molecular cloning, phylogenetic analysis, expressional profiling and in vitro studies of TINY2 from Arabidopsis thaliana. J Biochem Mol Biol 38: 440–446. [DOI] [PubMed] [Google Scholar]
- Wilczek AM, Roe JL, Knapp MC, Cooper MD, Lopez-Gallego C, Martin LJ, Muir CD, Sim S, Walker A, Anderson J, et al. 2009. Effects of genetic perturbation on seasonal life history plasticity. Science 323: 930–934. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Kalay G. 2011. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet 13: 59–69. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. 2004. Evolutionary changes in cis and trans gene regulation. Nature 430: 85–88. [DOI] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA. 2003. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol 20: 1377–1419. [DOI] [PubMed] [Google Scholar]
- Wu TD, Nacu S. 2010. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xianjun P, Xingyong M, Weihong F, Man S, Liqin C, Alam I, Lee B-H, Dongmei Q, Shihua S, Gongshe L. 2011. Improved drought and salt tolerance of Arabidopsis thaliana by transgenic expression of a novel DREB gene from Leymus chinensis. Plant Cell Rep 30: 1493–1502. [DOI] [PubMed] [Google Scholar]
- Yu LX, Nguyen HT. 1994. Genetic-variation detected with RAPD markers among upland and lowland rice cultivars (Oryza sativa L). Theor Appl Genet 87: 668–672. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.