

Abstract
Motivation: Identification of altered pathways that are clinically relevant across human cancers is a key challenge in cancer genomics. Precise identification and understanding of these altered pathways may provide novel insights into patient stratification, therapeutic strategies and the development of new drugs. However, a challenge remains in accurately identifying pathways altered by somatic mutations across human cancers, due to the diverse mutation spectrum. We developed an innovative approach to integrate somatic mutation data with gene networks and pathways, in order to identify pathways altered by somatic mutations across cancers.
Results: We applied our approach to The Cancer Genome Atlas (TCGA) dataset of somatic mutations in 4790 cancer patients with 19 different types of tumors. Our analysis identified cancer-type-specific altered pathways enriched with known cancer-relevant genes and targets of currently available drugs. To investigate the clinical significance of these altered pathways, we performed consensus clustering for patient stratification using member genes in the altered pathways coupled with gene expression datasets from 4870 patients from TCGA, and multiple independent cohorts confirmed that the altered pathways could be used to stratify patients into subgroups with significantly different clinical outcomes. Of particular significance, certain patient subpopulations with poor prognosis were identified because they had specific altered pathways for which there are available targeted therapies. These findings could be used to tailor and intensify therapy in these patients, for whom current therapy is suboptimal.
Availability and implementation: The code is available at: http://www.taehyunlab.org.
Contact: jhcheong@yuhs.ac or taehyun.hwang@utsouthwestern.edu or taehyun.cs@gmail.com
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
In the last few years, studies using high-throughput technologies have highlighted the fact that the development and progression of cancer hinges on somatic alterations. These somatic alterations may disrupt gene functions, such as activating oncogenes or inactivating tumor suppressor genes, and thus dysregulate critical pathways contributing to tumorigenesis. Therefore, precise identification and understanding of disrupted pathways may provide insights into therapeutic strategies and the development of novel agents. Many large-scale cancer genomics studies, such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC), have performed integrated analyses to draft an overview of somatic alterations in the cancer genome (Kandoth et al., 2013; Lawrence et al., 2013, 2014; Tamborero et al., 2013). Many of these studies have reported novel candidate cancer genes mutated at high and intermediate frequencies in specific cancers as well as across many cancer types (Lawrence et al., 2014). However, it is still a challenge to translate somatic mutations in tumors into the pathway model for clinical use (Baselga, 2011; Osmanbeyoglu et al., 2014). Recently, in order to improve the clinical relevance and utility of somatic mutation analyses, Hopfree et al. (2013) proposed integrating somatic mutation data with molecular interaction networks for patient stratification. They demonstrated that inclusion of prior knowledge, captured in molecular interaction networks, could improve identification of patient subgroups with significantly different histological, pathological or clinical outcomes and discover novel cancer-related pathways or subnetworks. In a similar manner, other network-based methods have demonstrated that incorporating molecular networks and/or biological pathways can improve accuracy in identifying cancer-related pathways (Cerami et al., 2010; Hwang et al., 2013; Vandin et al., 2011; Vaske et al., 2010).
One limitation of these network-based methods is that they are not designed to fully utilize large-scale somatic mutation data from multiple cancer types to determine which particular pathways are altered by somatic mutations across a range of human cancers. In addition, due to the incomplete knowledge of existing gene sets and/or pathway databases, these methods are limited in detecting pathways based on a number of altered genes annotated in existing gene set and pathway databases. Alternatively, the methods that build pathways de novo without incorporating biological prior knowledge can be applicable to detecting altered pathways, but these methods were also not designed to detect cancer-type specific or commonly altered pathways. To address these, we developed an algorithm named NTriPath (Network regularized sparse non-negative TRI matrix factorization for PATHway identification) to integrate somatic mutation, gene–gene interaction networks and gene set or pathway databases to discover pathways altered by somatic mutations in 4790 cancer patients with 19 different types of cancers. Incorporating existing gene set or pathway databases enables NTriPath to report a list of altered pathways across cancers, and make it easy to determine/compare which particular pathways are altered in a particular cancer type(s). In particular, the use of the large-scale genome-wide somatic mutations from 4790 cancer patients enables NTriPath to explore modular structures of mutational data within a cancer type and/or across multiple cancer types (using matrix factorization) to identify cancer-type-specific or commonly altered pathways. In addition, the use of gene–gene interaction networks with somatic mutation and pathway databases enables NTriPath to classify genes, which were not annotated in existing pathway databases, as new member genes of the identified altered pathways based on connectivity in the gene–gene interaction networks.
The questions that we investigate here are:
whether large-scale integrative somatic mutation analysis that integrates somatic mutations across many cancer types with the gene–gene interaction networks and pathway database can reliably identify cancer-type-specific or common pathways altered by somatic mutations across cancers;
whether the identified pathways can be used as a prognostic biomarker for patient stratification—with the assumption that the altered pathways contribute to cancer development and progression and, thus, impact survival.
In these experiments, we demonstrated that the cancer-type-specific and commonly altered pathways identified by NTriPath are biologically relevant to the corresponding cancer type and are associated with patient survival outcomes. We also showed that cancer-specific altered pathways are enriched with many known cancer-relevant genes and targets of available drugs, including those already FDA-approved. These results imply that the cancer-specific altered pathways can guide therapeutic strategy to target the altered pathways that are pivotal in each cancer type.
2 Methods
In this section, we first describe the notations for the data. We then review non-negative matrix tri-factorization (NMTF) and introduce the framework of network regularized sparse non-negative tri-matrix factorization for pathway identification.
2.1 Notations
We construct a binary data matrix from the mutation data, where n is the number of patients, m is the number of genes and the element of the matrix , is 1 if the ith patient has a mutation on the jth gene, 0 otherwise. We construct a binary matrix denoting a patient cluster, where k1 indicates the number of cancer types and indicates the ith patient has jth cancer type. We derive the adjacency matrix from the human gene–gene interaction networks and denote it as , where if the ith gene is interacting with the jth genes in the networks and 0 otherwise. We define the graph Laplacian matrix by , where each diagonal element in the diagonal matrix is given by . We construct a binary matrix from the specific pathway database denoting pathway information, where k2 is the number of pathways and if the ith gene is annotated in the jth pathway as a member in the pathway database, otherwise 0. Since the current pathway database annotation is still incomplete, we define a matrix denoting newly updated pathway information, including newly added member genes by NTriPath. We define a matrix denoting cancer type and pathway associations, where each element of represents the associations between the ith cancer type with the jth pathway. Higher values of elements indicate stronger associations between cancer types and pathways. The objective is to derive newly updated pathway information and cancer-type and pathway associations based on and (Fig. 1).
Fig. 1.
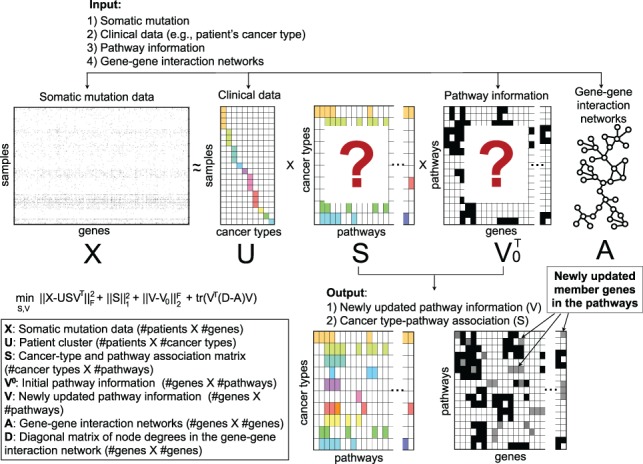
Network regularized sparse non-negative tri-matrix factorization for pathway identification. The binary mutation matrix is factorized into products of three matrices, patient cluster , pathway information and cancer-type and pathway association . Prior knowledge is introduced from gene–gene interaction networks
2.2 Non-negative matrix tri-factorization
NMTF aims to approximate a data matrix by the product of three matrices, such that , where and . Since the aim of our study is to discover altered pathways by somatic mutations across cancers, we can define the objective function to estimate the factor matrix , as
| (1) |
where is the Frobenius norm of matrix , and , and denote somatic mutation data from patients, patient’s cancer type, cancer-type and pathway associations, and cancer-related pathways, respectively.
A limitation of this approach in Equation (1) is the sparsity of somatic mutation matrix (>98% of entries are 0) used to predict cancer-type and pathway associations. Thus, the Frobenius norm in Equation (1) might not be appropriate to evaluate the goodness of the decomposition models since it is dominated by the errors on 0 entries when the data matrix is sparse. In addition, due to the incompleteness of current pathway database annotation, the predicted cancer-type and pathway associations might be biased toward pathways containing mutated genes like those currently annotated in existing pathway databases. A recent study suggests that incorporating biological prior knowledge, such as gene–gene interaction networks, as a regularization term to NMTF could help to more accurately identify new member genes in the existing pathways, as well as an association matrix (Hwang et al., 2012).
2.3 Network regularized sparse non-negative tri-matrix factorization model
To address the above problems, we developed an approach called NTriPath (Network regularized sparse non-negative TRI matrix factorization for PATHway identification) to handle the sparsity of the somatic mutation matrix and the incompleteness of current pathway database annotation by incorporating the prior knowledge from human gene–gene interaction networks.
We define a weighted loss function to deal with the sparseness of the somatic mutation data matrix . The weighted loss function enables us to focus on approximating errors at nonzero entries (i.e. somatic mutation). We introduce the graph Laplaican and the initial pathway information , derived from human gene–gene interaction networks and current pathway databases, respectively.
Then we define a new objective function as follows:
| (2) |
where is a weight matrix where if otherwise 0. The operator represents the element-wise multiplication. A regularization term Ω for and is as follows:
| (3) |
where denotes user-specific parameters and denotes the -norm of , which is equal to the sum of the absolute values of all the entries of . The third term in the regularization term (3) is introduced as a supervised way of minimizing the squared loss between the predicted newly updated pathway information and the initial pathway information . The fourth term introduces the graph Laplacian derived from the gene–gene interaction networks as prior knowledge to guide the clustering of the genes. This term is called the smoothness term, which encourages the connected nodes (genes) in a graph to be assigned to the same cluster (pathway).
Algorithm 1 NTriPath
1: procedure NTriPath(,λS,λV,)
2: Initialization Set and , where is a matrix with all ones. Set the user specified parameters . (In our experiments, we used and .)
3: while not converged do (Iteratively update and .)
| (4) |
| (5) |
where is set to κ if and , otherwise 0, and
4: end while
5: return .
6: end procedure
To estimate the optimal solutions of our minimization problem, we adapt the multiplicative update method (Lee and Seung, 2001), which can be derived from the gradient of the objective function (2) with respect to each factor matrix. The following are the update rules for the factor matrices, and :
| (6) |
| (7) |
where ).
However, one of the issues of the multiplicative update rules is that when an entry in the factors becomes 0, it is forever stuck at 0. In theory, the entries should never become 0 provided that all the entries in the factors are initialized with positive values. However, due to the finite precision of calculations, 0 entries may well appear in practice. To avoid this inadmissible zeros problem, we examine the Karush–Kuhn–Tucker (KKT) conditions for the solution in each update and then replace the inadmissible zeros entries with a small positive number κ (Chi and Kolda, 2012; Seung-Jun et al., 2012). Note that the KKT conditions for the factor matrix in Equation (2) can be written in an element-wise form:
| (8) |
where and are the numerator and the denominator of the multiplicative factor in Equation (6), respectively:
| (9) |
| (10) |
The KKT conditions state that if Sij > 0, the multiplicative factor should be equal to 1; otherwise it should be ≤1. Thus, we replace the 0 entry whose corresponding multiplicative factor is >1 with κ to prevent the inadmissible 0 from occurring. The complete NTriPath algorithm with the choice of parameters used in the experiments is outlined in Algorithm 1. We have also proven the convergence of the algorithm. See Supplementary Information for the detailed proof of algorithm correctness and convergence. Empirically, the algorithm converges fast within 50 iterations in the experiments.
3 Results
We conducted simulation experiments using synthetic datasets to investigate the performance of NTriPath to discover cancer-type-specific altered pathways and identify new member genes in the pathways. Then we performed experiments with TCGA mutation profiles to identify cancer-type-specific altered pathways across cancers. In the experiments using TCGA datasets, we first ran NTriPath to identify cancer-type-specific altered pathways across cancers. To investigate the clinical relevance of the identified cancer-type-specific pathways, we collected gene expression data from TCGA and independent datasets and performed consensus clustering using the member genes in the identified cancer-type specific pathways for patient stratification.
3.1 Simulation
We generated synthetic mutation datasets and performed experiments using NTriPath. Specifically, we generated synthetic mutation data containing five patient subgroups and 10 pathways. Each subgroup included between one and seven altered pathways. We generated the gene–gene interaction networks and member genes in the pathway were densely connected with each other in the networks. We introduced a higher mutation rate to one of the subgroups to investigate whether different mutation rates for each subgroup would affect the performance of NTriPath to discover cancer-type-specific altered pathways. Experimental results indicated that NTriPath could discover subgroup-specific altered pathways and new member genes in the altered pathways (Supplementary Figs. S1 and S3). Additional experiments using large-scale experiments (e.g. 12 000 genes) and with different mutation rates also indicated that NTriPath could accurately identify subgroup-specific altered pathways (Supplementary Table S1). We summarize the results of the simulation in Supplementary information.
3.2 TCGA somatic mutation profiles, pathway database and gene–gene interaction networks preparation
3.2.1 Somatic mutation, gene–gene interaction networks and pathway database
We collected the somatic mutation data (e.g. Mutation Annotation Format files) for 4790 patients and 19 different cancer types from the TCGA data portal on May 19, 2013 (strel’tsov et al., 2001) (see Supplementary Table S1). Then we generated a binary matrix of 4790 patients × 25168 genes, with 1 indicating a mutation and 0 no mutation. We constructed a matrix representing the gene–gene interaction networks by combining networks from Zhang et al. (2011), the Human Protein Reference Database (December 2013) (Keshava Prasad et al., 2009) and Rossin et al. (2011). The matrix contains 63898 binary undirected interactions between 12456 genes. We collected four sets of pathways: (1) 4620 conserved subnetworks from the human gene–gene interaction network (Suthram et al., 2010), (2) 186 KEGG, (3) 217 Biocarta and (4) 430 Reactome gene sets from MsigDB (September 2010) (Subramanian et al., 2005) to generate a matrix representing the initial pathway information. After preprocessing (i.e. removing genes not present in both the somatic mutation and the gene–gene interaction networks), we generated a dataset for 4970 patients in 19 cancer types with 11089 genes.
3.3 Cancer-type-specific altered pathway identification
We applied NTriPath to somatic mutation data from TCGA for 4790 patients and 19 different cancer types. NTriPath produced two matrices as output; (1) newly updated pathways altered by mutated genes and (2) altered pathways by cancer type matrix . We used matrix to identify cancer-type-specific altered pathways across cancers. Specifically, we ranked pathways based on values of elements of the matrix for each cancer type (e.g. rank pathways based on all values of the ith row which indicate association scores between the ith cancer type and all pathways). In addition, to measure the statistical significance of cancer-type and pathway associations, we performed a permutation test (e.g. we randomly permuted somatic mutation data and repeated the experiments 5000 times to calculate empirical P-values) and defined cancer-type-specific altered pathways based on the following strict criteria: (1) pathways must be ranked within the top Kth compared with other pathways in each cancer type based on their association scores in matrix S; and (2) pathways must have significant BH-adjusted P-values (Benjamini-Hochberg-adjusted P-values using a false discovery rate cutoff of 0.1) (see Supplementary Table S2).
3.4 Biological interpretation for the cancer-type-specific altered pathways
In each cancer type, we selected the top Kth ranked altered pathways by statistical significance from NTriPath to generate cancer-type-specific altered pathways. NTriPath accurately identified cancer-type-specific altered pathways that are biologically relevant for each type of cancer using KEGG, Biocarta and Reactome pathway datasets (Supplementary Table S3). However, those current pathway databases cover only a small fraction of human genes (e.g. there are 1267, 5267 and 4159 genes in Biocarta, KEGG and Reactome pathway databases from MSigDB Sept. 2010). To address this problem, we used 4620 conserved subnetworks which cover 8470 genes as additional pathway data. Thus, we reported the results of NTriPath using 4620 conserved subnetworks for further analysis. In each cancer type, we selected the top three ranked altered pathways by statistical significance from NTriPath with the 4620 subnetwork modules to generate cancer-type-specific altered pathways (Supplementary Table S4).
Interestingly, NTriPath was able to find altered pathways containing not only genes that were frequently mutated but also genes that were mutated in a small subset of patients in each cancer type (Supplementary Table 3 and Fig. S1). Gene set enrichment analysis using the genes from the top three altered pathways showed that the altered pathways are significantly enriched with well-known cancer-related genes from the COSMIC database (Forbes et al., 2015) and known drug target genes as well as cancer-relevant biological processes (Supplementary Tables S5 and S6).
Focusing on kidney renal clear cell carcinoma (KIRC) as a proof of concept, NTriPath identified the pathway consisting of VHL, USP33, DIO2, TCEB1 and TCEB2 as the top-ranked altered pathway in KIRC (Fig. 2a). The VHL (von-Hippel Lindau) gene is a well-known tumor suppressor associated with KIRC, and is frequently mutated in patients with KIRC (Nickerson et al., 2008; Peña-Llopis and Brugarolas, 2013; Peña-Llopis et al., 2012; Sato et al., 2013). VHL was the most frequently mutated gene in TCGA KIRC, with 55.7% of patients harboring mutations in the gene. TCEB1 is mutated at very low frequency in the TCGA KIRC cohort. A recent study found that TCEB1 is mutated in ∼3% of the KIRC patients without VHL inactivation, and found TCEB1 preventing the binding of Elongin C to VHL, which inactivates the VHL pathway (Sato et al., 2013). The second highest ranked pathway contained EP300 and TP53. EP300 and TP53 were mutated in 8.1 and 5.2% of patients, respectively. EP300 has been identified as a co-activator of hypoxia-inducible factor 1 alpha, whose activation is a hallmark of KIRC tumors. TP53 was previously found to be associated with poor outcome in TCGA KIRC (The Cancer Genome Atlas Research Network, 2013). The third highest ranked pathway contains LRP1 and matrix metalloproteinases (MMPs) (MMP1, MMP7, MMP9, MMP26). LRP1 is mutated in 10% of the TCGA KIRC cohort, but matrix MMPs were not mutated in the TCGA KIRC cohort. Biological and clinical relevance of LRP1 mutation in KIRC has not been previously reported. MMPs have been implicated in different types of cancer progression, including the acquisition of invasive and metastatic properties in many cancer types. The aberrant expression of MMPs has been associated with poor patient survival and prognosis in KIRC patients (Gialeli et al., 2011; Hwang et al., 2013). Interestingly, recent studies suggested that LRP1 induces the expression of matrix MMPs and thus promotes cancer cell invasion and metastasis in many cancers, including KIRC (Duan et al., 1995; Langlois et al., 2010; Sato et al., 2013; Staudt et al., 2013).
Fig. 2.
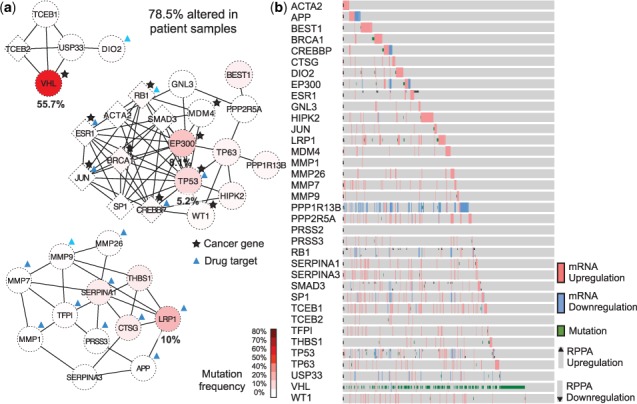
KIRC-specific altered pathways. (a) Diagrams of the top three ranked altered pathways in patients with KIRC. Red color indicates genes that are frequently mutated. A circular-shaped node represents the original member genes annotated in the pathway database, and a diamond-shaped node represents newly identified member genes of the pathways by NTriPath. (b) Protein and mRNA expression and mutation status for all genes identified in the top three KIRC altered pathways. Each row represents a member gene in the TCGA KIRC-specific altered pathway, and each column represents a patient sample in the TCGA KIRC cohort
NTriPath identified many new member genes in the top ranked pathways, including TCEB2, JUN and SP1, as well as other tumor suppressors such as CREBBP, SMAD3, BRCA1 and RB1. These newly identified member genes by NTriPath were mutated at a very low frequency or not mutated at all in TCGA KIRC patients. Instead, these genes interacted with many frequently mutated genes in the networks and were often dysregulated at the mRNA and protein levels in many KIRC patients (Fig. 2b). For example, TCEB2, SP1 and JUN were not yet mutated but their expression was dysregulated in 7, 10 and 2% of TCGA KIRC patients, respectively. Previous studies have shown that dysregulation in TCEB2 is expected to disrupt the protein complex that ubiquitinates HIF1α, resulting in the same phenotype as VHL inactivation by mutation or promoter hypermethylation (Duan et al., 1995; Ohh et al., 2000; Tanimoto et al., 2000). In addition, SP1 and JUN were previously identified as major transcriptional regulators associated with signaling circuits to promote tumor growth and invasion in KIRC (Nickerson et al., 2008). Taken together, these results show the feasibility of NTriPath to identify altered pathways that are biologically relevant to KIRC, including known cancer genes mutated at a high or intermediate frequency in the patients, as well as genes mutated at a very low frequency or not mutated at all yet may be fundamental role in the development and/or progression of KIRC.
3.5 Patient stratification using cancer-type-specific altered pathways identified by NTriPath
We hypothesized that altered mRNA expression of the member genes in the cancer-type-specific altered pathways reflect the molecular basis underlying the patient clinical outcomes. This would allow us to use mRNA expression profiles of member genes in the altered pathways as gene signatures to stratify patients into subgroups with different clinical outcomes for each type of cancer. We first collected a dataset consisting of gene expression profiles from 3656 patients with their survival information from TCGA cohorts. Specifically, we collected TCGA RNA-seq data for 10 different cancer types, including KIRC, HNSC, SKCM and lower grade glioma, from cBioPortal using the CGDS MATLAB toolbox with RNA Seq V2 RSEM option (Gao et al., 2013). We collected microarray gene expression profiles for TCGA GBM from the TCGA dataportal (strel’tsov et al., 2001) and TCGA OV and two others from Zhang et al. (2013). We then used member genes in the top three ranked cancer-type-specific altered pathways to perform consensus clustering for each cancer type. We generated Kaplan–Meier (KM) curves based on the groups produced by consensus clustering and found that patient survival was significantly different among the groups (Fig. 3 and Supplementary Fig. S3). In TCGA KIRC, we found three patient subgroups (A–C), with Group C having the poorest survival. A log-rank test indicated that Groups A and C had significantly different survival outcomes (P-value = 1.840e − 08, Hazard ratio = 2.94) with median survival times of 41.9 months for Group A compared with 30.8 months for Group C (Fig. 3a). Experiments with other TCGA datasets, including those for BLCA, HNSC and SKCM, consistently showed that the use of member genes in cancer-type-specific altered pathways could serve as a prognostic biomarker for patient stratification (Fig. 3b–d, and Supplementary Figs S2 and S3). For comparison, we also attempted to cluster patients using significant frequently mutated genes previously identified by the TCGA Pan-Cancer study (Kandoth et al., 2013). The results of consensus clustering using the NTriPath-derived pathway signatures and the TCGA Pan-Cancer-derived mutated gene signatures showed that the results from NTriPath-derived pathway signatures had higher significance levels (based on P-value measured by the log-rank test across different K groups for consensus clustering) for BLAC, BRCA and KIRC, and comparable results for the GBM, HNSC and LUAD cancer types (Fig. 4). These findings suggested that NTriPath-derived altered pathways could be used as prognostic biomarkers for better patient stratification.
Fig. 3.
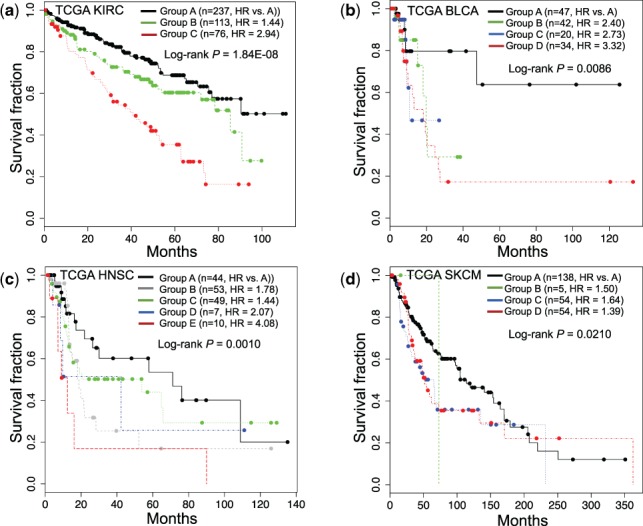
Cancer-type-specific altered pathways across cancers correlate with survival outcomes. KM survival plots based on patient subgroups defined by consensus clustering using genes from the top three altered pathways for (a) kidney renal cell carcinoma (KIRC), (b) bladder urothelial carcinoma (BLCA), (c) head and neck squamous carcinoma (HNSC) and (d) skin cutaneous melanoma (SKCM)
Fig. 4.

Comparing signatures with mutation-frequency-based signatures from TCGA Pan-Cancer. This figure compares patient stratification using signatures derived from NTriPath and mutation frequency reported in TCGA Pan-Cancer (Kandoth et al., 2013). In each subplot, the x-axis represents the number of cluster groups for consensus clustering (e.g. 3 means that we set cluster number as three groups and ran consensus clustering) and the y-axis represents −(P-value) calculated by the log-rank test. Higher values indicate more significant patient subgroup identification
3.6 Independent cohorts for the validation of the cancer-type-specific altered pathways.
We performed multiple validations to evaluate the robustness and the reproducibility of NTriPath. First, we evaluated the robustness of the cancer-type-specific altered pathways identified in the TCGA cohort for prognostic stratification. We generated gene expression profiles of 102 HNSC patients from our institution and used the member genes of the top three HNSC cancer-type-specific altered pathways in the TCGA cohort for patient stratification. In addition, we also used publically available gene expression data from two ovarian cancer datasets, one lung cancer dataset and three colon cancer datasets for a total of 1484 patients, and used the top three cancer-type specific altered pathways for the corresponding cancer type for independent validation. In the HNSC cohorts, we found six patient subgroups (A through F), with Group F patients having the poorest survival times (Fig. 5a). A log-rank test indicated that groups A and F had significantly different survival outcomes (P-value = 0.038, Hazard ratio = 1.88) with median survival times of 78.1 months for Group A and 26.7 months for Group F. Similarly, we found patient subgroups having significantly different survival outcomes in lung cancer, ovarian cancer and colorectal cancer (Fig. 5b–d and Supplementary Fig. S4). Second, we verified the reproducibility of NTriPath for the identification of the cancer-type-specific altered pathways. We collected the level 2 somatic mutation data from 19 human cancer types; those were updated after we collected the initial dataset used in the original experiments from the TCGA data portal. We found that there are 1891 newly updated patients’ mutation data from 15 cancer types (see Supplementary Table S6). We re-ran NTriPath to identify cancer-type-specific pathways across 19 cancers using 6681 patients’ somatic mutation data, including those of newly updated patients’ mutation data. Interestingly, we found that many top ranked pathways identified by NTriPath in the original experiments were consistently highly ranked in the new experiments (see Supplementary Table S7). These results may reassure the feasibility of the use of the altered pathways identified by NTriPath as potential prognostic signatures for identifying patient subgroups with different survival outcomes across multiple cancer types.
Fig. 5.
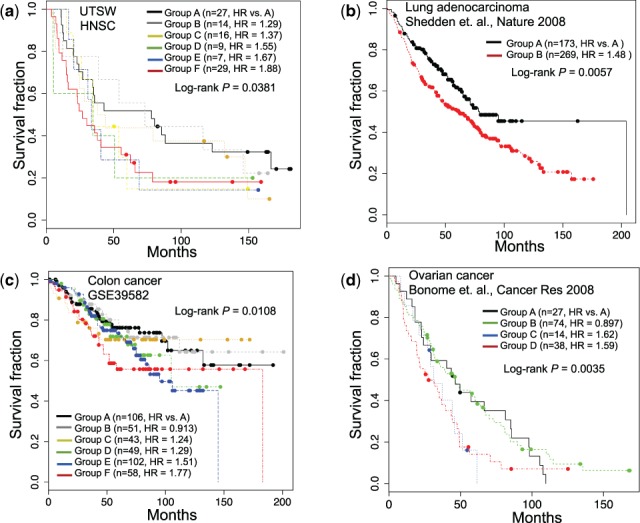
Independent cohorts for patient stratification using cancer-type-specific altered pathways. KM survival plots based on patient subgroups defined by consensus clustering using genes from the top three altered pathways for (a) UTSW HNSC, (b) lung adenocarcinoma, (c) colon cancer and (d) ovarian cancer
3.7 Identification of potential therapeutic targets in poor prognosis patient subgroups
We further investigated whether we could identify potential targets for therapy for the identified poor prognosis patient subgroups. Interestingly, we found that many known drug targets in the cancer-type specific altered pathways are often up-regulated in poor prognosis patient subgroups across cancers (Supplementary Table S8). For example, in TCGA KIRC cohort, MMP9, a target of the FDA-approved drug Captopril, was significantly up-regulated in the poor prognosis group compared with the good prognosis group (FDR-adjusted P-value < 0.05 with t-test). Captopril, an angiotensin-converting enzyme inhibitor, inhibits MMP9 expression (Jones et al., 2004; Okada et al., 2008; Williams et al., 2005; Yamamoto et al., 2008). Therefore, the use of Captopril might be recommended for renal cell carcinoma patients with high MMP9 levels. Another notable example includes DNA Topoisomerase I (TOP1), a target of well-known FDA-approved anticancer drugs such as Irinotecan and Topotecan, identified by NTriPath as a new member gene in the pathway containing TP53, EP300, AUKRA and CDK5. We found that TOP1 was up-regulated in poor prognosis subgroups in HNSC from both TCGA and UTSW cohorts. In addition, we also found that some patients with overexpression of TOP1 in the TCGA HNSC poor prognosis subgroup have developed therapy resistance against single chemotherapeutic agent such as Cisplatin. Interestingly, there is an ongoing trial in advanced HNSC showing efficacy of TOP1 inhibitor Irinotecan with Cisplatin in a poor prognosis patient subgroup (Gilbert et al., 2008). These observations may suggest the feasibility of TOP1 inhibitors-based combinations as an effective treatment option for HNSC patients with poor prognosis and/or therapy resistance, which should be evaluated further with multiple clinical samples.
4 Discussion
Systematic understanding of how somatic mutations influence clinical outcomes is essential for the development and application of personalized therapies. In particular, organizing alterations at the individual gene level and in the molecular pathways can correlate altered pathways and vulnerabilities with specific genetic lesions, and provide novel insights into cancer biology, biomarkers for patient stratification in clinical trials and potential targeted drug development (Garraway and Lander, 2013). Here, we systematically identified biological and clinical relevant cancer-type-specific pathways altered across multiple cancer types. In particular, the integration of somatic mutation with biological prior knowledge led to the identification of altered pathways that contain recurrently mutated genes as a hallmark of specific cancer types. We found that single gene expression analysis, in particular those of frequently mutated genes (e.g. TP53, EP300, BRCA1, etc.), in the commonly top-ranked pathways across many cancer types do not show clear separation into patient subgroups with different survival outcome. We also performed KM survival analysis based on mutation status of each of frequently mutated genes but did not find clear separation into patient subgroups with different survival outcomes either. Interestingly, we found that several genes, while not frequently mutated or not mutated at all in patients, were part of cancer-type-specific altered pathways that have been causally implicated in the development of corresponding cancer types, and significantly associated with clinical outcomes (Supplementary Fig. S5). For example, no mutation of MMP7 has been reported, but high expression of MMP7 [P-value = 0.00191, HR = 1.7 (95% CI 1.21–2.38)] is significantly associated with poor survival in TCGA KIRC patients. Other examples include CABLES1 [P-value = 0.00272, HR = 0.486 (95% CI 0.301–0.787)] in TCGA HNSC and LUAD, and GCH1 [P-value = 0.0000528, HR = 0.52 (95% CI 0.367–0.763)] in TCGA SKCM are not frequently or not mutated, but low or high expression of those genes are significantly associated with poor survival. In addition, we found that known drug targets are not frequently mutated but often up-regulated in poor prognosis patient subgroups across many cancers. These results further corroborate that the integrative analysis of somatic mutations with additional biological prior knowledge may elucidate potential candidate genes associated with clinical outcomes and could be potentially used to design targeted therapy, which cannot be readily identified by somatic mutation analysis alone. We performed additional experiments using member genes present only in the original pathway annotation to stratify patients and compared the performance of patient stratification to identify patient subgroups with different survival outcomes with pathway signatures containing new member genes identified by NTriPath. Interestingly, we found that the use of member genes in the pathways with additional new member genes identified by NTriPath improved patient stratification results overall (Supplementary Fig. S6). In our analysis, we did not remove synonymous mutations or further select a shorter list of recurrent mutated genes in cohorts with stringent criteria (Dees et al., 2012; Lawrence et al., 2013). We performed additional experiments exclusively using non-synonymous mutations to identify cancer-type-specific altered pathways to stratify patients into subgroups. Experimental results using pathway signatures identified by NTriPath with non-synonymous mutations only and with synonymous mutations to stratify patient subgroups showed comparable performance (Supplementary Fig. S7). These findings may suggest that NTriPath-derived altered pathways could lead to potential prognostic biomarkers for better patient stratification. However, the clinical utility of the altered pathways need to be rigorously validated independently in multiple clinical samples.
To evaluate the impact of different network resources, we used networks from the HPRD (Keshava Prasad et al., 2009) and Rossin et al. (2011) and repeated experiments. We summarize the results of altered pathways and patient stratification using different network resources and provide them on our supplement website. Lastly, NTriPath is a general computational algorithm and can be applied to other data types such as gene expression, copy number alteration and methylation to identify altered pathways by different types of genomic aberrations. NTriPath can also be used to find altered pathways across associated with other cancer-related phenotypes (e.g. patient groups having therapy resistance versus sensitivity, metastatic versus non-metastatic).
5 Conclusions
We have described an integrative somatic mutation analysis for discovering altered pathways in human cancers. NTriPath integrates somatic mutation data and prior biological knowledge from the pathway database and molecular networks to identify significantly altered pathways and their associations with specific cancer types. Specifically, NTriPath effectively utilizes mutation patterns that exist in only a subset of samples (or specific cancer types), thus revealing pathways altered by complex mutation patterns across cancer types. Furthermore, using gene–gene interaction networks and the pathway database provide the potential to identify altered pathways enriched with genes harboring mutations at high/intermediate frequencies, as well as those not mutated per se but nevertheless playing critical roles in tumorigenesis in network and pathway contexts. Thus, NTriPath is uniquely suited to provide a global analysis of altered pathways by somatic mutation across cancer types. We applied NTriPath to somatic mutation data from 19 types of cancers, and discovered cancer-type-specific altered pathways based on these mutations in human cancers. Functional enrichment analysis of cancer-type-specific pathways demonstrated that the identified cancer-type-specific altered pathways are biologically meaningful to each cancer type. It also provided unique pathway views of key biological processes underlying each cancer type. Of particular significance, we identified a patient subgroup with poor survival by cancer-type-specific altered pathway signatures from TCGA cohorts, which in independent cohorts. These results implied the potential utility of cancer-type-specific altered pathway signatures to serve as a guide to tailored treatment in a patient subgroup.
Funding
S.P., M.S., T.H.H. were supported by CPRIT grant number RP120840. T.H.H. and S.P. were supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award Number UL1TR001105. S.P., J.H.C., T.H.H. were supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare (HI13C2162), Korea. D.Y. was supported by the National Research Foundation of Korea grant funded by the Ministry of Science, ICT and Future Planning (NRF-2015R1C1A1A02036312). M.C. was supported by 5K25AR063761-03. X.W. was supported by R15GM113157. T.K.S. was supported by NIH funding grant 5R00CA151672-04, NIH Institutional Shared Resource grant to the Masonic Cancer Center P30-CA77598, Masonic Cancer Center SP3 grant, Mezin-Koats Colorectal Cancer Research Fund. The results published here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Conflict of interest: The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agency. T.H.H., S.P. and J.H.C. disclose pending patent intellectual proprietary interest as the inventor of NTriPath. A provisional patent application (US 62/217 417) relating to the identification of cancer-type specific pathways across cancers has been filed by the University of Texas Southwestern Medical Center with S.P., J.H.C. and T.H.H. listed as inventors.
Supplementary Material
References
- Baselga J. (2011) Targeting the phosphoinositide-3 (pi3) kinase pathway in breast cancer. Oncologist, 16(Suppl 1), 12–19. [DOI] [PubMed] [Google Scholar]
- Cerami E., et al. (2010) Automated network analysis identifies core pathways in glioblastoma. PLoS One, 5, e8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi E.C., Kolda T.G. (2012) On tensors, sparsity, and nonnegative factorizations. SIAM J. Matrix Anal. Appl., 33, 1272–1299. [Google Scholar]
- Dees N.D., et al. (2012) Music: identifying mutational significance in cancer genomes. Genome Res., 22, 1589–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D., et al. (1995) Inhibition of transcription elongation by the vhl tumor suppressor protein. Science, 269, 1402–1406. [DOI] [PubMed] [Google Scholar]
- Forbes S.A., et al. (2015) Cosmic: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res., 43(Database issue), D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal., 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway L.A., Lander E.S. (2013) Lessons from the cancer genome. Cell, 153, 17–37. [DOI] [PubMed] [Google Scholar]
- Gialeli C., et al. (2011) Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J., 278, 16–27. [DOI] [PubMed] [Google Scholar]
- Gilbert J., et al. (2008) Phase ii trial of irinotecan plus cisplatin in patients with recurrent or metastatic squamous carcinoma of the head and neck. Cancer, 113, 186–192. [DOI] [PubMed] [Google Scholar]
- Hofree M., et al. (2013) Network-based stratification of tumor mutations. Nat. Methods, 10, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T.H., et al. (2013) Large-scale integrative network-based analysis identifies common pathways disrupted by copy number alterations across cancers. BMC Genomics, 14, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T.H., et al. (2012) Co-clustering phenomegenome for phenotype classification and disease gene discovery. Nucleic Acids Res., 40, e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.H., et al. (2004) Combination antiangiogenesis therapy with marimastat, captopril and fragmin in patients with advanced cancer. Br. J. Cancer, 91, 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C., et al. (2013) Mutational landscape and significance across 12 major cancer types. Nature, 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava Prasad T.S., et al. (2009) Human protein reference database2009 update. Nucleic Acids Res., 37(Suppl 1), D767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.J., et al. (2012) Sparse robust matrix tri-factorization with application to cancer genomics. In: Proceeding of 3rd International workshop on Cognitive Information Processing, Baiona, Spain. May 28 - 30, 2012, pp. 1–6. [Google Scholar]
- Langlois B., et al. (2010) Lrp-1 promotes cancer cell invasion by supporting erk and inhibiting jnk signaling pathways. PLoS One, 5, e11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S., et al. (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature, 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S., et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature, 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.D., Seung H.S. (2001) Algorithms for non-negative matrix factorization. In: Leen T., et al., (eds.) Advances in Neural Information Processing Systems 13. Denver, CO, pp. 556–562. [Google Scholar]
- Nickerson M.L., et al. (2008) Improved identification of von hippel-lindau gene alterations in clear cell renal tumors. Clin. Cancer Res., 14, 4726–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohh M., et al. (2000) Ubiquitination of hypoxia-inducible factor requires direct binding to the [bgr]-domain of the von hippel-lindau protein. Nat. Cell Biol., 2, 423–427. [DOI] [PubMed] [Google Scholar]
- Okada M., et al. (2008) Captopril attenuates matrix metalloproteinase-2 and -9 in monocrotaline-induced right ventricular hypertrophy in rats. J. Pharmacol. Sci., 108, 487–494. [DOI] [PubMed] [Google Scholar]
- Osmanbeyoglu H.U., et al. (2014) Linking signaling pathways to transcriptional programs in breast cancer. Genome Res., 24, 1869–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Llopis S., Brugarolas J. (2013) Simultaneous isolation of high-quality dna, rna, mirna and proteins from tissues for genomic applications. Nat. Protocol., 8, 2240–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Llopis S., et al. (2012) Bap1 loss defines a new class of renal cell carcinoma. Nat. Genet., 44, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin E.J., et al. (2011) Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet., 7, e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y., et al. (2013) Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet., 45, 860–867. [DOI] [PubMed] [Google Scholar]
- Staudt N.D., et al. (2013) Myeloid cell receptor lrp1/cd91 regulates monocyte recruitment and angiogenesis in tumors. Cancer Res., 73, 3902–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- strel’tsov S.A., et al. (2001) Interaction of topotecan—a dna topoisomerase I inhibitor–with dual-stranded polydeoxyribonucleotides. ii. Formation of a complex containing several dna molecules in the presence of topotecan. Mol. Biol. (Mosk), 35, 442–450. [PubMed] [Google Scholar]
- Subramanian A., et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. U. S. A., 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthram S., et al. (2010) Network-based elucidation of human disease similarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput. Biol., 6, e1000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D., et al. (2013) Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep., 3, 2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto K., et al. (2000) Mechanism of regulation of the hypoxia-inducible factor-1a by the von Hippel-Lindau tumor suppressor protein. EMBO J., 19, 4298–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. (2013) Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature, 499, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandin F., et al. (2011) Algorithms for detecting significantly mutated pathways in cancer. J. Comput. Biol., 18, 507–522. [DOI] [PubMed] [Google Scholar]
- Vaske C.J., et al. (2010) Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using paradigm. Bioinformatics, 26, i237–i245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.N., et al. (2005) Inhibition of matrix metalloproteinase activity and growth of gastric adenocarcinoma cells by an angiotensin converting enzyme inhibitor in in vitro and murine models. Eur. J. Surg. Oncol., 31, 1042–1050. [DOI] [PubMed] [Google Scholar]
- Yamamoto D., et al. (2008) Inhibitory profiles of captopril on matrix metalloproteinase-9 activity. Eur. J. Pharmacol., 588, 277–279. [DOI] [PubMed] [Google Scholar]
- Zhang S., et al. (2011) A novel computational framework for simultaneous integration of multiple types of genomic data to identify microrna-gene regulatory modules. Bioinformatics, 27, i401–i409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., et al. (2013) Network-based survival analysis reveals subnetwork signatures for predicting outcomes of ovarian cancer treatment. PLoS Comput. Biol., 9, e1002975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.