

Abstract
Objective:
To report atypical MRI patterns associated with POLR3A and POLR3B mutations.
Methods:
This was a multicenter retrospective study to collect neuroradiologic, clinical, and molecular data of patients with mutations in POLR3A and POLR3B without the classic MRI phenotype, i.e., diffuse hypomyelination associated with relative T2 hypointensity of the ventrolateral thalamus, globus pallidus, optic radiation, corticospinal tract at the level of the internal capsule, and dentate nucleus, cerebellar atrophy, and thinning of the corpus callosum.
Results:
Eight patients were identified: 6 carried mutations in POLR3A and 2 in POLR3B. We identified 2 novel MRI patterns: 4 participants presented a selective involvement of the corticospinal tracts, specifically at the level of the posterior limbs of the internal capsules; 4 patients presented moderate to severe cerebellar atrophy. Incomplete hypomyelination was observed in 5 participants.
Conclusion:
Diffuse hypomyelination is not an obligatory feature of POLR3-related disorders. Two distinct patterns, selective involvement of the corticospinal tracts and cerebellar atrophy, are added to the MRI presentation of POLR3-related disorders.
POLR3-related leukodystrophy is a rare autosomal recessive disease characterized by hypomyelination often accompanied by dental abnormalities and hypogonadotropic hypogonadism.1–5 In its classical form, the association of these features is referred to as 4H syndrome.1,2 Mutations in the POLR3A and POLR3B genes, which encode for the 2 largest subunits of the RNA polymerase III (POLR3) complex, as well as in POLR1C, also encoding a POLR3 subunit, are responsible for this disease.6–11 With the identification of the causative genes, patients with suggestive clinical or MRI picture can undergo genetic testing, confirming the diagnosis.12 The MRI pattern of POLR3-related leukodystrophy is suggestive and characterized by diffuse hypomyelination associated with relative T2 hypointensity of the ventrolateral thalamus, globus pallidus, optic radiation, corticospinal tract at the level of the internal capsule and dentate nucleus, cerebellar atrophy, and thinning of the corpus callosum.12–14 Recognition of this pattern was proven effective in detecting patients with 4H leukodystrophy caused by POLR3A-B or POLR1C mutations and is therefore used to orient the diagnostic process.12–14 Patients without this pattern showing nonspecific hypomyelination are unlikely to carry mutations in POLR3A or POLR3B.15
METHODS
We performed a multi-institutional cross-sectional observational study of the clinical, radiologic, and molecular data of patients who fulfilled the following inclusion criteria: presence of recessive POLR3A or POLR3B mutations and absence of typical POLR3-related MRI features.13,14
We identified 8 patients from 7 nonconsanguineous families, all of Caucasian ethnicity, fulfilling these criteria. Five participants underwent POLR3A and POLR3B sequencing because they presented suggestive clinical features (hypodontia or dental abnormalities, short stature, and myopia, either associated or not). In 3 participants from 2 families, POLR3A and POLR3B mutations were identified by whole-exome sequencing (WES). For all families for which DNA was available, segregation was verified. Analysis for the potential pathogenicity of novel mutations was performed, including in silico analysis. For the novel splicing mutations, we sequenced cDNA when RNA was available.
MRI were reviewed collegially by our team. At least axial T2-weighted and sagittal T1- or T2-weighted images were available.
Standard protocol approvals, registrations, and patient consents.
The institutional review boards of each participating institution approved the use of clinical data for the study.
RESULTS
Mutation findings.
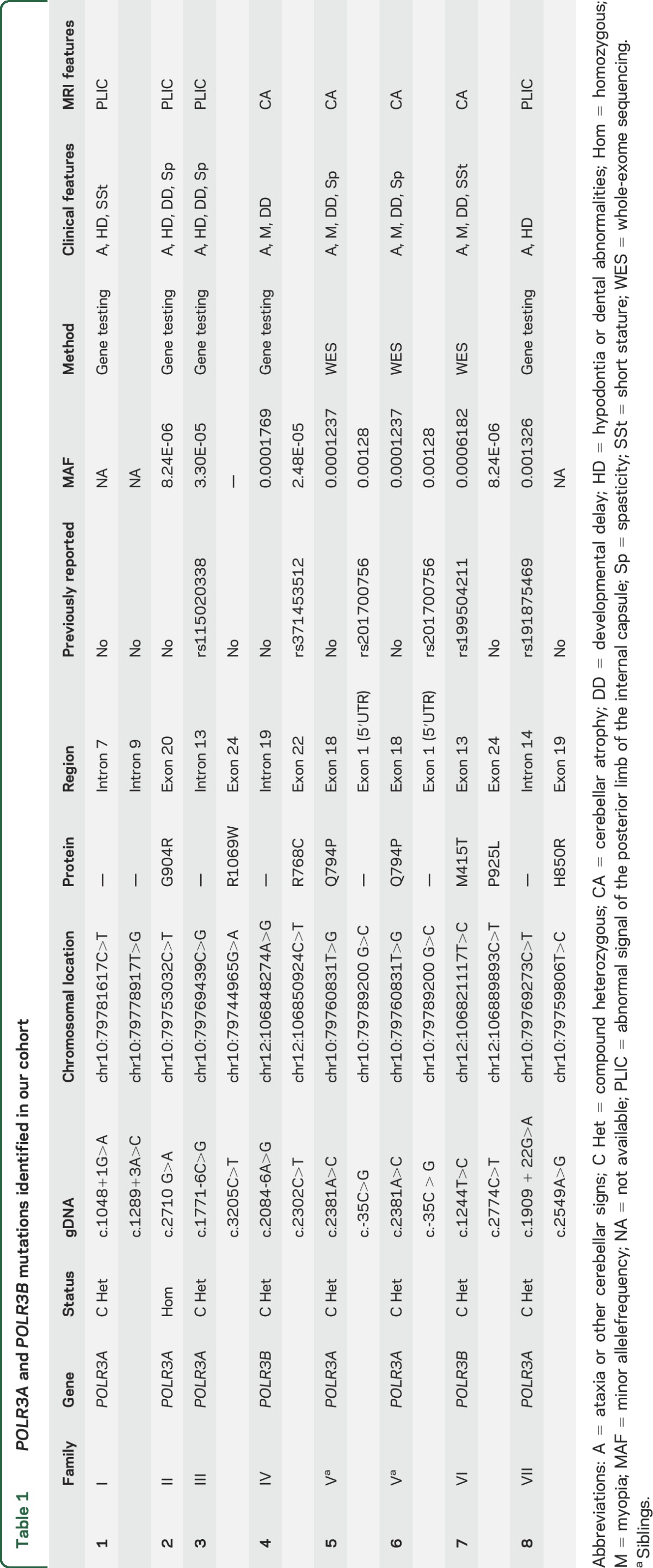
Six participants carried mutations in POLR3A and 2 in POLR3B (table 1). Among the 13 mutations identified, 8 were novel, and not reported in public databases (Exome Variant Server, NHLBI GO Exome Sequencing Project [ESP], Seattle, WA [http://evs.gs.washington.edu/EVS/] [June 2015]; Exome Aggregation Consortium [ExAC], Cambridge, MA [http://exac.broadinstitute.org] [June 2015]). Five mutations were intronic and affected a splice site or induced a new donor site. All new mutations were rare and predicted to be pathogenic by in silico tools16–18 (table e-1 on the Neurology® Web site at Neurology.org) besides for the c.-35C>G change, which had been reported in homozygous state in 2 out of over 60,000 participants (ExAC [http://exac.broadinstitute.org] [July 2015]) in patients 5 and 6. Segregation analysis in this family revealed that each of the parents carried one variant, and the healthy brother the paternal variant. WES failed to uncover other possible causal variants. Table 1 reports detailed information about the genetic status and mutations found in our cohort.
Table 1.
POLR3A and POLR3B mutations identified in our cohort
Clinical findings.
Age at onset ranged from 6 weeks to 10 years (mean age at onset 52.3 months). The symptoms at disease onset were gait ataxia, dysarthria, and tremor in 3 participants (cases 1, 4, and 8). Two participants (cases 2 and 3) presented with spasticity and diplegic gait. The patient with the earliest disease onset, at 1.5 months, presented with failure to thrive (case 7). Clinical examination revealed ataxia of variable severity in all patients; cerebellar tremor was documented in 4 participants (cases 1, 2, 3, and 4); pyramidal signs and spasticity were present in 4 participants (cases 2, 3, 5, and 6). One patient had severe dystonic tremor (case 3) (table 1).
Extraneurologic features were found in 6 patients. Specifically, 4 participants had hypodontia, delayed dentition, or other dental abnormalities (cases 1, 2, 3, and 8) suggestive of POLR3-related leukodystrophy. Two participants presented short stature (cases 1 and 7), and 4 had myopia (cases 4, 5, 6, and 7), both frequent findings in patients with mutations in POLR3A or POLR3B.
MRI findings.
Mean age at the last MRI was 15 years (range 4–35 years). We identified 2 distinct MRI patterns (figures 1, 2, e-1, and e-2). Four participants, all with POLR3A mutations, presented a selective involvement of the corticospinal tracts, which was particularly evident at the level of the posterior limb of the internal capsule as T2-hyperintense signal (cases 1, 2, 3, and 8) (figures 1 and e-1). In the other 4 patients (2 with POLR3A and 2 with POLR3B mutations), moderate to severe cerebellar atrophy was variably associated with nonspecific T2-hyperintense white matter abnormalities, as specified below, or thinning of the corpus callosum (figures 2 and e-2) (cases 4, 5, 6, and 7). We documented focal, partially confluent, T2-hyperintense white matter abnormalities located in the deep frontal and parietal white matter, suggesting partial hypomyelination, in 5 participants (cases 2, 3, 4, 5, and 6), while the remaining 3 presented adequate myelination for age.
Figure 1. Involvement of the corticospinal tracts.
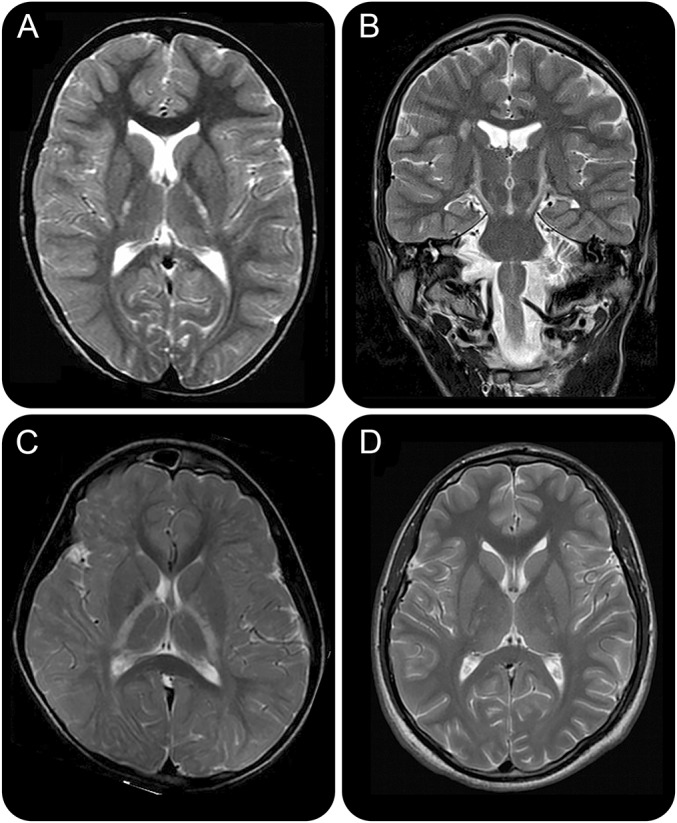
Axial (A, C, D) and coronal (B) T2-weighted images in patients 1 (A, B), 1 (C), and 8 (D) with POLR3A mutations show the presence of bilateral and symmetric T2-hyperintense signal at the level of the posterior limb of the internal capsules. Incomplete hypomyelination is seen in (B).
Figure 2. Cerebellar atrophy.
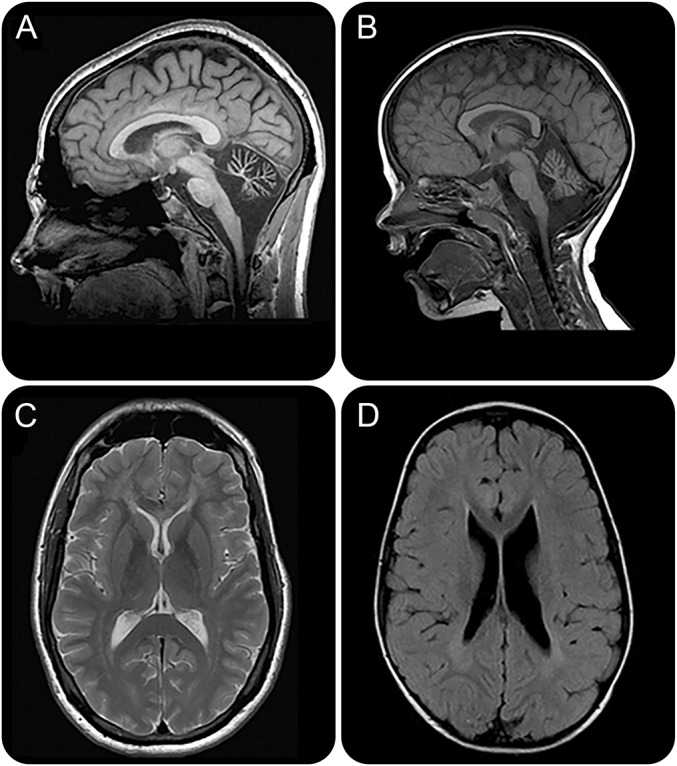
(A, B) Sagittal T1-weighted images from participant 4 with POLR3B (A) and participant 5 with POLR3A (B) mutations show the presence of severe (A) and moderate cerebellar atrophy. (C, D) Axial T2-weighted and T2–fluid-attenuated inversion recovery images of the patients presented respectively in (A, B) show the presence of partial hypomyelination.
DISCUSSION
Our work broadens the MRI spectrum of POLR3-related leukodystrophy by describing 2 new MRI patterns in this disease that has been known as a hypomyelinating disorder. Our results indicate that diffuse hypomyelination is not an obligatory feature. We also documented the presence of 6 POLR3A and 2 POLR3B mutations not reported before in public databases. Interestingly, 5 of the 13 mutations in our cohort were noncoding, 4 predicted to affect splicing. Pathogenicity of the variant in the 5′ untranslated region in patients 5 and 6 could not be unambiguously resolved as it had been reported in homozygous state in 2 participants in a large database. However, this variant is situated in a terminal oligopyrimidine tract (TOP). The change of a pyrimidine (C) for a purine (G) shortens the TOP, and it has been shown that deletions or substitutions in this region result in unregulated translation.19,20 Its effect might be mild, and homozygous carriers indeed might be unaffected, but in combination with a pathogenic mutation this variant could lead to disease. Segregation analysis and the absence of other possible causal variants in WES in this family indeed support a causal role for this variant, as does the identification of other families with isolated cerebellar atrophy.
A specific involvement of the corticospinal tracts, particularly evident at the level of the internal capsule as T2-hyperintense signal, was the most striking finding in a subgroup of patients. Interestingly, in typical cases, more commonly with POLR3B mutations,12 the corticospinal tracts are usually one of the better myelinated structures.
The second pattern is the presence of cerebellar atrophy in the absence of diffuse hypomyelination. Cerebellar atrophy was previously known to be associated with POLR3A or POLR3B mutations in more than 80% of the participants, always in combination with diffuse hypomyelination.12–14,21
Focal, partly confluent T2-hyperintense white matter changes were present in some participants of both groups and located in the deep frontal and parietal white matter. The signal intensity of the abnormal areas corresponds to the one seen in hypomyelination, focal hypomyelination therefore being the most likely interpretation. These changes are obviously reminiscent of the classical MRI of 4H leukodystrophy, but sufficiently different to make a straightforward diagnosis challenging. Our results confirm white matter involvement when POLR3A or POLR3B is mutated; however, different pathogenic processes could be responsible for the variable and expanded MRI phenotypes of brain abnormalities associated with POLR3A or POLR3B mutations. Further insight into the role of POLR3 in myelin formation and maintenance as well as in axonal integrity is needed to explain the heterogeneity of the radiologic patterns.
The diagnosis of POLR3-related leukodystrophy relies both on MRI findings and clinical signs. In our cohort, the presence of typical extraneurologic features oriented the clinicians towards the testing of POLR3A and POLR3B genes in 5 participants. Therefore, our study confirms the importance of the classical clinical criteria—particularly hypodontia and hypogonadotropic hypogonadism—in the diagnostic process of POLR3-related disorders, especially when cardinal MRI features are lacking. WES allowed the discovery of POLR3A and POLR3B mutations in the remaining cases, thus highlighting the role of next-generation sequencing in expanding the phenotypes of already known disorders.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants and their families for their support and cooperation; and the McGill University and Genome Quebec Innovation Center.
GLOSSARY
- ExAC
Exome Aggregation Consortium
- TOP
terminal oligopyrimidine tract
- WES
whole-exome sequencing
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Roberta La Piana collected, analyzed, and interpreted patient data, reviewed the article, and wrote the draft of the manuscript. Ferdy K. Cayami collected, analyzed, and interpreted patient data, reviewed the article, and wrote the draft of the manuscript. Luan T. Tran collected, analyzed, and interpreted patient data and reviewed the article. Kether Guerrero was responsible for molecular genetic analysis and reviewed the article. Rosalina van Spaendonk was responsible for molecular genetic analysis and reviewed the article. Katrin Õunap collected, analyzed, and interpreted patient data and reviewed the article. Sander Pajusalu collected, analyzed, and interpreted patient data, reviewed the article. Tobias Haack was responsible for molecular genetic analysis, reviewed the article. Evangeline Wassmer collected, analyzed, and interpreted patient data and reviewed the article. Dagmar Timmann collected, analyzed, and interpreted patient data and reviewed the article. Hanna Mierzewska collected, analyzed, and interpreted patient data and reviewed the article. Bwee T. Poll-Thé collected, analyzed, and interpreted patient data and reviewed the article. Chirag Patel collected, analyzed, and interpreted patient data and reviewed the article. Helen Cox collected, analyzed, and interpreted patient data and reviewed the article. Tahir Atik collected, analyzed, and interpreted patient data and reviewed the article. Huseyin Onay collected, analyzed, and interpreted patient data and reviewed the article. Ferda Ozkınay collected, analyzed, and interpreted patient data and reviewed the article. Adeline Vanderver collected, analyzed, and interpreted patient data and reviewed the article. Marjo S. van der Knaap collected, analyzed, and interpreted patient data and reviewed the article. Nicole I. Wolf designed and supervised the study, collected data, and reviewed the article. Genevieve Bernard designed and supervised the study, collected data, and reviewed the article.
STUDY FUNDING
R.L.P. has received a Doctoral Award from the Fonds de recherche du Québec–Santé (FRQS) and has been supported by the Fondation Monaco. F.K.C. is a recipient of the Directorate of Higher Education Overseas Scholarship–Dikti Scholarship, Ministry of National Education, Republic of Indonesia. G.B. has received a Research Scholar Junior 1 award from the Fonds de Recherche du Québec en Santé (FRQS). N.I.W. and M.S.v.d.K. are supported by ZonMw (TOP grant 91211005). This work was supported by Canadian Institute for Health Research grant MOP-G-287547 and the Réseau de Médecine Génétique Appliquée du FRQS to G.B. T.H. was supported by the BMBF through the Juniorverbund in der Systemmedizin “mitOmics” (FKZ 01ZX1405C).
DISCLOSURE
R. La Piana, F. Cayami, L.T. Tran, K. Guerrero, R. van Spaendonk, K. Ounap, S. Pajusalu, and T. Haack report no disclosures relevant to the manuscript. E. Wassmer received honoraria for speaking/travel costs from BiogenIdec, Teva, Genzyme, Shire, UCB, and Merck Serono. She is an investigator in trials with BiogenIdec, Sanofi, and Novartis. Her MS research projects have been funded by the UK MS Society, Action. D. Timmann sits on the editorial board of The Cerebellum and Cerebellum & Ataxias (unpaid positions). H. Mierzewska, B. Poll-The, C. Patel, H. Cox, T. Atik, H. Onay, and F. Ozkinay report no disclosures relevant to the manuscript. A. Vanderver has acted on an advisory board for Shire Pharmaceuticals as well as an unpaid consultant for Stem Cells Inc. M.S. van der Knaap reports no disclosures relevant to the manuscript. N.I. Wolf serves as communicating editor of the Journal of Inherited Metabolic Disease and as editor for Neuropediatrics, and is a member of the editorial board of Neurology®, all without payment. G. Bernard has received compensation from Actelion Pharmaceuticals, Genzyme, Shire, and Santhera Pharmaceuticals (speaker's honoraria and for serving on scientific advisory boards). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Wolf NI, Harting I, Boltshauser E, et al. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology 2005;64:1461–1464. [DOI] [PubMed] [Google Scholar]
- 2.Wolf NI, Harting I, Innes AM, et al. Ataxia, delayed dentition and hypomyelination: a novel leukoencephalopathy. Neuropediatrics 2007;38:64–70. [DOI] [PubMed] [Google Scholar]
- 3.Bernard G, Thiffault I, Tetreault M, et al. Tremor-ataxia with central hypomyelination (TACH) leukodystrophy maps to chromosome 10q22.3-10q23.31. Neurogenetics 2010;11:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atrouni S, Daraze A, Tamraz J, Cassia A, Caillaud C, Megarbane A. Leukodystrophy associated with oligodontia in a large inbred family: fortuitous association or new entity? Am J Medgeneta 2003;118A:76–81. [DOI] [PubMed] [Google Scholar]
- 5.Sasaki M, Takanashi J, Tada H, Sakuma H, Furushima W, Sato N. Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 2009;31:582–587. [DOI] [PubMed] [Google Scholar]
- 6.Daoud H, Tetreault M, Gibson W, et al. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J Medical Genetics 2013;50:194–197. [DOI] [PubMed] [Google Scholar]
- 7.Gutierrez M, Thiffault I, Guerrero K, et al. Large exonic deletions in POLR3B gene cause POLR3-related leukodystrophy. Orphanet Journal Rare Diseases 2015;10:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernard G, Chouery E, Putorti ML, et al. Mutations of POLR3A encoding a Catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet 2011;89:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tetreault M, Choquet K, Orcesi S, et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am Journal Human Genetics 2011;89:652–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saitsu H, Osaka H, Sasaki M, et al. Mutations in POLR3A and POLR3B encoding RNA Polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am Journal Human Genetics 2011;89:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiffault I, Wolf NI, Forget D, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Communications 2015;6:7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf NI, Vanderver A, van Spaendonk RM, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014;83:1898–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010;133:2971–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.La Piana R, Tonduti D, Gordish Dressman H, et al. Brain magnetic resonance imaging (MRI) pattern recognition in Pol III-related leukodystrophies. J Child Neurol 2014;29:214–220. [DOI] [PubMed] [Google Scholar]
- 15.Cayami FK, La Piana R, van Spaendonk RM, et al. Polr3a and POLR3B mutations in Unclassified hypomyelination. Neuropediatrics 2015;46:221–228. [DOI] [PubMed] [Google Scholar]
- 16.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361–362. [DOI] [PubMed] [Google Scholar]
- 17.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Research 2001;11:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardinali B, Di Cristina M, Pierandrei-Amaldi P. Interaction of proteins with the mRNA for ribosomal protein L1 in Xenopus: structural characterization of in vivo complexes and identification of proteins that bind in vitro to its 5'UTR. Nucleic Acids Research 1993;21:2301–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaspar RL, Kakegawa T, Cranston H, Morris DR, White MW. A regulatory cis element and a specific binding factor involved in the mitogenic control of murine ribosomal protein L32 translation. J Biol Chem 1992;267:508–514. [PubMed] [Google Scholar]
- 21.Takanashi J, Osaka H, Saitsu H, et al. Different patterns of cerebellar abnormality and hypomyelination between POLR3A and POLR3B mutations. Brain Development 2014;36:259–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.