

Schibler et al. show that both Set1 and H3K4 mutants display a benomyl resistance phenotype that requires components of the spindle assembly checkpoint (SAC), including Bub3 and Mad2. Interactions between Mad2 and H3K4 regulate resolution of the SAC by limiting closed Mad2 availability for Cdc20 inhibition.
Keywords: chromatin, lysine methylation, spindle assembly checkpoint, Set1, H3K4, HORMA domain
Abstract
Histone H3 methylation on Lys4 (H3K4me) is associated with active gene transcription in all eukaryotes. In Saccharomyces cerevisiae, Set1 is the sole lysine methyltransferase required for mono-, di-, and trimethylation of this site. Although H3K4me3 is linked to gene expression, whether H3K4 methylation regulates other cellular processes, such as mitosis, is less clear. Here we show that both Set1 and H3K4 mutants display a benomyl resistance phenotype that requires components of the spindle assembly checkpoint (SAC), including Bub3 and Mad2. These proteins inhibit Cdc20, an activator of the anaphase-promoting complex/cyclosome (APC/C). Mutations in Cdc20 that block Mad2 interactions suppress the benomyl resistance of both set1 and H3K4 mutant cells. Furthermore, the HORMA domain in Mad2 directly binds H3, identifying a new histone H3 “reader” motif. Mad2 undergoes a conformational change important for execution of the SAC. We found that the closed (active) conformation of both yeast and human Mad2 is capable of binding methylated H3K4, but, in contrast, the open (inactive) Mad2 conformation limits interaction with methylated H3. Collectively, our data indicate that interactions between Mad2 and H3K4 regulate resolution of the SAC by limiting closed Mad2 availability for Cdc20 inhibition.
Lysine methylation within histone H3 is commonly associated with gene regulation. H3K4 trimethylation in particular is associated with gene activation and is localized to gene promoters. In contrast, H3K4 dimethylation is present across the gene body of both active and repressed genes (Bernstein et al. 2002; Santos-Rosa et al. 2002). Lysine methylation also contributes to other cellular processes, such as DNA damage repair (Sollier et al. 2004) and telomere maintenance (Krogan et al. 2002). H3K4 methylation alters chromatin structure and also serves to recruit or exclude binding of nonhistone proteins to chromatin.
A single lysine methyltransferase, Set1, mediates all H3K4 methylation events in Saccharomyces cerevisiae (Briggs et al. 2001). Set1 is incorporated into a multiprotein complex termed COMPASS (Miller et al. 2001; Roguev et al. 2001; Krogan et al. 2002) and is homologous to human MLL1, which also methylates H3K4 and is incorporated into a COMPASS-like complex (Shilatifard 2012). Translocations of MLL1 are associated with leukemogenesis (Zeleznik-Le et al. 1994) and a poor prognosis (Pui et al. 1994). However, how mutations in MLL1 contribute to leukemogenesis and an unfavorable outcome is not clear. Identifying novel pathways regulated by SET1 in yeast can help elucidate cellular functions regulated by MLL1 in humans (Schneider et al. 2005; Milne et al. 2010; Shilatifard 2012).
COMPASS also methylates Dam1, a kinetochore protein (Zhang et al. 2005). Like H3K4 methylation, Dam1 methylation is regulated in trans by histone H2B ubiquitination (Latham et al. 2011). Moreover, Dam1 dimethylation on K233 inhibits phosphorylation of surrounding serines by the Aurora kinase Ipl1. Accordingly, mutations that eliminate Set1 expression or activity suppress the effects of conditional mutations in Ipl1 (Zhang et al. 2005).
Ipl1 phosphorylates a number of kinetochore proteins in response to inappropriate microtubule–kinetochore interactions that lead to triggering of the spindle assembly checkpoint (SAC) (Biggins and Murray 2001). The SAC guards against aberrant chromosome segregation during mitosis by preventing progression to anaphase until defective microtubule–kinetochore attachments are resolved and mitotic spindle tension is established (Rieder et al. 1994; Li and Nicklas 1995).
S. cerevisiae has provided a useful model to identify several SAC components, including Mad1 (mitotic arrest defect 1), Mad2, Mad3 (Li and Murray 1991), Bub1 (budding uninhibited by benomyl 1), and Bub3 (Hoyt et al. 1991). The SAC involves an orchestration of protein–protein interactions and phosphorylation events that ultimately prevent activation of the anaphase-promoting complex/cyclosome (APC/C) (Jia et al. 2013). The APC is an E3 ubiquitin ligase that requires Cdc20 to recruit proteins for ubiquitination and subsequent degradation. SAC proteins bind directly to Cdc20, preventing proteolysis of key APC substrates such as Securin (Pds1).
As our previous studies indicated that Set1 opposes the functions of Ipl1 (Zhang et al. 2005), we reasoned that Set1 might have additional functions during mitosis. Here we report that the highly conserved HORMA domain in Mad2 is a novel H3K4 methyl reader and that this modification and Set1 play an important role in regulating the release of the SAC through Mad2 interactions.
Results
Loss of SET1 induces benomyl resistance
To further address how lysine methylation might regulate mitosis, we subjected cells bearing deletions in SET1 or in other SET domain-encoding genes to growth in the presence of the microtubule depolymerizing agent benomyl, which interferes with mitotic spindle stability and antagonizes mitotic progression. Mutations in genes required for formation of the mitotic spindle, in components of the kinetochore, or in the activation and maintenance of mitotic checkpoints are characteristically sensitive to microtubule depolymerizing drugs (Spencer et al. 1990; Stearns et al. 1990; Hoyt et al. 1991; Li and Murray 1991).
We discovered that set1 mutant cells are highly resistant, rather than sensitive, to high levels of benomyl (Fig. 1A; Supplemental Fig. S1A). set1 mutant cells grow similarly to wild-type cells on rich medium or in the presence of dimethyl sulfoxide (DMSO) alone but display continued growth in levels of benomyl (30–40 µg/mL) that completely block growth of wild-type cells. Benomyl resistance was not observed upon deletion of any other SET domain-containing gene, indicating a unique function for Set1 in responding to microtubule poisons (Fig. 1A). Interestingly, since SET2 encodes the lysine methyltransferase required for histone H3K36 mono-, di-, and trimethylation, which is also associated with active transcription, our results suggest that defective transcription is not sufficient to confer benomyl resistance.
Figure 1.
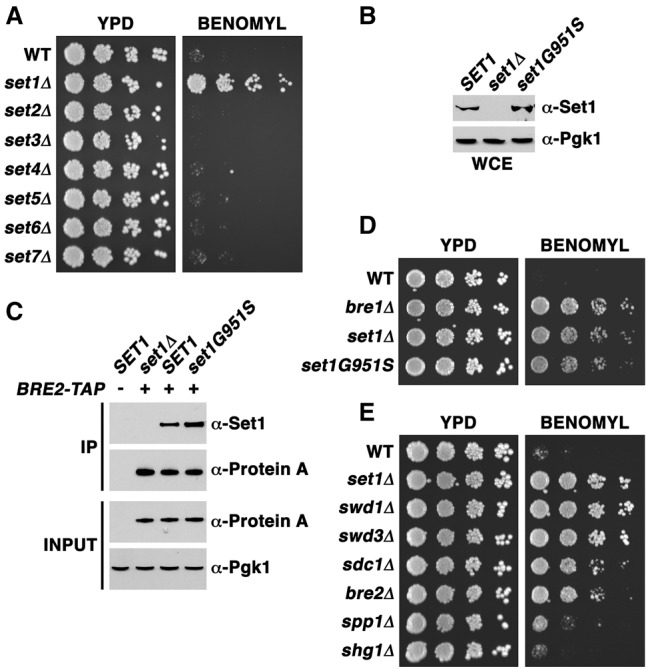
Loss of COMPASS-mediated lysine methylation results in benomyl resistance. (A) A serial fivefold dilution assay of yeast strains with the indicated genotypes was performed to visualize growth phenotypes. Cells were placed onto YPD plates (YPD) with or without 30 µg/mL benomyl, and these plates were placed for 2 d at 30°C. (B) Whole-cell extracts (WCEs) were isolated from strains with the indicated genotypes. The amount of total Set1 protein was assessed by immunoblot analysis using an antibody specific for Set1 (1:100; Santa Cruz Biotechnology, SC-101858). Pgk1 protein levels were used as a loading control. (C) Both wild-type Set1 and Set1G951S proteins similarly associate with the COMPASS component Bre2. Immunoprecipitation (IP) assays were used to pull down Bre2, and the associated levels of Set1 were assessed in the wild-type (WT) and set1G951S strains. Bre2-TAP (α-protein A) and Pgk1 (α-Pgk1) were used as loading controls. (D) bre1Δ, set1Δ, and the catalytically inactive mutant set1G951S were subjected to the same assay as in A. (E) COMPASS mutants were assessed for growth phenotypes using the same serial fivefold dilution assay as in A and D.
To address whether the benomyl resistance phenotype reflects loss of Set1 catalytic activity, we subjected cells bearing a mutation in the Set1 catalytic site, G951S (Nagy et al. 2002), to growth in the presence of the drug. Although some Set1 mutations that affect methyltransferase activity result in lower amounts of total Set1 protein (Soares et al. 2014), we detected equal amounts of wild-type Set1 and Set1G951S protein in immunoblots of whole-cell extracts (Fig. 1B). Moreover, we found equal incorporation of wild-type and mutant Set1 into the COMPASS complex, isolated via affinity purification of Bre2, another COMPASS component (Fig. 1C). As expected, the G951S mutation abrogated the ability of Set1 to methylate histone H3K4 (Supplemental Fig. S1B). Importantly, cells bearing the set1G951S mutation displayed benomyl resistance equivalent to that caused by SET1 deletion (Fig. 1D), indicating that Set1 catalytic activity is required for a normal response to microtubule depolymerization.
Our results are consistent with a previous large-scale screen that indicated that BRE1 mutants display resistance to benomyl (Rieger et al. 1999). Bre1 is an E3 ubiquitin ligase and a targeting factor for the E2 enzyme Rad6. Both Bre1 and Rad6 are required for histone H2BK123 monoubiquitination (Nakanishi et al. 2009), an event required upstream of Set1-mediated methylation of both H3K4 and Dam1 K233 (Lee et al. 2007; Latham et al. 2011).
Mutations in different components of the COMPASS complex (Fig. 1E) affect distinct methylation states of Set1 substrates (Schneider et al. 2005). We found that deletion of genes encoding subunits required for all three methylation states, such as SWD1 and SWD3, also results in benomyl resistance (Fig. 1E). Deletion of BRE2 or SDC1 inhibits dimethylation and trimethylation of H3K4 (Schneider et al. 2005; Latham et al. 2011), and these mutations also induce substantial resistance to benomyl, although not quite to the same degree as caused by SET1 mutations. Deletion of SPP1 eliminates trimethylation of H3K4 without affecting mono- and dimethylation (Schneider et al. 2005). Interestingly, spp1 mutants display only a slight benomyl resistance, indicating that loss of trimethylation of COMPASS substrates is not sufficient to trigger resistance. Deletion of SHG1 increases di- and trimethylation of H3K4 (Dehe et al. 2006) but does not affect benomyl resistance. Together, our results indicate that mono- and dimethylation of substrates by Set1, working within the COMPASS complex, are required for proper response to microtubule poisons.
Benomyl resistance of set1G951S cells requires an intact SAC and Cdc20 inhibition
Resistance to spindle poisons likely reflects failure in the regulation of the SAC, which maintains genome integrity by preventing anaphase progression in the presence of defective microtubule–kinetochore attachments (Fig. 2A). Importantly, although the components and functions of the SAC are highly conserved across evolution, kinetochore–microtubule interactions are maintained throughout the yeast cell cycle, with only a short disruption during early S phase (Kitamura et al. 2007). Unlike mammalian cells, the SAC becomes essential in S. cerevisiae only upon active damage to the spindle, such as upon exposure to benomyl (Hoyt et al. 1991; Li and Murray 1991). Consistent with those findings, set1Δ cells do not display any cell cycle aberrations, as measured by flow cytometry (Supplemental Fig. S1C) or a budding index assay (Supplemental Fig. S1D). Our set1Δ cells did grow at a marginally slower rate than wild-type cells (Supplemental Fig. S1E), as previously reported (Miller et al. 2001), correlating with a slight reduction in colony-forming units (Supplemental Fig. S1F), but no abnormal cellular morphologies were observed (Supplemental Fig. S1G). Therefore, the benomyl resistance phenotype displayed by set1 mutants is unlikely to be a downstream effect of other cell cycle anomalies.
Figure 2.

The set1G951S mutant requires the SAC and Cdc20 inhibition for benomyl resistance. (A) An illustration of Cdc20 associated with the APC complex in both an active and an inactive state. Mad2 and Mad3 bind to Cdc20 to inhibit the ubiquitination of APC substrates. (B–D) Serial fivefold dilution assay of yeast strains with the indicated genotypes grown on normal YPD plates (YPD) or plates containing 30 µg/mL benomyl for 2 d at 30°C.
To determine whether activation of the SAC is required for the benomyl resistance of set1 mutants, we deleted genes encoding SAC components in the set1G951S strain. Deletion of BUB1 and BUB3 eliminated the resistance of set1G951S cells to benomyl (Fig. 2B). These data indicate that these SAC components are required for the resistance phenotype and also that the set1 mutation does not prevent uptake of the drug, as the double mutants displayed a sensitivity to benomyl similar to that of bub1 and bub3 single mutants. Deletion of MAD1, MAD2, and MAD3 also suppressed the benomyl resistance phenotype of set1G951S cells (Fig. 2C). Collectively, these data indicate that the SAC must be activated and intact for the benomyl resistance to occur in the set1 mutant cells.
SAC component proteins inhibit Cdc20 to block entry into anaphase upon disruption of microtubule–kinetochore interactions. To further define the role of SAC activation in the benomyl resistance phenotype, we introduced a Cdc20 mutation that blocks inhibition into set1G951S cells. The dominant cdc20-127 allele disrupts binding of Mad2 and Mad3, thereby inhibiting SAC-mediated cell cycle arrest upon exposure to microtubule poisons (Hwang et al. 1998). We found that set1G951S cdc20-127 double mutants are sensitive to benomyl compared with set1G951S mutants alone, indicating that binding of SAC components to Cdc20 is required for benomyl resistance (Fig. 2D) and that lysine methylation events mediated by Set1 function to limit Cdc20 inhibition.
set1 mutants display a thick mitotic spindle and a defect in the release of the SAC
One phenotype reported previously for cdc20-1, a temperature-sensitive mutant of Cdc20, is a thick mitotic spindle (Sethi et al. 1991), although this phenotype appears to be variable (Lim et al. 1998). Confocal analyses indicated that the mitotic spindles in set1G951S mutant cells are thicker than in wild-type cells (Fig. 3A). Indeed, although thick spindles were rarely observed in wild-type cells (e.g., three of 34 cells examined [8.8%]), they occurred in a majority (10 of 13 [76.9%]) of set1 mutant cells (Fig. 3A). As Cdc20 recruits proteins to the APC/C for ubiquitination and degradation, the thick mitotic spindles in cdc20 mutants likely reflect a failure in the degradation of microtubule-bundling proteins (Sethi et al. 1991; Juang et al. 1997). To determine whether the thick mitotic spindles in set1G951S mutants also reflect Cdc20 inhibition, we monitored destruction of a Cdc20–APC/C substrate, Pds1 (Securin), after SAC activation and release. Pds1 ubiquitination and proteolysis are required for the separation of sister chromatids and progression through anaphase. Both wild-type and set1G951S cells were treated with nocodazole to activate the SAC. Flow cytometry indicated that both wild-type and set1G951S cells arrested in G2/M, as expected. The cells were then released into fresh medium without drugs, and mitotic progression was monitored by assessing Pds1 stability coupled with cell cycle analysis. Immunoblots indicate that Pds1 is degraded at a slower rate in set1 mutant cells (Fig. 3B, cf. lanes labeled 45′ and 60′). These results indicate that set1 mutants arrest normally after SAC activation but exhibit prolonged inhibition of the APC after checkpoint release.
Figure 3.
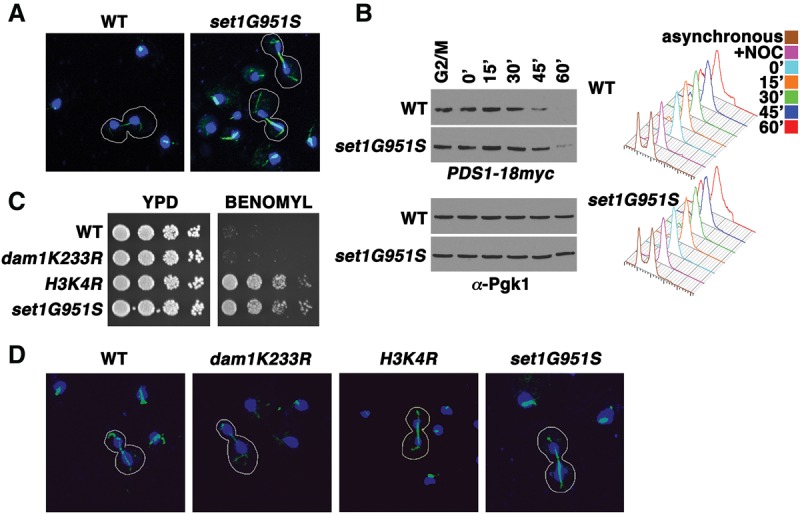
Loss of H3K4 methylation results in a thick mitotic spindle. (A) Cells bearing a SET1 catalytic mutant allele (set1G951S) display a more robust mitotic spindle staining compared with wild-type cells (WT). Immunofluorescent confocal images of wild-type and set1G951S mitotic cells are shown. Tubulin (green) and DAPI (blue) stainings were used to identify cells undergoing mitosis in an asynchronous culture. Mitotic cells are outlined in white. (B) Cell cycle arrest and release experiments reveal that Pds1 is more stable in SET1 mutants (set1G951S) than in wild-type cells. Cells were arrested in G2/M and released into fresh medium. Samples were taken at the indicated time points, and whole-cell extracts and immunoblots were prepared and probed with α-Myc antibody to assess the protein levels of Myc-tagged Pds1. Immunoblots of Pgk1 served as a loading control. Cells from the indicated time points were taken for analysis by flow cytometry. Histograms of DNA content reveal the cell cycle profile of the indicated strains at specific times after cell cycle arrest and release. (C) Serial fivefold dilution assays of wild-type and mutants with the indicated genotype placed onto either control plates (YPD) or plates containing 30 µg/mL benomyl as in previous figures. (D) Immunofluorescent confocal images of wild type and mutants with the indicated genotypes. Staining and selection of cells were preformed as in A.
Loss of H3K4 methylation results in benomyl resistance and a thick mitotic spindle
Two COMPASS substrates have been identified so far: H3K4 (Briggs et al. 2001) and Dam1 K233 (Zhang et al. 2005). We created genomic mutations in these methylation sites in order to determine whether either of these COMPASS substrates plays a role in regulating cellular responses to mitotic poisons. We found that dam1K233R cells are sensitive to benomyl, much like wild-type cells. However, cells bearing mutations in H3K4 (H3K4R) display benomyl resistance very similar to that seen in set1G951S cells (Fig. 3C). The yeast genome contains two alleles encoding H3: HHT1 and HHT2. To determine whether partial loss of H3K4 methylation was sufficient to induce benomyl resistance, we compared growth of wild-type cells, cells bearing single mutations (hht1K4R or hht2K4R), and cells harboring mutations in both H3 alleles (labeled here as H3K4R for simplicity). We found that only the double mutation conferred benomyl resistance (Supplemental Fig. S2A). Moreover, the loss of SET1 in H3K4R cells did not confer any further resistance, suggesting that H3K4 is the primary Set1 substrate regulating normal cellular response to mitotic poisons (Supplemental Fig. S2B). Importantly, we confirmed that Set1 protein levels and incorporation into the COMPASS complex were not affected by the H3K4R mutation (Supplemental Fig. S2C,D).
We also examined the mitotic spindle in wild-type, dam1K233R, H3K4R, and set1G951S cells (Fig. 3D). The mitotic spindle in dam1K233R cells appeared similar to that in wild-type cells. Only one in 16 (6.2%) displayed a thick spindle. In contrast, a majority (24 of 28 cells [85.7%]) of H3K4R cells displayed a thick mitotic spindle phenotype resembling that of set1G951S cells (Fig. 3D). These data indicate that methylation of H3K4 plays a novel role in regulating the response to microtubule depolymerizing drugs, related to increased amounts of polymerized tubulin or increased stability of the mitotic spindle, consistent with inhibition of Cdc20 function.
We next determined whether an intact SAC is required for benomyl resistance of H3K4R cells. As seen in set1G951S cells (Fig. 2), deletion of SAC component genes suppressed the benomyl resistance phenotype caused by H3K4R mutations (Fig. 4A,B). Immunoblots confirmed that loss of SAC components such as Mad2 had no effect on H3K4 methylation events, as H3K4 di- and trimethylation were unaltered in mad2Δ cells (Fig. 4C). Eliminating SAC function through deletion of MAD2 in H3K4R cells also resulted in suppression of the thick mitotic spindle phenotype (two of 18 [11.1%] for the double mutant and one of 18 [5.5%] for the MAD2 deletion alone, compared with 24 of 28 [85.7%] as noted above) (Fig. 4D; multiple panels in Supplemental Fig. S2E). These data further link the mitotic spindle phenotype with the benomyl resistance observed in the H3K4R cells. To test whether the H3K4R mutant also regulates the SAC through Cdc20 inhibition, we introduced the Cdc20-127 mutation into the H3K4R strain. As in set1G951S cells, H3K4R cells require Cdc20 inhibition for benomyl resistance (Fig. 4E), supporting a model of an overactive SAC when H3K4 methylation is inhibited.
Figure 4.
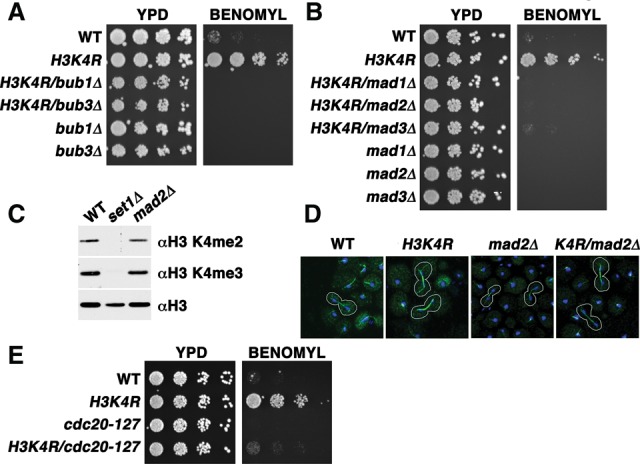
The H3K4R mutant requires the SAC and Cdc20 inhibition for benomyl resistance. (A,B) Serial fivefold dilution assay of wild-type (WT) and mutants with the indicated genotypes placed onto either control plates (YPD) or plates containing 30 µg/mL benomyl, as in previous figures. (C) Immunoblots of whole-cell extracts from wild-type, set1Δ, and mad2Δ cells to compare di- and trimethylation of histone H3 levels. Total H3 was used as a loading control. (D) Immunofluorescent confocal images of wild-type and mutants with the indicated genotypes. Tubulin (green) and DAPI (blue) were used to identify cells undergoing mitosis from an asynchronous culture. Mitotic cells are outlined in white, as in previous figures. (E) Serial fivefold dilution assay of wild-type and mutants with the indicated genotypes placed onto either control plates (YPD) or plates containing 30 µg/mL benomyl, as above.
Since H3K4 methylation is involved in transcriptional regulation, we compared previously published microarray data (Venkatasubrahmanyam et al. 2007) with known loss-of-function mutants that cause benomyl resistance (Brown et al. 2006). Loss of SET1 results in 127 significantly down-regulated genes, none of which are associated with mitosis or the SAC. We confirmed that expression of SAC components is not down-regulated upon loss of H3K4 methylation by comparing levels of Bub1, Bub3, Mad1, Mad2, and Mad3 RNA in both wild-type and set1Δ cells (Supplemental Fig. S2F). We also evaluated H3K4 methylation (H3K4me) events at pericentric regions through chromatin immunoprecipitation (ChIP) followed by sequencing (ChIP-seq) but found little enrichment of H3K4 dimethylation (H3K4me2) or trimethylation (H3K4me3) in locations proximal to the centromere (Supplemental Fig. S2G). These results indicate that the presence of H3K4me2/me3, but not a pericentromeric location of this mark, is important for normal cellular response to benomyl.
Mad2 binds methylated histone H3K4 in a conformation-specific manner
A number of protein domains “read” the methylation state of H3K4, including PHDs, WD40 domains, and Tudor domains (Huyen et al. 2004; Wysocka et al. 2005; Kim et al. 2006; Shi et al. 2006). Mad2 lacks those domains but does contain a HORMA domain, which is found in several chromatin-associated proteins (Aravind and Koonin 1998; Muniyappa et al. 2014). To test whether Mad2 can bind directly to histones, we performed pull-down experiments using recombinant GST-yMad2 fusion proteins and calf thymus histones. We found that GST-yMad2 binds directly to H3, but not to H2A or H2B, in vitro (Fig. 5A). Since Mad2 is composed almost entirely of a single HORMA domain, our data indicate that the HORMA domain likely acts as an H3 interaction domain.
Figure 5.
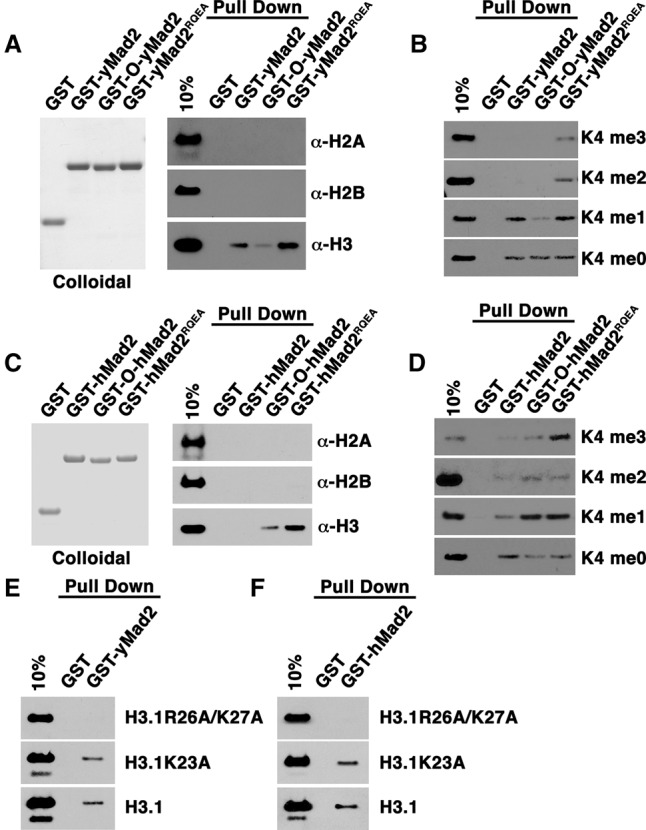
Mad2 binds H3 methylated at K4 in a conformation-specific manner. (A,C) Colloidal staining of purified recombinant proteins. Pull-down assays of the indicated recombinant proteins to identify direct binding with calf thymus histones, assayed using immunoblots of membranes probed with antibodies against H2A, H2B, or H3. (B,D) Pull-down assay with recombinant GST-Mad2 fusion proteins and unmodified H3 (K4me0) or MLA full-length histones generated to mimic H3K4 monomethylation (K4me1), H3K4 dimethylation (K4me2), or H3K4 trimethylation (K4me3). Direct binding was assayed using immunoblots of membranes probed with an antibody recognizing histone H3. (E,F) GST pull-down assays using recombinant GST-yMad2 and GST-hMad2 with wild-type histone H3 or H3 with various point mutations. Direct binding was assessed by using immunoblots of membranes probed with an antibody against H3.
Mad2 adopts two conformations—an inactive, open state (O-Mad2) and an active, closed state (C-Mad2)—in both yeast and human cells (Luo et al. 2000; Sironi et al. 2002; Nezi et al. 2006). After SAC activation, Mad2 assumes the C-Mad2 conformation upon binding to Mad1 at the kinetochore. C-Mad2 then binds and inhibits Cdc20. Recombinant wild-type Mad2 can adopt both O-Mad2 and C-Mad2 conformations and can dimerize in solution (Fang et al. 1998; Luo et al. 2004). Specific mutations limit dimerization and structurally constrain Mad2 to the O-Mad2 form or promote the C-Mad2 conformation (Luo et al. 2004; Nezi et al. 2006). Mutations that delete the C-terminal tail of Mad2 result exclusively in a monomeric O-Mad2 conformation (Fang et al. 1998). In contrast, the Mad2RQEA mutation, which also blocks dimerization, results in a high ratio of C-Mad2 to O-Mad2 monomers (Luo et al. 2004; Nezi et al. 2006). To determine whether Mad2 conformational changes affect binding to H3, we generated GST fusions to yeast wild-type, O-yMad2, and yMad2RQEA proteins. We found that recombinant O-yMad2 interacted less well with H3 than did wild-type yMad2. In contrast, yMad2RQEA pulled down more H3 than did either the wild-type or O-yMad2 conformations. None of the yMad2 proteins bound to H2A or H2B, confirming specificity for H3 interactions (Fig. 5A).
Importantly, O-yMad2 and yMad2RQEA displayed strikingly different profiles binding to modified and unmodified forms of H3. yMad2RQEA clearly bound to all methylation states of H3, whereas O-yMad2 bound best to H3K4me0 and displayed limited interaction with H3K4me1 (Fig. 5B). Both O-yMad2 and yMad2RQEA are monomeric, and yMad2RQEA –H3K4me binding indicates that C-Mad2, rather than O-Mad2, interacts with H3K4me. These findings identify Mad2 as a conformation-sensitive reader of lysine methylation states.
Yeast and human Mad2 structure and function are strikingly conserved. To determine whether human Mad2 (hMad2) also selectively binds H3 in a conformation-specific manner, we repeated the histone-binding assays with GST-hMad2 proteins. We purified GST fusions of wild-type hMad2, monomeric O-hMad2, and monomeric hMad2RQEA. Wild-type hMad2 did not bind well to any histone, but both O-hMAD2 and hMAD2RQEA bound H3 but not H2A or H2B. Like yMad2, hMad2RQEA pulled down more H3 than did O-hMad2 (Fig. 5C), indicating that the conformation sensitivity of Mad2 interactions with H3 is conserved from yeast to humans. Failure of the wild-type hMad2 to bind H3 suggests that hMad2 dimerization may limit its interactions with histones. Importantly, recombinant hMad2RQEA interacted with methylated H3K4 much like yMad2RQEA (Fig. 5D), indicating that closed hMad2 also serves as a reader of H3K4me states.
A sequence highly similar to a known consensus Mad2-binding site is found in amino acids 23–30 of H3. To test whether yMad2 requires this consensus sequence to directly bind histone H3, we generated two recombinant histone H3 mutant proteins K23A (H3.1K23A) and a second containing two mutations to both R26A and K27A (H3.1R26A/K27A). Wild-type histone H3 and the K23A mutant both bound to yMad2, while the R26A/K27A mutant H3 did not (Fig. 5E). Use of a GST-hMad2 construct confirmed that hMad2 also does not interact with the R26A/K27A H3 mutant (Fig. 5F). These results demonstrate that both yMad2 and hMad2 share a conserved H3-binding sequence.
The interactions of Mad2 with H3 suggest that the binding is bipartite, involving both the consensus sequence (amino acids 23–30) and the region around K4 in H3. To further test this idea, we tested previously described alanine scanning mutations in H3 (Dai et al. 2008) within the consensus Mad2 interaction motif (Luo et al. 2002; Sironi et al. 2002) for benomyl resistance (Supplemental Fig. S3A; Dai et al. 2008). As expected, H3K4A mutant cells displayed a striking benomyl resistance phenotype, as did H3R2A mutants. Previous reports indicated a functional like between R2 methylation and K4 methylation (Kirmizis et al. 2007). In addition, mutations to amino acids 23–30 also caused a moderate resistance to benomyl, consistent with the data above, indicating that this region also contributes to Mad2 interactions (Supplemental Fig. S3A).
set1G951S and the H3K4R mutations affect Mad2 localization in dividing cells
To further assess the consequences of loss of H3K4me on Mad2 function in vivo, we examined Mad2 localization in wild-type and set1G951S and H3K4R mutant cells. As we did not have an antibody for native Mad2, we sought to use established epitope-tagged Mad2 alleles. However, we found that C-terminal tags (both GFP and TAP tags) introduced into the MAD2 locus suppressed the benomyl resistance of set1Δ cells, further emphasizing the importance of the Mad2 C-terminal region in interactions with H3K4 (Supplemental Fig. S3B). Therefore, we used an inducible, N-terminally tagged GST-Mad2 fusion protein construct to assess the subcellular localization of Mad2. As expected, we found that GST-Mad2 localizes to both the nucleus and the cytoplasm in wild-type and set1G951S and H3K4R mutant cells (Supplemental Fig. S4A). However, in large budded G2/M cells, Mad2 was enriched around the bud neck (Supplemental Fig. S4A). This staining pattern is consistent with that reported previously for Kel2, a negative regulator of mitotic exit and Mad2-binding protein that localizes to the same region (Wong et al. 2007). Interestingly, this localization pattern was enriched roughly twofold in set1G951S and H3K4R mutant cells (Supplemental Fig. S4B), consistent with disruption of Mad2 localization upon loss of H3K4me.
We propose a model in which methylated H3K4 acts as a negative regulator of the SAC by sequestering active C-Mad2, thereby limiting Cdc20–Mad2 interactions and Cdc20 inhibition. In set1 mutants, loss of H3K4me releases C-Mad2, allowing greater inhibition of Cdc20 and also possibly favoring binding of O-Mad2 to chromatin (Fig. 6).
Figure 6.
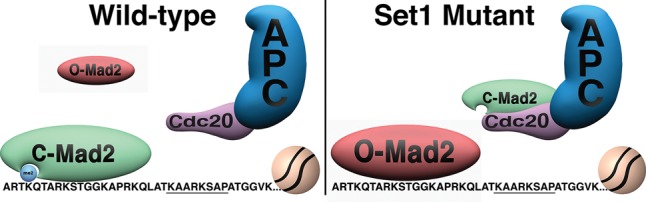
A model showing that methylated H3K4 binds to C-Mad2. An H3 sequence required for Mad2 binding, which is similar to the Mad2-binding consensus sequence, is underlined. Our in vivo and in vitro data indicate that H3K4me acts as a negative regulator of the SAC through direct binding of methylated H3K4 to an active C-Mad2, thereby limiting C-Mad2–Cdc20 interactions.
Discussion
Our data reveal a new function for H3K4me in regulating the SAC and identify the Mad2 HORMA domain as a conformation-specific reader of H3K4me. These studies illustrate how specific chromatin states can provide critical regulatory scaffolds for regulation of essential cellular processes where the HORMA domain likely acts as a histone interaction domain in several important, evolutionarily conserved processes.
We found that Mad2RQEA binds dimethylated and trimethylated H3K4, whereas O-Mad2 does not. In wild-type cells, O-Mad2 dimerizes to C-Mad2 at the kinetochore, where it undergoes a structural change resulting in an active form of Mad2 that binds to and inhibits Cdc20 (Nasmyth 2005; Yu 2006). Cells that cannot form an O-Mad2–C-Mad2 dimer or in which Mad2 is locked into an open conformation fail to arrest in mitosis after treatment with microtubule poisons (Nezi et al. 2006).
Mad2 binds to Cdc20 after SAC activation by adopting a conformation that inhibits ubiquitination of APC substrates, such as Pds1. Our data indicate that binding of C-Mad2 to H3K4me2 inhibits Cdc20 inhibition, likely by sequestering C-Mad2 and blocking O-Mad2 interactions with H3 (Figs. 4E, 5B). After tension is achieved by kinetochore attachments to the mitotic spindle, the SAC is deactivated, allowing APC activation and ubiquitination of APC substrates. We propose that loss of H3K4me allows continued inhibition of Cdc20 through the C-Mad2–Cdc20 interaction, leading to a delay in the release of the SAC, as indicated by the prolonged stability of Pds1.
Our model is similar to that proposed for negative regulators that act to silence the SAC, such as p31comet (MAD2L1BP), in vertebrate cells (Xia et al. 2004). Like H3K4me, p31comet interacts directly with C-Mad2 but not O-Mad2. We propose that H3K4me2 and H3K4me3 may function similarly to p31comet to sequester C-Mad and inhibit binding to Cdc20. Loss of this histone modification leads to excessive availability of active C-Mad2, which in turn leads to heightened inhibition of Cdc20 functions and delayed release of the SAC. p31comet is not expressed in S. cerevisiae, so H3K4me may serve as the primary regulator of C-Mad2 and O-Mad2 interactions in yeast. Intriguingly, p31comet is itself a HORMA domain protein, raising the question of whether H3K4me may regulate its function as well.
Multiple chromatin-associated proteins contain HORMA domains in mammals, including not only p31comet and MAD2 but also REV7, HORMAD1, and HORMAD2. Like Mad2, both HORMAD proteins and REV7 are thought to undergo a conformational switch, although such structural changes have yet to be studied in detail (Hara et al. 2010; Kim et al. 2014). REV7 is required for proper DNA translesion synthesis and double-strand break repair (Prakash et al. 2005; Kolas and Durocher 2006; Sale 2015). Loss of REV7 results in PARP inhibitor resistance in cancer, highlighting the importance of its functions (Xu et al. 2015). HORMAD proteins align meiotic chromosomes for efficient homologous recombination (Handel and Schimenti 2010). It will be interesting to determine how binding of H3 by these proteins influences their functions. Perhaps, for example, loss of H3–REV7 interactions might also contribute to PARP inhibitor resistance.
H3 is subject to lysine methylation at a number of other sites, and lysines are subject to multiple types of post-translational modifications (PTMs) in addition to methylation. Lysine methylation and acetylation are also subject to regulation by other modifications, such as phosphorylation or ubiquitination, at other sites (Latham and Dent 2007). Given the multiple kinases and phosphorylation events required for proper execution of the SAC, it will be important to determine whether such cross-talk occurs between H3K4me and these events and how such cross-talk impacts binding to Mad2 with histone H3.
Misregulation of histone modifications or the SAC is associated with severe human disease conditions, including cancer (Sharma et al. 2010; Fang and Zhang 2011). Several MLL proteins highly homologous to yeast Set1 exist in humans. Translocations of Mll1, in particular, that result in fusion of MLL to various members of the superelongation complex are associated with human leukemias (Smith et al. 2011). Although Mll1 functions in both normal and disease cells are usually thought of in terms of transcription, our work indicates that these proteins may also be important to maintain normal chromosome ploidy through regulation of the SAC.
Materials and methods
Yeast strains, media, and culture conditions
All strains generated for this study are isogenic to BY4741 (Supplemental Table S1). Cells were grown in YPD medium at 30°C under standard conditions unless otherwise stated.
Benomyl resistance assays
Plates were prepared with the indicated amounts of benomyl dissolved in DMSO. Wild-type and mutant strains were grown overnight in rich medium and placed onto either control plates (YPD or YPD with DMSO) or plates containing increasing amounts of benomyl (10, 20, 30, and 40 µg/mL). Plates were placed for 2 d at 30°C unless otherwise stated.
Flow cytometry
Five milliliters of culture of mid-log phase cells was pelleted and resuspended in 3 mL of dH2O. Seven milliliters of 95% ethanol was added to the culture and incubated overnight at 4°C. Cells were pelleted and washed in 5 mL of sodium citrate (pH 7.4). Cells were pelleted, resuspended in 1 mL 50 mM sodium citrate with 0.25 mg/mL RNase, and incubated for 1 h at 50°C. Proteinase K was then added for a final concentration of 1 mg/mL and incubated for an additional hour at 50°C. One milliliter of sodium citrate containing 16 µg/mL propidium iodide was added for a final concentration of 8 µg/mL and incubated for 30 min before flow cytometry analysis. Data were collected on a BD LSRFortessa (Becton Dickinson) and analyzed using FlowJo 7.6 software. The protocol was courtesy of Stephen P. Bell and can be found at http://web.mit.edu/flowcytometry/www.
Immunofluorescence
Immunofluorescence experiments were performed according to standard techniques (Burke et al. 2000). To visualize the mitotic spindle and nuclei, we used an anti-tubulin antibody (1:500; Millipore, CBL270) and 1 µM DAPI (Molecular Probes). To visualize GST-Mad2, cells were grown in YEP + 2% raffinose until mid-log phase. GST-Mad2 protein expression was induced by the addition of galactose to a final concentration of 2% for 30 min. We used an anti-GST antibody (1:500; GE, 27-4577-01) for immunofluorescence staining. Following staining procedures, images of wild-type and mutant yeast cells were acquired with a LSM510-Meta confocal microscope (Carl Zeiss, Germany) using a 100× PlanApochromat/1.4 N.A. objective. Images were processed using ImageJ software (National Institutes of Health) and Adobe Photoshop.
Nocodazole arrest and release
Cells were diluted into YPD medium from overnight cultures and allowed to grow until a density of 107 cells per milliliter. Nocodazole was added to a final concentration of 15 µg/mL, and the cells were allowed to arrest for 2 h. Cells were released from the G2/M arrest by washing twice in dH2O, resuspended into fresh YPD medium, and collected at the indicated time points.
Recombinant protein generation and purification
Budding yeast genes were cloned into a pGEX4T1 vector to generate an N-terminally tagged GST fusion proteins. The plasmids used in this study are listed in Supplemental Table S2. Mutations were generated by using the QuikChange XL kit (Stratagene) and sequenced. Proteins were expressed in the Escherichia coli strain Rosetta 2 (Novagen) by the addition of 0.2 mM IPTG for 20 h at 14°C. After protein expression, cells were collected and suspended in lysis buffer (50 mM Tris at pH 7.5, 150 mM NaCl, 0.05% NP-40, 1 mM PMSF, protease inhibitors, 0.5 mg/mL lysozyme solution). The cells were incubated for 45 min at 4°C and lysed using a EpiShear Probe sonicator (Active Motif). Lysed cells were centrifuged, and the supernatant was removed and incubated with glutathione sepharose 4B (GE). Recombinant GST fusion proteins were eluted by the addition of 15 mg/mL L-glutathione (Sigma) dissolved in 100 mM Tris (pH 8.0). Recombinant proteins were run on a 4%–12% BisTris gel and analyzed with colloidal staining.
Calf thymus histone-binding assays
Calf thymus histone-binding assays were performed by incubating 50 µg of calf thymus histones (Worthington) with 10 µg of recombinant GST fusion proteins in buffer (50 mM Tris at pH 7.5, 1 M NaCl, 1% NP-40) overnight. GST fusion protein histone interactions were assessed by adding glutathione sepharose 4B (GE) beads for 1 h and washing five times in buffer. The beads were resuspended in 5× SDS sample buffer, run on a 4%–12% Tris-Bis gel, and transferred to a nitrocellulose membrane followed by Western blot analysis. We used the following antibodies: anti-H3 (1:5000; Active Motif, 39163), anti-H2A (1:2000; Millipore, 07-146), and anti-H2B (1:2500; Millipore, 07-371).
MLA histone-binding assays
Similar to the calf thymus histone-binding assay, 2 µg of recombinant GST fusion proteins was incubated with 1 µg of full-length recombinant histone H3 with either no methylation on K4 or methylated lysine analogs (K4me1, K4me2, and K4me3) (Active Motif). GST-yMad2 fusion proteins were incubated in 50 mM Tris (pH 7.5), 1 M NaCl, and 1% NP-40, and GST-hMad2 proteins were incubated in 50 mM Tris (pH 7.5), 750 mM NaCl, and 1% NP-40 overnight. Glutathione sepharose 4B (GE) beads were added for 1 h, washed five times with the indicated buffer, and resuspended in 5× SDS sample buffer. Proteins were resolved on a gel, and interactions were assessed by transferring the proteins to a nitrocellulose membrane followed by Western blot analysis.
Histone H3.1 purification and binding assay
A gene encoding human H3.1 was cloned into the pET-30a(+) vector to create H3.1 C-terminally tagged with a 6xHIS epitope. Mutations were generated by using the QuikChange XL kit (Stratagene) and sequenced. Plasmids used in this assay are listed in Supplemental Table S2. Proteins were expressed in the E. coli strain Rosetta 2 (Novagen) by the addition of 0.2 mM IPTG for 16 h at 20°C. After protein expression, cells were collected and suspended in buffer (50 mM Tris at pH 8.0, 500 mM NaCl, 5% glycerol, 1 mM PMSF). The cells were disrupted using an EpiShear Probe sonicator (Active Motif). Cells were then pelleted and resuspended in denaturing buffer (50 mM Tris at pH 8.0, 500 mM NaCl, 5% glycerol, 6 M urea). The mixture was centrifuged, and the supernatant was removed and incubated with Ni-NTA agarose beads (Qiagen). Recombinant H3.1-6xHIS fusion proteins were eluted by the addition of 150 mM imidazole added to buffer containing 50 mM Tris (pH 8.0), 500 mM NaCl, and 5% glycerol. Eluates were run on a 4%–12% BisTris gel and evaluated with colloidal staining. Purified recombinant proteins were dialyzed overnight into buffer containing 20 mM Tris (pH 8.0), 5 mM DTT, 1 mM PMSF, 1 mM EDTA, 500 mM NaCl, and 5% glycerol to remove urea. Purified proteins were then used in binding assays. Two micrograms of recombinant GST fusion protein was incubated with 2 µg of full-length recombinant histone H3 in buffer (50 mM Tris at pH 7.5, 750 mM NaCl, 0.1%NP-40) overnight. Glutathione sepharose 4B (GE) beads were added to the binding assay for 1 h, washed five times with buffer, and resuspended in 5× SDS sample buffer. Proteins were resolved on a gel, and interactions were analyzed by transferring the proteins to a nitrocellulose membrane followed by Western blot analysis.
Supplementary Material
Acknowledgments
We thank Wendy Schoeber and Pamela Whitney for their help with flow cytometry and cell cycle analysis assays at the Science Park FACS core, partially supported by the Center for Environmental and Molecular Carcinogenesis. We are also thankful to Dr. Collene Jeter for her expertise in confocal microscopy, and Joi Holcomb for generating the illustrations. Last, we thank Dr. Sue Biggins for providing yeast strains, and Dr. Mark Bedford, Leah Gates, Dr. Bert W. O'Malley, Dr. Guillermo Garcia-Manero, and Dr. Toshiyuki Habu for providing reagents. We thank the Genetics Department Microscopy Core at University of Texas M.D. Anderson Cancer Center for the use of imaging equipment, and Henry Adams for his expertise in microscopy. We are grateful to the Schissler family and Foundation for supporting this research, and the Andrew Sowell-Wade Huggins Scholarship Fund. This work was supported by a Cancer Prevention Research Institute of Texas research training award (RP140106 to A.S.), a National Institutes of Health training award (5T32HD007325-23 to A.S.), National Institutes of Health grants CA132840 (to R.D.W.) and CA09717 (to R.D.W.), the Grady F Saunders, Ph.D. Distinguished Research Professorship (R.D.W.), the Centers of Environmental Carcinogenesis, and the Center for Cancer Epigenetics of the M.D. Anderson Cancer Center Institute of Basic Sciences. This study also made use of the Science Park Next-Generation Sequencing Core, supported by Cancer Prevention Research Institute of Texas grant RP120348.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.278887.116.
References
- Aravind L, Koonin EV. 1998. The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem Sci 23: 284–286. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL. 2002. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci 99: 8695–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Murray AW. 2001. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev 15: 3118–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD. 2001. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev 15: 3286–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Sherlock G, Myers CL, Burrows NM, Deng C, Wu HI, McCann KE, Troyanskaya OG, Brown JM. 2006. Global analysis of gene function in yeast by quantitative phenotypic profiling. Mol Syst Biol 2: 2006 0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke D, Dawson D, Stearns T, Cold Spring Harbor Laboratory. 2000. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Dai J, Hyland EM, Yuan DS, Huang H, Bader JS, Boeke JD. 2008. Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell 134: 1066–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehe PM, Dichtl B, Schaft D, Roguev A, Pamblanco M, Lebrun R, Rodriguez-Gil A, Mkandawire M, Landsberg K, Shevchenko A, et al. 2006. Protein interactions within the Set1 complex and their roles in the regulation of histone 3 lysine 4 methylation. J Biol Chem 281: 35404–35412. [DOI] [PubMed] [Google Scholar]
- Fang X, Zhang P. 2011. Aneuploidy and tumorigenesis. Semin Cell Dev Biol 22: 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. 1998. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev 12: 1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel MA, Schimenti JC. 2010. Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat Rev Genet 11: 124–136. [DOI] [PubMed] [Google Scholar]
- Hara K, Hashimoto H, Murakumo Y, Kobayashi S, Kogame T, Unzai S, Akashi S, Takeda S, Shimizu T, Sato M. 2010. Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase ζ and REV1. J Biol Chem 285: 12299–12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT. 1991. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66: 507–517. [DOI] [PubMed] [Google Scholar]
- Huyen Y, Zgheib O, Ditullio RA Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. 2004. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432: 406–411. [DOI] [PubMed] [Google Scholar]
- Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. 1998. Budding yeast Cdc20: a target of the spindle checkpoint. Science 279: 1041–1044. [DOI] [PubMed] [Google Scholar]
- Jia L, Kim S, Yu H. 2013. Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem Sci 38: 302–311. [DOI] [PubMed] [Google Scholar]
- Juang YL, Huang J, Peters JM, McLaughlin ME, Tai CY, Pellman D. 1997. APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science 275: 1311–1314. [DOI] [PubMed] [Google Scholar]
- Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. 2006. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep 7: 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Rosenberg SC, Kugel CL, Kostow N, Rog O, Davydov V, Su TY, Dernburg AF, Corbett KD. 2014. The chromosome axis controls meiotic events through a hierarchical assembly of HORMA domain proteins. Dev Cell 31: 487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmizis A, Santos-Rosa H, Penkett CJ, Singer MA, Vermeulen M, Mann M, Bahler J, Green RD, Kouzarides T. 2007. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature 449: 928–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura E, Tanaka K, Kitamura Y, Tanaka TU. 2007. Kinetochore microtubule interaction during S phase in Saccharomyces cerevisiae. Genes Dev 21: 3319–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolas NK, Durocher D. 2006. DNA repair: DNA polymerase ζ and Rev1 break in. Curr Biol 16: R296–R299. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A. 2002. COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277: 10753–10755. [DOI] [PubMed] [Google Scholar]
- Latham JA, Dent SY. 2007. Cross-regulation of histone modifications. Nat Struct Mol Biol 14: 1017–1024. [DOI] [PubMed] [Google Scholar]
- Latham JA, Chosed RJ, Wang S, Dent SY. 2011. Chromatin signaling to kinetochores: transregulation of Dam1 methylation by histone H2B ubiquitination. Cell 146: 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, Bhaumik SR, Shilatifard A. 2007. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell 131: 1084–1096. [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. 1991. Feedback control of mitosis in budding yeast. Cell 66: 519–531. [DOI] [PubMed] [Google Scholar]
- Li X, Nicklas RB. 1995. Mitotic forces control a cell-cycle checkpoint. Nature 373: 630–632. [DOI] [PubMed] [Google Scholar]
- Lim HH, Goh PY, Surana U. 1998. Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr Biol 8: 231–234. [DOI] [PubMed] [Google Scholar]
- Luo X, Fang G, Coldiron M, Lin Y, Yu H, Kirschner MW, Wagner G. 2000. Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat Struct Biol 7: 224–229. [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Rizo J, Yu H. 2002. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell 9: 59–71. [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H. 2004. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol 11: 338–345. [DOI] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A. 2001. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci 98: 12902–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb SJ, Wang Z, Ruthenburg AJ, Elenitoba-Johnson KS, Roeder RG, et al. 2010. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell 38: 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniyappa K, Kshirsagar R, Ghodke I. 2014. The HORMA domain: an evolutionarily conserved domain discovered in chromatin-associated proteins, has unanticipated diverse functions. Gene 545: 194–197. [DOI] [PubMed] [Google Scholar]
- Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML. 2002. A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci 99: 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S, Lee JS, Gardner KE, Gardner JM, Takahashi YH, Chandrasekharan MB, Sun ZW, Osley MA, Strahl BD, Jaspersen SL, et al. 2009. Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J Cell Biol 186: 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K. 2005. How do so few control so many? Cell 120: 739–746. [DOI] [PubMed] [Google Scholar]
- Nezi L, Rancati G, De Antoni A, Pasqualato S, Piatti S, Musacchio A. 2006. Accumulation of Mad2–Cdc20 complex during spindle checkpoint activation requires binding of open and closed conformers of Mad2 in Saccharomyces cerevisiae. J Cell Biol 174: 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, Johnson RE, Prakash L. 2005. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 74: 317–353. [DOI] [PubMed] [Google Scholar]
- Pui CH, Behm FG, Downing JR, Hancock ML, Shurtleff SA, Ribeiro RC, Head DR, Mahmoud HH, Sandlund JT, Furman WL, et al. 1994. 11q23/MLL rearrangement confers a poor prognosis in infants with acute lymphoblastic leukemia. J Clin Oncol 12: 909–915. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Schultz A, Cole R, Sluder G. 1994. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol 127: 1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger KJ, El-Alama M, Stein G, Bradshaw C, Slonimski PP, Maundrell K. 1999. Chemotyping of yeast mutants using robotics. Yeast 15: 973–986. [DOI] [PubMed] [Google Scholar]
- Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF. 2001. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J 20: 7137–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE. 2015. REV7/MAD2L2: the multitasking maestro emerges as a barrier to recombination. EMBO J 34: 1609–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. 2002. Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411. [DOI] [PubMed] [Google Scholar]
- Schneider J, Wood A, Lee JS, Schuster R, Dueker J, Maguire C, Swanson SK, Florens L, Washburn MP, Shilatifard A. 2005. Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol Cell 19: 849–856. [DOI] [PubMed] [Google Scholar]
- Sethi N, Monteagudo MC, Koshland D, Hogan E, Burke DJ. 1991. The CDC20 gene product of Saccharomyces cerevisiae, a β-transducin homolog, is required for a subset of microtubule-dependent cellular processes. Mol Cell Biol 11: 5592–5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA. 2010. Epigenetics in cancer. Carcinogenesis 31: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, et al. 2006. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442: 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81: 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi L, Mapelli M, Knapp S, De Antoni A, Jeang KT, Musacchio A. 2002. Crystal structure of the tetrameric Mad1–Mad2 core complex: implications of a ‘safety belt’ binding mechanism for the spindle checkpoint. EMBO J 21: 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Lin C, Shilatifard A. 2011. The super elongation complex (SEC) and MLL in development and disease. Genes Dev 25: 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares LM, Radman-Livaja M, Lin SG, Rando OJ, Buratowski S. 2014. Feedback control of Set1 protein levels is important for proper H3K4 methylation patterns. Cell Rep 6: 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Lin W, Soustelle C, Suhre K, Nicolas A, Geli V, de La Roche Saint-Andre C. 2004. Set1 is required for meiotic S-phase onset, double-strand break formation and middle gene expression. EMBO J 23: 1957–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer F, Gerring SL, Connelly C, Hieter P. 1990. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics 124: 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns T, Hoyt MA, Botstein D. 1990. Yeast mutants sensitive to antimicrotubule drugs define three genes that affect microtubule function. Genetics 124: 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubrahmanyam S, Hwang WW, Meneghini MD, Tong AH, Madhani HD. 2007. Genome-wide, as opposed to local, antisilencing is mediated redundantly by the euchromatic factors Set1 and H2A.Z. Proc Natl Acad Sci 104: 16609–16614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Nakajima Y, Westermann S, Shang C, Kang JS, Goodner C, Houshmand P, Fields S, Chan CS, Drubin D, et al. 2007. A protein interaction map of the mitotic spindle. Mol Biol Cell 18: 3800–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. 2005. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121: 859–872. [DOI] [PubMed] [Google Scholar]
- Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H. 2004. Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. EMBO J 23: 3133–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. 2015. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521: 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. 2006. Structural activation of Mad2 in the mitotic spindle checkpoint: the two-state Mad2 model versus the Mad2 template model. J Cell Biol 173: 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeleznik-Le NJ, Harden AM, Rowley JD. 1994. 11q23 translocations split the ‘AT-hook’ cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc Natl Acad Sci 91: 10610–10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Lin W, Latham JA, Riefler GM, Schumacher JM, Chan C, Tatchell K, Hawke DH, Kobayashi R, Dent SY. 2005. The Set1 methyltransferase opposes Ipl1 aurora kinase functions in chromosome segregation. Cell 122: 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.