

Summary
LAGLIDADG meganucleases are DNA cleaving enzymes used for genome engineering. While their cleavage specificity can be altered using several protein engineering and selection strategies, their overall ‘targetability’ is limited by highly specific indirect recognition of the central four basepairs within their recognition sites. In order to examine the physical basis of indirect sequence recognition and to expand the number of such nucleases available for genome engineering, we have determined the target sites, DNA-bound structures and ‘central four’ cleavage fidelities of 9 related enzymes. Subsequent crystallographic analyses of a meganuclease bound to two noncleavable target sites, each containing a single inactivating basepair substitution at its center, indicates that a localized slip of the mutated basepair causes a small change in the DNA backbone conformation that results in a loss of metal occupancy at one binding site, eliminating cleavage activity.
Graphical Abstract
Introduction
Homing endonucleases, also termed ‘meganucleases’, are highly specific DNA cleaving enzymes, encoded by mobile open reading frames, that are present within all microbial life as well as in mitochondria and chloroplasts (Stoddard, 2014). Meganuclease genes are mobilized as a result of the DNA cleavage activity of their gene products. When a meganuclease gene is embedded within an intron or intein, it can invade conserved coding sequences within a host’s genome without disrupting the production of the host gene product.
At least six families of meganucleases are known to exist, including the ‘LAGLIDADG’ family found in eukarya and archaea. Properties of LAGLIDADG meganucleases (hereafter referred to as ‘meganucleases’) include small size, long DNA target sites and high DNA cleavage specificity (Paques and Duchateau, 2007). Their DNA recognition profiles result from a complex network of direct and water-mediated contacts (Chevalier et al., 2003).
Meganucleases are one of 4 different classes of gene targeting nucleases, alongside zinc finger nucleases, TAL effector nucleases and CRISPR/Cas9 nucleases (Segal and Meckler, 2013). Meganucleases are rarely the first choice for such experiments, because redesigning them to cleave new target sites requires considerable experimental effort. Nonetheless, engineered meganucleases have been used for a wide variety of genome engineering and editing applications (Antunes et al., 2012; Arnould et al., 2007; Boissel et al., 2013; Chan et al., 2013; D’Halluin et al., 2013; Djukanovic et al., 2013; Gao et al., 2010; Izmiryan et al., 2016; Menoret et al., 2013; Osborn et al., 2015; Sather et al., 2015; Takeuchi et al., 2011; Wang et al., 2014). The addition of engineered TAL effector repeats to the N-termini of engineered meganucleases allows the generation of highly specific and active gene targeting nucleases that still correspond to monomeric protein scaffolds (Boissel et al., 2013; Osborn et al., 2015; Sather et al., 2015; Takeuchi et al., 2014; Wang et al., 2014).
While the reengineering of meganuclease specificity involves considerable effort that generally preclude their routine use, their small size and highly specific DNA cleavage activities make them desirable enzymes for gene targeting. As protein engineering methods have become more powerful, the utility of meganucleases for genome engineering is limited not by technical barriers, but rather by limitations in the ‘targetability’ of the proteins themselves. Similar to TAL nucleases (which require 5′ thymines at each end of their DNA target sites) (Mak et al., 2013) and CRISPR nucleases (which require unique ‘PAM’ sequences at the 3′ ends of their DNA targets) (Pennisi, 2013; Ran et al., 2013), meganucleases are constrained to targets that contain a nearly invariant 4 basepair sequence (termed their ‘central four’ recognition motif) at the center of their target sites (Scalley-Kim et al., 2007; Takeuchi et al., 2014; Thyme et al., 2009). These nucleotide positions are not directly contacted by the enzyme, yet they limit the DNA sequences an enzyme can recognize. While the basis of this type of ‘indirect readout’ is not precisely known, it is often correlated with enzyme-induced DNA bending (leading to the alternative term ‘shape-based readout’), because variable amounts of energy are required to unstack or otherwise remove different DNA basepairs from their immediate neighbors within a DNA duplex (Curuksu et al., 2009; Yakovchuk et al., 2006; Zakrzewska, 2003).
To further understand the effect of indirect sequence specificity on enzymatic activity, we have conducted a series of bioinformatic, structural and functional studies on 9 closely related meganucleases. Although these enzymes display similar structures and mechanisms for DNA recognition, they recognize a wide variety of DNA target sites and display disparate behaviors toward the ‘central 4’ basepair sequences in their respective DNA targets. Structural analyses of one such enzyme, bound to substrates containing a single inactivating basepair substitution within the center of its target site, demonstrates how basepair substitutions that are not directly read out by the enzyme cause a significant alteration of active site composition that eliminates catalytic activity.
Results
Identification and validation of meganuclease genes and target sites
34 putative LAGLIDADG meganuclease genes were identified within 22 separate fungal hosts (Table S1). These genes and their surrounding introns were embedded within 12 different mitochondrial host genes, encoding small and large ribosomal RNA subunits, the S5 ribosomal protein, multiple subunits of NADH dehydrogenase and NADH-ubiquinone oxidoreductase, and cytochrome oxidase. The putative meganucleases were comprised of single peptide chains ranging in length from 276 to 349 amino acids, and displayed 12% to 85% sequence identity against I-OnuI. Of the 34 meganucleases genes tested, 22 could be expressed on the yeast surface.
Of the 22 enzymes that successfully expressed on the yeast surface as full-length protein chains, 18 were could cleave predicted DNA substrates corresponding to the uninterrupted host genes at positions spanning the insertion site of the endonuclease gene and its surrounding intron (see Supplemental Experimental Procedures for an example of a full analysis). Sequencing the cleaved DNA products allowed precise mapping of the target sites and strand-specific cleavage events for all 18 enzymes. The centers of the target sites were generally found to be located within 4 basepairs of the genomic insertion site for the corresponding meganuclease. Across each target site, G:C or A:T basepairs are observed frequently at each position with the exception of the immediate center (positions −1 and +1), where G:C basepairs are almost never observed.
Solution behavior, stability and crystal structures
The enzymes that displayed high levels of expression and validated cleavage activities on their DNA target sites were subjected to additional biophysical analyses. Seven of those enzymes (I-AabMI, I-CpaMI, I-GpeMI, I-GzeII, I-LtrWI, I-PanMI and I-SmaMI) expressed at high levels, purified to homogeneity, displayed highly cooperative unfolding transitions in denaturation analyses via circular dichroism, and were crystallized in complex with their DNA targets. Those 7 proteins (along with the previously studied enzymes I-OnuI and I-LtrI (Takeuchi et al., 2011)) displayed a broad range of denaturation temperatures that spanned approximately 15° C ( Figure S1).
The structures of all 7 meganuclease-DNA complexes were determined (Figure 1 and Table 1), at resolutions ranging from 3.2 Å (I-AabMI) to 2.0 Å (I-SmaMI). The structure of one of these enzymes (I-SmaMI) was reported in a separate manuscript (Shen et al., 2016). When combined with the previously determined crystal structures of I-OnuI and I-LtrI, structures of 9 closely related meganucleases were available for detailed comparisons.
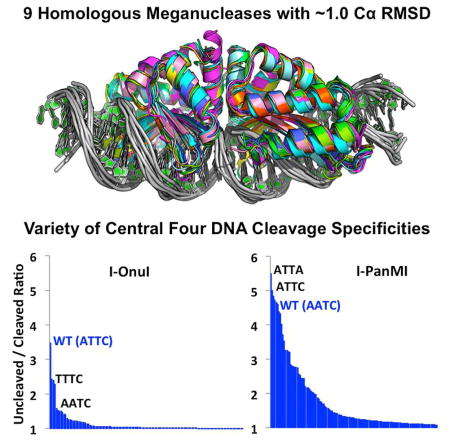
Figure 1.

Superimposed and individual crystal structures of the nine meganuclease-DNA complexes in this study. Data collection and refinement statistics are provided in Table 1. A structure-based sequence alignment, overall sequence identities and comparable backbone atom RMSD values are provided in Figure 2 and Table 2, respectively. See also Table S1 and Figure S1.
Table 1.
| I-AabMI | I-CpaMI | I-GpeMI | I-GzeII | I-LtrI | I-LtrWI | I-OnuI | I-PanMI | I-SmaMI (WT) |
I-SmaMI (TTGT) |
I-SmaMI (TTCT) |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB Code | 4YIT | 4YIS | 4YHX | 4Z1X | 3R7P | 4LQ0 | 3QQY | 5ESP | 5E5O | 4Z20 | 4Z1Z |
| Data Collection | |||||||||||
| Space group | P1 | P 212121 | C2 | C2 | C2 | P 212121 | P 212121 | P2 | P 21 21 21 | P21 | P21 |
| Cell dimensions | |||||||||||
| a, b, c (Å) | 41.96, 61.536, 88.621 | 75.87, 91.80, 139.64 | 151.99, 39.90, 73.99 | 125.83, 61.18, 128.88 | 113.09, 42.60, 103.20 | 40.55, 71.96, 168.65 | 37.95, 73.93, 166.93 | 69.56, 78.42, 101.02 | 61.33, 67.99, 97.74 | 45.7, 172.3, 60.0 | 41.9, 179.3, 65.4 |
| α, β, γ (°) | 97.18, 99.10, 101.89 | 90.00, 90.00, 90.00 | 90.00, 90.43, 90.00 | 90.00, 116.30, 90.00 | 90.00, 110.63, 90.00 | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 92.38, 90.00 | 90.00, 90.00, 90.00 | 90.0, 92.4, 90.0 | 90.0, 95.5, 90.0 |
| Resolution (Å) | 86.33–3.24 | 76.71–2.98 | 75.99–2.15 | 38.51–2.79 | 48.29–2.70 | 44.34–2.68 | 44.46–2.40 | 30.9–2.995 | 26.88 – 2.36 | 44.13–3.2 | 44.82–3.2 |
| Rmerge | 0.101 (0.175) | 0.092 (0.549) | 0.084 (0.432) | 0.091 (0.340) | 0.065 (0.240) | 0.092 (0.586) | 0.080 (0.140) | 0.157 (0.850) | 0.122 (0.809) | 0.064 (0.229) | 0.228 (0.283) |
| I/σI | 11.9 (6.26) | 8.74 (2.02) | 11.28 (2.61) | 22.9 (2.76) | 13.2 (6.1) | 20.0 (4.20) | 18.6 (9.62) | 13.2 (2.6) | 14.8 (2.0) | 16.9 (6.0) | 10.9 (5.05) |
| Completeness (%) | 96.8 (91.2) | 93.32 | 99.3 (97.6) | 98.7 (90.0) | 98.6 | 95.3 (95.0) | 96.8 (91.0) | 98.6 (97.7) | 99.2 (93.3) | 98.9 (98.3) | 98.5 (96.7) |
| Redundancy | 3.7 (3.6) | 3.2 (3.0) | 3.5 (3.0) | 6.50 (4.10) | 6.8 (6.5) | 11.3 (11.3) | 6.7 (4.8) | 5.8 (5.8) | 6.9 (5.6) | 3.8 (3.7) | 13.8 (11.8) |
| Refinement | |||||||||||
| No. Reflections | 12412 | 22357 | 23084 | 19780 | 12060 | 13100 | 17525 | 21715 | 16388 | 14420 | 16120 |
| Rwork (Rfree) | 22.53(29.88) | 21.73 (27.37) | 18.12 (24.40) | 17.49 (23.70) | 19.6 (27.0) | 19.7 (27.1) | 19.1 (24.0) | 22.6(26.9) | 19.06 (26.89) | 23.77 (28.61) | 24.19(28.71) |
| No. Complex in ASU | 2 | 2 | 1 | 2 | 1 | 1 | 1 | 2 | 1 | 2 | 2 |
| No. Atoms | |||||||||||
| Protein | 4129 | 4589 | 2326 | 4643 | 2357 | 2421 | 2419 | 4512 | 2392 | 4340 | 4335 |
| DNA | 2009 | 2280 | 1101 | 2170 | 1107 | 1060 | 1060 | 2199 | 1024 | 2132 | 2214 |
| Active Site Cations | 2 | 2 | 3 | 2 | 3 | 3 | 1 | 2 | 3 | 1 | 1 |
| Water | 19 | 37 | 219 | 120 | 43 | 18 | 73 | 3 | 85 | 11 | 4 |
| B-factor | 48 | 38.85 | 36.19 | 22.67 | 32.27 | 57.57 | 30.56 | 55.91 | 41.7 | 74.6 | 49.7 |
| R.m.s deviations | |||||||||||
| Bond lengths (Å) | 0.008 | 0.009 | 0.013 | 0.017 | 0.008 | 0.011 | 0.018 | 0.011 | 0.018 | 0.01 | 0.01 |
| Bond angles (°) | 1.385 | 1.2528 | 1.627 | 1.519 | 1.337 | 1.646 | 2.217 | 0.455 | 2.041 | 1.324 | 1.426 |
| Ramachandran | |||||||||||
| Preferred (%) | 87.38 | 94.76 | 96.48 | 95.33 | 94.81 | 94.26 | 95.25 | 95.47 | 93.8 | 91.91 | 88.16 |
| Allowed (%) | 11.68 | 4.54 | 3.17 | 3.46 | 4.33 | 5.07 | 3.73 | 4.01 | 5.5 | 6.2 | 10.81 |
| Outliers (%) | 0.94 | 0.7 | 0.35 | 1.21 | 0.87 | 0.68 | 1.02 | 0.52 | 0.69 | 1.89 | 1.03 |
Note: The structures of I-OnuI, I-LtrI and I-SmaMI bound to their wild-type DNA targets have been previously reported (Shen et al., 2016; Takeuchi et al., 2011). The statistics for these structures are reiterated in this table for ease of comparison.
The overall sequence identity between various pairwise alignments of these 9 meganucleases are generally between 34 and 48% (Figure 2), with the exception of I-GpeMI and I-OnuI, that display 85% sequence identity (the physiological target sites for those two enzymes differ at only two basepairs). The structural similarity between various pairwise superposition of the meganuclease crystals structures (calculated as RMSD values for all directly comparable alpha-carbons atoms) ranges from 0.61 Å to 1.58 Å (Table 2). The regions of greatest divergence between the crystal structures correspond to (1) the N- and C-termini, (2) several loops that connect individual β-strands that comprise the DNA contacting surface, and (3) a flexible linker that links the N- and C-terminal domains of each enzyme.
Figure 2.

Structure-based sequence alignment. Elements of secondary structure are designated above the sequence as cartoon shapes. Blue text highlights positions that are completely conserved across all nine structures. A black dot above the sequence alignment designates a position which contacts the bound DNA in at least one of the nine structures. The inset box provides the DNA target sequence for each meganuclease, with the central four basepairs highlighted red. See also Figures S2 and S3, which provides a corresponding ‘contact map’ of all superimposable positions on the nine protein backbones that are involved in DNA contacts.
Table 2.
Percent Identity (amino acid)/comparable Cα RMSD
| AabMI | CpaMI | GpeMI | GzeII | LtrI | LtrWI | OnuI | PanMI | SmaMI | |
|---|---|---|---|---|---|---|---|---|---|
| AabMI | 100% | 44%/1.20 | 42%/0.73 | 37%/0.82 | 45%/0.69 | 36%/0.92 | 41%/0.93 | 43%/0.90 | 40%/0.86 |
| CpaMI | 100% | 43%/1.47 | 37%/1.42 | 43%/1.21 | 34%/1.45 | 42%/1.58 | 41%/1.13 | 40%/1.20 | |
| GpeMI | 100% | 39%/0.70 | 48%/0.83 | 37%/0.91 | 85%/0.51 | 41%/0.77 | 46%/0.85 | ||
| GzeII | 100% | 40%/0.74 | 35%/0.95 | 43%/0.61 | 39%/0.53 | 40%/0.67 | |||
| LtrI | 100% | 38%/0.85 | 47%/0.92 | 44%/0.75 | 45%/0.67 | ||||
| LtrWI | 100% | 41%/1.09 | 39%/0.92 | 41%/0.91 | |||||
| OnuI | 100% | 44%/0.77 | 46%/0.74 | ||||||
| PanMI | 100% | 44%/0.64 | |||||||
| SmaMI | 100% |
Structure-based alignment of all 9 DNA-bound meganuclease structures allowed us to create a ‘contact map’ in which equivalent DNA-contacting positions within each meganuclease scaffold are assigned a common nomenclature that describes that residue’s position within the meganuclease scaffolds (Figures S2 and S3). These positions comprise all residues, in any of the structures, that make a direct or water-mediated contact to a nucleotide base or to the DNA backbone, and place at least one protein atom within 4 Å of any DNA atom.
Collectively, 99 positions across the entire group of structures (corresponding to about 1/3 of all residues within the aligned structures) are located near the DNA target in one or more of the enzyme/DNA complexes. Even though the structures are similar, there is variability between the individual DNA recognition surfaces: only 21 out of the 99 identified contact positions across the aligned protein scaffolds are found within close proximity to the DNA target in all 9 structures. The total number of DNA-contacting residues (as defined above) for each enzyme ranges from a low of 58 (for I-AabMI and I-GzeII) to a high of 76 (for I-Onu). Much of this disparity is attributed to differences in the length and conformation of flexible loops at each end of the DNA contacting surfaces.
Fifteen of the DNA-contacting residues are completely conserved across all 9 enzymes (Figure 2). These contacts correspond to contacts to the DNA backbone, and therefore appear to be constrained by pressure to maintain overall affinity of the protein-DNA interaction. In contrast, the residues that make contacts to nucleotide bases exhibit a greater level of diversity between the protein scaffolds, as might be expected for a collection of highly similar DNA binding proteins that display considerable differences in their sequence specificity.
Recognition fidelity across the ‘central four’ basepairs of the DNA target sites
The most constraining feature of DNA recognition by meganucleases is their preference for unique DNA sequences spanning the ‘central 4’ basepair positions within their target sites, even though each enzyme makes few contacts to those nucleotides (illustrated for I-OnuI in Figure 3). The majority of the enzymes do not make any observable direct or indirect contacts to any of the central four basepairs in their targets.
Figure 3.

DNA binding and cleavage by the I-OnuI meganuclease. (A) The structure of the DNA-binding surface of the meganuclease. The atoms of all side chains involved in DNA contacts are shown as light blue spheres. The atoms of the central four DNA basepairs are shown as multi-colored spheres (with the major groove basepair edges pointing away from the enzyme, toward the reader). Note the lack of contacts to these basepairs. (B) Binding curve for I-OnuI to its wild-type DNA target, produced using the flow cytometric binding assay with surface-expressed I-OnuI combined with increasing concentrations of a fluorescently labeled DNA duplex harboring the wild-type target site. The raw flow cytometric data is shown with its corresponding DNA substrate concentration above the binding curve, with increasing concentrations of DNA substrate leading to increased binding signal (A647). The rectangular gates shown were used for quantifying the data. A sample flow binding plot illustrates the bound DNA signal (A647) on the y-axis and the C-terminal protein stain (FITC) on the x-axis. Experiments of this type were conducted for each enzyme with its wild-type target in order to determine a DNA concentration corresponding to the approximate KD of the interaction. This DNA concentration was then used for subsequent comparative analyses of binding to variant DNA targets harboring single basepair substitutions. (C) Relative binding of 12 DNA target site variants, each harboring a single basepair substitution in the ‘central four’ region of the target site. The wild-type target, which contains an ‘ATTC’ sequence in the ‘central four’ region, is shown at the left, and binding signal from the flow cytometric binding assay for all the variant targets are normalized to wildtype. (D) Relative cleavage of the same panel of DNA substrates, measured using the tethered cleavage assay. See FIgures S4 and S5 and the Supplemental Experimental Procedures for a description of the flow cytometric binding and cleavage assays and the Supplemental Data File S1 for the raw data used to produce these graphs.
In order to systematically assess recognition fidelity at these basepair positions, we examined the effect of all possible single basepair substitutions within the central four region, for all nine meganucleases, on DNA binding and cleavage. Three different experiments were performed, in duplicate, across a total of 117 unique combinations of enzyme and substrate (9 enzymes × 13 DNA substrates for each):
Comparative binding assays conducted with a single DNA concentration (100–400 pM) corresponding to the approximate KD for each wild-type enzyme-DNA interaction (experimental conditions that are highly sensitive to changes in DNA binding affinity, as illustrated in Figure 3B).
‘Non-tethered’ in vitro DNA cleavage assays using 20 nM substrate concentrations (which requires high affinity binding in order for cleavage to occur).
Additional ‘tethered’ cleavage assays with the DNA substrate physically localized in close proximity to surface-displayed enzyme (which can result in ‘rescue’ of cleavage when activity is compromised due to reduced binding affinity).
Schematics and additional details of the experiments performed using yeast surface display and flow cytometry are shown in Figures S4 and S5, and described in Supplemental Experimental Procedures.
A comparison of the activities measured in these 3 assays for each enzyme allowed us to identify reductions in DNA cleavage that appear to be caused primarily by reduced binding affinity, versus reductions in cleavage that appear to be caused by compromised catalytic activity. For both cleavage assays, a single time-point was used corresponding to substantial (but not complete) digestion of the wild-type substrate, so that decreases in cleavage efficiency caused by single basepair substitutions would be easily detected. Summaries of the results for these experiments are provided in Figure 4; detailed examples of raw and tabulated data for all of the enzyme/DNA combinations are provided in the Supplemental Data File S1.
Figure 4.

The cleavage specificity and DNA distortion by meganucleases across the ‘central four’ target basepairs is variable. (A) Summary of “central four” DNA sequences cleavable by each of the nine meganucleases in this study. Bold, blue sequences correspond to the original wild-type targets for each enzyme. Bold, black sequences correspond to targets that are both bound and cleaved at near (≥90%) wild-type efficiency. Grey sequences correspond to targets that are either (1) bound less efficiently than the wild-type sequence, but still cleaved at ≥50% of wild-type efficiency, either by tethering to the enzyme, and/or by digesting higher concentrations of DNA (denoted with one asterisk, “ * ”) or (2) bound as tightly as wild-type, but cleaved with reduced efficiencies, 50–90% relative to the wild-type target (denoted with two asterisks, “ ** ”). Red boxes highlight the places where a G or C is tolerated at the −1 or +1 position of the central four. See FIgures S4 and S5 and the Supplemental Experimental Procedures for a description of the flow cytometric binding and cleavage assays and the Supplemental Data File S1 for the raw data used to produce this summary. (B) DNA bending analysis for all nine meganuclease/DNA complexes calculated by the 3DNA structural analysis software. Note that while the overall bend of the 22 basepair target sites is similar for each enzyme (see also Figure 1), that I-PanMI achieves that bend with a considerably reduced helical rise between the two central basepairs. (C) Superposition of the central four basepairs from the I-PanMI (red) and I-CpaMI (blue) crystal structures. This view highlights the unique distortion of the bound DNA by the I-PanMI enzyme, leading to a compression of the central two basepairs (arrow).
The most general observation from these experiments is that the ability to bind DNA substrates containing various basepair substitutions in the central four positions of the target is relatively broad, whereas the ability to bind and cleave the same DNA sequences is more limited. This is typified for the I-OnuI enzyme (Figure 3C and 3D), which binds at least eight alternative DNA substrates, each harboring a single basepair substitution at one of its central four positions, with relatively minor reductions in apparent binding affinity. In contrast, a limited subset of those same DNA targets can be efficiently cleaved by the same enzyme.
This observation extends across the panel of meganucleases examined: most of the enzymes display robust binding for more than half of the altered substrates that were tested. Five of the nucleases (I-GpeMI, I-GzeII, I-LtrI, I-LtrWI and I-OnuI) fail to bind and cleave any alternative substrates at near wild-type efficiency (≥90% relative to wild-type), while another three enzymes (I-AabMI, I-CpaMI and I-SmaMI) display near wild-type binding and cleavage of only one additional substrate. Several of the enzymes can cleave additional substrates efficiently, but only when the DNA is tethered in the proximity of surface-displayed enzyme (shown by a single asterisk, “*”, in Figure 4A). As well, some altered substrates are bound tightly but are cleaved inefficiently (denoted by two asterisks “**” in Figure 4A).
One enzyme (I-PanMI) is considerably more tolerant of basepair substitutions in its ‘central four’ region than its counterparts. I-PanMI binds nearly all variant target sites with little reduction in apparent affinity, binds and cleaves two alternative DNA target sequences with wild-type efficiency, and tolerates G:C basepairs at the immediate center of those two target sites. It also cleaves an additional six targets with an efficiency ≥50% of wild-type activity (Figure 4A). A structural analysis using webserver 3DNA (Zheng et al., 2009) was used to compare the DNA-bound crystal structure of I-PanMI to the other eight structures in this study. This analysis indicated that I-PanMI bends its DNA target to a similar degree as the other enzymes, but with a notable difference at the center of the DNA target (Figures 4B and 4C). The bound DNA duplex in the I-PanMI complex displays a significant reduction in the helical rise between its two central basepairs that may reduce the sequence-specific energetic penalty associated with DNA bending and central basepair unstacking by the enzyme (see Discussion).
We next asked two related questions:
Can single basepair substitutions that are individually well-tolerated be combined without an additional loss of catalytic activity?
Does combination of individual basepairs substitutions that are each not tolerated by an enzyme ever result in restoration of cleavage activity?
To do this, the specificities of the highly specific I-OnuI enzyme, and of the much less specific I-PanMI enzyme, were each examined using large panels of substrates that harbored multiple basepair substitutions throughout the central four positions of their target sites. This experiment was performed using a DNA cleavage assay in which the DNA substrate and the meganuclease are tethered together on the surface of yeast, followed by flow cytometric analysis of cleavage activity (Figure 5).
Figure 5.

Analysis of central four specificity for I-OnuI and I-PanMI using the tethered DNA flow cytometric cleavage assay. (A) Schematic of the tethered DNA cleavage assay. Details of the method are illustrated in Figures S4 and S5 and further described in Materials and Methods and in the Supplemental Experimental Procedures. (B) Ranked array of cleavage activity by I-OnuI against DNA target substrates containing basepair combinations spanning positions −2 to +2. Only three substrates are cleavable at an efficiency of ≥50% that of the wild-type central four target; the remainder are very inefficiently cleaved in this assay. (C) The same type of ranked array of cleavage activity of DNA substrates by the I-PanMI enzyme. Note the much broader tolerance of variation in the central four target sequence. See also Figure S6 for the raw flow cleavage assay data for the I-PanMI enzyme.
The I-OnuI enzyme displays tight specificity in these experiments, displaying near wild-type cleavage efficiency towards only three additional DNA sequences (Figure 5B). Two of those DNA sequences contain either a single A-->T substitution at the first position in the ‘central four’ sequence (‘TTTC’) or a single T-->A substitution in the third position (‘ATAC’). Combining both basepair substitutions, thereby generating a substrate with a ‘TTAC’ sequence comprising the central four region, did not reduce cleavage activity. No ‘cryptic’ combinations of basepair substitutions were identified that contain substitutions which reduce cleavage individually, but are tolerated when combined.
In contrast, the I-PanMI meganuclease is much more promiscuous with respect to the central four basepair target region. At least 30 substrates (out of 256 candidates containing all possible sequences at the ‘central four’ nucleotide positions) were found to be efficiently cleaved by the enzyme, while many more are cleaved with reduced, but still significant, activity (Figure 5C and Figure S6). Several of the most well-cleaved substrates contain a basepair substitution (a ‘T’ at position −2) that is, by itself, inhibitory in both types of cleavage assays used in this study.
The effect of indirect sequence preference on endonuclease structure and activity
Finally, in order to visualize a mechanism by which the substitution of a DNA basepair that is not in contact with the endonuclease can cause a complete reduction in cleavage activity, the I-SmaMI endonuclease was crystallized in the presence of two DNA substrates that each harbor a single basepair substitution (at position +1) that hinders cleavage, but does not significantly reduce binding affinity.
A comparison of those two cocrystal structures with uncleavable ‘central four’ sequences (corresponding to 5′ TTGT 3′ and 5′ TTCT 3′) to the original crystal structure (with a cleavable ‘central four’ sequence of 5′ TTAT 3′) demonstrates that the overall structure of the protein-DNA complex, and the nature and magnitude of DNA bending, is comparable across all three structures (Figure S7). However, closer examination and comparison of the three structures indicates that substitution of the A:T basepair at position +1 in the wild-type target with either a G:C or a C:G basepair results in a localized slip of that basepair, relative to its immediate neighbors, in a direction that is orthogonal to the DNA duplex axis and towards one of the DNA backbones (Figures 6A and 6B). This motion results in a slight widening of the minor groove immediately upstream of the mutated base (with the distance between opposing phosphates increasing by approximately 1.5 Å). While these changes in distance are obviously at the threshold of significance at the 3.1 Å resolution of these structures, the resulting effect on the composition of the active site is unambiguous: a loss of one of the two bound metal ions in the enzyme active site (Figure 6C–E). Thus, the substitution of a single basepair, in the absence of direct contacts to the enzyme, causes a series of very small structural movements that result in the loss of an essential cofactor (an active site metal ion) and inactivation of the enzyme.
Figure 6.

Structural analysis of I-SmaMI bound to its wild-type DNA target (which contains a ‘TTAT’ central four sequence) and to two variant DNA targets that each contain a single basepair substitution in the central four region (corresponding to ‘TTCT’ and ‘TTGT’). (A) 3DNA analysis of ‘Y-displacement’ across the three DNA target sites. Note the movement of the substituted basepair in the two variant target sites. See also Figure S7 for the same analysis for ‘twist’ and ‘roll’ parameters. (B) Superposition of the central four DNA region of the wild-type (‘TTAT’) structure (teal) and the variant ‘TTCT’ structure (blue). Distances between calcium-coordinating phosphate oxygens are indicated in angstroms. (C)–(E): Anomalous difference Fourier maps for I-SmaMI bound to wild-type and variant target sites, shown from the side (left) and looking down at the active site from above (right). Anomalous electron density from the bound calcium is shown as blue mesh (using a contour level cutoff of 5.0). See also Figure S8 for data demonstrating the ability of Mn2+ to rescue cleavage of the ‘TTAT’ and ‘TTCT’ containing substrates by the same enzyme.
This observation caused us to wonder if the substitution of magnesium with manganese (which is less discriminating with respect to the identity and distance to its coordinating ligand partners (Bock et al., 1999)) would rescue I-SmaMI activity in the presence of the same basepair substitutions that inactivate magnesium-dependent cleavage. DNA digests using the same set of substrates (Figure S8) verified this hypothesis, and appear to support the concept that indirect cleavage specificity at these positions is largely a function of catalytic metal binding.
Discussion
In this study, we examined the effect of DNA basepair substitutions in a series of target sites at positions that are not in direct (or water-mediated) contact with their enzyme, but nonetheless impart considerable sequence restraints on the ability of those enzymes to cleave DNA. Many of these basepair substitutions reduce binding affinity, while others instead have the effect of rendering the substrate non-cleavable by the enzyme.
The effect of basepair composition and sequence on the energetics of DNA binding affinity, particularly when DNA is bent by the protein have been well-studied. Several approaches have demonstrated that the correlation between protein-DNA binding energy and the overall long-range bending of an entire DNA target site (of six basepairs or more in length) is quite weak. Instead, short-range changes in structure corresponding to the alteration of nonbonded contacts (such as perturbation of base pairing and base stacking) and neighboring bonding interactions (such as rotations around dihedral bond angles) contribute significantly to the free energy change associated with DNA bending (Shrader and Crothers, 1989; Zakrzewska, 2003). Additional analyses, conducted using a variety of solution-based studies (augmented by molecular dynamic analyses) indicate that various forms of basepair unstacking are an especially significant contributor to sequence-specific free-energy changes that arise from protein-DNA binding (Curuksu et al., 2009; Yakovchuk et al., 2006).
Systematic analyses of the energy required to fully unstack two sequential basepairs has demonstrated that the corresponding free energy change (ΔGst) varies in a highly sequence-specific manner by as much as 2.5 to 3 kcal/mol per base step (Protozanova et al., 2004). Dinucleotide sequences comprised solely of G:C basepairs are usually the most energetically costly to unstack, while those consisting solely of A:T basepairs are less costly. The LAGLIDADG meganucleases studied here consistently bend their DNA targets in a manner that alters the inter-basepair roll angle across the central DNA basepair step (spanning positions −1 and +1) by up to 20 degrees, significantly unstacking the central basepairs from one another (Figure 4B). The observation that G:C basepairs appear to be strongly disfavored from positions −1 and +1 appears to correlate well with this observation.
The I-PanMI enzyme appears to transcend the effect of this DNA bending event by producing a significantly smaller helical rise between its central two basepairs (1.6 Å compared to a normal 3.3 Å) in the bound DNA complex (Figure 4C) while still altering the roll angle between those same basepairs. This appears to be attributable to a broadly distributed series of central basepair slides (‘x-displacement’) spanning several adjacent basepairs across the center of the target site that compensate for basepair unrolling. This subtle difference may reduce the energetic cost of unstacking by I-PanMI in a manner that allows the presence of individual G:C basepairs at the center of that enzyme’s target.
While the effect of indirect (or ‘shape-based) readout on protein-DNA binding affinity is relatively well-understood, it effect of on DNA cleavage activity is less clear. The analysis provided in the final crystallographic experiments in this paper indicates one type of mechanism that can eliminate cleavage activity. The DNA bend and unrolling at the central basepair step imparted by the I-SmaMI enzyme is tolerated for the wild-type DNA substrate, which contains a central ‘TpA’ dinucleotide sequence. When that sequence is replaced by a central ‘TpG’ or ‘TpC’ dinucleotide sequence, two results are noteworthy: binding affinity is relatively unaffected, while DNA cleavage is greatly reduced (Supplemental Data). In the crystallographic analyses of these structures, the energetic impact of basepair unrolling of these mutated dinucleotide sequences on binding affinity appears to be reduced by a small slip of the G:C or C:G basepair along the DNA duplex y-axis (toward the DNA backbone, in a direction that retreats from the opening and unstacking of the DNA strands) and a corresponding movement of the adjacent phosphates that leads to additional widening of the minor groove. While this localized motion might result in a more energetically favorable conformation for the protein-bound DNA duplex, it comes at a significant price: the very small motion of the basepair and its adjoining backbone phosphates causes the complete loss of a catalytically essential divalent cation.
This results in this study are similar to previous examinations of the mechanism of indirect readout for restriction endonucleases such as EcoRV (Martin et al., 1999) and HincII (Babic et al., 2008; Joshi et al., 2006). In those studies, the effect of substituted bases or protein side chains was more deleterious toward DNA cleavage than binding. These effects were described as an example of “structural adaptability” of the enzyme:DNA complex, because substitutions of either individual DNA basepairs or their protein contacts led to changes in binding that were accompanied by changes in structure that minimized effects on affinity, but at the cost of causing mispositonings in the active site that resulted in significant reductions in DNA cleavage activity (because catalysis requires more precise positioning of active site groups).
The studies reported in this paper shed light on an additional mechanism, beyond simply increasing the energy required to bind and bend DNA, that promotes indirect sequence specificity by nucleic acid enzymes, and also impact the continued development of meganucleases for use in targeted genome engineering. Because engineered meganucleases can not only be used as stand-alone reagents for such purposes, but (perhaps more importantly) as highly specific nuclease domains when combined with easily engineerable DNA binding platforms such as TAL effectors, the characterization of the details of their activity and specificity remains an important area of research for biotechnology.
Experimental Procedures
1. Identification of putative meganucleases and their target sites
A collection of related meganucleases were initially identified by submitting the sequence of the I-OnuI protein (Sethuraman et al., 2009) to the NCBI BLAST server as a query against its entire protein collection. Homologous sequences were aligned to I-OnuI and inspected for comparable lengths as well as the presence of two LAGLIDADG active sites spaced similarly to those in I-OnuI. Open reading frames were truncated at their N- and C-termini to match I-OnuI, but full-length proteins were also tested. A small number of individual point mutations (distal to the DNA-binding interface), as provided in the Supplemental Experimental Procedures, were incorporated on several protein surfaces to increase solubility. A general criterion to classify a candidate enzyme as part of the I-OnuI family was a minimum of at least 20% amino acid identity to I-OnuI, but initial candidate sequences included some with as low as 12% identity.
The DNA recognition sequence for each candidate enzyme was predicted using exon boundary annotations of the host gene (when available) or through alignments to homologous gene sequences. See the Supplemental Experimental Procedures for an extended example of target prediction (for I-GpeMI) using this sequence alignment strategy.
2. Assaying protein expression, DNA binding and cleavage via yeast surface display
Yeast expression constructs
All protein-coding DNA sequences were subcloned into the pETCON yeast surface expression vector (Addgene #41522). The pETCON vector is a modified version of the commonly used pCTCON2 vector (Chao et al., 2006), with altered restriction sites and a ‘G4S’ (Gly-Gly-Gly-Gly-Ser) linker between the C-terminus of the cloned enzyme and the Myc epitope tag.
Yeast transformation and induction
Yeast expression constructs were transformed into frozen competent EBY100 Saccharomyces cerevisiae (Invitrogen) using the lithium acetate method (Gietz and Schiestl, 2007a, b) and plated onto selective culture (SC) + 2% w/v glucose plates. Single colonies were transferred to growth in 1.5 mL SC media + 2% glucose at 30°C with shaking overnight. The next day, 20 million cells from the glucose cultures were transferred to 1.5 mL SC + 2% raffinose + 0.1% glucose media and grown at 30°C with shaking until reaching a density of 80–120 million cells/mL (approximately 7 hours). Finally, 30 million cells were washed with water and transferred to 1.5 mL SC + 2% galactose media for overnight induction on the benchtop at room temperature (14–16 hours).
Assays to characterize enzyme expression, DNA target binding, and DNA target cleavage using yeast surface displayed constructs and flow cytometric analyses are illustrated in Figures S4 and S5 and described in detail in Supplemental Experimental Procedures.
Enzyme surface expression assays
Enzymes expressed on the surface of yeast were analyzed by flow cytometry as previously described (Baxter et al., 2012; Jarjour et al., 2009). Successful surface expression was confirmed by staining of the C-terminal Myc epitope tag using an anti-Myc fluorescein isothiocyanate (FITC) antibody (ICL Labs, CMYC-45F). Observation of FITC signal indicates stable, full-length expression of the desired protein. See figure S4a.
Generation of Labeled DNA target substrates
Double-stranded DNA oligonucleotide substrates for flow cytometry and in vitro cleavage assays were prepared as previously described (Baxter et al., 2012; Jarjour et al., 2009). Briefly, 54-basepair double-stranded oligonucleotides were generated by PCR using Platinum Taq High Fidelity DNA polymerase (Invitrogen) with biotin- and AlexaFluor647-labeled primers (see the Supplemental Experimental Procedures for sequences of primers and PCR templates). Contaminating single-stranded template and/or primers were removed with a six hour Exonuclease I (NEB) digest at 37°C followed by purification on a G-100 Sephadex (GE Healthcare) column. The final labeled substrates were analyzed for purity on a 15% acrylamide gel.
DNA binding assays
In a V-bottom 96-well plate, 500,000 induced yeast cells were washed with “oligo cleavage buffer” (OCB): 150 mM KCl, 10 mM NaCl, 5 mM K-glutamate, 0.05% BSA, and 10 mM HEPES, pH 8.25, supplemented with 5 mM CaCl2 (to prevent cleavage activity). The surface-expressed enzyme was incubated for 2.5 hours at 4°C in a 50 μL volume containing OCB, 5 mM CaCl2, 100–400 pM A647-labeled DNA target substrate, and 1:100 anti-Myc-FITC antibody. Decreasing amounts of DNA substrate were tested for each meganuclease until reaching a concentration that displayed sufficient sensitivity for discrimination between good and poor binding. After incubation, cells were washed with OCB + 5 mM CaCl2 and run on a BD LSRII flow cytometer (BD Biosciences). See Supplemental Experimental Procedures and Figure S4b for details of the analysis of the flow binding data. Each dataset was collected in duplicate.
DNA cleavage assays
In a V-bottom 96-well plate, two million induced yeast were washed with “yeast staining buffer” (YSB): 180 mM KCl, 10 mM NaCl, 0.2% bovine serum albumin (BSA), 0.1% galactose, and 10 mM HEPES, pH 7.5. The cells were stained in a 50 μL volume for 2 hours at 4°C with 1:250 biotinylated anti-HA antibody (Covance) and 1:100 anti-Myc-FITC antibody (ICL Labs). A second stain (1 hour at 4°C) with 5 nM streptavidin -phycoerythrin (SAV-PE) (BD Biosciences, 554061) and 40 nM biotin/A647-labeled double-stranded DNA target substrate led to the formation of a physical biotin-streptavidin tethering of the DNA substrate to the N-terminus of the surface-expressed enzyme. A high-salt staining buffer (YSB + an additional 400 mM KCl) was used in the presence of DNA to discourage undesired binding of substrates tethered to neighboring cells. With the tethered DNA substrate in place, the cells were washed with OCB (see composition above), and divided equally into two separate wells. To each well was added 50 μL OCB + 5 mM CaCl2 (supports DNA binding without cleavage) or 5 mM MgCl2 (supports DNA cleavage), and the samples were incubated at 37°C for 30 minutes. After final washing in YSB, the yeast cells were run on a BD LSRII cytometer (BD Biosciences) and the data was analyzed with FloJo software (Tree Star, Inc.). See Supplemental Experimental Procedures, Figure S4c and Figure S5 for details of the analysis of flow cleavage data. The cleavage pattern for active enzymes were experimentally confirmed to correspond to 4-base, 3′ overhangs as described previously (Takeuchi et al. 2011). Each dataset was collected in duplicate.
3. Biochemical and structural studies using purified meganucleases
Overexpression in E. coli
Reading frames encoding meganucleases were subcloned into commercially available bacterial pET expression vectors (Novagen, Inc.) for protein production. For expression of poly-histidine tagged proteins, the meganuclease reading frames were cloned into a derivative of pET15b as previously described in (Mak et al., 2010). For expression of untagged proteins, reading frames were cloned into the commercially available expression vector pET21d to create T7-inducible constructs with no affinity purification tags. Details of enzyme purification, in vitro biochemical assays and crystallization are found in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Meganucleases display similar structures but large differences in DNA specificity
Indirect, shape based readout of DNA sequence is realized during binding or cleavage
Small structure shifts reduce the cost of bending DNA but also reduce enzyme activity
Acknowledgments
This work was supported by research grants from the NIH to BLS (R01 GM105695) and to AMS (RL1 CA133832), as well as funding by Bluebird Bio, Inc.
Footnotes
Author contributions. Bioinformatic analyses were conducted by ARL and AMS. Crystallographic analyses were conducted by ARL, JPH, BWS, JKC, JMB, NK, LIR and BLS. Biochemical experiments including binding and cleavage assays were conducted by ARL, JPH, BKK, JJ, and KH. All authors contributed to subsequent analyses, interpretation and manuscript preparation.
Data deposition. The x-ray diffraction data and refined coordinates for all newly determined meganuclease crystal structures have been deposited in the RCSB protein database for immediate release. All PDB ID codes are provided in Table 1.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antunes MS, Smith JJ, Jantz D, Medford JI. Targeted DNA excision in Arabidopsis by a re-engineered homing endonuclease. BMC biotechnology. 2012;12:86. doi: 10.1186/1472-6750-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnould S, Perez C, Cabaniols JP, Smith J, Gouble A, Grizot S, Epinat JC, Duclert A, Duchateau P, Paques F. Engineered I-CreI derivatives cleaving sequences from the human XPC gene can induce highly efficient gene correction in mammalian cells. J Mol Biol. 2007;371:49– 65. doi: 10.1016/j.jmb.2007.04.079. [DOI] [PubMed] [Google Scholar]
- Babic AC, Little EJ, Manohar VM, Bitinaite J, Horton NC. DNA distortion and specificity in a sequence-specific endonuclease. J Mol Biol. 2008;383:186–204. doi: 10.1016/j.jmb.2008.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter S, Lambert AR, Kuhar R, Jarjour J, Kulshina N, Parmeggiani F, Danaher P, Gano J, Baker D, Stoddard BL, et al. Engineering domain fusion chimeras from I-OnuI family LAGLIDADG homing endonucleases. Nucleic Acids Res. 2012;40:7985–8000. doi: 10.1093/nar/gks502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock CW, Katz AK, Markham GD, Glusker JP. Manganese as a Replacement for Magnesium and Zinc: Functional Comparison of the Divalent Ions. J Am Chem Soc. 1999;121:7360–7372. [Google Scholar]
- Boissel SJ, Astrakhan A, Jarjour J, Adey A, Shendure J, Stoddard BL, Certo M, Baker D, Scharenberg AM. MegaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt1224. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YS, Takeuchi R, Jarjour J, Huen DS, Stoddard BL, Russell S. The Design and In Vivo Evaluation of Engineered I-OnuI-Based Enzymes for HEG Gene Drive. PloS one. 2013;8:e74254. doi: 10.1371/journal.pone.0074254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protocols. 2006;1:755– 768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- Chevalier B, Turmel M, Lemieux C, Monnat RJ, Stoddard BL. Flexible DNA target site recognition by divergent homing endonuclease isoschizomers I-CreI and I-MsoI. J Mol Biol. 2003;329:253– 269. doi: 10.1016/s0022-2836(03)00447-9. [DOI] [PubMed] [Google Scholar]
- Curuksu J, Zacharias M, Lavery R, Zakrzewska K. Local and global efffects of strong DNA bending induced during molecular dynamics simulations. Nucleic Acids Res. 2009;37:3766– 3773. doi: 10.1093/nar/gkp234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Halluin K, Vanderstraeten C, Van Hulle J, Rosolowska J, Van Den Brande I, Pennewaert A, D’Hont K, Bossut M, Jantz D, Ruiter R, et al. Targeted molecular trait stacking in cotton through targeted double-strand break induction. Plant biotechnology journal. 2013;11:933–941. doi: 10.1111/pbi.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukanovic V, Smith J, Lowe K, Yang M, Gao H, Jones S, Nicholson MG, West A, Lape J, Bidney D, et al. Male-sterile maize plants produced by targeted mutagenesis of the cytochrome P450-like gene (MS26) using a re-designed I-CreI homing endonuclease. Plant J. 2013 doi: 10.1111/tpj.12335. [DOI] [PubMed] [Google Scholar]
- Gao H, Smith J, Yang M, Jones S, Djukanovic V, Nicholson MG, West A, Bidney D, Falco SC, Jantz D, et al. Heritable targeted mutagenesis in maize using a designed endonuclease. Plant J. 2010;61:176–187. doi: 10.1111/j.1365-313X.2009.04041.x. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. FrozFrozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG methoden competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat Protocols. 2007a;2:1– 4. doi: 10.1038/nprot.2007.17. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protocols. 2007b;2:31– 34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- Izmiryan A, Danos O, Hovnanian A. Meganuclease-Mediated COL7A1 Gene Correction for Recessive Dystrophic Epidermolysis Bullosa. J Invest Dermatol. 2016 doi: 10.1016/j.jid.2015.11.028. [DOI] [PubMed] [Google Scholar]
- Jarjour J, West-Foyle H, Certo MT, Hubert CG, Doyle L, Getz MM, Stoddard BL, Scharenberg AM. High-resolution profiling of homing endonuclease binding and catalytic specificity using yeast surface display. Nucleic Acids Res. 2009;37:6871–6880. doi: 10.1093/nar/gkp726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi HK, Etzkorn C, Chatwell L, Bitinaite J, Horton NC. Alteration of sequence specificity of the type II restriction endonuclease HincII through an indirect readout mechanism. J Biol Chem. 2006;281:23852–23869. doi: 10.1074/jbc.M512339200. [DOI] [PubMed] [Google Scholar]
- Mak AN, Bradley P, Bogdanove AJ, Stoddard BL. TAL effectors: function, structure, engineering and applications. Current opinion in structural biology. 2013;23:93–99. doi: 10.1016/j.sbi.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak ANS, Lambert AR, Stoddard BL. Folding, DNA recognition, and function of GIY-YIG endonucleases: crystal structures of R.Eco29kI. Structure. 2010;18:1– 11. doi: 10.1016/j.str.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AM, Sam MD, Reich NO, Perona JJ. Structural and energetic origins of indirect readout in site-specific DNA cleavage by a restriction endonuclease. Nat Struct Biol. 1999;6:269–277. doi: 10.1038/6707. [DOI] [PubMed] [Google Scholar]
- Menoret S, Fontaniere S, Jantz D, Tesson L, Thinard R, Remy S, Usal C, Ouisse LH, Fraichard A, Anegon I. Generation of Rag1-knockout immunodeficient rats and mice using engineered meganucleases. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27:703–711. doi: 10.1096/fj.12-219907. [DOI] [PubMed] [Google Scholar]
- Osborn MJ, Webber BR, Knipping F, Lonetree CL, Tennis N, DeFeo AP, McElroy AN, Starker CG, Lee C, Merkel S, et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Molecular therapy : the journal of the American Society of Gene Therapy. 2015 doi: 10.1038/mt.2015.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Duchateau P. Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Current Gene Therapy. 2007;7:49– 66. doi: 10.2174/156652307779940216. [DOI] [PubMed] [Google Scholar]
- Pennisi E. The CRISPR craze. Science. 2013;341:833– 836. doi: 10.1126/science.341.6148.833. [DOI] [PubMed] [Google Scholar]
- Protozanova E, Yakovchuk P, Frank-Kamenetskii MD. Stacked-unstacked equilibrium at the nick site of DNA. J Mol Biol. 2004;342:775– 785. doi: 10.1016/j.jmb.2004.07.075. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather BD, Romano Ibarra GS, Sommer K, Curinga G, Hale M, Khan IF, Singh S, Song Y, Gwiazda K, Sahni J, et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med. 2015;7:307ra156. doi: 10.1126/scitranslmed.aac5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalley-Kim M, McConnell-Smith A, Stoddard BL. Coevolution of homing endonuclease specificity and its host target sequence. J Mol Biol. 2007;372:1305– 1319. doi: 10.1016/j.jmb.2007.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal DJ, Meckler JF. Genome engineering at the dawn of the golden age. Ann Rev Genomics Hum Genet. 2013 doi: 10.1146/annurev-genom-091212-153435. advance access. [DOI] [PubMed] [Google Scholar]
- Sethuraman J, Majer A, Friedrich NC, Edgell DR, Hausner G. Genes within genes: multiple LAGLIDADG homing endonucleases target the ribosomal protein S3 gene encoded within an rnl group I intron of Ophiostoma and related taxa. Mol Biol Evol. 2009;26:2299– 2315. doi: 10.1093/molbev/msp145. [DOI] [PubMed] [Google Scholar]
- Shen BW, Lambert A, Walker BC, Stoddard BL, Kaiser BK. The structural basis of asymmetry in DNA binding and cleavage as exhibited by the I-SmaMI LAGLIDADG meganuclease. J Mol Biol. 2016 doi: 10.1016/j.jmb.2015.12.005. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrader T, Crothers D. Artificial nucleosome positioning sequences. PNAS USA. 1989;86:7418– 7422. doi: 10.1073/pnas.86.19.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard BL. Homing endonucleases from mobile group I introns: discovery to genome engineering. Mobile DNA. 2014;5:1–15. doi: 10.1186/1759-8753-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi R, Choi M, Stoddard BL. Efficient engineering of meganucleases and MegaTALs using bioinformatics and in vitro compartmentalization for targeted gene modification. PNAS USA. 2014 submitted. [Google Scholar]
- Takeuchi R, Lambert A, Mak A, Jacoby K, Dickson R, Gloor G, Scharenberg A, Edgell D, Stoddard B. Tapping natural reservoirs of homing endonucleases for targeted gene modification. PNAS USA. 2011;108:13077– 13082. doi: 10.1073/pnas.1107719108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyme SB, Jarjour J, Takeuchi R, Havranek JJ, Ashworth J, Scharenberg AM, Stoddard BL, Baker D. Exploitation of binding energy for catalysis and design. Nature. 2009;461:1300–1304. doi: 10.1038/nature08508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Khan IF, Boissel S, Jarjour J, Pangallo J, Thyme S, Baker D, Scharenberg AM, Rawlings DJ. Progressive engineering of a homing endonuclease genome editing reagent for the murine X-linked immunodeficiency locus. Nucleic Acids Res. 2014;42:6463–6475. doi: 10.1093/nar/gku224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovchuk P, Protozanova E, Frank-Kamenetskii MD. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 2006;34:564– 574. doi: 10.1093/nar/gkj454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewska K. DNA deformation energetic and protein binding. Biopolymers. 2003;70:414– 423. doi: 10.1002/bip.10476. [DOI] [PubMed] [Google Scholar]
- Zheng G, Lu XJ, Olson WK. Web 3DNA—a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res. 2009;37:W240– W246. doi: 10.1093/nar/gkp358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.