

Abstract
Purpose
Recent studies suggest that SIRT1 activating compounds (STACs) are a promising class of anti-cancer drugs, although their mechanism of action remains elusive. The main goal of this study is to determine the role of STACs as a potential therapy for pancreatic cancer. In addition, we also explored the mechanism by which these compounds affect pancreatic cancer.
Experimental design
Using in vitro (cell culture experiments) and in vivo (xenograft experiments) approaches we studied the role of SIRT1 agonists (STACs) in human pancreatic cancer cell viability and growth.
Results
We show that SIRT1 is highly expressed in pancreatic cancer cells and that the STACs SRT1720, SRT1460, and SRT3025 inhibited cell growth and survival of pancreatic cancer cells. STACs enhanced the sensitivity of pancreatic cells to gemcitabine and paclitaxel, indicating that these drugs could be used in combination with other chemotherapy drugs. We also show that STACs were very effective in inhibiting tumor xenograft growth. In mechanistic studies we observed that STACs activated a SIRT1-lysosomal-dependent cell death. Furthermore, the effect of STACs on cell viability was also dependent on the expression of the endogenous SIRT1 inhibitor DBC1.
Conclusions
Taken together our results reveal an essential role for SIRT1 and lysosomes in the death pathway regulated by STACs in pancreatic cancer cells.
Keywords: SIRT1, pancreatic cancer, lysosome, xenograft, SRT1720
Introduction
Pancreatic cancer is a leading cause of cancer-related death. The prognosis of pancreatic cancer patients is very poor. Chemotherapy is the primary treatment for most patients with pancreatic cancer; however, it has had limited impact on patient survival (1–2). Therefore, new therapeutic options are highly needed.
Sirtuins are nicotinamide adenine dinucleotide (NAD)-dependent deacetylases that are involved in physiological processes such as aging and cancer (3–6). To date, seven mammalian sirtuins have been identified (SIRT1-SIRT7) (5,6). The sirtuin SIRT1 deacetylates histones and non-histone proteins that are involved in metabolism, cell growth, apoptosis, and senescence (4). The role of SIRT1 in tumorigenesis and cancer progression is not clear, and data suggest that its role is tissue-type and context specific (7). On one hand, SIRT1 inhibition leads to growth arrest and apoptosis in lymphoma, breast, lung, and epithelial cancer (8–10). On the other hand, studies support the role of SIRT1 as a tumor suppressor (11,12). SIRT1 has been shown to be significantly up-regulated in acute myeloid leukemia (AML), prostate, gastric, and colon cancer, but down-regulated in glioma and ovarian cancers (12–16). Thus, SIRT1 may modulate a delicate balance between suppression and promotion of oncogenesis, depending on its level of activity, spatial and temporal distribution, and the stage of tumorigenesis (17).
Recent studies provided evidence for the potential clinical use of SIRT1-activating compounds (STACs) (18–20). STACs are potent small molecule activators of SIRT1 (21,22), and their use is being evaluated for metabolic syndrome, age-related diseases, and cancer (23–26). These compounds have been shown to induce apoptosis of myeloma cells (19) and necrosis of breast cancer cells (27). In contrast, SRT1720 promoted tumor cell migration and lung metastasis in mice (28), suggesting that the effects of STACs are complex and need to be evaluated in a cancer-specific manner. Specifically, one major question is which effects of these compounds are SIRT1-dependent, since some studies did not evaluate (19,29), or described SIRT1-independent effects of STACs (28,30).
In the present study, we show that STACs inhibited cell viability and growth of pancreatic cancer cell lines and also tumor growth in vivo. We also show that STACs induced apoptosis via a mechanism that involves SIRT1 and lysosomes. Our findings indicate that STACs may serve as a potential therapy for pancreatic cancer.
Material and methods
Cell lines
The source, characterization, authentication and culture conditions for the cell lines PaTu8988t, Panc-1, SU86.86, and HPDE were previously described (31). Cells were provided by Dr. D. Billadeau or from ATCC. Cultures used for experiments were reinitiated every 4–6 months from the cryopreserved stocks. The pancreatic cancer cells lines possess K-ras and/or p53 mutations that were authenticated/validated by DNA sequence analysis using published primers flanking each mutated exon. Cells were plated and maintained in media containing 1% FBS for 24–48 hours before experiments. All experiments were performed in media containing 1% FBS since, in these conditions, STACs were more effective than in 10% FBS (Supplementary Fig. S1).
Drugs, inhibitors and antibodies
SRT1720, SRT1460, and SRT3025 were from Sirtris Pharmaceuticals. Paclitaxel, 3-methyl-adenine (3-MA), bafilomycin A1, chloroquine, and ammonium chloride were purchased from Sigma-Aldrich. Glycyl-L-phenylalanine-beta-naphthylamide (GPN) was from Cayman Chemical Company. SIRT1 polyclonal (for immunoblotting) and Beta-tubulin antibodies were from Abcam, Beta-Actin antibody was from Sigma-Aldrich and DBC1 antibody was from Bethyl Laboratories. PARP-1, cleaved caspase-3, LC3, p62, and SIRT1 mouse monoclonal (for immunofluorescence) antibodies were from Cell Signaling Technology. Cathepsin B antibody was from Santa Cruz Biotechnology.
Cell viability assay
Cells were trypsinized and cell suspensions were mixed with Trypan blue dye. Cells were scored as live or dead based on Trypan blue exclusion.
MTT assay
Cells were plated in 96 well plates (3–5x103/well). 24 hours later, cells were treated with the drugs, and incubated for 24–72 hours at 37 °C. Then cells were incubated with 20 μl of 5 mg/ml MTT for 2 hours and the resulting formazan crystals were dissolved in dimethyl sulfoxide (200 μl). Absorbance was measured at 560 nm. The effect of the drugs on cell viability was assessed as percentage cell viability compared to vehicle-treated control cells, which were assigned 100 % viability. IC50 were calculated using CalcuSyn software (Biosoft).
Short interfering RNA
Non-targeting siRNA (Dharmacon # D001210-03-20) was used as control. SIRNAs to knockdown human SIRT1 were: ON-TARGETplus SMARTpool siRNA from Dharmacon (#1) and siRNA with the sense strand 5′-AGAGUUGCCACCCACACCUUU (#2). The siRNA duplex against DBC1 was: DBC1 siRNA sense strand, 5′-AAACGGAGCCUACUGAACAUU. Transfections were performed with 50 nM of siRNA using Lipofectamine RNAiMAX reagent (Life Technologies) according to the manufacturer’s instruction. 24 hours after transfection the cells were re-plated and allowed to attach for 24 hours. Cells were then treated with drugs and used for specific assays.
Western blot Analysis
Cells were lysed in 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, 5 mM NaF, 50 mM 2-glycerophosphate, and protease inhibitors (Roche). Lysates were centrifuged at 12,000 rpm at 4 °C for 10 minutes. Samples were separated through a SDS-PAGE, transferred to Immobilon P membranes, and immunoblotting was performed with specific antibodies. Blots are representative of at least three experiments.
Soft agar colony formation assay
Cells were seeded in 6-well plates (10,000/well) in 0.35% agar over 0.6% bottom agar layer in growth media containing 5% FBS and SRT1720 or SRT1460. Colonies measuring ≥ 50 μm were counted after 7–10 days of culture using a cell colony counter (Gelcount, Oxford Optronix).
ATP measurements
ATP levels were measured using ATPlite Luminescence assay system from PerkinElmer according to the manufacturer’s instructions.
Tumor xenograft study
Female athymic nu/nu mice were obtained from the National Cancer Institute (NCI). The experimental protocol was reviewed and approved by the Institutional Animal Care and Use Comittee at Mayo Clinic (protocol A39511). Subconfluent Panc-1 cells were harvested by trypsinization. Viability of cells was verified by Trypan blue exclusion. Only suspensions with 90% cell viability were used. Panc-1 cells were injected subcutaneously in both flanks of 5–6 week old female athymic nu/nu mice (4x106 cells in 100 μl of PBS:matrigel (1:1)/site). After 14 days of implantation, when the tumor volume reached ~60 mm3, mice were randomized in two groups: (i) untreated control (vehicle only, PBS containing 1% Hydroxypropyl)-β-cyclodextrine and 12% propylene glycol); (ii) SRT1720 or SRT3025 (50–200 mg/kg, daily for 10 days by oral gavage). Tumor volumes were measured weekly for an additional 10 days with a caliper and calculated using the formula V=4/3π(l x w x d), where l is the length, w is the width and d is the depth.
Immunofluorescence, LysoTrackerRed staining and Confocal Microscopy
For immunofluorescence analysis, non-transfected or transfected SU86.86 cells were plated on cover-slips. Cells were fixed with 3% paraformaldehyde for 10–12 minutes, permeabilized with 0.2% TritonX-100 for 15 minutes, pre-incubated with blocking buffer (4% BSA in PBS) and incubated with primary antibodies (LC3-1:1000; p62-1: 800; SIRT1-1:1000; Cathepsin B 1:800; LAMP-2 1:800) overnight. Cover-slips were washed with PBS and incubated with fluorescence-tagged secondary antibodies (Alexa Fluor 488 and/ or 568, Molecular probes, Invitrogen) in blocking buffer for 2 hours followed by counterstaining with Vectashield containing DAPI (Vector Labs). Cells were imaged using a Zeiss LSM780 confocal microscope with a 100x objective. For LysoTrackerRed staining, cells were incubated with 100 nM LysoTrackerRed (Molecular Probes) at 37 °C for 15 minutes. Cells were then fixed with 3% paraformaldehyde for 5–7 minutes followed by counterstaining with Vectashield containing DAPI and imaged immediately.
SIRT1 activity measurement
SIRT1 activity was measured with a fluorometric assay (Enzo Life Sciences, catalogue number BML-AK555-0001) as described before (32). The proteins used were recombinant SIRT1 from bacteria (BIOMOL) and GST-DBC1 purified from baculovirus (33). 1 μM recombinant SIRT1 was incubated in vitro with 5 μM DBC1. SRT1720 was used at 1 μM.
Quantification of mRNA
mRNAs from human biospecimens were obtained from the Mayo Clinic Tissue Registry under an approved IRB protocol. RNA was isolated from a set of pancreatic adenocarcinoma patient samples for which frozen, paired tumor and non-tumor pancreas tissue was available. RNA was isolated from pancreatic cell lines using the RNeasy kit (Qiagen). mRNA abundance was analyzed by quantitative PCR using an Applied Biosystems 7900HT thermal cycler and TaqMan Gene Expression Assay probe sets (Applied Biosystems) were: 18s (Hs99999901_s1), SIRT1 (Hs01009005_m1), DBC1 (Hs00221777_m1), and GAPDH (Hs02758991_g1). Gene expression in primary pancreatic cancers was normalized to 18S. In cells, the relative expression of target genes was normalized to GAPDH. The changes in expression were calculated relative to control pancreatic cells (HPDE).
Results
STACs reduce cell viability and growth of pancreatic cancer cells
To explore the role of STACs as a therapy for pancreatic cancer, we first examined their effect on the cell viability of pancreatic cancer cell lines. Treatment of these cells with SRT1720 inhibited cell viability in a dose-dependent manner (Fig 1A). All pancreatic cancer cells were more sensitive to this drug than control HPDE cells. There was a significant difference in the sensitivity of cells to SRT1720, with PaTu8988t and SU86.86 cells being the most sensitive. When different lengths of treatment with SRT1720 were analyzed, we observed the majority of decrease in cell viability in the first 24 hours (Fig 1B), indicating that SRT1720 quickly decreases cell viability.
Figure 1. STACs decrease pancreatic cancer cell survival and growth.

A, Cells were treated with vehicle (control) or different concentrations of SRT1720 and submitted to MTT analysis 72 hours after treatment. IC50s (μM) were: Patu8988t, 1.2 ± 0.07; SU86.86, 1.13 ± 0.04; Panc-1, 2.1 ± 0.04. B, SU86.86 and Patu8988t cells were treated with vehicle or 2 μM SRT1720 for different time periods. Cells were submitted to MTT analysis. C, Patu8988t cells were treated with vehicle (control) or SRT1720 for 72 hours and cells were counted by Trypan blue dye exclusion assay. D, ATP levels were measured in pancreatic cancer cells after treatment with vehicle (control) or 2.5 μM SRT1720 for 48 hours. E, Patu8988t cells were grown in soft agar containing vehicle or different concentrations of SRT1720 for 7 days. Graph shows quantitative analysis of colony number. F, Cells were treated with vehicle or different concentrations of SRT1460 and submitted to MTT analysis 72 hours after treatment. IC50s (μM) were: Patu8988t, 1.62 ± 0.13; SU86.86, 2.31 ± 0.23; Panc-1, 0.66 ± 0.02; HPDE, 2.39 ± 0.29. G, Cells were treated with vehicle or different concentrations of SRT3025 for 72 hours and submitted to MTT analysis. SRT3025 IC50 (μM) for SU86.86 cells was 0.98 ± 0.13. Vehicle-treated cells (control) were assigned as 100 % cell viability. Values are mean ± SEM of at least three independent experiments.* indicates p<0.05, compared to vehicle.
The effect of SRT1720 on cell viability and growth was confirmed by additional assays (Fig. 1C, 1D and 1E). In Patu8988t cells there was a dose-dependent decrease in the number of viable cells counted by Trypan blue exclusion assay after treatment with SRT1720 (Fig 1C). ATP can be used as a marker of cell viability, as ATP levels rapidly decline when cells undergo necrosis or apoptosis. There was a marked decrease in ATP levels in cells incubated with SRT1720 (Fig. 1D). Since colony formation of human pancreatic cancer cells in soft agar is one of the best in vitro indicators of malignant behavior (34), we measured the number of colonies of Patu8988t cells in soft agar during treatment with SRT1720. Treatment with this compound resulted in significantly fewer colonies than vehicle treatment, and this effect was dose-dependent (Figure 1E).
We also investigated if other STACs had similar effects on pancreatic cancer cells in vitro. Treatment with different concentrations of SRT1460 (24) inhibited cell viability in a dose-dependent manner (Fig. 1F), with all pancreatic cancer cells being more sensitive than the control HPDE cell. SRT1460 was also shown to inhibited cell growth by the Trypan blue exclusion and in the anchored-independent cell growth assays in Patu8988t (Supplementary Fig. S2A and S2B). In contrast, SRT3025 (35–36), a STAC that has been tested in humans (37), decreased viability mainly of SU86.86 cells in culture. Patu8988t and Panc-1 cells were not as sensitive to the effect of the drug (Fig. 1G). Our observations strongly demonstrate that STACs inhibit cell viability and growth of pancreatic cancer cells, with SRT1720 and SRT1460 being the most effective STACs.
Since conventional therapeutic agents for pancreatic carcinoma produce minimal survival benefit as single agents, numerous efforts have focused on drug combinations (2). We tested STACs in combination with gemcitabine and paclitaxel, drugs that have been used as therapies for pancreatic cancer. Our results show that, in both Patu8988t and SU86.86 cells, combinations of STACs with gemcitabine or paclitaxel caused a greater decrease in cell survival than addition of the STACs alone (Supplementary Fig. S3). The only exception was combination of gemcitabine and SRT1460 in Patu8988t, which was not more effective than SRT1460 alone. These results together indicate that STACs have the potential be used in combination with other drugs in pancreatic cancer.
STACs prevent pancreatic tumor growth in vivo
We next tested the in vivo effect of STACs using a xenograft animal model of pancreatic cancer. SRT1720 treatment markedly decreased tumor size in comparison with the vehicle-treated tumors (Fig. 2A). When different doses of SRT1720 were compared, there was a dose-dependent effect on tumor growth, with both 200 mg/kg and 100 mg/kg being significantly different from vehicle treatment (Fig. 2B). As shown in Figure 2C and 2D, tumor size and weight were greatly decreased in SRT1720-treated mice. During the treatment period, no obvious treatment-related toxicity was observed. The food intake and body weight of both groups of animals remained similar (Supplementary Fig. S4). We also performed in vivo experiments with Panc-1 xenografts using SRT3025. Similar to SRT1720, SRT3025 treatment also inhibited tumor growth in vivo (Fig 2E), even though it was not as effective in inhibiting the viability of Panc-1 cells in culture. SRT1460 was toxic to animals and could not be tested in vivo. These data show that STACs are also effective in inhibiting pancreatic tumor growth in vivo.
Figure 2. STACs prevent pancreatic tumor growth in vivo.

Panc1 cells were used in an in vivo subcutaneous xenograft mouse model. Starting at 14 days after tumor implantation the animals were treated with daily doses of vehicle or STACs by oral gavage for 10 days. Tumors were measured for an additional 10 days. A, Effect of 200mg/kg SRT1720 on tumor growth in vivo (n=17 tumors/treatment). B, The effect of different doses of oral SRT1720 on tumor growth in vivo (n=12 tumors/treatment). Graph shows relative tumor sizes at the end of the experiment. C, Representative images of the tumors in situ in animals treated with vehicle or 200mg/kg SRT1720 at the end of the study. D, Representative images (left) and average weight (right) of the dissected tumors from animals treated with vehicle or 200mg/kg of SRT1720. E, Effect of 200mg/kg SRT3025 on tumor growth in vivo (n=7 tumors/treatment). Values are mean ± SEM. * indicates p<0.05, compared to vehicle.
STAC-induced cell death is lysosomal dependent, but lysosomal permeabilization is not sufficient to cause cell death
Next, we investigated the mechanism underlying the effect of STACs in pancreatic cancer cells. In breast cancer cells, it was proposed that SRT1720 promotes cell death through a SIRT1-independent induction of lysosomal membrane permeabilization (LMP) (27). To determine whether STACs regulate lysosomal function in pancreatic cancer cells, SU86.86 cells were treated with SRT1720 and then incubated with LysoTracker Red. The LysoTracker Red dye concentrates in the lysosomal compartment and upon changes in lysosomal acidification or LMP, it diffuses out of the lysosome (38). 5 hours treatment with SRT1720 markedly decreased lysosome staining with LysoTracker Red (Fig. 3A). To distinguish between changes in lysosome acidification or LMP, we performed staining with cathepsin B antibody. Translocation of cathepsins from lysosomes to cytosol is a distinctive sign of LMP (38,39). Cathepsin B localized to lysosomes in vehicle-treated cells and showed a punctate staining. However, upon treatment with SRT1720, cathepsin B staining decreased and became diffuse (Fig. 3A). Immunofluorescence staining with an antibody for Lysosome-associated membrane protein 2 (LAMP-2) shows that STACs did not completely disrupt the lysosomes (Supplementary Fig. S5). It is then likely that STACs are promoting LMP.
Figure 3. STACs-induced cell death is dependent on lysosomes.

A, SU86.86 cells were incubated with vehicle or 5 μM SRT1720 for 5 hours for LysoTracker Red labeling and 16 hours for cathepsin B immunofluorescence, followed by DAPI staining. B, SU86.86 and C, Patu8988t were treated for 24 hours with 10 μM chloroquine (CQ), 2.5 mM NH4Cl, 1 nM bafilomycin A1 (BafA1) alone and in combination with 2 μM SRT1720, and submitted to MTT analysis. D, SU86.86 were treated for 24 hours with 10 μM CQ, 2.5 mM NH4Cl, or 1 nM BafA1 alone and in combination with 7.5 μM SRT1460 or SRT3025, and submitted to MTT analysis. E, SU86.86 cells were incubated with vehicle or 50 μM GPN for 16 hours. Immunofluorescence analysis was performed with cathepsin B antibody or cells were labeled with LysoTracker Red, followed by DAPI staining. F, SU86.86 cells were treated with vehicle, 50 and 100 μM GPN, and 2 μM SRT1720 for 48 hours and then submitted to MTT analysis. G, SU86.86 cells were incubated with vehicle or 5 μM SRT1720 for 16 hours. Immunofluorescence analysis was performed with LC3-II and p62 antibodies and cells were stained with DAPI. H, Patu8988t cells were treated for 16 hours with 5 μM SRT1720, SRT1460 or SRT3025. Expression levels of LC3-II and p62 were determined by immunoblotting. I, SU86.86, Patu8988t, and Panc-1 cells were treated for 24 hours with 3-methyladenine (3-MA, 2 mM for SU86.86 and 1.5 mM for Patu8988t and Panc-1) alone and in combination with 2 μM SRT1720 (SU86.86 and Patu8988t) or 2.3 μM SRT1720 (Panc-1), and submitted to MTT analysis. Graphs show mean ± SEM of at least three independent experiments. * indicates p<0.05, significantly different from STACs alone.
To determine whether the effect of STACs on the lysosomes was critical for regulation of pancreatic cancer cell viability, we used the lysosomotropic agents bafilomicyn A1, chloroquine, and ammonium chloride to inhibit lysosomal acidification and function (40). None of the three drugs decreased SU86.86 or Patu8988t cell viability (Fig. 3B and 3C), indicating that blocking lysosome acidification for 24 hours does not inhibit pancreatic cancer cell viability. Interestingly, these agents significantly reversed the effect of SRT1720 on cell viability (Fig. 3B and 3C). Similar results were also obtained when lysosome inhibitors were added in combination with SRT1460 and SRT3025 (Fig 3D). To confirm that lysosome inhibitors were indeed reducing lysosome acidification, we tested the effect of bafilomycin, ammonium chloride, and chloroquine on LysoTracker Red staining. Similar to SRT1720, these drugs decreased LysoTracker staining (Supplementary Fig. S6). To determine whether lysosome permeabilization was sufficient to regulate pancreatic cancer cell viability, we used glycyl-L-phenylalanine-beta-naphthylamide (GPN), a drug that osmotically permeabilize the lysosomes (41). GPN quickly decreased LysoTracker and cathepsin B staining, indicating lysosomal permeabilization, but did not inhibit pancreatic cell viability (Fig. 3E and 3F). We concluded that the inhibition of pancreatic cancer cell viability induced by STACs clearly depends on lysosomal function, but in contrast to breast cancer cells, permeabilization of the lysosomes alone is not sufficient to decrease pancreatic cancer cell viability.
It has been shown that pancreatic cancers require autophagy for tumor growth, and that inhibition of autophagy leads to tumor regression (42). Because lysosomes are important for autophagy (39), we investigated the expression of autophagy markers in cells treated with STACs. All three STACs increased expression of the autophagy marker LC3-II, detected by both immunofluorescence punctate staining and immunoblotting (Fig 3G, 3H, and Supplementary Fig. S7). Since an increase in LC3-II could be an indicator of increased autophagy or inhibition of autophagy flux (43), we measured levels of p62/SQSTM1. p62 is a ubiquitin binding protein that binds LC3-II and is degraded after fusion of autophagosome and lysosome, being considered a marker for autophagy flux (44). Because we observed an increase in p62 levels with SRT1720 treatment (Fig 3G, 3H, and Supplementary Fig. S7), it implies that STACs inhibit the autophagy flux in pancreatic cancer cells.
To study the role of autophagy in the pathway regulated by STACs, we blocked autophagy using the PI3K inhibitor 3-MA (40). Treatment with 3-MA alone for 24 hours did not decrease cell viability (Fig. 3I). When 3-MA was added in combination with SRT1720 in Patu8988t cells, there was an increase in cell survival compared to SRT1720 alone (Fig. 3I). However, in SU86.86 and Panc-1 cells, combination of SRT1720 and 3-MA caused an even greater inhibition of cell viability than SRT1720 alone (Fig 3I). Because 3-MA has different effects in these cell lines, it appears that the block in autophagy induced by STACs is not the main mechanism involved in the STAC-induced cell death. This conclusion is also supported by the observation that multiple lysosomal inhibitors did not cause cell death in pancreatic cancer cells at the time course analyzed (Fig 3B and 3C), but they did impair autophagy, as shown by an increase in LC3-II and p62 levels (Supplementary Fig S8). These results also indicate that the PI3K pathway, and possibly autophagy, has different roles in the pathway regulated by SRT1720 in distinct pancreatic cancer cells. In Patu8988t, the PI3K pathway appears to regulate the effect of SRT1720. In contrast, the PI3K inhibitor 3-MA was very effective in sensitizing SU86.86 and Panc-1cells to SRT1720, suggesting that combination of STACs and PI3K inhibitors could be an effective treatment for some pancreatic tumors.
SIR1720 induces apoptosis in pancreatic cancer cells, which is prevented by lysosomal inhibitors
To further explore the role of STACs in pancreatic cancer cells, we analyzed other downstream signaling pathways activated by STACs. Treatment of Patu8988t and SU86.86 cells with SRT1720 and SRT1460 increased the cleavage of PARP-1 and caspase-3, indicating an induction of apoptosis (Fig. 4A and 4B). These increases were both time- and dose-dependent (Fig 4A and Supplementary Fig. S9). Since lysosomal acidification is required for pancreatic cancer cell death, we tested the effect of lysosomal inhibitors in the apoptosis induced by SRT1720. SU86.86 cells were treated for different time periods with SRT1720 in the absence and presence of bafilomycin A1. When added in combination with SRT1720, bafilomycin A1 further enhanced the increases in LC3-II and p62 caused by SRT1720, detected by both immunoblotting (Fig. 4C) and immunofluorescence (Supplementary Fig. S10), implying that the combination of bafilomycin A1 and SRT1720 cause an even higher block in autophagy than the drugs alone. In contrast, when bafilomycin A1 and SRT1720 were added together, there was less cleavage of caspase-3 and PARP-1 than in the presence of SRT1720 alone (Fig. 4C), suggesting that bafilomycin A1 inhibits the effect of SRT1720 on apoptosis. Similar results were observed when SU86.86 cells were treated with SRT1720 in combination with chloroquine and ammonium chloride (Supplementary Fig. S11). Taken together, these data suggest that lysosomal function is important for the effect of STACs in cell viability and induction of apoptosis in pancreatic cancer cells, via an autophagy-independent pathway.
Figure 4. SRT1720-induced apoptosis is prevented by lysosomal inhibitors.
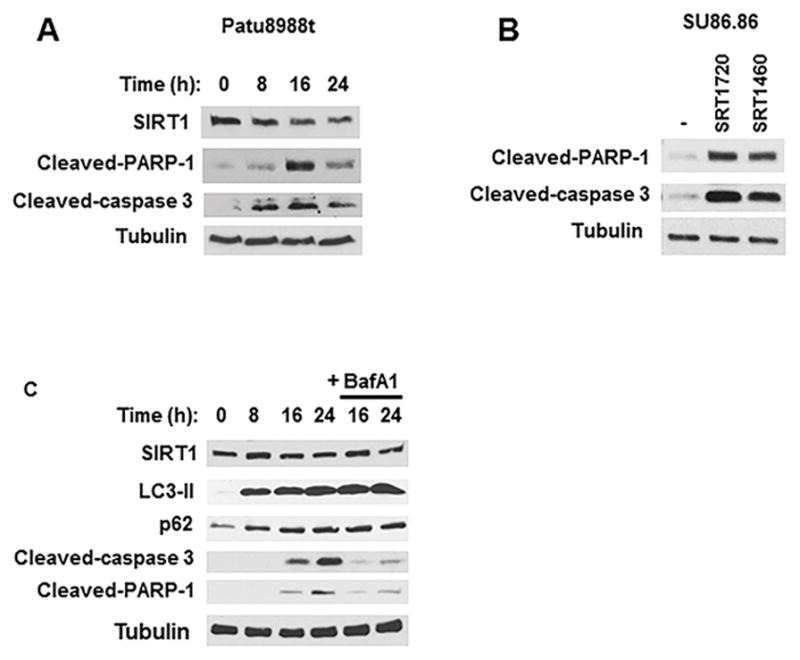
A, Patu8988t cells were treated with vehicle or 5 μM SRT1720 for different time periods. Expression levels of SIRT1, cleaved caspase-3, and cleaved PARP-1 were determined by immunoblotting. B, SU86.86 cells were incubated with vehicle, 5 μM SRT1720 or 5 μM SRT1460 for 24 hours, and expression of PARP-1 and cleaved caspase-3 were determined by immunoblotting. C, SU86.86 cells were treated for different time periods with 5 μM SRT1720 alone and in combination with 1 nM bafilomycin A1 (BafA1). Expression levels of SIRT1, LC3-II, p62, cleaved caspase-3, and cleaved PARP-1 were determined by immunoblotting.
SIRT1 and DBC1 regulate the effect of STACs on cell viability
It is inconclusive from the literature whether the effects of STACs are dependent on SIRT1. To establish the role of SIRT1 in the effect of STACs in pancreatic cancer cells, we first evaluated the protein expression of SIRT1 and its endogenous inhibitor, DBC1 (45), in the pancreatic cancer cell lines and HPDE. SIRT1 protein levels were higher, while DBC1 expression was lower, in all pancreatic cancer cells than in HPDE cells (Fig. 5A). Therefore, the ratio SIRT1/DBC1 was higher in pancreatic cancer cells than in HPDE cells (Figure 5A). This suggests that pancreatic cancer cells may have a higher amount of free SIRT1, which may be more easily activated by STACs. Indeed, our in vitro experiments show that SRT1720 activates SIRT1 better in the absence of DBC1 (Supplementary Fig. S12). To determine if the mRNA expression of SIRT1 and DBC1 relate to their protein levels, we measured the mRNA levels of both proteins in the different cell lines. Similar to the protein expression, the ratio of SIRT1/DBC1 mRNA expression was higher in the pancreatic cells than in the control HPDE cell (Fig 5B and Supplementary S13).
Figure 5. SIRT1 and DBC1 regulate the effect of SRT1720 on cell viability.
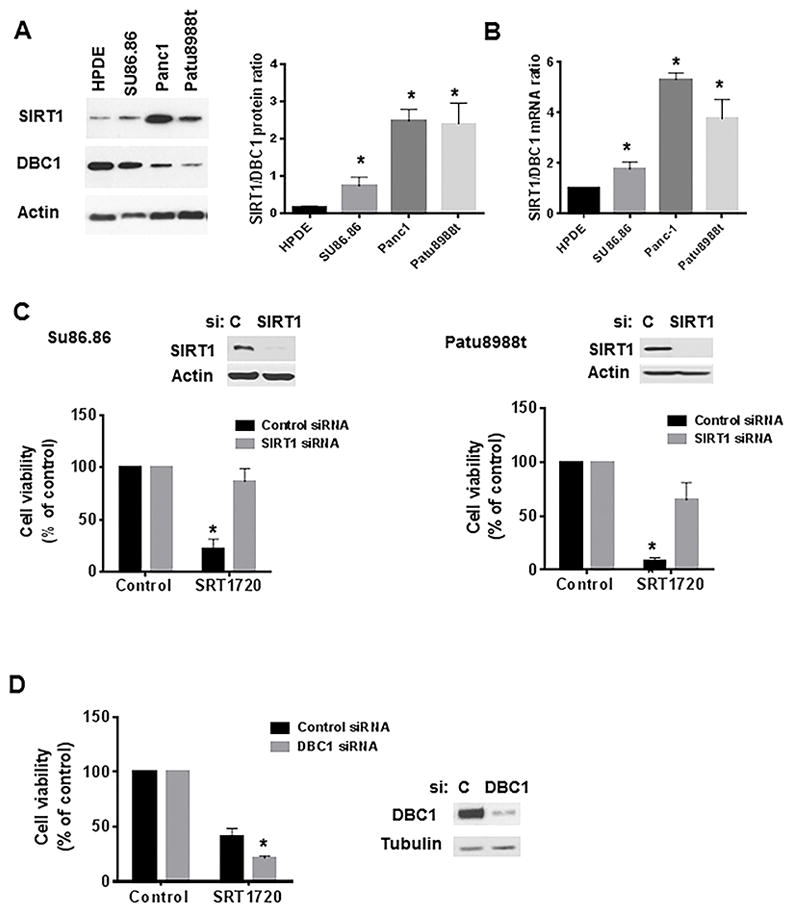
A, Cell lysates from different pancreatic cancer cell lines were immunoblotted for SIRT1, DBC1 and actin. Graph shows the average of three experiments measuring the ratio of SIRT1/DBC1 protein expression. B, Graph shows the average of three experiments measuring the ratio of SIRT1/DBC1 mRNA expression in pancreatic cells. * indicates p<0.05, compared to HPDE cells. C, SU86.86 cells (left) were transfected with SIRT1 siRNA#1 or a control siRNA. PaTu8988t cells (right) were transfected with SIRT1 siRNA#2 or a non-target siRNA (control). The efficiency of knockdown was analyzed by immunoblotting 72 hours after transfection. 24 hours after transfection, cells were re-plated and allowed to attach for 24 hours. Cells were then treated with vehicle (control) or 2 μM SRT1720 and submitted to MTT analysis 48 hours after treatment. Graph shows the cell viability in the absence and presence of SRT1720, in control siRNA and SIRT1 siRNA-transfected cells. Vehicle-treated cells were assigned as 100% viability. D, SU86.86 cells were transfected with control and DBC1 siRNA. Cells were re-plated 24 hours after transfection, allowed to attach for 24 hours, and then treated with vehicle or 2 μM SRT1720 for 48 hours. Graph shows mean ± SEM of three independent experiments measuring cell viability in the absence and presence of SRT1720, in control and DBC1 siRNA-transfected cells. Vehicle-treated cells were considered 100% cell viability. * indicates p<0.05, compared to vehicle.
To evaluate whether the effect of STACs on pancreatic cancer cell viability depends on SIRT1, we tested the effect of these compounds in cells knocked down for SIRT1. A strong reduction in SIRT1 levels was observed in cells upon transfection with SIRT1 siRNA compared with control siRNA (Fig. 5B). SU86.86 and Patu8988T cells transfected with SIRT1 siRNAs were considerably less sensitive to SRT1720 than control siRNA-transfected cells (Fig. 5B). In addition, SRT1460-inhibition of cell viability was also SIRT1-dependent (Supplementary Fig. S14). These results show that the anti-survival effect of STACs in pancreatic cancer cells is SIRT1-dependent.
Among the pancreatic cancer cells, SU86.86 had the highest levels of DBC1 and the smallest SIRT1/DBC1 ratio. To test whether DBC1 regulates the effect of STACs in pancreatic cancer cells we knocked down DBC1 in SU86.86 cells and measured cell viability in the absence and presence of SRT1720. While 2 μM SRT1720 decreased cell viability by 60% in the control siRNA transfected cells, there was an even further decrease (80%) in cells transfected with DBC1 siRNA (Figure 5C). These results confirm our hypothesis that DBC1 is a negative regulator of the SRT1720 effect in pancreatic cancer cells, and further supports a SIRT1-dependent effect of STACs.
We further investigated the expression of SIRT1 and DBC1 in pancreatic cancer tumor samples from patients. There was a significant variability in the expression of SIRT1 and DBC1 in pancreatic cancer samples, but in general, the expression of SIRT1 was higher and the expression of DBC1 was lower in samples from pancreatic tumors than in normal tissue (Supplementary Fig. S15). Our data together suggest that the high ratio of SIRT1/DBC1 in pancreatic cancer cells could explain why these cells are more sensitive to SIRT1 activators than normal HPDE cells. The relative expression of SIRT1 and DBC1 may serve as molecular markers for the responsiveness to SIRT1 activators in vivo.
SIRT1 regulates the effect of STACs in the lysosomes and in apoptosis
Immunofluorescence of SIRT1 in SU86.86 cells showed that in vehicle-treated cells SIRT1 is present in the cell nuclei. However, upon SRT1720 treatment, a fraction of SIRT1 translocated to the cytosol, increasing the ratio of cytoplasmic to nuclear SIRT1 (Fig. 6A). This increase in cytoplasmic SIRT1 is consistent with the hypothesis that SIRT1 may be regulating lysosomes in the cytosol. Thus, we investigated whether the effects of SRT1720 on lysosomes were SIRT1-dependent. SIRT1 siRNA transfection abolished SIRT1 staining, while control siRNA cells showed nuclear localization of SIRT1 (Fig. 6A). Treatment with SRT1720 quickly decreased LysoTracker Red staining by 3 hours in control siRNA-transfected cells, but was not as effective in SIRT1 siRNA-transfected cells (Fig. 6B). In addition, the cathepsin B distribution after SRT1720 treatment was also dependent on SIRT1. While cathepsin B shows a diffuse distribution after SRT1720 in control siRNA transfected cells, it still shows punctate staining in SIRT1 siRNA-transfected cells (Fig. 6C). These results collectively show that the effects of SRT1720 on LMP are SIRT1-dependent.
Figure 6. The effect of SRT1720 on lysosomes and apoptosis is SIRT1-dependent.

A–C, SU86.86 cells were transfected with SIRT1 siRNA#1 or the control siRNA. 24 hours after transfection, cells were re-plated and allowed to attach for 24 hours. Cells were then treated with vehicle (control) or 5 μM SRT1720. A, Cells were treated for 16 hours before immunofluorescence analysis with SIRT1 antibody and DAPI staining. Quantitation of nuclear and cytoplasmic SIRT1 was performed by measuring average fluorescent signals from the nucleus and cytoplasm of vehicle or SRT1720-treated cells (n=75 for each condition) by using Image-J software. Graph shows the average of the ratio of cytoplasmic to nuclear SIRT1 intensities (* indicates p<0.05, compared to vehicle). B, Cells were treated for 1.5 and 3 hours before LysoTracker Red labeling and DAPI staining. C, Cells were treated for 16 hours before immunofluorescence analysis with cathepsin B antibody and staining with DAPI. D, SU86.86 cells were transfected with control siRNA, SIRT1 siRNA#1, or DBC1 siRNA. 24 hours after transfection, cells were re-plated and allowed to attach for 24 hours. Cells were then treated with vehicle (control) or 5 μM SRT1720 for 16 hours. Expression levels of LC3-II, p62, cleaved Caspase-3, and cleaved PARP-1 were determined by immunoblotting.
To further explore the role of SIRT1 in the mechanism of cell death induced by STACs, we transfected SU86.86 cells with control, SIRT1, and DBC1 siRNAs. 48 hours after transfection, cells were treated with SRT1720 for 16 hours and the expression of proteins involved in apoptosis and autophagy was analyzed. In control siRNA-treated cells there was an increase in cleavage of caspase-3 and PARP-1 induced by SRT1720, and also an increase in levels of LC3-II and p62, confirming the induction in apoptosis and the inhibition of autophagy (Fig. 6D). In the presence of SIRT1 siRNA, there was a marked decrease in the cleavage of PARP-1 and caspase-3 induced by SRT1720 (Fig. 6D). However, the increase in expression of LC3-II and p62 induced by SRT1720 was not influenced by absence of SIRT1. In the presence of DBC1 siRNA there was still cleavage of caspase-3 and PARP-1, and an increase in LC3-II and p62 (Fig. 6D). These results together demonstrate that SIRT1 is required for regulation of lysosomal function and apoptosis induced by SRT1720, but does not appear to be involved in the inhibition of autophagy flux by this drug.
Discussion
The present study clearly shows a role for STACs as a potential therapy for pancreatic cancer. Here we describe that STACs inhibit cell survival of a panel of pancreatic cell lines, reduce anchorage-dependent and-independent pancreatic cancer cell growth, and inhibit xenograft tumor growth. STACs have been shown to differ in pharmacokinetics, potency and toxicity (24). The most effective STACs in pancreatic cancer cell lines were SRT1720 and SRT1460. However, even though SRT3025 was less effective in vitro, it did inhibit Panc-1 tumor growth in vivo. This indicates that although all STACs inhibit survival and growth of pancreatic cancer cells, different types of pancreatic tumors may have different sensitivities to specific STACs. Moreover, we and others show that SRT1720 and SRT3025 are well tolerated in mice and have the potential to be used in several diseases (19,26,27,35,36).
One of the main questions in STAC research at this time is whether the biological effects of STACs are mediated by SIRT1 activation. The original study describing the development and effects of STACs did not show a clear SIRT1 dependency for its effects (24). Furthermore, another study questioned the direct binding and activation of SIRT1 by STACs (30). In tumor biology studies, the dependency on SIRT1 was only investigated in one report which found confounding results, with the effect of STACs being SIRT1-dependent in vivo but not in vitro (27). Our study is the first to demonstrate a clear SIRT1-dependent biological effect of multiple STACs in several parameters in cancer cells. Importantly, our findings highly suggest that the effects of STACs in pancreatic cell growth, apoptosis, and lysosomal function are SIRT1-dependent. Our data are in agreement with at least two recent studies demonstrating that STACs indeed bind and activate SIRT1 directly, at least in vitro (46,47). Interestingly, we also observed that STACs had SIRT1-independent effects, such as the blockage of autophagy.
Our data together supports the idea that SIRT1 plays a key role in the survival and proliferation of pancreatic cancer cells. Previous reports show that inhibition of SIRT1 by EX527 or SIRT1 knockdown in pancreatic cancer cells reduces cell proliferation, and induces apoptosis and senescence (48). However, SIRT1 inhibition in vivo by EX527 was shown to promote xenograft tumor growth (49), indicating that the effect of SIRT1 in pancreatic cancer is complex, and it is possible that activation or inhibition of SIRT1 disturbs the growth of pancreatic cancer cells.
Although studies have explored the role of SIRT1 in cancer progression and the mechanisms involved in this regulation, little is known about the signaling pathways regulated by STACs in cancer. In multiple myeloma cells SRT1720 was shown to increase apoptosis, presumably through a pathway dependent on SIRT1-AMPK (19). In breast cancer cells, SRT1720 induces cell death through a lysosomal-dependent pathway, involving LMP and necrosis, and there is no induction of apoptosis (27). In addition, the SRT1720-induced cell death in breast cancer cells occurred irrespective of SIRT1. In contrast, in pancreatic cancer cells, lysosomes are required for SRT1720-induced cell death, and this pathway appears to involve induction of apoptosis and is highly dependent on SIRT1. In fact, accumulating data support the role of cathepsins as effectors of LMP-initiated cell death pathways, including LMP-induced apoptosis (39). This suggests that the lysosomal cell death pathway is connected with other death pathways, like apoptosis. Our findings indicate that STACs act by a coordinated pathway involving multiple components including SIRT1, lysosomes, and apoptotic components. A key factor in determining the type of cell death mediated by lysosomes (necrosis or apoptosis) seems to be the magnitude of lysosomal permeabilization and consequently the amount of proteolytic enzymes released into the cytosol (50). It will be important to explore in the future how STACs regulate lysosomal permeabilization and function, and ultimately the cell death process in pancreatic cancer cells. One possibility is that STACs are lysosomotropic agents that are being sequestered in the lysosomal compartment causing an increase in lysosomal permeability (51). This hypothesis is supported by the fact that STACs are basic, they displace LysoTracker Red from inside the lysosomes, and their effect is blocked by inhibitors of lysosome acidification. It is also possible that SIRT1 activation by STACs may lead to SIRT1 translocation to the cytoplasm and targeting of lysosomal proteins and function. However, at this point, except for the fact that STACs lead to increases in cytoplasmic SIRT1, we do not have mechanistic demonstration that this translocation is necessary for the cell killing induced by STACs.
The role that autophagy plays in the mechanism of cell death induced by STACs it is still not clear. All STACs appeared to block autophagy. However, autophagy inhibitors did not promote cell death when added for the same duration as STACs. Additionally, SIRT1 knockdown did not affect the expression of autophagy markers induced by STACs, supporting the notion that inhibition of autophagy is not the primary mechanism involved in the cell death. Nevertheless, it is possible that the autophagy block contributes to the effect of STACs in pancreatic cancer cells.
Human pancreatic tumors are hypovascular in nature and large areas of tumor survive under nutrient and oxygen starvation. However, human pancreatic tumor cells show extraordinary ability to tolerate extreme states through the modulation of energy metabolism. Therefore, agents that retard the tolerance of cancer cells to nutrient starvation represent a novel approach in anticancer drug discovery (52). Our study shows that pancreatic cancer cells are more sensitive to STACs in low serum conditions, implying that STACs may be a good therapy under nutrient starved conditions in the pancreatic tumor microenvironment.
The STAC doses that were effective in our animal studies have been shown to lead to micro-molar plasma levels (24). We observed that these concentrations were pharmacologically relevant in killing pancreatic cancer cells in vitro. Our findings also reveal that SIRT1 is a key molecular target for pancreatic cancer treatment. As new STACs, with high safety and efficacy, are developed it will be of great importance to test them as therapies for pancreatic cancer.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Pancreatic cancer incidence is on the rise. Currently this cancer continues to have extremely poor prognosis, and therapeutic options are limited. Novel therapies are urgently needed for this disease. Here we described pre-clinical evidences that SIRT1-agonists (STACs) induce pancreatic cancer cell death via a SIRT1/ lysosomal dependent process. Our data are extremely novel and presents one of the few examples of well documented SIRT1-mediated effects of STACs. In addition, the effects of these specific SIRT1 agonists have never been reported in pancreatic cancer. We believe that our findings justify further studies on the role of STACs as a therapeutic option in pancreatic cancer. We propose that in the future STACs may improve the therapeutic outcome of pancreatic cancer patients.
Acknowledgments
Grant support:
This work was supported by the Pancreatic Cancer SPORE project form NIH/NCI to Dr. ENC (Grant: CA102701-08), the Mayo Clinic Center for Cell Signaling in Gastroenterology (NIDDK P30DK084567). JMN is a recipient of a grant from from Coordenacão de Aperfeicoamento de Pessoal de Nível Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Authors would like to thank Gina Warner for editing the manuscript.
Footnotes
Conflict of interest: Dr. Eduardo Chini receives a sponsor research agreement from Sirtris Pharmaceuticals, Inc
References
- 1.Fryer RA, Galustian C, Dalgleish AG. Recent advances and developments in treatment strategies against pancreatic cancer. Curr Clin Pharmacol. 2009;4:102–12. doi: 10.2174/157488409788185007. [DOI] [PubMed] [Google Scholar]
- 2.Rossi ML, Rehman AA, Gondi CS. Therapeutic options for the management of pancreatic cancer. World J Gastroenterol. 2015;20:11142–59. doi: 10.3748/wjg.v20.i32.11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imai SC, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez-Pastor B, Mostoslavsky R. Sirtuins, metabolism, and cancer. Front Pharmacol. 2012;3:22. doi: 10.3389/fphar.2012.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–91. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takashi Nakagawa T, Guarente L. SnapShot: Sirtuins, NAD, and Aging. Cell Metab. 2014;20:192–192e1. doi: 10.1016/j.cmet.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Yuan H, Su L, Chen WY. The emerging and diverse roles of sirtuins in cancer: a clinical perspective. OncoTargets and Therapy. 2013;6:1399–416. doi: 10.2147/OTT.S37750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005;65:10457–63. doi: 10.1158/0008-5472.CAN-05-1923. [DOI] [PubMed] [Google Scholar]
- 9.Nihal M, Ahmad N, Wood GS. SIRT1 is upregulated in cutaneous T-cell lymphoma, and its inhibition induces growth arrest and apoptosis. Cell Cycle. 2014;13:632–40. doi: 10.4161/cc.27523. [DOI] [PubMed] [Google Scholar]
- 10.Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006;25:176–85. doi: 10.1038/sj.onc.1209049. [DOI] [PubMed] [Google Scholar]
- 11.Yuan J, Minter-Dykhouse K, Lou Z. A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol. 2009;185:203–11. doi: 10.1083/jcb.200809167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–23. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19:1751–9. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 14.Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–8. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 15.Feng AN, Zhang LH, Fan XS, Huang Q, Ye Q, Wu HY, et al. Expression of SIRT1 in gastric cardiac cancer and its clinicopathologic significance. Int J Surg Pathol. 2011;19:743–50. doi: 10.1177/1066896911412181. [DOI] [PubMed] [Google Scholar]
- 16.Lv L, Shen Z, Zhang J, Zhang H, Dong J, Yan Y, et al. Clinicopathological significance of SIRT1 expression in colorectal adenocarcinoma. Med Oncol. 2014;31:965. doi: 10.1007/s12032-014-0965-9. [DOI] [PubMed] [Google Scholar]
- 17.Bosch-Presegue L, Vaquero A. The dual role of sirtuins in cancer. Genes Cancer. 2011;2:648–62. doi: 10.1177/1947601911417862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harper CE, Patel BB, Wang J, Arabshahi A, Eltoum IA, Lamartiniere CA. Resveratrol suppresses prostate cancer progression in transgenic mice. Carcinogenesis. 2007;28:1946–53. doi: 10.1093/carcin/bgm144. [DOI] [PubMed] [Google Scholar]
- 19.Chauhan D, Bandi M, Singh AV, Ray A, Raje N, Richardson P, et al. Preclinical evaluation of a novel SIRT1 modulator SRT1720 in multiple myeloma cells. Br J Haematol. 2011;155:588–98. doi: 10.1111/j.1365-2141.2011.08888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harikumar KB, Kunnumakkara AB, Sethi G, Diagaradjane P, Anand P, Pandey MK, et al. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. Int J Cancer. 2010;127:257–68. doi: 10.1002/ijc.25041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinclair DA, Guarente L. Small-molecule allosteric activators of sirtuins. Ann Rev Pharmacol Toxicol. 2014;54:363–80. doi: 10.1146/annurev-pharmtox-010611-134657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014;35:146–54. doi: 10.1016/j.tips.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, et al. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–58. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 24.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–6. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, et al. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;70:1–10. doi: 10.1038/srep00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitchell SJ, Martin-Montalvo A, Mercken EM, Palacios HH, Ward TM, Abulwerdi G, et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014;13:836–43. doi: 10.1016/j.celrep.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lahusen TJ, Deng CX. SRT1720 induces lysosomal-dependent cell death of breast cancer cells. Mol Cancer Therap. 2015;14:183–92. doi: 10.1158/1535-7163.MCT-14-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki K, Hayashi R, Ichikawa T, Imanishi S, Yamada T, Inomata M, et al. SRT1720, a SIRT1 activator, promotes tumor cell migration, and lung metastasis of breast cancer in mice. Oncol Rep. 2012;27:1726–32. doi: 10.3892/or.2012.1750. [DOI] [PubMed] [Google Scholar]
- 29.Shin DH, Choi YJ, Park JW. SIRT1 and AMPK mediate hypoxia-induced resistance of non–small cell lung cancers to cisplatin and doxorubicin. Cancer Res. 2013;74:298–308. doi: 10.1158/0008-5472.CAN-13-2620. [DOI] [PubMed] [Google Scholar]
- 30.Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285:8340–51. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chini CC, Guerrico AM, Nin V, Camacho-Pereira J, Escande C, Barbosa MT, et al. Targeting of NAD metabolism in pancreatic cancer cells. Clin Cancer Res. 2014;20:120–30. doi: 10.1158/1078-0432.CCR-13-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nin V, Escande C, Chini CC, Giri S, Camacho-Pereira J, Matalonga J, et al. Role of Deleted in Breast Cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J Biol Chem. 2012;287:23489–501. doi: 10.1074/jbc.M112.365874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chini CC, Escande C, Nin V, Chini EN. HDAC3 is negatively regulated by the nuclear protein DBC1. J Biol Chem. 2010;285:40830–7. doi: 10.1074/jbc.M110.153270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffman RM. In vitro sensitivity assays in cancer: a review, analysis, and prognosis. J Clin Lab Anal. 1991;5:133–43. doi: 10.1002/jcla.1860050211. [DOI] [PubMed] [Google Scholar]
- 35.Artsi H, Cohen-Kfir E, Gurt I, Shahar R, Bajayo A, Kalish N, et al. The Sirtuin1 Activator SRT3025 Down-Regulates Sclerostin and Rescues Ovariectomy-Induced Bone Loss and Biomechanical Deterioration in Female Mice. Endocrinol. 2014;155:3508–15. doi: 10.1210/en.2014-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miranda MX, van Tits LJ, Lohmann C, Arsiwala T, Winnik S, Tailleux A, et al. The Sirt1 activator SRT3025 provides atheroprotection in Apoe−/− mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr expression. Eur Heart J. 2015;36:51–9. doi: 10.1093/eurheartj/ehu095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobson EW, Haddad J, Ellis J. A randomized, placebo-controlled, single blind, dose escalation, first-time- in-human study to assess the safety and pharmacokinetics of single and repeat doses of srt3025 in normal healthy volunteers. J Diabetes Metab. 2013;4:6. [Google Scholar]
- 38.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–51. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 39.Mrschtik M, Ryan KM. Lysosomal proteins in cell death and autophagy. FEBS J. 2015;282:1858–70. doi: 10.1111/febs.13253. [DOI] [PubMed] [Google Scholar]
- 40.Yang YP, Hu LF, Zheng HF, Mao CJ, Hu WD, Xiong KP, et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sinica. 2013;34:625–35. doi: 10.1038/aps.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berg TO, Strømhaug E, Løvdal T, Seglen O, Berg T. Use of glycyl-L-phenylalanine 2-naphthylamide, a lysosome-disrupting cathepsin C substrate, to distinguish between lysosomes and prelysosomal endocytic vacuoles. Biochem J. 1994;300:229–36. doi: 10.1042/bj3000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:17–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 44.Mizushima N, Yoshima T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Escande C, Chini CC, Nin V, Dykhouse KM, Novak CM, Levine J, et al. Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J Clin Invest. 2010;120:545–58. doi: 10.1172/JCI39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science. 2013;339:1216–9. doi: 10.1126/science.1231097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai H, Case AW, Riera TV, Considine T, Lee JE, Hamuro Y, et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat Commun. 2015;6:7645. doi: 10.1038/ncomms8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao G, Cui J, Zhang JG, Qin Q, Chen Q, Yin T, et al. SIRT1 RNAi knockdown induces apoptosis and senescence, inhibits invasion and enhances chemosensitivity in pancreatic cancer cells. Gene Ther. 2011;18:920–8. doi: 10.1038/gt.2011.81. [DOI] [PubMed] [Google Scholar]
- 49.Oon C, Strell C, Yeong KY, Ostman A, Prakash J. SIRT1 inhibition in pancreatic cancer models: Contrasting effects in vitro and in vivo. Eur J Pharmacol. 2015;757:59–67. doi: 10.1016/j.ejphar.2015.03.064. [DOI] [PubMed] [Google Scholar]
- 50.Aitas S, Jaattela M. Lysosomal cell death at a glance. J Cell Sci. 2013:1905–12. doi: 10.1242/jcs.091181. [DOI] [PubMed] [Google Scholar]
- 51.Nadanaciva S, Lu S, Gebhard DF, Jessen BA, Pennie WD, Will Y. A high content screening assay for identifying lysosomotropic compounds. Toxicology in vitro. 2011;25:715–23. doi: 10.1016/j.tiv.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 52.Izuishi K, Kato K, Ogura T, Kinoshita T, Esumi H. Remarkable tolerance of tumor cells to nutrient deprivation: possible new biochemical target for cancer therapy. Cancer Res. 2000;60:6201–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.