

Abstract
Due to the overexpression of somatostatin receptors in neuroendocrine (NE) cancers, drug nanocarriers conjugated with somatostatin analogs, such as octreotide (OCT), for targeted NE cancer therapy may offer increased therapeutic efficacies and decreased adverse effects. In this study, OCT-functionalized unimolecular micelles were prepared using individual hyperbranched polymer molecules consisting of a hyperbranched polymer core (Boltorn® H40) and approximately 25 amphiphilic polylactide-poly(-ethlyene glycol) (PLA-PEG) block copolymer arms (H40-PLA-PEG-OCH3/OCT). The resulting micelles, exhibiting a uniform core-shell shape and an average hydrodynamic diameter size of 66 nm, were loaded with thailandepsin-A (TDP-A), a relatively new naturally produced histone deacetylase (HDAC) inhibitor. In vitro studies using flow cytometry and confocal laser scanning microscopy (CLSM) demonstrated that OCT conjugation enhanced the cellular uptake of the unimolecular micelles. Consequently, TDP-A-loaded and OCT-conjugated micelles exhibited the highest cytotoxicity and caused the highest reduction of NE tumor markers. Finally, the in vivo studies on NE cancer bearing nude mice demonstrated that TDP-A-loaded and OCT-conjugated micelles possessed superior anticancer activity in comparison with other TDP-A formulations or drug alone, while showing no detectable systemic toxicity. Thus, these TDP-A-loaded and OCT-conjugated micelles offer a promising approach for targeted NE cancer therapy.
Keywords: Targeted drug delivery, Unimolecular micelles, Octreotide, Neuroendocrine cancer, Thailandepsin A, Histone deacetylase inhibitor
1. Introduction
Neuroendocrine (NE) cancers are malignant transformations of cells of the diffuse endocrine system. These cancers are comprised of heterogeneous groups ranging from benign to highly aggressive and originating in various anatomic locations, including gastrointestinal tract, lung, pancreas and thyroid [1–4]. Neuroendocrine neoplasms, in response to a signal from the nervous system, release higher-than-normal amounts of hormones into the blood, such as chromogranin A (CgA), serotonin or 5-hydroxytryptamine (5-HT), synaptophysin, somatostatin, and neuron-specific enolase (NSE), which cause debilitating symptoms such as uncontrollable diarrhea, flushing, skin rashes, and heart failure. Although NE cancers are uncommon in incidence, they represent a significant clinical challenge because at the time of the diagnosis they are often associated with liver metastases [3,5–7]. As a consequence of widespread metastases or the degree of hepatic involvement by the NE cancers, surgical resection is often non-curative [8]. The recently completed phase 3 trials, including those evaluating octreotide, lanreotide, sunitinib, and everolimus did not demonstrate sufficient evidence to be recommended as a routine use [9,10]. These treatment options for NE cancers can stabilize the disease, but patients become refractory to the therapy. Thus, identification of alternative clinical targets and development of drug delivery technologies to address tumor burden and symptom control are in need.
Our group has an ongoing interest in developing an approach of reactivating the gene expression of tumor suppressor pathways and altering malignant NE phenotype. We have shown that inhibition of histone deacetylases (HDACs) triggered activation of Notch signaling and induced cell cycle arrest and apoptosis, leading to NE cancer cell death [11–13]. We and others have shown that thailandepsin A (TDP-A), a recently reported potent class I HDAC inhibitor isolated from the culture of Burkholderia thailandensis E264, possesses a promising therapeutic potential at low nanomolar concentrations [14,15]. Herein we describe a novel unimolecular micelle nanoparticle as a nanocarrier for TDP-A, which can enhance the drug’s aqueous solubility while improving its anticancer efficacy due to the tumor-targeting abilities of the drug nanocarrier.
Nanovectors are desirable anticancer drug delivery vehicles, mainly due to their unique passive (due to the enhanced permeation and retention (EPR) effect exhibited by the solid tumors) and active (via conjugation of specific tumor-targeting ligands) targeting abilities towards cancerous tissues/cells, which are able to eradicate cancer cells without significantly harming healthy tissues/cells and, concomitantly, reducing the systemic toxicity of the anticancer drug [16,17]. Moreover, using judiciously designed nanovectors, anticancer drugs can be delivered in a controlled and time-released manner.
Polymeric micelles are among the most widely studied nanoplatforms as efficient nanocarriers for cancer treatment due to their unique core shell structure, small size, and fairly narrow size distribution, modifiable surface and high drug-loading capacity. However, conventional multi-molecular polymer micelles formed by a large number of amphiphilic block copolymers have the risk of premature disassociation during in vivo circulation owing to their poor stability, which may cause burst release of the payloads thereby leading to the potential systemic toxicity and compromise of the tumor-targeting ability of the micelles [18–23]. Unimolecular micelles formed by single hyperbranched amphiphilic block copolymer molecules could overcome the disadvantages of conventional multi-molecular polymeric micelles [24–30]. More importantly, the loading capacity and release kinetics of drugs in the micelle can be conveniently modulated by adjusting the chemistry of the block copolymers.
In this study, a multifunctional unimolecular micelle has been developed for targeted NE cancer therapy. The micelle was constructed by hyperbranched amphiphilic block copolymers consisting of a hyperbranched Boltorn® H40 core, a hydrophobic poly(L-lactide) (PLA) inner shell, and a hydrophilic poly(ethylene glycol) (PEG) outer shell (Boltorn H40-poly(L-lactide)-poly(ethylene glycol)/octreotide, H40-PLA-PEG-OCH3/OCT). Boltorn H40 (H40), a hyperbranched aliphatic polyester, has been considered as a desirable core material for unimolecular micelles due to its biodegradability, biocompatibility, globular architecture, and chemical versatility [19,31,32]. To achieve active NE tumor targeting, octreotide (OCT), a somatostatin analog, with a strong binding affinity to somatostatin receptors (SSTRs), was conjugated to the PEG outer shell. TDP-A, a recently reported potent HDAC inhibitor produced by Burkholderia thailandensis isolated from the rice field of Thailand [33], was encapsulated to the H40-based nanocarriers at an optimum drug loading level. Their potential for targeted NE cancer therapy were evaluated by both in vitro studies using a human carcinoid cell line (BON cells) and in vivo studies against NE cancer-bearing mice.
2. Materials and methods
2.1. Materials
Boltron® H40 (a hyperbranched aliphatic polyester with 64 hydroxyl terminal groups; Mn: 2833 Da) was provided by Perstorp Polyols Inc., USA, and purified by fractional precipitation with acetone and tetrahydrofuran (THF). L-Lactide was purchased from Sigma-Aldrich (Milwaukee, WI, USA) and purified by recrystallization from ethyl acetate before use. The heterobifunctional PEG derivatives, succinimidyl (NHS)-PEG114-OH (NHS-PEG-OH) or methoxy (−OCH3))-PEG114-OH (MPEG-OH), both of which having a Mw of 5000 Da, were purchased from JenKem Technology (Allen, TX, USA). Doxorubicin hydrochloride (DOX-HCl) (Tecoland Corporation, Irvine, CA, USA), and octreotide acetate (Aroz Technologies, USA) are commercially available. Succinic anhydride, 4- dimethylamino pyridine (DMAP), 1,3-dicyclohexylcarbodiimide (DCC), and dichloromethane (DCM) were purchased from Sigma-Aldrich (Milwaukee, WI, USA) and used without further purification. Solvents used for reactions including THF, triethylamine (TEA), dimethyl sulfoxide (DMSO), and dimethylformamide (DMF) were purchased from Sigma-Aldrich (Milwaukee, WI, USA) and were distilled before use. Thailandepsin-A (TDP-A) was obtained from Dr. YiQiang Cheng’s laboratory [33].
2.2. Synthesis of H40-PLA
H40-PLA was prepared by ring-opening polymerization (ROP) of L-lactide in the presence of H40 as a macroinitiator and Sn(Oct)2 as a catalyst. In a typical polymerization procedure, a 50 mL Schlenk flask was charged with 400 mg of H40 (9.04 mmol of hydroxyl groups) under an inert atmosphere. In order to facilitate the mixing of H40 and L-lactide, the flask was first placed in an oil bath at 120 °C. After the melting of H40, 4.00 g (27.80 mmol) of L-lactide was slowly introduced into the flask and a catalytic amount ([catalyst]/[monomer] of 1:1000) of Sn(Oct)2(9 μL, 27.80 μmol) was added. The polymerization reaction was carried out for 24 h. The crude polymer was dissolved in THF and passed through a neutral alumina column. Next, the mixture was concentrated and poured into excess cold diethyl ether to precipitate the product. The final product was dried in vacuo for 24 h and H40-PLA was obtained as white powders.
2.3. Synthesis of carboxyl-functionalized H40-PLA (H40-PLA-COOH)
The hydroxyl groups of H40-PLA was converted to carboxylic acid groups by the reaction with succinic anhydride in the presence of 4-dimethylaminopyridine (DMAP) as the catalyst. The reaction was conducted by reacting H40-PLA (1.00 g, 38.10 μmol) with succinic anhydride (0.38 g, 3.80 mmol) and catalyzed by DMAP (0.70 g, 5.70 mmol) in anhydrous DCM (10 mL) at room temperature. After 48 h, the crude product was precipitated with cold diethyl ether and dried under vacuum. Afterward, dialysis against deionized water by using cellulose tubing (molecular weight cutoff of 3500 Da) was performed for 48 h in order to remove the impurities and unreacted materials. Finally, H40-PLA-COOH was obtained by lyophilization.
2.4. Synthesis of H40-PLA-PEG-OCH3 (used to prepare the nontargeted micelles)
MPEG-OH (175 mg, 35 μmol) was conjugated to H40-PLA-COOH (40 mg, 1.4 μmol) in the presence of DCC (7.2 mg, 35 μmol) and DMAP (0.43 mg, 3.5 μmol). They were dissolved in 10 mL anhydrous DMF at around 0 °C followed by reacting at room temperature for 24 h under constant stirring. The reaction mixture was first filtrated to remove the by-product, dicyclohexylcarbodiurea (DCU) and then precipitated with cold diethyl ether. The final products were obtained by freeze drying after dialyzing against DMF for 48 h.
2.5. Synthesis of H40-PLA-PEG-OCH3/NHS
In order to synthesize the hyperbranched amphiphilic copolymer conjugated with OCT (i.e., H40-PLA-PEG-OCH3/OCT), two kinds of PEG derivatives including MPEG-OH (122.5 mg, 24.5 μmol) and NHS-PEG-OH (52.2 mg, 10.5 μmol) were conjugated onto H40-PLA-COOH (40 mg, 1.4 μmol) in the presence of DCC (7.2 mg, 34.8 μmol) and DMAP(0.43 mg, 3.5 μmol). They were first dissolved in 10 mL anhydrous DMF and mixed well at about 0 °C. Then the reaction was performed at room temperature for 24 h. For purification of the product, filtration was carried out to get rid of the byproduct, DCU followed by precipitating with an excess amount of cold diethyl ether. Afterward, the products were dialyzed against DMF and freeze dried to get the final product.
2.6. Synthesis of H40-PLA-PEG-OCH3/OCT (used to prepare the targeted micelles)
H40-PLA-PEG-OCH3/OCT (targeted micelles) was prepared by reacting H40-PLA-PEG-OCH3/NHS (100 mg) with Octreotide (5.0 mg, 4.9 μmol). After stirring for 24 h, the solution will be purified by dialysis against DI water for 24 h, followed by freeze-drying.
2.7. Preparation of DOX-loaded H40-PLA-PEG-OCH3/OCT micelles
In order to evaluate the effect of the OCT targeting ligand on the cellular uptake, both targeted and non-targeted micelles were loaded with DOX to take advantage of DOX’s fluorescent properties. These DOX-loaded micelles were used only for the cellular uptake studies. For the formation of the DOX-loaded targeted micelles, 2.5 mg of DOX · HCl was firstly treated with 2 mol excess of triethylamine (2.16 μl). Then, 15 mg of polymer (H40-PLA-PEG-OCH3/OCT) and 3 mL of DMF were added under stirring followed by the addition of 9 mL of deionized water dropwise. The resulting micelle solution was purified by dialysis against deionized water using a dialysis tubing with a molecular weight cut-off of 2 kDa for 24 h followed by freeze-drying. DOX-loaded non-targeted micelles were prepared likewise by using H40-PLA-PEG-OCH3 polymers as the nanocarriers.
2.8. Preparation of TDP-A-loaded micelles
For the preparation of the TDP-A-loaded targeted micelles, 60 mg of the H40-PLA-PEG-OCH3/OCT polymers and 8 mg of TDP-A were dissolved in 10 mL of acetonitrile (CH3CN) under stirring. Micelles were formed by dropwise adding of 30 mL of deionized water as a non-solvent. The micelle solution was stirred for 4 h in darkness at room temperature. After removing the remaining CH3CN by rotary evaporation, the micelles were purified by centrifugation at 4000 g for 10 min. The TDP-A-loaded micelles were obtained by collecting the supernatant after centrifugation and lyophilization. TDP-A-loaded non-targeted micelles were prepared following a similar procedure by using H40-PLA-PEG-OCH3 as the nanocarriers.
2.9. Characterization of micelles
Various techniques were used to characterize the hyperbranched amphiphilic block copolymers and the resulting unimolecular micelles according to our previous reports [19]. The structures of the intermediate and final polymer products were confirmed by 1H NMR recorded at 25 °C on a Bruker DPX 300 spectrometer using CDCl3 or DMSO as the solvents. Gel permeation chromatography (GPC) equipped with a refractive index detector, a viscometer detector, and a light scattering detector (Viscotek, USA) was used to measure the molecular weight of the polymers. The mobile phase consists of DMF (with 10 mmol/L LiBr) at a flow rate of 1 mL/min. The particle size and size distribution of the micelles were determined by dynamic light scattering (DLS, ZetaSizer Nano ZS90, Malvern Instrument, USA) at a polymer concentration of 0.05 mg/mL Transmission electron microscope (TEM) images were taken on Hitachi H-600 (75 kV) for observing the morphology and size of the dried micelles. TEM Samples were prepared by drying a dispersion of the micelle solution with 0.8 wt% of phosphotungstic acid onto a copper grid coated with amorphous carbon. Extra solution in the copper grid was gently blotted with filter paper. The sample was dried adequately at room temperature before TEM observation. High performance liquid chromatography (HPLC, Hitachi) equipped with a UV detector and a reverse-phase C18 column (150 × 4.6 mm) was used to measure the drug loading content of TDP-A, defined as the weight percentage of TDP-A in the TDP-A-loaded unimolecular micelles, and drug loading efficiency, defined as the ratio between loaded and feed amount of drug, respectively. Water and acetonitrile (v/v 60/40) at a constant flow rate of 1 mL/min was used as a non-gradient mobile phase. The TDP-A peak was measured at a wavelength of 200 nm and quantitatively determined by comparing with a standard curve of TDP-A. The DOX loading content in the DOX-loaded unimolecular micelles was quantified with a Varian Cary 300 Bio UV–visible spectrophotometer by comparing with a standard curve of DOX. Prior to the UV measurements, a weighed quantity (25 mg) of DOX-loaded micelles was extracted in ethanol at room temperature for 48 h under uniform stirring. After centrifugation of the micelle solution, the supernatant was collected and the amount of DOX was assayed by a UV–visible spectrophotometer at a wavelength of 485 nm. All of the HPLC and UV measurements were performed in triplicates.
2.10. Cell culture
Human gastrointestinal (GI) carcinoid (BON) cells were obtained from Drs. B. Mark Evers and Courtney M. Townsend Jr. (University of Texas Medical Branch, Galveston, TX). The cells were maintained in Dulbecco’s modified Eagle medium-nutrient mixture Ham’s F-12 K 1:1 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Sigma, St Louis, MO) and a combination of 100 IU/mL penicillin and 100 g/mL streptomycin (Invitrogen) in a humidified atmosphere of 5%CO2 in air at 37 °C.
2.11. Cellular uptake of micelles
Based on DOX auto-fluorescence (488 nm and 575 nm, excitation and emission, respectively), the cellular uptake behaviors of the micelles in BON cells were analyzed using both flow cytometry and confocal laser scanning microscopy (CLSM). For flow cytometry, BON cells were seeded in 6-well plates at a density of 3 × 105 cells/ml and incubated overnight in a DMEM medium with 10% FBS under standard conditions. Then, the cells were treated with DOX-loaded either targeted or non-targeted micelles at a DOX concentration of 1 μg/mL and further incubated for 120 min. The micelle-free culture medium was used as blank control. Thereafter, the cells were washed with PBS to remove unincorporated micelles and lifted using a nonenzymatic cell dissociation solution and stained with DAPI (10 ng/mL). DOX uptake was analyzed using a FACS Calibur four-color analysis cytometer (Becton Dickinson, San Jose, CA) and FlowJo analysis software (Tree Star, Inc., Ashland, OR). For each sample at least 1 × 105 cells were analyzed and DOX fluorescence was displayed on a linear scale. The cellular uptake behavior of the micelles in WI-38 (pulmonary fibroblasts), a cell line that does not over-express SSTRs, was studied following similar procedures using flow cytometry and was used as a negative control. For the CLSM studies, BON cells were seeded (10 × 105) onto 96-well high optical quality plates and grown overnight. Cells were treated with DOX-loaded either targeted or non-targeted micelles at a DOX concentration of 1 (μg/mL followed by incubation for 120 min. The cells were then washed with warm PBS, fixed with 1.5% formaldehyde for 15 min, and washed again with double-distilled H2O. Then, the cells in the optical plate were mounted with Prolong Gold antifade reagent with DAPI (Life Technologies, Carlsbad, CA) and DOX uptake was observed with a Nikon A1R-Si high speed spectral laser scanning confocal inverted microscope (Nikon, Melville, NY) with emission filters detecting fluorescent light at 575 nm. Digital monochromatic images were acquired using NIS-Elements BR Software.
2.12. Cell proliferation assay
The ability of TDP-A-loaded micelles to suppress BON cell proliferation was measured using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assays. BON cells were plated in quadruplicate on 96-well plates and incubated overnight to allow cell adhesion. The following day, the media was replaced with fresh media containing TDP-A-loaded targeted and non-targeted micelles, and four controls: empty targeted and empty nontargeted micelles, free TDP-A, and medium. In free TDP-A and TDP-A-loaded micelles, the concentration of TDP-A was 5 nM. The cells were further incubated for 24, 48 or 72 h and the MTT assay was performed at each time point by aspirating the treatment media and adding 25 μl of serum-free media containing 0.5 mg/mL MTT reagent and incubating it at 37 °C for 4 h. Following incubation, 75 μl of DMSO was added to each well and mixed thoroughly. The multi-well plates were then measured at 570 nm using a spectrophotometer (Quant, Bio-Tek Instruments, Winooski, VT), and the average absorbance and percent of cell viability compared to the control (pure medium) were calculated.
2.13. Tumor marker expression assay
The Western blot analysis was used to assess the NE marker expression, including achaete-scute complex-like1 (ASCL1) and chromogranin A (CgA). BON cells were seeded into 100 mm culture plates and incubated overnight. The culture medium was removed and replaced with aliquots of TDP-A-loaded non-targeted and targeted micelles, empty targeted micelles and empty non-targeted, free TDP-A, and medium at a concentration of 5 nM of TDP-A. The plates were incubated for 48 h and protein was harvested according to a previously described protocol (Sippel 2003). Protein concentrations were determined by a bicinchoninic acid assay (Thermo Scientific, Waltham, MA) and protein lysates (20 μg) were denatured and resolved on 10% SDS-PAGE gels. Protein bands in the gel were then transferred to a nitrocellulose membrane and incubated with the appropriate primary antibody with dilutions as follows: 1:2000 for ASCL1,1:3000 for CgA and 1:10000 for GAPDH. Membranes were incubated overnight at 4 °C and washed the next day with buffer (1 × PBS, 0.05% Tween 20). Horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibodies were used for secondary incubation. Proteins were then visualized with chemiluminescent substrates Immunstar (GAPDH and CgA) or Femto (ASCL1).
2.14. In vivo study
Four-week-old male athymic nude mice were obtained from Charles Rivers (Wilmington, Maryland, USA). They were allowed to acclimate one week in the animal facility to reduce stress after arrival. Mice were maintained under specific pathogen-free conditions. The subcutaneous tumor xenograft model was established by subcutaneous implanting 107 BON cells in 200 μl of Hanks Balanced Salt Solution (Mediatech, Inc, Manassas, Virginia, USA) into the left flank of mouse. Two weeks after inoculation, mice with palpable tumors were randomized into four treatment groups receiving: PBS (control), free TDP-A, TDP-A-loaded non-targeted micelles, and TDP-A-loaded targeted micelles. Each treatment group, consisting of 7 mice, received five intravenous injections every 4 days. Free TDP-A and TDP-A-loaded micelles were injected at the equivalent dose of TDP-A 3.125 mg/kg BW, below the maximum tolerated dose (12.5 mg/kg BW, unpublished data; performed by NCI-DTP). Tumor volumes were measured by an external caliper every four days and then were calculated by the modified ellipsoidal formula: Tumor volume = 1/2 (length × width2). Body weight change was monitored at the same time. At the end of the experiment, mice were sacrificed and postmortem examination of the lungs, liver, kidneys, and spleen were performed to confirm that there was no evidence of metastases or tumor growth outside of the inoculation site. Moreover, different organs (liver, brain, heart, and leg muscles) of mice treated with TDP-A-loaded targeted micelles were collected, fixed in 10% formalin and embedded in paraffin blocks to prepare hematoxylin and eosin (H&E) stained sections for the pathological assessment under the optical microscope. All experimental procedures were done in compliance with our animal care protocol approved by the University of Wisconsin-Madison Animal Care and Use Committee.
2.15. Statistical analysis
Weights and volumes were compared between groups using an analysis of variance (ANOVA), with pair-wise comparisons performed using Fisher’s protected least significant difference tests. Data were log-transformed prior to analyses in order to better meet the assumptions of ANOVA. P-values less than 0.05 were considered to be statistically significant.
3. Results and discussion
3.1. Synthesis and characterization of H40-PLA-PEG-OCH3/OCT
The synthesis scheme for the hyperbranched amphiphilic H40-PLA-PEG-OCH3/OCT block copolymer used to form the targeted nanocarriers is shown in Fig. 1. First, H40-PLA was prepared by the ring-opening polymerization of L-lactide initiated by H40 and catalyzed by Sn(Oct)2. The obtained polymer H40-PLA was characterized by 1H NMR and GPC. Fig. 2A shows the 1H NMR spectrum of H40-PLA. The peaks located at 1.54–1.56 ppm (a) and 5.10–5.16 ppm (b) were ascribed to protons of the methyl and methane groups in the PLA main chains, respectively. The peaks of the terminal methyl and methine protons of PLA (HOCHCH3) in H40-PLA were found at 1.45 ppm (c) and 4.32–4.35 ppm (d). In addition, Fig. 2A also displayed clear characteristic peaks of H40 at 1.18–1.22 ppm and around 4.20 ppm corresponding to the protons of the methyl groups and methylene groups of H40, respectively. By comparing the integral ratio between resonances at 1.45 ppm (attributed to the methyl group adjacent to the hydroxyl end group of PLA) and 1.56 ppm (ascribed to methyl groups present in the PLA polymer chain), the molecular weight (Mn) and degree of polymerization (DP) of the PLA arms were calculated to be about 936 Da and 13, respectively. Based on the Mn of the PLA obtained through the NMR analysis and the molecular weights of H40 and H40-PLA measured by the GPC data shown in Table 1, revealed that the average number of arms per H40-PLA molecule was approximately 25. This result is very consistent with earlier report by Kreutzer et al. as well as our previous studies [32,34,35].
Fig. 1.
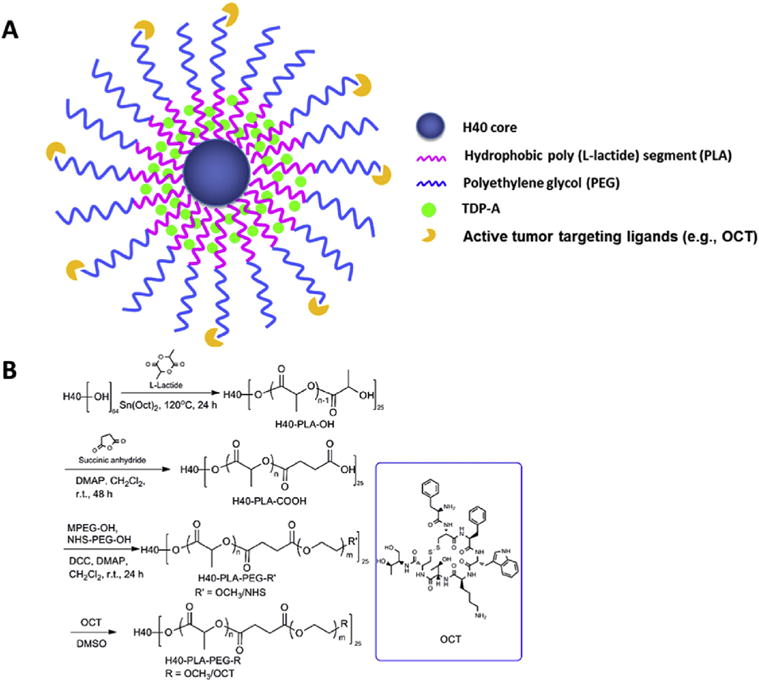
A) A schematic illustration of the H40-PLA-PEG-OCT nanocarriers for NET-targeted drug delivery. B) The synthesis scheme of the hyperbranched H40-PLA-PEG-OCT amphiphilic block copolymers.
Fig. 2.
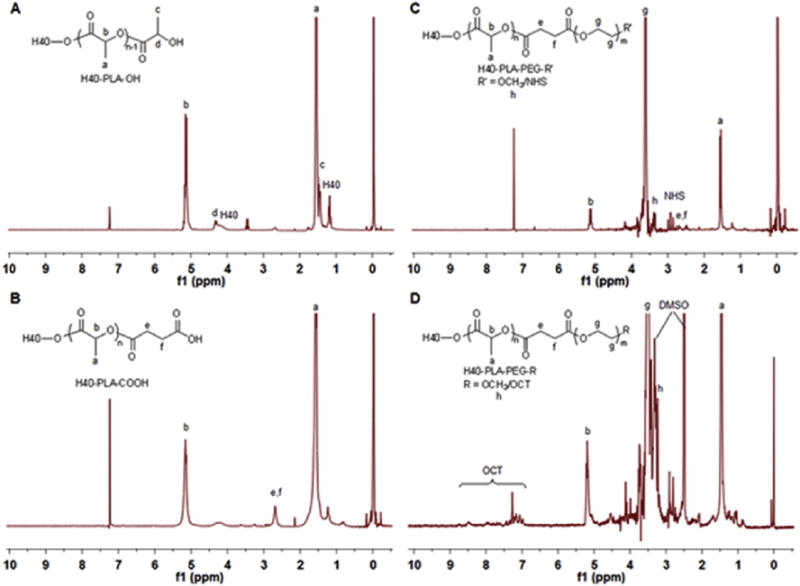
1H NMR spectra of (A) H40-PLA, (B) H40-PLA-COOH, (C) H40-PLA-PEG-OCH3/NHS, and (D) H40-PLA-PEG-OCT.
Table 1.
Gel permeation chromatography of H40, H40–PLA and H40–PLA-PEG-OCT.
| Sample | Mn | Mw | Mw/Mn |
|---|---|---|---|
| H40a | 2833 | 5100 | 1.80 |
| H40–PLA | 26208 | 36456 | 1.39 |
| H40–PLA-PEG/OCT | 160265 | 248643 | 1.55 |
In the second step, in order to conjugate PEG to H40-PLA, carboxylation of the hydroxyl terminal groups of H40-PLA were first carried out by reacting H40-PLA with succinic anhydride using DMAP as a catalyst. The 1H NMR spectrum shown in Fig. 2B confirmed the successful synthesis of H40-PLA-COOH since a new peak at 2.65 ppm attributed to the methylene groups of succinic anhydride (e,f) was detected.
The third reaction step was the conjugation of two kinds of hydrophilic PEG derivatives, namely MPEG-OH and NHS-PEG-OH, onto the hydrophobic hyperbranched polymer of H40-PLA-COOH at the feed molar ratio of 15:10:1. Comparing the 1H NMR spectrum of the hyperbranched amphiphilic block copolymer H40-PLA-PEG-OCH3/NHS (Fig. 2C) with that of the hyperbranched polymer H40-PLA-COOH, a series of new proton signals appeared in the spectrum of the H40-PLA-PEG-OCH3/NHS copolymer including the peaks around 2.81 ppm assigned to the ethylene group of the succinimidyl groups (NHS), peaks around 3.65 ppm (g) and 3.38 ppm (h) ascribed to the methylene protons of oxyethylene units and methyl protons of MPEG, respectively, indicating the successful conjugation of the two kinds of PEGs to the H40-PLA-COOH. The average number of PEG arms per H40-PLA-PEG molecule was approximately 25 based on the GPC results as shown in Table 1, which is consistent with the number of PLA arms estimated for H40-PLA calculated earlier.
Lastly, the OCT for NE tumor specific targeting was conjugated onto the surface of the hyperbranched amphiphilic copolymer H40-PLA-PEG-OCH3/NHS. The NHS groups of H40-PLA-PEG-OCH3/NHS reacted with the NH2 groups of OCT, where the feed molar ratio of OCT:H40-PLA-PEG-OCH3/NHS was set at 7.5:1. The 1H NMR spectrum of H40-PLA-PEG-OCH3/OCT shown in Fig. 2D indicated the successful conjugation of OCT to H40-PLA-PEG-OCH3/NHS due to the appearance of a group of NMR peaks ranging from 6.82 to 8.50 ppm attributed to the protons of the OCT.
3.2. Micellar properties of the hyperbranched amphiphilic block copolymer H40-PLA-PEG-OCH3/OCT
The hyperbranched amphiphilic block copolymer H40-PLA-PEG-OCH3/OCT can form unimolecular micelles in aqueous solutions due to their amphiphilicity and globular architecture. The resulting unimolecular micelle consists of H40-PLA as an inner hydrophobic core and a PEG layer as an outer hydrophilic shell. The hydrophobic drug TDP-A was easily encapsulated into the hydrophobic core of the micelles via hydrophobic interactions. The morphology and stability of the nanoparticles have strong influence on the cellular internalization and in vivo performance of the drug nanocarriers [16]. The size and size distribution of the TDP-A-loaded targeted micelles were studied by both TEM and DLS. The micelles exhibited a core-shell structure with a size ranging from 23 to 35 nm under TEM (Fig. 3A).
Fig. 3.
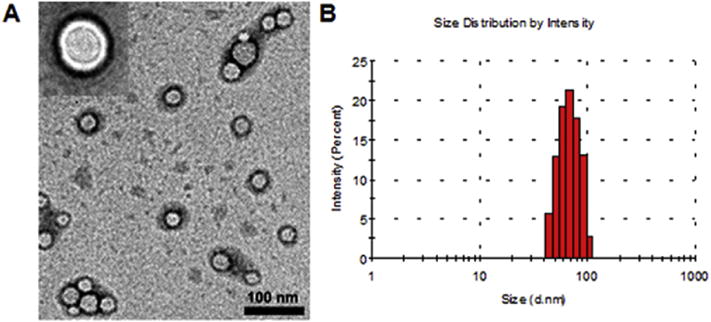
The (A) morphology and (B) size distribution of unimolecular micelles as measured by TEM and DLS, respectively.
The average hydrodynamic diameter measured by DLS was 66 nm (Fig. 3B). In addition, the drug loading content and drug loading efficiency were quantified to be 11.2 wt.% and 97% by HPLC, respectively.
3.3. In vitro cellular uptake studies
Based on DOX’s auto-fluorescence properties, the effects of the OCT-targeting ligand on the cellular uptake and localization of the micelles were measured by flow cytometry and CLSM. Quantitative flow cytometry analysis revealed that the intracellular uptake of the DOX-loaded and OCT-conjugated micelles (2 h incubation, DOX concentration: 1 μg/mL) was 4 times higher than the DOX-loaded nontargeted micelles (Fig. 4A) in the BON cells, a human carcinoid cell line overexpressing SSTRs. However, in the negative control experiment as shown in Fig. S1, no differences were observed in the levels of cellular uptake between the targeted and non-targeted micelles in WI-38 cells which do not overexpress SSTRs. These findings indicate that NE cancer cells overexpressing SSTRs on their surface exhibited an increased intracellular uptake of the OCT-conjugated micelles through the SSTR-mediated endocytosis process. ACLSM analysis was applied to further compare the cellular uptake of targeted and non-targeted micelles in BON cells. Based on DOX localization, this assay revealed that OCT conjugated micelles had faster cellular uptake after 2 h incubation with stronger fluorescent signals. Moreover, DOX auto-fluorescence was observed mainly in the cytoplasm of the cells treated with either targeted or non-targeted micelles (Fig. 4B). In contrast, the fluorescence signals in cells treated with free DOX were predominantly shown in the nuclear region of the cells. This difference in DOX localization might be due to the distinctively different mechanisms of the cellular uptake of free DOX and DOX-loaded micelles. While free DOX has a direct access to the nuclear region through cellular membranes by a passive diffusion mechanism [36,37], the DOX-loaded micelles were taken up by the tumor cells via endocytosis. Due to these different uptake mechanisms, free DOX was accumulated more rapidly in the nucleus where it was bound to the chromosomes [38], while the fluorescent signals from DOX-loaded micelles were more intense in the cytoplasm. Overall, the greater uptake of the targeted micelles in comparison with the nontargeted ones indicates that the OCT targeting ligand was effective in promoting SSTR-mediated endocytosis. In contrast, nontargeted micelles are taken up by the cells through nonspecific endocytosis [19,25,39]. Notably, OCT-conjugated nanoparticles are effective drug delivery vehicles against several other types of cancers overexpressing SSTRs including hepatocellular carcinoma, breast cancer, and melanoma [40,41].
Fig. 4.
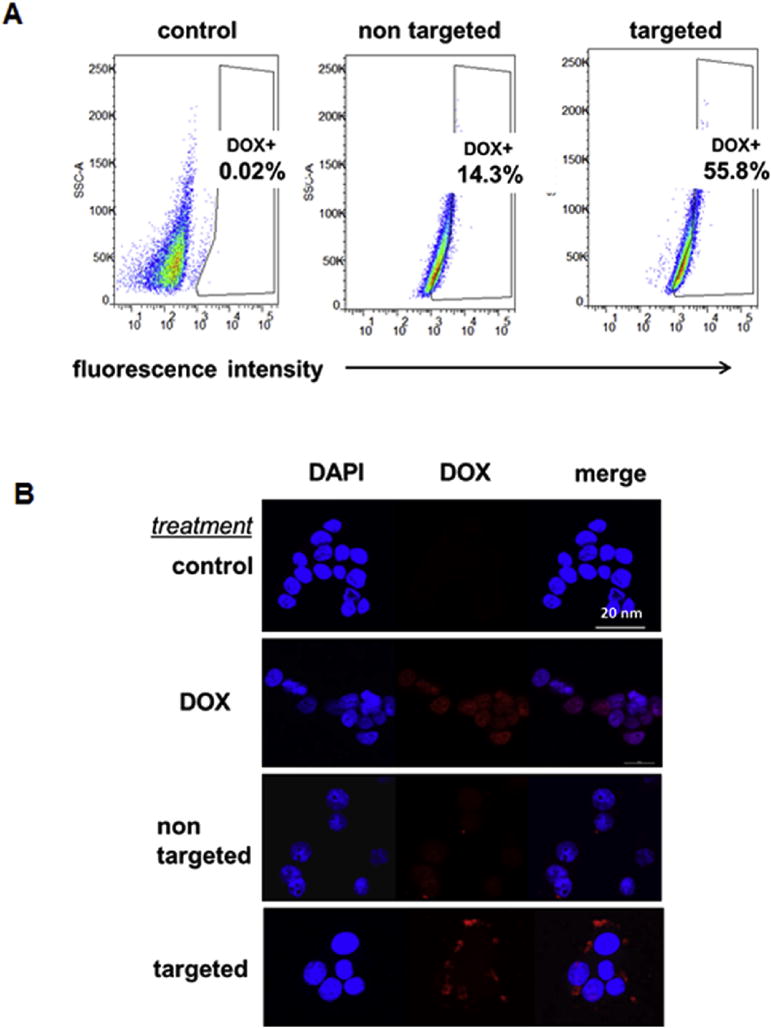
Ligand enhanced cellular uptake in BON cells measured by flow cytometry and CLSM. (A) Flow cytometry revealed that intracellular uptake of micelles labeled with DOX (2 h incubation, DOX concentration: 1 μg/mL), was 4 times higher for OCT-conjugated micelles comparing to their non-targeted controls. (B) CLSM images showed that OCT conjugated nanocarriers had faster cellular uptake after 2 h incubation (DOX concentration: 1 μg/mL).
3.4. In vitro evaluation of the anticancer efficacy of the TDP-A-loaded micelles
Anti-proliferative activities of TDP-A-loaded micelles were evaluated in BON cells using an MTT assay after 24, 48 and 72 h treatment. The levels of the cytotoxicity of the TDP-A-loaded and empty micelles (both targeted and nontargeted) were compared to free TDP-A and control medium (an equivalent of TDP-A concentration of 5 nM). This cytotoxicity assay revealed that at each time point TDP-A-loaded targeted micelles (4th bar) were much more effective in suppressing tumor cell proliferation than other types of micelles containing fewer components (Fig. 5). These observations indicate that the enhanced cytotoxicity of TDP-A-loaded targeted micelles may be attributed to their enhanced cellular uptake in the BON cells with overexpressed SSTRs via receptor-mediated endocytosis. Interestingly, we also observed that cells treated with TDP-A-loaded targeted micelles were more sensitive compared to free TDP-A at all incubation periods. These differences in cytotoxicity may be a result of the different mechanism of cellular uptake of the free TDP-A and TDP-A formulated in targeted micelles. As the cellular uptake of free TDP-A occurs through a passive diffusion mechanism, the drug may be trapped at the P-gap junction in the cell membrane and expelled from the cancer cells while in the case of TDP-A-loaded targeted micelles, the drug is released in a time dependent manner from the endosomes what may overcome the P-gap pumping action [42]. To further prove that the enhanced cellular uptake can increase the anticancer efficacy of TDP-A-loaded targeted micelles, we performed Western blot analysis on the NE marker expression of BON cells treated with various micelles (with an equivalent TDP-A concentration of 5 nM). ASCL-1 and CgA have been well characterized as markers of NE malignancy and high levels of ASCL-1 and CgA are both linked to poor prognosis of NE cancers [43,44]. Therefore, we used these markers to assess the hormone–producing activity of the BON cells treated with TDP-A-loaded micelles. As shown in Fig. 6, the TDP-A-loaded targeted micelles (lane 4) were much more effective in reducing the expression of both NE markers, ASCL1 and CgA, than all other types of micelles containing fewer components as well as TDP-A alone. These results demonstrate that TDP-A-loaded targeted micelles optimally inhibit NE cancer cell proliferation and bioactive hormone secretion in vitro by altering the malignant neuroendocrine phenotype.
Fig. 5.
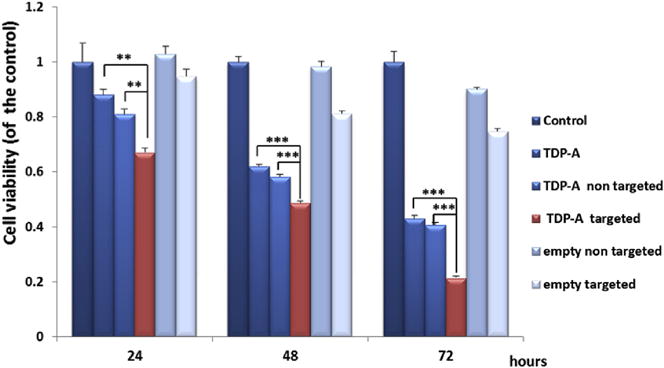
MTT assay of BON cells treated with various micelles (TDP-A concentration 5 nM). TDP-A-loaded targeted micelles (4th bar) were much more effective in suppressing tumor cell proliferation than other types of micelles containing fewer components. All values are presented as a mean SD (n = 4); **, P < 0.01; ***, P < 0.001.
Fig. 6.
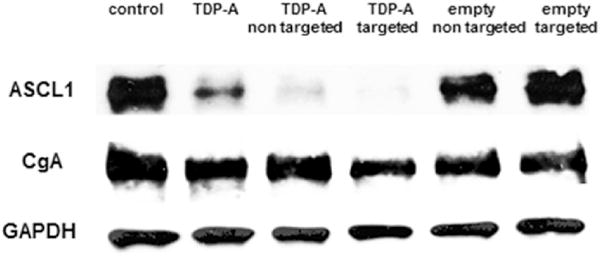
Western blot analysis for ASCL1 and CgA of BON carcinoid cells treated with various micelles (TDP-A concentration 5 nM). TDP-A-loaded targeted micelles (lane 4) was much more effective in reducing NET markers ASCL1 and CgA than other types of micelles containing fewer components. GAPDH was used as a loading control.
3.5. In vivo evaluation of the antitumor effects of TDP-A-loaded micelles
To further investigate the anti-tumor effects of the TDP-A-loaded and OCT-conjugated (i.e., targeted) unimolecular micelles and a number of other related groups including free TDP-A, TDP-A-loaded non-targeted micelles, as well as no treatment (i.e., the control group), in vivo studies were performed using a NE cancer xenograft model. As shown in Fig. 7A, all treatments significantly decreased the tumor burden when compared with the control group (p < 0.05 for TDP-A-loaded non-targeted micelles and pure TDP-A; p < 0.01 for TDP-A-loaded targeted micelles). Furthermore, TDP-A-loaded targeted micelles induced the better antitumor efficacy compared to either TDP-A-loaded non-targeted micelles or free TDP-A (p < 0.05), and did not cause any significant changes in body weight (Fig. 7B) or survival. In contrast, while free TDP-A did also lead to a slower tumor growth rate as compared to the control, its severe toxicity in mice was demonstrated by both body weight loss and decreased survival (2 mice out of 7 died before terminating the experiment). Moreover, pathological assessment of H&E–stained sections of different organs (brain, heart, liver and leg muscles) of mice treated with TDP-A-loaded targeted micelles did not indicate any signs of acute or chronic inflammation, or apoptotic or necrotic regions (Fig. 7C). The initial pathological assessment indicates that the TDP-A-loaded NET-tumor targeting unimolecular micelle delivery system was safe for organs other than the NE cancerous tissues, and that the potential off-target uptake of micelles by normal tissues did not cause any detectable damage. Overall, TDP-A-loaded targeted micelles were the most effective in retarding tumor growth among the various groups studied including TDP-A-loaded non-targeted micelles and free TDP-A. This observation can be largely contributed to the fact that the TDP-A-loaded, OCT-conjugated NET-targeting unimolecular micelles have both passive (via the EPR effect [45,46]) and active (via OCT conjugation) tumor targeting abilities [47–49]. In contrast, the TDP-A-loaded non-targeted unimolecular micelles only have passive tumor targeting ability, while free TDP-A does not have any specific tumor targeting ability resulting in high in vivo systemic toxicity. These data suggest that the growth inhibitory effects of TDP-A-loaded targeted micelles observed in vitro can be replicated in vivo with i.v. injections. Moreover, our data confirms that SSTRs abundant in the NE tumors are responsible for the increased uptake of OCT-conjugated micelles in an animal xenograft model [40,50,51].
Fig. 7.
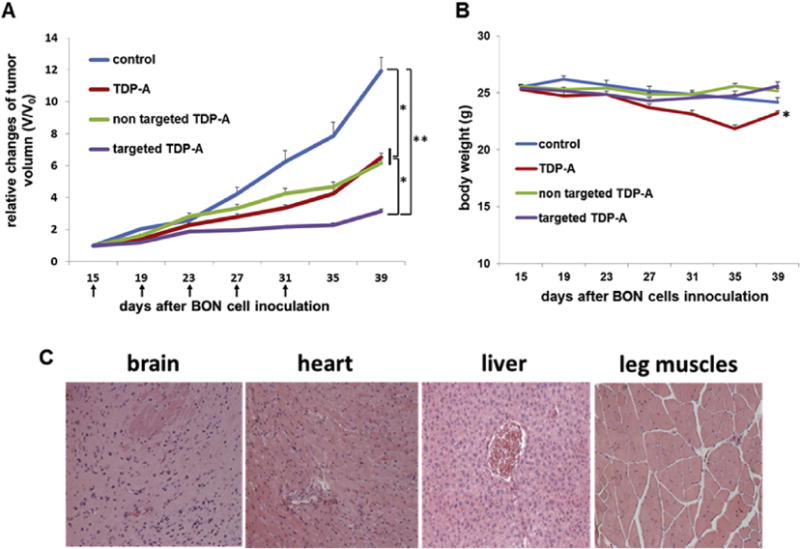
Therapeutic effects of TDP-A-loaded micelles in the subcutaneous BON cell xenografts. (A) In vivo anti-cancer efficacy of different TDP-A formulations in BON tumor xenografts. Each mouse received five intravenous injections (3.125 mg/kgBW/dose) every 4 days (indicated by arrows). (B) Change in the body weight of animals as a function of time. (C) Representative H&E-stained sections of brain, heart, liver, and leg muscles of the mouse treated with TDP-A-loaded targeted micelles. No signs of apoptotic or necrotic areas were observed. All values are presented as a mean SD (n = 7); *, P < 0.05; **, P < 0.01.
4. Conclusions
OCT-conjugated unimolecular micelles made of hyperbranched amphiphilic block copolymers were synthesized and characterized for targeted NE cancer therapy. By using octreotide as the NET-tumor targeting moiety, we determined that the cellular uptake of the targeted micelles by gastrointestinal NE cancer cells overexpressing SSTRs was significantly higher than the non-targeted micelles. Moreover, we confirmed that TDP-A-loaded OCT-conjugated unimolecular micelles were more effective in suppressing NE cancer cell growth and hormone production than other types of micelles or free TDP-A. More importantly, the in vivo investigations on NE cancer xenografts further confirmed that the TDP-A-loaded OCT-conjugated micelles possessed superior tumor-targeting ability and antitumor activity over other TDP-A formulations and significantly reduced systemic cytotoxicity in comparison with free TDP-A. These findings may support the development of TDP-A-loaded NET-targeting unimolecular micelles in the treatment and palliation of patients with NE cancers, which can significantly enhance the therapeutic outcome of cancer therapy while minimizing undesirable side-effects.
Supplementary Material
Acknowledgments
This project was financially supported by grants NIH (R01 CA121115 to H. Chen and 1K25CA166178 to S. Gong), American Cancer Society (MEN2 Thyroid Cancer Professorship 120319-RPM-11-080-01-TBG to H. Chen and Research Scholar Award RSGM TBE-121413 to H. Chen), Layton F. Rikkers, MD, Chair in Surgical Leadership Professorship (H. Chen), and Caring for Carcinoids Foundation and AACR (14-60-33-CHEN). The authors thank Dr. Ricardo Lloyd for the pathological assessment of the mice tissues and Dr. Glen Leverson for the statistical analysis.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2016.03.010.
References
- 1.Roy M, Chen H, Sippel RS. Current understanding and management of medullary thyroid cancer. Oncologist. 2013;18:1093–1100. doi: 10.1634/theoncologist.2013-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinchot SN, Kunnimalaiyaan M, Sippel RS, Chen H. Medullary thyroid carcinoma: targeted therapies and future directions. J Oncol. 2009;2009 doi: 10.1155/2009/183031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncol. 2008;13:1255–1269. doi: 10.1634/theoncologist.2008-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic neuroendocrine tumours. lancet Oncol. 2008;9:61–72. doi: 10.1016/S1470-2045(07)70410-2. [DOI] [PubMed] [Google Scholar]
- 5.Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after “Carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 6.Isozaki T, Kiba T, Numata K, Saito S, Shimamura T, Kitamura T, et al. Medullary thyroid carcinoma with multiple hepatic metastases: treatment with transcatheter arterial embolization and percutaneous ethanol injection. Intern Med. 1999;38:17–21. doi: 10.2169/internalmedicine.38.17. [DOI] [PubMed] [Google Scholar]
- 7.Brown KT, Koh BY, Brody LA, Getrajdman GI, Susman J, Fong Y, et al. Particle embolization of hepatic neuroendocrine metastases for control of pain and hormonal symptoms. J Vasc Interv Radiol. 1999;10:397–403. doi: 10.1016/s1051-0443(99)70055-2. [DOI] [PubMed] [Google Scholar]
- 8.Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology. 135:1469–1492. doi: 10.1053/j.gastro.2008.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunz PL, Reidy-Lagunes D, Anthony LB, Bertino EM, Brendtro K, Chan JA, et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 2013;42:557–577. doi: 10.1097/MPA.0b013e31828e34a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan J, Kulke M. Targeting the mTOR signaling pathway in neuroendocrine tumors. Curr Treat Options Oncol. 2014;15:365–379. doi: 10.1007/s11864-014-0294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ning L, Jaskula-Sztul R, Kunnimalaiyaan M, Chen H. Suberoyl bishydroxamic acid activates Notch1 signaling and suppresses tumor progression in an animal model of medullary thyroid carcinoma. Ann Surg Oncol. 2008;15:2600–2605. doi: 10.1245/s10434-008-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaskula-Sztul R, Eide J, Tesfazghi S, Dammalapati A, Harrison AD, Yu XM, et al. Tumor-suppressor role of Notch3 in medullary thyroid carcinoma revealed by genetic and pharmacological induction. Mol Cancer Ther. 2015;14:499–512. doi: 10.1158/1535-7163.MCT-14-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenblatt DY, Vaccaro AM, Jaskula-Sztul R, Ning L, Haymart M, Kunnimalaiyaan M, et al. Valproic acid activates notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. Oncologist. 2007;12:942–951. doi: 10.1634/theoncologist.12-8-942. [DOI] [PubMed] [Google Scholar]
- 14.Wilson AJ, Cheng YQ, Khabele D. Thailandepsins are new small molecule class I HDAC inhibitors with potent cytotoxic activity in ovarian cancer cells: a preclinical study of epigenetic ovarian cancer therapy. J Ovarian Res. 2012;5 doi: 10.1186/1757-2215-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinlander E, Somnay Y, Harrison AD, Wang C, Cheng YQ, Jaskula-Sztul R, et al. The novel histone deacetylase inhibitor thailandepsin A inhibits anaplastic thyroid cancer growth. J Surg Res. 2014;190:191–197. doi: 10.1016/j.jss.2014.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nano. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 17.Cabral H, Nishiyama N, Kataoka K. Supramolecular nanodevices: from design validation to theranostic nanomedicine. Accounts Chem Res. 2011;44:999–1008. doi: 10.1021/ar200094a. [DOI] [PubMed] [Google Scholar]
- 18.Zhao B, Zhao Y, Huang Y, Luo L, Song P, Wang X, et al. The efficiency of tumor-specific pH-responsive peptide-modified polymeric micelles containing paclitaxel. Biomaterials. 2012;33:2508–2520. doi: 10.1016/j.biomaterials.2011.11.078. [DOI] [PubMed] [Google Scholar]
- 19.Xiao Y, Hong H, Javadi A, Engle JW, Xu W, Yang Y, et al. Multifunctional unimolecular micelles for cancer-targeted drug delivery and positron emission tomography imaging. Biomaterials. 2012;33:3071–3082. doi: 10.1016/j.biomaterials.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao L, Xiong X, Sun X, Zhu Y, Yang H, Chen H, et al. Role of cellular uptake in the reversal of multidrug resistance by PEG-b-PLA polymeric micelles. Biomaterials. 2011;32:5148–5157. doi: 10.1016/j.biomaterials.2011.03.071. [DOI] [PubMed] [Google Scholar]
- 21.Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 2007;24:1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 22.Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther. 2006;112:630–648. doi: 10.1016/j.pharmthera.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 23.Gong C, Deng S, Wu Q, Xiang M, Wei X, Li L, et al. Improving antiangiogenesis and anti-tumor activity of curcumin by biodegradable polymeric micelles. Biomaterials. 2013;34:1413–1432. doi: 10.1016/j.biomaterials.2012.10.068. [DOI] [PubMed] [Google Scholar]
- 24.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong S. Folate-conjugated amphiphilic hyperbranched block copolymers based on Boltorn® H40, poly(l-lactide) and poly(ethylene glycol) for tumor-targeted drug delivery. Biomaterials. 2009;30:3009–3019. doi: 10.1016/j.biomaterials.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Xu WJ, Burke JF, Pilla S, Chen H, Jaskula-Sztul R, Gong SQ. Octreotide-functionalized and resveratrol-loaded unimolecular micelles for targeted neuroendocrine cancer therapy. Nanoscale. 2013;5:9924–9933. doi: 10.1039/c3nr03102k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu W, Siddiqui IA, Nihal M, Pilla S, Rosenthal K, Mukhtar H, et al. Aptamer-conjugated and doxorubicin-loaded unimolecular micelles for targeted therapy of prostate cancer. Biomaterials. 2013;34:5244–5253. doi: 10.1016/j.biomaterials.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pang XC, Zhao L, Han W, Xin XK, Lin ZQ. A general and robust strategy for the synthesis of nearly monodisperse colloidal nanocrystals. Nat Nanotechnol. 2013;8:426–431. doi: 10.1038/nnano.2013.85. [DOI] [PubMed] [Google Scholar]
- 28.Guo JT, Hong H, Chen GJ, Shi SX, Zheng QF, Zhang Y, et al. Image-guided and tumor-targeted drug delivery with radiolabeled unimolecular micelles. Biomaterials. 2013;34:8323–8332. doi: 10.1016/j.biomaterials.2013.07.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo JT, Hong H, Chen GJ, Shi SX, Nayak TR, Theuer CP, et al. Theranostic unimolecular micelles based on brush-shaped amphiphilic block copolymers for tumor-targeted drug delivery and positron emission tomography imaging. ACS Appl Mater Inter. 2014;6:21769–21779. doi: 10.1021/am5002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen G, Wang L, Cordie T, Vokoun C, Eliceiri KW, Gong S. Multi-functional self-fluorescent unimolecular micelles for tumor-targeted drug delivery and bioimaging. Biomaterials. 2015;47:41–50. doi: 10.1016/j.biomaterials.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Qian Y, Liu T, Hu X, Zhang G, You Y, et al. Amphiphilic multiarm star block copolymer-based multifunctional unimolecular micelles for cancer targeted drug delivery and MR imaging. Biomaterials. 2011;32:6595–6605. doi: 10.1016/j.biomaterials.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 32.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong S. Amphiphilic multi-arm-block copolymer conjugated with doxorubicin via pH-sensitive hydra-zone bond for tumor-targeted drug delivery. Biomaterials. 2009;30:5757–5766. doi: 10.1016/j.biomaterials.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 33.Wang C, Henkes LM, Doughty LB, He M, Wang D, Meyer-Almes FJ, et al. Thailandepsins: bacterial products with potent histone deacetylase inhibitory activities and broad-spectrum antiproliferative activities. J Nat Prod. 2011;74:2031–2038. doi: 10.1021/np200324x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang XQ, Grailer JJ, Pilla S, Steeber DA, Gong SQ. Tumor-targeting, pH-responsive, and stable unimolecular micelles as drug nanocarriers for targeted cancer therapy. Bioconjugate Chem. 2010;21:496–504. doi: 10.1021/bc900422j. [DOI] [PubMed] [Google Scholar]
- 35.Kreutzer G, Ternat C, Nguyen TQ, Plummer CJG, Månson JAE, Castelletto V, et al. Water-soluble, unimolecular containers based on amphiphilic multiarm star block copolymers. Macromolecules. 2006;39:4507–4516. [Google Scholar]
- 36.Speelmans G, Staffhorst RWHM, Dekruijff B, Dewolf FA. Transport studies of doxorubicin in model membranes indicate a difference in passive diffusion across and binding at the outer and inner leaflets of the plasma-membrane. Biochemistry. 1994;33:13761–13768. doi: 10.1021/bi00250a029. [DOI] [PubMed] [Google Scholar]
- 37.Lee Y, Park SY, Mok H, Park TG. Synthesis, characterization, antitumor activity of pluronic mimicking copolymer micelles conjugated with doxorubicin via acid-cleavable linkage. Bioconjugate Chem. 2008;19:525–531. doi: 10.1021/bc700382z. [DOI] [PubMed] [Google Scholar]
- 38.Gillies ER, Fréchet JMJ. pH-responsive copolymer assemblies for controlled release of doxorubicin. Bioconjugate Chem. 2005;16:361–368. doi: 10.1021/bc049851c. [DOI] [PubMed] [Google Scholar]
- 39.Xiao Y, Jaskula-Sztul R, Javadi A, Xu W, Eide J, Dammalapati A, et al. Codelivery of doxorubicin and siRNA using octreotide-conjugated gold nanorods for targeted neuroendocrine cancer therapy. Nanoscale. 2012;4:7185–7193. doi: 10.1039/c2nr31853a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun MJ, Wang Y, Shen J, Xiao YY, Su ZG, Ping QN. Octreotide-modification enhances the delivery and targeting of doxorubicin-loaded liposomes to somatostatin receptors expressing tumor in vitro and in vivo. Nanotechnology. 2010;21 doi: 10.1088/0957-4484/21/47/475101. [DOI] [PubMed] [Google Scholar]
- 41.Huo M, Zou A, Yao C, Zhang Y, Zhou J, Wang J, et al. Somatostatin receptor-mediated tumor-targeting drug delivery using octreotide-PEG-deoxycholic acid conjugate-modified N-deoxycholic acid-O, N-hydroxyethylation chitosan micelles. Biomaterials. 2012;33:6393–6407. doi: 10.1016/j.biomaterials.2012.05.052. [DOI] [PubMed] [Google Scholar]
- 42.Kamba SA, Ismail M, Hussein-Al-Ali SH, Ibrahim TAT, Zakaria ZA. In vitro delivery and controlled release of doxorubicin for targeting osteosarcoma bone cancer. Molecules. 2013;18:10580–10598. doi: 10.3390/molecules180910580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oberg K, Janson ET, Eriksson B. Tumour markers in neuroendocrine tumours. Ital J Gastroenterol Hepatol. 1999;31:S160–S2. [PubMed] [Google Scholar]
- 44.Chen H, Udelsman R, Zeiger MA, Ball DW. Human achaete-scute homolog-1 is highly expressed in a subset of neuroendocrine tumors. Oncol Rep. 1997;4:775–778. doi: 10.3892/or.4.4.775. [DOI] [PubMed] [Google Scholar]
- 45.Ogawara K-i, Yoshizawa Y, Un K, Araki T, Kimura T, Higaki K. Nanoparticle-based passive drug targeting to tumors: considerations and implications for optimization. Biol Pharm Bull. 2013;36:698–702. doi: 10.1248/bpb.b13-00015. [DOI] [PubMed] [Google Scholar]
- 46.Bertrand N, Wu J, Xu XY, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Del Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 48.MacEwan SR, Callahan DJ, Chilkoti A. Stimulus-responsive macromolecules and nanoparticles for cancer drug delivery. Nanomedicine (Lond) 2010;5:793–806. doi: 10.2217/nnm.10.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang J, Sawa T, Maeda H. Factors and mechanism of “EPR” effect and the enhanced antitumor effects of macromolecular drugs including SMANCS. Adv Exp Med Biol. 2003;519:29–49. doi: 10.1007/0-306-47932-X_2. [DOI] [PubMed] [Google Scholar]
- 50.Wängberg B, Nilsson O, Johanson V, Kölby L, Forssell-Aronsson E, Andersson P, et al. Somatostatin receptors in the diagnosis and therapy of neuroendocrine tumors. Oncologist. 1997;2:50–58. [PubMed] [Google Scholar]
- 51.Ono K, Suzuki T, Miki Y, Taniyama Y, Nakamura Y, Noda Y, et al. Somatostatin receptor subtypes in human non-functioning neuroendocrine tumors and effects of somatostatin analogue SOM230 on cell proliferation in cell line NCI-H727. Anticancer Res. 2007;27:2231–2239. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.