

Abstract
Rotaviruses are the single leading cause of life-threatening diarrhea affecting children under 5 years of age. Rotavirus entry into the host cell seems to occur by sequential interactions between virion proteins and various cell surface molecules. The entry mechanisms seem to involve the contribution of cellular molecules having binding, chaperoning and oxido-reducing activities. It appears to be that the receptor usage and tropism of rotaviruses is determined by the species, cell line and rotavirus strain. Rotaviruses have evolved functions which can antagonize the host innate immune response, whereas are able to induce endoplasmic reticulum (ER) stress, oxidative stress and inflammatory signaling. A networking between ER stress, inflammation and oxidative stress is suggested, in which release of calcium from the ER increases the generation of mitochondrial reactive oxygen species (ROS) leading to toxic accumulation of ROS within ER and mitochondria. Sustained ER stress potentially stimulates inflammatory response through unfolded protein response pathways. However, the detailed characterization of the molecular mechanisms underpinning these rotavirus-induced stressful conditions is still lacking. The signaling events triggered by host recognition of virus-associated molecular patterns offers an opportunity for the development of novel therapeutic strategies aimed at interfering with rotavirus infection. The use of N-acetylcysteine, non-steroidal anti-inflammatory drugs and PPARγ agonists to inhibit rotavirus infection opens a new way for treating the rotavirus-induced diarrhea and complementing vaccines.
Keywords: Rotaviruses, Oxidative stress, Inflammatory signaling, Antioxidant treatment, Anti-inflammatory treatment
Core tip: Rotavirus entry into the host cell requires cell surface molecules providing binding, chaperoning and oxido-reducing functions. Sialic acid/integrin α2β1, heat shock cognate protein 70 and protein disulfide isomerase (PDI) seem to perform these functions. Recently, the cell surface oxido-reduction activity based at least on PDI has been highlighted as a potential determinant of the conformational changes that are required by viral structural proteins in order to facilitate virus entry. The rotavirus-induced oxidative stress and inflammatory signaling is an attractive target for therapeutic intervention as antioxidant and anti-inflammatory treatment has proved to efficiently inhibit rotavirus infection.
INTRODUCTION
Rotaviruses are the major cause of severe, acute, and dehydrating diarrhea in children under 5 years of age worldwide. World Health Organization estimates that more than 25 million outpatient visits and 2 million hospitalizations attributable to rotavirus infections occurred each year[1]. Child deaths caused by rotaviruses were estimated at more than 453000 in 2008 globally[2]. Rotaviruses belong to the family Reoviridae and their 11-segmented double-stranded RNA (dsRNA) genome is encapsidated within a non-enveloped virion composed by three concentric protein layers [triple-layered particle (TLP)][3]. The outer layer is made of two structural proteins, VP4 and VP7. The middle layer is composed of VP6 that surrounds the core shell. The inner layer is composed of the core shell (VP2), which encloses VP1, VP3 and the genomic RNA[4,5].
Rotavirus entry into the host cell seems to be mediated by the sequential interaction of virions with various cell surface molecules including sialic acid (SA)[6], heat shock cognate protein 70 (Hsc70)[7,8], integrins[9-11] and protein disulfide isomerase (PDI)[12]. Virion penetration into the host cell involves the loss of VP4 and VP7 converting the TLP into a double-layered particle (DLP), which becomes transcriptionally active by generating positive-strand RNAs (mRNAs)[13]. Besides translation into viral proteins, positive-strand RNAs also serve as template for synthesizing the new dsRNA genomic segments. Electron-dense structures, named viroplasms, appear early in the cytoplasm of rotavirus-infected infected cells. The dsRNA synthesis and the initial steps of virion assembly occur in viroplasms[14]. Several structural and non-structural virus-encoded proteins accumulate in viroplasms for participating in the formation of viroplasms and contributing to dsRNA synthesis and viral replication[15,16]. The newly assembled DLPs bud into the endoplasmic reticulum (ER) lumen where a transiently acquired envelope is later replaced by an outer protein layer consisting of VP4 and VP7[17]. Releasing of mature virions from infected cells take place by either cell lysis or by a non-classical, Golgi apparatus-independent, vesicular transport pathway[16,17].
Here, we review the current knowledge on the oxidative stress and inflammation responses induced by rotavirus infection and the contribution of these responses to viral pathogenesis. The analysis of the implication of cellular proteins having oxidoreductase, thiol isomerization and chaperone activities is also emphasized in the context of rotavirus entry into the host cell.
OXIDATIVE STRESS
Balancing oxidation-reduction (redox) status in cells seems to be a crucial event for maintaining life[18,19]. Molecular oxygen has the ability to form free radicals which are highly reactive species having a single unpaired electron in their outermost shell. Reactive oxygen species (ROS) include the superoxide anion (O2-) that is transformed into H2O2 through the reaction catalyzed by superoxide dismutase (SOD). H2O2 may interact with transition metals such as iron and cupper to form the hydroxyl radical (OH-). Reactive nitrogen species (RNS) are initially produced in cells by the reaction of nitric oxide (NO) and O2- that produces peroxynitrite (ONOO-), whereas NO is biosynthesized by various nitric oxide synthases. ROS and RNS are normally generated by cellular metabolism and at low or moderate concentrations play physiological roles including cellular response to infectious agents, cellular signaling, induction of mitogenic response, neurotransmission, blood pressure regulation, smooth muscle relaxation, and immune regulation[20]. Oxidative stress occurs when the production of ROS and other reactive species overwhelm the capacity of cellular antioxidant defenses to detoxify these potentially injurious species. Redox imbalance can be produced through an increased generation of ROS, depletion of cellular antioxidant molecules and decrease in antioxidant molecules[21]. Harmful effects of ROS are represented by oxidative damage to proteins, lipids and DNA, whereas RNS can cause protein nitrosylation, lipid oxidation and DNA fragmentation[20]. On the other hand, excessive ROS and RNS have been linked to pathogenesis of cancer, cardiovascular disease, atherosclerosis, hypertension, ischemia/reperfusion injury, diabetes mellitus, neurodegenerative diseases, rheumatoid arthritis, pulmonary disease, and ageing[20,22]. Oxidative stress has been implicated in pathological conditions associated with different human inflammatory diseases[23]. Cells have developed mechanisms to deal with damaging oxidative environments. These mechanisms include intracellular redox systems such as GSH/GSSG (the glutathione system), NADH/NAD+, NADPH/NADP+ and Trx(SH)2/Trx(S-S) (the thioredoxin system). GSH is the most abundant and ubiquitous intracellular antioxidant in cells from higher organisms and oxidative stress is commonly associated with decreased GSH or increased GSSG levels. However, cellular oxidative stress has been defined in terms of the disruption of biological redox signaling events rather than a simple imbalance between pro- and ant-oxidant systems[24,25]. Increasing data suggest that oxidative stress is involved in the pathogenesis of many diseases and disorders, including infectious diseases caused by viruses affecting the gastrointestinal tract.
INTESTINAL REDOX BALANCE
The mammalian gastrointestinal epithelium, the largest surface area contacting the external environment, consists of five major cell types (enterocytes, mucus-secreting goblet cells, hormone-secreting enteroendocrine cells, Paneth cells, and tuft cells)[26]. Normally, the villus tip enterocytes at 4-5 d post-differentiation spontaneously undergo anoikis (apoptosis) before being shed into the gut lumen[27]. Rotaviruses proliferate in the non-dividing mature enterocytes localized near the tips of the villi causing alterations in the small intestinal epithelium leading to diarrhea[28]. Homeostatic control of the intestinal redox environment seems to be a critical factor for maintaining intestine functions. Cells from intestinal epithelium must face the challenge not only of endogenously generated ROS but also of oxidant agents, mutagens and carcinogens accessing the luminal environment. Mucosal integrity is ensured by the luminal redox balance of the GSH/GSSG and cysteine/cystine (Cys/CySS) couples, that are also involved in maintaining luminal nutrient absorption, mucus fluidity, and microbiota[27,29]. Normal intestinal cell transition from proliferative state to non-dividing differentiated state or apoptosis has been associated with increasing oxidation of intracellular GSH/GSSG or extracellular Cys/CySS redox systems[29]. The homeostasis of the mucosal GSH is maintained through GSH uptake[30], regeneration from GSSG[31], and de novo synthesis[32]. Nevertheless, the extracellular/luminal redox environment is predominantly maintained by the Cys/CySS couple, with contributions from the GSH system[33]. Recent advances on intestinal redox biology suggest that the loss of intestinal homeostasis caused by oxidative stress in the mucosal and adjacent tissues can alter nutrient digestion and absorption, stem cell proliferation, enterocyte apoptosis, and immune response[27]. Understanding the mechanisms by which rotaviruses alter the intestinal homeostasis through the induction of oxidative stress open the way for designing new strategies based on the use of antioxidants as therapeutic tools for treating the severe and dehydrating rotavirus-induced diarrhea.
INNATE IMMUNE RESPONSE
Innate immunity, the first arm of the host immunity system, plays an important role in immediately controlling the pathogen invasion before induction of the mechanisms leading to an adaptive immune response. Innate immune system activation occurs through the recognition of pathogens by the germ-line-encoded pattern-recognition receptors (PRR). These receptors recognize specific structures present in pathogens, such as bacterial wall components or viral dsRNA. PRRs function by recognizing conserved pathogen-associated molecular patterns (PAMP) that are expressed by the invading pathogens. PRRs include toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs) and AIM2-like receptors. Ten different TLRs have been identified in humans, whereas there are 12 functional TLRs known in mice[34]. TLR9 is activated upon stimulation with viral DNA, TLR7 and TLR8 are activated by viral single-stranded RNA, while TLR3 activation is produced by viral double-stranded RNA[35]. Following receptor activation by virus associated molecular patterns and recruitment of several adaptor proteins, signaling pathways are activated resulting in the induction of cytokine production in virus-infected cells. Activation of TLRs stimulates nuclear factor-κB (NF-κB) and IRF3/7 signaling leading to the expression of type I interferons (IFNs) IFN-α and IFN-β, the production of pro-inflammatory cytokines, such as pro-interleukin (IL)-1β, and the activation of natural killer cells[36]. RLRs, including RNA helicases such as retinoic acid inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA-5), and double stranded RNA-dependent protein kinase (PKR) are particularly important in viral infections[37]. The NLR family consists of 22 proteins in humans and 34 in mice[38]. NLRs are involved in various innate immunity-associated functions including their assembly into multimeric protein complexes named as inflammasomes which are in charge of processing precursors of cytokines IL-1β and IL-18[39]. NLRP1, NLRP3, NLRP6, NLRP12 and NLRC4 have been found in distinct inflammasomes which participate in the recognition of different stimuli such as bacteria and viruses, among others[39].
Host cells response to viral infections through an early innate response consisting in the expression and secretion of type I, II and III IFN which, in turn, stimulate the expression of numerous IFN-stimulated gene (ISG) products having antiviral activities[40]. The IFN-regulatory factor (IRF) family of transcription factors comprises nine members (IRF1 to IRF9) which play crucial roles in activating innate and adaptive immune responses to viral infection[41]. IRF3, IRF5, and IRF7 are particularly important for inducing the expression of type I IFN[42]. The activation of the NF-κB by virus infection plays an important role during the induction of innate immune responses[43]. Transcription of type I IFN is induced by activation of RLRs RIG-I and MDA-5 following recognition of cytoplasmic RNA[44]. NF-κB plays a role in the expression control of over 500 genes involved in immune inflammatory responses, acute-phase inflammatory responses, angiogenesis, oxidative stress responses, cell adhesion, differentiation, apoptosis, AIDS, atherosclerosis, asthma, arthritis and metastasis[45,46]. The central role played by NF-κB signal pathway in physiological and pathological conditions has made it a potential target for pharmacological intervention[45,47].
Rotavirus infection stimulates early antiviral gene expression and IFN-β via a signaling pathway that involves the participation of IFN-β promoter stimulator 1 which is recruited to signaling complexes after activation of RIG-I or MDA-5[48,49]. However, rotavirus PAMPs have not been exactly characterized and some rotavirus replication products have been suggested as activators of RIG-I and MDA-5[48,49]. The exact identification of rotavirus PAMPs that are recognized by RLRs have been judged to be critical for understanding of rotavirus-host cell interactions[50]. Endosomal and cell surface membrane-associated PRRs, including TLR3, TLR7 and TLR9, have been implicated in rotavirus recognition for stimulating innate immune response to infection[51-53]. An increased level of type I and II IFNs has been found in children and animals as a consequence of rotavirus recognition by host PRRs[54,55]. However, some studies have suggested that whereas rotaviruses are able to trigger IFN production, they also can suppress the IFN effects[56]. Evidence has been provided that both IFN-α/β and IFN-γ play an important role in host response to rotavirus infection. However, their relative contribution may depend on the nature of rotavirus strain, site of replication, synergistic effects of IFN-γ, sustained replication and host age[50]. It has been shown that type II IFNs have a relatively modest effect in restricting early replication of homologous rotavirus strains in comparison with a higher effect on heterologous strains[57]. Further studies are needed to assess the roles of TLRs and IFNs during the early infection by homologous and heterologous rotavirus strains.
CELLULAR PROTEINS CONTRIBUTING REDOX AND CHAPERONE ACTIVITIES
Cellular proteins having oxidoreduction and/or chaperone activities have been shown to be essential for successful replication of many viruses. In this context, PDI and Hsc70, and other related cellular proteins, deserve to be highlighted. The PDI family of dithiol-disulfide oxidoreductases comprises at least 17 members in mammalian cells[58] and up to 21 members including other organisms[59]. PDI is mostly present in the ER where it catalyzes the oxidative formation of disulfide bonds in nascent proteins entering the secretory pathway[60,61]. Conversely, PDI acts as a reductase on cell membrane surface, thereby reducing cell membrane-bound protein disulfide bonds[59,62]. Erp57, a protein disulfide isomerase chaperone similar to PDI, has been found to be involved in ER quality control of newly-synthesized glycoproteins[63]. Erp57 is located in the ER but it is also present on the cell surface and plasma membrane lipid microdomains (rafts) from some cells[64]. PDI family proteins catalyze the introduction, reduction and isomerization of disulfides bonds and are also enzymatic chaperones reconstructing misfolded proteins. Human PDI is a 57 kDa protein containing four characteristic thioredoxin-like domains, two of which containing the common structural motif CXXC in the active site[65].
PDI redox activity can be inhibited by cell membrane-impermeant thiol/disulfide-reactive agents such as DTNB [5, 5-dithio-bis-(2-nitrobenzoic acid)] and bacitracin[66,67]. Recent studies have shown that Bak, a pro-apoptotic Bcl-2 protein, mediates the pro-apoptotic function previously reported for several PDI members. This Bak-dependent function of PDI is performed by inducing mitochondrial outer membrane permeabilization, linking in this way ER chaperone proteins and apoptotic signalling[68]. NADPH oxidase complex (Nox) is the major contributor of ROS in cells. PDI has been shown to interact with Nox within the ER and also in the citosol[69,70]. The PDI overexpression has been shown to produce an increase in NADPH oxidase activity, leading to increased levels of cellular ROS[71].
In the context of cellular chaperone activity, Hsc70 has been shown to play an important role in the virus life cycle by modulating infectivity[72,73], serving as a receptor molecule[7,8] or participating in viral assembly and morphogenesis[74,75]. Hsc70 is a constitutively expressed molecular chaperone belonging to the Hsp70 family. Hsc70 has been reported to be involved in protection from several forms of cellular stress performing multiple cellular functions including assistance in folding of nascent polypeptides, prevention of protein aggregation, translocation of proteins across membranes, chaperone mediation of autophagy, survival of cancer cells, and disassembly of clathrin-coated vesicles[76]. Hsc70 has been reported to protect cells from oxidative stress and apoptosis[77]. Although Hsc70 has not been reported as a cell surface receptor facilitating attachment of Japanese encephalitis virus (JEV) virions, it has been found to be associated with virus penetration via clathrin-mediated endocitosis[78]. There is evidence showing that NF-κB p65-induced cell proliferation is dependent on a NF-κB p65-mediated decrease of Hsc70 levels[79]. The above-mentioned evidences indicate that chaperone and oxidoreduction activities are present at different subcellular locations which can be used by viruses during their life cycle stages. Further studies must be conducted in order to better understand the specific implications of chaperone and oxidoreduction activities in both physiological and pathophysiological conditions.
PDI IMPLICATION IN VIRUS ENTRY
PDI redox function has been found to be needed for entry of some viruses into the host cell. Early studies demonstrated that human immunodeficiency virus (HIV) entry was inhibited by membrane-impermeant thiol/disulfide-reactive agents through inhibiting PDI redox function[66] or other cell-surface molecules showing redox activity[80,81]. PDI and thioredoxin-1 have been shown to reduce the disulfide bonds present on HIV glycoprotein gp120 facilitating the virus entry[82]. It has been suggested that endothelial PDI reduces integrins β1 and β3 causing the internalization of dengue virus[83]. Avian leukosis virus[84] and Sindbis virus[85] entry has been found to be dependent on the generation of free thiols in their fusion protein. The conserved cysteine residues from the hepatitis B virus (HBV) envelope protein coating hepatitis delta virus particles have been shown to be essential for virus entry[86]. Generation of free thiols in Newcastle disease virus fusion (F) protein have been shown to be required for virus entry into cells and cell fusion[87,88]; it has been suggested that PDI family isomerases could be responsible for such thiol generation[89]. Cell surface PDI has been found to facilitate the infection of HeLa cells by mouse polyoma virus[90]. Studies have identified novel functions of PDI that are relevant for various diseases including virus infections[91-93].
Rotavirus infectivity inhibition has been reported to be caused by treatment of MA104 cells with DTNB, bacitracin or anti-PDI antibodies[12]. It was suggested that thiol/disulfide exchange activity on cell membrane surface was involved in rotavirus infection as DTNB can modified thiol-containing cell surface proteins and bacitracin can react with proteins containing the tetra-peptide motif CXXC. The cell surface PDI implication in rotavirus entry was concluded from results showing a physical in vitro interaction between PDI and TLPs and a significant rotavirus inhibition caused by cell pre-treatment with anti-PDI monoclonal antibodies (mAbs)[12]. In the same study, it was observed that infectivity of rotavirus TLPs was reduced by pre-treating them with DTNB, whereas pre-treatment of TLPs with bacitracin or anti-PDI mAb did not affect TLP infectivity. These findings suggested that rotavirus virions contain thiol groups that are required for virus infectivity. From this study, it was concluded that membrane-impermeant thiol/disulfide-reactive agents and anti-PDI mAbs inhibit rotavirus infectivity at entry but during a post-binding step[12]. The implication of PDI during the rotavirus entry process has been further studied using synthetic peptides derived from rotavirus structural protein amino acid sequences potentially mediating cell surface PDI–substrate interactions[94]. Cysteine-containing VP4 and VP7 peptides were observed to cause a significant inhibitory effect of infectivity when added to MA104 cells by competing with infectious virions. It was also found that antibodies against these cysteine-containing VP7 or VP4 peptides significantly inhibited rotavirus infectivity suggesting that PDI can use at least these viral amino acid sequences for interacting with rotavirus structural proteins[94]. Interestingly, antibodies to VP7-derived amino acid sequences inhibited virus infectivity only after virions were attached to host cell surface membrane. These finding allowed authors to suggest that these VP7 amino acid sequences were exposed after a cell surface interaction-dependent conformational change occurred[94]. From these findings it can be summarized that a thiol/disulfide exchange is contributing to rotavirus entry to MA104 cells and that cell-surface PDI is a potential target for DTNB and bacitracin-induced infectivity inhibition as cell surface thiol/disulfide exchange blockade prevented at least viral structural proteins from being modified by cell surface proteins catalyzing thiol/disulfide exchange (i.e., PDI). Incubation of TLPs, VP5, VP6 or VP7 with rPDI or PDI in membrane-enriched fractions resulted in redox changes in viral proteins as such proteins reacted with maleimide, a thiol reactive moiety (Rivera M, Guerrero CA, Acosta O. Manuscript in preparation). Taken together, the above described findings suggest that cell surface PDI reducing activity is implicated during rotavirus entry. This fact opens the way for the rational design of membrane-impermeant thiol/disulfide compounds able to specifically inhibit the virus entry into the host cell.
ROTAVIRUS ENTRY INTO HOST CELL
Several cell-surface molecules have been involved in the early interactions between rotavirus virions and host cells. Rotavirus entry seems to occur by sequential interactions between virion proteins and various cell surface molecules[95,96]. After these initial interactions, the internalization of rotaviruses into the host cell takes place through distinct endocytic pathways that are determined by the viral structural protein VP4[97-99]. The rotavirus spike protein VP4 is cleaved by trypsin into N-terminal VP8* and C-terminal VP5* fragments to prime TLPs for efficient infectivity[100]. The structural characterization of an infectious rotavirus particle has allowed authors to propose a model involving a sequence of conformational changes in VP4 leading to the distortion of host cell membrane during entry[4,101,102]. However, the complete understanding of the mechanisms by which rotavirus enter cells is still lacking. Rotavirus structural proteins VP4 (VP5* and VP8*), VP6 and VP7[96,103] have been involved in different interactions with cell surface molecules during entry. Experimental results indicated that N-acetyl neuraminic (sialic) acid (SA)-dependent/neuraminidase-sensitive strains bind first through VP8* to SA before interacting with integrin α2β1 whereas this integrin is directly bound by SA-independent/neuraminidase-insensitive strains through VP5*[104-106]. Available evidence has indicated that SA is a crucial determinant for the binding of both neuraminidase-sensitive and neuraminidase-insensitive rotavirus strains[6]. Most commonly occurring human VP4 serotypes use their VP8* subunit to interact with cell surface GM1 ganglioside containing the internal N-acetylneuraminic acid, the most common SA[107,108]. This is in contrast with most animal rotaviruses that bind terminal sialic acids without using GM1 for VP4 cell binding or infection[107]. It has also been shown that VP8* of a human rotavirus strains specifically recognizes histo-blood group antigens[109-112]. After the initial binding to SA and integrin α2β1, post-binding studies have led to conclude that that rotavirus interacts with cell surface Hsc70[7,8]. Similar studies have also shown that rotavirus virions interact with integrins α4β1, αxβ2 or αvβ3 after their binding to α2β1[9,10,113]. Recently, it has been reported that rotaviruses also interact with reducing cell surface PDI during entry[12,94], most probably through their structural proteins VP5*, VP6 and VP7 which are potential substrates of PDI (Rivera M, Guerrero CA, Acosta O, manuscript in preparation). Post-binding interactions of VP5 and VP6 with Hsc70 has been well documented[7,8,103], whereas post-binding interactions with integrins α4β1, αxβ2 or αvβ3 have involved VP7[9,10,113,114]. However, the sequence in which these post-binding interactions occur has not been yet established. Interactions of cell surface molecules and rotavirus structural proteins are summarized and schematized in Figure 1.
Figure 1.
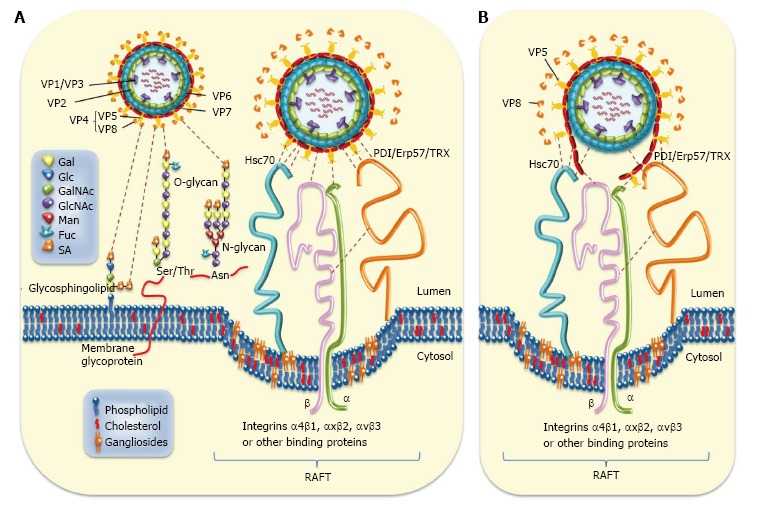
Rotavirus-cell surface interactions during entry. A: The rotavirus particle-associated proteins (VP1/2/3/4/5/6/7) that enclose the viral genome are represented. Cell surface molecules including sialic acid (SA), Hsc70, PDI, Erp57, thioredoxin (TRX), and integrins α4β1, αxβ2, and αvβ3 are also represented. Infection is initiated by the VP8*-mediated binding (attachment) of virion to terminal or non-terminal (neuraminidase -resistant) SAs located on cell surface glycolipids including gangliosides or to SAs located on cell surface glycoproteins. The N- and O-substituted derivatives of neuraminic acid (SAs) are indicated. Neuraminidase-resistant rotavirus strains can bind directly to integrin α2β1 through VP5* DGE sequence. SA-dependent strains bind first through VP8* to SA before interacting with integrin α2β1 through VP5*. A putative caveolae containing raft-associated cell surface receptors is depicted. The sequence of virion-cell interactions taking place after binding to α2β1 has not yet been established. However, several interactions involving rotavirus structural protein (VP5*, VP7 and VP6) and raft-associated cell surface receptors (Hsc70, PDI and integrins α4β1, αxβ2 and αvβ3) have been documented. Interactions between rotavirus structural proteins and cell surface molecules are illustrated; B: Disruption of rotavirus proteins (VP5*, VP7 and VP6) caused by cell surface-associated chaperone (Hsc70, PDI) and oxido-reductase activities (PDI, integrin αvβ3) is depicted. Hsc70: Heat shock cognate protein 70; PDI: Protein disulfide isomerase.
Crystallographic studies of VP5* have suggested that the trypsin cleavage of VP4 is determinant in generating conformational changes priming the VP8* and VP5* cleavage products for interacting with their corresponding cell surface receptors[101,102]. It has been hypothesized that a conformational transition from a dimer to a folded-back trimer of VP5* would facilitate the interaction of VP5* with the lipid bilayer membrane, resembling the fusogenic conformational changes in enveloped-virus fusion proteins[101]. Regarding the functional identity of Hsc70, this protein could be a candidate contributing to such conformational transition. Hsc70 has been proposed as a penetration receptor mediating JEV entry into cell by generating conformational changes in the envelope glycoprotein E of JEV, the protein responsible for receptor binding and membrane fusion[115]. Most likely, Hsc70 not only plays a role in anchoring rotavirus virions to cell membrane but also generating conformational transitions in VP5* to facilitate its transition from a dimeric to trimeric conformation. Other studies have suggested that VP5* bound to integrin α2β1 could undergo conformational changes associated to its trimerization[10,116]. Integrin α2β1 has also been shown to undergo conformational changes and activation that may facilitate binding of VP5* to cell membrane[10].
Chaperones such as Hsc70 commonly interact with hydrophobic regions of target proteins to perform ATP-dependent protein complex disassembly[117]. Although Hsc70 interacts with VP4 through the domains aa 642-658[8] and aa 531-554[103], the potentially fusogenic domain of VP5* (aa 385-404) could be a Hsc70 substrate. Hsc70-TLP interaction in solution seemed to induce conformational changes in VP5* and VP7[118]. Moreover, there are studies showing that DLPs interact physically with Hsc70 at least through the VP6 sequence aa 280-297 and that cell treatment with a synthetic peptide comprising this sequence was able to inhibit infection by animal and human rotavirus strains[103]. In the same study, the presence of antibodies to the VP6 synthetic peptide was shown to also inhibit rotavirus infectivity, suggesting that DLPs interact with Hsc70 during the entry process. Overall, it is not unlikely that post-binding interactions of rotavirus virions with Hsc70 might facilitate the generation of conformational changes in VP5* leading to the trimeric conformation able to destabilize de lipid bi-layer of cell membrane or endocytic vesicle[119,120].
Despite the identification of these potential receptors, there is no known single cell surface protein whose reaction with specific antibodies leads to an almost complete abolition of rotavirus infectivity. For instance, partial inhibition of rotavirus infectivity by anti-Hsc70 or anti-integrin antibodies might be reflecting the existence of alternative entry routes[96,99,121,122] or “dead-end” pathways[123]. Partial inhibition of rotavirus infectivity by anti-PDI mAbs might be suggesting that rotaviruses use alternative entry paths or that the anti-PDI mAbs used partially inhibited PDI activity[12]. These mAbs have been shown to inhibit PDI activity by 49% to 90%, depending on the assay system used[66,124-126]. The finding that bacitracin greatly inhibit PDI-TLP interaction in vitro suggested that the CXXC motif in the PDI catalytic domain was required for this interaction rather than the presence of free thiols in virion proteins, as shown by the insensitivity of this interaction to DTNB treatment[12]. Obviously, the PDI’s chaperone activity implication in PDI-TLP interaction cannot be ruled out because such activity has been reported to have become notably reduced by bacitracin treatment.
PDI, Hsc70 and integrin αvβ3 have been found to interact in lipid microdomains (“rafts”)[12,127], which have been proposed as being essential platforms facilitating efficient interaction between virus particles and cellular receptors[96,128]. On the other hand, some reports have indicated that PDI forms complexes with integrins α2β1 and αvβ3[129,130] that have been identified as cell surface rotavirus receptors in MA104 cells. Since integrin β3 is known to be an endothelial cell-surface PDI substrate[129], it would be interesting to determine whether free thiol generation in this integrin is required for its activation and interaction with rotavirus during entry. Evidence has been provided that IL-1α-mediated innate response of macrophages to adenovirus implicating the interaction of virus RGD motif with integrin β3 for triggering the activation of pro-inflammatory responses to the virus[131]. Interestingly, results have been presented that a specific inhibitor of integrin β3 (a secondary adenoviral receptor) attenuated the cytokine release and the inflammatory hepatic toxicity induced by an oncolytic adenovirus without interfering with its infectivity and oncolytic properties[132]. Integrin β3 expression has been shown to be required and up-regulated by classical swine fever virus (CSFV) infection[133]. However, PDI expression has been found to be inhibited in the heart, liver, spleen, lung, kidney and mesenteric lymph node tissue from a CSFV-positive pig[134]. Evidence has been provided that dengue virus serotype 2 (DV2) induce up-regulation of integrin β3 which is also required for DV2 entry into the cell[135]. However, studies using intestinal cell lines showed that rotavirus infection up-regulated the expression of integrins α2β1 and β2, whereas down-regulated that of integrins αvβ3, αvβ5, and α5β1[136]. It would be interesting to examine whether cell surface PDI activates integrin β3 to facilitate rotavirus infection since PDI expression has been up-regulated by rotavirus infection[127]. It has been found that dengue virus infection increases cell surface PDI expression for activating integrins β1 and β3 and facilitating virus entry into epithelial cells[83]. Chaperone and thiol-disulfide exchange activities are schematized in Figure 2.
Figure 2.
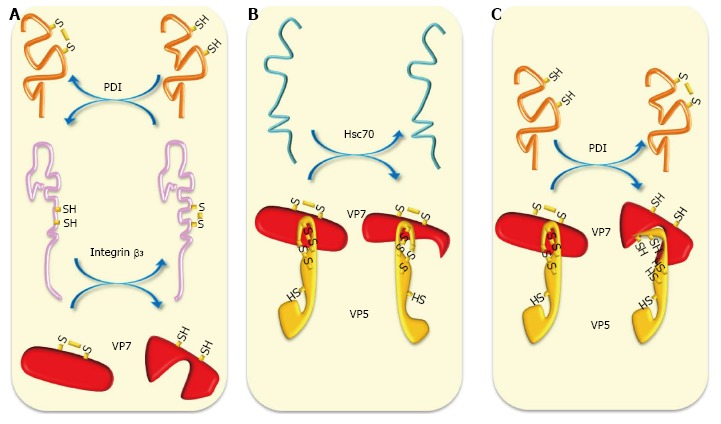
Schematic representation of chaperone and oxidoreduction activities during rotavirus entry. A: Cell surface reducing PDI has been shown to form complexes with integrins α2β1 and αvβ3 to generate free thiols in these integrins. Reducing integrin β3 can reduce thiol-disulfide bonds present in VP7; B: Chaperone activity of Hsc70 can induce conformational changes in VP5* and VP7 priming them for further interactions; C: Cell surface reducing PDI can reduce thiol-disulfide-containing VP5* and VP7 generating in them conformational changes needed for further interactions and entry. Hsc70: Heat shock cognate protein 70; PDI: Protein disulfide isomerase.
Rotavirus virion binding to the cell surface and the subsequent post-binding events seem to involve conformational changes and oxidoreduction reactions in the virus structural proteins. Regarding the properties of cell surface proteins interacting with virus structural proteins, it can be proposed that conformational changes could be produced by the chaperone activity characterizing Hsc70 and PDI, whereas redox status changes involving also conformational changes could be induced by oxidoreductase and thiol/disulfide isomerase activities present in PDI and integrin αvβ3[129,137]. On the other hand, there is evidence suggesting that thiol isomerases such as PDI and Erp57 bind to β3 subunit of integrins αIIbβ3 and αvβ3 for regulating their function during thrombus formation and that αIIbβ3 also has an endogenous thiol isomerase activity[138]. These results have led to propose that integrin β3 function might be regulated by both exogenous and endogenous thiol isomerase activity and that PDI inhibitors could be useful therapeutic tools for treating integrin-associated diseases[138].
Rotavirus structural proteins VP4, VP6 and VP7 have been reported to contain cysteine residues able to form intramolecular disulfide bonds[139-141]. However, crystal structure studies of VP6[142] VP8*[108] and VP5*[101,143] have shown that these proteins lack disulfide bonds. Rotavirus VP4 from many SA-dependent animal strains contains five conserved cysteines at positions 203, 216, 318, 380 and 774. It has been shown for simian RRV and SA11 that their VP4 contains two disulfide bonds residing in the VP8* (Cys-203/Cis-216) and VP5* (Cys-318/Cis-380) domains[139]. A SA-independent variant of RRV was reported to have an additional cysteine at position 267 that was able to form an alternative disulfide bond implicating Cys-318 while co-existing with the disulfide bond Cys-318/Cys-380[144]. The presence of highly conserved disulfide bonds in VP5* has been suggested to facilitate bringing together the trypsin cleavage sites, the integrin binding site and the putative fusogenic peptide into intimate proximity[121,139]. The mutant VP5* containing mixed species of disulfide bonds was supposed to have an altered conformation explaining its ability interact with the host cell surface independently from SA interaction[144]. In this context, the concept of functional disulfide bonds[145] could be extended to the interactions between disulfide bond-containing proteins of rotavirus virions and the cell surface proteins having thiol isomerase activity including PDI and integrins. Interactions of rotavirus structural proteins and cell surface molecules during entry are summarized in Table 1.
Table 1.
Cell surface molecules interacting with rotavirus structural proteins during entry
| Cellular molecule | Rotavirus protein | Activity | Viral protein motif involved | Ref. |
| Sialic acid | VP8* | Binding | Carbohydrate binding site | [108] |
| α2β1 | VP5* | Post-binding | DGE (VP5*) | [106] |
| α4β1 | VP7, VP5* | Post-binding | YGL (VP5*); LDV o LDI (VP7) | [11,121] |
| αxβ2 | VP7 | Post-binding | GPR (VP7) | [114,121] |
| αvβ3 | VP7 | Post-binding, oxidoreduction | 161NEWLCNPMD169 | [10,113] |
| Hsc70 | VP5*, VP7, VP6 | Chaperoning | aa 642-658 (VP5*); aa 280-296 (VP6); aa 531-554 (VP5*) | [8,103] |
| PDI | VP5*, VP7, VP6 | Chaperoning, oxidoreduction | aa 200-219 (VP4); aa 189-210 and aa 243-263 (VP7) | [12,94, Rivera M, Guerrero CA, Acosta O, manuscript in preparation] |
| HBGAs | VP8* | Binding | Carbohydrate binding site | [111] |
Hsc70: Heat shock cognate protein 70; PDI: Protein disulfide isomerase; HBGAs: Histo-blood group antigens.
Research aims at unraveling the mechanisms involved in rotavirus entry is very critical for understanding versatility of rotaviruses in using different cell surface receptors. However, the accumulated findings on rotavirus entry mechanisms suggest that in addition to the initial attachment to SA-containing molecules, rotavirus structural proteins undergo conformational changes mediated by cell surface chaperone and thiol-disulfide activities. Clearly more research is needed to fully understand if rotavirus certainly use alternative entry pathways or at least partially shared pathways that finally lead to the conversion of TLPs into transcriptionally active DLPs[146].
DLP-TLP INTER-CONVERSION
Rotavirus entry process contributes to convert TLPs into transcriptionally active DLPs, whereas newly formed DLPs are converted into new TLPs. Although the mechanisms involved in this inter-conversion are not entirely composed by the same reactions in opposite directions, it sounds illustrative to compare some of these membrane-associated reactions: Those aimed at removing VP4 and VP7 from TLPs to generate DLPs and those aimed at coating DLPs to generate TLPs. Cell membrane must possess molecular systems capable of inducing the necessary conformational changes facilitating viral proteins to disturb cell membrane for penetration[147]. In the case of rotaviruses, potential receptors having chaperone and oxidoreductase activity has been identified[7,12]. The ER has been described as complex membranous network that is used by many viruses during infection[148]. ER participates in rotavirus assembly especially during the late steps of the morphogenesis events converting DLPs into TLPs. NSP4 recruits VP4 and DLP to the ER membrane before budding into the ER lumen where a transient membrane layer is removed and replaced by VP7 to generate mature TLPs budding from the ER[16,149]. The mechanisms involved in removing the ER-derived transient membrane layer are unclear, except that unassembled VP7 has been reported to have a membrane lytic activity[150]. Although formation of virus-induced ER-derived structures is considered critical for viral replication and assembly[148], viral infections induce ER stress and interferon responses that are interfered by viruses to ensure viral replication or pathogenesis[151].
Despite the advances in structural characterization, the sequence of events occurring during uncoating for generating and releasing DLPs into cytoplasm is still unknown[4,97]. These events involve removing of structural proteins VP4 and VP7 to produce DLPs. To this step, the general event could be assumed as a set of reactions proceeding in the opposite direction to those reported for the morphogenesis of TLPS from DLPs during the ER budding. Removing VP4 and VP7 led to generating a hydrophobic surface that might facilitate the translocation of DLPs into the cytoplasm through cellular or endosomal membrane. However, the sequence of VP4 and VP7 assembly did not explain the mechanisms of entry-associated uncoating[123]. Recoating experiments in which rotavirus DLPs are recoated in vitro with recombinant outer proteins have been useful in approaching the sequence of virion assembly in vivo. These experiments allow obtaining an efficient in vitro coating of DLPs that favors the hypothesis that in vivo VP4 assembly precedes that of VP7[123]. Reversing the coating assembly during entry means that VP7 should be removed before VP4, except that VP4 underwent a previous trypsin cleavage that generates VP8* and VP5*. However, VP8* is supposed to be released from VP5* before cell membrane destabilization[101].
Studies using cysteine-containing synthetic peptides derived from VP4 and VP7 suggested that VP4 and VP7 probably are PDI substrates as pre-treatment of cells with these peptides inhibited rotavirus infection[94]. Similarly, pre-treatment of TLPs with antibodies against these peptides led to decreased infectivity. These findings allow hypothesizing that the disulfide bonds present in VP8* or VP5* could be reduced by PDI facilitating the TLP uncoating. The fact that PDI can produce in vitro modifications in the redox status of VP4 and VP7 (Rivera M, Guerrero CA, Acosta O, manuscript in preparation) gives support to this hypothesis. It would be interesting to know whether the mechanism causing the release of VP8* from VP5* involves redox reactions. Disulfide bond reduction could be a plausible candidate mechanism for ensuring the conformational changes needed for detachment of VP5* from integrin α2β1, contributing in this way to make the entry process irreversible. VP7 maturation in the ER lumen has been reported to involve oxidation reactions caused by the oxidant PDI to generate intra-molecular disulfide bonds and a proper conformation to ensure its assembly on DLPs[140,152,153]. The contribution of disulfide bonds to VP7 conformation seems to be crucial for the structural and functional roles of this protein during assembly and interaction of virions with cell surface receptors. Since virus entry leads to uncoating of TLPs by losing VP4 and VP7 to release the transcriptionally active DLPs into cytoplasm, it is tempting to propose that the reducing cell surface PDI could contribute to destabilize VP7 by reducing its disulfide bonds beside the contributions of the decreased Ca2+ concentration and acidification reported to occur in the endocytic environment[154-156]. DLPs in the absence of VP4 and VP7 have been shown to be able to bud into the ER lumen. Taking into account that the reduced VP7 seem to have low affinity for DLPs, it is not unlikely that DLPs present in the endosomal vesicles can bud into the cytoplasm through the permeabilized endosomal membrane[156]. In the opposite redox reaction, oxidized VP7 has been shown to be required to remove the transient lipid envelope in a calcium-dependent process to generate mature TLPs in the ER lumen[16]. Research aims at knowing whether the reducing PDI is modifying the redox status of VP7 in the endosomal membrane would be useful to determine the potential participation of redox reactions during generation of DLPs in vivo. Interaction between the outer capsid proteins (VP4 and VP7) and PDI has been demonstrated in vitro and also the generation of free thiols in these proteins after this interaction. Similarly, thiol groups are generated in the outer capsid proteins after TLPs contact the cell surface, suggesting that PDI or other related thioredoxins are able to reduce disulfide bonds in viral proteins (Rivera M, Guerrero CA, Acosta O, manuscript in preparation). Studies characterizing potential cell surface receptors for rotavirus infection of small intestinal villus cells from mice showed that raft-associated Hsc70, PDI and integrin β3 played an important role in the rotavirus entry process as previously shown for MA104 cells[127]. It has been reported that integrin subunit β3 and integrin α2β1 are present on the cell surface of murine and human enterocytes[157,158], and that rotavirus-susceptible MA104, COS7 and Caco-2 cells also contain cell surface receptors including αvβ3 and Hsc70[113,128]. The colocalization of PDI, integrin β3, Hsc70 and rotavirus particles in lipid microdomains (rafts) from MA104 and intestinal villus cells[12,127] suggest that PDI reducing function at cell surface can activate either integrins or VP7 to interact each other during entry[94].
The role of thiol-disulfide exchange during rotavirus infection is well documented, but the detailed processes of this implication still remain incompletely elucidated. Although PDI has emerged as a significant contributor for generating thiol-disulfide-associated conformational changes in rotavirus structural proteins during uncoating and assembly of viral particles, contributions from integrins and other thioredoxins cannot be ruled out. Again, a better understanding about the involvement of thiol-disulphide exchange in the rotavirus infection process could facilitate the identification of potential targets of therapeutic strategies.
OXIDATIVE STRESS AND ROTAVIRUS INFECTION
Several studies have demonstrated the implication of redox balance disruption in the establishing of viral infection and the progression of virus-induced diseases[159]. The oxidants induced by viral infections include superoxide anion (O2-)[160], which can be transformed into hydroxyl radical (OH-), nitric oxide radical (NO), H2O2 or peroxynitrite (ONOO-) through enzymatic and non-enzymatic reactions. The findings showing pathogenic interactions between ROS and HIV stimulated research into the role these interactions may play in the pathogenesis of many viruses, opening the way for novel antioxidant-based antiviral therapeutic strategies[161,162]. ROS may modulate the viral replication and cellular response, and also contribute to viral pathogenesis[163,164]. Virus-induced oxidative stress has been reported during HIV[165], influenza virus[166], HBV[167], hepatitis C virus[168], encephalomyocarditis virus (EMCV)[169], respiratory syncytial virus (RSV)[170], dengue virus[171,172], and JEV[173] infections.
Early studies on rotavirus infection showed decreased SOD and glutathione peroxidase activities in whole intestine homogenates from infant mice[174]. A more recent study reported that rotavirus infection was able to induce an increase in inducible nitric oxide synthase (iNOS) mRNA in murine ileum and iNOS expression also in murine ileum upon exposure to NSP4[175]. NSP4-induced release of NO metabolites was reported in cultured human intestinal epithelial cells incubated with purified NSP4[176]. Increased NO metabolites were also observed in mice infected with murine rotavirus EDIM beside upregulation of iNOS mRNA in ileum, but not in duodenum or jejunum[176]. A prospective clinical study including acutely rotavirus-infected children showed that viral infection stimulated NO production[176]. However, studies using Caco-2 cells infected with SA11 rotavirus showed that viral infection increased the expression of the mitochondrial superoxide dismutase (MnSOD) within the first 48 h.p.i. This increased SOD expression was correlated with a decrease in ROS generation during the early phase of infection (8 h.p.i.) and a lack of cellular glutathione (GSH) depletion[177]. Despite the increase in enzyme activity was not directly proportional to the rise in protein expression level in the cell lysates studied, during the later post-infection times ROS returned to the control levels even in the presence of increased MnSOD protein expression. This fact was interpreted as being due to an overproduction of mitochondrial ROS that overwhelmed the activity of the MnSOD[177]. Induction of MnSOD overexpression has been shown to occur as a consequence of increased production of ROS through a pathway involving inflammatory cytokines such as tumor necrosis factor-α (TNF-α) or IL-1[178,179] and activation of the NF-κB factor by ROS[180]. ER stress was found to increase ROS[181] and also induce MnSOD through nuclear factor NF-κB and AP-1 activation after exposure of HeLa cells to various agents interfering with ER functions[182].
Acute gastroenteritis in piglets has been associated with increased levels of high-mobility group box 1 (HMGB1) protein (a nuclear DNA-binding protein), and serum haptoglobin and ceruloplasmin which suggest an acute phase response[183]. A significant decrease of total antioxidant capacity and antioxidant enzyme activities has been found in serum from piglets affected with acute enteritis. Increased values of oxidative stress indices, including the malondialdehyde (MDA) and NO concentrations in serum have also been associated with pathological condition[183]. However, HMGB1 protein, acute phase response and oxidative stress indices were even more prominent in the cases in which porcine rotavirus infection took place.
Although there is a large body of information available about the involvement of oxidative stress in viral infection and its effects on cell functions leading to cell death, the extent to which oxidative stress is part of a natural defense response of cells to virus infection or a mechanism by which viruses induce cell injury is still unknown. Advances in the understanding of the role of oxidative stress in rotavirus infection might contribute to improved treatment strategies of rotavirus-induced diarrhea. Interestingly, rotavirus infection of cultured cell lines, and in vivo conditions using animals and human patients has been shown to be inhibited by anti-oxidant therapy[184-186]. These findings encourage research to clarify the role of virus induced-oxidative stress as a damaging by-product of infection or a condition required for a successful viral life cycle.
ER STRESS AND ROTAVIRUS INFECTION
Disruption of protein folding homeostasis in the ER leads to unfolded or misfolded protein accumulation in the ER lumen and alteration in the calcium homeostasis. Protein misfolding in the ER contributes to the pathogenesis of many diseases. ER stress of intestinal epithelial cells activates signaling pathways known as unfolded protein response (UPR) which have been associated with inflammatory bowel disease[187]. Alterations in ER homeostasis are normally sensed and followed by activation of the UPR pathway in order to restore homeostasis by activating genes implicated in protein folding. Failure to resolve ER stress causes activation of apoptotic pathways that lead to cell death[188]. Misfolded proteins in the ER activate UPR and induce oxidative stress and apoptosis in vitro and in vivo in mice, whereas antioxidant treatment counteracts UPR activation, oxidative stress, and apoptosis[189]. Release of calcium from the ER increases the generation of mitochondrial ROS leading to toxic accumulation of ROS within ER and mitochondria. On the other hand, sustained ER stress has been found to potentially stimulate inflammatory response through UPR pathways. Moreover, ROS produced as a consequence of inflammation or mitochondrial dysfunction could aggravate ER malfunction[190]. This picture suggests that a networking occurs between ER stress, inflammation and oxidative stress. Dysfunctional UPR pathways have been associated with numerous diseases including several neurodegenerative diseases, stroke, metabolic disorders, cancer, inflammatory disease, diabetes mellitus, cardiovascular disease, among others[190]. A crosstalk between ROS generation and ER stress response has been proposed as the ER-stress-associated redox status may be correlated with ER-stress-associated ROS[191]. Although the production of ROS has been correlated with ER stress in many pathological states, the detailed mechanisms on how changes in the protein-folding environment in the ER lumen cause oxidative stress are still unclear[192].
Proper protein folding and disulfide bond formation that take place in the ER are critically dependent on the redox status of the ER lumen. This compartment is highly oxidizing showing a high ratio of GSSG/GSH, which contrasts with the citosol environment[193]. The oxidizing environment of ER lumen is required to ensure disulfide bond formation and avoid aggregation or unfolded protein accumulation in this compartment[194]. Resident enzymes of the ER lumen contribute to regulate redox status and facilitate disulfide bond formation and isomerization[195]. The oxidative folding of proteins is facilitated by a family of ER oxidoreductases including ERp57 and PDI among others[196]. Oxidative folding catalyzed by ER oxidoreductases leads to their reduction, whereas their reoxidation is performed by ERO-1, an enzyme that can use molecular oxygen as an electron acceptor[191,197]. Then, disulfide bond formation catalyzed by ERO-1 is a significant source of the total generation of ROS in the cell as the incomplete oxygen reduction leads to the anion superoxide formation[198].
Given that in the ER occurs the major synthesis and folding of secreted and transmembrane proteins, alterations in the protein flux into the ER have been associated ER stress. Recent studies have shown that viroporins, small hydrophobic virus-encoded proteins that oligomerize to form aqueous pores through cellular membranes, play important role in virus replication by affecting normal physiology of host cell and contributing to viral pathogenesis[199,200]. Since replication of most RNA viruses occurs in intimate interaction with the ER and causes ER stress in the infected cells, its underlying mechanisms are a central issue of the research about virus-host interactions. Many viroporins localize to the ER where alter the membrane potential of the ER and modulate the ER stress response and autophagy induction[201]. Viral infections can act as stress signals that alter ER homeostasis affecting negatively ER functions[151,202]. Many viruses have been shown to cause ER stress and induce one or more branches of the UPR in the infected cells[199,203]. Some studies have shown that several viruses induce ER stress and UPR signaling but also modulate UPR for protecting the infected cells from ER stress-mediated death to ensure virus replication[204-206]. Several properties of the viroporins suggest that they might also modulate the virus-induced ER stress response[207]. The relatively high concentration of Ca2+ in the ER lumen is needed for proper functioning of many calcium dependent chaperones and enzymes including PDI[208]. Many viroporins, including rotavirus NSP4, induce leakage of ER luminal calcium into the cytosol[209,210] affecting the calcium-dependent protein folding machinery and consequentially inducing ER stress. Translocation of NSP4 to mitochondria has been observed to dissipate mitochondrial membrane potential and induce apoptosis during the early infection. However, the pro-apoptotic activity of NSP4 was counteracted by NSP1, which activates PI3K/AKT[211]. In addition, autophagy could be induced by the increased Ca2+ concentration in the cytosol as it has been shown for foot and mouth disease virus[212]. Rotavirus NSP4, a protein inducing diarrhea in young mice, has been shown to anchor to the ER through its N-terminus, where its domain spanning amino acids 47-90 has been found to insert into ER membrane and show structural characteristics of viroporins[209]. NSP4 has also been shown to modulate autophagy induction in the virus-infected cells as cellular autophagy is required by rotaviruses to ensure their successful replication[213].
Rhesus rotavirus (RRV) has shown to induce ER stress in the rhesus monkey epithelial cell line MA104 and also activate two components of the UPR pathway[214]. However, this ER-mediated signaling was interrupted at the transcription level by the non-structural protein 3 (NSP3). Specific virus-encoded proteins have been identified as inducers of UPR during infection in the case of coronavirus[215], dengue 2 virus[216], human cytomegalovirus[217] and West Nilo virus[218]. In contrast, a single specific virus protein in RRV-infected MA104 cells did not trigger the activation of UPR. It was supposed that a multifactorial event involving either the budding of the DLPs into the ER, the formation of viroplasms, or the activation of genome replication could be the inducer of the UPR[214].
Rotavirus infection has been shown to induce ER stress leading to disturbances in the cellular calcium compartments and generation of ROS. Rotavirus-induced diarrhea involves a series of secretory and osmotic mechanisms[219] where NSP4 plays a key role by inducing release of intracellular deposits of calcium from enterocytes[220,221] and altering ion secretion[222]. It has been reported a NSP4-dependent chloride secretion in human enterocytes[223], which has also been demonstrated in Caco-2 cells infected with SA11 rotavirus[224]. In this case, NSP4-dependent chloride secretion was associated with an increase in ROS and a decreased reduced (GSH) to oxidized (GSSG) ratio. The same effects were observed when Caco-2 cells were treated with purified NSP4, whereas the increase in ROS and the GSH imbalance were strongly inhibited by N-acetylcysteine. These findings suggested an association between oxidative stress and rotavirus-induced diarrhea[224]. There are data supporting the hypothesis that ROS can induce intestinal epithelial cell apoptosis in mice through the Fas and Fas-L expression[225].
A number of stimuli and insults, including pathogen invasions such as virus infections have been found to induce ER stress affecting protein folding function and other disturbances including alterations in calcium homeostasis and increase of ROS. Then, the ER-induced UPR signaling has emerged as a central subject in the context of pathological processes including virus infections. However, the UPR-associated molecular mechanisms leading to minimize the accumulation and aggregation of misfolded proteins in response to virus infections need further investigation to be completely understood. The knowledge gained from UPR mechanisms could provide basis for antiviral development.
N-ACETYLCYSTEINE IN THE TREATMENT OF VIRAL INFECTIONS
N-acetylcysteine (NAC) is an amino acid that functions as a cysteine pro-drug and glutathione (GSH) precursor, the most powerful cellular antioxidant[226]. NAC is readily deacetylated primarily in the liver to yield L-cysteine thereby promoting intracellular GSH synthesis[227]. It has been used during several decades as mucolytic agent and also for the treatment of various disorders including paracetamol intoxication[228]. NAC has been also used for treatment of numerous disorders linked to oxidative stress including gastrointestinal[229], renal[230], cardiovascular[231], pulmonary[232], hepatic[233], psychiatric and neurological disorders. The mucolytic activity of NAC is due to its ability to break up the disulfide bonds of the high molecular weight glycoproteins present in the mucus. NAC functions in cells as a free radical scavenger antioxidant agent as it reacts with ROS such as H2O2 and OH-[234].
NAC has been used in the treatment numerous infectious diseases, including virus infections. A significant reduction of the incidence of clinical symptoms and improvement of cell-mediated immunity were reported after treatment with NAC[235]. Similarly, GSH has been reported to inhibit infection by influenza virus in both cultured cells and mice[236]. High doses of NAC have proven to be synergistic with oseltamivir treatment in protecting mice from fatal influenza infection[237], whereas a synergistic combination of NAC and ribavirin was also effective in preventing mice from lethal influenza virus infection[238]. A long-term NAC administration attenuated influenza symptoms in elderly patients with chronic degenerative disease[235]. In addition, a patient infected with the A/H1N1 influenza virus improved rapidly after treatment with a high-dose NAC therapy in combination with antiviral medication[239]. NAC has been shown to reduce H5N1-induced cytopathic effects, virus-induced apoptosis and the production of some pro-inflammatory molecules whereas it inhibited the activation of oxidant sensitive pathways including NF-κB and mitogen activated protein kinase p38[240]. However, a universal inhibitory activity against influenza A viruses has not yet been demonstrated[241]. A successful outcome was reported following early administration of NAC to children affected with dengue hemorrhagic fever or dengue shock syndrome complicated by acute liver failure[242]. Before highly active antiretroviral therapy (HAART), NAC was tested to replenish GSH levels in HIV-infected patients since cysteine and GSH levels decrease as the HIV disease progresses[243,244]. However, NAC has been offered as a useful adjunct therapy to increase protection against oxidative stress, improve immune system function and increase detoxification of acetaminophen and other drugs in patients treated with HAART[245].
The sensitivity of rotavirus infection to NAC has recently been demonstrated. A study that screened for drugs with the potential ability to interfere with cellular redox reactions, found that infection of MA104 and Caco-2 cells with several rotavirus strains was significantly inhibited by NAC in both cell systems[184]. On the other hand, the rotavirus NSP4-induced chloride secretion has been shown to be inhibited by pre-treating Caco-2 cell with NAC, suggesting that the enterotoxic effect of NSP4 is stress oxidative-dependent[224]. Inhibition of rotavirus infection by NAC was further demonstrated in ICR mice infected with rotavirus ECwt as the percentage of viral antigen-positive villus cells was significantly decreased by NAC treatment[185]. The use of NAC as a therapeutic tool for treatment of rotavirus disease in children was also demonstrated. Administration of NAC after the first diarrheal episode was shown to decrease the number of diarrheal episodes, excretion of fecal rotavirus antigen, and resolution of symptoms after 2 d of treatment[186].
There is demonstration that TNF-α stimulates HIV transcription through activation of NF-κB[246] and that this stimulation is inhibited by NAC treatment[247]. It has been also found that intracellular thiols regulate NF-κB activation since low thiol levels lead to its activation whilst high thiols levels inhibit its activation[248]. NAC has been shown to be a potent inhibitor of NF-κB activation in terms of inhibiting its nuclear migration and DNA binding activity in vascular endothelial cells[249]. NAC and other antioxidants have been reported to inhibit hydrogen peroxide-induced NF-κB activation[45]. Moreover, NAC has been reported to block NF-κB activation by interfering with IκB kinase (IKK) activation and inhibitor of κB phosphorylation, which suggested that ROS could be ubiquitous mediators of NF-κB activation[250,251]. However, it has been reported that NAC inhibits NF-κB activation in Hela and L929 cells independently of its anti-oxidative function. NAC seems to block selectively TNF-induced signaling by decreasing the affinity of receptor for TNF[252]. The NAC inhibitory effect on NF-κB activation appears to be a well established fact.
It has been reported that NAC inhibits the expression of VCAM-1 by interfering with the binding of NF-κB to the VCAM-1 κB motif[253,254]. Many harmful effects of TNF-α associated to endothelial dysfunction have been partially prevented by increasing GSH through NAC treatment[255]. NAC has also been reported to completely inhibit ROS, JNK and NF-κB activation induced by leptin, suggesting that hyperleptinemia is sensitive to redox signaling[256]. Attenuation of TNF-α-induced p38 mitogen-activated protein kinase (MAPK) activity in pulmonary vascular endothelial cells was obtained by NAC treatment, suggesting that p38 MAPK pathway is regulated by redox environment[257]. Activation of NF-κB in response to various signals, including IL-1, TNF and H2O2 can be inhibited by NAC treatment, suggesting that ROS are common signaling modulators[258]. Moreover, NAC was found to enhance the effect of IFN-α on liver tumor cells through inhibition of NF-κB[259]. On the other hand, some studies suggested that NAC inhibits the upstream IKK activation induced by TNF-α[260].
The NAC antiviral activity has mainly been associated with inhibition of pro-inflammatory molecules including those belonging to the NF-κB pathway and its associated generation of ROS. These findings suggest that inflammatory and oxidative stress pathways are intimately involved in the virus infection-associated pathogenesis. Nevertheless, the underlying mechanisms of NAC treatment of virus infections need further research in order to differentiate the direct and indirect effects associated with its antioxidant ability. A probable direct effect on disulfide bonds harbored in cellular and virus-encoded proteins cannot be excluded.
ROTAVIRUS INFECTION AND INFLAMMATORY SIGNALING
RNAs from rotavirus replication are sensed by RIG-I and MDA-5, which result in induction of an IFN-mediated innate immune response involving the activation of IRF3[48,49]. However, NSP1 from group A rotavirus is involved in evading innate immune response by antagonizing the induction of IFN and IFN-stimulated gene (ISG) products[261,262]. Rotavirus NSP1 has been shown to be involved in the evasion of innate immune response by interfering with the induction of IFN via induction of the degradation of IRF-3, IRF-5 and IRF-7. NSP1 from several rotavirus strains has been shown to target IRF3 for proteasome degradation during early post-infection[263-265]. Recent studies indicated that NSP1 can induce degradation of IRF proteins (IRF3 to IRF9) by targeting their IRF association domains needed for their dimerization and nuclear translocation[261]. Nevertheless, there is evidence indicating that NSP1 from some rotavirus strains such as OSU is inefficient in degrading IRF-3[266]. It has been reported that IRF3 is activated and remains stable in cells following infection with porcine rotavirus strain OSU. An alternative mechanism for blocking induction of IFN-β by rotavirus strain OSU has been recently reported[266]. Results from this work showed that NF-κB activation was blocked in cells infected with rotavirus strain OSU due in part to stabilization of phosphorylated IκBα. It was found that the SCFβ-TrCP E3 ligase was targeted for proteosome degradation by NSP1, which provided an explanation for the IκBα stabilization and the consequent absence of NF-κB activation in virus-infected cells. Most human group A rotaviruses encode NSP1s that contain a C-terminal recognition motif (DSGxS) for β-transducing repeat-containing protein (β-TrCP)[267]. This feature allows NSP1 to inhibit NF-κB activation by inducing proteasome-dependent degradation of β-TrCP[266]. Many NSP1s from group A rotaviruses that lack the β-TrCP recognition motif are then able to induce the degradation of IRF3, IRF5 and IRF7[261,263].
Regarding that rotaviral RNAs have the potential of triggering activation of IFN, it has been also suggested that sequestering of viral RNAs in the viroplasms and in the progeny capsids could contribute to delay the antiviral innate response in rotavirus-infected cells[50]. Moreover, it has been shown that infection of MA104 cells with rotavirus RRV is able to block expression of NF-κB-dependent gene expression without reducing NF-κB activation. This suggested that rotavirus can efficiently activate NF-κB in MA104 cells although this activated transcription factor was not functional in enhancing gene expression[268]. On the other hand, the role of NSP1 has been studied in modulation of apoptosis and it has been found that NSP1 contributes to the establishment and replication of bovine rotavirus wild type A5-13 in MA104 cells by inhibiting apoptosis through the activation of the pro-survival pathways PI3K/Akt and NF-κB during early infection stages[269].
Studies on malnutrition and concomitant rotavirus infection in neonatal piglets have suggested an inflammatory response during rotavirus infection. It has been shown that concentrations of intestinal prostaglandin E2 (PGE2) were elevated early after rotavirus infection regardless of nutritional state[270]. However, malnutrition increased PGE2 response to rotavirus infection while prolonged diarrhea in rotavirus infected and malnourished piglets was found to be associated with more intense and sustained expression of local mediators or markers of intestinal inflammation[270]. Rotavirus pro-inflammatory actions have been suggested based on studies in which the rotavirus infection of cultured cells or mice was significantly inhibited by treatment with various peroxisome proliferator-activated receptor gamma (PPARγ) agonists and nonsteroidal antiinflammatory drugs (NSAIDs)[184,185,271]. Cyclooxygenase-2 (COX-2), which is responsible for increased synthesis of prostaglandins[272], seems to be mainly regulated by various MAPKs and transcription factors such as NF-κB[273,274]. Moreover, PKA-mediated ERK1/2 and NF-κB pathways have been shown to be involved in the COX activity induction during rotavirus infection[271]. The nonspecific COX inhibitor indomethacin has been shown to significantly reduced rotavirus Wa infection of Caco-2 cells. Similarly, inhibition of the ERK1/2 and p38 MAPK pathways resulted in a significant decrease of rotavirus infection of Caco-2 cells[271]. Antiviral effects have been obtained by treatment with COX-2 inhibitors[275,276]. PPARγ ligands have been found to downregulate the transcriptional activation of COX-2 through multiple mechanisms[277], including the inhibition of multiple steps of the NF-κB pathway[278]. Evidence has been provided that rotavirus infectivity in MA104 and Caco-2 cells and mice is significantly inhibited not only by NAC, but also by pioglitazone and rosiglitazone which are drugs affecting the NF-κB pathway involved in the COX-2 transcriptional activation[184,185]. PPAR-γ agonists have been highlighted as potential therapeutic tools due to their ability to down-regulate the inflammatory responses to respiratory virus-related pulmonary inflammation[279]. PPARs participate antagonizing oxidant and inflammatory pathways such as NF-κB, AP1, and STAT[280,281]. Down-regulation of these signaling pathways by thiazolidine-2-4-diones (TZDs), including pioglitazone and rosiglitazone, has led to reduced levels of oxidative products in monocyte/macrophages[282]. PPARγ has emerged as an anti-inflammatory and antioxidant gene since its encoded product may directly modulate the expression of several antioxidant and pro-oxidant genes in response to oxidative stress[283-285]. However, it should be noted that oxidants such as ROS could interact with NF-κB signaling pathways in many ways. The transcription of genes depending on NF-κB influences the ROS levels, and in turn, the ROS levels also regulate the NF-κB activity levels. It has been argued that ROS influence is context-dependent and even cell-type specific being either positive or negative for NF-κB signaling[286].
Clearly rotavirus NSP1 has been implicated in down-regulating interferon expression being a key factor in the evasion of host innate immune response. However, the NSP1 mechanism for anti-interferon activity seems to be rotavirus strain-dependent. A more comprehensive understanding of the rotavirus pro-inflammatory actions could lead to identification of potential targets of anti-inflammatory therapeutics. Cellular innate response to rotavirus infection is schematized in Figure 3.
Figure 3.
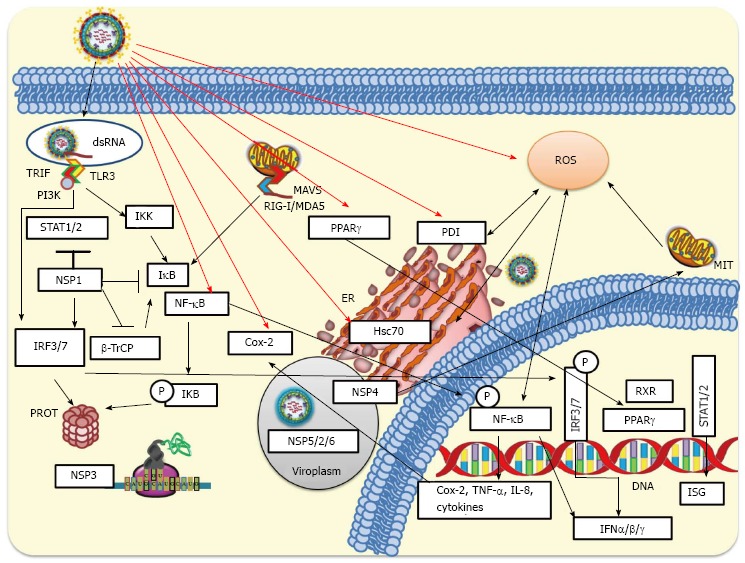
Cellular innate response to rotavirus infection. During rotavirus internalization viral nucleic acid may be exposed and recognized by either Toll-like receptors (TLR3) or intracellular RIG-I-like receptors (RLRs). Activated RLRs can bind and activate mitochondrial antiviral-signaling protein (MAVS), which recruits a signaling complex needed to activate cytoplasmic transcription factors including interferon regulatory factor 3 (IRF3) and nuclear factor-κB (NF-κB). On the other hand, activation of endosomal TLR3 facilitates the adaptor TRIF recruitment, which allows the recruitment of signaling molecules such as IKKs that phosphorylate IRF3 or NF-κB. Phosphorylated IRF3 is dimerized and then translocated to the nucleus. Signaling pathways induced by rotavirus infection produce phosphorylation of IκB (inhibitor of NF-κB) and its subsequent ubiquitination and proteasomal degradation mediated by SCFβ-TrCP E3 ligase. This signaling pathway leads to NF-κB translocation to the nucleus where, jointly with IRF3 and IRF7, binds to the interferon (IFN)-β promoter for transcription of IFN-β mRNA. Rotavirus can early counteract signaling pathways of innate response by NSP1-mediated degradation of IRF3 and IRF7. NSP1 encoded by some rotavirus strains can target SCFβ-TrCP for proteasomal degradation, whereas NSP1 from other strains has been implicated in the direct inhibition of the IFN-mediated STAT1 activation. NSP3 can interfere with the translation of cellular-encoded proteins including those induced by the IFN signaling. The viroplasm, which includes some viral non-structural proteins (NSP2/5/6), can protect viral RNAs from being recognized by some pattern-recognition receptors (RIG-I, MDA-5, among others) involved in antiviral response. MIT, ER and PROT are indicated. IKK: IκB kinase; MIT: Mitochondria; ER: Endoplasmic reticulum; PROT: Proteasome; TNF: Tumor necrosis factor.
ROTAVIRUS INFECTION AND PROTEIN SYNTHESIS
Viruses are fully dependent on the host cell translation machinery to produce their proteins needed for viral replication. Viruses take control of host ribosomes, translation factors and signaling pathways involved in protein synthesis. This control ensures the production of virus-encoded proteins and the inhibition of cellular innate defenses[287]. Most cellular mRNAs use a cap-dependent mechanism for their translation that involves the binding of a complex termed eIF4F comprised of eukaryotic initiation factors eIF4G, eIF4E and eIF4A to cap structure located at the 5’ end of the mRNA. However, some cellular and many viral mRNAs use a cap-independent mechanisms for initiating translation that involves an internal ribosome-entry site located in the 5’untranslated region of mRNAs that is use during ER stress[288]. Viruses have evolved a wide range of strategies for exploiting and controlling the cellular translation machinery. Several virus-encoded functions are dedicated to controlling the cellular translation machinery including its initiation, elongation and termination steps[289].
Early in the infection process rotaviruses takes over the host cell translation machinery, inducing a shut off of host cell-directed protein synthesis although not all cellular proteins stop being synthesized[290]. Rotavirus NSP3 has been implicated in the inhibition of cellular mRNA translation by binding to eIF4G or interfering with the shuttling of nascent cellular mRNAs[291,292]. Binding of NSP3 to eIF4G disturbs its interaction with poly(A)-binding protein which is required for the initiation of cellular mRNA translation. However, siRNA-mediated knockdown of NSP3 expression and a NSP3 defective mutant failed to interfere rotavirus-directed synthesis and its replication[293,294]. Rotavirus-induced phosphorylation of eIF2α in a double-stranded PKR-dependent manner has been reported to inhibit cellular translation[295]. However, the presence of naked RNA in rotavirus infected cells as part of the viral cycle is an unresolved question[50].
Increase of jejunal protein synthesis in rotavirus-infected piglets has been reported[296,297]. It was hypothesized that this increased protein synthesis was mainly due to actively proliferating enterocytes differentiating and migrating up the villus. However, it was suggested that rotavirus activates mTOR signaling through p70S6K since rotavirus-induced mobilization of calcium has been shown to be a stimulator of p70S6K[298]. However, in these studies it was not specified whether the increased protein synthesis was either cell or virus-directed. On the other hand, expression of some specific cellular proteins is increased during viral infections[299,300]. COX-2, an enzyme induced by pro-inflammatory agents, has been reported to be increased in infections caused by RSV[301], gammaherpesvirus 68[302], influenza virus[275], herpes simplex virus[301], and EMCV[303]. Despite Hsc70 is a constitutively expressed protein, it has been shown to be increased following infection with SV40[304], Autographa californica multiple nucleopolyhedrovirus[305], JEV[78], and white spot syndrome virus[306]. Evidence has been shown that rotavirus ECwt infection of mice induce cellular proteins COX-2, ERp57, Hsc70, NF-κB, Hsp70, PDI and PPARγ in intestinal villus cells, whereas NAC treatment of infected cells reduced Hsc70 and PDI to expression levels similar to those observed in villi from uninfected control mice[185]. The virus-associated increased expression of these cellular proteins adds evidence suggesting that rotavirus infection benefits from inducing oxidative stress and activating pro-inflammatory signaling in villus cells since treatment of rotavirus infected mice with NAC, NSAIDs or PPARγ agonists led to significantly reduced infection[185]. Inhibition of rotavirus infection by treatment with antioxidants, NSAIDs and PPARγ agonists are schematized in Figure 4.
Figure 4.
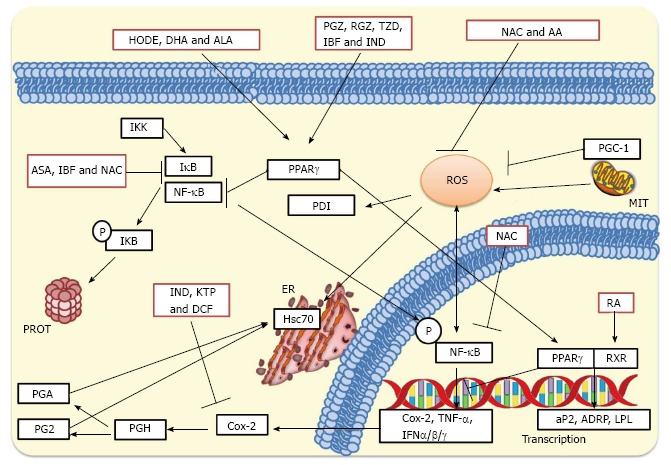
Inhibition of rotavirus infection by treatment with antioxidants, nonsteroidal antiinflammatory drugs and peroxisome proliferator-activated receptor gamma agonists. NAC and AA can inhibit the production of ROS, whereas NAC can also affect IκB preventing the cytoplasmic activation of NF-κB. NAC can further inhibit nuclear phosphorylated NF-κB preventing the transcription of pro-inflammatory genes. NSAIDs such as KTP, IND and DCF inhibit Cox-2 leading to a significant inhibition of prostaglandin accumulation. On the other hand, ASA and IBF inhibit activation of NF-κB suppressing the transcription of IFN-α, IFN-β and IFN-γ, cytokines and interleukins. These NSAID treatments significantly inhibit rotavirus infections in cultured cells and mice. PPARγ agonists such as 13(S)-hydroxyoctadecadienoic acid (HODE), ALA and DHA, and thiazolidinediones such as PGZ, RGZ, and 2, 4-thiazolidinedione (TZD) activate PPARγ leading to inhibition of cytoplasmic NF-κB. PPARγ can heterodimerize with the RA-activated RXR for promoting transcription of anti-inflammatory genes. This complex can also cause inhibition of phosphorylated NF-κB which in turn leads to decreased transcription of pro-inflammatory genes. MIT, ER, and PROT are indicated. NAC: N-acetylcysteine; NSAIDs: Nonsteroidal antiinflammatory drugs; PPARγ: Peroxisome proliferator-activated receptor gamma; AA: Ascorbic acid; ROS: Reactive oxygen species; NF-κB: Nuclear factor-κB; KTP: Ketoprofen; IND: Indomethacin; DCF: Diclofenac; Cox-2: Cyclooxygenase-2; ASA: Acetylsalicylic acid; IBF: Ibuprofen; IFN-α: Interferon-α; ALA: Alpha-linolenic acid; DHA: Docosahexaenoic acid; PGZ: Pioglitazone; RGZ: Rosiglitazone; RA: Retinoic acid; RXR: Retinoid X receptor; MIT: Mitochondria; ER: Endoplasmic reticulum; PROT: Proteasome.
The dependence of viruses on the host translation machinery imposes the recruitment of ribosomes for the translation of their functions and inhibition of the cellular innate defenses. More studies are needed to understand the detailed mechanisms involved in the strategy by which rotaviruses induce the shutoff of host protein synthesis machinery.
CONCLUSION
Despite the advances made over the past decade in the understanding of mechanisms explaining rotavirus infection, there are many unanswered questions regarding entry and internalization processes of rotavirus. A relevant question is whether rotaviruses have alternative entry pathways since inhibition of any of the proposed receptors failed to interfere completely with the viral infectivity. It is tempting to hypothesize that rotaviruses seem to have evolved to enter the target cell using three different types of cell surface molecules: (1) binding molecules represented by SA and some integrins; (2) chaperoning molecules including Hsc70 and other heat shock proteins; and (3) redox molecules such as PDI, Erp57 and other related thioredoxins. The current proposed receptors for rotavirus fall into these major categories of molecules supporting entry mechanisms. It should not be excluded that other molecules, as yet undiscovered, could also perform the same functions in other cell types and for other rotavirus strains. Within this line of reasoning, it appears to be a universal mechanism for rotavirus entry, but the receptor molecules executing the entry mechanism might differ partially or wholly depending on the species, cell line and rotavirus strain. The receptor usage and tropism of rotaviruses would be determined by the relative abundance and physical proximity of the receptors in the host cell surface. Rotavirus structural proteins implicated in the early steps of the rotaviral life cycle are likely to be substrates of the cell surface molecules having oxidoreductase, thiol isomerase and chaperone activities which would be responsible for the conformational changes these viral interacting proteins need for ensuring internalization. Future research should emphasize the elucidation of the reason why many receptors are used by rotaviruses. The fact that rotaviruses induce oxidative stress and inflammatory signaling offers an opportunity for the development of novel therapeutic strategies aimed at interfering with rotavirus infection. The use of NAC, NSAIDs and PPARγ agonists to inhibit rotavirus infection opens a new way for treating the life-threatening rotavirus diarrhea and complementing vaccines. However, a major gap in the understanding of the rotavirus infectious strategy is the fact that rotavirus seems to antagonize the pro-inflammatory signaling in order to ensure replication but anti-inflammatory treatment inhibits virus infection. This gap poses a substantial challenge because a more detailed characterization of the molecular mechanisms underpinning rotavirus-induced inflammatory signaling is needed. Another unsolved issue is that the rotavirus-induced oxidative stress, seemingly at the same time, influences positively and negatively NF-κB signaling, whereas antioxidant treatment inhibits virus infection.
ACKNOWLEDGMENTS
The authors are very grateful to Ana I Ramos-Murillo and Dory Gómez for their assistance in designing and preparing the figures.
Footnotes
Conflict-of-interest statement: The authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 13, 2015
First decision: September 28, 2015
Article in press: January 29, 2016
P- Reviewer: Davis DA, Roohvand F, Tugizov SM S- Editor: Gong XM L- Editor: A E- Editor: Lu YJ
References
- 1.Parashar UD, Hummelman EG, Bresee JS, Miller MA, Glass RI. Global illness and deaths caused by rotavirus disease in children. Emerg Infect Dis. 2003;9:565–572. doi: 10.3201/eid0905.020562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tate JE, Burton AH, Boschi-Pinto C, Steele AD, Duque J, Parashar UD. 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. Lancet Infect Dis. 2012;12:136–141. doi: 10.1016/S1473-3099(11)70253-5. [DOI] [PubMed] [Google Scholar]
- 3.Yeager M, Dryden KA, Olson NH, Greenberg HB, Baker TS. Three-dimensional structure of rhesus rotavirus by cryoelectron microscopy and image reconstruction. J Cell Biol. 1990;110:2133–2144. doi: 10.1083/jcb.110.6.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC. Atomic model of an infectious rotavirus particle. EMBO J. 2011;30:408–416. doi: 10.1038/emboj.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McClain B, Settembre E, Temple BR, Bellamy AR, Harrison SC. X-ray crystal structure of the rotavirus inner capsid particle at 3.8 A resolution. J Mol Biol. 2010;397:587–599. doi: 10.1016/j.jmb.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haselhorst T, Fleming FE, Dyason JC, Hartnell RD, Yu X, Holloway G, Santegoets K, Kiefel MJ, Blanchard H, Coulson BS, et al. Sialic acid dependence in rotavirus host cell invasion. Nat Chem Biol. 2009;5:91–93. doi: 10.1038/nchembio.134. [DOI] [PubMed] [Google Scholar]
- 7.Guerrero CA, Bouyssounade D, Zárate S, Isa P, López T, Espinosa R, Romero P, Méndez E, López S, Arias CF. Heat shock cognate protein 70 is involved in rotavirus cell entry. J Virol. 2002;76:4096–4102. doi: 10.1128/JVI.76.8.4096-4102.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zárate S, Cuadras MA, Espinosa R, Romero P, Juárez KO, Camacho-Nuez M, Arias CF, López S. Interaction of rotaviruses with Hsc70 during cell entry is mediated by VP5. J Virol. 2003;77:7254–7260. doi: 10.1128/JVI.77.13.7254-7260.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zárate S, Romero P, Espinosa R, Arias CF, López S. VP7 mediates the interaction of rotaviruses with integrin alphavbeta3 through a novel integrin-binding site. J Virol. 2004;78:10839–10847. doi: 10.1128/JVI.78.20.10839-10847.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham KL, Halasz P, Tan Y, Hewish MJ, Takada Y, Mackow ER, Robinson MK, Coulson BS. Integrin-using rotaviruses bind alpha2beta1 integrin alpha2 I domain via VP4 DGE sequence and recognize alphaXbeta2 and alphaVbeta3 by using VP7 during cell entry. J Virol. 2003;77:9969–9978. doi: 10.1128/JVI.77.18.9969-9978.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham KL, Fleming FE, Halasz P, Hewish MJ, Nagesha HS, Holmes IH, Takada Y, Coulson BS. Rotaviruses interact with alpha4beta7 and alpha4beta1 integrins by binding the same integrin domains as natural ligands. J Gen Virol. 2005;86:3397–3408. doi: 10.1099/vir.0.81102-0. [DOI] [PubMed] [Google Scholar]
- 12.Calderon MN, Guerrero CA, Acosta O, Lopez S, Arias CF. Inhibiting rotavirus infection by membrane-impermeant thiol/disulfide exchange blockers and antibodies against protein disulfide isomerase. Intervirology. 2012;55:451–464. doi: 10.1159/000335262. [DOI] [PubMed] [Google Scholar]
- 13.Lawton JA, Estes MK, Prasad BV. Three-dimensional visualization of mRNA release from actively transcribing rotavirus particles. Nat Struct Biol. 1997;4:118–121. doi: 10.1038/nsb0297-118. [DOI] [PubMed] [Google Scholar]
- 14.Petrie BL, Greenberg HB, Graham DY, Estes MK. Ultrastructural localization of rotavirus antigens using colloidal gold. Virus Res. 1984;1:133–152. doi: 10.1016/0168-1702(84)90069-8. [DOI] [PubMed] [Google Scholar]
- 15.Campagna M, Eichwald C, Vascotto F, Burrone OR. RNA interference of rotavirus segment 11 mRNA reveals the essential role of NSP5 in the virus replicative cycle. J Gen Virol. 2005;86:1481–1487. doi: 10.1099/vir.0.80598-0. [DOI] [PubMed] [Google Scholar]
- 16.López T, Camacho M, Zayas M, Nájera R, Sánchez R, Arias CF, López S. Silencing the morphogenesis of rotavirus. J Virol. 2005;79:184–192. doi: 10.1128/JVI.79.1.184-192.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Estes M, Kapikian A. Rotaviruses. In: Knipe D, Griffin D, Lamb R, Martin M, Roizman B, et al., editors. Fields of Virology. 5th ed. Philadelphia: Kluwer/Lippincott Williams and Wilkins; 2007. pp. 1917–1975. [Google Scholar]
- 18.Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct. 2012;2012:646354. doi: 10.1155/2012/646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol. 2007;292:R18–R36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 20.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Jorgenson TC, Zhong W, Oberley TD. Redox imbalance and biochemical changes in cancer. Cancer Res. 2013;73:6118–6123. doi: 10.1158/0008-5472.CAN-13-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oyinloye BE, Adenowo AF, Kappo AP. Reactive oxygen species, apoptosis, antimicrobial peptides and human inflammatory diseases. Pharmaceuticals (Basel) 2015;8:151–175. doi: 10.3390/ph8020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 25.Sies H. Oxidative stress: Introductory remarks. In: Sies H, et al., editors. Oxidative Stress. London: Academic Press; 1985. pp. 1–8. [Google Scholar]
- 26.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 27.Circu ML, Aw TY. Intestinal redox biology and oxidative stress. Semin Cell Dev Biol. 2012;23:729–737. doi: 10.1016/j.semcdb.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramig RF. Pathogenesis of intestinal and systemic rotavirus infection. J Virol. 2004;78:10213–10220. doi: 10.1128/JVI.78.19.10213-10220.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones DP, Go YM. Redox compartmentalization and cellular stress. Diabetes Obes Metab. 2010;12 Suppl 2:116–125. doi: 10.1111/j.1463-1326.2010.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aw TY, Williams MW. Intestinal absorption and lymphatic transport of peroxidized lipids in rats: effect of exogenous GSH. Am J Physiol. 1992;263:G665–G672. doi: 10.1152/ajpgi.1992.263.5.G665. [DOI] [PubMed] [Google Scholar]
- 31.Shan XQ, Aw TY, Jones DP. Glutathione-dependent protection against oxidative injury. Pharmacol Ther. 1990;47:61–71. doi: 10.1016/0163-7258(90)90045-4. [DOI] [PubMed] [Google Scholar]
- 32.Aw TY, Wierzbicka G, Jones DP. Oral glutathione increases tissue glutathione in vivo. Chem Biol Interact. 1991;80:89–97. doi: 10.1016/0009-2797(91)90033-4. [DOI] [PubMed] [Google Scholar]
- 33.Dahm LJ, Jones DP. Rat jejunum controls luminal thiol-disulfide redox. J Nutr. 2000;130:2739–2745. doi: 10.1093/jn/130.11.2739. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem. 2007;141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 35.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 36.Katze MG, He Y, Gale M. Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 37.Thompson AJ, Locarnini SA. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol Cell Biol. 2007;85:435–445. doi: 10.1038/sj.icb.7100100. [DOI] [PubMed] [Google Scholar]
- 38.Mason DR, Beck PL, Muruve DA. Nucleotide-binding oligomerization domain-like receptors and inflammasomes in the pathogenesis of non-microbial inflammation and diseases. J Innate Immun. 2012;4:16–30. doi: 10.1159/000334247. [DOI] [PubMed] [Google Scholar]
- 39.Gram AM, Frenkel J, Ressing ME. Inflammasomes and viruses: cellular defence versus viral offence. J Gen Virol. 2012;93:2063–2075. doi: 10.1099/vir.0.042978-0. [DOI] [PubMed] [Google Scholar]
- 40.O’Neill LA, Bowie AG. Sensing and signaling in antiviral innate immunity. Curr Biol. 2010;20:R328–R333. doi: 10.1016/j.cub.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 41.Yanai H, Mizutani T, Inuzuka T, Honda K, Takaoka A, Taniguchi T. IRF family transcription factors in type I interferon induction. International Congress. 2005;1285:104–113. [Google Scholar]
- 42.Yanai H, Chen HM, Inuzuka T, Kondo S, Mak TW, Takaoka A, Honda K, Taniguchi T. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc Natl Acad Sci USA. 2007;104:3402–3407. doi: 10.1073/pnas.0611559104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santoro MG, Rossi A, Amici C. NF-kappaB and virus infection: who controls whom. EMBO J. 2003;22:2552–2560. doi: 10.1093/emboj/cdg267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Onoguchi K, Yoneyama M, Fujita T. Retinoic acid-inducible gene-I-like receptors. J Interferon Cytokine Res. 2011;31:27–31. doi: 10.1089/jir.2010.0057. [DOI] [PubMed] [Google Scholar]
- 45.Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799:775–787. doi: 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 47.Magné N, Toillon RA, Bottero V, Didelot C, Houtte PV, Gérard JP, Peyron JF. NF-kappaB modulation and ionizing radiation: mechanisms and future directions for cancer treatment. Cancer Lett. 2006;231:158–168. doi: 10.1016/j.canlet.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 48.Broquet AH, Hirata Y, McAllister CS, Kagnoff MF. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J Immunol. 2011;186:1618–1626. doi: 10.4049/jimmunol.1002862. [DOI] [PubMed] [Google Scholar]
- 49.Sen A, Pruijssers AJ, Dermody TS, García-Sastre A, Greenberg HB. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J Virol. 2011;85:3717–3732. doi: 10.1128/JVI.02634-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnold MM, Sen A, Greenberg HB, Patton JT. The battle between rotavirus and its host for control of the interferon signaling pathway. PLoS Pathog. 2013;9:e1003064. doi: 10.1371/journal.ppat.1003064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deal EM, Jaimes MC, Crawford SE, Estes MK, Greenberg HB. Rotavirus structural proteins and dsRNA are required for the human primary plasmacytoid dendritic cell IFNalpha response. PLoS Pathog. 2010;6:e1000931. doi: 10.1371/journal.ppat.1000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lopez-Guerrero DV, Meza-Perez S, Ramirez-Pliego O, Santana-Calderon MA, Espino-Solis P, Gutierrez-Xicotencatl L, Flores-Romo L, Esquivel-Guadarrama FR. Rotavirus infection activates dendritic cells from Peyer’s patches in adult mice. J Virol. 2010;84:1856–1866. doi: 10.1128/JVI.02640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pott J, Stockinger S, Torow N, Smoczek A, Lindner C, McInerney G, Bäckhed F, Baumann U, Pabst O, Bleich A, et al. Age-dependent TLR3 expression of the intestinal epithelium contributes to rotavirus susceptibility. PLoS Pathog. 2012;8:e1002670. doi: 10.1371/journal.ppat.1002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Azim T, Zaki MH, Podder G, Sultana N, Salam MA, Rahman SM, Sefat-e-Khuda DA. Rotavirus-specific subclass antibody and cytokine responses in Bangladeshi children with rotavirus diarrhoea. J Med Virol. 2003;69:286–295. doi: 10.1002/jmv.10280. [DOI] [PubMed] [Google Scholar]
- 55.Jiang B, Snipes-Magaldi L, Dennehy P, Keyserling H, Holman RC, Bresee J, Gentsch J, Glass RI. Cytokines as mediators for or effectors against rotavirus disease in children. Clin Diagn Lab Immunol. 2003;10:995–1001. doi: 10.1128/CDLI.10.6.995-1001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vanden Broecke C, Schwers A, Dagenais L, Goossens A, Maenhoudt M, Pastoret PP, Werenne J. Interferon response in colostrum-deprived newborn calves infected with bovine rotavirus: its possible role in the control of the pathogenicity. Ann Rech Vet. 1984;15:29–34. [PubMed] [Google Scholar]
- 57.Broome RL, Vo PT, Ward RL, Clark HF, Greenberg HB. Murine rotavirus genes encoding outer capsid proteins VP4 and VP7 are not major determinants of host range restriction and virulence. J Virol. 1993;67:2448–2455. doi: 10.1128/jvi.67.5.2448-2455.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jessop CE, Watkins RH, Simmons JJ, Tasab M, Bulleid NJ. Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J Cell Sci. 2009;122:4287–4295. doi: 10.1242/jcs.059154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 60.Mamathambika BS, Bardwell JC. Disulfide-linked protein folding pathways. Annu Rev Cell Dev Biol. 2008;24:211–235. doi: 10.1146/annurev.cellbio.24.110707.175333. [DOI] [PubMed] [Google Scholar]
- 61.Oka OB, Yeoh HY, Bulleid NJ. Thiol-disulfide exchange between the PDI family of oxidoreductases negates the requirement for an oxidase or reductase for each enzyme. Biochem J. 2015;469:279–288. doi: 10.1042/BJ20141423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benham AM. The protein disulfide isomerase family: key players in health and disease. Antioxid Redox Signal. 2012;16:781–789. doi: 10.1089/ars.2011.4439. [DOI] [PubMed] [Google Scholar]
- 63.Coe H, Michalak M. ERp57, a multifunctional endoplasmic reticulum resident oxidoreductase. Int J Biochem Cell Biol. 2010;42:796–799. doi: 10.1016/j.biocel.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 64.Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol. 2002;193:154–163. doi: 10.1002/jcp.10172. [DOI] [PubMed] [Google Scholar]
- 65.Kozlov G, Määttänen P, Thomas DY, Gehring K. A structural overview of the PDI family of proteins. FEBS J. 2010;277:3924–3936. doi: 10.1111/j.1742-4658.2010.07793.x. [DOI] [PubMed] [Google Scholar]
- 66.Ryser HJ, Levy EM, Mandel R, DiSciullo GJ. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc Natl Acad Sci USA. 1994;91:4559–4563. doi: 10.1073/pnas.91.10.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karala AR, Ruddock LW. Bacitracin is not a specific inhibitor of protein disulfide isomerase. FEBS J. 2010;277:2454–2462. doi: 10.1111/j.1742-4658.2010.07660.x. [DOI] [PubMed] [Google Scholar]
- 68.Zhao G, Lu H, Li C. Proapoptotic activities of protein disulfide isomerase (PDI) and PDIA3 protein, a role of the Bcl-2 protein Bak. J Biol Chem. 2015;290:8949–8963. doi: 10.1074/jbc.M114.619353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laurindo FR, Araujo TL, Abrahão TB. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid Redox Signal. 2014;20:2755–2775. doi: 10.1089/ars.2013.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Santos CX, Stolf BS, Takemoto PV, Amanso AM, Lopes LR, Souza EB, Goto H, Laurindo FR. Protein disulfide isomerase (PDI) associates with NADPH oxidase and is required for phagocytosis of Leishmania chagasi promastigotes by macrophages. J Leukoc Biol. 2009;86:989–998. doi: 10.1189/jlb.0608354. [DOI] [PubMed] [Google Scholar]
- 71.Fernandes DC, Manoel AH, Wosniak J, Laurindo FR. Protein disulfide isomerase overexpression in vascular smooth muscle cells induces spontaneous preemptive NADPH oxidase activation and Nox1 mRNA expression: effects of nitrosothiol exposure. Arch Biochem Biophys. 2009;484:197–204. doi: 10.1016/j.abb.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 72.Parent R, Qu X, Petit MA, Beretta L. The heat shock cognate protein 70 is associated with hepatitis C virus particles and modulates virus infectivity. Hepatology. 2009;49:1798–1809. doi: 10.1002/hep.22852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sullivan CS, Pipas JM. The virus-chaperone connection. Virology. 2001;287:1–8. doi: 10.1006/viro.2001.1038. [DOI] [PubMed] [Google Scholar]
- 74.Chromy LR, Pipas JM, Garcea RL. Chaperone-mediated in vitro assembly of Polyomavirus capsids. Proc Natl Acad Sci USA. 2003;100:10477–10482. doi: 10.1073/pnas.1832245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watanabe K, Fuse T, Asano I, Tsukahara F, Maru Y, Nagata K, Kitazato K, Kobayashi N. Identification of Hsc70 as an influenza virus matrix protein (M1) binding factor involved in the virus life cycle. FEBS Lett. 2006;580:5785–5790. doi: 10.1016/j.febslet.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 76.Liu T, Daniels CK, Cao S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol Ther. 2012;136:354–374. doi: 10.1016/j.pharmthera.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 77.Dastoor Z, Dreyer J. Nuclear translocation and aggregate formation of heat shock cognate protein 70 (Hsc70) in oxidative stress and apoptosis. J Cell Sci. 2000;113(Pt 16):2845–2854. doi: 10.1242/jcs.113.16.2845. [DOI] [PubMed] [Google Scholar]
- 78.Chuang CK, Yang TH, Chen TH, Yang CF, Chen WJ. Heat shock cognate protein 70 isoform D is required for clathrin-dependent endocytosis of Japanese encephalitis virus in C6/36 cells. J Gen Virol. 2015;96:793–803. doi: 10.1099/jgv.0.000015. [DOI] [PubMed] [Google Scholar]
- 79.Lim JW, Kim KH, Kim H. NF-kappaB p65 regulates nuclear translocation of Ku70 via degradation of heat shock cognate protein 70 in pancreatic acinar AR42J cells. Int J Biochem Cell Biol. 2008;40:2065–2077. doi: 10.1016/j.biocel.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 80.Ou W, Silver J. Role of protein disulfide isomerase and other thiol-reactive proteins in HIV-1 envelope protein-mediated fusion. Virology. 2006;350:406–417. doi: 10.1016/j.virol.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 81.Auwerx J, Isacsson O, Söderlund J, Balzarini J, Johansson M, Lundberg M. Human glutaredoxin-1 catalyzes the reduction of HIV-1 gp120 and CD4 disulfides and its inhibition reduces HIV-1 replication. Int J Biochem Cell Biol. 2009;41:1269–1275. doi: 10.1016/j.biocel.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 82.Reiser K, François KO, Schols D, Bergman T, Jörnvall H, Balzarini J, Karlsson A, Lundberg M. Thioredoxin-1 and protein disulfide isomerase catalyze the reduction of similar disulfides in HIV gp120. Int J Biochem Cell Biol. 2012;44:556–562. doi: 10.1016/j.biocel.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 83.Wan SW, Lin CF, Lu YT, Lei HY, Anderson R, Lin YS. Endothelial cell surface expression of protein disulfide isomerase activates β1 and β3 integrins and facilitates dengue virus infection. J Cell Biochem. 2012;113:1681–1691. doi: 10.1002/jcb.24037. [DOI] [PubMed] [Google Scholar]
- 84.Smith JG, Cunningham JM. Receptor-induced thiolate couples Env activation to retrovirus fusion and infection. PLoS Pathog. 2007;3:e198. doi: 10.1371/journal.ppat.0030198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abell BA, Brown DT. Sindbis virus membrane fusion is mediated by reduction of glycoprotein disulfide bridges at the cell surface. J Virol. 1993;67:5496–5501. doi: 10.1128/jvi.67.9.5496-5501.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abou-Jaoudé G, Sureau C. Entry of hepatitis delta virus requires the conserved cysteine residues of the hepatitis B virus envelope protein antigenic loop and is blocked by inhibitors of thiol-disulfide exchange. J Virol. 2007;81:13057–13066. doi: 10.1128/JVI.01495-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jain S, McGinnes LW, Morrison TG. Thiol/disulfide exchange is required for membrane fusion directed by the Newcastle disease virus fusion protein. J Virol. 2007;81:2328–2339. doi: 10.1128/JVI.01940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jain S, McGinnes LW, Morrison TG. Role of thiol/disulfide exchange in newcastle disease virus entry. J Virol. 2009;83:241–249. doi: 10.1128/JVI.01407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jain S, McGinnes LW, Morrison TG. Overexpression of thiol/disulfide isomerases enhances membrane fusion directed by the Newcastle disease virus fusion protein. J Virol. 2008;82:12039–12048. doi: 10.1128/JVI.01406-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gilbert J, Ou W, Silver J, Benjamin T. Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J Virol. 2006;80:10868–10870. doi: 10.1128/JVI.01117-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ali Khan H, Mutus B. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front Chem. 2014;2:70. doi: 10.3389/fchem.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Parakh S, Atkin JD. Novel roles for protein disulphide isomerase in disease states: a double edged sword? Front Cell Dev Biol. 2015;3:30. doi: 10.3389/fcell.2015.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stegmann M, Metcalfe C, Barclay AN. Immunoregulation through membrane proteins modified by reducing conditions induced by immune reactions. Eur J Immunol. 2013;43:15–21. doi: 10.1002/eji.201242849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Calderón MN, Guzmán F, Acosta O, Guerrero CA. Rotavirus VP4 and VP7-derived synthetic peptides as potential substrates of protein disulfide isomerase lead to inhibition of rotavirus infection. Int J Pept Res Ther. 2012;18:373–382. [Google Scholar]
- 95.Lopez S, Arias CF. Early steps in rotavirus cell entry. Curr Top Microbiol Immunol. 2006;309:39–66. doi: 10.1007/3-540-30773-7_2. [DOI] [PubMed] [Google Scholar]
- 96.Isa P, Gutierrez M, Arias CF, Lopez S. Rotavirus cell entry. Future Virol. 2008;3:135–146. [Google Scholar]
- 97.Díaz-Salinas MA, Silva-Ayala D, López S, Arias CF. Rotaviruses reach late endosomes and require the cation-dependent mannose-6-phosphate receptor and the activity of cathepsin proteases to enter the cell. J Virol. 2014;88:4389–4402. doi: 10.1128/JVI.03457-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Díaz-Salinas MA, Romero P, Espinosa R, Hoshino Y, López S, Arias CF. The spike protein VP4 defines the endocytic pathway used by rotavirus to enter MA104 cells. J Virol. 2013;87:1658–1663. doi: 10.1128/JVI.02086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gutiérrez M, Isa P, Sánchez-San Martin C, Pérez-Vargas J, Espinosa R, Arias CF, López S. Different rotavirus strains enter MA104 cells through different endocytic pathways: the role of clathrin-mediated endocytosis. J Virol. 2010;84:9161–9169. doi: 10.1128/JVI.00731-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trask SD, Kim IS, Harrison SC, Dormitzer PR. A rotavirus spike protein conformational intermediate binds lipid bilayers. J Virol. 2010;84:1764–1770. doi: 10.1128/JVI.01682-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dormitzer PR, Nason EB, Prasad BV, Harrison SC. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature. 2004;430:1053–1058. doi: 10.1038/nature02836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rodríguez JM, Chichón FJ, Martín-Forero E, González-Camacho F, Carrascosa JL, Castón JR, Luque D. New insights into rotavirus entry machinery: stabilization of rotavirus spike conformation is independent of trypsin cleavage. PLoS Pathog. 2014;10:e1004157. doi: 10.1371/journal.ppat.1004157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gualtero DF, Guzmán F, Acosta O, Guerrero CA. Amino acid domains 280-297 of VP6 and 531-554 of VP4 are implicated in heat shock cognate protein hsc70-mediated rotavirus infection. Arch Virol. 2007;152:2183–2196. doi: 10.1007/s00705-007-1055-5. [DOI] [PubMed] [Google Scholar]
- 104.Isa P, Arias CF, López S. Role of sialic acids in rotavirus infection. Glycoconj J. 2006;23:27–37. doi: 10.1007/s10719-006-5435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zárate S, Espinosa R, Romero P, Méndez E, Arias CF, López S. The VP5 domain of VP4 can mediate attachment of rotaviruses to cells. J Virol. 2000;74:593–599. doi: 10.1128/jvi.74.2.593-599.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zárate S, Espinosa R, Romero P, Guerrero CA, Arias CF, López S. Integrin alpha2beta1 mediates the cell attachment of the rotavirus neuraminidase-resistant variant nar3. Virology. 2000;278:50–54. doi: 10.1006/viro.2000.0660. [DOI] [PubMed] [Google Scholar]
- 107.Fleming FE, Böhm R, Dang VT, Holloway G, Haselhorst T, Madge PD, Deveryshetty J, Yu X, Blanchard H, von Itzstein M, et al. Relative roles of GM1 ganglioside, N-acylneuraminic acids, and α2β1 integrin in mediating rotavirus infection. J Virol. 2014;88:4558–4571. doi: 10.1128/JVI.03431-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dormitzer PR, Sun ZY, Wagner G, Harrison SC. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 2002;21:885–897. doi: 10.1093/emboj/21.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Venkataram Prasad BV, Shanker S, Hu L, Choi JM, Crawford SE, Ramani S, Czako R, Atmar RL, Estes MK. Structural basis of glycan interaction in gastroenteric viral pathogens. Curr Opin Virol. 2014;7:119–127. doi: 10.1016/j.coviro.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hu L, Crawford SE, Czako R, Cortes-Penfield NW, Smith DF, Le Pendu J, Estes MK, Prasad BV. Cell attachment protein VP8* of a human rotavirus specifically interacts with A-type histo-blood group antigen. Nature. 2012;485:256–259. doi: 10.1038/nature10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu Y, Huang P, Tan M, Liu Y, Biesiada J, Meller J, Castello AA, Jiang B, Jiang X. Rotavirus VP8*: phylogeny, host range, and interaction with histo-blood group antigens. J Virol. 2012;86:9899–9910. doi: 10.1128/JVI.00979-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang P, Xia M, Tan M, Zhong W, Wei C, Wang L, Morrow A, Jiang X. Spike protein VP8* of human rotavirus recognizes histo-blood group antigens in a type-specific manner. J Virol. 2012;86:4833–4843. doi: 10.1128/JVI.05507-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guerrero CA, Méndez E, Zárate S, Isa P, López S, Arias CF. Integrin alpha(v)beta(3) mediates rotavirus cell entry. Proc Natl Acad Sci USA. 2000;97:14644–14649. doi: 10.1073/pnas.250299897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hewish MJ, Takada Y, Coulson BS. Integrins alpha2beta1 and alpha4beta1 can mediate SA11 rotavirus attachment and entry into cells. J Virol. 2000;74:228–236. doi: 10.1128/jvi.74.1.228-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ren J, Ding T, Zhang W, Song J, Ma W. Does Japanese encephalitis virus share the same cellular receptor with other mosquito-borne flaviviruses on the C6/36 mosquito cells? Virol J. 2007;4:83. doi: 10.1186/1743-422X-4-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Graham KL, Takada Y, Coulson BS. Rotavirus spike protein VP5* binds alpha2beta1 integrin on the cell surface and competes with virus for cell binding and infectivity. J Gen Virol. 2006;87:1275–1283. doi: 10.1099/vir.0.81580-0. [DOI] [PubMed] [Google Scholar]
- 117.Morimoto RI. Dynamic remodeling of transcription complexes by molecular chaperones. Cell. 2002;110:281–284. doi: 10.1016/s0092-8674(02)00860-7. [DOI] [PubMed] [Google Scholar]
- 118.Pérez-Vargas J, Romero P, López S, Arias CF. The peptide-binding and ATPase domains of recombinant hsc70 are required to interact with rotavirus and reduce its infectivity. J Virol. 2006;80:3322–3331. doi: 10.1128/JVI.80.7.3322-3331.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Delmas O, Durand-Schneider AM, Cohen J, Colard O, Trugnan G. Spike protein VP4 assembly with maturing rotavirus requires a postendoplasmic reticulum event in polarized caco-2 cells. J Virol. 2004;78:10987–10994. doi: 10.1128/JVI.78.20.10987-10994.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dowling W, Denisova E, LaMonica R, Mackow ER. Selective membrane permeabilization by the rotavirus VP5* protein is abrogated by mutations in an internal hydrophobic domain. J Virol. 2000;74:6368–6376. doi: 10.1128/jvi.74.14.6368-6376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Coulson BS, Londrigan SL, Lee DJ. Rotavirus contains integrin ligand sequences and a disintegrin-like domain that are implicated in virus entry into cells. Proc Natl Acad Sci USA. 1997;94:5389–5394. doi: 10.1073/pnas.94.10.5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.López S, Arias CF. Multistep entry of rotavirus into cells: a Versaillesque dance. Trends Microbiol. 2004;12:271–278. doi: 10.1016/j.tim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 123.Trask SD, Dormitzer PR. Assembly of highly infectious rotavirus particles recoated with recombinant outer capsid proteins. J Virol. 2006;80:11293–11304. doi: 10.1128/JVI.01346-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mandel R, Ryser HJ, Ghani F, Wu M, Peak D. Inhibition of a reductive function of the plasma membrane by bacitracin and antibodies against protein disulfide-isomerase. Proc Natl Acad Sci USA. 1993;90:4112–4116. doi: 10.1073/pnas.90.9.4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Couët J, de Bernard S, Loosfelt H, Saunier B, Milgrom E, Misrahi M. Cell surface protein disulfide-isomerase is involved in the shedding of human thyrotropin receptor ectodomain. Biochemistry. 1996;35:14800–14805. doi: 10.1021/bi961359w. [DOI] [PubMed] [Google Scholar]
- 126.Orlandi PA. Protein-disulfide isomerase-mediated reduction of the A subunit of cholera toxin in a human intestinal cell line. J Biol Chem. 1997;272:4591–4599. [PubMed] [Google Scholar]
- 127.Santana AY, Guerrero CA, Acosta O. Implication of Hsc70, PDI and integrin αvβ3 involvement during entry of the murine rotavirus ECwt into small-intestinal villi of suckling mice. Arch Virol. 2013;158:1323–1336. doi: 10.1007/s00705-013-1626-6. [DOI] [PubMed] [Google Scholar]
- 128.Isa P, Realpe M, Romero P, López S, Arias CF. Rotavirus RRV associates with lipid membrane microdomains during cell entry. Virology. 2004;322:370–381. doi: 10.1016/j.virol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 129.Swiatkowska M, Szymański J, Padula G, Cierniewski CS. Interaction and functional association of protein disulfide isomerase with alphaVbeta3 integrin on endothelial cells. FEBS J. 2008;275:1813–1823. doi: 10.1111/j.1742-4658.2008.06339.x. [DOI] [PubMed] [Google Scholar]
- 130.Lahav J, Wijnen EM, Hess O, Hamaia SW, Griffiths D, Makris M, Knight CG, Essex DW, Farndale RW. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood. 2003;102:2085–2092. doi: 10.1182/blood-2002-06-1646. [DOI] [PubMed] [Google Scholar]
- 131.Di Paolo NC, Miao EA, Iwakura Y, Murali-Krishna K, Aderem A, Flavell RA, Papayannopoulou T, Shayakhmetov DM. Virus binding to a plasma membrane receptor triggers interleukin-1 alpha-mediated proinflammatory macrophage response in vivo. Immunity. 2009;31:110–121. doi: 10.1016/j.immuni.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Browne A, Tookman LA, Ingemarsdotter CK, Bouwman RD, Pirlo K, Wang Y, McNeish IA, Lockley M. Pharmacological Inhibition of β3 Integrin Reduces the Inflammatory Toxicities Caused by Oncolytic Adenovirus without Compromising Anticancer Activity. Cancer Res. 2015;75:2811–2821. doi: 10.1158/0008-5472.CAN-14-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li W, Wang G, Liang W, Kang K, Guo K, Zhang Y. Integrin β3 is required in infection and proliferation of classical swine fever virus. PLoS One. 2014;9:e110911. doi: 10.1371/journal.pone.0110911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ning P, An L, Liang W, Zhang Y. Identification of inhibition of protein disulphide isomerase expression related to classical swine fever virus infection by using real-time PCR analysis. Biotechnol Equip. 2015;29:564–569. [Google Scholar]
- 135.Zhang JL, Wang JL, Gao N, Chen ZT, Tian YP, An J. Up-regulated expression of beta3 integrin induced by dengue virus serotype 2 infection associated with virus entry into human dermal microvascular endothelial cells. Biochem Biophys Res Commun. 2007;356:763–768. doi: 10.1016/j.bbrc.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 136.Halasz P, Holloway G, Turner SJ, Coulson BS. Rotavirus replication in intestinal cells differentially regulates integrin expression by a phosphatidylinositol 3-kinase-dependent pathway, resulting in increased cell adhesion and virus yield. J Virol. 2008;82:148–160. doi: 10.1128/JVI.01980-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang C, Li W, Ren J, Fang J, Ke H, Gong W, Feng W, Wang CC. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid Redox Signal. 2013;19:36–45. doi: 10.1089/ars.2012.4630. [DOI] [PubMed] [Google Scholar]
- 138.Mor-Cohen R. Disulfide Bonds as Regulators of Integrin Function in Thrombosis and Hemostasis. Antioxid Redox Signal. 2016;24:16–31. doi: 10.1089/ars.2014.6149. [DOI] [PubMed] [Google Scholar]
- 139.Patton JT, Hua J, Mansell EA. Location of intrachain disulfide bonds in the VP5* and VP8* trypsin cleavage fragments of the rhesus rotavirus spike protein VP4. J Virol. 1993;67:4848–4855. doi: 10.1128/jvi.67.8.4848-4855.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Svensson L, Dormitzer PR, von Bonsdorff CH, Maunula L, Greenberg HB. Intracellular manipulation of disulfide bond formation in rotavirus proteins during assembly. J Virol. 1994;68:5204–5215. doi: 10.1128/jvi.68.8.5204-5215.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Aoki ST, Settembre EC, Trask SD, Greenberg HB, Harrison SC, Dormitzer PR. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science. 2009;324:1444–1447. doi: 10.1126/science.1170481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mathieu M, Petitpas I, Navaza J, Lepault J, Kohli E, Pothier P, Prasad BV, Cohen J, Rey FA. Atomic structure of the major capsid protein of rotavirus: implications for the architecture of the virion. EMBO J. 2001;20:1485–1497. doi: 10.1093/emboj/20.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yoder JD, Trask SD, Vo TP, Binka M, Feng N, Harrison SC, Greenberg HB, Dormitzer PR. VP5* rearranges when rotavirus uncoats. J Virol. 2009;83:11372–11377. doi: 10.1128/JVI.01228-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cuadras MA, Méndez E, Arias CF, López S. A new cysteine in rotavirus VP4 participates in the formation of an alternate disulfide bond. J Gen Virol. 1998;79(Pt 11):2673–2677. doi: 10.1099/0022-1317-79-11-2673. [DOI] [PubMed] [Google Scholar]
- 145.Butera D, Cook KM, Chiu J, Wong JW, Hogg PJ. Control of blood proteins by functional disulfide bonds. Blood. 2014;123:2000–2007. doi: 10.1182/blood-2014-01-549816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Arias CF, Silva-Ayala D, López S. Rotavirus entry: a deep journey into the cell with several exits. J Virol. 2015;89:890–893. doi: 10.1128/JVI.01787-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Inoue T, Moore P, Tsai B. How viruses and toxins disassemble to enter host cells. Annu Rev Microbiol. 2011;65:287–305. doi: 10.1146/annurev-micro-090110-102855. [DOI] [PubMed] [Google Scholar]
- 148.Inoue T, Tsai B. How viruses use the endoplasmic reticulum for entry, replication, and assembly. Cold Spring Harb Perspect Biol. 2013;5:a013250. doi: 10.1101/cshperspect.a013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cuadras MA, Bordier BB, Zambrano JL, Ludert JE, Greenberg HB. Dissecting rotavirus particle-raft interaction with small interfering RNAs: insights into rotavirus transit through the secretory pathway. J Virol. 2006;80:3935–3946. doi: 10.1128/JVI.80.8.3935-3946.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Charpilienne A, Abad MJ, Michelangeli F, Alvarado F, Vasseur M, Cohen J, Ruiz MC. Solubilized and cleaved VP7, the outer glycoprotein of rotavirus, induces permeabilization of cell membrane vesicles. J Gen Virol. 1997;78(Pt 6):1367–1371. doi: 10.1099/0022-1317-78-6-1367. [DOI] [PubMed] [Google Scholar]
- 151.He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13:393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 152.Mirazimi A, Svensson L. Carbohydrates facilitate correct disulfide bond formation and folding of rotavirus VP7. J Virol. 1998;72:3887–3892. doi: 10.1128/jvi.72.5.3887-3892.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Maruri-Avidal L, López S, Arias CF. Endoplasmic reticulum chaperones are involved in the morphogenesis of rotavirus infectious particles. J Virol. 2008;82:5368–5380. doi: 10.1128/JVI.02751-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chemello ME, Aristimuño OC, Michelangeli F, Ruiz MC. Requirement for vacuolar H+ -ATPase activity and Ca2+ gradient during entry of rotavirus into MA104 cells. J Virol. 2002;76:13083–13087. doi: 10.1128/JVI.76.24.13083-13087.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gerasimenko JV, Tepikin AV, Petersen OH, Gerasimenko OV. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr Biol. 1998;8:1335–1338. doi: 10.1016/s0960-9822(07)00565-9. [DOI] [PubMed] [Google Scholar]
- 156.Ruiz MC, Abad MJ, Charpilienne A, Cohen J, Michelangeli F. Cell lines susceptible to infection are permeabilized by cleaved and solubilized outer layer proteins of rotavirus. J Gen Virol. 1997;78(Pt 11):2883–2893. doi: 10.1099/0022-1317-78-11-2883. [DOI] [PubMed] [Google Scholar]
- 157.Beaulieu JF. Differential expression of the VLA family of integrins along the crypt-villus axis in the human small intestine. J Cell Sci. 1992;102(Pt 3):427–436. doi: 10.1242/jcs.102.3.427. [DOI] [PubMed] [Google Scholar]
- 158.Hamilton TE, McClane SJ, Baldwin S, Burke C, Patel H, Rombeau JL, Raper SE. Efficient adenoviral-mediated murine neonatal small intestinal gene transfer is dependent on alpha(v) integrin expression. J Pediatr Surg. 1997;32:1695–1703. doi: 10.1016/s0022-3468(97)90508-x. [DOI] [PubMed] [Google Scholar]
- 159.Beck MA, Handy J, Levander OA. The role of oxidative stress in viral infections. Ann N Y Acad Sci. 2000;917:906–912. doi: 10.1111/j.1749-6632.2000.tb05456.x. [DOI] [PubMed] [Google Scholar]
- 160.Djordjević VB. Free radicals in cell biology. Int Rev Cytol. 2004;237:57–89. doi: 10.1016/S0074-7696(04)37002-6. [DOI] [PubMed] [Google Scholar]
- 161.Schwarz KB. Oxidative stress during viral infection: a review. Free Radic Biol Med. 1996;21:641–649. doi: 10.1016/0891-5849(96)00131-1. [DOI] [PubMed] [Google Scholar]
- 162.Zhang Y, Wang Z, Chen H, Chen Z, Tian Y. Antioxidants: potential antiviral agents for Japanese encephalitis virus infection. Int J Infect Dis. 2014;24:30–36. doi: 10.1016/j.ijid.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 163.Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science. 2013;340:701–706. doi: 10.1126/science.1233028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Akaike T. Role of free radicals in viral pathogenesis and mutation. Rev Med Virol. 2001;11:87–101. doi: 10.1002/rmv.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dobmeyer TS, Findhammer S, Dobmeyer JM, Klein SA, Raffel B, Hoelzer D, Helm EB, Kabelitz D, Rossol R. Ex vivo induction of apoptosis in lymphocytes is mediated by oxidative stress: role for lymphocyte loss in HIV infection. Free Radic Biol Med. 1997;22:775–785. doi: 10.1016/s0891-5849(96)00403-0. [DOI] [PubMed] [Google Scholar]
- 166.Knobil K, Choi AM, Weigand GW, Jacoby DB. Role of oxidants in influenza virus-induced gene expression. Am J Physiol. 1998;274:L134–L142. doi: 10.1152/ajplung.1998.274.1.L134. [DOI] [PubMed] [Google Scholar]
- 167.Dikici I, Mehmetoglu I, Dikici N, Bitirgen M, Kurban S. Investigation of oxidative stress and some antioxidants in patients with acute and chronic viral hepatitis B and the effect of interferon-alpha treatment. Clin Biochem. 2005;38:1141–1144. doi: 10.1016/j.clinbiochem.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 168.Korenaga M, Wang T, Li Y, Showalter LA, Chan T, Sun J, Weinman SA. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem. 2005;280:37481–37488. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- 169.Ano Y, Sakudo A, Kimata T, Uraki R, Sugiura K, Onodera T. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci Lett. 2010;469:39–43. doi: 10.1016/j.neulet.2009.11.040. [DOI] [PubMed] [Google Scholar]
- 170.Mochizuki H, Todokoro M, Arakawa H. RS virus-induced inflammation and the intracellular glutathione redox state in cultured human airway epithelial cells. Inflammation. 2009;32:252–264. doi: 10.1007/s10753-009-9128-0. [DOI] [PubMed] [Google Scholar]
- 171.Olagnier D, Peri S, Steel C, van Montfoort N, Chiang C, Beljanski V, Slifker M, He Z, Nichols CN, Lin R, et al. Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS Pathog. 2014;10:e1004566. doi: 10.1371/journal.ppat.1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gullberg RC, Jordan Steel J, Moon SL, Soltani E, Geiss BJ. Oxidative stress influences positive strand RNA virus genome synthesis and capping. Virology. 2015;475:219–229. doi: 10.1016/j.virol.2014.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liao SL, Raung SL, Chen CJ. Japanese encephalitis virus stimulates superoxide dismutase activity in rat glial cultures. Neurosci Lett. 2002;324:133–136. doi: 10.1016/s0304-3940(02)00236-7. [DOI] [PubMed] [Google Scholar]
- 174.Sodhi CP, Katyal R, Rana SV, Attri S, Singh V. Study of oxidative-stress in rotavirus infected infant mice. Indian J Med Res. 1996;104:245–249. [PubMed] [Google Scholar]
- 175.Borghan MA, Mori Y, El-Mahmoudy AB, Ito N, Sugiyama M, Takewaki T, Minamoto N. Induction of nitric oxide synthase by rotavirus enterotoxin NSP4: implication for rotavirus pathogenicity. J Gen Virol. 2007;88:2064–2072. doi: 10.1099/vir.0.82618-0. [DOI] [PubMed] [Google Scholar]
- 176.Rodríguez-Díaz J, Banasaz M, Istrate C, Buesa J, Lundgren O, Espinoza F, Sundqvist T, Rottenberg M, Svensson L. Role of nitric oxide during rotavirus infection. J Med Virol. 2006;78:979–985. doi: 10.1002/jmv.20650. [DOI] [PubMed] [Google Scholar]
- 177.Gac M, Bigda J, Vahlenkamp TW. Increased mitochondrial superoxide dismutase expression and lowered production of reactive oxygen species during rotavirus infection. Virology. 2010;404:293–303. doi: 10.1016/j.virol.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 178.Warner BB, Stuart L, Gebb S, Wispé JR. Redox regulation of manganese superoxide dismutase. Am J Physiol. 1996;271:L150–L158. doi: 10.1152/ajplung.1996.271.1.L150. [DOI] [PubMed] [Google Scholar]
- 179.Nogae C, Makino N, Hata T, Nogae I, Takahashi S, Suzuki K, Taniguchi N, Yanaga T. Interleukin 1 alpha-induced expression of manganous superoxide dismutase reduces myocardial reperfusion injury in the rat. J Mol Cell Cardiol. 1995;27:2091–2099. doi: 10.1016/s0022-2828(95)91155-3. [DOI] [PubMed] [Google Scholar]
- 180.Jones PL, Ping D, Boss JM. Tumor necrosis factor alpha and interleukin-1beta regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NF-kappaB. Mol Cell Biol. 1997;17:6970–6981. doi: 10.1128/mcb.17.12.6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004;15:767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 182.Kaneko M, Takahashi T, Niinuma Y, Nomura Y. Manganese superoxide dismutase is induced by endoplasmic reticulum stress through IRE1-mediated nuclear factor (NF)-kappaB and AP-1 activation. Biol Pharm Bull. 2004;27:1202–1206. doi: 10.1248/bpb.27.1202. [DOI] [PubMed] [Google Scholar]
- 183.Kumar De U, Mukherjee R, Nandi S, Patel BH, Dimri U, Ravishankar C, Verma AK. Alterations in oxidant/antioxidant balance, high-mobility group box 1 protein and acute phase response in cross-bred suckling piglets suffering from rotaviral enteritis. Trop Anim Health Prod. 2014;46:1127–1133. doi: 10.1007/s11250-014-0616-3. [DOI] [PubMed] [Google Scholar]
- 184.Guerrero CA, Murillo A, Acosta O. Inhibition of rotavirus infection in cultured cells by N-acetyl-cysteine, PPARγ agonists and NSAIDs. Antiviral Res. 2012;96:1–12. doi: 10.1016/j.antiviral.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 185.Guerrero CA, Paula Pardo VR, Rafael Guerrero OA. Inhibition of rotavirus ECwt infection in ICR suckling mice by N-acetylcysteine, peroxisome proliferator-activated receptor gamma agonists and cyclooxygenase-2 inhibitors. Mem Inst Oswaldo Cruz. 2013;108:741–754. doi: 10.1590/0074-0276108062013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Guerrero CA, Torres DP, García LL, Guerrero RA, Acosta O. N-Acetylcysteine treatment of rotavirus-associated diarrhea in children. Pharmacotherapy. 2014;34:e333–e340. doi: 10.1002/phar.1489. [DOI] [PubMed] [Google Scholar]
- 187.Luo K, Cao SS. Endoplasmic reticulum stress in intestinal epithelial cell function and inflammatory bowel disease. Gastroenterol Res Pract. 2015;2015:328791. doi: 10.1155/2015/328791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kaufman RJ. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem Sci. 2004;29:152–158. doi: 10.1016/j.tibs.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 189.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci USA. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. 2014;8:213. doi: 10.3389/fncel.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bhandary B, Marahatta A, Kim HR, Chae HJ. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci. 2012;14:434–456. doi: 10.3390/ijms14010434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014;21:396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 194.van der Vlies D, Makkinje M, Jansens A, Braakman I, Verkleij AJ, Wirtz KW, Post JA. Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid Redox Signal. 2003;5:381–387. doi: 10.1089/152308603768295113. [DOI] [PubMed] [Google Scholar]
- 195.Sideraki V, Gilbert HF. Mechanism of the antichaperone activity of protein disulfide isomerase: facilitated assembly of large, insoluble aggregates of denatured lysozyme and PDI. Biochemistry. 2000;39:1180–1188. doi: 10.1021/bi992246q. [DOI] [PubMed] [Google Scholar]
- 196.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 197.Gross E, Kastner DB, Kaiser CA, Fass D. Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell. 2004;117:601–610. doi: 10.1016/s0092-8674(04)00418-0. [DOI] [PubMed] [Google Scholar]
- 198.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 199.Fung TS, Liu DX. Coronavirus infection, ER stress, apoptosis and innate immunity. Front Microbiol. 2014;5:296. doi: 10.3389/fmicb.2014.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nieva JL, Madan V, Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol. 2012;10:563–574. doi: 10.1038/nrmicro2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Crawford SE, Hyser JM, Utama B, Estes MK. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase-β signaling is required for rotavirus replication. Proc Natl Acad Sci USA. 2012;109:E3405–E3413. doi: 10.1073/pnas.1216539109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhang L, Wang A. Virus-induced ER stress and the unfolded protein response. Front Plant Sci. 2012;3:293. doi: 10.3389/fpls.2012.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jheng JR, Ho JY, Horng JT. ER stress, autophagy, and RNA viruses. Front Microbiol. 2014;5:388. doi: 10.3389/fmicb.2014.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol. 2005;79:6890–6899. doi: 10.1128/JVI.79.11.6890-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Pavio N, Romano PR, Graczyk TM, Feinstone SM, Taylor DR. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J Virol. 2003;77:3578–3585. doi: 10.1128/JVI.77.6.3578-3585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Tardif KD, Mori K, Siddiqui A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J Virol. 2002;76:7453–7459. doi: 10.1128/JVI.76.15.7453-7459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Fung TS, Torres J, Liu DX. The Emerging Roles of Viroporins in ER Stress Response and Autophagy Induction during Virus Infection. Viruses. 2015;7:2834–2857. doi: 10.3390/v7062749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol. 2011;3:pii: a004317. doi: 10.1101/cshperspect.a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hyser JM, Collinson-Pautz MR, Utama B, Estes MK. Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. MBio. 2010;1:pii: e00265–10. doi: 10.1128/mBio.00265-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Guo HC, Sun SQ, Sun DH, Wei YQ, Xu J, Huang M, Liu XT, Liu ZX, Luo JX, Yin H, et al. Viroporin activity and membrane topology of classic swine fever virus p7 protein. Int J Biochem Cell Biol. 2013;45:1186–1194. doi: 10.1016/j.biocel.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 211.Bhowmick R, Halder UC, Chattopadhyay S, Chanda S, Nandi S, Bagchi P, Nayak MK, Chakrabarti O, Kobayashi N, Chawla-Sarkar M. Rotaviral enterotoxin nonstructural protein 4 targets mitochondria for activation of apoptosis during infection. J Biol Chem. 2012;287:35004–35020. doi: 10.1074/jbc.M112.369595. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 212.Ao D, Guo HC, Sun SQ, Sun DH, Fung TS, Wei YQ, Han SC, Yao XP, Cao SZ, Liu DX, et al. Viroporin Activity of the Foot-and-Mouth Disease Virus Non-Structural 2B Protein. PLoS One. 2015;10:e0125828. doi: 10.1371/journal.pone.0125828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Berkova Z, Crawford SE, Trugnan G, Yoshimori T, Morris AP, Estes MK. Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J Virol. 2006;80:6061–6071. doi: 10.1128/JVI.02167-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Trujillo-Alonso V, Maruri-Avidal L, Arias CF, López S. Rotavirus infection induces the unfolded protein response of the cell and controls it through the nonstructural protein NSP3. J Virol. 2011;85:12594–12604. doi: 10.1128/JVI.05620-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Chan CP, Siu KL, Chin KT, Yuen KY, Zheng B, Jin DY. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2006;80:9279–9287. doi: 10.1128/JVI.00659-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yu CY, Hsu YW, Liao CL, Lin YL. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J Virol. 2006;80:11868–11880. doi: 10.1128/JVI.00879-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Xuan B, Qian Z, Torigoi E, Yu D. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J Virol. 2009;83:3463–3474. doi: 10.1128/JVI.02307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ambrose RL, Mackenzie JM. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J Virol. 2011;85:2723–2732. doi: 10.1128/JVI.02050-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Field M. Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest. 2003;111:931–943. doi: 10.1172/JCI18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tian P, Estes MK, Hu Y, Ball JM, Zeng CQ, Schilling WP. The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+ from the endoplasmic reticulum. J Virol. 1995;69:5763–5772. doi: 10.1128/jvi.69.9.5763-5772.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ball JM, Tian P, Zeng CQ, Morris AP, Estes MK. Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science. 1996;272:101–104. doi: 10.1126/science.272.5258.101. [DOI] [PubMed] [Google Scholar]
- 222.Ousingsawat J, Mirza M, Tian Y, Roussa E, Schreiber R, Cook DI, Kunzelmann K. Rotavirus toxin NSP4 induces diarrhea by activation of TMEM16A and inhibition of Na+ absorption. Pflugers Arch. 2011;461:579–589. doi: 10.1007/s00424-011-0947-0. [DOI] [PubMed] [Google Scholar]
- 223.Lorrot M, Vasseur M. How do the rotavirus NSP4 and bacterial enterotoxins lead differently to diarrhea? Virol J. 2007;4:31. doi: 10.1186/1743-422X-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Buccigrossi V, Laudiero G, Russo C, Miele E, Sofia M, Monini M, Ruggeri FM, Guarino A. Chloride secretion induced by rotavirus is oxidative stress-dependent and inhibited by Saccharomyces boulardii in human enterocytes. PLoS One. 2014;9:e99830. doi: 10.1371/journal.pone.0099830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Denning TL, Takaishi H, Crowe SE, Boldogh I, Jevnikar A, Ernst PB. Oxidative stress induces the expression of Fas and Fas ligand and apoptosis in murine intestinal epithelial cells. Free Radic Biol Med. 2002;33:1641–1650. doi: 10.1016/s0891-5849(02)01141-3. [DOI] [PubMed] [Google Scholar]
- 226.Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine--a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol. 2007;7:355–359. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Cotgreave IA. N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv Pharmacol. 1997;38:205–227. [PubMed] [Google Scholar]
- 228.Aitio ML. N-acetylcysteine -- passe-partout or much ado about nothing? Br J Clin Pharmacol. 2006;61:5–15. doi: 10.1111/j.1365-2125.2005.02523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sparks B, Kesavan A. Treatment of a gastric lactobezoar with N-acetylcysteine. Case Rep Gastrointest Med. 2014;2014:254741. doi: 10.1155/2014/254741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Song JW, Shim JK, Soh S, Jang J, Kwak YL. Double-blinded, randomized controlled trial of N-acetylcysteine for prevention of acute kidney injury in high risk patients undergoing off-pump coronary artery bypass. Nephrology (Carlton) 2015;20:96–102. doi: 10.1111/nep.12361. [DOI] [PubMed] [Google Scholar]
- 231.Mahmoud KM, Ammar AS. Effect of N-acetylcysteine on cardiac injury and oxidative stress after abdominal aortic aneurysm repair: a randomized controlled trial. Acta Anaesthesiol Scand. 2011;55:1015–1021. doi: 10.1111/j.1399-6576.2011.02492.x. [DOI] [PubMed] [Google Scholar]
- 232.Sakamoto S, Muramatsu Y, Satoh K, Ishida F, Kikuchi N, Sano G, Sugino K, Isobe K, Takai Y, Homma S. Effectiveness of combined therapy with pirfenidone and inhaled N-acetylcysteine for advanced idiopathic pulmonary fibrosis: a case-control study. Respirology. 2015;20:445–452. doi: 10.1111/resp.12477. [DOI] [PubMed] [Google Scholar]
- 233.El Rahi C, Thompson-Moore N, Mejia P, De Hoyos P. Successful use of N-acetylcysteine to treat severe hepatic injury caused by a dietary fitness supplement. Pharmacotherapy. 2015;35:e96–e101. doi: 10.1002/phar.1572. [DOI] [PubMed] [Google Scholar]
- 234.Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 235.De Flora S, Grassi C, Carati L. Attenuation of influenza-like symptomatology and improvement of cell-mediated immunity with long-term N-acetylcysteine treatment. Eur Respir J. 1997;10:1535–1541. doi: 10.1183/09031936.97.10071535. [DOI] [PubMed] [Google Scholar]
- 236.Cai J, Chen Y, Seth S, Furukawa S, Compans RW, Jones DP. Inhibition of influenza infection by glutathione. Free Radic Biol Med. 2003;34:928–936. doi: 10.1016/s0891-5849(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 237.Garozzo A, Tempera G, Ungheri D, Timpanaro R, Castro A. N-acetylcysteine synergizes with oseltamivir in protecting mice from lethal influenza infection. Int J Immunopathol Pharmacol. 2007;20:349–354. doi: 10.1177/039463200702000215. [DOI] [PubMed] [Google Scholar]
- 238.Ghezzi P, Ungheri D. Synergistic combination of N-acetylcysteine and ribavirin to protect from lethal influenza viral infection in a mouse model. Int J Immunopathol Pharmacol. 2004;17:99–102. doi: 10.1177/039463200401700114. [DOI] [PubMed] [Google Scholar]
- 239.Lai KY, Ng WY, Osburga Chan PK, Wong KF, Cheng F. High-dose N-acetylcysteine therapy for novel H1N1 influenza pneumonia. Ann Intern Med. 2010;152:687–688. doi: 10.7326/0003-4819-152-10-201005180-00017. [DOI] [PubMed] [Google Scholar]
- 240.Geiler J, Michaelis M, Naczk P, Leutz A, Langer K, Doerr HW, Cinatl J. N-acetyl-L-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem Pharmacol. 2010;79:413–420. doi: 10.1016/j.bcp.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 241.Garigliany MM, Desmecht DJ. N-acetylcysteine lacks universal inhibitory activity against influenza A viruses. J Negat Results Biomed. 2011;10:5. doi: 10.1186/1477-5751-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Senanayake MP, Jayamanne MD, Kankananarachchi I. N-acetylcysteine in children with acute liver failure complicating dengue viral infection. Ceylon Med J. 2013;58:80–82. doi: 10.4038/cmj.v58i2.5684. [DOI] [PubMed] [Google Scholar]
- 243.Herzenberg LA, De Rosa SC, Dubs JG, Roederer M, Anderson MT, Ela SW, Deresinski SC, Herzenberg LA. Glutathione deficiency is associated with impaired survival in HIV disease. Proc Natl Acad Sci USA. 1997;94:1967–1972. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Dröge W, Breitkreutz R. N-acetyl-cysteine in the therapy of HIV-positive patients. Curr Opin Clin Nutr Metab Care. 1999;2:493–498. doi: 10.1097/00075197-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 245.De Rosa SC, Zaretsky MD, Dubs JG, Roederer M, Anderson M, Green A, Mitra D, Watanabe N, Nakamura H, Tjioe I, et al. N-acetylcysteine replenishes glutathione in HIV infection. Eur J Clin Invest. 2000;30:915–929. doi: 10.1046/j.1365-2362.2000.00736.x. [DOI] [PubMed] [Google Scholar]
- 246.Duh EJ, Maury WJ, Folks TM, Fauci AS, Rabson AB. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc Natl Acad Sci USA. 1989;86:5974–5978. doi: 10.1073/pnas.86.15.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Roederer M, Staal FJ, Raju PA, Ela SW, Herzenberg LA, Herzenberg LA. Cytokine-stimulated human immunodeficiency virus replication is inhibited by N-acetyl-L-cysteine. Proc Natl Acad Sci USA. 1990;87:4884–4888. doi: 10.1073/pnas.87.12.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Staal FJ, Roederer M, Herzenberg LA, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci USA. 1990;87:9943–9947. doi: 10.1073/pnas.87.24.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Schubert SY, Neeman I, Resnick N. A novel mechanism for the inhibition of NF-kappaB activation in vascular endothelial cells by natural antioxidants. FASEB J. 2002;16:1931–1933. doi: 10.1096/fj.02-0147fje. [DOI] [PubMed] [Google Scholar]
- 250.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 251.Flohé L, Brigelius-Flohé R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- 252.Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K. Evidence that reactive oxygen species do not mediate NF-kappaB activation. EMBO J. 2003;22:3356–3366. doi: 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, Medford RM. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest. 1993;92:1866–1874. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Faruqi RM, Poptic EJ, Faruqi TR, De La Motte C, DiCorleto PE. Distinct mechanisms for N-acetylcysteine inhibition of cytokine-induced E-selectin and VCAM-1 expression. Am J Physiol. 1997;273:H817–H826. doi: 10.1152/ajpheart.1997.273.2.H817. [DOI] [PubMed] [Google Scholar]
- 255.Toborek M, Barger SW, Mattson MP, McClain CJ, Hennig B. Role of glutathione redox cycle in TNF-alpha-mediated endothelial cell dysfunction. Atherosclerosis. 1995;117:179–188. doi: 10.1016/0021-9150(95)05568-h. [DOI] [PubMed] [Google Scholar]
- 256.Bouloumie A, Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB J. 1999;13:1231–1238. [PubMed] [Google Scholar]
- 257.Hashimoto S, Gon Y, Matsumoto K, Takeshita I, Horie T. N-acetylcysteine attenuates TNF-alpha-induced p38 MAP kinase activation and p38 MAP kinase-mediated IL-8 production by human pulmonary vascular endothelial cells. Br J Pharmacol. 2001;132:270–276. doi: 10.1038/sj.bjp.0703787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kretzmann NA, Chiela E, Matte U, Marroni N, Marroni CA. N-acetylcysteine improves antitumoural response of Interferon alpha by NF-kB downregulation in liver cancer cells. Comp Hepatol. 2012;11:4. doi: 10.1186/1476-5926-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Li YQ, Zhang ZX, Xu YJ, Ni W, Chen SX, Yang Z, Ma D. N-Acetyl-L-cysteine and pyrrolidine dithiocarbamate inhibited nuclear factor-kappaB activation in alveolar macrophages by different mechanisms. Acta Pharmacol Sin. 2006;27:339–346. doi: 10.1111/j.1745-7254.2006.00264.x. [DOI] [PubMed] [Google Scholar]
- 261.Arnold MM, Barro M, Patton JT. Rotavirus NSP1 mediates degradation of interferon regulatory factors through targeting of the dimerization domain. J Virol. 2013;87:9813–9821. doi: 10.1128/JVI.01146-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hu L, Crawford SE, Hyser JM, Estes MK, Prasad BV. Rotavirus non-structural proteins: structure and function. Curr Opin Virol. 2012;2:380–388. doi: 10.1016/j.coviro.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Barro M, Patton JT. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol. 2007;81:4473–4481. doi: 10.1128/JVI.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Feng N, Sen A, Nguyen H, Vo P, Hoshino Y, Deal EM, Greenberg HB. Variation in antagonism of the interferon response to rotavirus NSP1 results in differential infectivity in mouse embryonic fibroblasts. J Virol. 2009;83:6987–6994. doi: 10.1128/JVI.00585-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Sen A, Feng N, Ettayebi K, Hardy ME, Greenberg HB. IRF3 inhibition by rotavirus NSP1 is host cell and virus strain dependent but independent of NSP1 proteasomal degradation. J Virol. 2009;83:10322–10335. doi: 10.1128/JVI.01186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Graff JW, Ettayebi K, Hardy ME. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 2009;5:e1000280. doi: 10.1371/journal.ppat.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Mansur DS, Maluquer de Motes C, Unterholzner L, Sumner RP, Ferguson BJ, Ren H, Strnadova P, Bowie AG, Smith GL. Poxvirus targeting of E3 ligase β-TrCP by molecular mimicry: a mechanism to inhibit NF-κB activation and promote immune evasion and virulence. PLoS Pathog. 2013;9:e1003183. doi: 10.1371/journal.ppat.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Holloway G, Truong TT, Coulson BS. Rotavirus antagonizes cellular antiviral responses by inhibiting the nuclear accumulation of STAT1, STAT2, and NF-kappaB. J Virol. 2009;83:4942–4951. doi: 10.1128/JVI.01450-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Bagchi P, Dutta D, Chattopadhyay S, Mukherjee A, Halder UC, Sarkar S, Kobayashi N, Komoto S, Taniguchi K, Chawla-Sarkar M. Rotavirus nonstructural protein 1 suppresses virus-induced cellular apoptosis to facilitate viral growth by activating the cell survival pathways during early stages of infection. J Virol. 2010;84:6834–6845. doi: 10.1128/JVI.00225-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zijlstra RT, McCracken BA, Odle J, Donovan SM, Gelberg HB, Petschow BW, Zuckermann FA, Gaskins HR. Malnutrition modifies pig small intestinal inflammatory responses to rotavirus. J Nutr. 1999;129:838–843. doi: 10.1093/jn/129.4.838. [DOI] [PubMed] [Google Scholar]
- 271.Rossen JW, Bouma J, Raatgeep RH, Büller HA, Einerhand AW. Inhibition of cyclooxygenase activity reduces rotavirus infection at a postbinding step. J Virol. 2004;78:9721–9730. doi: 10.1128/JVI.78.18.9721-9730.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Marnett LJ, Kalgutkar AS. Cyclooxygenase 2 inhibitors: discovery, selectivity and the future. Trends Pharmacol Sci. 1999;20:465–469. doi: 10.1016/s0165-6147(99)01385-1. [DOI] [PubMed] [Google Scholar]
- 273.Bartlett SR, Sawdy R, Mann GE. Induction of cyclooxygenase-2 expression in human myometrial smooth muscle cells by interleukin-1beta: involvement of p38 mitogen-activated protein kinase. J Physiol. 1999;520 Pt 2:399–406. doi: 10.1111/j.1469-7793.1999.00399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Subbaramaiah K, Hart JC, Norton L, Dannenberg AJ. Microtubule-interfering agents stimulate the transcription of cyclooxygenase-2. Evidence for involvement of ERK1/2 AND p38 mitogen-activated protein kinase pathways. J Biol Chem. 2000;275:14838–14845. doi: 10.1074/jbc.275.20.14838. [DOI] [PubMed] [Google Scholar]
- 275.Lee SM, Gai WW, Cheung TK, Peiris JS. Antiviral effect of a selective COX-2 inhibitor on H5N1 infection in vitro. Antiviral Res. 2011;91:330–334. doi: 10.1016/j.antiviral.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 276.Carey MA, Bradbury JA, Rebolloso YD, Graves JP, Zeldin DC, Germolec DR. Pharmacologic inhibition of COX-1 and COX-2 in influenza A viral infection in mice. PLoS One. 2010;5:e11610. doi: 10.1371/journal.pone.0011610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Subbaramaiah K, Lin DT, Hart JC, Dannenberg AJ. Peroxisome proliferator-activated receptor gamma ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. J Biol Chem. 2001;276:12440–12448. doi: 10.1074/jbc.M007237200. [DOI] [PubMed] [Google Scholar]
- 278.Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci USA. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Bassaganya-Riera J, Song R, Roberts PC, Hontecillas R. PPAR-gamma activation as an anti-inflammatory therapy for respiratory virus infections. Viral Immunol. 2010;23:343–352. doi: 10.1089/vim.2010.0016. [DOI] [PubMed] [Google Scholar]
- 280.Liu J, Xia Q, Zhang Q, Li H, Zhang J, Li A, Xiu R. Peroxisome proliferator-activated receptor-gamma ligands 15-deoxy-delta(12,14)-prostaglandin J2 and pioglitazone inhibit hydroxyl peroxide-induced TNF-alpha and lipopolysaccharide-induced CXC chemokine expression in neonatal rat cardiac myocytes. Shock. 2009;32:317–324. doi: 10.1097/SHK.0b013e31819c374c. [DOI] [PubMed] [Google Scholar]
- 281.Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol. 2000;20:4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 283.Polvani S, Tarocchi M, Galli A. PPARγ and Oxidative Stress: Con(β) Catenating NRF2 and FOXO. PPAR Res. 2012;2012:641087. doi: 10.1155/2012/641087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Okuno Y, Matsuda M, Miyata Y, Fukuhara A, Komuro R, Shimabukuro M, Shimomura I. Human catalase gene is regulated by peroxisome proliferator activated receptor-gamma through a response element distinct from that of mouse. Endocr J. 2010;57:303–309. doi: 10.1507/endocrj.k09e-113. [DOI] [PubMed] [Google Scholar]
- 285.Ren Y, Sun C, Sun Y, Tan H, Wu Y, Cui B, Wu Z. PPAR gamma protects cardiomyocytes against oxidative stress and apoptosis via Bcl-2 upregulation. Vascul Pharmacol. 2009;51:169–174. doi: 10.1016/j.vph.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 286.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Walsh D, Mohr I. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol. 2011;9:860–875. doi: 10.1038/nrmicro2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 289.Walsh D, Mathews MB, Mohr I. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol. 2013;5:a012351. doi: 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Firth AE, Brierley I. Non-canonical translation in RNA viruses. J Gen Virol. 2012;93:1385–1409. doi: 10.1099/vir.0.042499-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Piron M, Vende P, Cohen J, Poncet D. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J. 1998;17:5811–5821. doi: 10.1093/emboj/17.19.5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Deo RC, Groft CM, Rajashankar KR, Burley SK. Recognition of the rotavirus mRNA 3’ consensus by an asymmetric NSP3 homodimer. Cell. 2002;108:71–81. doi: 10.1016/s0092-8674(01)00632-8. [DOI] [PubMed] [Google Scholar]
- 293.Montero H, Arias CF, Lopez S. Rotavirus Nonstructural Protein NSP3 is not required for viral protein synthesis. J Virol. 2006;80:9031–9038. doi: 10.1128/JVI.00437-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Arnold MM, Brownback CS, Taraporewala ZF, Patton JT. Rotavirus variant replicates efficiently although encoding an aberrant NSP3 that fails to induce nuclear localization of poly(A)-binding protein. J Gen Virol. 2012;93:1483–1494. doi: 10.1099/vir.0.041830-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Rojas M, Arias CF, López S. Protein kinase R is responsible for the phosphorylation of eIF2alpha in rotavirus infection. J Virol. 2010;84:10457–10466. doi: 10.1128/JVI.00625-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Corl BA, Odle J, Niu X, Moeser AJ, Gatlin LA, Phillips OT, Blikslager AT, Rhoads JM. Arginine activates intestinal p70(S6k) and protein synthesis in piglet rotavirus enteritis. J Nutr. 2008;138:24–29. doi: 10.1093/jn/138.1.24. [DOI] [PubMed] [Google Scholar]
- 297.Rhoads JM, Corl BA, Harrell R, Niu X, Gatlin L, Phillips O, Blikslager A, Moeser A, Wu G, Odle J. Intestinal ribosomal p70(S6K) signaling is increased in piglet rotavirus enteritis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G913–G922. doi: 10.1152/ajpgi.00468.2006. [DOI] [PubMed] [Google Scholar]
- 298.Graves LM, He Y, Lambert J, Hunter D, Li X, Earp HS. An intracellular calcium signal activates p70 but not p90 ribosomal S6 kinase in liver epithelial cells. J Biol Chem. 1997;272:1920–1928. doi: 10.1074/jbc.272.3.1920. [DOI] [PubMed] [Google Scholar]
- 299.Kirkby NS, Zaiss AK, Wright WR, Jiao J, Chan MV, Warner TD, Herschman HR, Mitchell JA. Differential COX-2 induction by viral and bacterial PAMPs: Consequences for cytokine and interferon responses and implications for anti-viral COX-2 directed therapies. Biochem Biophys Res Commun. 2013;438:249–256. doi: 10.1016/j.bbrc.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Steer SA, Corbett JA. The role and regulation of COX-2 during viral infection. Viral Immunol. 2003;16:447–460. doi: 10.1089/088282403771926283. [DOI] [PubMed] [Google Scholar]
- 301.Richardson JY, Ottolini MG, Pletneva L, Boukhvalova M, Zhang S, Vogel SN, Prince GA, Blanco JC. Respiratory syncytial virus (RSV) infection induces cyclooxygenase 2: a potential target for RSV therapy. J Immunol. 2005;174:4356–4364. doi: 10.4049/jimmunol.174.7.4356. [DOI] [PubMed] [Google Scholar]
- 302.Symensma TL, Martinez-Guzman D, Jia Q, Bortz E, Wu TT, Rudra-Ganguly N, Cole S, Herschman H, Sun R. COX-2 induction during murine gammaherpesvirus 68 infection leads to enhancement of viral gene expression. J Virol. 2003;77:12753–12763. doi: 10.1128/JVI.77.23.12753-12763.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Steer SA, Moran JM, Maggi LB, Buller RM, Perlman H, Corbett JA. Regulation of cyclooxygenase-2 expression by macrophages in response to double-stranded RNA and viral infection. J Immunol. 2003;170:1070–1076. doi: 10.4049/jimmunol.170.2.1070. [DOI] [PubMed] [Google Scholar]
- 304.Sainis I, Angelidis C, Pagoulatos G, Lazaridis I. The hsc70 gene which is slightly induced by heat is the main virus inducible member of the hsp70 gene family. FEBS Lett. 1994;355:282–286. doi: 10.1016/0014-5793(94)01210-5. [DOI] [PubMed] [Google Scholar]
- 305.Lyupina YV, Zatsepina OG, Timokhova AV, Orlova OV, Kostyuchenko MV, Beljelarskaya SN, Evgen’ev MB, Mikhailov VS. New insights into the induction of the heat shock proteins in baculovirus infected insect cells. Virology. 2011;421:34–41. doi: 10.1016/j.virol.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 306.Xu H, Yan F, Deng X, Wang J, Zou T, Ma X, Zhang X, Qi Y. The interaction of white spot syndrome virus envelope protein VP28 with shrimp Hsc70 is specific and ATP-dependent. Fish Shellfish Immunol. 2009;26:414–421. doi: 10.1016/j.fsi.2009.01.001. [DOI] [PubMed] [Google Scholar]