

Abstract
Oxidative damage represents the most significant insult to organisms because of continuous production of the reactive oxygen species (ROS) in vivo. Oxidative damage in DNA, a critical target of ROS, is repaired primarily via the base excision repair (BER) pathway which appears to be the simplest among the three excision repair pathways. However, it is now evident that although BER can be carried with four or five enzymes in vitro, a large number of proteins, including some required for nucleotide excision repair (NER), are needed for in vivo repair of oxidative damage. Furthermore, BER in transcribed vs. nontranscribed DNA regions requires distinct sets of proteins, as in the case of NER. We propose an additional complexity in repair of replicating vs. nonreplicating DNA. Unlike DNA bulky adducts, the oxidized base lesions could be incorporated in the nascent DNA strand, repair of which may share components of the mismatch repair process. Distinct enzyme specificities are thus warranted for repair of lesions in the parental vs. nascent DNA strand. Repair synthesis may be carried out by DNA polymerase β or replicative polymerases δ and ε. Thus, multiple subpathways are needed for repairing oxidative DNA damage, and the pathway decision may require coordination of the successive steps in repair. Such coordination includes transfer of the product of a DNA glycosylase to AP-endonuclease, the next enzyme in the pathway. Interactions among proteins in the pathway may also reflect such coordination, characterization of which should help elucidate these subpathways and their in vivo regulation.
Keywords: oxidative DNA damage, reactive oxygen species, base excision repair, replication-associated repair, coordination of repair pathway
REMINISCENCE ABOUT RICHARD B. SETLOW BY SANKAR MITRA
I considered it a great privilege to get to know Dick Setlow, who influenced my research career in a profound way, both directly and indirectly. After completing postdoctoral studies at Stanford University in Palo Alto, CA, I returned to India in 1966 and joined the faculty of the Bose Institute, Calcutta. Although I always had a broad interest in molecular biology, and nucleic acids, I knew very little until then about DNA repair. The classic paper of Dick Setlow and Bill Carrier [Setlow and Carrier, 1964] (as well as that of Boyce and Howard-Flanders, [1964]) on the mechanism of excision repair of UV-damaged DNA in E. coli appeared when I was still at Stanford University. I read those papers and marveled at the simplicity of the mode of repair only because the experiments were so elegantly and convincingly carried out.
Although my students and I dabbled in studies of DNA damage induced by UV and X rays during my tenure at the Bose Institute, we could not carry out any in-depth studies with the limited experimental facilities available at that time. I eventually became tired of the many nonscientific problems and decided to return to the U.S. The first person I thought of applying to was Dick Setlow, then at Oak Ridge National Laboratory (ORNL). It was actually Salil Niyogi, who has been one of my closest friends since my college days in India, who got to know Dick very well because he and his wife Audrey Stevens joined the Biology Division of ORNL in 1967, a few years after Dick moved to Oak Ridge from Yale. The Biology Division, under the stewardship of Alexander Hollaender, was a remarkably vibrant place in those days, with an extraordinary collection of outstanding scientists in genetics and molecular and radiation biology.
With Salil’s encouragement, I wrote to Dick to explore the possibility of spending a couple of years in his laboratory as a visiting scientist. I mentioned in my letter that I had been interested in DNA repair and wanted to acquire experience in his quantitative approach to measuring DNA repair. I was pleasantly surprised to receive an encouraging response from Dick, who suggested that I could meet with him at the Biophysics Congress to be held at MIT campus in Cambridge in August 1969. I had a suspicion that Dick had a hand in my receiving a travel award to attend that meeting. Then, for the first time, I had an opportunity to meet Dick in person during the meeting. My preconceived idea that a giant in science should be giant in size was obviously demolished when I noticed that Dick and I could exchange clothes. Later, after I joined ORNL, Dick’s then wife Jane once cracked a joke about this.
Dick told me that while he would have been very happy to have me in his group, he felt that I would be better off by joining the Biochemistry Section, under the leadership of Ken Volkin. Dick was the head of the Biophysics Section of the Biology Division at that time. I found out later that the Administration was not particularly anxious to enhance the size of Dick’s own research group.
In any event, although I joined Ken Volkin’s section, I got to know Dick quite well, particularly after he took over the administration of the Graduate School of Biomedical Sciences, as its Director. Although Dick was already well recognized for his seminal discovery of nucleotide excision repair, the final accolade in the form of induction in the U.S. National Academy of Sciences was given to him after I joined the Oak Ridge National Laboratory, and I remember the champagne party that followed the announcement (alcohol was not prohibited in any U.S. Department of Energy facilities at that time). Alex Hollaender was still alive at that time, and Dick was a regular companion of Alex’s in his fossil hunting forays on Sunday mornings.
What has always impressed me the most about Dick is his discipline and compartmentalization of efforts. While he wore several administrative hats simultaneously at Oak Ridge, he was still performing experiments with his own hands. During the 1970s, he made seminal observations regarding repair patch size for UV damage in mammalian cells by developing an ultracentrifugation assay for DNA after photolytic cleavage of the 5-bromouracil-containing repaired strand. In fact, he preferred to work by himself and left members of his research group to their own devices. At the same time, he was always on top of their latest results.
While I myself never worked in the area of nucleotide excision repair, my research interests nearly converged with Dick’s in the 1980s after Dick moved to Brookhaven National Laboratory in Upton, Long Island. He became interested in chemical carcinogenesis and repair of procarcinogenic DNA adducts. Evelyn Waldstein joined Dick’s lab as a visiting scientist from Israel, and they investigated the “kamikaze” protein O6-methylguanine-DNA methyltransferase by a direct reversal reaction via in situ dealkylation of the base. My group and Tomas Lindahl’s laboratory independently discovered this repair protein [Foote et al., 1980; Olsson and Lindahl, 1980], while Waldstein et al. [1982] developed a new assay for this protein.
In later years, both Dick and I became interested in oxidative DNA damage, which I have branded the “mother of all damage.” Dick’s lifetime interest has been photobiology. He became interested in oxidative DNA damage that could result from singlet oxygen species generated by long wavelength UV-A (and B), which is present in sunlight. He had been studying a fish model for carcinogenesis induced by UV light.
Before concluding, I would like to point out that, with the availability of the total sequence information of the human genome, and the development of various techniques and materials for sophisticated studies at the molecular level, some of the early work of Dick Setlow and other pioneers in molecular biology may not generate much excitement for the young generation today. However, the brilliance of Dick’s early studies should be judged by his ability to push the frontiers of science profoundly by using contemporaneous experimental tools and mostly by dint of his scientific acuity and insight.
INTRODUCTION
Cellular damage due to reactive oxygen species (ROS) is currently a hot topic in biomedical research. The mutagenic DNA lesions generated by these oxidizing species appear to be responsible for sporadic carcinogenesis, in addition to a plethora of other pathophysiological states. In this article, we will review our current state of understanding of the mechanism of repair of oxidized base lesions and will venture speculation regarding the future direction of this area of research in the postgenomic era. The genomes of all organisms are continuously exposed to a wide variety of insults.
Some of these are unavoidable because they are endogenously generated, while others, e.g., UV light of the sun, are exogenous and can thus be avoided. Because they are generated as by-products of respiration, reactive oxygen species constitute the major class of endogenous toxic agents in aerobic organisms. These include all partially reduced oxygen species, namely, the O2−• anion radical, H2O2, and •OH radicals. Both •NO and HOCl are also reactive species and are included in the ROS group. The primary function of •NO is to act as a second messenger in signaling processes, whereas HOCl, generated by myeloperoxidase in activated neutrophils from Cl− and H2O2, may be involved in inflammatory signaling and in preventing infection. Although all of these reactive molecules have a beneficial role in the activation of signaling processes, they are also genotoxic and oxidize various cellular components, including DNA. ROS-induced cellular changes have been implicated in a multitude of diseases, including cardiovascular dysfunction, arthritis, and cancer, as well as in the aging process. ROS induces many oxidized base lesions and abasic (AP) sites, many of which are mutagenic. Critical mutations induced by ROS in oncogenes and/or tumor suppressor genes result from misreplication and could lead to sporadic cancer. Widespread acceptance of this scenario is underscored by the current exhortation for dietary intake of large doses of a variety of antioxidants.
The oxidized bases and AP sites, as well as DNA single-strand breaks induced by ROS with 3′ phosphoglycolate blocked ends, are repaired predominantly by the DNA base excision repair (BER) pathway, which until recently was believed to be the simplest and most thoroughly defined of all repair processes. Nevertheless, the BER pathway never received as much attention as the other excision repair pathways, i.e., nucleotide excision repair (NER) and DNA mismatch repair (MMR), presumably because deficiencies of NER and MMR have been linked to cancer and other diseases, whereas no disease phenotype has been known to be linked to BER deficiency so far. However, compared to the other two DNA excision repair pathways, BER appears to be rather simple and requires five distinct enzymatic activities in the basic reaction steps.
In fact, four enzymes provide these five activities for carrying out repair of DNA containing AP sites or base damage. As outlined in Fig. 1, these are as follows: a DNA glycosylase, AP-endonuclease I (APE1), DNA polymerase β (Polβ), and a DNA ligase [Mitra et al., 1997]. After removal of the damaged base, e.g., 8-oxoguanine (G*) by 8-oxoguanine-DNA glycosylase (OGG), the resulting AP site is cleaved by APE, to generate 3′ OH and 5′ deoxyribose phosphate (dRP). Polβ has an intrinsic dRPase activity as well as the DNA polymerase activity [Matsumoto and Kim, 1995; Prasad et al., 1998]. As was shown in a series of elegant studies, the dRPase activity cleaves the dRP residue generating a nucleotide gap with 3′ OH and 5′ phosphate. Polβ fills in the single nucleotide gap, and the resulting nick is finally sealed by DNA ligase. The whole pathway has been demonstrated in vitro with DNA oligonucleotide duplexes with a single base lesion [Kubota et al., 1996; Singhal et al., 1995].
Fig. 1.
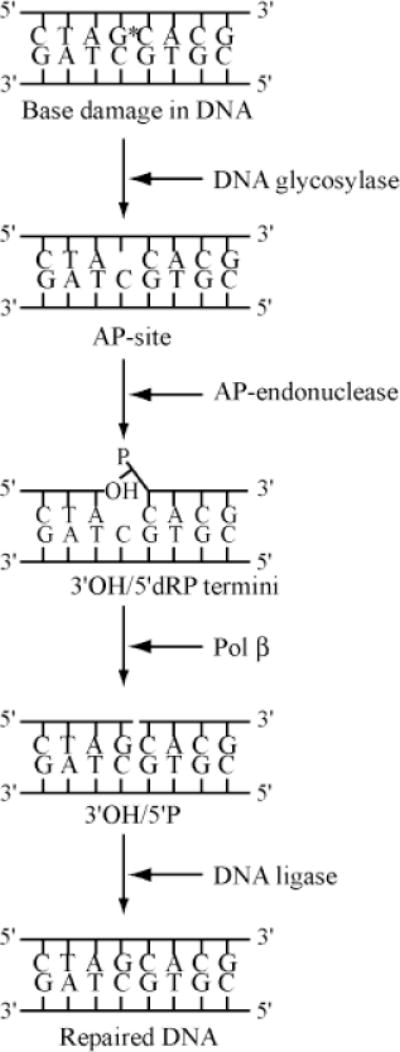
Schematic outline of the basic steps in DNA base excision repair in mammalian cells. The damaged base is represented by G* in duplex DNA, and the AP site by the missing base. Other details are discussed in the text.
The other types of DNA lesions repaired by the BER pathway are single-strand breaks with 3′ blocking residues at these cleavage sites. These could be induced directly because of an attack of sugar residues by ROS and include 3′ phosphate, 3′ phosphoglycolaldehyde, or 3′ phosphoglycolate [Breen and Murphy, 1995]. DNA glycosylases specific for oxidized base lesions have associated AP lyase activity that causes cleavage of the AP site produced because of excision of damaged bases. β or βδ elimination reaction generates 3′-phospho-α, β-unsaturated aldehyde or 3′ phosphate. In all cases when the 3′ terminus is blocked at the site of strand cleavage, the 5′ terminus is not blocked and contains a phosphate residue. Thus, unlike in the case of APE cleavage product of an AP site, where the 5′ terminus is blocked, the 3′ terminus of ROS-induced DNA strand breaks needs to be cleaned. The intrinsic 3′ phosphodiesterase activity of all APEs is required for removing the 3′ blocking group. Thus APE plays a central role in repair of all types of ROS-damaged DNA.
Recent Evidence for Complexities in the BER Pathway
Several recent observations have raised questions about this apparent simplicity of the BER pathway, particularly for the in vivo repair of oxidative DNA damage. These are summarized as follows.
Involvement of Non-BER Proteins in BER
Seminal studies by Priscilla Cooper, Tony Leadon, and their collaborators have shown that the repair of thymine glycol in DNA of H2O2-treated cells preferentially occurs in transcriptionally active regions, and particularly in the transcribed DNA strand [Cooper et al., 1997; Gowen et al., 1998]. Transcription-coupled repair (TCR) was discovered in Hanawalt’s laboratory as a distinct subpathway of NER [Bohr et al., 1985; Mellon et al., 1987]. The bulky DNA adducts, such as UV-photoproducts, block transcription and replication. Even in quiescent G0/G1 cells, which do not undergo DNA replication, transcription of critical genes occurs continuously, and the transcription complex stops at bulky adduct sites. It thus makes sense that prompt repair is required in these transcriptionally active regions, and specifically of adducts in the transcribed strand relative to the bulk of the chromatin, which is transcriptionally inactive. Thus, repair occurs in the transcribed strand at a much higher rate than in the chromatin overall. In fact, global repair of UV damage in rodents is quite inefficient relative to that in human cells. In contrast, similar TCR activity is present in both rodent and human cells.
The observation of a similar distinction between TCR and global repair of thymine glycol and possibly other oxidative lesions in DNA was unexpected, because most such lesions do not block transcription or replication. It is therefore possible that although the overall processes of TCR of bulky adducts and oxidative lesions are similar, these may be mechanistically distinct, particularly in regards to recruitment of the repair machines.
More recently, Cooper and collaborators showed that several NER proteins, in particular XPG, an essential endonuclease in NER, are also involved in repair of thymine glycol in vivo, even though its nuclease activity is dispensable. This supports the idea that TCR of oxidized bases does not function via NER, and BER is still the pathway of choice [Cooper et al., 1997].
Leadon and collaborators showed that the BRCA1 and its associated protein BAP1, which have been discovered from their role in the suppression of breast cancer, are also involved in the TCR of thymine glycol [Gowen et al., 1998; S. A. Leadon, personal communication]. BRCA1 and BRCA2 have also been implicated in TCR of 8-oxoguanine [Le Page et al., 2000c]. Finally, p53, arguably the most important tumor suppressor protein, recently has been shown to activate repair of DNA AP sites in an in vitro assay [Offer et al., 2001].
These observations have posed a paradox that the in vivo repair process appears to be far more complicated than the in vitro reaction pathway. However, this could be explained by the possibility that a large array of accessory proteins is required for in vivo repair of ROS-induced DNA damage, which may be involved in various steps starting with lesion recognition and extending to coordination of the repair process.
Inherent Problems in Repair of Chromatin DNA
While almost all in vitro repair studies have utilized naked oligonucleotides or plasmid DNA, DNA exists in vivo only in a chromatinized form. Early efforts to repair DNA in chromatin form showed that histones inhibit repair. Thus, the next level of challenge lies in determining how lesions, both bulky adducts and oxidatively modified bases in chromatin, are repaired by NER or BER processes. Recent in vitro studies on repair of DNA lesions in reconstituted chromatin form are promising [Hara et al., 2000]. However, it appears that in both BER and NER, additional components are needed to allow access of repair machineries to the lesion site which may be in intimate contact with nucleosome core histones and nonhistone chromosomal proteins. In view of the recent observations on the need for chromatin remodeling during transcription, one obvious possibility is that acetylation, as well as other posttranslational modifications of histones, and possibly other processes, are needed to loosen the nucleosome core from the wrapped DNA for repair as much as for transcription and replication. It was shown recently that the transcriptional activator CBP/p300, with histone acetylase activity, may be required for chromatin remodeling via histone acetylation. It also interacts with PCNA, which is an essential component of the DNA replication machinery, and may thus be needed for nascent DNA synthesis after UV irradiation [Hasan et al., 2001]. Thus CBP/p300 may also participate in chromatin remodeling during repair of oxidized base lesions.
Rapid Repair of Oxidized Bases In Vivo
Finally, there appears to be a discrepancy between the in vivo rate of repair of oxidized base lesions via the BER pathway in mammals and the in vitro reaction rate of mammalian DNA glycosylases specific for these lesions (namely, 8-oxoguanine-DNA glycosylase or OGG1, and endonuclease III homolog or NTH1). These enzymes, purified to homogeneity in recombinant form, are noted by their extremely low turnover under optimum conditions when used alone with their DNA substrates [Hill et al., 2001; Ikeda et al., 1998]. In fact, a hallmark of all DNA glycosylases except uracil-DNA glycosylase (UDG) is their low specific activity. At least the early fast phase of in vivo repair of oxidized bases in nuclear DNA occurs in minutes, whereas the in vitro turnover rate of a human OGG1 molecule is ~0.1/min at 37°C. Furthermore, we need to bear in mind that the lesions in chromatin DNA are not as easily accessible as in naked DNA, and in vivo repair has to be carried out in the presence of a huge excess of undamaged DNA which usually inhibits DNA glycosylases. Thus, the high efficiency of in vivo repair is indeed amazing and cannot be reconciled with the in vitro reaction rate of the early enzymes in the repair process.
Coordination of the Steps in the Repair of Oxidized Bases via the BER Pathway
Taken together, these observations paint a much more complex picture of in vivo repair than the minimum requirement observed for in vitro repair [Mitra et al., 1997]. Although all of the steps in this process are not understood as yet, a general pattern of sequential steps has emerged which could account for rapid repair in vivo and is summarized as follows.
Damage-Induced Nuclear Targeting of DNA Glycosylases and APEs
All of the early enzymes in the BER pathway have putative nuclear localization signals (NLS), but they are not exclusively localized in the nucleus, rather distributed more or less randomly throughout the cell. However, within 15–30 min after the cells in culture were exposed to a subtoxic dose of oxidative stress (including ionizing radiation), many of these proteins (specifically OGG1 and APE1, which were examined immunocytochemically) were found to be concentrated in the nucleus (Fig. 2). Because of the rapidity of this response, it is unlikely that the nuclear accumulation of these proteins represents their de novo synthesis. This suggests that a signaling pathway is induced by oxidative stress, which causes transfer of the repair enzymes to the nucleus, which is their site of action.
Fig. 2.
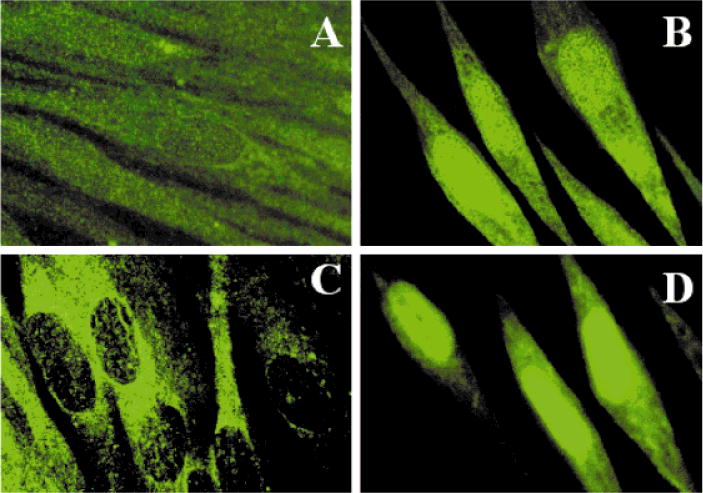
Nuclear accumulation of APE1 and OGG1 induced by ionizing radiation in primary human fibroblast cells. Control and γ-irradiated (0.1 Gy) WI38 cells were fixed and treated with rabbit anti-APE1 or anti-OGG1 antibodies and then visualized by treatment with fluorescein-conjugated anti-rabbit IgG. A and B, anti-OGG1 antibody; C and D, anti-APE1 antibody; A and C, control cells; B and D, 30 min after irradiation.
It now has become clear that the BER pathway is active in mitochondria [LeDoux et al., 1999], and mitochondrial isoforms of several DNA glycosylases have been identified [Nishioka et al., 1999; Ohtsubo et al., 2000]. Although we will not discuss the complexities of oxidative DNA damage repair in mitochondria, our preliminary results suggest that oxidative stress induces targeting of DNA glycosylases to the mitochondria, just as we have observed for nuclear accumulation of these enzymes (I. Boldogh, unpublished observations).
Such nuclear import (and mitochondrial targeting) may be a part of the general adaptive response of cells and organisms for enhanced efficiency of repair, particularly of exogenously induced oxidative DNA damage, which may be more critical than other types of damage.
Hands-Off Mode of Enzyme Reactions for Sequential Steps in the BER Pathway
Tainer’s laboratory, in collaboration with us, recently used X-ray crystallography to elucidate the structure of human APE1 complexed with the substrate AP-containing duplex oligonucleotide, as well as with the cleaved product [Mol et al., 2000]. The cleaved phosphodeoxyribose residue was found to be located in close proximity to an arginine residue (Arg 177). Mutating this residue to alanine increased the turnover of the enzyme about threefold. This and other related observations led to the hypothesis that both DNA glycosylases and APEs do not conform to ideal enzyme behavior. These enzymes tend to bind to the product and may be subject to product inhibition. In fact, substrate concentration vs. velocity plots for the DNA glycosylases (as well as for APE1) show non-Michaelis–Menten kinetics and are indicative of product inhibition [Hill et al., 2001]. Additionally, DNA binding studies showed that although both-wild type and R177A APE mutants bind the AP-containing substrate DNA, only the wild-type protein binds the product efficiently. Even more interestingly, the first 60 amino acid residues in the 36-kDa APE1 polypeptide were found to be dispensable for APE activity, although comparative kinetics of the wild-type and deletion mutant enzymes have not been investigated carefully [Izumi and Mitra, 1998]. We have now found that some residues in the N-terminal domain, as well as Arg177, are involved in the binding of the cleaved DNA (T. Izumi, unpublished observations).
More direct evidence for the general phenomenon of product inhibition and binding was provided by experiments on DNA glycosylases. Earlier studies showed that the G • T-specific thymine-DNA glycosylase (TDG) did not turn over very efficiently and that APE1 stimulated its activity [Waters et al., 1999]. Similarly, UDG was also found to be stimulated by APE1 [Parikh et al., 1998]. We and others have made similar observations with the turnover of human OGG1 [Hill et al., 2001; Vidal et al., 2001]. We have shown specifically that OGG1 has a higher affinity for its product AP site than for its substrate [Hill et al., 2001; Izumi et al., 1999]. Furthermore, although both APE1 and OGG1 have comparable affinity for the AP site, APE1 is able to compete for this lesion because of its much higher turnover. Unlike in the case of UDG and TDG, only molar equivalence of APE1 is required for near maximum stimulation of OGG1 [Hill et al., 2001]. Furthermore, at least in the absence of DNA, no direct physical interaction has been detected so far between APE1 and OGG1 in the absence of DNA (G. Roy, unpublished observations).
These results indicate that at least the early enzymes of BER carry out synchronized reaction steps when the glycosylase holds on to the AP site reaction product until the APE1 is recruited in a hands-off fashion, as proposed earlier [Mol et al., 2000; Wilson and Kunkel, 2000].
Long Patch vs. Short Patch Repair Synthesis in BER
DNA polymerase β (Polβ) has long been known to be the main repair polymerase in mammalian cells. DNA repair by definition involves unscheduled DNA synthesis in the absence of DNA replication in S-phase cells. Unlike replicative DNA polymerases α, δ, and ε, the cell cycle-dependent levels of which are the highest in S phase, the Polβ level does not change significantly during cell cycle progression. Thus, repair of the APE cleavage product 5′ dRP, which occurs in all phases of the cell cycle, should be carried out by Polβ. As shown in a series of elegant studies, the intrinsic dRPase activity of Polβ is localized in its N-terminal 8-kD domain, whereas the DNA polymerase activity requires the 31-kD C-terminal domain [Prasad et al., 1998]. The 5′ dRPase activity is an AP lyase rather than a 5′ endonuclease and involves the formation of a covalent Schiff base intermediate between the C1′ aldehyde group of the dRP and a Lys residue in the 8-kD domain [Piersen et al., 1996]. Subsequent β-elimination reaction causes breakage of the phosphodiester bond with the next deoxynucleotide, resulting in a free α, β-unsaturated phosphoglycolal-dehyde and 5′-phosphate terminus in the DNA strand. The one-nucleotide gap thus generated in the DNA strand could then be filled in by Polβ in a concerted reaction with dRP removal [Singhal et al., 1995]. The observations that Polβ is most efficient in filling a one-nucleotide gap and interacts with the 5′-phosphate terminus upstream of the gap are consistent with this function of Polβ in BER. The one nucleotide repair patch observed in this case is the shortest possible. After the gap filling, which generates a single-strand nick, DNA ligase seals the nick to complete the repair process.
Physical Interaction Among BER Proteins
Early studies show that Polβ forms a complex with DNA ligase I [Dimitriadis et al., 1998]. It also was shown independently that such short patch pair could involve DNA ligase III, which exists as a tightly bound complex with XRCC1 [Kubota et al., 1996]. XRCC1 deficiency causes cellular sensitivity to ionizing radiation (or oxidative stress) [Thompson and West, 2000]. It has been shown recently that XRCC1 forms a complex with polynucleotide kinase (PNK), which has intrinsic 5′ kinase and 3′-phosphatase activities for DNA, along with Polβ and DNA ligase III [Whitehouse et al., 2001]. PNK removes the phosphate residue from the replication blocking 3′-phosphate terminus generated after radiation or ROS-induced strand cleavage. Thus, a transient complex of APE1, and/or PNK, with XRCC1, Polβ, and DNA ligase III may be responsible for repair of ionizing radiation (IR)-induced DNA single-strand breaks with 3′ blocking groups. Physical association between Polβ and APE1 in the presence of DNA was observed earlier [Bennett et al., 1997].
All of these interactions paint a picture of transient complexes being formed to carry out repair of DNA damage via the BER pathway. That DNA glycosylases are the only components of this pathway [with the possible exception of MutY homolog (MYH)], which may not interact directly with APE1, could be explained by the scenario that multiple DNA glycosylases react with widely diverse lesions, and all generate an AP site for which repair is then carried out by a common set of proteins.
Long Patch Repair by Replicative DNA Polymerases
Several lines of investigation have recently suggested that BER may also be carried out by the replicative DNA polymerases [Mitra et al., 1997]. First, although Polβ knockout mouse mutants could not be generated because of embryonic lethality, fibroblast lines were developed from the mutant embryos. While these cells are more sensitive to alkylating mutagens than the wild-type cells, the mutation did not affect their sensitivity to oxidative stress [Ochs et al., 1999; Pascucci et al., 1999; Sobol et al., 1996]. These results indicate that Polβ is dispensable for repair of oxidative DNA damage, presumably because replicative polymerases can carry out such repair. Second, repair of misincorporated bases in the nascent DNA strand is carried out by the DNA mismatch repair process. For example, MutY (or its human ortholog, MYH) removes A when present opposite 8-oxoG (or G) [Grollman and Moriya, 1993; Michaels and Miller, 1992]. This suggests that A is specifically removed by this DNA glycosylase when it is incorporated during misreplication of 8-oxoG in the template strand. While such replication-associated repair could be carried out by Polβ, it is easier to visualize that the nascent strand-specific repair is carried out by replicative DNA polymerases. Recent studies showing interaction of MYH with RPA and PCNA, which are components of the DNA replication machinery, strongly support the prediction that MYH is specific for replicating DNA [Parker et al., 2001]. However, we cannot exclude the possibility that the Polβ-mediated repair is the primary mechanism for BER and that replicative DNA polymerases substitute for Polβ in its absence in Polβ null mutant cells [Dianov et al., 1998].
Independent studies during the last 2–3 years have provided compelling evidence that replicative DNA polymerases δ and ε (Polδ and Polε) can be utilized during repair of AP sites after cleavage with APE1 [Klungland and Lindahl, 1997; Matsumoto et al., 1994]. Because these polymerases do not have intrinsic dRPase activity, the 5′ terminus at the cleavage site needs to be cleaned up by an endo (or exo) nuclease. One obvious candidate is the flap endonuclease (FEN1), a paralog of the 5′-exonuclease activity intrinsic to Escherichia coli DNA polymerase I. FEN1 is normally involved in removal of the 5′-RNA primers of nascent Okazaki fragments during DNA replication [Murante et al., 1998]. In vitro reconstitution experiments showed that FEN1 can remove the dRP residue, along with several additional residues, from the 5′ terminus [Gary et al., 1999]. This will leave a multinucleotide gap in the duplex DNA to be filled in subsequently by Polδ (or Polε). Such multinucleotide repair patches (two to six nucleotides) indeed have been observed in vitro in assays for AP site repair with cell-free extracts or a reconstituted system with purified proteins [Fortini et al., 2000; Pascucci et al., 1999]. It is interesting that the replicative polymerases are stimulated by PCNA, which is primarily expressed during S phase and binds tightly to FEN1. In fact, PCNA also binds to DNA ligase I, suggesting that the replication machinery involves physical interaction among the key enzymes.
Physical Interaction Between APE1 and FEN1/PCNA
The long patch vs. short patch pathways during the BER process for AP site repair are distinguished by the mechanism of 5′-end cleaning after the APE reaction. Because APE1 was found to interact with Polβ, leading to short patch repair, it was expected that APE1 can similarly interact with FEN1 for long patch repair. In fact, that was exactly what we observed serendipitously. During purification of endogenous APE1 from cultured HeLa cells, we observed routinely the presence of several contaminating proteins which were coeluted with APE1 during early stages of purification. We identified one of these contaminants to be FEN1 by determining the partial sequence of its N-terminal region. Subsequently we showed by coimmunoprecipitation that APE1 was present in the immunoprecipitates of both FEN1 and PCNA (G. Roy et al., unpublished observations). The presence of FEN1 and PCNA in the same immunoprecipitate was entirely expected because of the tight complex between the proteins [Chen et al., 1996].
To test whether the observed interaction between APE1 and PCNA is mediated by FEN1, which interacts with both proteins, we performed coimmunoprecipitation studies for testing in vitro complex formation of recombinant APE1 with FEN1 and PCNA in a pairwise fashion. We used human O6-methylguanine-DNA methyltransferase (MGMT) as a negative control. MGMT is an unusual repair protein which acts in a stoichiometric suicide reaction to restore the original guanine by dealkylation of O6-alkylguanine, a mutagenic DNA lesion induced by alkylating agents [Mitra and Kaina, 1993]. Because MGMT acts alone and does not require any BER proteins, it is not expected to interact with them. Our results show clearly that APE1 forms binary complexes with PCNA and FEN1, whereas MGMT was not present in the immunocomplexes of FEN1, PCNA, or APE1 (Roy et al., unpublished observations).
Taken together, these results suggest that APE1 first binds to the AP site and then may interact with Polβ, leading to single-nucleotide gap synthesis. Alternatively, APE1 interacts with FEN1, which removes multiple nucleotides as well as the dRP residue, leading to repair synthesis to fill in a multinucleotide gap. We have observed that although FEN1 does not increase APE1 activity, APE1 increases the specific activity of FEN1 by about threefold. In an analogous situation, Polβ was activated by APE1, but not vice versa [Bennett et al., 1997]. We should point out that these preliminary observations have raised more questions than provided answers. For example, what is the biochemical basis of the enhancement of FEN1 activity by APE1, and what recognition motifs of these proteins are needed for their interaction? We have carried out our enzymatic assays with purified FEN1 and APE1 in the absence of PCNA and the DNA replication machinery. It is possible that these activities and interactions would be modulated significantly in the presence of other proteins, in particular, PCNA and Polδ. All of these proteins are expected to be present during long patch repair synthesis, as depicted in Fig. 3. It also should be evident that other additional participants, not indicated in the figure, are likely to be involved in repair pathways. This figure is intended to underscore the presence of a point of divergence in the BER process after the APE1 reaction. Another point of divergence may be in the final step of ligation. Because the roles of both DNA ligase I and DNA ligase III/XRCC1 in BER have been shown by in vitro studies, further experiments may elucidate the mechanism of selection of the DNA ligase in vivo.
Fig. 3.

A model of pathway decision for repair of an AP site. After cleavage of the AP site with APE1, either Polβ or FEN1 is recruited at the lesion site because of their interaction with APE1. Multiple factors, including relative abundance of these enzymes, location of the lesion in the genome, cell cycle phase, as well as chromatin organization, may determine which enzyme takes over from APE1 and thus decide the pathway. Polβ acts as both a dRPase and a DNA polymerase to perform a single base-filling reaction as shown on the left side. In the pathway outlined on the right side, FEN1 cooperates with PCNA/Polδ(ε) or Polβ to carry out a multibase-filling reaction. No sequential order in recruitment of these proteins in completing repair is implied. The roles of XPG, BRCA1, MMR, and other accessory proteins are not indicated in these repair pathways.
Finally, we should recognize the possibility of an alternative pathway for long patch repair which has been proposed to involve Polβ. In this model, Polβ does not interact with FEN1 directly but in an indirect fashion fills in the long patch gap generated by FEN1 [Prasad et al., 2000]. We need to recognize that all of the experimental evidence so far, which has led to various models, involves only in vitro studies. In vivo experiments, although rather difficult, are required to establish unequivocally the role of various BER proteins in short patch vs. long patch repair.
Pathway Decision and DNA Replication-Associated Base Excision Repair
It may be interesting to speculate about how a cell chooses to utilize the Polβ vs. FEN1-dependent repair pathway when carrying out repair of AP sites. In spite of the observed interactions among the BER proteins, it is likely that these proteins do not form stable complexes. In fact, we postulate that distinct transient complexes of BER proteins are responsible for repair of lesions in a cell cycle and transcription-dependent fashion. Most mammalian cells are postmitotic and do not undergo further multiplication. However, although these are all transcriptionally active, much of the genomic DNA in any cell is not transcribed at all and even the transcriptionally competent regions are not transcribed in all cell types. The existence of distinct TCR for bulky DNA adducts in eukaryotic cells could be rationalized by the fact that such adducts block transcription, which is essential for survival even for nonreplicating G0/G1 cells [Mellon et al., 1987]. Thus, although a nonreplicating cell can tolerate a significant load of persistent lesions in the nontranscribed strand, and more globally in the nontranscribed regions, a specialized process has evolved to carry out repair of the lesions in the transcriptionally active region, and specifically, the transcribed strand [Cooper et al., 1997]. The recent discovery of similar TCR in vivo for oxidized base lesions, which do not completely inhibit transcription, suggests that complete inhibition of transcription may not be an essential signal for this TCR process [Le Page et al., 2000a,b). It is also possible that the mechanism of TCR of oxidized base lesions differs in specific details from that of TCR of bulky adducts which are subject to nucleotide excision repair.
Unlike transcription, DNA replication occurs only during the S phase of dividing cells. Because bulky DNA adducts block not only transcription but also replication, whether a specialized replication-coupled nucleotide excision repair process is utilized to remove the replication-blocking adducts in pre-S-phase cells in vivo is not known. However, postreplication repair has to be active in dividing cells to correct misincorporation of nucleotides during DNA synthesis. Although it has not been demonstrated directly, DNA mismatch repair is predicted to be specific for the nascent strand and is thus a replication-associated process.
The BER process is also required for repairing U misincorporated in place of T or A opposite 8-oxoG in the nascent strand. In fact, among multiple UDGs identified in mammalian cells, UNG2 appears to repair U in the nascent strand because its expression increases during S phase and was found to be associated with replication foci in the nucleus [Nilsen et al., 2000]. Similarly, MYH, which repairs A opposite 8-oxoG, has to be nascent strand-specific, which we had predicted earlier [Hazra et al., 1998]; recent results showing its overexpression during S phase and its association with the replication foci support this prediction [Boldogh et al., 2001; Parker et al., 2001].
We and others have proposed that unlike bulky DNA lesions, oxidized base lesions such as 8-oxoG and 5-hydroxyuracil are incorporated by DNA polymerases into the nascent DNA strand from the deoxynucleotide pool. Thus 8-oxoG can be incorporated opposite A in the template strand. In this situation, repair of A by MYH would be mutagenic, so excision of 8-oxoG is required to prevent mutation. Because OGG1, the predominant DNA glycosylase specific for 8-oxoG, is inactive with an 8-oxoG • A pair, our identification of a second OGG, OGG2, which has a significant activity in excising 8-oxoG from an 8-oxoG • A pair, both in human cells and in E. coli, led to the model of bipartite antimutagenic processing of 8-oxoguanine [Hazra et al., 1998, 2000]. We have proposed that OGG2 (similar to MYH) is specific for the nascent DNA strand. Recently OGG1 homozygous null mouse mutants (and embryo fibroblast lines therefrom) have been generated. A high level of 8-oxoG was observed in the DNA of mutant cells, indicating that OGG1 is largely responsible for global repair of 8-oxoG in the genome [Minowa et al., 2000; Klungland et al., 1999]. However, using a transfected plasmid rescue assay, two groups have recently shown that 8-oxoG repair was quite efficient in the transcribed strand of the transfected plasmid. This provides strong support for the existence not only of TCR of 8-oxoG, which does not block transcription or replication, but also for the hypothesis that such transcribed strand-specific repair does not require OGG1 [Le Page et al., 2000a,b). Whether such repair is carried out by OGG2 or via some other process remains to be elucidated. Nevertheless, we propose that the BER process for oxidized bases can be subdivided into three types (Table I).
TABLE I.
A Model of Repair Subpathways for Oxidized Bases
| Global genome repair | Transcription coupled repair | Replication-associated repair
|
||
|---|---|---|---|---|
| Prereplication (parental strand) | Postreplication (nascent strand) | |||
| Cell cycle | Both proliferating and postmitotic cells | Both proliferating and postmitotic cells | Proliferating cells in S phase | |
| Strand specificity of repair | Both transcriptionally active and inactive regions in both strands | Transcribed strand in transcriptionally active region | Total genome | |
| OGG1 | UNG2 MYH OGG2 |
|||
| Specific repair Proteins | Pol β, XRCC1, DNA ligase III p53 (?) |
FEN1, PCNA, Pol δ (ε) BRCA1, BRCA2, TFIIH, CSB, XPG, and MMR proteins DNA ligase I | FEN1, PCNA, Pol δ (ε), Pol β (?)XPG and MMR proteins (?)DNA ligase I | |
| Repair patch size | Single nucleotide | Multiple nucleotides | Multiple nucleotides | |
These three subpathways differ in the requirement of various enzymes and other proteins. The details in this list are speculative and based on our current understanding of the established or predicted roles of the participants. We should stress that we have not included in the list all likely participants of the repair pathway. The purpose of presenting the model in this review is to stimulate discussions regarding its feasibility and to design experiments to test its predictions.
Concluding Remarks
In this brief review, we hope to have provided a glimpse of the complexities of in vivo repair processes for oxidative DNA damage, and our speculations to explain the experimental results. One prediction we can make with absolute certainty is that the models and hypotheses proposed here and elsewhere will require modification in the near future as more knowledge accumulates regarding the mechanisms and functions of proteins such as XPG, BRCA tumor suppressor proteins, and DNA mismatch repair proteins, which have no obvious BER activity, and their interactions during repair of oxidized bases and AP sites. One aspect we have left untouched is the impact of chromatin structure on BER, including its remodeling as observed during transcription. The excitement in unraveling the mysteries of mammalian DNA repair, particularly of oxidative damage repair, is still gaining momentum in the postgenomic era and is far from reaching crescendo.
Acknowledgments
The research carried out in our laboratories have been funded by U.S. Public Health Services Grants CA81063, CA53791, ES08457 (S.M.), and CA 84461 (I.B.). We thank several colleagues, particularly Dr. Rabindra Roy, Ms. Gargi Roy, Dr. Kishor Bhakat, and Ms. Julie Lock for carrying out many of the studies discussed in this review, and Dr. David Konkel for critically reading the manuscript.
Contract grant sponsor: U.S. Public Health Services; Contract grant number: CA81063, CA53791, ES08457, and CA 84461.
References
- Bennett RA, Wilson DM, III, Wong D, Demple B. Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc Natl Acad Sci USA. 1997;94:7166–7169. doi: 10.1073/pnas.94.14.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr VA, Smith CA, Okumoto DS, Hanawalt PC. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell. 1985;40:359–369. doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- Boldogh I, Milligan D, Lee MS, Bassett H, Lloyd RS, McCullough AK. hMYH cell cycle dependent expression, subcellular localization, and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 2001;29:2802–2809. doi: 10.1093/nar/29.13.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce RP, Howard-Flanders P. Release of ultraviolet light-induced thymine dimmers from DNA in E. coli K-12. Proc Natl Acad Sci USA. 1964;51:293–300. doi: 10.1073/pnas.51.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen AP, Murphy JA. Reactions of oxyl radicals with DNA. Free Radic Biol Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- Chen U, Chen S, Saha P, Dutta A. p21Cip1/Waf1 disrupts the recruitment of human Fen1 by proliferating-cell nuclear antigen into the DNA replication complex. Proc Natl Acad Sci USA. 1996;93:11597–11602. doi: 10.1073/pnas.93.21.11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper PK, Nouspikel T, Clarkson SG, Leadon SA. Defective transcription coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science. 1997;275:990–993. doi: 10.1126/science.275.5302.990. [DOI] [PubMed] [Google Scholar]
- Dianov G, Bischoff C, Piotrowski J, Bohr VA. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J Biol Chem. 1998;273:33811–33816. doi: 10.1074/jbc.273.50.33811. [DOI] [PubMed] [Google Scholar]
- Dimitriadis EK, Prasad R, Vaske MK, Chen L, Tomkinson AE, Lewis MS, Wilson SH. Thermodynamics of human DNA ligase I trimerization and association with DNA polymerase beta. J Biol Chem. 1998;273:20540–20550. doi: 10.1074/jbc.273.32.20540. [DOI] [PubMed] [Google Scholar]
- Fortini P, Pascucci B, Belisario F, Dogliotti E. DNA polymerase beta is required for efficient DNA strand break repair induced by methyl methane sulfonate but not by hydrogen peroxide. Nucleic Acids Res. 2000;28:3040–3046. doi: 10.1093/nar/28.16.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote RS, Mitra S, Pal BC. Demethylation of O6-methylguanine in a synthetic DNA polymer by an inducible activity in Escherichia coli. Biochem Biophys Res Commun. 1980;97:654–659. doi: 10.1016/0006-291x(80)90314-9. [DOI] [PubMed] [Google Scholar]
- Gary R, Kim K, Cornelius HL, Park MS, Matsumoto Y. Proliferating cell nuclear antigen facilitates excision in long-patch base excision repair. J Biol Chem. 1999;274:4354–4363. doi: 10.1074/jbc.274.7.4354. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Avrutskaya AV, Latour AM, Koller BH, Leadon SA. BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science. 1998;281:1009–1012. doi: 10.1126/science.281.5379.1009. [DOI] [PubMed] [Google Scholar]
- Grollman AP, Moriya M. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- Hara R, Mo J, Sancar A. DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol Cell Biol. 2000;20:9173–9181. doi: 10.1128/mcb.20.24.9173-9181.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan S, Hassa PO, Imhof R, Hottiger MO. Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature. 2001;410:387–391. doi: 10.1038/35066610. [DOI] [PubMed] [Google Scholar]
- Hazra TK, Izumi T, Maidt L, Floyd RA, Mitra S. The presence of two distinct 8-oxoguanine repair enzymes in human cells: their potential complementary roles in preventing mutation. Nucleic Acids Res. 1998;26:5116–5122. doi: 10.1093/nar/26.22.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra TK, Izumi I, Venkataraman R, Kow YW, Dizdaroglu M, Mitra S. Characterization of a novel 8-oxoguanine-DNA glycosylase activity in Escherichia coli and identification of the enzyme as endonuclease VIII. J Biol Chem. 2000;275:27762–27767. doi: 10.1074/jbc.M004052200. [DOI] [PubMed] [Google Scholar]
- Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Biswas T, Roy R, Izumi T, Boldogh I, Kurosky A, Sarker AH, Seki S, Mitra S. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease direct identification of Lys-212 as the active nucleophilic residue. J Biol Chem. 1998;273:21585–21593. doi: 10.1074/jbc.273.34.21585. [DOI] [PubMed] [Google Scholar]
- Izumi T, Mitra S. Deletion analysis of human AP-endonuclease: minimum sequence required for the endonuclease activity. Carcinogenesis. 1998;19:525–527. doi: 10.1093/carcin/19.3.525. [DOI] [PubMed] [Google Scholar]
- Izumi T, Malecki J, Chaudhry MA, Weinfeld M, Hill JH, Lee JC, Mitra S. Intragenic suppression of an active site mutation in the human apurinic/apyrimidinic endonuclease. J Mol Biol. 1999;287:47–57. doi: 10.1006/jmbi.1999.2573. [DOI] [PubMed] [Google Scholar]
- Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- LeDoux SP, Driggers WJ, Hollensworth BS, Wilson GL. Repair of alkylation and oxidative damage in mitochondrial DNA. Mutat Res. 1999;434:149–159. doi: 10.1016/s0921-8777(99)00026-9. [DOI] [PubMed] [Google Scholar]
- Le Page F, Klungland A, Barnes DE, Sarasin A, Boiteux S. Transcription coupled repair of 8-oxoguanine in murine cells: the Ogg1 protein is required for repair in nontranscribed sequences but not in transcribed sequences. Proc Natl Acad Sci USA. 2000a;97:8397–8402. doi: 10.1073/pnas.140137297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Page F, Kwoh EE, Avrutskaya A, Gentil A, Leadon SA, Sarasin A, Cooper P. Transcription-coupled repair of 8-oxoguanine: requirement for XPG, TFIIH, and CSB and implications for Cockayne syndrome. Cell. 2000b;101:159–171. doi: 10.1016/s0092-8674(00)80827-2. [DOI] [PubMed] [Google Scholar]
- Le Page F, Randrianarison V, Marot D, Cabannes J, Perricaudet M, Feunteun J, Sarasin A. BRCA1 and BRCA2 are necessary for the transcription-coupled repair of the oxidative 8-oxoguanine lesion in human cells. Cancer Res. 2000c;60:5548–5552. [PubMed] [Google Scholar]
- Matsumoto Y, Kim K, Bogenhagen DF. Proliferating cell nuclear antigen-dependent abasic site repair in Xenopus laevis oocytes: an alternative pathway of base excision DNA repair. Mol Cell Biol. 1994;14:6187–6197. doi: 10.1128/mcb.14.9.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- Mellon I, Spivak G, Hanawalt PC. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 1987;51:241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- Michaels ML, Miller JH. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) J Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa O, Arai T, Hirano M, Monden Y, Nakai S, Fukuda M, Itoh M, Takano H, Hippou Y, Aburatani H, Masumura K, Nohmi T, Nishimura S, Noda T. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc Natl Acad Sci USA. 2000;97:4156–4161. doi: 10.1073/pnas.050404497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Kaina B. Regulation of repair of alkylation damage in mammalian genomes. Prog Nucleic Acid Res Mol Biol. 1993;44:109–142. doi: 10.1016/s0079-6603(08)60218-4. [DOI] [PubMed] [Google Scholar]
- Mitra S, Hazra TK, Roy R, Ikeda S, Biswas T, Lock J, Boldogh I, Izumi T. Complexities of DNA base excision repair in mammalian cells. Mol Cells. 1997;7:305–312. [PubMed] [Google Scholar]
- Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- Murante RS, Henricksen LA, Bambara RA. Junction ribonuclease: an activity in Okazaki fragment processing. Proc Natl Acad Sci USA. 1998;95:2244–2249. doi: 10.1073/pnas.95.5.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cells. 2000;5:1059–1065. doi: 10.1016/s1097-2765(00)80271-3. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Ohtsubo T, Oda H, Fujiwara T, Kang D, Sugimachi K, Nakabeppu Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol Biol Cell. 1999;10:1637–1652. doi: 10.1091/mbc.10.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs K, Sobol RW, Wilson SH, Kaina B. Cells deficient in DNA polymerase beta are hypersensitive to alkylating agent-induced apoptosis and chromosomal breakage. Cancer Res. 1999;59:1544–1551. [PubMed] [Google Scholar]
- Offer H, Zurer I, Banfalvi G, Reha’k M, Falcovitz A, Milyavsky M, Goldfinger N, Rotter V. p53 modulates base excision repair activity in a cell cycles-pecific manner after genotoxic stress. Cancer Res. 2001;61:88–96. [PubMed] [Google Scholar]
- Ohtsubo T, Nishioka K, Imaiso Y, Iwai S, Shimokawa H, Oda H, Fujiwara T, Nakabeppu Y. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000;28:1355–1364. doi: 10.1093/nar/28.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M, Lindahl T. Repair of alkylated DNA in Escherichia coli. Methyl group transfer from O6-methylguanine to a protein cysteine residue. J Biol Chem. 1980;255:10569–10571. [PubMed] [Google Scholar]
- Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker A, Gu Y, Mahoney W, Lee SH, Singh KK, Lu AL. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J Biol Chem. 2001;276:5547–5555. doi: 10.1074/jbc.M008463200. [DOI] [PubMed] [Google Scholar]
- Pascucci B, Stucki M, Jonson ZO, Dogliotti E, Hubscher U. Long patch base excision repair with purified human proteins. J Biol Chem. 1999;274:33696–33702. doi: 10.1074/jbc.274.47.33696. [DOI] [PubMed] [Google Scholar]
- Piersen CE, Prasad R, Wilson SH, Lloyd RS. Evidence for an imino intermediate in the DNA polymerase beta deoxyribose phosphate excision reaction. J Biol Chem. 1996;271:17811–17815. doi: 10.1074/jbc.271.30.17811. [DOI] [PubMed] [Google Scholar]
- Prasad R, Beard WA, Chyan JY, Maciejewski MW, Mullen GP, Wilson SH. Functional analysis of the amino-terminal 8-kDa domain of DNA polymerase beta as revealed by site-directed mutagenesis. DNA binding and 5′-deoxyribose phosphate lyase activities. J Biol Chem. 1998;273:11121–11126. doi: 10.1074/jbc.273.18.11121. [DOI] [PubMed] [Google Scholar]
- Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase beta mediates an excision step in mammalian long patch base excision repair. J Biol Chem. 2000;275:4460–4466. doi: 10.1074/jbc.275.6.4460. [DOI] [PubMed] [Google Scholar]
- Setlow RB, Carrier WL. The disappearance of thymine dimmers from DNA: An error correcting mechanism. Proc Natl Acad Sci USA. 1964;51:226–231. doi: 10.1073/pnas.51.2.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal RK, Prasad R, Wilson SH. DNA polymerase beta conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J Biol Chem. 1995;270:949–957. doi: 10.1074/jbc.270.2.949. [DOI] [PubMed] [Google Scholar]
- Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerasebeta in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- Thompson LH, West MG. XRCC1 keeps DNA from getting stranded. Mutat Res. 2000;459:1–18. doi: 10.1016/s0921-8777(99)00058-0. [DOI] [PubMed] [Google Scholar]
- Vidal AE, Hickson ID, Boiteux S, Radicella JP. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP-endonuclease: bypass of the AP lyase activity step. Nucleic Acids Res. 2001;29:1285–1292. doi: 10.1093/nar/29.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldstein EA, Cao EH, Setlow RB. Direct assay for O6-methylguanine-acceptor protein in cell extracts. Anal Biochem. 1982;126:268–272. doi: 10.1016/0003-2697(82)90514-0. [DOI] [PubMed] [Google Scholar]
- Waters TR, Gallinari P, Jiricny J, Swann PF. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J Biol Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nature Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]