

Abstract
Peripheral membrane proteins are structurally diverse proteins that are involved in fundamental cellular processes. Their activity of these proteins is frequently modulated through their interaction with cellular membranes, and as a result techniques to study the interfacial interaction between peripheral proteins and the membrane are in high demand. Due to the fluid nature of the membrane and the reversibility of protein–membrane interactions, the experimental study of these systems remains a challenging task. Molecular dynamics simulations offer a suitable approach to study protein–lipid interactions; however, the slow dynamics of the lipids often prevents sufficient sampling of specific membrane–protein interactions in atomistic simulations. To increase lipid dynamics while preserving the atomistic detail of protein–lipid interactions, in the highly mobile membrane-mimetic (HMMM) model the membrane core is replaced by an organic solvent, while short-tailed lipids provide a nearly complete representation of natural lipids at the organic solvent/water interface. Here, we present a brief introduction and a summary of recent applications of the HMMM to study different membrane proteins, complementing the experimental characterization of the presented systems, and we offer a perspective of future applications of the HMMM to study other classes of membrane proteins.
Keywords: Membrane, Peripheral Proteins, Molecular Dynamics, Lipids
1. Complementing Experiment with Simulation at the Membrane Interface
Biological membranes are an essential component of all living cells, forming a dynamic barrier that demarcates the boundaries of the cell and the compartments within them. Due to this compartmentalization process, membranes are a fundamental signature of life, arising early in evolutionary history to allow for gradients to be generated that can be exploited in metabolism [1–3]. Control of this metabolism is orchestrated by a host of membrane-associated proteins, which together account for ~ 30% of the human proteome [4, 5] and accounts for ~ 60% of current drug targets [6], making these membrane-associated proteins a perpetually popular topic of investigation.
Over the past decade, it has become very clear that the membrane itself is not a passive, fluid substrate as once thought [7], but rather is an active participant in regulating the activity of membrane-associated proteins through its composition or spatial organization [8, 9]. Nowhere is this clearer than for peripheral membrane proteins, a subset of membrane proteins whose principal membrane interactions to the headgroups of membrane lipids. These structurally diverse peripheral proteins[10] are indispensable to cellular signaling [11, 12]. Beyond coupling, peripheral proteins also detoxify small molecules [13], and initiate important biological processes to human health such as the blood coagulation cascade [14] or viral fusion [15]. The centrality of membranes in the activity modulation of peripheral membrane proteins is now well established, and the future of the field is in demonstrating the cause and effect between specific membrane-protein interactions and observable phenotypes.
Despite the rapidly growing interest in characterizing lipid-protein interactions in peripheral proteins, the fluid nature of the lipid bilayer makes experimental studies on peripheral proteins, especially the characterization of the membrane bound structure, extremely challenging. In cases where a crystal structure can be solved, dynamic properties, such as ligand binding and large-scale conformational change upon membrane binding, cannot be characterized based on resulting static structures. Furthermore, due to the transient nature of the interaction between peripheral proteins and membrane lipids, crystallizing the membrane-bound complexes of peripheral proteins for X-ray analyses is exceedingly difficult. Other techniques, such as SAXS [16–18], EPR [19–21], NMR [22–24] including high–resolution field–cycling NMR [25–28], FRET [29, 30], fluorescence correlation spectroscopy [31–33], x–ray reflectivity [34, 35], neutron reflectometry [36, 37], and mutagenesis studies can bridge the gap, and provide low-resolution information on protein-lipid interactions such as determination of the binding face of the protein as it interacts with a membrane interface. However, without the guidance of a structural model of the protein-membrane complex, even when structures of constituent proteins are resolved, hypothesis-driven experimental investigations into the biological mechanisms are limited by the uncertainties inherent in missing the contribution of the membrane. Knowledge of structures of protein-membrane complexes at atomic level of detail is thus of critical importance.
The use of molecular dynamics (MD) simulations to resolve detailed models capable of capturing specific lipid-protein interactions is an established method and can be fruitfully employed to guide experiment [38–46]. However, one of the drawbacks of conventional MD is one of timescale. Due to the relatively slow lateral diffusion of lipids (D ~ 8 × 10−8 CM2S−1) [47, 48], over the course of 100 ns for a typical atomistic MD trajectory, individual lipids may only exchange with their neighbors once. Thus the resulting membrane representation in conventional atomistic simulations is often effectively static, preventing the description of lipid motion and lateral diffusion necessary for lipid mixing and adequate sampling of lipid-protein interactions in mixed bilayers found in vivo [10, 49], complicating our understanding of specific lipid-protein interaction.
Over the years, a number of computational approaches were developed to circumvent the problem of slow lipid dynamics including coarse-grained [50–56] and implicit membrane models [57–61]. Coarse-grained models work by mapping groups of atoms into beads, e.g., four heavy atoms and their associated hydrogens are represented as a single bead, which increases simulation timescales by allowing for longer timesteps (due to the increased mass of each particle) that occur more frequently (due to the reduced number of particles) [50]. Coarse-grained approaches can handle mixed lipids and probe membrane complex formation [56, 62, 63]. Implicit membrane models work by reducing the membrane to a continuum representation, e.g., a spatial region within the system with a different dielectric constant [64]. More recent implicit membrane models continue to be developed [65–67], and are successfully used to study membrane protein dynamics [68] due to the reduced particle count that enhances simulation rate. By design, these approaches have none or only limited ability to resolve specific atomic lipid-protein interactions, either due to the absence of lipids in the implicit membrane case or the bundling together of atoms in coarse-grained systems.
Another approach to enhance lipid dynamics is based on the replica-exchange MD (REMD) method [69]. In conventional REMD, multiple replicas of the system of interest are prepared and simulated at different temperatures [69]. During the REMD simulation, the temperatures of different replicas are exchanged periodically according to a monte carlo metropolis algorithm, thus allowing the system to overcome energy barriers more readily, effectively resulting in enhanced sampling of the conformational space. Although REMD has been employed to study atomistic membrane systems [70, 71], its applicability is limited due to small temperature range at which lipid bilayer structures are stable. To overcome this limitation, an alternative REMD method in which, instead of the temperature, the membrane surface tension is exchanged between different replicas has been recently proposed [72]. This approach has successfully resulted in enhanced lateral diffusion of lipid molecules [72].
An alternative approach towards overcoming slow lipid exchange and sampling is to re-engineer the lipid representation to increase its sampling. This is the approach taken by the Highly Mobile Membrane Mimetic (HMMM) model, where the long acyl tails in the bilayer center are replaced by an organic solvent, typically 1,1-dicholorethane (DCLE), designed to mimic the membrane interior (Fig. 1) [73, 74]. The short-tailed, yet fully atomistic lipids, placed at the organic solvent/water interface to capture the chemistry inherent in protein-lipid interactions, have a self-diffusion constant that is at least an order of magnitude higher than conventional lipids [45, 74]. As a result, insertion processes on the membrane interface will be significantly accelerated [75] while accurately reproducing the energetics of molecular interactions at the interface [76]. Membrane-bound forms of peripheral proteins, peptides and other membrane–associated molecular species, can be captured using an ensemble of relatively short but convergent MD simulations [74, 77–83]. Thus the HMMM can reveal specific protein-membrane interactions at atomic detail with a substantially reduced computational cost, making it a great companion to experimental techniques aiming at understanding membrane–bound configurations of proteins, and, in particular, lipid dependence of their binding.
Figure 1.
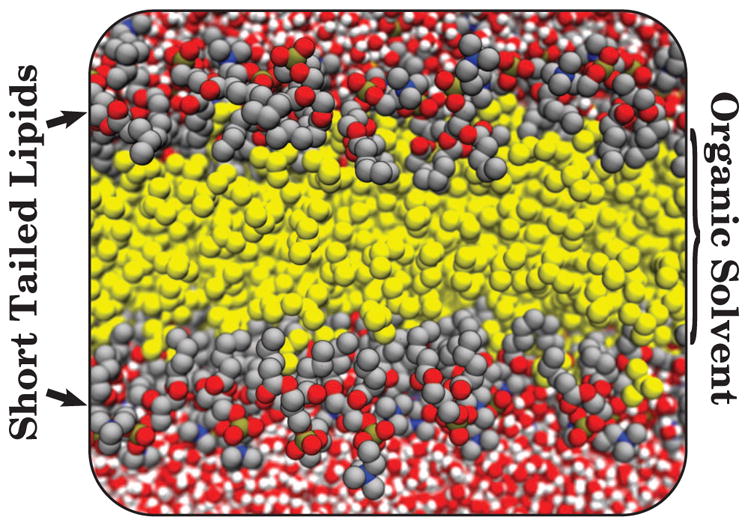
The HMMM model is constructed using a combination of short-tailed lipids (foreground grey, red, blue, and brown spheres), representing in full atomic detail the lipid head groups that are often the main protein-interacting elements, with a liquid representation of the membrane core (yellow spheres - the 1,1-dichloroethane organic solvent).
The applicability of the HMMM as a complement to experiment is at this point no longer just a theoretical construct. There are many examples of the application of the HMMM to real biological systems now in the literature by both our own lab [74, 77–81, 83, 84] and others [85, 86], and we have chosen a subset to review here that particularly highlight the potential impact of the HMMM to react to as well as guide experiment. Specifically, we will highlight recent work on identifying the binding face of hemagglutinin fusion peptide [81] and human cytochrome P450 [77, 84], conformational changes induced by membrane binding in talin [80] and α-Synuclein [78], as well as ongoing work in assembling blood coagulation factors on the membrane surface [74, 83].
2. Membrane Binding of Hemagglutinin Fusion Peptide
The infection of a cell by the influenza virus starts with the fusion of the viral and cellular membranes [87]. This process is mediated by hemagglutinin (HA), a protein located on the virus surface [88]. HA is composed of two major domains: HA1, anchored in the viral membrane, and HA2, the domain that mediates attachment to the host membrane. During the fusion process, the N-terminal 23 residues of HA2, known as the HA fusion peptide (HAfp), are the only part of the virus in direct contact with the host membrane [89]. Although the detailed mechanism by which HA mediates membrane fusion remains elusive, it is established that the process relies on the direct interaction and insertion of HAfp into the host membrane. [90, 91].
Due to its key role in the fusion process, HAfp and its interaction with lipid environments have been investigated in its isolated form (i.e., in the absence of the remainder of HA2) in numerous experimental biophysical studies [92–96]. Binding experiments have indicated that all variants of HAfp insert into lipid bilayers and promote membrane fusion [92–94]. The micelle–bound, full–length structure of HAfp was resolved recently by NMR [97], in an attempt to get insight into the membrane–bound form of the peptide. Moreover, subsequent NMR studies aiming at characterization of the structure and dynamics of HAfp revealed pH-and length-dependence of both the conformation and fusogenic activity of the peptide [95, 96]. However, because of the highly dynamic nature of the peptide–membrane interactions, the details of the binding and insertion mechanism of HAfp into a lipid bilayer remained difficult to elucidate experimentally.
The HMMM model was employed to characterize the membrane–bound form of HAfp [81]. Taking advantage of the enhanced lipid dynamics of the HMMM membrane, the NMR structure of micelle–bound HAfp [97] was employed to perform multiple independent 100- ns simulations where the unbiased membrane–binding of HAfp was captured. These simulations were not only unbiased in that they did not enforce any particular membrane–bound form (by placing HAfp in solution), but they also started with a wide range of different initial orientations of the peptide (7 orientations, labeled a, b, r, i, n, p, and s in Fig. 2A), with the peptide initially placed away from the membrane. In order to achieve better sampling, we performed multiple simulations to further increase the sampling of the peptide-membrane interactions at a relatively low computational cost.
Figure 2.

Developing a membrane–bound HAfp through multiple, independent, membrane binding simulations of the peptide captured with the HMMM model. (A) Diverse initial configurations of HAfp employed for the simulations. Helix A and B of HAfp are shown in red and blue, respectively. The letter in parantheses indicates the label identifying each system which was simulated in three independent replicates (1–3) for 100 ns each. (B) Membrane–bound profile of HAfp, characterized by the average z–position of individual side chains. The average was taken over the last 10 ns of the 100 ns trajectories. The dashed line represents the average position of the phosphorus (PO4) atoms of the membrane. The number after each system label indicates the simulation replicate. (C) Membrane-bound structures obtained from independent HMMM simulations, colored using the same color scheme as in (B), showing the side and top view of the peptide in the membrane. Reprinted (adapted) with permission from Baylon and Tajkhorshid [81]. Copyright 2015 American Chemical Society.
Despite employing a wide range of different initial orientations of the peptide for simulations, HAfp insertion into the membrane was reproducibly captured in the majority of the performed simulations (18 out of 21), resulting in a convergent membrane-bound form of the peptide (Fig. 2B and C). The structure of HAfp is characterized by two alpha helices (Helices A and B, Fig 2A). The simulations clearly show that in the membrane–bound form, Helix A is inserted in the membrane deeper than Helix B. The observed deeper insertion of Helix A in the simulations suggests that this helix is the main contributor to peptide insertion by establishing the majority of hydrophobic interactions with the membrane, while Helix B increases the affinity by providing amphipathic interactions at the interfacial region. After insertion, HAfp adopts an almost parallel orientation with respect to the membrane plane, similar to what has been suggested in NMR studies of micelle–bound HAfp [97, 98].
3. Molecular Insight into Membrane–Mediated Drug Recruitment by Human Cytochrome P450 Enzymes
Cytochrome P450 (CYP) enzymes constitute a large family of heme–containing proteins found in all domains of life [99], involved in the metabolism of a wide range of compounds. In the human body, CYPs are involved in the biotransformation of both xenobiotics and endogenous compounds [100], such as steroid hormones [101] and fatty acids [102], most of which partition substantially into lipid bilayers composing various membranes in the cell. Human CYPs are membrane–bound proteins anchored to the cellular membrane by an N-terminal transmembrane alpha helix [103, 104]. In addition, the catalytic domain of the enzyme can also interact with the surface of the membrane through specific hydrophobic elements found in the globular domain [104–106], increasing the probability of recruiting amphipatic and hydrophobic substrates, such as drugs or endogenous hormones, from the membrane where these molecular species concentrate.
It has been only recently that the importance of protein–membrane interactions that regulate different functions of CYPs, such as ligand binding [107] and cooperativity [108], has been appreciated. For example, fluorescence and absorbance spectroscopy, employed to monitor ligand binding to model membranes and to the enzyme respectively, have shown that the presence of the membrane has a significant effect in ligand binding [107], revealing preferential binding to either the membrane or the enzyme for different types of ligands. Therefore, in order to fully understand the mechanism of drug binding to CYPs, it is important to characterize their membrane–bound form, specifically, how deeply they penetrate the membrane, their orientation on the surface of the membrane, and whether or not any conformational changes are induced by membrane binding, particularly in the access tunnels connecting the buried active site of the enzyme to the membrane.
To gain insights into the structure and dynamics of membrane–bound CYPs, cytochrome P450 3A4 (CYP3A4) was studied with the HMMM model [77]. CYP3A4 is the most abundant CYP isoform in the human body [109] and is responsible for the metabolism of more than 50% of the drugs [13] that undergo biotransformation in the body before eliminated. To obtain the membrane–bound form of CYP3A4, the membrane–binding of the globular domain of the enzyme was captured with the HMMM model (Fig. 3A). Simulations were started by placing CYP3A4 away from a PC membrane in a wide range of different initial orientations. Independent simulations were performed, and the spontaneous insertion of the globular domain of the enzyme was captured within 20 ns, revealing some of the key structural elements involved in protein–lipid interactions, e.g., the heavy involvement of F’-G’ loop in membrane interaction (Fig. 3A). Furthermore, these interactions were preserved after the HMMM-derived model was converted to a full membrane system by extending the lipid tails, confirming the stability of the model.
Figure 3.

Membrane–bound form of CYP3A4 captured with the HMMM model. (A) Representative trajectory of spontaneous membrane binding of CYP3A4. Membrane binding was consistently observed in 5 independent simulations, permitting the identification of structural elements of CYP3A4 interacting with the membrane (e.g., A-anchor shown in orange stick representation, F’-G’ helices shown in magenta). (B) Insertion of the A-anchor into the membrane (i.e., at or below the PO4 level) was consistently observed in all 5 independent simulations. The average positions of the phosphorus (PO4) and the nitrogen (choline) atoms of the lipid head groups were employed to monitor membrane binding, and are shown using brown and gray lines, respectively. (C) Each simulation began from a different orientation relative to the membrane, as measured by the heme tilt angle. The orientation of the globular domain with respect to the membrane normal for each of the simulations converged to around an average value of 72.2 ± 3.2°. The orientation of the protein was also measured experimentally, with an average tilt angle of 59.7 ± 4.1° (thick and thin dotted lines for average and standard deviation in tilt angle plot, respectively), yielding excellent agreement between simulations and experiment. Adapted from Baylon et al. [77]. Copyright 2013 American Chemical Society.
The convergence of the simulations was then assessed by monitoring the orientation that the globular domain takes in the membrane, as characterized by the heme tilt angle with respect to the membrane normal (Fig. 3C), reaching an average value of 72.2 ± 3.2° in the HMMM membrane [77]. Linear dichroism measurements of nanodisc-bound CYP3A4 [77] were performed in parallel to characterize the orientation of the enzyme in the membrane. Experimentally, the average tilt angle was found to be 59.7 ± 4.1°, yielding excellent agreement between simulations and experiment.
The CYP3A4 simulations revealed that its binding to the membrane favors transient opening of small access tunnels leading to the active site by inducing conformational changes of side chains located at the protein–membrane interface, which might in turn facilitate the binding of amphiphilic and lipophilic drugs to CYP3A4. The resulting membrane-bound structures of CYP3A4 have been recently employed to further probe the mechanism of drug-drug interaction mediated by membrane–bound CYP3A4 with atomic detail [110]. Moreover, the general simulation approach to model membrane association of a peripheral protein has been employed for CYP2J2, another human CYP [82]. For CYP2J2, membrane–binding simulations revealed important membrane–interacting residues (Ile-236 and Phe-239), and their role in membrane–binding was examined experimentally using Trp-quenching fluorescence spectroscopy in nanodiscs. It was shown that mutation of these resiudes greatly affects activity of the enzyme, as well as its depth of insertion in nanodiscs, thus providing strong support for the computational model developed by the HMMM simulation [82].
4. Membrane–Induced Conformational Change of Talin in Integrin Activation
Integrins are a ubiquitous set of heterodimeric, transmembrane proteins that are involved in cell adhesion and motility [111]. These conformationally-labile proteins are integral to biological processes such as epithelial integrity, leukocyte extravasation, cell differentiation, and tumor metastasis [111–115]. Integrins themselves are usually expressed in a non-adhesive “off-state” [112, 114–122]. Upon receiving the appropriate signal, the peripheral protein, talin, binds to and separates the cytoplasmic domains of integrin [115–117, 120], exposing the extracellular adhesion domain. Biochemical experiments [123–125] have demonstrated the importance of a basic patch of residues in the F2 subdomain, termed the membrane orientation patch (MOP), in membrane association of talin to the anionic membrane surface. Additional biophysical [126–129] and mutagenesis [125] studies showed the presence of a hydrophobic interaction between talin and the membrane, as well as the simultaneous interaction of the F3 subdomain with integrin and the membrane. Following the crystallization of talin F2F3 [18, 130–132], several questions remained, however, including which comprises the membrane anchor of talin and how simultaneous interaction of the F2 and F3 subdomains is achieved.
Because talin occupies a significant surface area on the membrane [126, 127], significant lipid rearrangement is needed for complete membrane binding. The HMMM model offers significantly accelerated membrane remodeling over conventional MD simulations [74], providing a unique opportunity to observe spontaneous membrane binding of talin [80]. Starting from diverse initial orientations and distances from the membrane (see an example in Fig. 4A), 5 independent simulations of talin F2F3 binding to a PS HMMM were performed. Across all 5 simulations the talin F2F3 subdomain was observed to spontaneously and fully bind to the membrane [80]. As demonstrated by both experiment [123–125] and coarse-grained simulations [133, 134], the basic residue-rich MOP was seen to initiate interaction between talin and the membrane (Fig. 4B), binding ~12 lipids and providing the impetus for initial membrane association. Separate electrostatic calculations of the talin F2F3 subdomain show that the MOP creates a large positive electrostatic potential, which is strongly attracted to the anionic surface of the PS HMMM. Thus, the simulations showed that arrangement of basic residues in the MOP creates the initial attraction that draws talin to the membrane.
Figure 4.

Membrane-binding Sequence of Talin F2F3. (A) The initial configuration of talin F2F3 (green cartoon) above a PS HMMM model (PS lipids, brown sticks; DCLE, yellow van der Waals). The MOP (blue sticks), Phe-rich membrane anchor (red sticks), and FAP (gray sticks) are depicted. Water and ions are excluded for clarity. (B) The first step in membrane binding is the interaction between the positively charged MOP and the anionic surface of the membrane. Following this, local conformational changes induced by binding of the MOP, allow for insertion of the Phe-rich membrane anchor into the hydrophobic core of the membrane (C). Once the membrane anchor is inserted, a large, interdomain conformational change brings the basic residues of the FAP (in F3) into contact with the PS lipids (D). (E) A plot of the height of MOP (blue), Phe-rich membrane anchor (red), and FAP (gray) is shown as a function of time for a representative simulation, indicating a deep penetration of these elements into the membrane during the simulation. The dashed line indicates the position of the phosphate layer. Data was originally published by Arcario and Tajkhorshid [80], reprinted with permission of Elsevier B. V.
While binding of the MOP provides impetus for membrane association, water and/or ions can easily displace the Coulomb interactions formed between talin and the membrane in this configuration. Therefore, additional and more intimate interactions are necessary to stabilize the membrane–bound form. Upon association of the F2 subdomain, however, a local conformational change allows for exposure of a phenylalanine-rich (F261/F283) membrane anchor (Fig. 4C). With the phenylalanine exposed, they both rotate around the Cα-Cβ bond and insert into the membrane core, creating a hydrophobic interaction between talin and the membrane. The insertion of this Phe-rich membrane anchor creates a bound state of talin that will not be disrupted readily by water and/or ions and a model that is consistent with experimental observations [126, 127, 129]. Upon insertion of the membrane anchor, a large scale, interdomain conformational change brings the talin F3 subdomain into contact with the surface of the membrane (Fig. 4, transition from panel C to D) while the talin F2 subdomain remains bound to the membrane surface. The interdomain conformational change, not observed in solution simulations of talin, is driven by interaction between K325/K327 in the talin F3 subdomain and the anionic PS headgroups on the membrane surface. While the bound state model obtained by these HMMM simulations agrees with previous experimental data [125], the model also demonstrates the power of HMMM simulations to act as a bridge between experimental data and the static structural models obtained by crystallization.
5. Lipid-Specific Membrane-Induced Conformational Change in α-Synuclein
α-Synuclein (αS) is an intrinsically disordered protein that is thought to act as a component of the SNARE complex in neurotransmitter vesicle release [135]. αS misfolding and aggregation into Lewy Bodies are one of the hallmarks of Parkinson’s disease [136, 137], and as such has been extensively studied with a variety of experimental and computational techniques. It is known that upon binding to anionic phospholipids the N-termini of monomeric αS will fold into a helical structure [138, 139] that is resistant to aggregation [140], however the membrane-bound structure of αS was a matter of debate. NMR and EPR experiments of micelle-bound αS identified a horseshoe-like, broken-helix structure, with a kinked region in between two helical segments [141–143]. ESR and DEER experiments on αS bound to bilayers instead were consistent with an extended helix model without a kink [144–147]. Interconversion between these two states had been hypothesized [36, 148–150], with special emphasis on the role of specific lipid interactions in regulating the equilibrium between the two populations [36, 151, 152]. Coarse-grained simulations sampled this conformational diversity in part [153, 154], but due to their resolution limitations could not identify specific interactions that anionic membranes might disrupt to allow this transition to take place.
In another example of using computation to complement experimental techniques, simulations of αS using the HMMM model probed the specific interactions that control the interconversion process [78]. Starting from the micelle-bound NMR structure [143] where the αS monomer was in the broken helix state, 20 independent αS monomers were placed above a mixed HMMM membrane with a 1:1 ratio of anionic PS and zwitterionic PC headgroups such that there was no initial contact between αS and the membrane. The αS monomers rapidly bound to the anionic membrane, and began to respond to their membrane environment [78]. One of the key findings was the observation of two distinguishable states within the simulations. In a subset of the trajectories, αS transitioned to a semi-extended state where the N- and C-terminal helices were clearly separated from one another (Fig. 5A, Sims. 1–4 and 18–20). This semi-extended state represents an intermediate between the two experimentally observed extended and horseshoe-like structures. However in the majority of cases, αS remained in the initial horseshoe-like broken helix state. In these instances, the N-and C-terminal helices were held together by salt bridges and hydrogen bonds between the helices, and as a result the angle between the helices was observed to be small (Fig. 5B).
Figure 5.

Composite data of αS HMMM simulations. (A) A top view of all 20 membrane bound forms of αS, highlighting the heterogeneity observed in the relative orientation of the N- and C-helices. (B) Inter-helix angle (θh) of all 20 membrane-binding simulations, where θh is defined by the angle between the two vectors formed by a linear interpolation of the α carbons of the N- and C-helices. θh distinguishes between the broken horseshoe-like and semi-extended helix states: a broken helix is where θh never exceeds 47°, and a semi-extended intermediate state is where the inter-helix angle exceeds 47° and often increases up to approximately 90° . Data originally published in Vermaas and Tajkhorshid [78], used with permission from Elsevier B.V.
The formation of the semi-extended state appears to be related to specific interactions with anionic lipids formed upon membrane binding. In the simulations that yielded semi-extended states, PS aggregates around basic residues on the interior face of the αS monomer, disrupting the salt bridges that hold the two helices together [78]. Conversely, when PS did not intercalate between the two helices, αS remained in the horseshoe-like conformation. This is a clear example that specific membrane-protein interactions are critical to influencing peripheral protein structure, a motif that repeats across biology [155, 156], and demonstrates the value of the HMMM representation in finding specific interactions that are key to biological processes.
6. Towards Understanding Regulation and Lipid Specificity in Coagulation Factors
Coagulation factors are biomedically important enzymes which regulate the clotting cascade. Binding of coagulation factors to the membrane, which is highly lipid–dependent, is a critical step for their attraction. In response to vascular damage, anionic lipids from the inner leaflet of the cell membrane are exposed to the outer leaflet. Coagulation factors then bind to these anionic lipids and interact on the membrane surface. Membrane binding is a pivotal event, allowing coagulation factors, normally weak enzymes, to become activated in a sequence of steps resulting in generation of a sudden burst of thrombin [14]. Understanding the molecular basis of lipid specificity and details of the membrane–bound forms of coagulation factors is therefore critical to developing treatments for a wide variety of disease conditions, including hemophilia and thrombosis. Of particular interest is understanding the atomic-level interactions formed between coagulation factor binding domains and anionic lipids, which allow the clotting cascade to be initiated [14, 157]. Two common domains used by coagulation factors to anchor to the membrane include GLA domains and C2-like discoidin domains. GLA domains are rich in carboxy glutamic acid residues and calcium residues, which mediate phospholipid recognition, and are present in, e.g., factors VII, IX, X, and prothrombin. C2-like discoidin domains, found in factors V and VIII, are thought to accomplish membrane binding through a combination of interactions of their ”spiky” structural elements with lipids [14]. Many aspects of the binding process remain mysterious, including how lipid specificity and affinity is modulated in structurally and sequentially similar GLA domains, bestowing them a wide range of membrane affinities. Atomic-level experimental information regarding coagulation protein-lipid interactions has remained scarce, however.
An examination of the available structural information for coagulation factors VII (FVII) and VIII (FVIII) serves to illustrate the contemporary situation. While high-quality crystal structures have been solved for the FVII GLA and the two FVIII membrane-anchor C2-like domains, these structures provide no information on lipids and how they interact with the protein [158–160]. Prothrombin GLA domain crystal and NMR structures have been solved which included a single lipid–like ligand, a lysophosphatidylserine, which provided some insight into atomic-level possible interactions between GLA domains and PS lipids [161]. Even in this case, however, no information can be derived as to membrane binding orientation and insertion depth, and only limited information can be inferred about physiological protein-lipid interactions. NMR has also been used to study some aspects of the FVII activation process, such as allosteric activation of FVII by tissue factor, but these approaches have removed the membrane entirely as a factor by using soluble tissue factor [162]. Limited information on proximity to the membrane in some binding proteins has been inferred through such techniques as SAXS [16], EPR [29, 163], FRET [29, 30, 164], and mutagenesis studies [165, 166]. This information, however, is of insufficient detail to provide insight into atomic-level lipid-protein interactions.
In order to study the relevant yet experimentally elusive FVII protein-lipid interactions, an atomic level membrane-bound model of the FVII GLA domain was developed using MD simulations and the HMMM [74]. FVII GLAs spontaneously inserted into a PS HMMM in multiple independent simulations and converged in terms of insertion depth within 30 ns (Fig. 6). A structure of the membrane-bound FVII GLA domain had previously been developed through a combination of conventional lipid bilayers and steered MD simulations, in which the insertion process was induced by the application of externel forces [168]. Significantly less sampling of the bound state was possible in these simulations due to high computational cost [168].
Figure 6.

Spontaneous binding and insertion of the GLA domain to anionic membranes captured by HMMM. (A) The binding of GLA domains (purple trace) to PS headgroups is mediated by bound Ca2+ ions (purple spheres) and basic side chains (green licorice). Ohkubo and Tajkhorshid [44] and Ohkubo et al. [74] studied the binding process in both conventional (left) and HMMM (right) bilayers. (B) Penetration depth over time for 10 independent HMMM binding simulations, in which all GLA domains bind to the target membrane within 30 ns and converged to a single insertion depth. Figure originally published in Vermaas et al. [167], with data from Ohkubo et al. [74]. Reprinted with permission of Springer. Copyright Springer Science+Business Media New York 2015.
Using the HMMM, in 10 independent simulations the FVII GLA inserted into the bilayer and reached converged membrane-bound states without the need for any biased simulations. Rapid binding seen in HMMM permitted repeated simulations of the membrane insertion events (10 times) [74]. Subsequent to converged GLA penetration depth having been achieved, specific contacts between PS head-groups and the GLA domain were observed to form over a longer time-scale. The HMMM allowed for atomic-level observation of a process out of reach of both current experimental approaches and traditional full-tail membrane computational approaches.
Employing a similar approach, it was also possible to develop a model for the FVIII C2 and C1 membrane binding domains and propose possible membrane bound conformations for the FVIIa:FIXa complex which activates FX [83]. Converged membrane binding depth was reached within 15 ns for binding trajectories. It was found that FVIII C2 adopts a perpendicular membrane-bound configuration, and a potential lipid binding pocket was identified in between the hydrophobic spikes. Insertion depth relative to the membrane converged for both the C1 and C2 domains to similar values in different simulations. Using the resulting structures of bound C1 and C2 domains, a putative model was constructed for the FVIIIa:FIXa complex [83]. This complex has never been crystallized, but the homologous FVa:FXa complex has [169]. FIXa and FVIIa were aligned with their counterparts in this homologous structure, and the resulting structure was then overlayed onto the converged HMMM-bound structures of FVIII C1 and C2 domain. This preliminary model allowed for more informed speculation as to subunit arrangement in the FVIIIa:FIXa complex.
7. Perspectives on Future HMMM Application
The examples presented here illustrate the utility of the HMMM model to study the structure and dynamics of peripheral membrane proteins, addressing questions that are difficult to answer otherwise. The HMMM model has been successfully employed to describe membrane binding and insertion of proteins, while also providing increased sampling of important protein–lipid interactions due to the accelerated lipid dynamics. The proteins studied with the HMMM so far interact with the membrane periphery, and therefore the protein–membrane interactions are accurately captured by the HMMM model [76]. Given the increasing awareness of the importance of lipid–protein interactions in modulating protein function [170, 171] and the ubiquity of peripheral proteins on the membrane surface, simply applying the model to similar systems is an important contribution to the field, supplying mechanistic details to the broad body of evidence suggesting that the function of these proteins is tightly regulated by their interaction with the membrane [11, 171, 172].
However there is a need for the HMMM in new fields, specifically extending the model to characterizing the interaction of integral membrane proteins with lipids. In contrast to peripheral proteins where the main effect of the membrane appears to be the control of protein binding [10], various functional aspects of integral membrane proteins can be targeted and modulated by the change in lipid composition of the membrane [8, 170, 173–177]. Our preliminary work with integral membrane proteins suggests that extending the applicability of the HMMM representation to systems with a large transmembrane component requires significant modification of the organic solvent currently employed, as DCLE will intercalate too readily into transmembrane proteins and disrupt protein structure, in contrast to the effectiveness of the model in treating peripheral systems [76]. A comparison of amino acid translocation free energies across the membrane [76] to atomistic [178] or coarse grained [179] simulations as well as to experimental hydrophobicity scales [180–183] shows that the origins of this phenomena can be traced back to the choice of the inorganic solvent (DCLE), whose non-zero dipole can interact more strongly with protein side chains than native membrane constituents would. Furthermore, the liquid nature of the solvent naturally results in an overestimation of entropy of bulky amino acids in the core of the membrane [76]. Overcoming these challenges is an area of current research.
Thinking more broadly, the HMMM can be a boon to research whenever lipid mixing/diffusion is rate limiting, but certainly should only be used as appropriate to complement more realistic representations of a membrane. The HMMM is best employed during the initial phase of a simulation project, in order to accelerate lipid reorganization and capture slow processes, e.g., inhomogeneous lipid distributions that arise in response to insertion of a peptide or in the presence of an integral membrane protein. In this way, the HMMM is used to sample specific protein–lipid interactions more efficiently, establishing the most likely positions of the lipid head groups around the protein. These short-tailed lipids are then converted into full tail lipids for further study in an environment that more closely mimics experimental conditions. Using the HMMM in this way plays to its strengths as a model of the membrane periphery [76], and is very cost effective way of obtaining an unbiased pool of membrane-bound protein conformations. The publicly available HMMM building tools facilitate this workflow of obtaining a bound structure with the HMMM and then converting the bound state back into a conventional bilayer [184]. Used in this way, the HMMM is a valuable weapon in the arsenal of computational scientists to study the remarkable functional coupling of proteins to biological membranes, and is superior to placing the inserting protein into the bilayer.
Supplementary Material
Highlights.
The HMMM model is designed to enhance lipid dynamics in atomistic MD simulations.
HMMM facilitates studying protein-lipid interactions to complement experiments.
Recent applications of the HMMM model to study peripheral proteins are described.
Challenges, limitations, and future directions of the HMMM model are discussed.
Acknowledgments
This work was supported in part by the National Institutes of Health (Grants R01-GM101048, R01-GM086749, U54-GM087519, and P41-GM104601 to E.T.) and XSEDE compute resources (grant TG-MCA06N060 to E.T. and grant TG-MCB130112 to T.V.P.). J.V.V. acknowledges support from the Sandia National Laboratories Campus Executive Program, which is funded by the Laboratory Directed Research and Development (LDRD) Program. Sandia is a multi-program laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the US Department of Energy’s National Nuclear Security Administration under Contract No. DE-AC04-94AL85000. M.J.A. acknowledges past support from the NSF GRF Program. T.V.P. is grateful for the support from the Illinois Campus Research Board. T.V.P. was a Faculty Fellow of the National Center for Supercomputing Applications when this work was completed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Javier L. Baylon, Email: baylonc2@illinois.edu.
Josh V. Vermaas, Email: vermaas2@illinois.edu.
Melanie P. Muller, Email: mmulle23@illinois.edu.
Mark J. Arcario, Email: marcar2@illinois.edu.
Taras V. Pogorelov, Email: pogorelo@illinois.edu.
References
- 1.Chen IA, Walde P. From Self-Assembled Vesicles to Protocells. Cold Spring Harbor Perspect Biol. 2010;2(7):a002170. doi: 10.1101/cshperspect.a002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dzieciol AJ, Mann S. Designs for life: protocell models in the laboratory. Chem Soc Rev. 2012;41(1):79–85. doi: 10.1039/C1CS15211D. [DOI] [PubMed] [Google Scholar]
- 3.Georgiou CD, Deamer DW. Lipids as Universal Biomarkers of Extraterrestrial Life. Astrobio. 2014;14(6):541–549. doi: 10.1089/ast.2013.1134. [DOI] [PubMed] [Google Scholar]
- 4.Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10(6):1141–1149. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- 5.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480(7376):254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yildirim MA, Goh K-I, Cusick ME, Barabasi A-L, Vidal M. Drug–target network. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 7.Singer S, Nicolson G. The fluid mosaic model of the structure of cell membranes. Science. 1972;173:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 8.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vereb G, Szollosi J, Matko J, Nagy P, Farkas T, Vigh L, Matyus L, Waldmann TA, Damjanovich S. Dynamic, yet structured: The cell membrane three decades after the Singer-Nicolson model. Proc Natl Acad Sci USA. 2003;100(14):8053–8058. doi: 10.1073/pnas.1332550100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- 11.Whited A, Johs A. The interactions of peripheral membrane proteins with biological membranes. Chem Phys of Lipids. 2015;192:51–59. doi: 10.1016/j.chemphyslip.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 12.Kholodenko BN, Hancock JF, Kolch W. Signalling ballet in space and time. Nat Rev Mol Cell Biol. 2010;11:414–426. doi: 10.1038/nrm2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guengerich PF. Cytochrome P450 3A4: regulation and role in drug metabolism. Annual Review of Pharmacology and Toxicology. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 14.Zwaal RFA, Comfurius P, Bevers EM. Lipid-protein interactions in blood coagulation. Biochim Biophys Acta. 1998;1376:433–453. doi: 10.1016/s0304-4157(98)00018-5. [DOI] [PubMed] [Google Scholar]
- 15.Harrison SC. Viral membrane fusion. Virology. 2015;479–480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 17.Mosbaek CR, Nolan D, Persson E, Svergun DI, Bukrinsky JT, Vestergaard B. Extensive small-angle X-ray scattering studies of blood coagulation factor VIIa reveal interdomain felxibility. Biochemistry. 2010;49:9739–9745. doi: 10.1021/bi1011207. [DOI] [PubMed] [Google Scholar]
- 18.Elliott PR, Goult BT, Kopp PM, Bate N, Grossmann JG, Roberts GC, Critchley DR, Barsukov IL. The Structure of the Talin Head Reveals a Novel Extended Conformation of the FERM Domain. Structure. 2010;18:1289–1299. doi: 10.1016/j.str.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osterberg JR, Chon NL, Boo A, Maynard FA, Lin H, Knight JD. Membrane Docking of the Synaptotagmin 7 C2A Domain: Electron Paramagnetic Resonance Measurements Show Contributions from Two Membrane Binding Loops. Biochemistry. 2015;57(37):5684–5695. doi: 10.1021/acs.biochem.5b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Virág E, Belagy J, Gazdag Z, Vágvölgyi C, Pesti M. Direct in vivo interaction of the antibiotic primycin with the plasma membrane of Candida albicans: An EPR study. Biochim Biophys Acta Biomembr. 2012;1818:42–48. doi: 10.1016/j.bbamem.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 21.Dikiy I, Eliezer D. Folding and misfolding of alpha-synuclein on membranes. Biochim Biophys Acta Biomembr. 2012;1818:1013–1018. doi: 10.1016/j.bbamem.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epand RM, D’Souza K, Berno B, Schlame M. Membrane curvature modulation of protein activity determined by NMR. Biochim Biophys Acta Biomembr. 2015;1848(1):220–228. doi: 10.1016/j.bbamem.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Goult BT, Bate N, Anthis NJ, Wegener KL, Gingras AR, Patel B, Barsukov IL, Campbell ID, Roberts GC, Critchley DR. The Structure of an Interdomain Complex that Regulates Talin Activity. J Biol Chem. 2009;284:15097–15106. doi: 10.1074/jbc.M900078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maltsev AS, Chen J, Levine RL, Bax A. Site-Specific Interaction between α-Synuclein and Membranes Probed by NMR-Observed Methionine Oxidation Rates. J Am Chem Soc. 2013;135:2943–2946. doi: 10.1021/ja312415q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pu M, Orr A, Redfield AG, Roberts MF. Defining Specific Lipid Binding Sites for a Peripheral Membrane Protein in Situ Using Subtesla Field-cycling NMR. J Biol Chem. 2010;285(35):26916–26922. doi: 10.1074/jbc.M110.123083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng J, Goldstein R, Gershenson A, Stec B, Roberts MF. The Cation-π Box Is a Specific Phosphatidylcholine Membrane Targeting Motif. J Biol Chem. 2013;288(21):14863–14873. doi: 10.1074/jbc.M113.466532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai J, Guo S, Lomasney JW, Roberts MF. Ca2+-independent Binding of Anionic Phospholipids by Phospholipase C δ1 EF-hand Domain. J Biol Chem. 2013;288(52):37277–37288. doi: 10.1074/jbc.M113.512186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei Y, Stec B, Redfield AG, Weerapana E, Roberts MF. Phospholipid-binding Sites of Phosphatase and Tensin Homolog (PTEN): Exploring the Mechanism of Phosphatidylinositol 4,5-bisphosphate Activation. J Biol Chem. 2015;290(3):1592–1606. doi: 10.1074/jbc.M114.588590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohout SC, Corbalán-García S, Gómez-Fernández JC, Falke JJ. C2 domain of protein kinase Cα: Elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry. 2003;42:1254–1265. doi: 10.1021/bi026596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCallum CD, Su B, Neuenschwander PF, Morrissey JH, Johnson AE. Tissue Factor Positions and Maintains the Factor VIIa Active Site Far above the Membrane Surface Even in the Absence of the Factor VIIa Gla Domain — A Fluorescence Resonance Energy Transfer Study. J Biol Chem. 1997;272:30160–30166. doi: 10.1074/jbc.272.48.30160. [DOI] [PubMed] [Google Scholar]
- 31.Pu M, Roberts MF, Gershenson A. Fluorescence Correlation Spectroscopy of Phosphatidylinositol-Specific Phospholipase C Monitors the Interplay of Substrate and Activator Lipid Binding. Biochemistry. 2009;48(29):6835–6845. doi: 10.1021/bi900633p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grauffel C, Yang B, He T, Roberts MF, Gershenson A, Reuter N. Cation-π Interactions As Lipid-Specific Anchors for Phosphatidylinositol-Specific Phospholipase C. J Am Chem Soc. 2013;135(15):5740–5750. doi: 10.1021/ja312656v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang B, Pu M, Khan HM, Friedman L, Reuter N, Roberts MF, Gershenson A. Quantifying Transient Interactions between Bacillus Phosphatidylinositol-Specific Phospholipase-C and Phosphatidylcholine-Rich Vesicles. J Am Chem Soc. 2015;137(1):14–17. doi: 10.1021/ja508631n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J. Intrinsically disordered p53 extreme C-terminus binds to S100B (ββ) through fly-casting. J Am Chem Soc. 2009;131(6):2088–2089. doi: 10.1021/ja809547p. [DOI] [PubMed] [Google Scholar]
- 35.Tietjen GT, Vargas E, Chen C-H, Schlossman M, Lin B, Meron M, Adams E, Lee KY. Molecular Basis for Immune Recognition of Exposed Phosphatidylserine via the Tim Family of Proteins. Biophys J. 2012;102:495a. [Google Scholar]
- 36.Jiang Z, Hess SK, Heinrich F, Lee JC. Molecular Details of α-Synuclein Membrane Association Revealed by Neutrons and Photons. J Phys Chem B. 2015;119(14):4812–4823. doi: 10.1021/jp512499r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Brun AP, Haigh CL, Drew SC, James M, Boland MP, Collins SJ. Neutron Reflectometry Studies Define Prion Protein N-terminal Peptide Membrane Binding. Biophys J. 2014;107(10):2313–2324. doi: 10.1016/j.bpj.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindahl E, Sansom MSP. Membrane proteins: molecular dynamics simulations. Curr Opin Struct Biol. 2008;18:425–431. doi: 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Ayton GS, Voth GA. Systematic multiscale simulation of membrane protein systems. Curr Opin Struct Biol. 2009;19:138–144. doi: 10.1016/j.sbi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khalid S, Bond PJ. Multiscale Molecular Dynamics Simulations of Membrane Proteins. Methods in Molecular Biology. 2013;924:635–657. doi: 10.1007/978-1-62703-017-5_25. [DOI] [PubMed] [Google Scholar]
- 41.Braun AR, Lacy MM, Ducas VC, Rhoades E, Sachs JN. α-Synuclein-induced membrane remodeling is driven by binding affinity, partition depth, and interleaflet order asymmetry. J Am Chem Soc. 2014;136(28):9962–72. doi: 10.1021/ja5016958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai C-L, Landgraf KE, Voth GA, Falke JJ. Membrane Docking Geometry and Target Lipid Stoichiometry of Membrane-Bound PKC C2 Domain: A Combined Molecular Dynamics and Experimental Study. J Mol Biol. 2010;402:301–310. doi: 10.1016/j.jmb.2010.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bucher D, Hsu YH, Mouchlis VD, Dennis EA, McCammon JA. Insertion of the Ca2+-independent phospholipase A2 into a phospholipid bilayer via coarse-grained and atomistic molecular dynamics simulations. PLoS Comput Biol. 2013;9(7):e1003156. doi: 10.1371/journal.pcbi.1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohkubo YZ, Tajkhorshid E. Distinct Structural and Adhesive Roles of Ca2+ in Membrane Binding of Blood Coagulation Factors. Structure. 2008;16:72–81. doi: 10.1016/j.str.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 45.Vermaas JV, Baylon JL, Arcario MJ, Muller MP, Wu Z, Pogorelov TV, Tajkhorshid E. Efficient Exploration of Membrane-Associated Phenomena at Atomic Resolution. J Membr Biol. 2015;248(3):563–582. doi: 10.1007/s00232-015-9806-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai C-L, Srivastava A, Pilling C, Chase AR, Falke JJ, Voth GA. Molecular Mechanism of Membrane Binidng of the GRP1 PH Domain. J Mol Biol. 2013;425:3073–3090. doi: 10.1016/j.jmb.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klauda JB, Brooks BR, Pastor RW. Dynamical motions of lipids and a finite size effect in simulations of bilayers. J Chem Phys. 2006;125:144710. doi: 10.1063/1.2354486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wohlert J, Edholm O. Dynamics in atomistic simulations of phospholipid membranes: Nuclear magnetic resonance relaxation rates and lateral diffusion. J Chem Phys. 2006;125:204703. doi: 10.1063/1.2393240. [DOI] [PubMed] [Google Scholar]
- 49.Lee AG. Lipid-protein interactions in biological membranes: a structural perspective. Biochim Biophys Acta. 2003;1612:1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 50.Marrink SJ, Tieleman DP. Perspective on the MARTINI model. Chemical Society Reviews. 2013;42:6801–6822. doi: 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- 51.Shelley JC, Shelley MY, Reeder RC, Bandyopadhyay S, Moore PB, Klein ML. Simulations of phospholipids using a coarse grain model. J Phys Chem B. 2001;105:9785–9792. [Google Scholar]
- 52.Marrink SJ, de Vries AH, Mark AE. Coarse Grained Model for Semiquantitative Lipid Simulations. J Phys Chem B. 2004;108:750–760. [Google Scholar]
- 53.Izvekov S, Voth GA. Multiscale coarse graining of liquid-state systems. J Chem Phys. 2005;123:134105. doi: 10.1063/1.2038787. [DOI] [PubMed] [Google Scholar]
- 54.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 55.Ayton GS, Lyman E, Voth GA. Hierarchical coarse-graining strategy for protein-membrane systems to access mesoscopic scales. Faraday Discuss. 2010;144:347–357. doi: 10.1039/b901996k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ingólfsson HI, Lopez CA, Uusitalo JJ, de Jong DH, Gopal SM, Periole X, Marrink SJ. The power of coarse graining in biomolecular simulations. WIREs Comput Mol Sci. 2014;4(3):225–248. doi: 10.1002/wcms.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Im W, Feig M, Brooks CL., III An Implicit Membrane Generalized Born Theory for the Study of Structure, Stability, and Interactions of Membrane Proteins. Biophys J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins. 2003;52(2):176–192. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- 59.Chen J, Im W, Brooks CL., III Balancing Solvation and Intramolecular Interactions: Toward a Consistent Generalized Born Force Field. J Am Chem Soc. 2006;128:3728–3736. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bu L, Im W, Brooks CL., III Membrane Assembly of Simple Helix Homo-Oligomers Studied via Molecular Dynamics Simulations. Biophys J. 2007;92:854–863. doi: 10.1529/biophysj.106.095216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mondal J, Zhu X, Cui Q, Yethiraj A. Sequence-Dependent Interaction of β-Peptides with Membranes. J Phys Chem B. 2010;114:13585–13592. doi: 10.1021/jp1070242. [DOI] [PubMed] [Google Scholar]
- 62.Lee C-K, Pao C-W, Smit B. PSII-LHCII Supercomplex Organizations in Photosynthetic Membrane by Coarse-Grained Simulation. J Phys Chem B. 2015;119:3999–4008. doi: 10.1021/jp511277c. [DOI] [PubMed] [Google Scholar]
- 63.MacDermaid CM, Kashyap HK, DeVane RH, Shinoda W, Klauda JB, Klein ML, Fiorin G. Molecular dynamics simulations of cholesterol-rich membranes using a coarse-grained force field for cyclic alkanes. J Chem Phys. 2015;143(24):243144. doi: 10.1063/1.4937153. [DOI] [PubMed] [Google Scholar]
- 64.Grossfield A. Implicit modeling of membranes. Current Topics in Membranes. 2008;60:131–157. [Google Scholar]
- 65.Setzler J, Seith C, Brieg M, Wenzel W. SLIM: An improved generalized Born implicit membrane model. J Comp Chem. 2014;35:2027–2039. doi: 10.1002/jcc.23717. [DOI] [PubMed] [Google Scholar]
- 66.Kim BL, Schafer NP, Wolynes PG. Predictive energy landscapes for folding α-helical transmembrane proteins. Proc Natl Acad Sci USA. 2014;111:11031–11036. doi: 10.1073/pnas.1410529111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Truong HH, Kim BL, Schafer NP, Wolynes PG. Predictive energy landscapes for folding membrane protein assemblies. J Chem Phys. 2014;143:243101. doi: 10.1063/1.4929598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tanizaki S, Feig M. Molecular Dynamics Simulations of Large Integral Membrane Proteins with an Implicit Membrane Model. J Phys Chem B. 2006;110:548–556. doi: 10.1021/jp054694f. [DOI] [PubMed] [Google Scholar]
- 69.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 70.Nymeyer H, Woolf TB, García AE. Folding is not required for bilayer insertion: Replica exchange simulations of an -helical peptide with an explicit lipid bilayer. Proteins: Struct, Func, Bioinf. 2005;59(4):783–790. doi: 10.1002/prot.20460. [DOI] [PubMed] [Google Scholar]
- 71.Vogel A, Roark M, Feller SE. A reinterpretation of neutron scattering experiments on a lipdated Ras peptide using replica exchange molecular dynamics. Biochim Bio-phys Acta Biomembr. 2012;1818:219–224. doi: 10.1016/j.bbamem.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 72.Mori T, Jung J, Sugita Y. Surface-Tension Replica-Exchange Molecular Dynamics Method for Enhanced Sampling of Biological Membrane Systems. J Chem Theor Comp. 2013;9(12):5629–5640. doi: 10.1021/ct400445k. [DOI] [PubMed] [Google Scholar]
- 73.Arcario MJ, Ohkubo YZ, Tajkhorshid E. Capturing Spontaneous Partitioning of Peripheral Proteins using a Biphasic Membrane-Mimetic Model. J Phys Chem B. 2011;115:7029–7037. doi: 10.1021/jp109631y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ohkubo YZ, Pogorelov TV, Arcario MJ, Christensen GA, Tajkhorshid E. Accelerating Membrane Insertion of Peripheral Proteins with a Novel Membrane Mimetic Model. Biophys J. 2012;102:2130–2139. doi: 10.1016/j.bpj.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vermaas JV, Tajkhorshid E. A Microscopic View of Phospholipid Insertion into Biological Membranes. J Phys Chem B. 2014;118:1754–1764. doi: 10.1021/jp409854w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pogorelov TV, Vermaas JV, Arcario MJ, Tajkhorshid E. Partitioning of Amino Acids into a Model Membrane: Capturing the Interface. J Phys Chem B. 2014;118:1481–1492. doi: 10.1021/jp4089113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. Characterizing the Membrane-Bound State of Cytochrome P450 3A4: Structure, Depth of Insertion, and Orientation. J Am Chem Soc. 2013;135(23):8542–8551. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vermaas JV, Tajkhorshid E. Conformational heterogeneity of α-synuclein in membrane. Biochim Biophys Acta Biomembr. 2014;1838(12):3107–3117. doi: 10.1016/j.bbamem.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blanchard AE, Arcario MJ, Schulten K, Tajkhorshid E. A Highly Tilted Membrane Configuration for the Pre-Fusion State of Synaptobrevin. Biophys J. 2014;107:2112–2121. doi: 10.1016/j.bpj.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arcario MJ, Tajkhorshid E. Membrane-Induced Structural Rearrangement and Identification of a Novel Membrane Anchor in Talin F2F3. Biophys J. 2014;107(9):2059–2069. doi: 10.1016/j.bpj.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baylon JL, Tajkhorshid E. Capturing Spontaneous Membrane Insertion of the Influenza Virus Hemagglutinin Fusion Peptide. J Phys Chem B. doi: 10.1021/acs.jpcb.5b02135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McDougle DR, Baylon JL, Meling DD, Kambalyal A, Grinkova YV, Hammernik J, Tajkhorshid E, Das A. Incorporation of charged residues in the CYP2J2 F-G loop disrupts CYP2J2-lipid bilayer interactions. Biochim Biophys Acta. 2015;1848(10):2460–2470. doi: 10.1016/j.bbamem.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Madsen JJ, Ohkubo YZ, Peters GH, Faber JH, Tajkhorshid E. Membrane Interaction of the Factor VIIIa Discoidin Domains in Atomistic Detail. Biochemistry. 2015;54(39):6123–6131. doi: 10.1021/acs.biochem.5b00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McDougle DR, Baylon JL, Meling DD, Kambalyal A, Grinkova YV, Hammernik J, Tajkhorshid E, Das A. Incorporation of charged residues in the CYP2J2 FG loop disrupts CYP2J2–lipid bilayer interactions. Biochim Biophys Acta Biomembr. 2015;1848(10):2460–2470. doi: 10.1016/j.bbamem.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu Z, Schulten K. Synaptotagmin’s Role in Neurotransmitter Release Likely Involves Ca2+-induced Conformational Transition. Biophys J. 2014;107:1156–1166. doi: 10.1016/j.bpj.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rhéault JF, Gagné E, Guertin M, Lamoureux G, Auger M, Lagüe P. Molecular Model of Hemoglobin N from Mycobacterium tuberculosis Bound to Lipid Bilayers: A Combined Spectroscopic and Computational Study. Biochemistry. 2015;54(11):2073–2084. doi: 10.1021/bi5010624. [DOI] [PubMed] [Google Scholar]
- 87.Luo M. Influenza Virus Entry. Adv Exp Med Biol. 2012;726:201–221. doi: 10.1007/978-1-4614-0980-9_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wiley DC, Skehel JJ. The Structure and Function of the Haemagglutinin Membrane Glycoprotein of Influenza Virus. Annu Rev Biochem. 1987;56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- 89.Durrer P, Galli C, Hoenke S, Corti C, Glück R, Vorherr T, Brunner J. H+–induced Membrane Insertion of Influenza Virus Hemagglutinin Involves the HA2 Amino-terminal Fusion Peptide but not the Coiled Coil Region. J Biol Chem. 1996;271(23):13417–13421. doi: 10.1074/jbc.271.23.13417. [DOI] [PubMed] [Google Scholar]
- 90.Gruenke JA, Armstrong RT, Newcomb WW, Brown JC, White JM. New Insights into the Spring-Loaded Conformational Change of Influenza Virus Hemagglutinin. J Virol. 2002;76:4456–4466. doi: 10.1128/JVI.76.9.4456-4466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tamm LK. Hypothesis: Spring-Loaded Boomerang Mechanism of Influenza Hemagglutinin-Mediated Membrane Fusion. Biochim Biophys Acta. 2003;1614:14–23. doi: 10.1016/s0005-2736(03)00159-7. [DOI] [PubMed] [Google Scholar]
- 92.Han X, Tamm LK. A Host-Guest System to Study Structure-Function Relationships of Membrane Fusion Peptides. Proc Natl Acad Sci U S A. 2000;97(24):13097–13102. doi: 10.1073/pnas.230212097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Han X, Tamm LK. pH-Dependent Self-Association of Influenza Hemagglutinin Fusion Peptides in Lipid Bilayers. J Mol Biol. 2000;304(5):953–965. doi: 10.1006/jmbi.2000.4251. [DOI] [PubMed] [Google Scholar]
- 94.Han X, Bushweller JH, Cafiso DS, Tamm LK. Membrane Structure and Fusion-Triggering Conformational Change of the Fusion Domain from Influenza Hemagglutinin. Nat Struct Mol Biol. 2001;8(8):715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- 95.Lorieau JL, Louis JM, Bax A. The Impact of Influenza Hemagglutinin Fusion Peptide Length and Viral Subtype in its Structure and Dynamics. J Am Chem Soc. 2012;99(3):189–195. doi: 10.1002/bip.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lorieau JL, Louis JM, Schwieters CD, Bax A. pH-Triggered, Activated-State Conformations of the Influenza Hemagglutinin Fusion Peptide Revealed by NMR. Proc Natl Acad Sci U S A. 2012;109(49):19994–19999. doi: 10.1073/pnas.1213801109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lorieau JL, Louis JM, Bax A. The Complete Influenza Hemagglutinin Fusion Peptide Adopts a Tight Helical Hairpin Arrangement at the Lipid:Water Interface. Proc Natl Acad Sci U S A. 2010;107(25):11341–11346. doi: 10.1073/pnas.1006142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lorieau JL, Louis JM, Bax A. Whole-Body Rocking Motion of a Fusion Peptide in Lipid Bilayers from Size-Dispersed 15N NMR Relaxation. J Am Chem Soc. 2011;133:14184–14187. doi: 10.1021/ja2045309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, Gunsalus IC, Nebert DW. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6(1):1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 100.Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 58:5–6. doi: 10.1007/PL00000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine Reviews. 2011;32(1):81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, Dechend R, Konkel A, von Schacky C, Luft FC, Muller DN, Rothe M, Schunck W-H. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of omega-3 fatty acids. J Biol Chem. 2010;285:32720–32733. doi: 10.1074/jbc.M110.118406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sakaguchi M, Mihara K, Sato R. A short amino-terminal segment of microsomal cytochrome P-450 functions both as an insertion signal and as a stop-transfer sequence. EMBO J. 1987;6(8):2425–2431. doi: 10.1002/j.1460-2075.1987.tb02521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Black SD. Membrane topology of the mammalian P450 cytochromes. FASEB J. 1992;6(2):680–685. doi: 10.1096/fasebj.6.2.1537456. [DOI] [PubMed] [Google Scholar]
- 105.Scott EE, He YQ, Halpert JR. Substrate routes to the buried active site may vary among cytochromes P450: mutagenesis of the F-G region in P450 2B1. Chemical Research in Toxicology. 2002;15(11):1407–1413. doi: 10.1021/tx020057u. [DOI] [PubMed] [Google Scholar]
- 106.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5(1):121–31. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 107.Nath A, Grinkova YV, Sligar SG, Atkins WM. Ligand binding to cytochrome P450 3A4 in phospholipid bilayer nanodiscs: the effect of model membranes. J Biol Chem. 2007;282:28309–28320. doi: 10.1074/jbc.M703568200. [DOI] [PubMed] [Google Scholar]
- 108.Denisov IG, Baas BJ, Grinkova YV, Sligar SG. Cooperativity in P450 CYP3A4: Linkages in substrate binding, spin state, uncoupling and product formation. J Biol Chem. 2007;282:7066–7076. doi: 10.1074/jbc.M609589200. [DOI] [PubMed] [Google Scholar]
- 109.Guengerich PF. Cytochrome P450 and Chemical Toxicology. Chemical Research in Toxicology. 2008;21(1):70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 110.Denisov IG, Grinkova YV, Baylon JL, Tajkhorshid E, Sligar SG. Mechanism of Drug-Drug Interactions Mediated by Human Cytochrome P450 CYP3A4 Monomer. Biochemistry. 2015;54(13):2227–2239. doi: 10.1021/acs.biochem.5b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 112.Liddington RC, Ginsberg MH. Integrin Activation Takes Shape. J Cell Biol. 2002;158:833–839. doi: 10.1083/jcb.200206011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Felding-Habermann B, Lerner RA, Lillo A, Zhuang S, Weber MR, Arrues S, Gao C, Mao S, Saven A, Janda KD. Combinatorial Antibody Libraries from Cancer Patients Yield Ligand-Mimetic Arg-Gly-Asp-containing immunoglobulins that Inhibit Breast Cancer Metastasis. Proc Natl Acad Sci USA. 2004;101:17210–17215. doi: 10.1073/pnas.0407869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ginsberg MH, Partridge A, Shattil SJ. Integrin Regulation. Curr Opin Cell Biol. 2005;17:509–516. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 115.Ma Y, Qin J, Plow E. Platelet Integrin αIIbβ3 Activation Mechanisms. J Thromb Haem. 2007;5:1345–1352. doi: 10.1111/j.1538-7836.2007.02537.x. [DOI] [PubMed] [Google Scholar]
- 116.Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, dePereda JM, Ginsberg MH, Calderwood DA. Talin Binding to Integrin β Tails: A Final Common Step in Integrin Activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 117.Shattil SJ, Kim C, Ginsberg MH. The Final Steps of Integrin Activation: The End Game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Humphries MJ, McEwan PA, Barton SJ, Buckley PA, Bella J, Mould AP. Integrin Structure: Heady Advances in Ligand Binding, but Activation Still Makes the Knees Wobble. Trends Biochem Sci. 2003;28:313–320. doi: 10.1016/s0968-0004(03)00112-9. [DOI] [PubMed] [Google Scholar]
- 119.Wegener KL, Campbell ID. Transmembrane and Cytoplasmic Domains in Integrin Activation and Protein-Protein Interactions. Mol Membr Biol. 2008;25:376–387. doi: 10.1080/09687680802269886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ratnikov B, Partridge A, Ginsberg M. Integrin Activation by Talin. J Thromb Haem. 2005;3:1783–1790. doi: 10.1111/j.1538-7836.2005.01362.x. [DOI] [PubMed] [Google Scholar]
- 121.Campbell ID, Ginsberg MH. The Talin-Tail Interaction Places Integrin Activation on FERM Ground. Trends Biochem Sci. 2004;29:429–435. doi: 10.1016/j.tibs.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 122.Moser M, Legate KR, Zent R, Fässler R. The Tail of Integrins, Talin, and Kindlins. Science. 2009;324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 123.Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH. The talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. 1999;274:28071–28074. doi: 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- 124.Kim M, Carman CV, Springer TA. Bidirectional Transmembrane Signaling by Cytoplasmic Domain Separation of Integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 125.Saltel F, Mortier E, Hytönen VP, Jacquier M-C, Zimmermann P, Vogel V, Liu W, Wehrle-Haller B. New PI(4,5)P2- and Membrane Proximal Integrin-Binding Motifs in the Talin Head Control β3-integrin Clustering. J Cell Biol. 2009;187:715–731. doi: 10.1083/jcb.200908134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dietrich C, Goldmann W, Sackmann E, Isenberg G. Interaction of NBD-talin with Lipid Monolayers. FEBS Lett. 1993;324:37–40. doi: 10.1016/0014-5793(93)81527-7. [DOI] [PubMed] [Google Scholar]
- 127.Isenberg G, Goldmann WH. Peptide-Specific Antibodies Localize the Major Lipid Binding Sites of Talin Dimers to Oppositely Arranged N-Terminal 47 kDa Subdomains. FEBS Lett. 1998;426:165–170. doi: 10.1016/s0014-5793(98)00336-6. [DOI] [PubMed] [Google Scholar]
- 128.Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID. Structural Basis of Integrin Activation by Talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 129.Seelig A, Blatter XL, Frentzel A, Isenberg G. Phospholipid Binding of Synthetic Talin Peptides Provides Evidence for an Intrinsic Membrane Anchor of Talin. Biol Chem. 2000;275:17954–17961. doi: 10.1074/jbc.M002264200. [DOI] [PubMed] [Google Scholar]
- 130.Anthis NJ, Wegener KL, Ye F, Kim C, Goult BT, Lowe ED, Vakonakis I, Bate N, Critchley DR, Ginsberg MH, Campbell ID. The Structure of an Integrin/Talin Complex Reveals the Basis of Inside-Out Signal Transduction. EMBO J. 2009;28:3623–3632. doi: 10.1038/emboj.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.García-Alvarez B, dePereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, Ginsberg MH, Liddington RC. Structural Determinants of Integrin Recognition by Talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 132.Song X, Yang J, Hirbawi J, Ye S, Perera HD, Goksoy E, Dwivedi P, Plow EF, Zhang R, Qin J. A novel membrane–dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res. 2012;11:1533–1545. doi: 10.1038/cr.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kalli AC, Wegener KL, Goult BT, Anthis NJ, Campbell ID, Sansom MS. The Structure of the Talin/Integrin Complex at a Lipid Bilayer: An NMR and MD Simulation Study. Structure. 2010;18:1280–1288. doi: 10.1016/j.str.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kalli AC, Campbell ID, Sansom MS. Multiscale Simulations Suggest a Mechanism for Integrin Inside-Out Activation. Proc Natl Acad Sci USA. 2011;108:11890–11895. doi: 10.1073/pnas.1104505108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lees AJ, Hardy J, Revesz T. Parkinson’s Disease. Lancet. 2009;373:13–19. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 137.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 138.Robotta M, Gerding HR, Vogel A, Hauser K, Schildknecht S, Karreman C, Leist M, Subramaniam V, Drescher M. Alpha-Synuclein Binds to the Inner Membrane of Mitochondria in an α-Helical Conformation. ChemBioChem. 2014;15(17):2499–2502. doi: 10.1002/cbic.201402281. [DOI] [PubMed] [Google Scholar]
- 139.Jo E, McLaurin J, Yip CM, George-Hyslop PS, Fraser PE. α-Synuclein Membrane Interactions and Lipid Specificity. J Biol Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 140.Burre J, Sharma M, Sudhof TC. Definition of a Molecular Pathway Mediating α-Synuclein Neurotoxicity. J Neurosci. 2015;35(13):5221–5232. doi: 10.1523/JNEUROSCI.4650-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 142.Drescher M, Veldhuis G, van Rooijen BD, Milikisyants S, Subramaniam V, Huber M. Antiparallel Arrangement of the Helicies of Vesicle-Bound α-Synuclein. J Am Chem Soc. 2008;130:7796–7797. doi: 10.1021/ja801594s. [DOI] [PubMed] [Google Scholar]
- 143.Rao JN, Jao CC, Hegde BG, Langen R, Ulmer TS. A Combinatorial NMR and EPR Apporoach for Evaluating the Structural Ensemble of Partially Folded Proteins. J Am Chem Soc. 2010;132:8657–8668. doi: 10.1021/ja100646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound α-synuclein studied by site-directed spin-labeling. Proc Natl Acad Sci USA. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. Membrane-Bound Alpha-Synuclein Forms an Extended Helix: Long-Distance Pulsed ESR Measurements Using Vesicles, Bicelles and Rod-Like Micelles. J Am Chem Soc. 2008;130:12856–12857. doi: 10.1021/ja804517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci USA. 2008;105(50):19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Trexler AJ, Rhoades E. α-Synuclein Binds Large Unilamellar Vesicles as an Extended Helix. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. The Lipid-binding Domain of Wild Type and Mutant α-Synuclein: Compactness and Interconversion Between the Broken and Extended Helix Forms. J Biol Chem. 2010;285:28261–28274. doi: 10.1074/jbc.M110.157214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Robotta M, Braun P, van Rooijen B, Subramaniam V, Huber M, Drescher M. Direct Evidence of Coexisting Horseshoe and Extended Helix Conformations of Membrane-Bound Alpha-Synuclein. ChemPhysChem. 2011;12:267–269. doi: 10.1002/cphc.201000815. [DOI] [PubMed] [Google Scholar]
- 150.Lokappa SB, Ulmer TS. Alpha-synuclein populates both elongated and broken helix states on small unilamellar vesicles. J Biol Chem. 2011;286(24):21450–21457. doi: 10.1074/jbc.M111.224055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Borbat P, Ramlall TF, Freed JH, Eliezer D. Inter-Helix Distances in Lysophospholipid Micelle-Bound α-Synuclein from Pulsed ESR Measurements. J Am Chem Soc. 2006;128:10004–10005. doi: 10.1021/ja063122l. [DOI] [PubMed] [Google Scholar]
- 152.Middleton ER, Rhoades E. Effects of Curvature and Composition on α-Synuclein Binding to Lipid Vesicles. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Braun AR, Sevcsik E, Chin P, Rhoades E, Tristram-Nagle S, Sachs JN. α-Synuclein Induces both Positive Mean Curvature and Negative Gaussian Curvature in Membranes. J Am Chem Soc. 2012;134:2613–2620. doi: 10.1021/ja208316h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Braun AR, Sachs JN. α-Synuclein Reduces Tension and Increases Undulations in Simulations of Small Unilamella Vesicles. Biophys J. 2015;108(8):1848–1851. doi: 10.1016/j.bpj.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bucciantini M, Rigacci S, Stefani M. Amyloid Aggregation: Role of Biological Membranes and the Aggregate-Membrane System. J Phys Chem Lett. 2014;5(3):517–527. doi: 10.1021/jz4024354. [DOI] [PubMed] [Google Scholar]
- 156.Cho W, Stahelin RV. Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 157.Tavoosi N, Davis-Harrison RL, Pogorelov TV, Ohkubo YZ, Arcario MJ, Clay MC, Rienstra CM, Tajkhorshid E, Morrissey JH. Molecular determinants of phospholipid synergy in blood clotting. J Biol Chem. 2011;286:23247–23253. doi: 10.1074/jbc.M111.251769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Banner DW, D’Arcy A, Chène C, Winkler FK, Guha A, Konigsberg WH, Nemerson Y. The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature. 1996;380:41–46. doi: 10.1038/380041a0. [DOI] [PubMed] [Google Scholar]
- 159.Bajaj SP, Schmidt AE, Agah S, Bajaj MS, Padmanabhan K. High Resolution Structures of p-Aminobenzamidine- and Benzoamidine-VIIa/Soluble Tissue Factor -Unpredicted conformation of the 192–193 peptide bond and mapping of Ca2+, Mg2+, Na+,and Zn2+ sites in factor VIIa. J Biol Chem. 2006;281:24873–24888. doi: 10.1074/jbc.M509971200. [DOI] [PubMed] [Google Scholar]
- 160.Ngo JCK, Huang M, Roth DA, Furie BC, Furie B. Crystal Structure of Human Factor VIII: Implications for the Formation of the Factor IXa–Factor VIIIa Complex. Structure. 2008;16:597–606. doi: 10.1016/j.str.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 161.Huang M, Rigby AC, Morelli X, Grant MA, Huang G, Furie B, Seaton B, Furie BC. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat Struct Biol. 2003;10:751–756. doi: 10.1038/nsb971. [DOI] [PubMed] [Google Scholar]
- 162.Randles LG, Rounsevell RWS, Clarke J. Spectrin Domains Lose Cooperativity in Forced Unfolding. Biophys J. 2007;92:571–577. doi: 10.1529/biophysj.106.093690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lin Y, Nielsen R, Murray D, Hubbell WL, Mailer C, Robinson BH, Gelb MH. Docking Phospholipase A2 on Membranes Using Electrostatic Potential–Modulated Spin Relaxation Magnetic Resonance. Science. 1998;279:1925–1929. doi: 10.1126/science.279.5358.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Springer; New York: 1999. [Google Scholar]
- 165.Nelsestuen GL. Enhancement of Vitamin-K-Dependent Protein Function by Modification of the γ-Carboxyglutamic Acid Domain: Studies of Protein C and Factor VII. Trends Cardiovasc Med. 1999;9:162–167. doi: 10.1016/s1050-1738(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 166.Harvey SB, Stone MD, Martinez MB, Nelsestuen GL. Mutagenesis of the γ-Carboxyglutamic Acid Domain of Human Factor VII to Generate Maximum Enhancement of the Membrane Contact Site. J Biol Chem. 2003;278:8363–8369. doi: 10.1074/jbc.M211629200. [DOI] [PubMed] [Google Scholar]
- 167.Vermaas JV, Taguchi AT, Dikanov SA, Wraight CA, Tajkhorshid E. Redox Potential Tuning through Differential Quinone Binding in the Photosynthetic Reaction Center of Rhodobacter sphaeroides. Biochemistry. 2015;54(12):2104–2116. doi: 10.1021/acs.biochem.5b00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ohkubo YZ, Morrissey JH, Tajkhorshid E. Dynamical view of membrane binding and complex formation of human factor VIIa and tissue factor. J Thromb Haem. 2010;8:1044–1053. doi: 10.1111/j.1538-7836.2010.03826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lechtenberg B, Murray-Rust T, Johnson D, Adams T, Krishnaswamy S, Camire R, Huntington J. Crytal structure of the prothrombinase complex from the venom of Pseudonaja textilis. Blood. 2013;122:2777–2783. doi: 10.1182/blood-2013-06-511733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ, Robinson CV. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Moravcevic K, Oxley CL, Lemmon MA. Conditional peripheral membrane proteins: facing up to limited specificity. Structure. 2012;20(1):15–27. doi: 10.1016/j.str.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lee AG. Biological membranes: the importance of molecular detail. Trends Biochem Sci. 2011;36(9):493–500. doi: 10.1016/j.tibs.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 173.Soubias O, Walter E, Teague J, Hines KG, Mitchell DC, Gawrisch K. Contribution of Membrane Elastic Energy to Rhodopsin Function. Biophys J. 2010;99:817–824. doi: 10.1016/j.bpj.2010.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mondal S, Khelashvili G, Shan J, Andersen OS, Weinstein H. Quantitative modeling of membrane deformations by multihelical membrane proteins: application to G-protein coupled receptors. Biophys J. 2011;101:2092–2101. doi: 10.1016/j.bpj.2011.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Anishkin A, Kamaraju K, Sukharev S. Mechanosensitive Channel MscS in the Open State: Modeling of the Transition, Explicit Simulations, and Experimental Measurements of Conductance. J Gen Physiol. 2008;132:67–83. doi: 10.1085/jgp.200810000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Howery AE, Elvington S, Abraham SJ, Choi KH, Dworschak-Simpson S, Phillips S, Ryan CM, Sanford RL, Almqvist J, Tran K, Chew TA, Zachariae U, Andersen OS, Whitelegge J, Matulef K, Du Bois J, Maduke MC. A designed inhibitor of a CLC antiporter blocks function through a unique binding mode. Chem Biol. 2012;19(11):1460–1470. doi: 10.1016/j.chembiol.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Paradies G, Paradies V, Benedictis VD, Ruggiero FM, Petrosillo G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim Biophys Acta – Bioener. 2014;1837:408–417. doi: 10.1016/j.bbabio.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 178.MacCallum JL, Bennett WFD, Tieleman DP. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys J. 2008;94:3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ. The MARTINI coarse grained forcefield: Extension to proteins. J Chem Theor Comp. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 180.Radzicka A, Wolfenden R. Comparing the Polarities of the Amino Acids: Side Chain Distribution Coefficients between the Vapor Phase, Cyclohexane, 1-Octanol, nd Neutral Aqueous Solution. Biochemistry. 1988;27:1664–1670. [Google Scholar]
- 181.Wimley WC, Creamer TP, White SH. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry. 1996;35(16):5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- 182.Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 183.Moon CP, Fleming KG. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc Natl Acad Sci USA. 2011;108:10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Qi Y, Cheng X, Lee J, Vermaas JV, Pogorelov TV, Tajkhorshid E, Park S, Klauda JB, Im W. CHARMM-GUI HMMM Builder for Membrane Simulations with the Highly Mobile Membrane-Mimetic Model. Biophys J. 2015;109:2012–2022. doi: 10.1016/j.bpj.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.