

Summary
Oxidative stress and telomere attrition are considered the driving factors of aging. As oxidative damage to telomeric DNA favors the erosion of chromosome ends and, in turn, telomere shortening increases the sensitivity to pro‐oxidants, these two factors may trigger a detrimental vicious cycle. To check whether limiting oxidative stress slows down telomere shortening and related progeria, we have investigated the effect of p66SHC deletion, which has been shown to reduce oxidative stress and mitochondrial apoptosis, on late‐generation TERC (telomerase RNA component)‐deficient mice having short telomeres and reduced lifespan. Double mutant (TERC −/− p66SHC −/−) mice were generated, and their telomere length, fertility, and lifespan investigated in different generations. Results revealed that p66SHC deletion partially rescues sterility and weight loss, as well as organ atrophy, of TERC‐deficient mice, but not their short lifespan and telomere erosion.
Therefore, our data suggest that p66SHC‐mediated oxidative stress and telomere shortening synergize in some tissues (including testes) to accelerate aging; however, early mortality of late‐generation mice seems to be independent of any link between p66SHC‐mediated oxidative stress and telomere attrition.
Keywords: fertility, lifespan, oxidative DNA damage, telo‐meres
Introduction
Telomeres are conserved repetitive DNA sequences at the ends of chromatids that protect from chromosomal rearrangements and loss of genetic information (Blackburn et al., 2006). Shortening of telomeres occurs in somatic cells during aging (Daniali et al., 2013) due to DNA damage and incomplete end processing (von Zglinicki et al., 2000). Above a threshold, telomere erosion results in the arrest of cellular proliferation and the dysfunction of renewable tissues. For this reason, telomerase activity and recombination‐based processes that maintain telomere length are critical for tissue homeostasis (Sahin & Depinho, 2010). In fact, mutations in either the gene encoding the telomerase catalytic subunit (telomerase reverse transcriptase, TERT), or the telomerase RNA gene (TERC) are found in patients with dyskeratosis congenita who have short telomeres and show accelerated aging and reduced lifespan (Kirwan & Dokal, 2009). Consistently, late‐generation (G3 and beyond) TERT (Rudolph et al., 1999) or TERC‐deficient mice (TERT−/− and TERC−/−, respectively) are short living and show reduced fertility, early alopecia, kyphosis, anemia, and lymphopenia (Wong et al., 2003). Interestingly, TERC−/− mice present altered mitochondrial functions, including increased production of reactive oxygen species (ROS) from the electron transfer chain (ETC) (Passos et al., 2010; Sahin et al., 2011). Thereby, telomeres have been suggested to play a role in controlling mitochondrial ROS accumulation and oxidative stress during aging (Sahin & Depinho, 2010).
As telomere DNA is particularly sensitive to oxidative damage (Oikawa & Kawanishi, 1999) and single‐strand breaks or other stress‐induced lesions at telomeres are inefficiently repaired (Petersen et al., 1998), oxidative stress is considered to accelerate telomere erosion (von Zglinicki, 2002; Houben et al., 2008). Consistently, mice expressing a variant of the mitochondrial uncoupling protein 2 that increases ROS production by ETC, and therefore, oxidative stress shows short telomeres (Salpea et al., 2010). Moreover, it has been shown that treating mouse embryos with chemicals able to induce mitochondrial dysfunction and ROS accumulation, led to telomere loss and chromosomal instability, which was prevented by a concomitant treatment with N‐acetylcysteine, a compound that improves ROS scavenging (Liu et al., 2002). However, other mutations in genes involved in ROS metabolism, such as SOD2 haploinsufficiency, do not cooperate with telomere dysfunction (Guachalla et al., 2009), and the treatment with N‐acetylcysteine does not rescue progeric phenotypes of late‐generation TERT‐deficient mice (Sahin et al., 2011).
P66SHC protein is the largest isoform encoded by the ShcA locus, typical of vertebrates, and it sustains intracellular levels of ROS (Giorgio et al., 2005 Gertz & Steegborn, 2010) and regulates redox signaling pathways (Frijhoff et al., 2014), mitochondrial apoptosis (Migliaccio et al., 2006), and aging (Trinei et al., 2009). P66SHC null mice (p66SHC−/−) develop normally and resulted to be protected from aging‐associated diseases, such as atherosclerosis (Martin‐Padura et al., 2008), diabetes compliances (Menini et al., 2007), onset (Tomilov et al., 2011), cognitive decline (Berry et al., 2007), and neurodegeneration (Savino et al., 2013). Moreover, p66SHC null cells were shown to be resistant to apoptosis induced by a variety of different signals, including hydrogen peroxide, UV, staurosporine, taxol, growth factor deprivation, calcium ionophore, osmotic shock, and CD3–CD4 cross‐linking (as reviewed in Migliaccio et al., 2006). Similarly, different tissues of p66SHC−/− mice were found to be resistant to apoptosis induced by paraquat (Migliaccio et al., 1999), hypercholesterolemia (Napoli et al., 2003), hyperglycemia (Menini et al., 2007), immunotoxicity (Su et al., 2012), and ischemia/reperfusion injury (Carpi et al., 2009). At mechanistic level, substantial evidence indicates that p66SHC regulates redox balance and mitochondrial apoptosis by suppressing ROS scavenging and increasing ROS production from plasma membrane oxidases and mitochondria where, in particular, p66SHC favors H2O2 production by ETC (Gertz & Steegborn, 2010; Trinei et al., 2013). Accordingly, p66SHC−/− tissues showed a reduced level of oxidative damage to nuclear and mitochondrial DNA (Trinei et al., 2002), lipids (Napoli et al., 2003), carbohydrates (Menini et al., 2007), and proteins (Carpi et al., 2009), which can be either unspecifically or specifically (e.g., redox‐regulated phosphatases; Frijhoff et al., 2014) damaged by oxidative stress.
To investigate the link between endogenously generated oxidative stress, telomeres, and aging, we have investigated the effect of p66SHC deletion on late‐generation TERC‐deficient mice, by generating and studying double mutants TERC−/− P66SHC−/− mice.
Results
The deletion of p66SHC improves fertility of late‐generation TERC−/− mice
At first, we backcrossed TERC+/− male with C57Bl6/J wild‐type (WT) female mice to generate TERC+/− mice in pure C57Bl/6 background (100% C57BL/6 according to the Marker‐Assisted Accelerated Backcrossing, MAX‐BAX®, analysis). These mice were bred together to generate TERC−/− (p66SHC+/+) mice that were bred successfully, by crossing mice of the same generation, up to the fifth generation (G5), as already reported (Rudolph et al., 1999).
Then, we crossed C57Bl/6J TERC+/− female with pure C57Bl/6J p66SHC−/− male mice and selected TERC+/− p66SHC+/− double heterozygous mice among the resulting litters. These mice were then crossed together and litters genotyped for TERC and p66SHC mutations (Fig. 1A). The resulting TERC−/− p66SHC+/− mice were selected as the first generation (G1) of TERC null mice and were bred together to segregate into three genotypes: p66SHC−/−, p66SHC+/−, and p66SHC+/+ (Table 1). In this way, we obtained G2 TERC−/− p66SHC+/+ and G2 TERC−/− p66SHC−/− mice that were propagated as distinct colonies by breeding mice from the same generations (Fig. 1B). As observed for the other colonies of TERC−/− mice, at G5 TERC−/− p66SHC+/+ crosses were not fertile, as well as the crosses of males (or females) G5 TERC−/− p66SHC+/+ with WT female (or male) mice. On the opposite, half of the crosses of G5 and also approximately a third of the G6 TERC−/− p66SHC−/− mice were fertile, and thus, it was possible to generate G7 TERC−/− p66SHC−/− mice (Fig. 2A).
Figure 1.

Genealogy of TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice. (A) PCR genotype analysis for TERC (left panel) and p66SHC mutation (right panel). (B) Scheme of the crosses performed to generate the different generations of TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice.
Table 1.
Frequency of p66SHC+/+, p66SHC+/−, and p66SHC−/− genotypes in the progeny obtained crossing G1 TERC−/− p66SHC+/− mice
| GENOTYPE | Number | Males | Females | Expected (%) | Observed (%) |
|---|---|---|---|---|---|
| TERC−/−P66SHC+/− | 35 | 18 | 17 | 50 | 57.4 |
| TERC−/−P66SHC+/+ | 10 | 6 | 4 | 25 | 16.4 |
| TERC−/−P66SHC−/− | 16 | 8 | 8 | 25 | 26.2 |
| Total | 61 | 32 | 29 | 100 | 100 |
Figure 2.
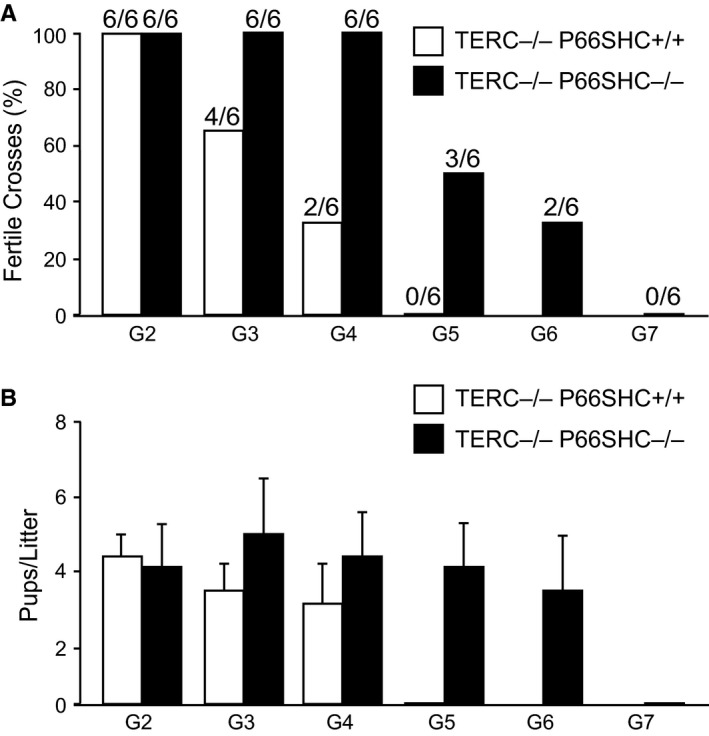
Fertility of different generations of TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice. (A) Percentage of productive crosses along generations (n = 6 couples per genotype per generation). (B) Average number ±SD of pups per litter along generations.
The average number of pups per dam was comparable between G2 TERC−/− p66SHC+/+ and G2 TERC−/− p66SHC−/− mice, as well as between WT and p66SHC−/− animals (Giorgio et al., 2012). At G3, TERC−/− p66SHC+/+, but not TERC−/− p66SHC−/−, mice showed a significant fertility reduction. Only, at G5, we observed a decrease in litter size of TERC−/− p66SHC−/− mice (Fig. 2B).
Furthermore, we crossed G3 TERC−/− p66SHC+/+ females with TERC−/− p66SHC−/− male mice to produce G4 TERC−/− p66SHC+/− mice. By crossing these mice, we could segregate other G5 TERC−/− p66SHC+/+ animals. Interestingly, one of four crosses of these G5 TERC−/− p66SHC+/+ was fertile and two males of four of the resulting G6 TERC−/− p66SHC+/+ litter were able to generate offspring when crossed with WT females.
These data clearly suggest that p66SHC deletion improves fertility of late‐generation TERC−/− mice.
The deletion of p66SHC prevents DNA oxidative damage but not telomere shortening in TERC−/− mice
Eight‐hydroxy‐2‐deoxy guanosine (8‐OH‐dG) and isoprostanes are recurrent oxidized DNA base and fatty acids products, respectively, that marks oxidative stress to DNA and lipids and were found reduced in p66SHC−/− mice (Trinei et al., 2002; Napoli et al., 2003; Lunghi et al., 2015). We have measured the content of 8‐OH‐dG in the genomic DNA extracted from liver, spleen, lung, and testis of G0 and G3; and liver and spleen of G5 TERC−/− p66SHC+/+ or TERC−/− p66SHC−/− 6‐month‐old mice using a competitive ELISA assay with specific anti‐8‐OH‐dG antibody and of 8‐iso‐PGF2α (8‐isoprostane) by immunohistochemistry analysis of tissue sections from testis, lung, and liver of G0 and G3, TERC−/− p66SHC+/+, and TERC−/− p66SHC−/− mice. Results showed that the deletion of p66SHC reduced the amount of 8‐OH‐dG (Fig. 3A) and of 8‐isoprostane (Fig. 3B) in TERC−/− genetic background as well.
Figure 3.
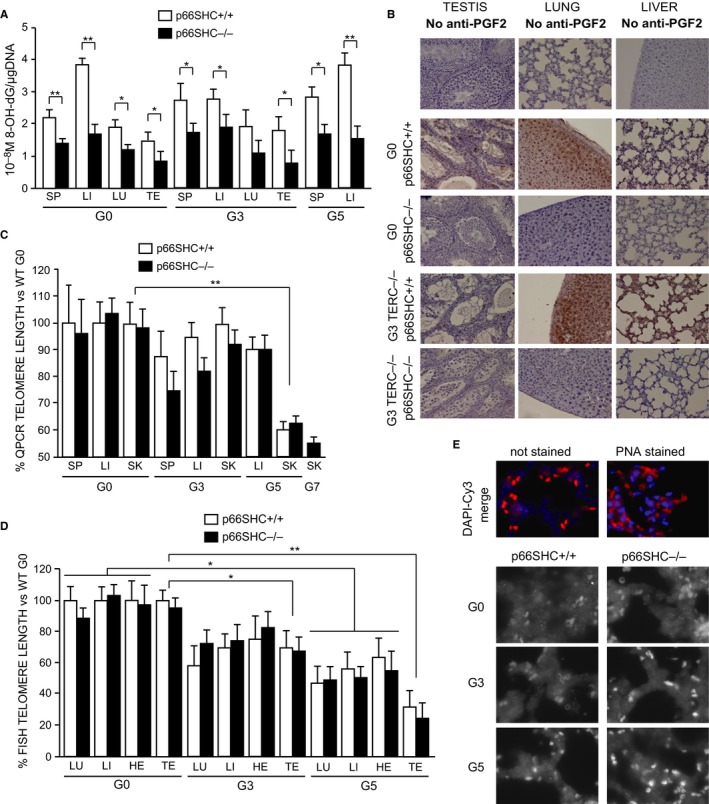
Oxidative stress and telomere length in TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice. (A) Levels of 8‐OH‐dG per μg of DNA in TERC −/− p66SHC +/+ (white bars) and TERC −/− p66SHC −/− (black bars) spleen (SP), liver (LI), lung (LU), and testis (TE) measured by competitive enzyme immunoassay. Average values ±SD from n = 5 individuals for the G0 group and n = 3 individuals for the G3 and G5 groups are reported: * for P < 0.05 and ** for P < 0.01. (B) 8‐isoprostane IHC representative images of testis, lung, and liver from G0 and G3 TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice. Level of background signal is shown in the first three unstained slices. (C and D) Telomere length (expressed as a percentage versus the telomere length of C57Bl/6J WT mice) in spleen (SP), liver (LI), lung (LU), heart (HE), testis (TE) and skin (SK) from TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice of different generation as evaluated by qPCR (C) where the average values ±SD from n = 3 individuals per group, measured in quadruplicates, are reported or Q‐FISH (D) where the average values ±SD from n = 3 individuals per group, each sample evaluated from two different sections for a total of around 40 nuclei, are reported. * for P < 0.05, ** for P < 0.01. (E) Representative FISH images (Cy3 alone grayscale and DAPI‐Cy3 merged color) obtained from the lung of different generation TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice.
To determine telomere length, we performed qPCR analysis of telomeric sequences in genomic DNA extracted from skin, spleen, and liver of 6‐month‐old G3–G5 TERC−/− p66SHC+/+, G3‐G7 TERC−/− p66SHC−/−, and WT mice (Fig. 3C), and quantitative fluorescent in situ hybridization (Q‐FISH) analysis on tissue sections from liver, lung, heart, and testis of 6‐month‐old G0, G3, and G5 TERC−/− p66SHC+/+ or TERC−/− p66SHC−/− mice (Figs 3 D and E and S1 in Supporting information). As expected, we observed a progressive telomere shortening in successive generations of TERC−/− mice, particularly in the skin by the qPCR analysis and more evident in all the tissues by Q‐FISH. However, the TERC−/− p66SHC+/+ and TERC−/−p66SHC−/− showed a similar telomere decline, suggesting that p66SHC does not contribute to telomere erosion.
The deletion of p66SHC prevents weight loss and organ shrinkage in late‐generation TERC−/− mice
Along with reduced fertility, starting from G3, C57Bl6/J TERC−/− mice showed signs of premature aging as already reported (Wong et al., 2003; Siegl‐Cachedenier et al., 2007). At first sight, young adult (2–3 months old) TERC−/− p66SHC+/+ mice exhibited alopecia on the back and kyphosis and were of smaller size compared to G3 TERC−/− p66SHC−/− mice that appeared bigger and healthier (Fig. 4A). At G5, TERC−/− p66SHC+/+ mice were significantly smaller compared to age‐matched TERC−/− p66SHC−/− mice (Fig. 4B). We observed a reduction in the size and weight (Fig. 4C) of a number of organs including liver, spleen, kidneys, and testis, in late‐generation TERC−/− p66SHC+/+ mice; in particular, starting from G3 these mice presented testis atrophy that, however, was not observed in TERC−/− p66SHC−/− mice (Fig. 5A). Notably, the frequency of apoptotic cells as evaluated by IHC analysis with anti‐activated caspase‐3 antibody appeared significantly reduced in the testis from p66SHC−/− background (Fig. S2). Aplastic anemia has been also associated to telomere shortening in both humans and mouse models (Siegl‐Cachedenier et al., 2007); however, blot tests and bone marrow examination did not evidence severe aplastic anemia either in TERC−/− p66SHC+/+ or TERC−/− p66SHC−/− mice (Fig. 5B). No clear signs of muscle degeneration but rare loss of the striations, vacuolization, and infiltrations in the G3 TERC−/− p66SHC+/+, as shown in the Fig. S3, were observed. Abnormal glycaemia was not revealed in all generations as well. Lung emphysema was observed in late‐generation TERC−/− regardless the p66SHC genotype, whereas lung fibrosis was markedly evident in p66SHC−/− mice (Fig. 5C).
Figure 4.
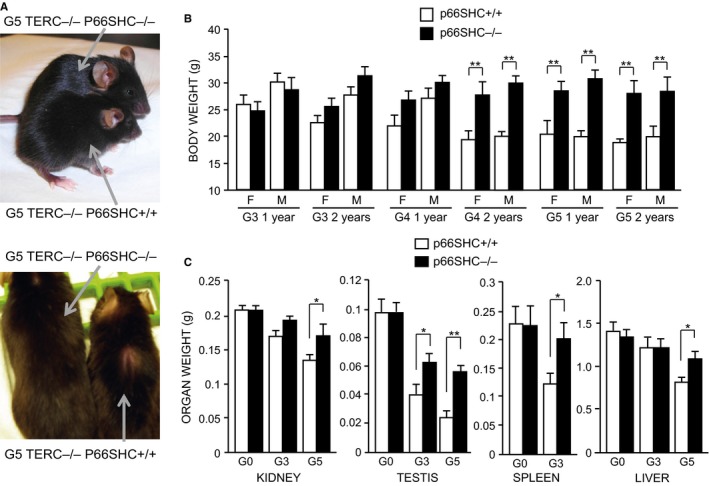
Appearance and weight of TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice. (A) Representative images of 3 months old G5 TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice. (B) Mean body weight of TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice at 1 and 2 years of age (F, female; M, male), *for p < 0.05 ** for P < 0.01. (C) Kidney, testis, spleen, and liver weight of G0–G5 TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice.
Figure 5.
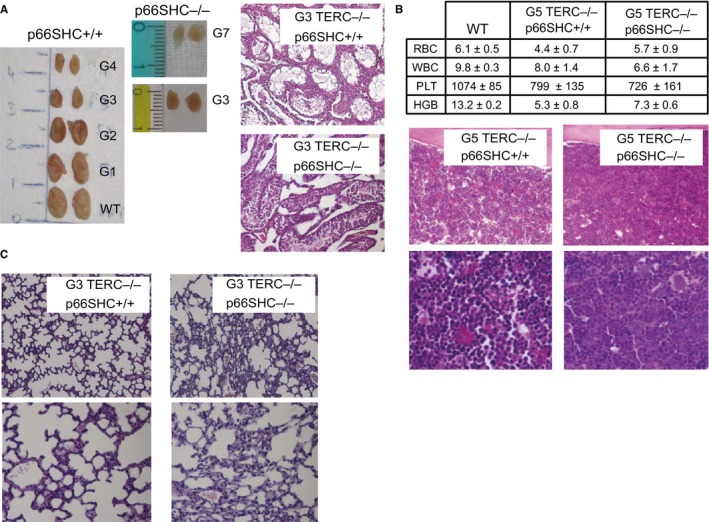
Phenotypic characterization of TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice. (A) Representative images of morphology (left) and histology (right) of testes from TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice. (B) Blood counts (upper table: WBC 103/μL, RBC 106/μL, PLT 103/μL, HGB mg dL−1; n = 6 mice per group) and bone marrow histology of TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice. (C) Histological examination of lung tissues from G5 TERC −/− p66SHC +/+ and TERC −/− p66SHC −/− mice.
P66SHC does not affect the lifespan of late‐generation TERC−/− mice
G3 C57Bl/6 TERC−/− mice were shown to be short living (Rudolph et al., 1999). The comparison of survival curves of WT and G3 TERC−/− p66SHC+/+ mice confirmed this observation (Fig. 6A). In particular, mortality risk (Fig. 6A, right upper panel) was comparable for the first 2 years, whereas starting from the third year it increases much more for TERC−/− mice than for WT; G3 TERC−/− p66SHC+/+ males showed the shortest survival.
Figure 6.
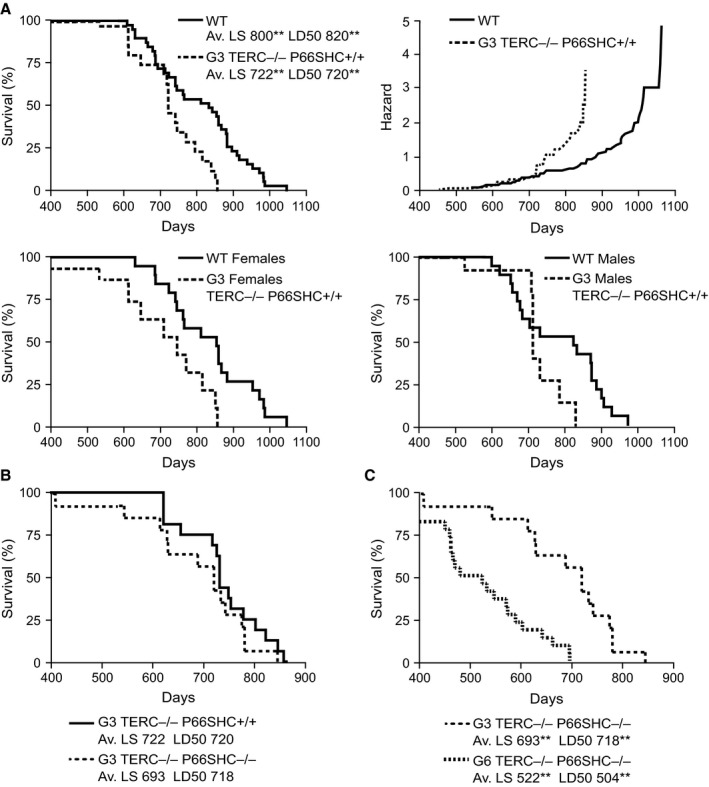
Survival curves of WT, TERC −/− p66SHC +/+, and TERC −/− p66SHC −/− mice. (A) Left upper graph, WT (n = 40) and G3 TERC −/− p66SHC +/+ (n = 18) male and female mice (Av. LS, average lifespan; LD50, median life time; **P < 0.01 for the effect of TERC genotype); right upper graph, cumulative hazard function for WT and G3 TERC −/− p66SHC +/+ male and female mice only; left lower graph, WT (n = 20) and G3 TERC −/− p66SHC +/+ (n = 10) female mice; right lower graph, WT (n = 20) and G3 TERC −/− p66SHC +/+ (n = 8) male mice only. (B) G3 TERC −/− p66SHC +/+ (n = 18) and G3 TERC −/− p66SHC −/− (n = 14) male and female mice. (C) G3 TERC −/− p66SHC −/− (n = 14) and G6 TERC −/− p66SHC −/− (n = 22) male and female mice (Av. LS and LD50 **P < 0.01 for the effect of generation).
The survival rate of G3 TERC−/− p66SHC−/− was identical to that of G3 TERC−/−p66SHC+/+ mice (Fig. 6B). They showed the same reduction in survival compared to the WT and p66SHC−/− (TERC+/+) mice. We could also determine the lifespan of G6 TERC−/− p66SHC−/− mice, revealing a further shortening of lifespan with respect to the G3 (Fig. 6C). Necroscopic examination of spontaneously dead G3 and G6 mice revealed the presence of visible tumor masses only in few cases (12 of 100 mice), equally distributed with respect to p66SHC mutation, rare enlargement of bladder and fecal prolapse, no splenomegaly or enlarged lymph nodes, and no hemorrhagic tissues or ascites.
In particular, the average body weight of G6 TERC−/− p66SHC−/− mice was 18.5 ± 0.4 g at 3 months of age and 23.1 ± 1.8 g at 1 year of age. They showed tremor and lethargy because 6–8 months of age and frequently hair loss around the nose and the limb, no abnormalities in the blood formula but a mild anemia, normal glycaemia and lipidic profile, and small lungs and thymus at death.
Discussion
Consecutive crosses of TERC−/− mice originate individuals with insufficient telomere length, organ dysfunctions, low fertility, and short lifespan (Wong et al., 2003 and Fig. 6A).
Oxidative stress shortens telomeres (von Zglinicki, 2002), and the deletion of p66SHC reduces oxidative stress (Trinei et al., 2002 and Fig. 3A and B). However, in the double mutants (TERC−/− p66SHC−/−) mice, we do not observe differences of telomere length with respect to the TERC−/− p66SHC+/+ (Fig. 3 C and D). This result indicates that the effect of reducing oxidative damage, by deleting p66SHC, on telomere shortening is less important with respect to other reasons of telomere erosion such as the incomplete replication of chromosomal ends.
The function of p66SHC appears instead relevant for the downstream consequences of dysfunctional chromosome ends, as p66SHC deletion improved fertility and reduce weight and shrinkage of different organs including testis of late‐generation TERC−/− mice (Fig. 4). Short telomeres of late‐generation TERC−/− mice induce senescence and apoptosis (Wong et al., 2003) that determine the detrimental effects of telomere erosion ultimately. P66SHC triggers mitochondrial apoptosis upon a variety of stresses (Migliaccio et al., 2006). At mechanistic level, p66SHC is plausible to mediate cell loss following telomere erosion rather than having a role in telomere shortening.
Regardless of the recovered fertility, weight, and healthier appearance, the p66SHC deletion does not improve longevity of G3 TERC−/− mice (Fig. 6B). Indeed, lung emphysema and fibrosis are frequent in both G3 and later generations of TERC−/− independently by the p66SHC deletion, although we could not establish that lung dysfunction is the cause of death of these mice. Histiocytic sarcoma was reported to be the most frequent tumor lesion in C57Bl/6 mice, including p66SHC−/− (Ramsey et al., 2014) and TERC−/− (Khoo et al., 2007) models. In any case, the early mortality of TERC−/− mice is not determined by p66SHC suggesting that oxidative stress and telomere attrition do not cooperate to determine lifespan. In this view, the unification of mitochondrial (free radicals) and nuclear (telomere) theories of aging (Sahin & DePinho, 2012) appears uncertain since the peculiar reasons of death of laboratory C57BL/6 mice.
From an evolutionary point of view, short telomeres and repressed telomerase (as in human somatic tissues) have been suggested to co‐evolve with homeothermy and replicative aging in mammals (Gomes et al., 2011), while p66SHC with the metabolic adaptation to harsh energetic environments (i.e., food deprivation and cold temperature) (Giorgio et al., 2012). In this view, the relative healthier aging of TERC−/− p66SHC−/− compared to TERC−/− p66SHC+/+ mice suggests that the cost to pay for having a plastic metabolism (as ensured by p66SHC) is to amplify the negative effect of telomere erosion on body size and fertility.
Finally, results from the study of p66SHC/TERC support the notion that genetic factors control the response to telomere erosion of specific organs. Thus, the predictive value of telomere length on organ aging depends on genetic background. Then, p66SHC expression and activity is induced in several tissues by obesogenic diets (Giorgio et al., 2012) or hyperglycemia (Albiero et al., 2014) that are known to affect telomere length as well (Laimer et al., 2015). The interaction of aging and metabolic pathways, including p66SHC, SIRT1, or AMPK/mTOR/S6K (Fadini et al., 2011), with telomere erosion in the presence of diabetes or obesity is a promising field to reveal how metabolic disorders impact on aging.
Experimental procedures
Mice
P66SHC mice were generated in our laboratory, at the European Institute of Oncology (Milan, Italy); TERC+/− mice (Lee et al., 2001) originated from the laboratory of Ronald DePinho (Dana‐Farber Cancer Institute, 450 Brookline Ave. Boston, MA, USA), and WT C57Bl6/j mice were purchased from Charles River Laboratories, Italy. All mice were housed at the specific‐pathogen‐free FELASA‐certified mouse facility in a temperature‐controlled room (temperature 21 ± 1 °C, relative humidity 60 ± 10%) under a 12‐h light/12‐h dark cycle (lights on from 7:00 a.m. to 7:00 p.m.) and with ad libitum food availability (2018S Teklad Global 18% Protein Rodent Diet; Harlan Laboratories Lesmo, Italy) and drinking water (autoclaved tap water). Proper group housing (four animals per cage) was chosen to improve animal welfare. Home cages were Plexiglas boxes (42 × 27 × 14 cm) with sawdust as bedding.
Experiments have been carried out in accordance with institutional guidelines and the Italian law, which enforces EU directives regarding the protection of animals used for experimental and other scientific purpose.
TERC and p66SHC genotyping
The primers and reaction conditions used for PCR genotyping were as follows: for TERC mouse mutants, 5′ GGG GCT GCT AAA GCG CAT 3′; 5′ TTC TGA CCA CCA CCA ACT TCA AT 3′; 5′ CTA AGC CGG CAC TCC TTA CAA G 3′; and reactions: 95 °C 15 min, 94 °C 1 min, −55 °C 1 min, −72 °C 1 min (35 cycles), 72 °C 7 min. For p66SHC mouse mutants: 5′ CTC GTG TGG GCT TAT TGA CAA AG 3′; 5′ CCT CCC CAG GTC ATC TGT TAT CC 3′; 5′ GGG TGG AGA GGC TTT TTG CTT C 3′; and reactions: 95 °C 15 min, 94 °C 1 min, −64 °C 1 min, −72 °C 1 min (35 cycles), 72 °C 7 min.
Telomere length qPCR and Q‐FISH assays
All buffers were purged with nitrogen and supplemented with 50 μm phenyl‐tert‐butyl nitrone (Sigma‐Aldrich Milan, Italy Srl) to prevent oxidation and minimize oxidative damage to DNA, which may alter the efficiency of the PCR if abasic sites are generated. Genomic DNA was isolated from mouse tissues using the silica‐gel‐membrane‐based DNeasy Tissue Kit (QIAGEN, Italy Srl Milan, Italy), according to the manufacturer's protocol with minor modifications. To minimize abasic site generation, the initial high temperature lysis with proteinase K was replaced by 6 hours incubation at 37 °C. Following the elution of purified DNA, 1 mm DTT (dithiothreitol) was added to the DNA samples, which were stored at −80 °C until use. DNA was quantified in triplicate using a NanoDrop spectrophotometer (Thermo Fisher Scientific Inc. Waltham, MA USA). Each PCR (20 μL) was performed as follows: 20 ng DNA, 1 × SYBR® Green master mix (Life Technologies‐Applied Biosystems Italia Monza, Italy), 100 nm telomere forward primer (CGG TTT GTT TGG GTT TGG GTT TGG GTT TGG GTT TGG GTT), and 100 nm telomere reverse primer (GGC TTG CCT TAC CCT TAC CCT TAC CCT TAC CCT TAC CCT) (O'Callaghan et al., 2008). Results are from the measure of three individuals per group performed in quadruplicates.
Q‐FISH was performed on 5 μm deparaffinized sections treated with 0.1 mg mL−1 RNA A solution and then with 0.5 mg mL−1 proteinase K solution for 20 min each time at 37 oC. Slices were then prewarmed at 85 oC for 5 min and incubated with 200 nm solution of Cy3 conjugated PNA TelC probe (PNA Bio) in 60% formamide, 10 mm Tris–HCl pH 7.5, and 0.2 μg mL−1 salmon sperm first for 10 min at 85 oC and then for 2 h at RT. Then, the probe was washes twice with 2XSSC, 0.1% Tween‐20 solution for 10 min at 58 oC. DAPI staining was used for the staining of the nucleus.
Images were collected through an Olympus BX51 upright fluorescent microscope equipped with a cool snap EZ CCD camera (Photometrics Tucson, AZ, USA) and processed by Metamorph (Molecular Device Sunnyvale, CA, USA) and ImageJ software. The Cy3 fluorescence intensity of at least 40 nuclei observed in at least two different slices per experimental group was compared (Meeker et al., 2002).
8‐OH‐dG and 8‐isoprostane analysis
8‐OH‐dG was measured using the commercially available immunoassay kit (Abcam Cambridge, UK) on genomic DNA. One microgram of total genomic DNA was denatured 5 min at 95 °C and digested with nuclease P1 (Sigma‐Aldrich Srl), then dephosphorylated by incubation with Antarctic phosphatase (New England Biolabs; EuroClone S.P.A. Pero, Italy) and loaded in duplicate on the anti‐8‐OH‐dG antibody‐coated plate. The amount of 8‐OH‐dG in the samples was quantified by measuring competition with labeled 8‐OH‐dG for antibody binding.
8‐isoprostane was detected by immunohistochemistry on deparaffinized 10 μm sections upon unmasking treatment for 45 min at 90 oC with sodium citrate, 10 min of peroxide blocking at RT and decoration with anti‐8‐epi‐PGF2α (Oxford Biomedical Research Rochester Hills, MI, USA) diluted 1:500 as primary antibody, and then with biotinylated anti‐goat and VECTASTAIN ABC Kit (Vector laboratories Burlingame, CA, USA) followed by minimal counterstaining with hematoxylin.
Tissue analysis
Blood formula was obtained using the Haematological Analyzer Act 5 Diff (Beckman Dickinson Franklin Lakes, NJ, USA). Histological analysis was performed on fresh tissues that were rapidly washed in PBS and incubated in 4% formaldehyde‐buffered solution for 16 h at room temperature. Fixed samples were then dehydrated by increasing concentrations of ethanol and included in paraffin using the Leica ASP300 tissue processor (Leica Biosystems – Leica Microsystems Srl Milan, Italy). Then, 4 μm sections were cut from paraffin blocks and stained with hematoxylin–eosin. Anti‐active caspase 3 antibody (Abcam) was used to reveal apoptotic cells by IHC.
Statistical analysis
Data were analyzed using Student's t‐test and Fisher's exact test. Survival was estimated with Kaplan–Meier estimators and differences tested with the log rank test.
Author contributions
Marco Giorgio planned and performed the experiments, analyzed the results, and wrote the manuscript. Massimo Stendardo performed the experiments. Enrica Migliaccio analyzed the results. Pier Giuseppe Pelicci planned the experiments and wrote the manuscript.
Funding
No funding information provided.
Conflict of interest
None declared.
Supporting information
Fig. S1 Representative FISH images of sections of different organs from G0, G3 and G5 TERC−/−P66SHC+/+ and TERC−/−p66SHC−/− mice, stained with the Cy3 conjugated TelC PNA probe (grayscale panels), the colored panels show Cy3 and DAPI fluorescence pattern in a stained section of WT liver as control.
Fig. S2 IHC analysis of testis slices from G3 and G5 TERC−/− p66SHC+/+ and TERC−/− p66SHC−/− mice with anti‐activated caspase 3 antibody.
Fig. S3 HE stained sections of quadriceps from G0 and G3 TERC−/−p66SHC+/+ or p66SHC−/− mice.
Acknowledgments
We thank Prof. Ronald DePinho for having kindly provided us with the TERC+/− mice, Dr. Mauro Romanenghi for technical help with telomere qPCR assay, and Dr. Roberta Aina for writing assistance. This work was supported by grants from the Fondazione Umberto Veronesi (FUV) to MG and the National Institutes of Health (NIH) to PGP.
References
- Albiero M, Poncina N, Tjwa M, Ciciliot S, Menegazzo L, Ceolotto G, Vigili de Kreutzenberg S, Moura R, Giorgio M, Pelicci P, Avogaro A, Fadini GP (2014) Diabetes causes bone marrow autonomic neuropathy and impairs stem cell mobilization via dysregulated p66Shc and Sirt1. Diabetes 63, 1353–1365. [DOI] [PubMed] [Google Scholar]
- Berry A, Capone F, Giorgio M, Pelicci PG, de Kloet ER, Alleva E, Minghetti L, Cirulli F (2007) Deletion of the life span determinant p66Shc prevents age‐dependent increases in emotionality and pain sensitivity in mice. Exp. Gerontol. 422, 37–45. [DOI] [PubMed] [Google Scholar]
- Blackburn EH, Greider CW, Szostak JW (2006) Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 12, 1133–1138. [DOI] [PubMed] [Google Scholar]
- Carpi A, Menabò R, Kaludercic N, Pelicci P, Di Lisa F, Giorgio M (2009) The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim. Biophys. Acta 1787, 774–780. [DOI] [PubMed] [Google Scholar]
- Daniali L, Benetos A, Susser E, Kark JD, Labat C, Kimura M, Desai K, Granick M, Aviv A (2013) Telomeres shorten at equivalent rates in somatic tissues of adults. Nat. Commun. 4, 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadini GP, Ceolotto G, Pagnin E, de Kreutzenberg S, Avogaro A (2011) At the crossroads of longevity and metabolism: the metabolic syndrome and lifespan determinant pathways. Aging Cell 10, 10–17. [DOI] [PubMed] [Google Scholar]
- Frijhoff J, Dagnell M, Augsten M, Beltrami E, Giorgio M, Ostman A (2014) The mitochondrial reactive oxygen species regulator p66Shc controls PDGF‐induced signaling and migration through protein tyrosine phosphatase oxidation. Free Radic. Biol. Med. 68, 268–277. [DOI] [PubMed] [Google Scholar]
- Gertz M, Steegborn C (2010) The Lifespan‐regulator p66Shc in mitochondria: redox enzyme or redox sensor? Antioxid. Redox Signal. 13, 1417–1428. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122, 221–233. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Berry A, Berniakovich I, Poletaeva I, Trinei M, Stendardo M, Hagopian K, Ramsey JJ, Cortopassi G, Migliaccio E, Nötzli S, Amrein I, Lipp HP, Cirulli F, Pelicci PG (2012) The p66Shc knocked out mice are short lived under natural condition. Aging Cell 11, 162–168. [DOI] [PubMed] [Google Scholar]
- Gomes NM, Ryder OA, Houck ML, Charter SJ, Walker W, Forsyth NR, Austad SN, Venditti C, Pagel M, Shay JW, Wright WE (2011) Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell 10, 761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guachalla LM, Ju Z, Koziel R, von Figura G, Song Z, Fusser M, Epe B, Jansen‐Durr P, Rudolph KL (2009) Sod2 haploinsufficiency does not accelerate aging of telomere dysfunctional mice. Aging 1, 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben JMJ, Moonen HJJ, van Schooten FJ, Hageman GJ (2008) Telomere length assessment: biomarker of chronic oxidative stress? Free Radic. Biol. Med. 44, 235–246. [DOI] [PubMed] [Google Scholar]
- Khoo CM, Carrasco DR, Bosenberg MW, Paik JH, Depinho RA (2007) Ink4a/Arf tumor suppressor does not modulate the degenerative conditions or tumor spectrum of the telomerase‐deficient mouse. Proc. Natl Acad. Sci. USA 104, 3931–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwan M, Dokal I (2009) Dyskeratosis congenita, stem cells and telomeres. Biochim. Biophys. Acta 1792, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laimer M, Melmer A, Lamina C, Raschenberger J, Adamovski P, Engl J, Ress C, Tschoner A, Gelsinger C, Mair L, Kiechl S, Willeit J, Willeit P, Stettler C, Tilg H, Kronenberg F, Ebenbichler C (2015) Telomere length increase after weight loss induced by bariatric surgery: results from a 10 years prospective study. Int. J. Obes. 2015 Nov 26. [DOI] [PubMed] [Google Scholar]
- Lee KH, Rudolph KL, Ju YJ, Greenberg RA, Cannizzaro L, Chin L, Weiler SR, DePinho RA (2001) Telomere dysfunction alters the chemotherapeutic profile of transformed cells. Proc. Natl Acad. Sci. USA 98, 3381–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Trimarchi JR, Smith PJ, Keefe DL (2002) Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 1, 40–46. [DOI] [PubMed] [Google Scholar]
- Lunghi B, De Cunto G, Cavarra E, Fineschi S, Bartalesi B, Lungarella G, Lucattelli M (2015) Smoking p66Shc knocked out mice develop respiratory bronchiolitis with fibrosis but not emphysema. PLoS One 10, e0119797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Padura I, de Nigris F, Migliaccio E, Mansueto G, Minardi S, Rienzo M, Lerman LO, Stendardo M, Giorgio M, De Rosa G, Pelicci PG, Napoli C (2008) p66 Shc deletion confers vascular protection in advanced atherosclerosis in hypercholesterolemic apolipoprotein E knockout mice. Endothelium 15, 276–287. [DOI] [PubMed] [Google Scholar]
- Meeker AK, Gage WR, Hicks JL, Simon I, Coffman JR, Platz EA, March GE, De Marzo AM (2002) Telomere length assessment in human archival tissues: combined telomere fluorescence in situ hybridization and immunostaining. Am. J. Pathol. 160, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menini S, Iacobini C, Ricci C, Oddi G, Pesce C, Pugliese F, Block K, Abboud HE, Giorgio M, Migliaccio E, Pelicci PG, Pugliese G (2007) Ablation of the gene encoding p66Shc protects mice against AGE‐induced glomerulopathy by preventing oxidant‐dependent tissue injury and further AGE accumulation. Diabetologia 50, 1997–2007. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG (1999) The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402, 309–313. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Pelicci PG (2006) Apoptosis and aging: role of p66Shc redox protein. Antioxid. Redox Signal. 8, 600–608. [DOI] [PubMed] [Google Scholar]
- Napoli C, Martin‐Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P (2003) Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high‐fat diet. Proc. Natl Acad. Sci. USA 100, 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Callaghan N, Dhillon V, Thomas P, Fenech M (2008) A quantitative real‐time PCR method for absolute telomere length. Biotechniques 44, 807–809. [DOI] [PubMed] [Google Scholar]
- Oikawa S, Kawanishi S (1999) Site‐specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 368, 435–365. [DOI] [PubMed] [Google Scholar]
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB, von Zglinicki T (2010) Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen S, Saretzki G, von Zglinicki T (1998) Preferential accumulation of single‐stranded regions in telomeres of human fibroblasts. Exp. Lung Res. 239, 152–160. [DOI] [PubMed] [Google Scholar]
- Ramsey JJ, Tran D, Giorgio M, Griffey SM, Koehne A, Laing ST, Taylor SL, Kim K, Cortopassi GA, Lloyd KC, Hagopian K, Tomilov AA, Migliaccio E, Pelicci PG, McDonald RB (2014) The influence of Shc proteins on life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 69, 1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA (1999) Longevity, stress response, and cancer in aging telomerase‐deficient mice. Cell 96, 701–712. [DOI] [PubMed] [Google Scholar]
- Sahin E, Depinho RA (2010) Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464, 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E, DePinho RA (2012) Axis of ageing: telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 13, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost‐Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA (2011) Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpea K, Talmud PJ, Cooper JA, Maubaret CG, Stephens JW, Abelak K, Humphries SE (2010) Association of telomere length with type 2 diabetes, oxidative stress and UCP2 gene variation. Atherosclerosis 209, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savino C, Pelicci PG, Giorgio M (2013) The P66Shc/mitochondrial permeability transition pore pathway determines neurodegeneration. Oxid. Med. Cell. Longev. 2013, 719407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegl‐Cachedenier I, Flores I, Klatt P, Blasco MA (2007) Telomerase reverses epidermal hair follicle stem cell defects and loss of long‐term survival associated with critically short telomeres. J. Cell Biol. 179, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su KG, Savino C, Marracci G, Chaudhary P, Yu X, Morris B, Galipeau D, Giorgio M, Forte M, Bourdette D (2012) Genetic inactivation of the p66 isoform of ShcA is neuroprotective in a murine model of multiple sclerosis. Eur. J. Neurosci. 35, 562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomilov AA, Ramsey JJ, Hagopian K, Giorgio M, Kim KM, Lam A, Migliaccio E, Lloyd KC, Berniakovich I, Prolla TA, Pelicci P, Cortopassi GA (2011) The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell 10, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, Milia E, Padura IM, Raker VA, Maccarana M, Petronilli V, Minucci S, Bernardi P, Lanfrancone L, Pelicci PG (2002) A p53‐p66Shc signalling pathway controls intracellular redox status, levels of oxidation‐damaged DNA and oxidative stress‐induced apoptosis. Oncogene 21, 3872–3878. [DOI] [PubMed] [Google Scholar]
- Trinei M, Berniakovich I, Beltrami E, Migliaccio E, Fassina A, Pelicci P, Giorgio M (2009) P66Shc signals to age. Aging 1, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinei M, Migliaccio E, Bernardi P, Paolucci F, Pelicci P, Giorgio M (2013) p66Shc, mitochondria, and the generation of reactive oxygen species. Methods Enzymol. 528, 99–110. [DOI] [PubMed] [Google Scholar]
- Wong KK, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, Alt FW, DePinho RA. (2003) Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 421, 643–648. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T (2002) Oxidative stress shortens telomeres. Trends Biochem. Sci. 27, 339–344. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T, Pilger R, Sitte N (2000) Accumulation of single‐strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 28, 64–74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Representative FISH images of sections of different organs from G0, G3 and G5 TERC−/−P66SHC+/+ and TERC−/−p66SHC−/− mice, stained with the Cy3 conjugated TelC PNA probe (grayscale panels), the colored panels show Cy3 and DAPI fluorescence pattern in a stained section of WT liver as control.
Fig. S2 IHC analysis of testis slices from G3 and G5 TERC−/− p66SHC+/+ and TERC−/− p66SHC−/− mice with anti‐activated caspase 3 antibody.
Fig. S3 HE stained sections of quadriceps from G0 and G3 TERC−/−p66SHC+/+ or p66SHC−/− mice.